 Aminder A. Singh
Aminder A. Singh Deeti K. Shetty
Deeti K. Shetty Aishwarya G. Jacob
Aishwarya G. Jacob Sanjay Sinha
Sanjay Sinha- Wellcome-MRC Cambridge Stem Cell Institute, Jeffrey Cheah Biomedical Centre, Cambridge, United Kingdom
Thoracic aortic disease (TAD) is often silent until a life-threatening complication occurs. However, genetic information can inform both identification and treatment at an early stage. Indeed, a diagnosis is important for personalised surveillance and intervention plans, as well as cascade screening of family members. Currently, only 20% of heritable TAD patients have a causative mutation identified and, consequently, further advances in genetic coverage are required to define the remaining molecular landscape. The rapid expansion of next generation sequencing technologies is providing a huge resource of genetic data, but a critical issue remains in functionally validating these findings. Induced pluripotent stem cells (iPSCs) are patient-derived, reprogrammed cell lines which allow mechanistic insights, complex modelling of genetic disease and a platform to study aortic genetic variants. This review will address the need for iPSCs as a frontline diagnostic tool to evaluate variants identified by genomic discovery studies and explore their evolving role in biological insight through to drug discovery.
1 Introduction
Aortic wall structure reflects its unique function to provide vascular capacitance for every cardiac cycle and predisposes to thoracic aortic disease (TAD) which includes classical pathologies of aneurysm and dissection. These diseases are the end points of molecular failures within the wall, and occur when wall stress exceeds strength resulting in life threatening complications (1). Acute aortic dissection confers high mortality with an estimated incidence of 6 events per 100,000 population (2). Despite the devastating clinical presentation, clinical outcomes for this cohort have seen little improvement over the last 15 years (3). Diagnosis without cross sectional imaging is challenging and is compounded by the lack of a TAD specific biomarker (4). Importantly, there are no pharmaceutical treatments which prevent aneurysm formation or progression effectively, with current focus on reducing biomechanical loading of the aorta through antihypertensives. Patients require regular surveillance imaging and often surgical intervention. Presently, aortic diameter is the main indication for surgical intervention however an acute event can occur with a normal sized aorta. As such, there is need for additional molecular information that can be used in multimodal risk scoring to predict disease severity and determine the need, and timeframe, for intervention.
Rapid development of next generation sequencing technologies has led to genetic testing being increasingly used for Mendelian disease (5) but it remains difficult to functionally validate the huge wealth of data generated. Challenges in assigning causality to genetic variants and studying TAD include high inter- and intra-genic variability in risk and/or type of first aortic event (6), incomplete penetrance, private mutations, and lack of aortic tissue available for mechanistic study. The overarching rationale for genetic testing is critical because individuals can be identified as at-risk for TAD and associated mortality and morbidity are preventable, including for potentially at-risk family. Despite extensive genomic coverage in patients recruited through large databases, such as The 100,000 Genomes Project, many individuals are undiagnosed and uncertainty around assigning causality to variants remains (7), limiting clinical utility.
Induced pluripotent stem cells (iPSCs) are patient-derived, reprogrammed cell lines which allow mechanistic insights and the ability to create complex models of genetic disease. This review will begin by defining the genetic basis of TAD, key signalling pathways implicated in pathogenesis and discuss unifying theories to aid targeting of therapeutics. Particular focus is given to the latest advances in the use of iPSCs and genetic engineering which can be applied to studying aortic disease variants identified by next generation sequencing (Figure 1). These recent strategies allow for increased mechanistic exploration with a view to expanding personalised management in TAD.
