
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 05 February 2024
Sec. Cardiovascular Metabolism
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1353533
This article is part of the Research Topic New Discoveries on Calcium Handling in Cardiovascular Pathology View all 7 articles
 Bo Lu1,2,†
Bo Lu1,2,† Xiaozhe Chen1,3,†
Xiaozhe Chen1,3,† Yulong Ma1
Yulong Ma1 Mingtai Gui1
Mingtai Gui1 Lei Yao1
Lei Yao1 Jianhua Li1
Jianhua Li1 Mingzhu Wang1
Mingzhu Wang1 Xunjie Zhou1,3*Deyu Fu1,3*
Xunjie Zhou1,3*Deyu Fu1,3*
Mitochondria-associated membrane (MAM) serve as crucial contact sites between mitochondria and the endoplasmic reticulum (ER). Recent research has highlighted the significance of MAM, which serve as a platform for various protein molecules, in processes such as calcium signaling, ATP production, mitochondrial structure and function, and autophagy. Cardiac diseases caused by any reason can lead to changes in myocardial structure and function, significantly impacting human health. Notably, MAM exhibits various regulatory effects to maintain cellular balance in several cardiac diseases conditions, such as obesity, diabetes mellitus, and cardiotoxicity. MAM proteins independently or interact with their counterparts, forming essential tethers between the ER and mitochondria in cardiomyocytes. This review provides an overview of key MAM regulators, detailing their structure and functions. Additionally, it explores the connection between MAM and various cardiac injuries, suggesting that precise genetic, pharmacological, and physical regulation of MAM may be a promising strategy for preventing and treating heart failure.
The interplay among organelles is the fundamental framework for cellular signaling. Approximately 5%–20% of the outer mitochondrial membrane (OMM) intimately interfaces with the endoplasmic reticulum (ER), maintaining a distance within the range of 10–30 nm and forming a specialized structure termed the mitochondrial-associated endoplasmic reticulum membrane (MAM) (1). Proteomic analysis has unveiled the presence of approximately 1,000–2,000 distinct proteins within the MAM, highlighting its role as a multifaceted signaling platform (2, 3). The MAM orchestrates bidirectional regulation of organelle functions, thereby exerting influence over a spectrum of cellular processes, including energy metabolism, calcium (Ca2+) handling, lipid balance, and the intricate control of cell survival and apoptosis. Furthermore, it is a central hub (4) for signaling in regulating mitochondrial fission. Cardiac diseases due to any cause, such as diabetes, obesity, hypertension, and myocardial ischemia‒reperfusion (I/R) injury, can increase the risk of heart failure and cardiovascular mortality. Recent investigations have underscored the pivotal role played by MAM in these processes, and this article provides an overview of the latest developments in this field.
MAM, a highly dynamic structure, functions as a cellular bridge, coordinating the exchange of substances and signaling molecules. It plays a pivotal role in preserving the physiological functions and metabolic balance of cells. Complex regulatory mechanisms intricately govern these structures and functions under physiological conditions and stress responses.
Diverse proteins and associated complexes within the MAM play pivotal roles in maintaining the appropriate flow of Ca2+ and essential physiological functions between the ER and mitochondria. Precise Ca2+ transfer not only governs mitochondrial bioenergetics but also regulates processes such as mitochondria-mediated cell death, autophagy/mitophagy, dynamics of mitochondrial fusion and fission, generation of reactive oxygen species (ROS), and transmission of redox signals (5). In mammals, the ER-resident chaperone glucose-regulated protein 75 (GRP75) forms a complex with the ER transmembrane Ca2+ release channel, inositol 1,4,5-trisphosphate receptor (IP3R), and the OMM protein voltage-dependent anion channel (VDAC), establishing an efficient transfer of Ca2+ from the ER to mitochondria (6). Various stimuli can dynamically influence the stability of this complex, favoring the uptake of Ca2+ into mitochondria through direct release via the IP3R channel (6). Once Ca2+ traverses the OMM, it enters the mitochondrial matrix via the mitochondrial Ca2+ uniporter (MCU) complex (including MCU, MCUb and regulatory subunits), mitochondrial ryanodine receptor (RyR) or rapid mode Ca2+ uptake (7, 8). However, when external stimuli enhance the interplay between the ER and mitochondria through IP3R-GRP75-VDAC1 complex (6, 9), a substantial amount of Ca2+ is absorbed by mitochondria, stimulating the opening of the mitochondrial permeability transition pore (mPTP), which leads to the release of cytochrome C and a cascade of apoptotic events (10). Phosphofurin acidic cluster sorting protein 2 (PACS-2) within MAM facilitates Ca2+ transfer between the ER and mitochondria and plays a role in regulating lipid synthases and autophagy (11). The depletion of PACS-2 increases the separation between these two organelles and triggers apoptosis by promoting the cleavage of B-cell receptor-associated protein 31 (BAP31) (12). BAP31 promotes apoptosis by mobilizing ER calcium stores at MAM and regulates mitochondrial function via interaction with mitochondrial fission protein 1 (Fis1) (13) or mitochondrial membrane 40 (Tom40) (14), respectively. Mitochondrial fusion proteins (MFNs), including MFN1 and MFN2 (15), are involved in the regulation of mitochondrial Ca2+ (mCa2+). MFN1 is predominantly located in the OMM, while MFN2 is found in both the OMM and ER membranes (16, 17). MFN2 forms heterotypic or homotypic complexes with MFN1 or MFN2, facilitating the bridging of the ER and mitochondria (16). Additionally, protein complexes such as RyR 2 and VDAC2 are also present in cardiac MAM and play a role in regulating Ca2+ transfer (18).
Mitochondria, often called powerhouses, occupy approximately 40% of the volume in adult cardiomyocytes and generate approximately 90% of the cell's adenosine triphosphate (ATP) (19). MAM significantly influences mitochondrial respiratory function, thereby contributing to maintaining mitochondrial metabolic homeostasis (20). The concentration of mCa2+ serves as a central signaling mechanism that regulates mitochondrial metabolism (21, 22) by governing the activities of key enzymes such as pyruvate dehydrogenase/phosphatase, isocitrate dehydrogenase, alpha-ketoglutarate dehydrogenase, and ATP synthase. These enzymes promote the production of reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide, which, in turn, sustain the functionality of the electron transport chain (23). In addition, ER stress can induce changes in the conformation of the ER, thereby altering the distribution of mitochondria around the ER and increasing the contact points between these two organelles. Adaptive modifications of the MAM structure facilitate Ca2+ exchange and ATP production in response to stressors (24). However, loss of Ca2+ transfer results in impaired mitochondrial metabolism, leading to increased phosphorylation of enzymes, rendering them inactive, which also slows down the tricarboxylic acid cycle, ultimately compromising the production of mitochondrial ATP (25). When cellular ATP levels become insufficient to support basic metabolic processes, cell necrosis can occur (26). At rest, the relatively low activity of the MCU in the inner mitochondrial membrane (IMM) is adequate to maintain baseline ATP levels. However, when stressors trigger rapid heartbeats, there is an increase in MCU-dependent mCa2+ uptake (27), accompanied by intensified NADH fluorescence signals (28). These changes indicate an elevation in mitochondrial metabolism (29), followed by a significant increase in ATP synthesis after an initial 10% sharp decline. This indicates that the regulation of cellular energy homeostasis by MAM involves intricate and finely tuned mechanisms.
By 2050, it is estimated that more than 1.31 billion individuals will suffer from diabetes (30). This condition has sparked significant interest because of its serious complications, especially diabetic cardiomyopathy (DbCM). Numerous studies have highlighted that change in the structure of MAM and the consequent dysfunction of mitochondria are pivotal mechanisms of DbCM. This mechanism is complex and involves various cell types and proteins, and some research results are still under dispute (Figure 1).
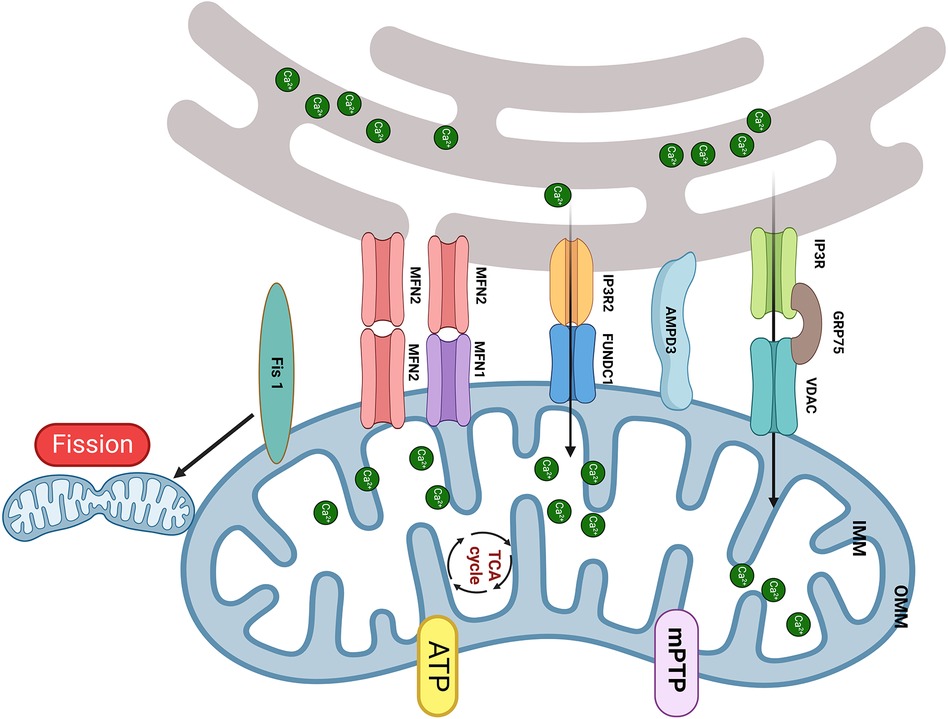
Figure 1. The MAM proteins involved in DbCM. (1) The upregulation of FUNDC1 and IP3R2 leads to mCa2+ overload results in mitochondrial fragmentation, mPTP opening, and cardiomyocyte apoptosis; (2) Increased expression of Fis1 in DbCM leads to excessive mitochondrial fission, causing myocardial injury; (3) Disruption of the interaction between IP3R and VDAC is accompanied by a decrease in mCa2+ and mitochondrial bioenergetics, results in myocardial injury; (4) The upregulation of MFN2 and AMPD shorten the distance of MAM and induced mitochondrial dysfunction. Created with BioRender.com.
