 Yiren Yao
Yiren Yao Hongyang Liu†
Hongyang Liu† Yang Gu
Yang Gu Xiaojin Xu
Xiaojin Xu- Department of Cardiology, The Affiliated Huaian No.1 People’s Hospital of Nanjing Medical University, Huai’an, China
Objectives: To look into the connection between amyotrophic lateral sclerosis (ALS) and atrial fibrillation (AF) using Mendelian randomization (MR).
Methods: Two-sample MR was performed using genetic information from genome-wide association studies (GWAS). Genetic variants robustly associated with ALS and AF were used as instrumental variables. GWAS genetic data for ALS (n = 138,086, ncase = 27,205) and AF (n = 1,030,836, ncase = 60,620), publicly available from IEU Open. The specific MR protocols were Inverse variance-weighted (IVW), Simple mode, MR Egger, Weighted mode, and Weight median estimator (WME). Subsequently, the MR-Egger intercept and Cochran Q examine were used to evaluate instrumental variables (IVs)' heterogeneity and multiplicative effects (IVs). In addition, MR-PRESSO analysis was conducted to exclude any potential pleiotropy.
Results: The IVW method demonstrated that ALS positively affected AF [OR: 1.062, 95% CI (1.004–1.122); P = 0.035]. Indeed, other MR methods were in accordance with the tendency of the IVW method (all OR > 1), and sensitivity testing verified the reliability of this MR result.
Conclusions: This MR study proves a positive causal connection between ALS and atrial fibrillation. Further studies are warranted to elucidate the mechanisms linking ALS and AF.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that causes the selective degeneration of upper and lower motor neurons in the motor cortex, brainstem, and spinal cord (1, 2). This causes progressive muscle weakness, atrophy, paralysis, and death, usually from respiratory failure, within 2–6 years of the onset of symptoms (1, 2). The average onset age is between 55 and 65 years, and the lifetime risk of ALS is approximately 1 in 300 (3). Most cases of ALS occur sporadically but about 5%–10% are familial, caused by inherited genetic mutations (3). The cause of sporadic ALS is still largely unknown. Nonetheless, suggested mechanisms include glutamate excitotoxicity and protein misfolding, oxidative damage, mitochondrial disease, and neuroinflammation (1, 4). As such, ALS remains an incurable disease with significant unmet medical needs.
Several cardiac arrhythmias can be observed among ALS. Such as supraventricular tachycardia, ventricular tachycardia, ventricular fibrillation, atrial fibrillation, long QT syndrome, and heart blocks (5). Epidemiological studies indicate that patients with ALS have higher rates of cardiovascular comorbidities, including coronary artery disease, stroke, and heart failure, compared to the general population (6). Studies have found that ALS can present with mild cardiovascular autonomic dysfunction in the early course of the disease, which increases the risk of arrhythmia and sudden death (7). Mandrioli et al. also discovered that cardiovascular diseases negatively associated with ALS included AF, heart failure, and hypertension, thus determining that cardiovascular disease may have a negative impact in the prognosis of ALS (8). Besides, Hoda et al. also found that AF is independent prognostic factors for the survival of ALS, and AF are independently associated with a shorter survival period of ALS (6).
AF is distinguished by rapid and disordered electrical activity in the atria, causing an irregular response from the heart (9). This leads to symptoms such as palpitations, fatigue, exercise intolerance, increased risk of heart failure, stroke, and death rates (9). In the general public, the lifetime risk of AF is about 25% (10).
While the association between ALS and AF has been documented, the nature and causality of this relationship remain unclear. One major limitation of previous observational studies is the inability to determine causality due to potential confounding and reverse causation biases. For instance, shared risk factors could predispose patients to ALS and AF. It is also plausible that AF could represent a consequence of ALS pathophysiology or that ALS may develop secondary to AF-related cerebral hypoperfusion and hypoxia (11). Therefore, elucidating the causal dynamics between ALS and AF has important implications. It may uncover novel etiological mechanisms and identify potential prognostic factors or therapeutic targets for ALS or AF.
Mendelian randomization (MR) has appeared as an effective technique to assess causal relationships between modifiable exposures and outcomes based on genetic variations as instrumental variables (IVs) (12). Given that different genetic variants are distributed at random during conception, MR is less susceptible to confounding (13, 14). Moreover, germline genetic variants precede disease onset. They are generally unaffected by the disease process itself, minimizing reverse causation. Two-sample MR utilizes summarizing genetic information from various genome-wide association studies (GWAS) of the exposure and outcome. This approach enhances statistical power by leveraging large GWAS datasets. MR techniques have been increasingly applied to investigate the causal relationships between various modifiable exposures and ALS or AF (15). For example, Xia et al. found a positive causal relationship between linoleic acid (LA) and ALS risk through MR analysis. At the same time, vitamin D was negatively correlated with ALS risk (16). Xia et al. also found a causal connection between increased telomere length and reduced hazard of ALS through MR analysis (17). Mohammadi-Shemirani et al. also found through MR analysis that Elevated Lipoprotein (a) is a potential causal mediator associated with increased risk of atrial fibrillation (15). However, MR studies examining the potential causal relationship between ALS and AF are lacking.