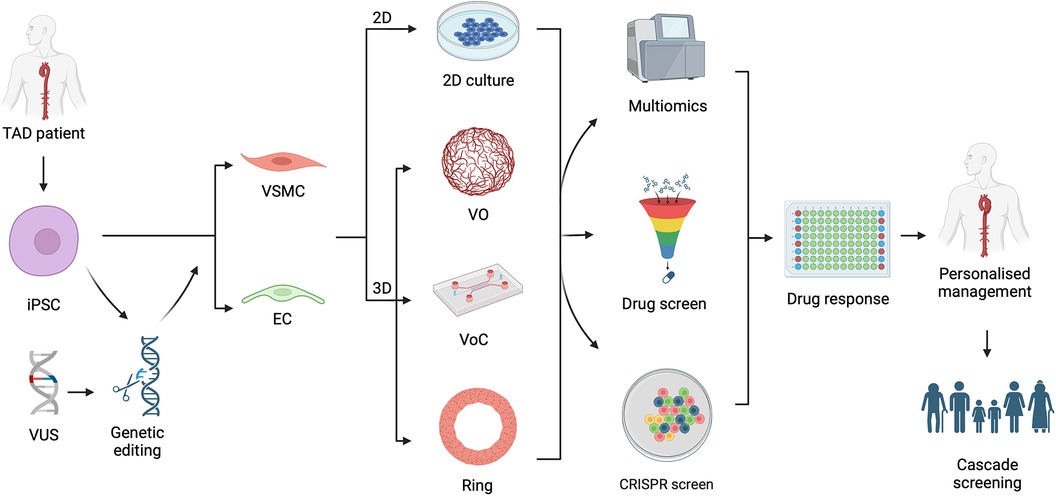
Figure 1. Graphical summary demonstrating the utility of induced pluripotent stem cells (iPSC) in understanding thoracic aortic disease (TAD) variants of unknown significance (VUS). iPSCs can be generated from a TAD patient or an established iPSC line genetically edited to carry a VUS. These can be differentiated in to vascular smooth muscle cell (VSMC) or endothelial cell (EC) in the form of a 2-dimensional (2D) culture or 3-dimensional (3D) culture system including vascular organoids (VO), vessel on a chip (VoC) and vascular ring. These models can proceed to multiomic evaluation, be used for drug or CRISPR screening, and then followed by assessment of drug response in high throughput manner. Such an approach allows for personalised management of the TAD patient and allow determination for need of cascade screening of family members. Figure created with BioRender.com. Publication licence granted by BioRender. Agreement number UH266AGKA0.
2 Vascular abnormalities in thoracic aortic disease
The aortic wall consists of three layers—tunica intima, tunica media and tunica adventitia. The intimal layer, on the luminal surface, is composed of endothelial cells and the basement membrane, separated from the tunica media with an elastic lamina. The tunica media is composed predominantly of concentrically arranged vascular smooth muscle cells (VSMCs), elastin and collagen. The adventitial layer is comprised of fibroblasts, pericytes, immune cells, nerves and lymphatics (8). Key to normal function is the high level of compliance within the aortic wall (9). Increased stiffness results in wall weakness and elevated cardiac afterload resulting in higher cardiovascular events (10) as well as increasing risk for type A aortic dissection when combined with high wall stress (11).
2.1 Vascular smooth muscle cell contractility required for vessel wall homeostasis
VSMCs are the most abundant cell type within the aortic wall with a substantial body of evidence, particularly from human genetic data, implicating them in pathogenesis of TAD (12–14). Similar abnormalities in their function and surrounding extracellular matrix (ECM) are observed in both syndromic and non-syndromic TAD, making them a strong candidate for mechanistic research and therapeutic strategy building. Specifically, loss of VSMC contractility is a common feature observed in many hereditary TADs (15, 16). Mutations in VSMC contractile genes are predicted to decrease the cells ability to generate the force needed to regulate the mechanical state of the wall (1). Altered force generation is one of the causes of TADs (17) with mutations in VSMC contractile protein coding genes ACTA2, MYH11 and MYLK responsible (18–20). Gain of function mutations in PRKG1 result in reduction of VSMC relaxation and predisposes to TAD, further implicating VSMC specific genes in pathogenesis (21). As such, contractility loss serves as a good readout in phenotypic assays (22).