Compared to nondiabetic individuals, diabetic patients exhibit a significant elevation in FUN14 domain containing 1 (FUNDC1) expression within their cardiac tissues (31). In vitro, high glucose (HG) can increase FUNDC1, IP3R2, and MAM levels in neonatal mouse cardiomyocytes, resulting in increased mCa2+ (31, 32). Elevated mCa2+ concentration adversely leads to mitochondrial fragmentation, prolongation of mPTP opening duration, and the initiation of cell apoptosis. The transgenic downregulation of FUNDC1 or IP3R2 effectively hindered the formation of MAM, decreased Ca2+ flux, and ameliorated mitochondrial dysfunction in cardiomyocytes exposed to HG. Additionally, the interaction between FUNDC1 and IP3R2 inhibits the ubiquitination and protease-mediated degradation of IP3R2. In vivo, cardiomyocyte-specific deficiency of FUNDC1 in diabetic mice abolishes MAM formation, mitigates the increase in mCa2+, and enhances both mitochondrial and cardiac function. Moreover, diabetes prompts the expression of Fis1 within cardiac mitochondria, leading to excessive mitochondrial fission and consequent disruption of cardiac structure and function. Knocking out FUNDC1 can maintain normal mitochondrial morphology and enhance cardiac structure and function by suppressing cardiomyocyte Fis1 expression. Consequently, the absence of FUNDC1 in cardiomyocytes ameliorates cardiac dysfunction by lowering mCa2+ levels and mitigating mitochondrial fission. Furthermore, FUNDC1 can interact with light chain 3 (LC3), serving as a mitophagy receptor. However, it is worth noting that mitophagy is not upregulated in DbCM, which suggests that FUNDC1 mediates DbCM through a pathway independent of mitophagy.
In contrast, diabetic mice fed a 16-week high-fat high-sugar diet (HFHSD) had a notable decrease in the interaction between IP3R and VDAC within the cardiac tissue (33). This alteration is accompanied by a decline in Ca2+ transfer and mitochondrial bioenergetics, resulting in reduced cardiomyocyte contractility (33). The expression of ER-mitochondrial junctions via adenovirus-mediated delivery could rescue this situation. Notably, the levels of ER Ca2+, cytosolic Ca2+ transients, and MCU function remained unchanged. In vitro, a reduction in the IP3R/GRP75/VDAC Ca2+ channel complex was reduced in HFHSD cardiomyocytes, along with a decrease in IP3R-stimulated Ca2+ transfer to mitochondria (33). An 8-week diet reversal can restore normal cardiac function and prevent the progression of DbCM by mitigating MAM-mediated mCa2+ dysfunction (33). Therefore, the diminished transfer of Ca2+ from the ER to mitochondria appears to be a reversible trigger for mitochondrial dysfunction in DbCM. Of note, Fauconnier et al. (34) reported that both HFHSD and ob/ob diabetic mice had a reduction in IP3-driven mCa2+ transport, which suggested that the duration of diabetes may exert a more significant influence on MAM function than the causes, emphasizing the need for further research into its potential mechanisms.
MFN2 also plays a pivotal role in the process of DbCM. In vitro, HG can accelerate ER stress (ERS) and mitochondrial oxidative stress and lead to the accumulation of MAM proteins, especially GRP75 and MFN2, within primary cardiomyocytes (35). The ERS inhibitor sodium 4-phenylbutyrate can counteract these changes. The distance of MAM is reduced when HL-1 cells are exposed to ERS, which is reversed by MFN2 siRNA (35). The disruption of MAM plays a crucial role in preventing mitochondrial dysfunction due to mCa2+ overload and then protects cells from ERS-induced apoptosis (35). Furthermore, HG also induces the formation of MAM in H9c2 cells, which results in a reduction in mitochondrial biogenesis, fusion, and oxidative phosphorylation. The downregulation of critical components in mitochondrial respiratory complexes resulted in ATP deficiency, leading to subsequent release of apoptotic proteins from mitochondria, along with a shortage of antiapoptotic proteins (32). These findings underscore that MAM-mediated mitochondrial impairments can induce apoptosis in cardiomyocytes.
Adenosine monophosphate deaminase (AMPD3) impairs diastolic function in pressure-overloaded type 2 diabetic hearts by reducing ATP production (36). The same group also found that the 90-kDa AMPD3 protein is present in the OMM/ER and MAM. In addition, its levels are significantly higher in the cardiac tissue of obese diabetic model rats (Otsuka Long-Evans Tokushima Fatty, OLETF)) than in control rats (Long-Evans Tokushima Otsuka, LETO). Following dobutamine-induced pressure overload, OLETF rats display diastolic dysfunction, a 57% increase in MAM area and a 47% elevation in mCa2+ level compared to LETO rats. Intriguingly, before pressure overload, the Ca2+ retention capacity in MAM-containing crude mitochondria of OLETF was 21% lower than that of LETO. Moreover, in vitro transfection of FLAG-AMPD3 in cells promotes the formation of MAM, resulting in mCa2+ overload, mitochondrial dysfunction and impaired mitochondrial respiration (37).
The function of T cells is also linked with myocardial fibrosis (38, 39). The ketogenic diet (KD) reduced regulatory T cells (Tregs) proportion in plasma and induced cardiac diastolic disfunction and fibrosis in db/db mice (40). Moreover, culture medium from ketone bodies (KBs)-treated Tregs can synergistically activate cardiac fibroblasts. KBs inhibited the differentiation and proliferation of naive CD4+ T cells into Tregs, and hampered the MAM, mitochondrial respiration, fatty acid oxidation within Tregs. Improving T-cell function will simultaneously enhance MAM and mitochondrial respiratory function (40). Therefore, MAM dysfunction of Tregs might play a indirectly role in KD induced diabetic myocardial fibrosis, and its direct effect on Tregs needs to futher clarified.
As obesity rates continue to rise, attention is increasingly focused on the associated myocardial injuries. Mice with metabolic syndrome (MS) induced by prolonged consumption of a Western diet often exhibit cardiac microvascular dysfunction, mitochondrial damage, and cardiac remodeling (41). The reshaping and deformation of mitochondria and the ER markedly reduce MAM size in MS mouse cardiomyocytes. Additionally, it has been noted that the expression of MFN2 in the hearts of MS mice is significantly decreased compared to the control group. Altogether, these data suggest that obesity can disrupt communication between the ER and mitochondria (41). Furthermore, it has been reported that the deletion of FUNDC1 aggravates cardiac remodeling, mitochondrial dysfunction, and cell death in high-fat diet (HFD)-fed mice. The absence of FUNDC1 also reduces ER Ca2+-regulating protein IP3R3 degradation by disrupting its interaction with F-Box and leucine rich repeat protein 2 (FBXL2), which exacerbates cardiac structural and functional abnormalities and mitochondrial dysfunction through Ca2+ overload (42). This study also reveals that both autophagy and mitophagy levels are suppressed in the hearts of obese mice. Systemic FUNDC1 knockout enhances the loss of autophagy induced by HFD but has little impact on other mitophagy proteins, which suggests that FUNDC1-mediated mitophagy uniquely affects HFD-induced cardiac abnormalities (42). Thus, FUNDC1 is a unique therapeutic target in obesity-induced cardiac abnormalities indirectly through the FBXL2-IP3R3 axis. In addition, syntaxin 17 (STX17), a scaffold protein located in MAM, acts as a central hub for physical and functional interactions between the ER and mitochondria and is also involved in autophagy (43) and mitophagy (44). The levels of STX17 are significantly elevated in the plasma of obese patients and in the heart tissues of mice fed with HFD. This elevation promotes mCa2+ overload, leading to obesity-related cardiomyopathy in a Parkin-dependent manner. Deletion and overexpression of STX17 mitigate and exacerbate HFD-induced cardiac oxidative stress, mitochondrial damage, and functional impairments, respectively (45). In summary, MAM may be involved in obesity-related cardiomyopathy either independently or through autophagy.
mCa2+ overload plays a pivotal role in myocardial I/R injury (46), giving rise to disruptions of oxidative phosphorylation, the generation of ROS, and the opening of the mPTP (47). mPTP is one of the crucial mediators to cell death in I/R (48), whereas the exact molecular composition of mPTP is still controversial (49), e.g., the double knock-out of adenine necleotide translocator (ANT)1/2 or triple ANT1, 2, 4 genes have different effects on mPTP activity (49, 50). Cyclophilin D (CYPD) within the mitochondrial matrix has been identified as a pivotal regulator of the pore opening (51), and its interaction with IP3R1 in cardiomyocytes is intensified under conditions of hypoxia-reoxygenation (H/R) (52). Notably, CYPD deficiency can confer protection to the heart against I/R injury and necrosis (53). The knockout of CYPD can induce anomalies in IP3R-mediated Ca2+ transport from the ER to the mitochondria (54). Furthermore, genetic deletion of the PPIF gene, which is responsible for encoding CYPD in cardiomyocytes, disrupts the interaction between CYPD and the VDAC1/GRP75/IP3R1 complex, mitigating the mCa2+ load induced by H/R (52). Similarly, pharmacological or genetic inhibition of CYPD, IP3R1, or GRP75 can shield cardiomyocytes from cell death and mCa2+ overload caused by H/R (52).