In this research, we used a two-sample MR analysis to look into the relationship between ALS and AF. utilizing GWAS data from large populations of European ancestry. Genetic instruments strongly associated with each trait were applied in bidirectional MR frameworks. Several sensitivity studies were carried out to assess and account for probable pleiotropy. Elucidating the possible causal relationship between ALS and AF will provide novel insights into disease mechanisms and may have important implications for patient prognosis, monitoring, and management.
Methods and material
Study design and data
Figure 1 shows the flowchart of the MR study. All of the genetic information used in this MR study was obtained from public sources, which are openly available on the IEU Open GWAS project, and specific ethical approvals were expressed in the original GWAS article. This study followed the three hypotheses of MR research: (i) genetic variation is strongly associated with risk factors; (ii) genetic variation is not associated with confounding factors; and (iii) genetic variation affects outcomes only through risk factors. GWAS data for ALS (ID: ebi-a-GCST90027164) and atrial fibrillation (ID: ebi-a-GCST006414) were found on the IEU Web site (https://gwas.mrcieu.ac.uk/), accessed on 2024-03-23. Table 1 provides details of the GWAS data sources. The pooled data for ALS included 138,086 participants (27,205 cases, 110,881 controls). The pooled data for AF consisted of 1,030,836 participants (60,620 cases, 970,216 controls). All participants were from the European population.
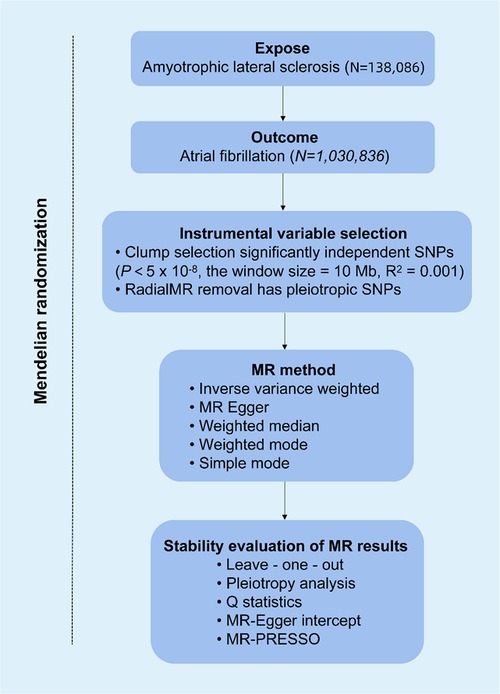
Figure 1. Overview of Mendelian randomization (MR) analysis of amyotrophic lateral sclerosis (ALS) and atrial fibrillation (AF).

Table 1. Phenotype source and ALS and AF.
Selection of instrumental variable (IVs)
In previous studies (18), significant Single nucleotide polymorphism (SNPs) were selected from the GWAS pooled data of ALS (selection criteria: P < 5 × 10−8, linkage disequilibrium R2 of 0.001, and linkage disequilibrium region width of 10,000 kb) to make sure each SNP was independent of each other and to omit the impact of genetic pleiotropy on the results. In addition, to quantify the level of gene variation, the F-statistic was calculated, and those SNPs with an F-statistic of 10 or higher were selected as the essential correlation criteria. The F statistic for specific individual SNPs was calculated as F = (N − 2)/K × R2/(1 − R2), where N refers to the total number of samples contained in the exposure database, K refers to the total number of IVs and R2 is the proportion of variants explained by SNPs in the exposure database. R2 was calculated as R2 = 2×EAF × (1 − EAF) × β2, where EAF stands for the effect allele frequency and β refers to the value of the allele's effect.
The above-filtered SNPs associated with ALS were then extracted from the GWAS pooled data of AF, and SNPs without suitable alternative loci were eliminated. Summarizing the information of the ALS and AF datasets, SNPs directly associated with AF were also eliminated (P < 5 × 10−8). Following the above steps, the remaining SNPs were finally used as IVs. A detailed description of the IVs used in this MR study is presented in Table 2.
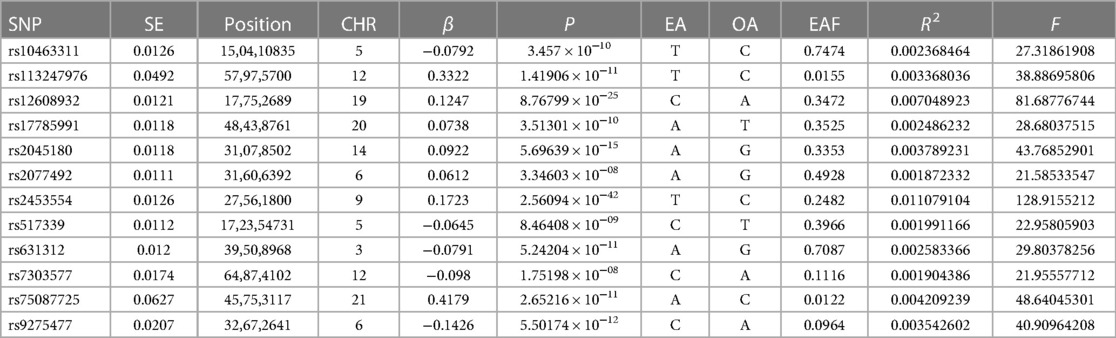
Table 2. Characteristics of the genetic IVs at the genome-wide significance level (P < 5 × 10−8).