2.2 Maladaptive extracellular matrix remodelling at the core of aortic tissue failure
Most major vessel groups have a characteristic ECM composition based on function and the aorta is typically is composed of elastin, glycoproteins, collagen and proteoglycans (23). The combination of elastic fibres and fibrillar collagens provides mechanical integrity as well as compliance. Indeed, pathogenic variants in ECM related genes such as LOX, encoding for an enzyme which crosslinks collagen and elastin, are causal in heritable TAD (24). There is constant remodelling of ECM in normal physiology through interplay between degrading proteins and their tissue inhibitors, in which VSMCs have a central role through a mechanotransductive feedback loop in response to mechanical load. VSMCs attach to the ECM through focal adhesions and integrins forming the elastin-contractile unit which is involved in force generation and mechanotransduction (25). A more plastic matrix would encourage the VSMCs to secrete fibrillar ECM components, such as collagens and fibrilins, to enable fibre formation. Mechano-adaptive remodelling is a well-orchestrated process entailing transcriptional, translational, and secretory pathway modulation required for normal functioning of the vessel wall (26–30). However, derangement of these processes results in maladaptive remodelling and an abnormal ECM predisposes to TAD. Ultimately, these changes impact cellular processes such as proliferation, cell migration, and contractility. Given this, understanding the VSMC-mediated ECM remodelling and turnover is critical in uncovering of TAD pathogenesis.
VSMC contractile machinery, ECM and TGF-β pathways together are thought to regulate mechanosensing (mechanical stress and strain sensing) by VSMCs and control overall aortic wall health (31). The central hypothesis is that if the sensing of these mechanical cues is abnormal then inappropriate remodelling predisposes to wall failure. Recent mouse studies have highlighted the need for having a mechanoprotective role of YAP and TAZ for homeostatic functioning of aorta whereby their depletion leads to aortic aneurysm (32). Having fewer functioning elastin-contractile units precludes appropriate mechanosensation and application of VSMC contractility, rendering the aortic wall susceptible to damage at high pressures (1, 33). Aside from contractile unit loss, failure of the structure leads to abnormal focal adhesion complexes and inappropriate sensing of the ECM by the VSMC (34–36).
While diseased human and mouse aortic tissue provides an endpoint view of compromised mechanical integrity, there is limited scope to study matrix dynamics in detail. Use of decellularized ECM for label-free quantification of ECM proteins and glycoproteins using MaxQuant mass spectrometry has been used to study iPSC derived VSMCs in arterial diseases (37). Individual proteins such as elastin and collagen deposition have been evaluated using human VSMCs (38, 39). Secretome forms a huge part of VSMC matrix modulatory phenotypic responses and number of studies have explored this using ELISA-based MMP assays and zymography for MMPs and tissue inhibitors of metalloproteinases (40, 41). The potential of quantifying nascent ECM secretion through BONCAT mass spectroscopy has been shown with other disease models but could be also applied to TAD (42). When it comes to studying the mechanics of VSMCs, measuring both the stiffness of cells and its secreted ECM is key, particularly in 3D models. By using tools such as Atomic force microscopy, uniaxial stress and strain measurements, temporal insights to how elasticity of substrates can influence VSMC cell migration and proliferation can be gained (43, 44). Evidence of ECM stiffness and strain influencing the phenotypic switching of VSMCs was obtained by culturing primary VSMCs in vitro (33, 45). As such, TAD patient iPSC VSMCs can also be evaluated for the tendency or potential for phenotypic switching and calcification (46–48).