In the hearts of mice, a close association is observed between NADPH Oxidase 4 (NOX4) and MAM-labeled FACL4 in the ER region proximate to mitochondria, implying that NOX4 may be localized to MAM (55). During I/R, Nox4 deficiency in cardiomyocytes increases cardiac troponin I release sevenfold, exacerbating myocardial damage. NOX4 can enhance Akt-dependent phosphorylation of IP3R1, consequently inhibiting Ca2+ flux and mPTP-dependent necrosis. Ultimately, NOX4 reduces the extent of myocardial infarction, underscoring the pro-survival effects of NOX4, which rely on spatially confined signal transduction within MAM.
Protein tyrosine phosphatase interacting protein 51 (PTPIP51) is a phospholipid transfer protein endowed with MAM-tethering capabilities. It engages with the OMM protein PTPIP51, forming a chain of MAM with the assistance of the integral ER vesicle-associated membrane protein-associated protein B (VAPB) (56). This tether contributes to forming MAM and regulating Ca2+ homeostasis and autophagy (57, 58). Furthermore, PTPIP51 is significantly upregulated in hearts affected by I/R. The adenovirus-mediated upregulation of PTPIP51 substantially enhances the interaction between mitochondria and the SR, thereby augmenting mCa2+ uptake via MCU. Conversely, MCU inhibition or genetic deletion within mitochondria can reverse the PTPIP51-induced elevation of mCa2+ and protect cardiomyocytes from PTPIP51-induced apoptosis. Most notably, the cardiac-specific knockout of PTPIP51 significantly reduces the extent of myocardial infarction and cardiac damage following I/R injury (59). These findings suggest that PTPIP51 may represent a therapeutic target for mitigating I/R injury.
Nolwenn Tessier et al. demonstrated for the first time that transient receptor potential vanilloid type 1 (TRPV1) is involved in regulating MAM-mediated Ca2+ exchange (60). RTX, a TPRV1 agonist, slowly increased the mitochondrial Ca2+ content, whereas iRTX had the opposite effect. This indicates that mitochondria gradually buffered Ca2+ ER leakage from TRPV1 channels both inside and outside MAM. Activation of TRPV1 by RTX resulted in an elevation of Ca2+ concentration in specific areas on the mitochondrial surface (Ca2+ hot spots), which could directly mobilize Ca2+ from the ER to mitochondria and trigger the remodeling of MAM. Apart from Ca2+ transfer, sustained activation of TRPV1 also reduced the contacts between the ER and mitochondria, which might mutually decrease the Ca2+ content within mitochondria (60). Sun et al. demonstrated that TRPV1 activation at the beginning of H/R is harmful to cell survival, and inhibiting it protects cells from death (61). In H/R conditions, the results showed a protective effect of RTX by reducing cell mortality by ∼18% compared to control H/R only when RTX was applied before hypoxia (60). The controversial effects of the pharmacological activation of TRPV1 may be related to the treatment window, which should be finely tuned with respect to ischemia (Figure 2).
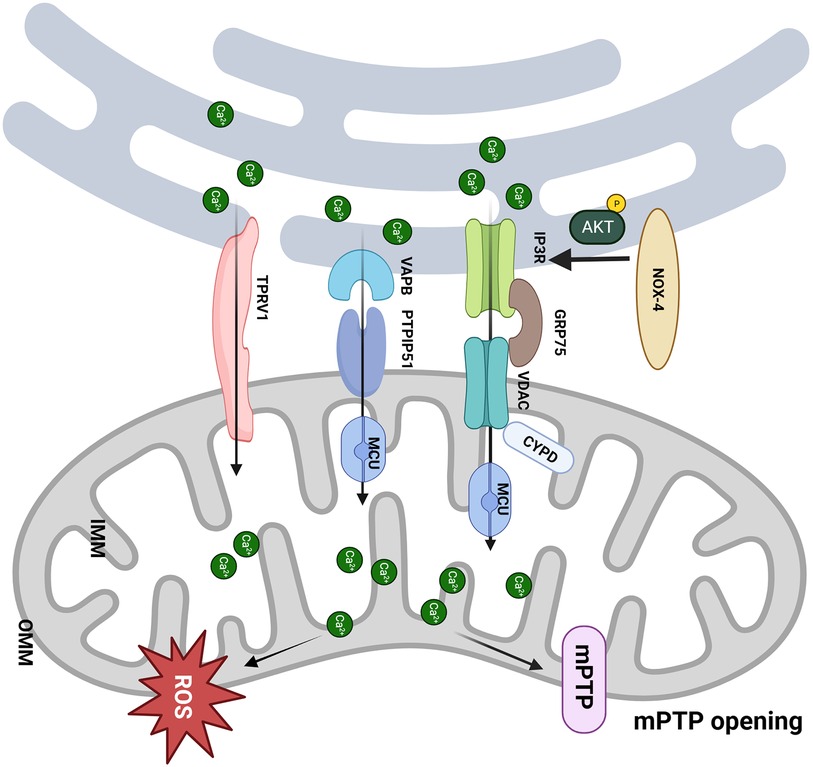
Figure 2. MAM participates in the pathological process of I/R. (1) The CYPD deficiency improves the mCa2+ load and mPTP opening via interacting with the VDAC1/GRP75/IP3R1 complex, which protects the cardiomyocytes from I/R; (2) NOX4 enhances the Akt-dependent phosphorylation of IP3R1, consequently inhibits Ca2+ flux and mPTP-dependent necrosis; (3) Upregulation of PTPIP51 promotes interaction between mitochondria and endoplasmic reticulum and induces cell apoptosis through increasing mCa2+ uptake by MCU; (4) Activation of TRPV1 mobilizes Ca2+ from ER to mitochondria and remodels MAM. Created with BioRender.com.
Sleep-disordered breathing (SDB), including obstructive and central sleep apnea, causes intermittent hypoxia (IH) and reduces the survival of cardiomyocytes (62, 63). The right atrial biopsies of SDB patients and IH-exposed mice revealed that IH can activate hypoxia-inducible factor 1 (HIF-1), ER stress, and cell apoptosis (64). IH failed to elevate glucose-regulated protein 78 (GRP78) and C/EBP homologous protein (CHOP) levels in HIF-1α+/− mice. Most significantly, IH-exposed HIF-1α+/− mice exhibited a remarkable inhibition of cell apoptosis compared to HIF-1α+/+ mice. These findings strongly indicate that HIF-1 is pivotal in mediating IH-induced ER stress and cell apoptosis. In addition, IH can injure MAM, Ca2+ homeostasis, mitochondrial respiration, and Ca2+ retention capacity. Persistent MAM disruption, evidenced by reduced interaction between VDAC and IP3R1, and disturbances in Ca2+ homeostasis are linked to myocardial cell death (64). Notably, IH did not affect the interaction between VDAC and IP3R1 in HIF-1α+/− mice. Both in vivo and in vitro experiments found no discernible impact of IH on MAM structure and mitochondrial function in HIF-1α+/− mouse myocardium, while overexpression of HIF-1α had the opposite effect. Hence, targeting HIF-1 may hold promise for mitigating alterations in MAM and mitochondrial dysfunction induced by IH, further improving cardiomyocyte apoptosis (64).
The hypobaric hypoxic (HH) environment is a significant challenge, and prolonged exposure to HH can impair right heart function. PACS2 is crucial in regulating mitophagy. Both PACS2 and mitophagy levels are downregulated in HH mouse hearts (65). Cardiac-specific knockout of PACS2 exacerbates the inhibition of mitophagy induced by HH (65). When cardiomyocytes are exposed to HH, the absence of PACS2 disrupts MAM formation and Ca2+ flux between the ER and mitochondria, further inhibiting mitophagy and mitochondrial energy metabolism. Conversely, overexpression of PACS2 reverses these effects to maintain right heart function (65). However, the precise molecular mechanisms underlying how PACS2 collaborates with proteins such as FUNDC1 to regulate Ca2+ flux, mitophagy, and cardiac function remain unclear. Böckler and Zou demonstrated that mitophagy requires Ca2+ flux through MAM, and artificially tethering the ER and mitochondria can rescue mitophagy defects (66, 67). Some researchers have found that decreased ATP production by inhibiting MCU-mediated Ca2+ influx enhances mitophagy and provides cardiac protection in heart failure (68). Thus, there is still debate regarding the impact of Ca2+ flux on mitophagy.