MR analysis approach
Several MR methods were utilized in this study to assess the causal relationship between ALS and AF. The primary analysis was carried out using the conventional Inverse variance weighted (IVW) method. The IVW method combines the estimates of each SNP using inverse-variance weighting to estimate the causal effect. IVW is a method used in MR to perform meta-analysis of the effects of multiple SNPs on multiple loci. The prerequisite for the application of IVW is that all SNPs are valid instrumental variables and independent of each other (13). To complement the IVW estimates, additional MR techniques such as MR-Egger regression, Weight median estimator (WME), Weighted mode and Simple mode were also carried out. These approaches can provide more robust causal inferences under varying scenarios. MR-Egger regression has the ability to identify and take into account heterogeneity and unbalanced pleiotropy (12). The WME approach gives consistent effect estimates in the event that at least fifty percent of the genetic variants are valid instruments (19). Simple mode is the simplest MR analysis strategy. Its calculation is straightforward and easy to use, and even if it contains a small number of weak instrumental variables, it can provide a relatively accurate estimation of causal effects (13).
Sensitivity evaluates
In order to evaluate the dependability of the MR estimates, a number of different sensitivity evaluates were carried out. The possible horizontal pleiotropy of the instruments was taken into account with MR-Egger regression. Leave-one-out cross-validation was conducted by iteratively removing each SNP from the instrument sets. Between-SNP heterogeneity was quantified using Cochran's Q statistic. In addition, funnel plots visually depicting SNP-specific effects against instrument strength were assessed to check for directional pleiotropy.
Statistical analysis
The R software's TwoSampleMR package was used to carry out the MR analyses (for more information, visit http://www.R-project.org). In each of the analyses, statistical significance was defined as a P-value that was lower than 0.05.
Results
ALS has a positive causal relationship with AF
Characteristics of the genetic IVs at the genome-wide significance level are presented in Table 2, including the SNP ID, effect allele, reference allele, SNP position, Chr, Beta, Se and P. Statistically, most of the F-statistics for the IVs were more than 10, reducing the likelihood that this study had weak IVs. Five MR methods were used in this study to examine the causal outcomes, with the IVW approach as the primary method to determine causality. As shown in Table 3, the IVW method demonstrated a positive causal relation of ALS on AF [OR: 1.062, 95% CI (1.004–1.122); P = 0.035]. Similarly, the Weighted median also demonstrated a positive causal relation of ALS on AF [OR: 1.097, 95% CI (1.030–1.167); P = 0.004]. The other three MR methods, such as MR Egger [OR: 1.167, 95% CI (1.032–1.320); P = 0.034], Simple mode [OR: 1.116, 95% CI (1.023–1.218); P = 0.032], and Weighted mode [OR: 1.102, 95% CI (1.026–1.183); P = 0.022], although they cannot separately display a causal relationship between ALS and AF, the OR direction remains consistent with IVW, which also supports the results of this IVW (Table 3, Figure 2). Specifically, the effect of each SNP in the MR analysis is shown in Figure 3. The results consistently showed a positive causal effect of ALS on AF risk.
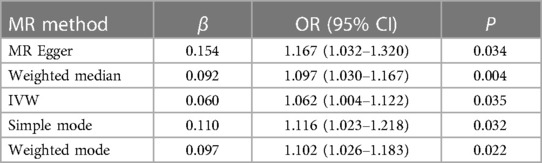
Table 3. The causal relationship between ALS and AF through MR analysis.