2.3 Controversial role of TGF-β signalling in aortopathies
In VSMC homeostasis regulation, transforming growth factor-β (TGF-β) signalling has a key role in cellular processes of proliferation, differentiation, and ECM remodelling (6). However, there remains considerable debate around whether TGF-β signalling is implicated in TAD or if it confers a protective effect. TGF-β is a VSMC differentiating factor and increases expression of smooth muscle actin, smooth muscle myosin, calponin and myocardin. Contributions of both canonical and non-canonical signalling are integral to embryonic VSMC lineage specification during development and VSMC cell fate (49).
Impaired TGF-β signalling can lead to a loss of VSMC contractility markers and gain of proliferative markers indicating a synthetic phenotype switch. As a result, degradation of ECM and abnormal aortic wall remodelling is seen (50). In Marfan Syndrome (MFS), increased activation of TGF-β is related in part to abnormal Fibrillin-1, which normally sequesters latent TGF-β within the ECM (51). In MFS mouse models, increased TGF-β signalling has been demonstrated (52) with a TGF-β neutralising antibody or an angiotensin II type 1 receptor blocker reversing the disease phenotype (53). However, the exact role of TGF-β is not fully understood with several studies demonstrating a protective role (54–56). Use of TGF-β neutralising antibody conversely resulted in a severe manifestation of aortic disease (57) although the timing of this appears important with a protective benefit demonstrated at a later stage (54). Loeys-Dietz Syndrome (LDS) is defined by mutations in the TGF-β receptor or SMAD2/3 where patients exhibit aortopathy and cardiovascular events are the leading cause of death. In LDS, despite predominantly loss of function mutations in the TGF-β pathway, a paradoxical increase in phosphorylated Smad2/Smad3 complex has been shown in the aortic wall (58, 59) suggesting that even with an abnormal TGF-β receptor, overactivity of TGF-β is implicated in disease. Subsequent studies have identified lineage-specific TGF-β events in distinct VSMC subsets as an explanation for this paradox (60, 61). Nevertheless, it appears the exact role of TGF-β in TAD pathogenesis remains controversial, compounded by its temporal role in multiple cellular processes and crosstalk with other implicated signalling pathways. As such, this is an area where future scientific endeavours should continue to focus.
2.4 Intersection of “unifying theories” lead to convergence of key druggable candidates from shared pathways
Given the pathological and morphological similarities between many distinct genetic and polygenic types of TAD, is there a unifying theory for a “final common pathway” leading to aneurysm or dissection? This argues that regardless of the underlying signalling pathway that is perturbed, the changes that result are common across TAD. This includes phenotypic switching of a subset of VSMCs to form fibroblast-like, macrophage-like or adipocyte-like cells, marked by progressive loss in contractility, increased apoptosis, abnormal ECM remodelling alongside inflammation and endothelial cell related oxidative stress (62). These changes are aligned with dysfunctional VSMC mechanosensing which further exacerbates loss of ECM homeostasis through abnormal remodelling (63). Such a theory is attractive as it suggests that regardless of the initiator, therapeutic development could focus on the shared pathways.
2.5 Other cell types important in vessel wall functioning and integrity
Although VSMCs appear the dominant cell type in disease, there is evidence of the role of other cell types in the aortic wall. Endothelial cells sense shear stress and crosstalk with VSMCs (64) and endothelial dysfunction has been demonstrated to contribute to aortic wall pathology (13, 65, 66). Endothelial cell function is impacted by dysfunctional ECM through altered distribution of junctional proteins, like occludin and claudins, which has consequence on blood vessel integrity (67). The adventitial layer has a role in remodelling the aortic wall in response to mechanical stress (68). In such conditions, fibroblasts undergo phenotype switching to myofibroblasts which deposit collagen and invoke an inflammatory response (69). Abnormal activation of this pathway contributes to abnormal ECM remodelling and can lead to reduced integrity. Endothelial cells, and particularly adventitial cells, are relatively understudied in comparison to VSMCs in TAD pathogenesis and an approach which encompasses all these cell types is preferable.