During pathological hypertrophy, significant changes occur in the heart's energy metabolism, accompanied by insulin resistance. mCa2+ uptake plays a pivotal role in insulin signaling and cardiomyocyte metabolism. Insulin triggers the release and transportation of Ca2+ from the ER to mitochondria (69). In the norepinephrine-induced cardiomyocyte hypertrophy model, an increased distance between the ER and mitochondria hampers insulin-induced mCa2+ uptake, Akt phosphorylation, glucose uptake, and oxygen consumption. This highlights the pivotal role of ER-mitochondrial communication in the pathogenesis of cardiac hypertrophy and related metabolic dysfunction (70). Moreover, pressure overload-induced cardiac remodeling mice exhibit elevated expression of TRPV1. TRPV1 promotes the formation of MAM, acting as a safeguard against cardiac hypertrophy. Capsaicin, a TRPV1 agonist, triggers the phosphorylation of AMP-activated protein kinase (AMPK) and upregulates MFN2 expression, facilitating MAM formation, which mitigates the cardiomyocyte size enlargement induced by adrenaline and enhances mitochondrial function. However, disruption of MAM via siMFN2 eliminates the protective effects of TRPV1 on mitochondria (71). In summary, maintaining MAM function has the potential to improve cardiomyocyte hypertrophy.
Nip3-like protein X (Nix), a protein that promotes cellular apoptosis, exhibits increased transcription levels during cardiac hypertrophy (72, 73). A recent study showed that Nix can initiate tethering of the ER to mitochondria, enhance Ca2+ transport, and establish “Ca2+ hot spots”. This mechanism leads to programmed apoptosis and necrosis in cardiomyocytes, directly contributing to the development of heart failure (26).
IP3R has three subtypes, and IP3R2 is the primary subtype in the heart (74). The interaction between FUNDC1 and IP3R2 within the MAM is crucial in maintaining mitochondrial dynamics and functionality. Wu et al. found that disrupting the FUNDC1-IP3R2 interactions within the MAM leads to decreased Ca2+ levels in both mitochondria and cytoplasm. This disruption ultimately results in mitochondrial dysfunction and contributes to heart failure (67). Heart-specific knockout of FUNDC1 impairs the structural integrity of MAM and reduces IP3R2 levels, causing mitochondrial elongation and mitochondrial dysfunction. Moreover, FUNDC1 regulates the expression of the Fis1 gene and controls mitochondrial fission through the Ca2+/cAMP response element-binding protein (CREB) pathway. Compared to healthy hearts, individuals with heart failure exhibit a more elongated morphology and reduced levels of MAM-associated proteins, including IP3R1, IP3R3, FUNDC1, phosphorylated CREB, and Fis1, resulting in a significant decrease in the proportion of adjacent ER to mitochondria. Paradoxically, these findings, in conjunction with the insights from the literature (42), highlight that the systemic and cardiac-specific knockout of FUNDC1 can yield different effects in different diseases. Further investigation into potential mechanisms is necessary, providing valuable paths for therapy.
Mitochondrial ATP-dependent Lon protease 1 (LonP1) (75) primarily resides within the mitochondrial matrix, where it acts as a crucial molecular chaperone, facilitating mitochondrial protein transport, protein folding, and unfolding processes. Additionally, LonP1 functions as a protease responsible for regulating mitochondrial protein balance, thereby playing a pivotal role in sustaining normal mitochondrial function. More recently, a study showed that LonP1 was a novel MAM tethering protein. Selectively knocking out LonP1 in mouse cardiomyocytes can disrupt the integrity of MAM, which substantially reduces interactions between mitochondria and the ER, marked by a significant decrease in IP3R3 expression levels (reduced to approximately 63% of the WT group). MAM dysfunction affects the ER unfolded protein response, although the precise mechanisms remain unclear (75). Moreover, a deficiency in LonP1 within the heart triggers metabolic reprogramming in cardiomyocytes, significantly upregulating genes associated with gluconeogenesis and amino acid metabolism, contributing to pathological heart failure. Targeting LonP1 presents a promising therapeutic approach for the treatment of heart failure.
Cardiotoxicity linked to anticancer agents is an important complication. Sorafenib (Sor), primarily used for renal cell carcinoma and hepatocellular carcinoma, stimulated the biogenesis of MAM and led to a significant increase in the essential MAM components PACS2 and FUNDC1 in cardiomyocytes. Remarkably, FUNDC1 knockdown impeded sorafenib-induced mitochondrial Ca2+ efflux, necrotic apoptosis, and Ca2+/calmodulin-dependent protein kinase II delta (CaMKIIδ) expression, highlighting the role of MAM-mediated Ca2+ efflux in initiating necrotic apoptosis of cardiomyocytes. Sor also reduces MFN2 protein expression in a concentration-dependent manner. MFN2 overexpression disrupts MAM components and mitochondria-ER connections, lowering activated CaMKII in cardiomyocytes, while silencing MFN2 enhances MAM components, shortens the distance, and activates CaMKII. Overall, both global and cardiac-specific MFN2 overexpression alleviate sorafenib-induced cardiac dysfunction and cardiomyocyte necrosis by inhibiting the MAM-CaMKIIδ-receptor-interacting protein kinase 3 (RIP3)/mixed-lineage kinase domain-like protein (MLKL) pathway (76). In addition, MAM is also involved in heavy metal-induced cardiotoxicity. Exposure to heavy metals such as molybdenum (Mo) and/or cadmium (Cd) can result in morphological myocardial damage, impaired oxidative function, significantly reduced mCa2+, and an increased distance of MAM. The disrupted MAM structure is accompanied by lower levels of MAM-related genes, including IP3R, FUNDC1, MFN2 and VDAC1 (77). Combined exposure to Mo and Cd exacerbates these effects (77). Controversially, GRP75 levels rise, which can facilitate reactions in mitochondrial energy metabolism (78). Moreover, Mo and Cd can induce autophagy in sheep hearts, which may be related to MAM.
Sepsis-induced myocardial dysfunction (SIMD) is a significant complication (79, 80), with a higher mortality rate compared to patients without SIMD (70%–90% vs. 20%) (81). FUNDC1 plays a pivotal role in lipopolysaccharide (LPS)-induced myocardial dysfunction, and LPS also increases phosphorylated signal transducer and activator of transcription 3 (p-STAT3) and FUNDC1 expression in H9c2 cells. Deleting FUNDC1 interferes with MAM formation, leading to lowered intracellular Ca2+ and ATP levels, mitochondrial dysfunction, and decreased ROS production in LPS-injured H9c2 cells. STAT3 serves as a potential transcription factor for FUNDC1, with siSTAT3 significantly inhibiting FUNDC1 levels, while STAT3 overexpression exerts the opposite effect (82). FUNDC1 deficiency inhibits LPS-induced phosphorylation of IP3R (Ser 1,756). Stattic (a STAT3 inhibitor) suppresses the levels of the MAM-related proteins FUNDC1 and p-IP3R induced by LPS in H9c2 cells. Likewise, the interleukin-6 (IL-6)/STAT3 inhibitor bazedoxifene reduces FUNDC1 and p-IP3R levels in the hearts of LPS-treated mice. Thus, the IL-6/STAT3 signaling pathway exacerbates SIMD by promoting MAM formation dependent on FUNDC1 (82). Moreover, FUNDC1-dependent MAM formation may influence intracellular Ca2+ levels, in part, by impacting Cav1.2 and RyR2 levels. Nonetheless, LPS administered at a dose of 5 mg/kg significantly reduces the formation of MAM in hearts (83). These varying study outcomes result from different disease stages in sepsis (12 h vs. 18 h), emphasizing the necessity to adjust interventions based on the progression of SIMD.
Emerging evidence suggests that exercise, or exercise preconditioning (EP), potentially contributes to cardiac protection by modulating MAM proteins and functions (84). For instance, EP exerts its protective effects by regulating various molecules, including MFN2, MFN1, FUNDC1, VDAC1, GRP75, IP3R, CYPD, and others, all impacting MAM function. Among these, MFN1 and MFN2 play roles in mediating mitochondrial fusion and mitophagy, and they accumulate significantly in failing hearts. Eight weeks of exercise reduces the accumulation of MFN1 and MFN2 in failing hearts, restoring the balance of mitochondrial fission and fusion and decreasing the accumulation of mitochondrial fragments, ultimately providing cardiac protection (85). Coincidentally, six weeks of exercise improved mitochondrial dynamics by decreasing phosphorylated dynamin-related peptide 1 (Drpl)/Drp1 levels and increasing MFN2/VDAC levels in obese hearts (86). Consequently, MFN2 or MFN1 may be implicated in exercise-induced cardioprotection, and the precise regulatory mechanisms require further elucidation.
The prolonged accumulation of aging cardiomyocytes can detrimentally impact cardiac function, yet the associated molecular mechanisms are complicated. Recent findings suggest a connection between alterations in MAM function and cardiac aging. CDGSH iron sulfur domain 2 (Cisd2), a gene that promotes longevity, is notably abundant within MAM (87). Elevated Cisd2 levels are crucial in preserving and dynamically regulating energy metabolism. They also reduce the accumulation of myocardial lipofuscin, ultimately preserving myocardial ultrastructure and improving cardiac function related to aging. Notably, the overexpression of Cisd2 can reverse age-related cardiac functional impairments. Conversely, Cisd2 knockout leads to premature aging phenotypes, a shortened lifespan, mitochondrial dysfunction, cellular solute Ca2+ homeostasis disruptions, increased ROS production, and impaired autophagy (87).