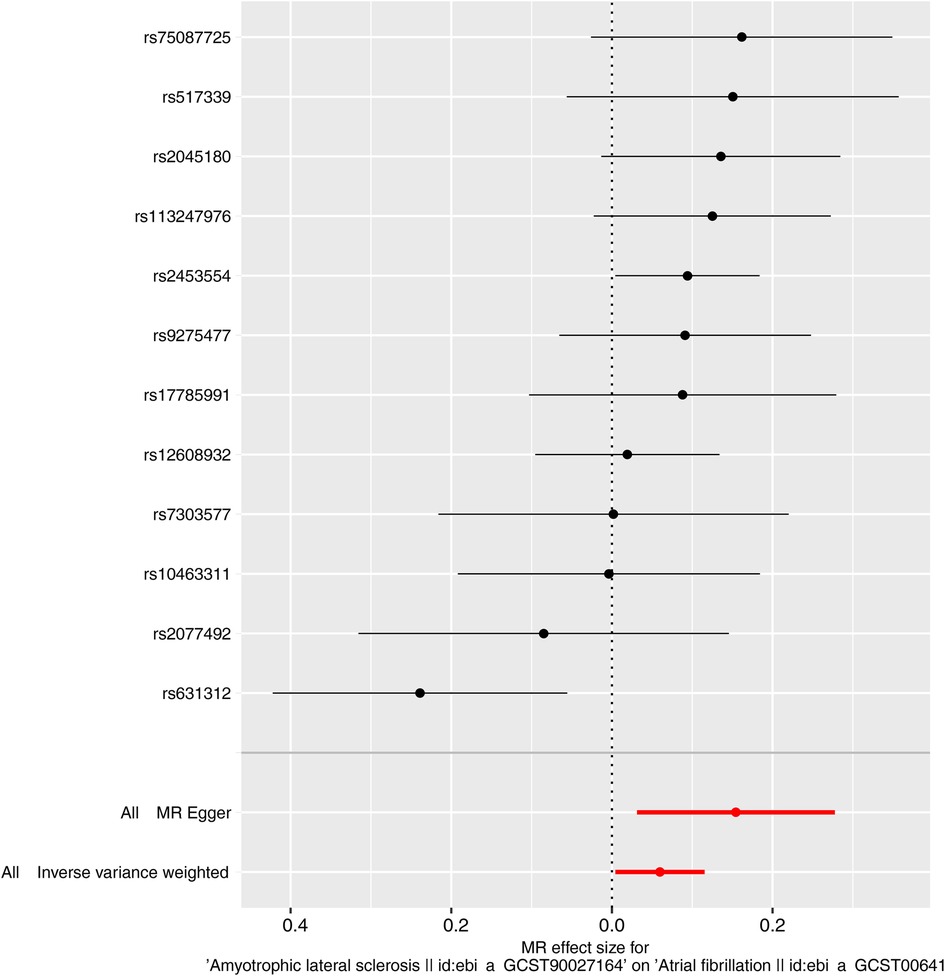
Figure 2. Forest plot of MR analysis of ALS and AF.
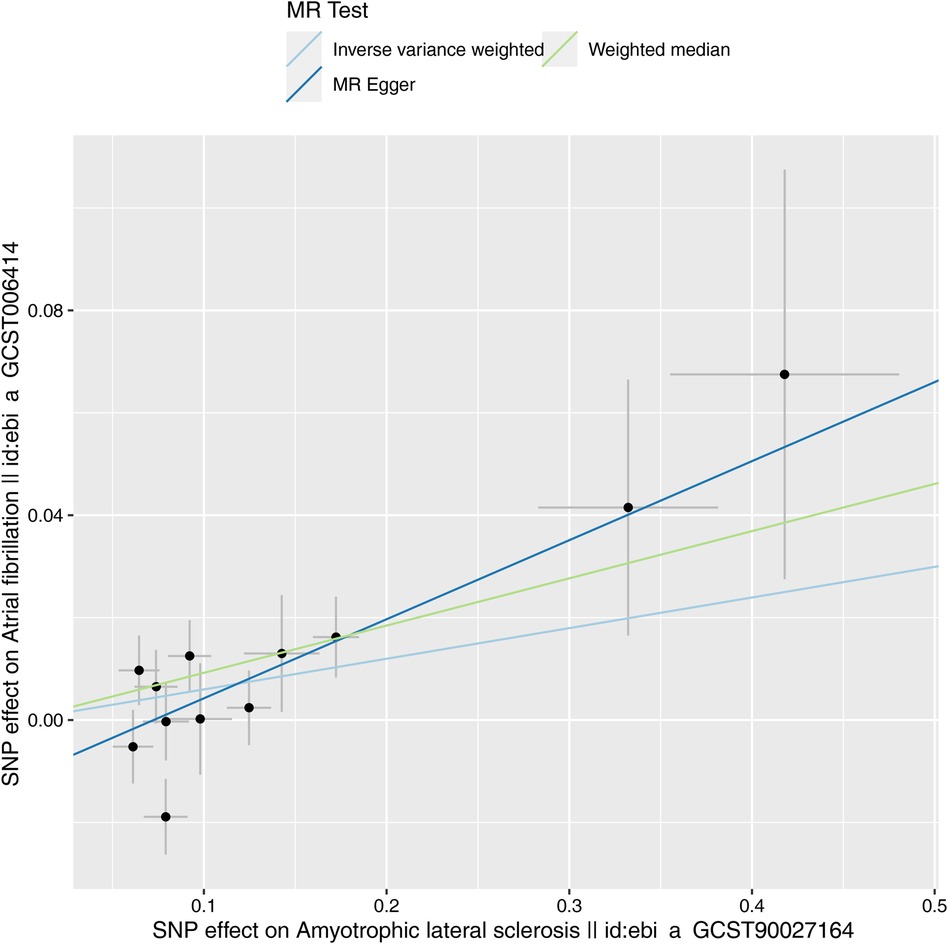
Figure 3. Scatter plot of MR analysis of ALS and AF.
Sensitivity analyses
Next, we evaluated the differences in IVs, and the funnel plot showed that all IVs were roughly evenly distributed without significant deviation (Figure 4). The leave-one-out examination of the remaining method indicates that the relation between ALS and AF does not stem from a single SNP (Figure 5). The MR-Egger intercept revealed no substantial horizontal pleiotropy for these IVs (P = 0.128). In addition, the Cochran Q test results based on the IVW method also showed no heterogeneity in IVs (P = 0.099). Finally, the MR-PRESSO analysis also showed no potential pleiotropic outliers (P = 0.129).