2.6 Regional aortic heterogeneity
The thoracic aorta can be classified based on embryological origin with the aortic root derived from lateral plate mesoderm, the ascending and aortic arch from neural crest and the descending and abdominal aorta from paraxial mesoderm (70). Abdominal aortic aneurysms (AAA) are associated with hypertension, smoking, and atherosclerosis and are rarely seen as part of a multisystem connective tissue disorder. Aneurysm in the abdominal segment is more common than in the thoracic but the underlying aetiology of these are divergent despite sharing some common abnormalities in VSMC apoptosis, ECM degradation and immune infiltration. Evolving evidence from single cell RNA sequencing demonstrates both molecular similarities and differences between abdominal and thoracic aortic aneurysm (71). Despite this, genetic abnormalities play a significantly less important role in aneurysmal degeneration of the abdominal vs. the thoracic segment, in particular there is no clear inheritance pattern in AAA. Recent endeavours have focussed on collating AAA implicated genes, such as in the AAAKB, to aid genetic study (72). No published iPSC models focus on AAA but VSMCs of paraxial mesoderm lineage (73) could be generated with view to establishing it. As such, this review will retain focus on TAD.
Regional heterogeneity within the thoracic aorta is thought to be due to differential lineage specific response due to diverse embryological origin on the constituent cells. A recent study of single cell sequencing of lineage traced secondary heart field (SHF) derived VSMCs from the aortic root of Marfan mice (C1039G) revealed a distinct transcriptomic profile from the neural crest derived aortic SMCs and the activation of specific pathways such as integrin α5 signalling (74). That combined with the preponderance of MFS aneurysms in the aortic root, imply that secondary heart field derived VSMCs are a critical cell type to be examined. Gong et al. (61) demonstrated abnormal SMC contractility and ECM deposition in lateral mesoderm derived VSMCs suggesting a lineage specific response to a SMAD3 variant. This differential response can be evaluated using specific iPSC differentiation protocols producing VSMCs from lateral mesoderm, neural crest, and paraxial mesoderm (73, 75, 76). Such an approach can provide mechanistic insight into disease distribution in the aorta given that LDS has higher propensity of aortic root aneurysm, where there is predominantly lateral mesoderm derived VSMCs (60), and can aid clinicians in assigning region-based size thresholds for intervention.
3 Disease modelling of TAD
The difficulty in obtaining early-stage tissues from patients with TAD and the challenges of clinical trials mean that we need to use suitable model systems to understand pathogenicity and identify and test novel therapies. The complexity of the aortic environment, with multiple cell types featuring complex inter- and intra-cellular signalling, extensive vessel wall and vascular niche remodelling along with varying haemodynamic loads in a three-dimensional space makes this a challenging endeavour. Nevertheless, we would do well to remember the observation by George Box that “All models are wrong but some are useful” (77). Consequently, we will briefly review common in vivo models of TAD before describing iPSC based in vitro systems in some depth and discuss their advantages and disadvantages.
3.1 In vivo models offer a window into studying spatial interactions but are restrictive in mimicking human disease complexity
While in vivo modelling using genetically engineered rodents offers an important solution for the spatial and systemic complexity of vascular diseases, they remain somewhat limited in their scope. The pathways affected in these models are not necessarily conserved with human disease. In monogenic aneurysmal disorders such as MFS and LDS, some mutant mouse models have captured aneurysm formation and dissections and have been useful in delineating the impact of these mutations in disease progression. However, these models are low throughput and capture only a small proportion of the mutational landscape seen in humans. For example, MFS has been reported to be associated with >1,500 mutations in FBN1 featuring missense mutations and splicing errors that lead to truncations (78). The two most widely used mouse models of MFS are the C1039G (79) and the MgR (80) which contain a missense and hypomorphic mutation in the FBN1 gene respectively. Further, these inbred models do not capture the heterogeneity arising from the genetic background of the affected human population and are not fully effective at predicting drug responses, a classic example being that of losartan that proved remarkably effective in vivo in the C1039G/+ model MFS (53) but did not perform as well in clinical trials (81).