The interaction between mitochondria and the ER has garnered significant attention in the past decade. However, our current understanding of the composition and function of MAM remains incomplete and subject to controversy. Paillard et al. reported that acute disruption of MAM (achieved by inhibiting GRP75) could protect the heart from I/R injury by preventing mCa2+ overload, mPTP opening, and cell apoptosis (52). The same team recently showed that MAM disruption, leading to reduced Ca2+ transfer, could induce early mitochondrial dysfunction and contribute to DbCM (33). In addition, Papanicolaou discovered that cardiomyocytes become more resistant to cell death when MFN2 is knocked out, as they exhibit increased tolerance to Ca2+-induced mPTP opening (88). Nevertheless, Filadi R reported that MFN2 gene knockout or silencing displayed increased tethering between the ER and mitochondria, enhanced Ca2+ transfer, and increased sensitivity to apoptotic stimuli associated with mCa2+ overload toxicity (89). MFN2 disruption additionally worsened ER/mitochondrial metabolic feedback, leaving ventricular cardiomyocytes from MFN2 knockout mice incapable of inducing ATP production through IP3-mediated ER Ca2+ release. Furthermore, MFN2-mediated ER-mitochondrial communication varied depending on mitochondrial position and receptor types, which may help explain the controversies in this field (90). In addition, FUNDC1 deficiency leads to heart failure due to reduced formation of MAM and decreased mCa2+ levels (67). In contrast, FUNDC1 KO mitigates cardiac function abnormalities in Akita and streptozotocin-induced diabetes, primarily due to reduced MAM formation and lower mCa2+ levels (31). Nevertheless, the absence of FUNDC1 exacerbates mitochondrial dysfunction and Ca2+ overload in obesity-related cardiomyopathy (42). The MAM regulation on mitochondrial dynamics is regulated by numerous functional regulatory pathways, forming a complex spatial and temporal regulatory network that is currently not fully understood. The combinations of different molecules exhibit distinctly different MAM regulatory effects, which may be influenced by specific disease models, injury time, MAM assessment methods, and the involved MAM molecules. Hence, comprehending the importance of diverse pathological stimuli, mitochondrial localization, MAM biogenesis, and other factors in inducing MAM changes is crucial for the precise regulation of MAM adaptive homeostasis (Figure 3 and Table 1).
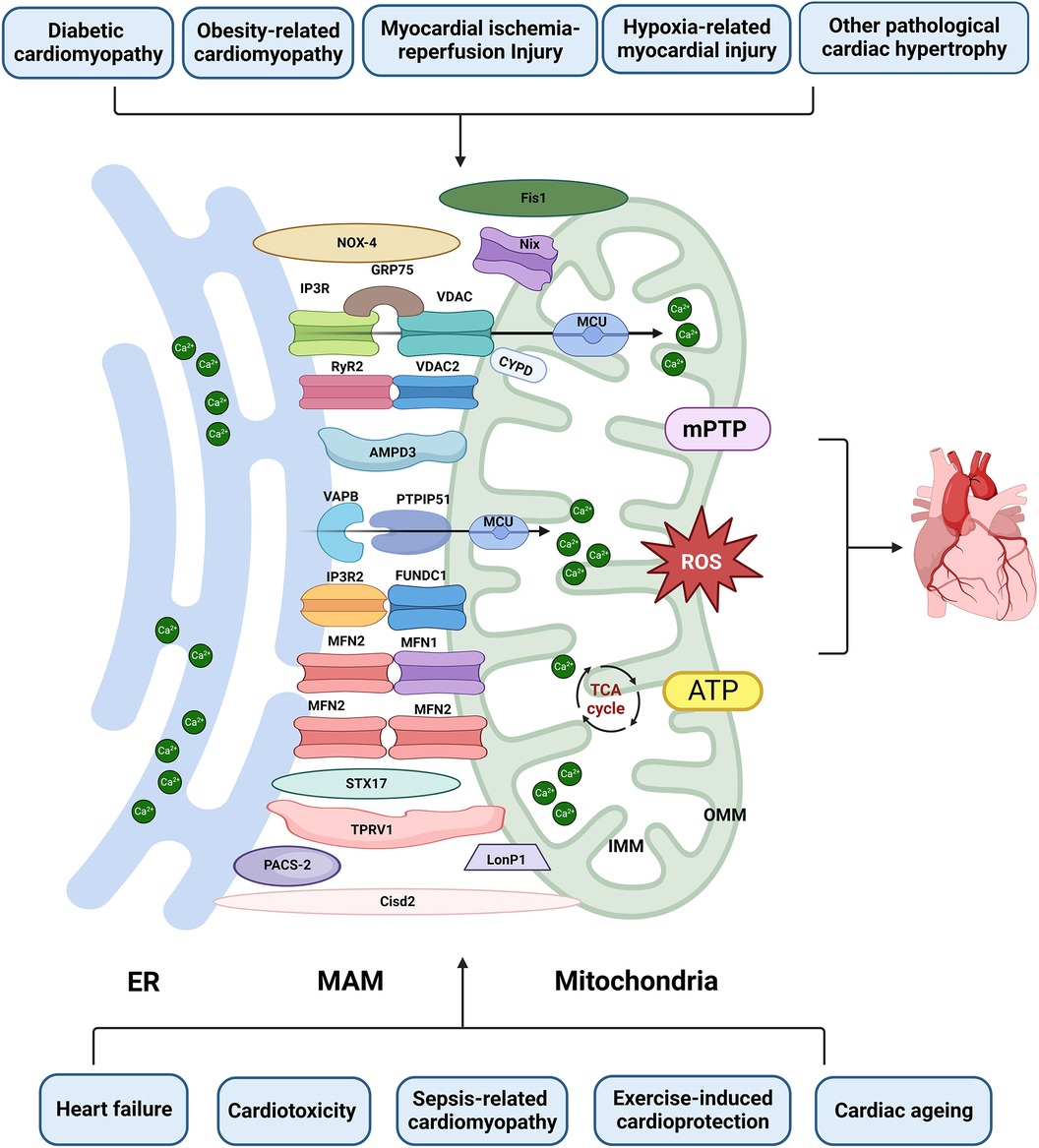
Figure 3. The role of MAM in cardiac disease. Proteins located on the ER surface, such as IP3R, VAPB, MFN2, and RyR2, interact with their counterparts on the OMM, including VDAC/FUNDC1, PTPIP5, MFN1/2, and VDAC2. These interactions form major tethering complexes at the MAM in cardiomyocytes. Additionally, PACS-2, NOX4, Nix, STX17, PACS-2, LonP1, TPRV1, Fis1 and Cisd1 are also involved in the structure and functions of MAM. Dysfunction of MAM can lead to Ca2+ overload, abnormal ROS and ATP production, subsequently inducing mPTP opening and apoptosis. This dysfunction is implicated in various types of heart injuries. Created with BioRender.com.
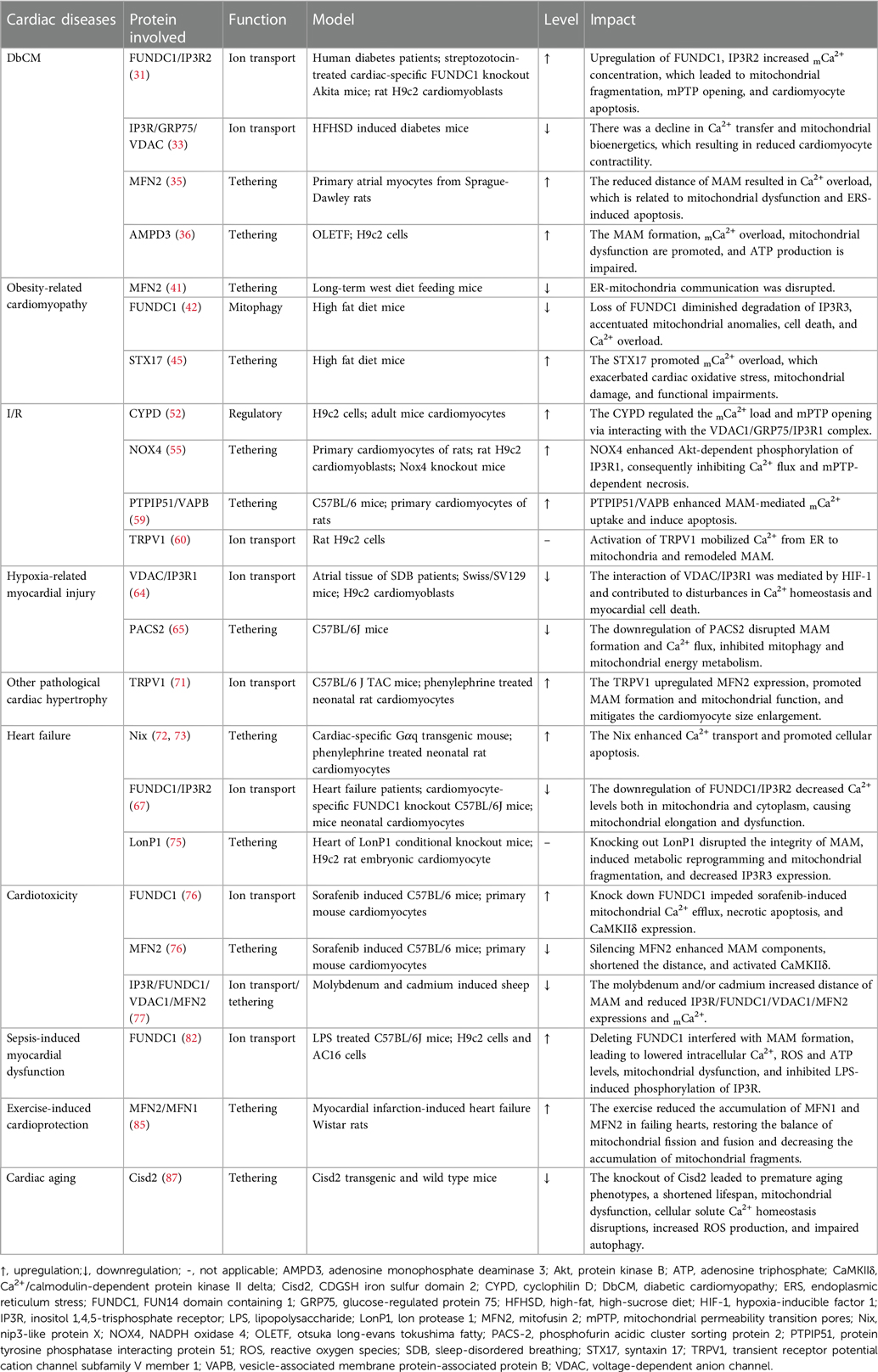
Table 1. The function and expression of MAM components in different cardiac diseases.