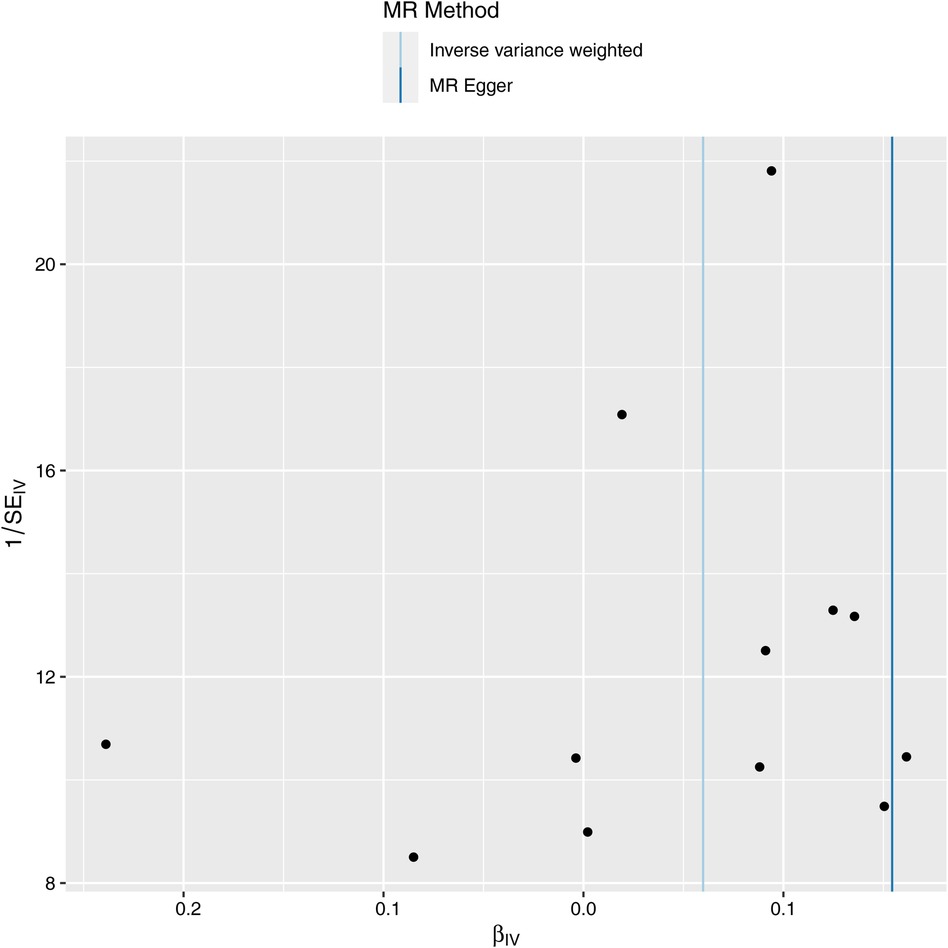
Figure 4. Funnel plot of MR analysis of ALS and AF.
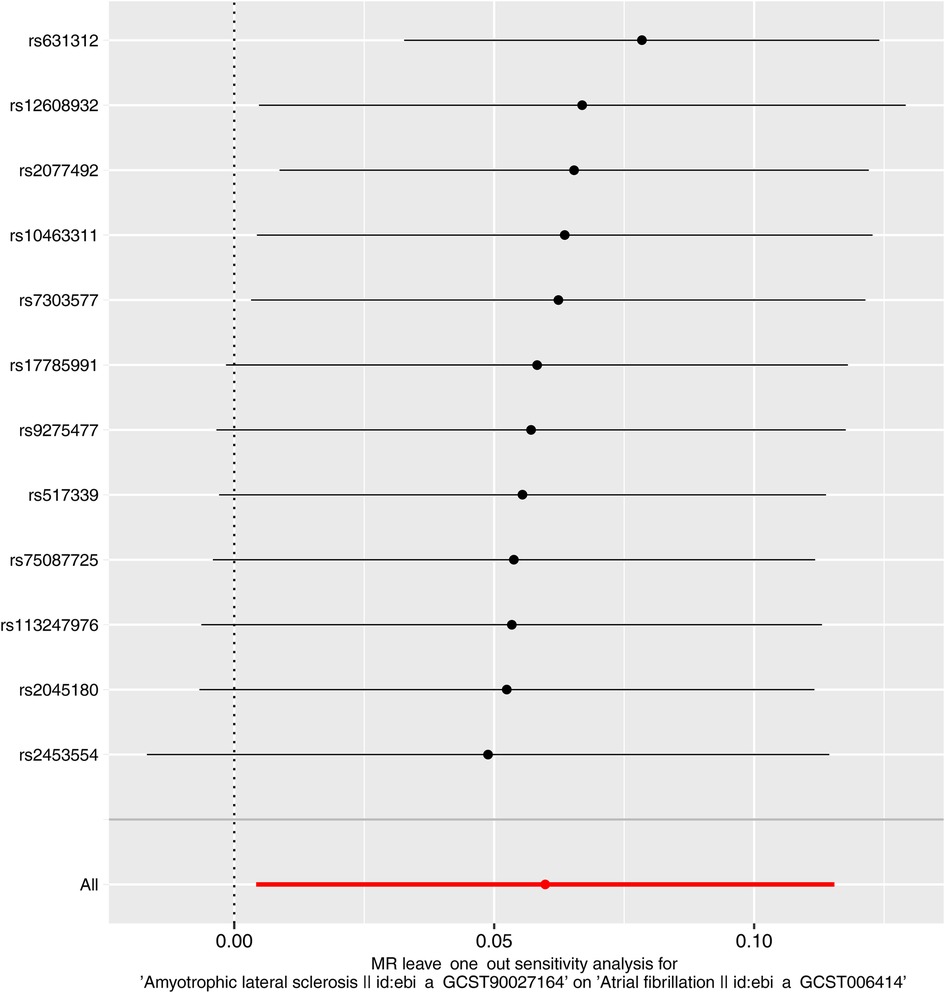
Figure 5. The leave-one-out plots for MR analysis of ALS and AF.