Therefore, to accurately model human genetic diseases it appears that a humanised model may have some advantages based around the conserved genetic background. In vitro models also have a wider scope than in vivo models for high throughput studies and allow detailed molecular characterisation, phenotype assessment, mechanistic exploration, and drug testing. In vitro models using primary vascular cells—smooth muscle or pericyte and endothelial—can be highly technically challenging. For example, VSMCs are phenotypically plastic and tend to lose their contractile properties and alter their molecular profiles once dissociated from tissue and cultured. Moreover, primary cultures display considerable variability making reproducibility a major challenge.
3.2 iPSCs based modelling addresses genetic diversity of the mutation landscape
The advent of iPSCs has been a game changer for disease modelling using direct patient-derived culture systems. They provide an ethical source of cells and disease models compared to in vivo systems, and even embryonic stem cell-based platforms. Typically, skin fibroblasts or peripheral blood mononuclear cells are collected from the patient and re-programmed directly to produce pluripotent stem cells that retain the genetics of the somatic cells they were derived from (82–86). iPSCs can then be efficiently differentiated into a variety of cell types, and in the context of disease these iPSC-derived cells can be used as effective patient-specific models (Table 1). This makes them an incredibly versatile and high-throughput platform for modelling disease states and capturing the heterogeneity between patients. Attempts to use iPSCs to model vascular cell types began in the late 2000s (93, 94) resulting in a mix of several cells including VSMCs and endothelial cells (ECs), in a form of co-culture. Since then, a large number of protocols have been described using highly defined media with small molecules or cytokine cocktails to drive differentiation [several of which are well summarised in these reviews (95–97)] that claim to have successfully produced VSMCs or ECs of high purities, organ-specific, developmentally pertinent lineages (73, 76) and fine-tuned phenotypic states including the contractile one (37, 98, 99).
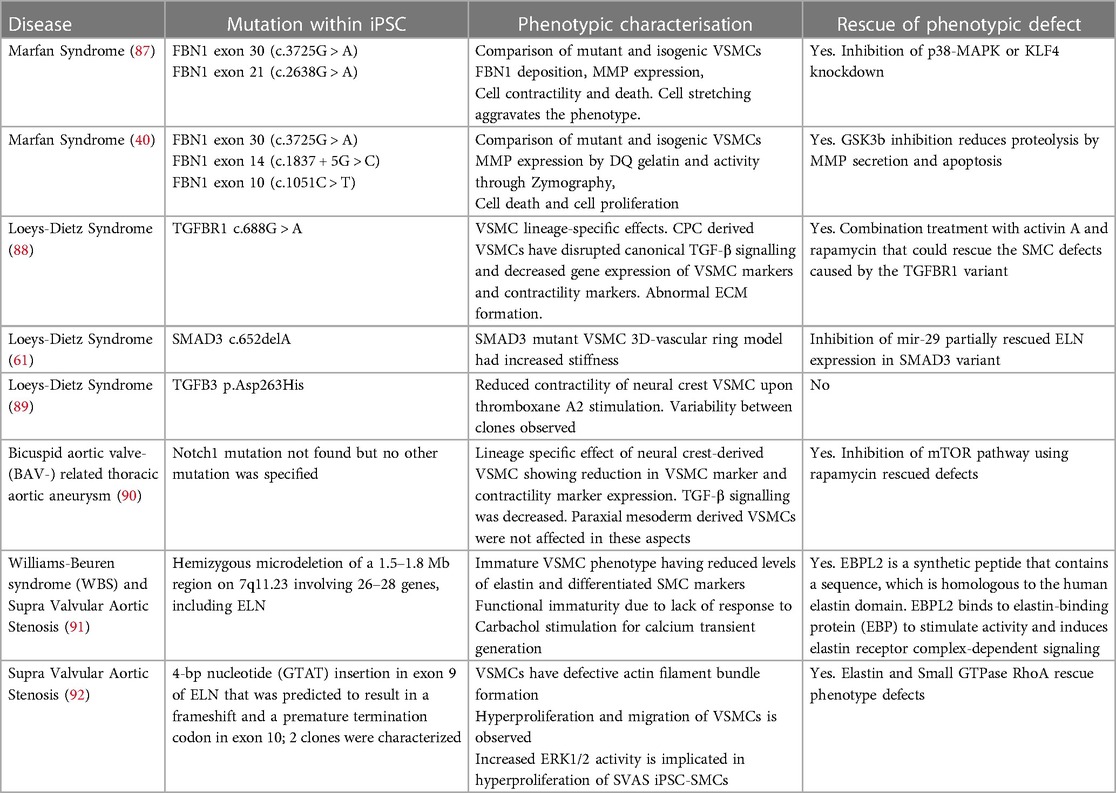
Table 1. iPSC models of thoracic aortic disease that have been phenotypically characterized and reported.