Apart from the proteins, the physical connection between the ER and mitochondria MAM, also known as the mitochondria-endoplasmic reticulum contact (MERC) tether, holds promise as a therapeutic target (91). Prior research has demonstrated that compounds such as arachidonic acid or melatonin, which intervene in the function of MERC, can ameliorate DbCM (32) and myocardial damage in type 3 cardiorenal syndrome (92). Since endogenous mitochondrial-ER tether proteins serve multiple functions, genetically targeting them is unsuitable for selectively investigating specific tether functions. A previous study showed that an artificial ER-mitochondria connector formed a single-protein bridge between the OMM and ER, resulting in a more robust and expansive organelle association (91). The heart-specific engineered tether in mice can enhance MERC, leading to long-term adaptations that sustain excitation-energy coupling. Concurrently, enhanced interactions lessened the susceptibility of female mice to β-adrenergic stress and alleviated myocyte death and myocardial dysfunction induced by mCa2+ overload during I/R injury (93). Therefore, the MERC tether not only facilitates the investigation of specific roles of mitochondrial-ER interactions in the heart but also offers the potential to examine their overall impact on tissues. Further exploration of the long-lasting effects of continuous enhancement of cardiac mitochondrial ER tethering opens up possibilities for utilizing structural preconditioning as a genetic therapeutic strategy.
In general, ER and mitochondrial networks undergo highly dynamic changes, which are capable of altering their shapes and/or distribution in response to various stimuli. The integrity of the MAM forms the foundation for effective signal transmission and uniquely regulates cardiomyocyte mitochondrial homeostasis, apoptosis, and autophagy (3). Therefore, analyzing MAM length, the distance between the two organelles, the number of contact points, communication types, and persistence at different stages of diseases can help elucidate the potential therapeutic value of targeting MAM in preventing cardiac disease. Despite recent achievements, there is still a long way to go.
BL: Conceptualization, Writing – review & editing, Funding acquisition. XC: Conceptualization, Writing – review & editing. YM: Writing – original draft. MG: Writing – original draft. LY: Writing – review & editing. JL: Writing – original draft. MW: Writing – original draft. XZ: Supervision, Writing – review & editing. DF: Funding acquisition, Writing – review & editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article.
This work was funded by the National Natural Science Foundation of China (grant no. 82274262; 82174130), the Shanghai Collaborative Innovation Center of Industrial Transformation of Hospital TCM Preparation, and the Program of Shanghai Municipal Health Commission (grant no. 202240053).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Paillusson S, Stoica R, Gomez-Suaga P, Lau DHW, Mueller S, Miller T, et al. There’s something wrong with my MAM; the ER–mitochondria axis and neurodegenerative diseases. Trends Neurosci. (2016) 39(3):146–57. doi: 10.1016/j.tins.2016.01.008
2. Poston CN, Krishnan SC, Bazemore-Walker CR. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J Proteomics. (2013) 79:219–30. doi: 10.1016/j.jprot.2012.12.018
3. Carpio MA, Means RE, Brill AL, Sainz A, Ehrlich BE, Katz SG. BOK controls apoptosis by Ca2+ transfer through ER-mitochondrial contact sites. Cell Rep. (2021) 34(10):108827. doi: 10.1016/j.celrep.2021.108827
4. Lopez-Crisosto C, Bravo-Sagua R, Rodriguez-Pena M, Mera C, Castro PF, Quest AF, et al. ER-to-mitochondria miscommunication and metabolic diseases. Biochim Biophys Acta. (2015) 1852(10 Pt A):2096–105. doi: 10.1016/j.bbadis.2015.07.011
5. Wei X, Wei X, Lu Z, Li L, Hu Y, Sun F, et al. A activation of TRPV1 channel antagonizes diabetic nephropathy through inhibiting endoplasmic reticulum-mitochondria contact in podocytes. Metab Clin Exp. (2020) 105:154182. doi: 10.1016/j.metabol.2020.154182
6. Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. (2006) 175(6):901–11. doi: 10.1083/jcb.200608073
7. Ryu SY, Beutner G, Dirksen RT, Kinnally KW, Sheu SS. Mitochondrial ryanodine receptors and other mitochondrial Ca2+ permeable channels. FEBS Lett. (2010) 584(10):1948–55. doi: 10.1016/j.febslet.2010.01.032
8. Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell. (2014) 53(5):726–37. doi: 10.1016/j.molcel.2014.01.013
9. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. (2012) 19(2):267–73. doi: 10.1038/cdd.2011.92
10. Rasola A, Bernardi P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium. (2011) 50(3):222–33. doi: 10.1016/j.ceca.2011.04.007
11. Li C, Li L, Yang M, Zeng L, Sun L. PACS-2: a key regulator of mitochondria-associated membranes (MAMs). Pharmacol Res. (2020) 160:105080. doi: 10.1016/j.phrs.2020.105080
12. Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and bid-mediated apoptosis. EMBO J. (2005) 24(4):717–29. doi: 10.1038/sj.emboj.7600559
13. Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the mitochondria–ER interface to establish a platform for apoptosis induction. EMBO J. (2011) 30(3):556–68. doi: 10.1038/emboj.2010.346
14. Namba T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci Adv. (2019) 5(6):eaaw1386. doi: 10.1126/sciadv.aaw1386
15. Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. (2001) 114(5):867–74. doi: 10.1242/jcs.114.5.867
16. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. (2008) 456(7222):605–10. doi: 10.1038/nature07534
17. Merkwirth C, Langer T. Mitofusin 2 builds a bridge between ER and mitochondria. Cell. (2008) 135(7):1165–7. doi: 10.1016/j.cell.2008.12.005
18. Min CK, Yeom DR, Lee KE, Kwon HK, Kang M, Kim YS, et al. Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem J. (2012) 447(3):371–9. doi: 10.1042/BJ20120705
19. Chen L, Liu B, Qin Y, Li A, Gao M, Liu H, et al. Mitochondrial fusion protein Mfn2 and its role in heart failure. Front Mol Biosci. (2021) 8:681237. doi: 10.3389/fmolb.2021.681237
20. Rieusset J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death Dis. (2018) 9(3):388. doi: 10.1038/s41419-018-0416-1
21. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. (2006) 99(2):172–82. doi: 10.1161/01.RES.0000232546.92777.05
22. Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. (2012) 51(14):2959–73. doi: 10.1021/bi2018909
23. Hansford RG. Physiological role of mitochondrial Ca2+ transport. J Bioenerg Biomembr. (1994) 26(5):495–508. doi: 10.1007/bf00762734
24. Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. (2011) 124(Pt 13):2143–52. doi: 10.1242/jcs.080762
25. Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, et al. MCUR1 Is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. (2012) 14(12):1336–43. doi: 10.1038/ncb2622
26. Dorn GW 2nd, Scorrano L. Two close, too close: sarcoplasmic reticulum-mitochondrial crosstalk and cardiomyocyte fate. Circ Res. (2010) 107(6):689–99. doi: 10.1161/CIRCRESAHA.110.225714
27. Bell CJ, Bright NA, Rutter GA, Griffiths EJ. ATP Regulation in adult rat cardiomyocytes: time-resolved decoding of rapid mitochondrial calcium spiking imaged with targeted photoproteins. J Biol Chem. (2006) 281(38):28058–67. doi: 10.1074/jbc.M604540200
28. Maack C, O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. (2007) 102(5):369–92. doi: 10.1007/s00395-007-0666-z
29. Rolf BD, Bers M. Simultaneous measurements of mitochondrial NADH and Ca2+ during increased work in intact rat heart trabeculae. Biophys J. (2002) 83:587–604. doi: 10.1016/S0006-3495(02)75194-1
30. Collaborators GBDD. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the global burden of disease study 2021. Lancet. (2023) 402(10397):203–34. doi: 10.1016/S0140-6736(23)01301-6
31. Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P, et al. Hyperglycemia-driven inhibition of AMP-activated protein kinase alpha2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes in vivo. Circulation. (2019) 139(16):1913–36. doi: 10.1161/CIRCULATIONAHA.118.033552