In summary, the ALS genetic instruments demonstrated indication for a potential positive causal effect on AF risk.
Discussion
This two-sample MR study supports a causal association between ALS and an increased risk of AF. The primary IVW analysis demonstrated a statistically significant positive causal effect of ALS on AF risk, with an odds ratio (OR) of 1.062 (95% CI 1.004–1.122, P = 0.035). This finding was corroborated by consistent directionality of effect estimates across several complementary MR approaches, including the WME, Simple mode, MR Egger, and Weighted mode models. Multiple sensitivity analyses showed no evidence of significant horizontal pleiotropy or heterogeneity among the genetic instruments. These results indicate that ALS may causally contribute to increased susceptibility to AF.
The potential mechanisms uncovering this association remain fully elucidated but may involve both central and peripheral pathophysiological processes related to ALS that predispose to cardiac arrhythmia (6). ALS is defined by the progressive degeneration and death of upper and lower motor neurons in the motor cortex or brainstem, which results in muscular atrophy and weakness (1). Autonomic nervous system dysfunction manifested as abnormal heart rate variability has been documented in ALS patients, even at early stages, indicative of cardiac autonomic imbalance (6). This autonomic neuropathy likely contributes to the increased risk of cardiac arrhythmias and sudden death observed in ALS (20, 21). At the tissue level, neurodegenerative changes in brain regions involved in central autonomic control, such as the hypothalamus, medulla, and intermediolateral cell columns of the spinal cord, may disrupt standard cardiovagal modulation and promote sympathoexcitation (22). Loss of inhibitory cortical inputs to the brainstem and spinal cord autonomic nuclei could further exacerbate autonomic dysfunction in ALS (6). Peripheral denervation of postganglionic sympathetic neurons innervating the heart may also directly alter electrophysiological properties and automaticity of cardiomyocytes, providing substrate for AF (23).
Furthermore, oxidative stress and mitochondrial dysfunction, key pathophysiological processes in ALS pathogenesis, likely also contribute to cardiac dysregulation and arrhythmogenesis (4, 24). Reactive oxygen species generation and antioxidant depletion intrinsically alter calcium handling, membrane excitability, and conduction in cardiomyocytes, facilitating triggered activity and re-entry (25). Impaired mitochondrial energetics and dysfunctional quality control further promote oxidative damage and protein aggregation in conduction system tissue (26, 27). Besides, neuroinflammation is another salient feature of ALS mediated by reactive astrocytes, microglia, and cytokines such as TGF-β, TNF-α, and IL-6 (28). Chronic inflammation is linked with AF susceptibility and commonly observed in atrial biopsy specimens from AF patients (29). Pro-inflammatory cytokines may contribute to fibrotic remodeling of atrial tissue as well as direct effects on ion channels and calcium handling proteins that influence arrhythmogenesis (29). The relationship between systemic inflammation and AF was further supported by Liu et al., showing IL-6 and TNF-α levels had causal effects on increased AF risk (29). Hence, neuroinflammation in ALS could potentially mediate AF via circulating cytokines.
Additional pathways by which ALS may lead to AF include respiratory compromise and acid-base abnormalities from progressive respiratory muscle weakness (30). Hypoxemia, hypercapnia, and acidosis can directly provoke arrhythmias and may exacerbate underlying cardiovascular dysfunction in ALS patients (31).
Our study has several strengths supporting the validity of its findings. MR circumvents biases from confounding and reverse causation inherent to conventional epidemiological studies by utilizing genetic instruments rather than directly measured phenotypes. Two-sample MR leverages massive GWAS datasets, enhancing statistical power to detect causal effects. The SNPs applied as instruments for ALS were selected based on stringent criteria and passed all prerequisite assumptions. The MR results were robust across several complementary analysis methods, and sensitivity analyses showed no significant issues with pleiotropy or heterogeneity.