3.3 Single cell transcriptomic studies have hailed the in-depth study and comparison of in vitro iPSC-derived cell types and in vivo counterparts
The rapid expansion of single cell transcriptomics has facilitated nuanced descriptions of individual vascular cells in different tissues and their developmental trajectories which, in turn, will pave the way for further advancements toward highly precise and effective differentiation protocols to achieve the exact cell state needed to model a specific condition. The ability of these protocols to produce vascular bed or niche-specific cells is important because some vascular diseases often show region-specific susceptibility to aneurysm formation e.g., aortic root aneurysms in LDS. Our own studies on MFS have provided huge insights into how patient iPSC-derived VSMCs are capable of clearly reflecting disease severity in vitro while uncovering the differential response to drugs between patients (22, 40) and hence are an invaluable platform for the development of personalised, tailored therapeutics.
Despite these advances in iPSC-based modelling of patient-specific disease, there are several problems associated with these systems. Personal insights on technical aspects and limitations of iPSC models of aortic disease have been extensively discussed previously (22). Heterogeneity can be a major issue when trying to decipher pathways commonly perturbed in patients. While this can be a strength in modelling varying genetic backgrounds, particularly for disease susceptibility and drug responses, it can pose several technical challenges that might undermine the findings from such models. Another problem is modelling the correct cell type i.e., the appropriate source—vascular bed, lineage and/or regional specification on the vessel with respect to hydrodynamic forces. Directed differentiation protocols now allow for specification of several of these features in VSMCs and ECs. The incorporation of specific chemical cues, mechanical strain and flow-induced shear stress have helped to further fine-tune cell phenotypic state e.g., contractile or synthetic or phenotypically switched. Work to increase the maturity of iPSC derived cells is ongoing with modification of differentiation protocols based on sequencing data. However, these disadvantages are far outweighed by the benefits of iPSC-based disease modelling and therapeutic targeting.
4 Genetic modulation for mechanistic insight
Utilising tools to manipulate cellular behaviour has advanced mechanistic understanding of disease. Particularly, DNA-level modulation offers stable and inheritable alterations, a vital tool in foundational research and therapeutic development, and has been useful in investigating cardiovascular disease. Progress in genome editing has augmented our toolkit, providing the ability to enact DNA modifications through numerous strategies across diverse frameworks, enabling either site-specific or random edits, and classifications based on modification nature (e.g., knock-ins, knock-outs) or timeliness (e.g., temporal, constitutive). Site specific edits allow manipulation of genetic information at endogenous genes despite presenting considerable challenges, such as hard-to-identify off-target effects. Moreover, technologies such as the Tet ON/Tet OFF systems allow temporal control over genetic modifications and their subsequent cellular impacts, thus widening the scope and potential applications of genome editing technologies (100, 101).