32. Raj PS, Nair A, Rani MRP, Rajankutty K, Ranjith S, Raghu KG. Ferulic acid attenuates high glucose-induced MAM alterations via PACS2/IP3R2/FUNDC1/VDAC1 pathway activating proapoptotic proteins and ameliorates cardiomyopathy in diabetic rats. Int J Cardiol. (2023) 372:101–9. doi: 10.1016/j.ijcard.2022.12.003
33. Dia M, Gomez L, Thibault H, Tessier N, Leon C, Chouabe C, et al. Reduced reticulum-mitochondria Ca2+ transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res Cardiol. (2020) 115(6):74. doi: 10.1007/s00395-020-00835-7
34. Fauconnier J, Lanner JT, Zhang SJ, Tavi P, Bruton JD, Katz A, et al. Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca2+ transients and reveal mitochondrial Ca2+ handling defects in cardiomyocytes of Ob/Ob mice. Diabetes. (2005) 54(11):3340. doi: 10.2337/diabetes.54.8.2375
35. Yuan M, Gong M, Zhang Z, Meng L, Tse G, Zhao Y, et al. Hyperglycemia induces endoplasmic reticulum stress in atrial cardiomyocytes, and mitofusin-2 downregulation prevents mitochondrial dysfunction and subsequent cell death. Oxid Med Cell Longev. (2020) 2020:6569728. doi: 10.1155/2020/6569728
36. Igaki Y, Tanno M, Sato T, Kouzu H, Ogawa T, Osanami A, et al. Xanthine oxidoreductase-mediated injury is amplified by upregulated AMP deaminase in type 2 diabetic rat hearts under the condition of pressure overload. J Mol Cell Cardiol. (2021) 154:21–31. doi: 10.1016/j.yjmcc.2021.01.002
37. Osanami A, Sato T, Toda Y, Shimizu M, Kuno A, Kouzu H, et al. Adenosine monophosphate deaminase in the endoplasmic reticulum-mitochondria interface promotes mitochondrial Ca2+ overload in type 2 diabetes rat hearts. J Diabetes Investig. (2023) 14(4):560–9. doi: 10.1111/jdi.13982
38. Blanton RM, Carrillo-Salinas FJ, Alcaide P. T-cell recruitment to the heart: friendly guests or unwelcome visitors? Am J Physiol Heart Circ Physiol. (2019) 317(1):H124–40. doi: 10.1152/ajpheart.00028.2019
39. Han Y, Lai J, Tao J, Tai Y, Zhou W, Guo P, et al. Sustaining circulating regulatory T cell subset contributes to the therapeutic effect of paroxetine on mice with diabetic cardiomyopathy. Circ J. (2020) 84(9):1587–98. doi: 10.1253/circj.CJ-19-1182
40. Tao J, Chen H, Wang YJ, Qiu JX, Meng QQ, Zou RJ, et al. Ketogenic diet suppressed T-regulatory cells and promoted cardiac fibrosis via reducing mitochondria-associated membranes and inhibiting mitochondrial function. Oxid Med Cell Longev. (2021) 2021:5512322. doi: 10.1155/2021/5512322
41. Liu IF, Lin TC, Wang SC, Yen CH, Li CY, Kuo HF, et al. Long-term administration of western diet induced metabolic syndrome in mice and causes cardiac microvascular dysfunction, cardiomyocyte mitochondrial damage, and cardiac remodeling involving caveolae and caveolin-1 expression. Biol Direct. (2023) 18(1):9. doi: 10.1186/s13062-023-00363-z
42. Ren J, Sun MM, Zhou H, Ajoolabady A, Zhou Y, Tao J, et al. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci Adv. (2020) 6(38):eabc8561. ARTN eabc856110.1126/sciadv.abc8561 32938669
43. Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell. (2015) 32(3):304–17. doi: 10.1016/j.devcel.2014.12.011
44. Xian H, Yang Q, Xiao L, Shen HM, Liou YC. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat Commun. (2019) 10(1):2059. doi: 10.1038/s41467-019-10096-1
45. Xu H, Yu W, Sun M, Bi Y, Wu NN, Zhou Y, et al. Syntaxin17 contributes to obesity cardiomyopathy through promoting mitochondrial Ca2+ overload in a parkin-MCUb-dependent manner. Metab Clin Exp. (2023) 143:155551. doi: 10.1016/j.metabol.2023.155551
46. Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. (2012) 94(2):168–80. doi: 10.1093/cvr/cvs116
47. Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. (1998) 258(2):729–35. doi: 10.1046/j.1432-1327.1998.2580729.x
48. Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol. (2015) 78:23–34. doi: 10.1016/j.yjmcc.2014.11.005
49. Hurst S, Hoek J, Sheu SS. Mitochondrial Ca2+ and regulation of the permeability transition pore. J Bioenerg Biomembr. (2017) 49(1):27–47. doi: 10.1007/s10863-016-9672-x
50. Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, et al. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci Adv. (2019) 5(8):eaaw4597. doi: 10.1126/sciadv.aaw4597
51. Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J. (2013) 77(5):1111–22. doi: 10.1253/circj.cj-13-0321
52. Paillard M, Tubbs E, Thiebaut PA, Gomez L, Fauconnier J, Da Silva CC, et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation. (2013) 128(14):1555–65. doi: 10.1161/CIRCULATIONAHA.113.001225
53. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. (2005) 434(7033):652–8. doi: 10.1038/nature03317
54. Rieusset J, Fauconnier J, Paillard M, Belaidi E, Tubbs E, Chauvin MA, et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia. (2016) 59(3):614–23. doi: 10.1007/s00125-015-3829-8
55. Beretta M, Santos CX, Molenaar C, Hafstad AD, Miller CC, Revazian A, et al. Nox4 regulates InsP3 receptor-dependent Ca2+ release into mitochondria to promote cell survival. EMBO J. (2020) 39(19):e103530. doi: 10.15252/embj.2019103530
56. Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF, et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun. (2014) 5:3996. doi: 10.1038/ncomms4996
57. De Vos KJ, Morotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet. (2012) 21(6):1299–311. doi: 10.1093/hmg/ddr559
58. Gomez-Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, Miller CCJ. The ER- the ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr Biol. (2017) 27(3):371–85. doi: 10.1016/j.cub.2016.12.038
59. Qiao X, Jia S, Ye J, Fang X, Zhang C, Cao Y, et al. PTPIP51 regulates mouse cardiac ischemia/reperfusion through mediating the mitochondria-SR junction. Sci Rep. (2017) 7:45379. doi: 10.1038/srep45379
60. Tessier N, Ducrozet M, Dia M, Badawi S, Chouabe C, Crola Da Silva C, et al. TRPV1 channels are new players in the reticulum-mitochondria Ca2+ coupling in a rat cardiomyoblast cell line. Cells. (2023) 12(18):2322. doi: 10.3390/cells12182322
61. Sun Z, Han J, Zhao W, Zhang Y, Wang S, Ye L, et al. TRPV1 activation exacerbates hypoxia/reoxygenation-induced apoptosis in H9C2 cells via calcium overload and mitochondrial dysfunction. Int J Mol Sci. (2014) 15(10):18362–80. doi: 10.3390/ijms151018362
62. Chen TI, Lai CJ, Hsieh CJ, Tsai KL, Yang KT. Differences in left ventricular cardiomyocyte loss induced by chronic intermittent hypoxia between spontaneously hypertensive and Wistar-Kyoto rats. Sleep Breath. (2011) 15(4):845–54. doi: 10.1007/s11325-010-0448-y
63. Zhou S, Yin X, Zheng Y, Miao X, Feng W, Cai J, et al. Metallothionein prevents intermittent hypoxia-induced cardiac endoplasmic reticulum stress and cell death likely via activation of Akt signaling pathway in mice. Toxicol Lett. (2014) 227(2):113–23. doi: 10.1016/j.toxlet.2014.03.011
64. Moulin S, Thomas A, Wagner S, Arzt M, Dubouchaud H, Lamarche F, et al. Intermittent hypoxia-induced cardiomyocyte death is mediated by HIF-1 dependent MAM disruption. Antioxidants. (2022) 11(8):1462. doi: 10.3390/antiox11081462
65. Yang J, Sun M, Chen R, Ye X, Wu B, Liu Z, et al. Mitochondria-associated membrane protein PACS2 maintains right cardiac function in hypobaric hypoxia. iScience. (2023) 26(4):106328. doi: 10.1016/j.isci.2023.106328
66. Bockler S, Westermann B. ER-mitochondria contacts as sites of mitophagosome formation. Autophagy. (2014) 10(7):1346–7. doi: 10.4161/auto.28981
67. Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, et al. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. (2017) 136(23):2248–66. doi: 10.1161/CIRCULATIONAHA.117.030235
68. Tang Y, Wu Y. Decreased ATP production during mitochondrial calcium uniporter inhibition enhances autophagy and mitophagy to provide cardioprotection in cardiac failure. Int J Cardiol. (2019) 282:67. doi: 10.1016/j.ijcard.2018.11.130
69. Contreras-Ferrat AE, Toro B, Bravo R, Parra V, Vasquez C, Ibarra C, et al. An inositol 1,4,5-triphosphate (IP3)-IP3 receptor pathway is required for insulin-stimulated glucose transporter 4 translocation and glucose uptake in cardiomyocytes. Endocrinology. (2010) 151(10):4665–77. doi: 10.1210/en.2010-0116