In brief, this two-sample MR study provides evidence that ALS may play a causal role in increasing susceptibility to AF, potentially mediated through autonomic dysfunction, oxidative stress, neuroinflammation, respiratory compromise, and medication effects. The findings add to prior observational data associating ALS with more significant cardiovascular morbidity and earlier mortality. This relationship appears biologically plausible given pathophysiological processes in ALS predisposing to cardiac dysregulation and arrhythmogenesis. AF may serve as a prognostic factor in ALS and portend more significant risks of complications that could hasten functional decline. Clinicians may need heightened vigilance for cardiac rhythm abnormalities when evaluating and managing ALS patients. Future research should continue exploring this association's underlying mechanisms and translational implications. Confirmation across other large cohorts and ancestries is warranted. Exploring potential druggable targets influencing both diseases could yield therapeutic advances. This study provides novel insights into the causal relationships between neurodegenerative and cardiovascular disorders.
However, some limitations should be acknowledged. Given the rarity of ALS, there is currently a lack of observational studies confirming a higher incidence of AF in ALS patients. Therefore, the relevance of assessing the impact of ALS on AF risk seems limited. Despite large sample sizes overall, the number of eligible genetic instruments for ALS was modest, restricting analytical power. The populations studied were of European ancestry, limiting generalizability to other ethnicities. Also, We cannot exclude the possibility of residual pleiotropy from unmeasured traits influencing the results. MR estimates represent the lifetime effects of genetically determined exposures, whereas acquired factors modifying disease risk may further mediate observed associations. As summary data were used, exploring gene-environment or gene-gene interactions was impossible. Finally, the specific biological mechanisms linking ALS and AF remain speculative based on our analysis. Further studies must elucidate the pathophysiological pathways involved and confirm reproducibility in other populations.
Conclusion
In conclusion, this two-sample MR study shows that ALS causally increases AF risk, though the biological mechanism requires further research. More studies are needed to confirm this association and uncover the underlying pathways.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://gwas.mrcieu.ac.uk/, ebi-a-GCST005647, ebi-a-GCST006414.
Ethics Statement
Given that the data for this study were obtained from open access IEU stored GWAS data, no additional clinical trial registration or ethical approval was required.
Author contributions
YY: Writing – original draft. HL: Writing – original draft. YG: Writing – review & editing. XX: Writing – review & editing. XZ: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to express our gratitude to the volunteers and investigators involved in IEU GWAS project for their contributions to this human medical research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Feldman E, Goutman S, Petri S, Mazzini L, Savelieff M, Shaw P, et al. Amyotrophic lateral sclerosis. Lancet (London, England). (2022) 400:1363–80. doi: 10.1016/S0140-6736(22)01272-7
2. van Es M, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp R, Veldink J, et al. Amyotrophic lateral sclerosis. Lancet (London, England). (2017) 390:2084–98. doi: 10.1016/S0140-6736(17)31287-4
3. Xu L, Liu T, Liu L, Yao X, Chen L, Fan D, et al. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol. (2020) 267:944–53. doi: 10.1007/s00415-019-09652-y
4. Motataianu A, Serban G, Barcutean L, Balasa R. Oxidative stress in amyotrophic lateral sclerosis: synergy of genetic and environmental factors. Int J Mol Sci. (2022) 23(16):9339. doi: 10.3390/ijms23169339
5. Ramphul K, Verma R, Kumar N, Ramphul Y, Kumari K, Sombans S, et al. Abstract 9912: cardiac events among amyotrophic lateral sclerosis patients in the United States; a fresh perspective from the 2019 national inpatient sample. Circulation. (2022) 146(Suppl_1). doi: 10.1161/circ.146.suppl_1.9912
6. Abdel Magid H, Topol B, McGuire V, Hinman J, Kasarskis E, Nelson L. Cardiovascular diseases, medications, and ALS: a population-based case-control study. Neuroepidemiology. (2022) 56:423–32. doi: 10.1159/000526982
7. Shimizu T. Sympathetic hyperactivity and sympathovagal imbalance in amyotrophic lateral sclerosis. Eur Neurol Rev. (2013) 8:46–50. doi: 10.17925/ENR.2013.08.01.46
8. Mandrioli J, Ferri L, Fasano A, Zucchi E, Fini N, Moglia C, et al. Cardiovascular diseases may play a negative role in the prognosis of amyotrophic lateral sclerosis. Eur J Neurol. (2018) 25:861–8. doi: 10.1111/ene.13620