70. Gutierrez T, Parra V, Troncoso R, Pennanen C, Contreras-Ferrat A, Vasquez-Trincado C, et al. Alteration in mitochondrial Ca2+ uptake disrupts insulin signaling in hypertrophic cardiomyocytes. Cell Commun Signal. (2014) 12:68. doi: 10.1186/s12964-014-0068-4
71. Wang YX, Li XC, Xu XL, Qu XM, Yang YJ. Transient receptor potential vanilloid type 1 protects against pressure overload-induced cardiac hypertrophy by promoting mitochondria-associated endoplasmic reticulum membranes. J Cardiovasc Pharm. (2022) 80(3):430–41. doi: 10.1097/Fjc.0000000000001301
72. Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, et al. Mitochondrial death protein nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med. (2002) 8(7):725–30. doi: 10.1038/nm719
73. Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, et al. Distinct pathways regulate proapoptotic nix and BNip3 in cardiac stress. J Biol Chem. (2006) 281(3):1442–8. doi: 10.1074/jbc.M509056200
74. Wojcikiewicz RJ. Type I, II, and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J Biol Chem. (1995) 270(19):11678–83. doi: 10.1074/jbc.270.19.11678
75. Li Y, Huang D, Jia L, Shangguan F, Gong S, Lan L, et al. Lonp1 links mitochondria-ER interaction to regulate heart function. Research. (2023) 6:0175. doi: 10.34133/research.0175
76. Song Z, Song H, Liu D, Yan B, Wang D, Zhang Y, et al. Overexpression of MFN2 alleviates sorafenib-induced cardiomyocyte necroptosis via the MAM-CaMKIIδ pathway in vitro and in vivo. Theranostics. (2022) 12(3):1267–85. doi: 10.7150/thno.65716
77. Peng C, Yang S, Yang F, Xiong Z, Liu Q, Liao S, et al. Crosstalk between Mfn2-mediated mitochondria associated membranes disorder and autophagy induced by molybdenum and cadmium in sheep heart. Food Chem Toxicol. (2023) 174:113660. doi: 10.1016/j.fct.2023.113660
78. Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, Chauvin MA, et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes. (2014) 63(10):3279–94. doi: 10.2337/db13-1751
79. Li Y, Feng YF, Liu XT, Li YC, Zhu HM, Sun MR, et al. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. (2021) 38:101771. doi: 10.1016/j.redox.2020.101771
80. Lin Y, Xu Y, Zhang Z. Sepsis-induced myocardial dysfunction (SIMD): the pathophysiological mechanisms and therapeutic strategies targeting mitochondria. Inflammation. (2020) 43(4):1184–200. doi: 10.1007/s10753-020-01233-w
81. Han D, Li X, Li S, Su T, Fan L, Fan WS, et al. Reduced silent information regulator 1 signaling exacerbates sepsis-induced myocardial injury and mitigates the protective effect of a liver X receptor agonist. Free Radic Biol Med. (2017) 113:291–303. doi: 10.1016/j.freeradbiomed.2017.10.005
82. Jiang T, Peng D, Shi W, Guo J, Huo S, Men L, et al. IL-6/STAT3 signaling promotes cardiac dysfunction by upregulating FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes formation in sepsis mice. Front Cardiovasc Med. (2021) 8:790612. doi: 10.3389/fcvm.2021.790612
83. Sun Y, Cai Y, Qian S, Chiou H, Zang QS. Beclin-1 improves mitochondria-associated membranes in the heart during endotoxemia. FASEB Bioadv. (2021) 3(3):123–35. doi: 10.1096/fba.2020-00039
84. Lv Y, Cheng L, Peng F. Compositions and functions of mitochondria-associated endoplasmic reticulum membranes and their contribution to cardioprotection by exercise preconditioning. Front Physiol. (2022) 13:910452. doi: 10.3389/fphys.2022.910452
85. Campos JC, Queliconi BB, Bozi LHM, Bechara LRG, Dourado PMM, Andres AM, et al. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy. (2017) 13(8):1304–17. doi: 10.1080/15548627.2017.1325062
86. Palee S, Minta W, Mantor D, Sutham W, Jaiwongkam T, Kerdphoo S, et al. Combination of exercise and calorie restriction exerts greater efficacy on cardioprotection than monotherapy in obese-insulin resistant rats through the improvement of cardiac calcium regulation. Metab Clin Exp. (2019) 94:77–87. doi: 10.1016/j.metabol.2019.02.003
87. Yeh CH, Chou YJ, Kao CH, Tsai TF. Mitochondria and calcium homeostasis: Cisd2 as a big player in cardiac ageing. Int J Mol Sci. (2020) 21(23):9238. doi: 10.3390/ijms21239238
88. Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. (2011) 31(6):1309–28. doi: 10.1128/MCB.00911-10
89. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci U S A. (2015) 112(17):E2174–81. doi: 10.1073/pnas.1504880112
90. Seidlmayer LK, Mages C, Berbner A, Eder-Negrin P, Arias-Loza PA, Kaspar M, et al. Mitofusin 2 is essential for IP(3)-mediated SR/mitochondria metabolic feedback in ventricular myocytes. Front Physiol. (2019) 10:733. doi: 10.3389/fphys.2019.00733
91. Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. (2006) 174(7):915–21. doi: 10.1083/jcb.200604016
92. Wang J, Toan S, Li R, Zhou H. Melatonin fine-tunes intracellular calcium signals and eliminates myocardial damage through the IP3R/MCU pathways in cardiorenal syndrome type 3. Biochem Pharmacol. (2020) 174:113832. doi: 10.1016/j.bcp.2020.113832
Keywords: mitochondria-associated membrane, cardiac disease, Ca2+ homeostasis, apoptosis, autophagy
Citation: Lu B, Chen X, Ma Y, Gui M, Yao L, Li J, Wang M, Zhou X and Fu D (2024) So close, yet so far away: the relationship between MAM and cardiac disease. Front. Cardiovasc. Med. 11:1353533. doi: 10.3389/fcvm.2024.1353533
Received: 10 December 2023; Accepted: 22 January 2024;
Published: 5 February 2024.
Edited by:
Andreas Rinne, University of Medicine and Pharmacy “Carol Davila”, RomaniaReviewed by:
Chi Keung Lam, University of Delaware, United States© 2024 Lu, Chen, Ma, Gui, Yao, Li, Wang, Zhou and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xunjie Zhou a2Vuc2hpbjIwMDBAc2h1dGNtLmVkdS5jbg== Deyu Fu eXkyNjA4QHNodXRjbS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Abbreviations ANT, adenine nucleotide translocator; ATP, adenosine triphosphate; AMPD3, adenosine monophosphate deaminase; AMPK, AMP-activated protein kinase; BAP31, B-cell receptor-associated protein 31; Cd, cadmium; CaMKIIδ, calcmodulin-dependent protein kinase II delta; Ca2+, calcium; CHOP, C/EBP homologous protein; CREB, CAMP response element-binding protein; CISD2, CDGSH iron sulfur domain 2; CYPD, cyclophilin D; DbCM, diabetic cardiomyopathy; Drp1, dynamin-related peptide 1; ER, endoplasmic reticulum; EP, exercise preconditioning; FBXL2, F-box and leucine rich repeat protein 2; Fis1, fission protein 1; FUNDC1, FUN14 domain containing 1; GRP75, glucose-regulated protein 75; GRP78, glucose-regulated protein 78; HFD, high-fat diet; HFHSD, high-fat high-sugar diet; HG, high glucose; HH, hypobaric hypoxic; HIF-1, hypoxia-inducible factor 1; H/R, hypoxia-reoxygenation; I/R, hypoxia-reperfusion; IL-6, interleukin-6; IP3R, inositol 1,4,5-trisphosphate receptor; IMM, inner mitochondrial membrane; IH, intermittent hypoxia; KD, ketogenic diet; KBs, ketone bodies; LC3, light chain 3; LPS, lipopolysaccharide; LETO, long-evans tokushima otsuka; MS, metabolic syndrome; MAM, mitochondria-associated membrane; MERCs, mitochondria-endoplasmic reticulum contacts; LonP1, mitochondrial ATP-dependent lon protease 1; mCa2+, mitochondrial Ca2+; MCU, mitochondrial Ca2+ uniporter; MFNs, mitochondrial fusion proteins; MLKL, mixed-lineage kinase domain-like protein; mPTP, mitochondrial permeability transition pore; Mo, molybdenum; NADH, reduced nicotinamide adenine dinucleotide; NOX4, NADPH oxidase 4; Nix, nip3-like protein X; OLETF, otsuka long-evans tokushima fatty; OMM, outer mitochondrial membrane; p-STAT3, phosphorylated signal transducer and activator of transcription 3; PACS-2, phosphofurin acidic cluster sorting protein 2; PTPIP51, protein tyrosine phosphatase interacting protein 51; ROS, reactive oxygen species; RIP3, receptor-interacting protein kinase 3; RyR, ryanodine receptor; SIMD, sepsis-induced myocardial dysfunction; SDB, sleep-disordered breathing; Sor, sorafenib; STX17, syntaxin 17; Tom40, mitochondrial membrane 40; Tregs, regulatory T cells; TRPV1, transient receptor potential vanilloid type 1; VDAC, voltage-dependent anion channel; VAPB, vesicle-associated membrane protein-associated protein B.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.