9. Zimetbaum P. Atrial fibrillation. Ann Intern Med. (2017) 166:ITC33–48. doi: 10.7326/AITC201703070
10. Kallistratos M, Poulimenos L, Manolis A. Atrial fibrillation and arterial hypertension. Pharmacol Res. (2018) 128:322–6. doi: 10.1016/j.phrs.2017.10.007
11. Hoshino T, Matsuzawa S, Takahashi R. 6-Deoxyjacareubin, a natural compound preventing hypoxia-induced cell death, ameliorates neurodegeneration in a mouse model of familial amyotrophic lateral sclerosis. Neurosci Res. (2021) 163:43–51. doi: 10.1016/j.neures.2020.02.011
12. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. (2015) 44:512–25. doi: 10.1093/ije/dyv080
13. Birney E. Mendelian randomization. Cold Spring Harbor Perspect Med. (2022) 12(4). doi: 10.1101/cshperspect.a041302
14. Bowden J, Holmes M. Meta-analysis and Mendelian randomization: a review. Res Synth Methods. (2019) 10:486–96. doi: 10.1002/jrsm.1346
15. Mohammadi-Shemirani P, Chong M, Narula S, Perrot N, Conen D, Roberts J, et al. Elevated lipoprotein(a) and risk of atrial fibrillation: an observational and Mendelian randomization study. J Am Coll Cardiol. (2022) 79:1579–90. doi: 10.1016/j.jacc.2022.02.018
16. Xia K, Wang Y, Zhang L, Tang L, Zhang G, Huang T, et al. Dietary-derived essential nutrients and amyotrophic lateral sclerosis: a two-sample Mendelian randomization study. Nutrients. (2022) 14(5):920. doi: 10.3390/nu14050920
17. Xia K, Zhang L, Zhang G, Wang Y, Huang T, Fan D. Leukocyte telomere length and amyotrophic lateral sclerosis: a Mendelian randomization study. Orphanet J Rare Dis. (2021) 16:508. doi: 10.1186/s13023-021-02135-2
18. Julian T, Boddy S, Islam M, Kurz J, Whittaker K, Moll T, et al. A review of Mendelian randomization in amyotrophic lateral sclerosis. Brain. (2022) 145:832–42. doi: 10.1093/brain/awab420
19. Bowden J, Davey Smith G, Haycock P, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. (2016) 40:304–14. doi: 10.1002/gepi.21965
20. Körner S, Kollewe K, Ilsemann J, Müller-Heine A, Dengler R, Krampfl K, et al. Prevalence and prognostic impact of comorbidities in amyotrophic lateral sclerosis. Eur J Neurol. (2013) 20:647–54. doi: 10.1111/ene.12015
21. Russo V, Rago A, Nigro G. Sudden cardiac death in neuromuscolar disorders: time to establish shared protocols for cardiac pacing. Int J Cardiol. (2016) 207:284–5. doi: 10.1016/j.ijcard.2016.01.175
22. Majewski J, Varma M, Davis G, Lelakowski J. Disease-specific aspects of management of cardiac arrhythmias in patients with muscular dystrophies. Pol Merkur Lekarski. (2019) 47:153–6.31760399
23. Wang SP, Wang LH. Disease implication of hyper-hippo signalling. Open Biol. (2016) 6(10):160119. doi: 10.1098/rsob.160119
24. Chen J, Bassot A, Giuliani F, Simmen T. Amyotrophic lateral sclerosis (ALS): stressed by dysfunctional mitochondria-endoplasmic Reticulum contacts (MERCs). Cells. (2021) 10(7):1789. doi: 10.3390/cells10071789
25. Jin J, Blackwood E, Azizi K, Thuerauf D, Fahem A, Hofmann C, et al. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res. (2017) 120:862–75. doi: 10.1161/CIRCRESAHA.116.310266
26. Kane K, Goldstein S, Mescher M. Class I alloantigen is sufficient for cytolytic T lymphocyte binding and transmembrane signaling. Eur J Immunol. (1988) 18:1925–9. doi: 10.1002/eji.1830181209
27. Ramachandra C, Hernandez-Resendiz S, Crespo-Avilan G, Lin Y, Hausenloy D. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. (2020) 57:102884. doi: 10.1016/j.ebiom.2020.102884
28. Garofalo S, Cocozza G, Bernardini G, Savage J, Raspa M, Aronica E, et al. Blocking immune cell infiltration of the central nervous system to tame neuroinflammation in amyotrophic lateral sclerosis. Brain Behav Immun. (2022) 105:1–14. doi: 10.1016/j.bbi.2022.06.004
29. Chen B, Collen LV, Mowat C, Isaacs KL, Singh S, Kane SV, et al. Inflammatory bowel disease and cardiovascular diseases. Am J Med. (2022) 135:1453–60. doi: 10.1016/j.amjmed.2022.08.012
30. Niedermeyer S, Murn M, Choi P. Respiratory failure in amyotrophic lateral sclerosis. Chest. (2019) 155:401–8. doi: 10.1016/j.chest.2018.06.035
Keywords: amyotrophic lateral sclerosis, atrial fibrillation, arrhythmia, causal relationship, Mendelian randomization
Citation: Yao Y, Liu H, Gu Y, Xu X and Zhang X (2024) A causal association between amyotrophic lateral sclerosis and atrial fibrillation: a two-sample Mendelian randomization study. Front. Cardiovasc. Med. 11:1351495. doi: 10.3389/fcvm.2024.1351495
Received: 24 January 2024; Accepted: 1 April 2024;
Published: 11 April 2024.
Edited by:
Panpan Hao, Shandong University, ChinaReviewed by:
Lazaros Lataniotis, Sana Biotechnology, Inc, United StatesAshish Kapoor, University of Texas Health Science Center at Houston, United States
© 2024 Yao, Liu, Gu, Xu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojin Xu bHVuYXIueHVAMTYzLmNvbQ== Xiwen Zhang emhhbmd4aXdlbjMwM0AxNjMuY29t
†These authors have contributed equally to this work