4.1 CRISPR systems for gene editing
The conventional CRISPR approach hinges on the Cas protein inducing a double-stranded break (DSB) in the DNA, where the guide RNA finds complementarity in the genome. Subsequent repair of this intentional DSB is executed via the cell's intrinsic replication machinery, chiefly through two primary pathways: Non-Homologous End Joining (NHEJ) and Homology-Directed Repair (HDR) (102, 103). The evolution of CRISPR-Cas technology also brought the emergence of diverse Cas proteins, base editors, and prime editors which contribute to enhanced specificity and versatility of genome editing techniques. Engineered variants of Cas proteins have been pivotal in reducing off-target editing (104, 105) and enabling access to a broader genomic region, thereby expanding the scope of potentially editable regions (106). Moreover, the development of base editors, which use catalytically repressed or inactive Cas9 fused to a DNA deaminase enzyme, provide a means to edit single base pairs without generating a double-stranded break (DSB) (107). Using base editing, a recent study investigated the causal variants implicated in blood pressure through VSMCs (108).
A more recent approach, prime editing, utilizes a Cas9 nickase fused to a reverse transcriptase, alongside pegRNA which is produced through the fusion of the repair template with the guide RNA (109). pegRNA therefore serves as a template for the reverse transcriptase to synthesize the edited strand, which then integrates directly into the genomic DNA, while guiding the Cas9 to the target site, collectively minimizing indel formation and off-target effects. While these newer technologies hold their own limitations, such as the inability to generate transversion mutations and potential inadvertent RNA edits with base editing, as well as the restriction on sizable insertions with prime editing, continuous improvements in methodologies have expanded the efficiency and toolkit of prime editing, propelling forward the evolution of CRISPR technology.
Modulating cellular behaviour extends beyond manipulations at the DNA level, encompassing various regulatory layers, including the epigenome and RNA. The CRISPR domain has been adeptly utilized for such purposes, especially through innovative strategies involving engineered proteins. For example, by coupling CRISPR with a catalytically deactivated Cas protein and subsequently fusing it with specific domain allows modulating epigenetic behaviour (110), adding a nuanced level of genetic control without altering the underlying DNA sequence. Additionally, different Cas proteins can be employed to orchestrate edits at the RNA level, providing a mechanism to regulate gene expression post-transcriptionally (111).
4.2 Genetic modifications in iPSCs for disease modelling
iPSCs themselves are amenable to genetic modulations including via CRISPR-Cas9, making them a powerful tool to characterise the mechanisms underlying disease aetiology. For instance, “CRISPR-correction” of patient iPSCs at mutated loci into wildtype sequences, allows for the generation of isogenic iPSC lines that preserve the genetic background of the patient while establishing causality of the mutation in question (87). What this also means is that patient-specific differences in disease susceptibility due to genetic background, for example the effect of accompanying SNPs, can be modelled in the absence of the confounding “main” mutation. Several studies have used iPSCs and derived vascular cells—VSMCs, pericytes and ECs—to model a variety of vascular diseases including, pulmonary arterial hypertension (112–119), Hutchison-Gilford progeria (120–124), atherosclerosis (47, 125, 126), neurodegenerative cerebro-vascular conditions like Moyamoya disease (127–131), CADASIL (132–135), diabetes and its associated conditions (136–138).
Coupling CRISPR gene editing technology with iPSCs has allowed for significant enhancement of aortic disease models such as syndromic aortopathies (22, 40, 87, 88) and aortopathy genetic variants (61, 139). Correction of a FBN1 mutation demonstrated rescue of in vitro MFS disease phenotype (87). iPSCs can determine causality of a disease variant of interest (61, 140) and provide information for precision medicine and need for cascade screening. Further, researchers have generated reporter lines of ACTA2 and MYH11, which are used in improving stem cell derived VSMC differentiations (99).
Despite challenges, each advancement in the CRISPR technology has gradually improved the specificity and applicability of these vital tools, enabling unprecedented insights into genetic mechanisms. This has facilitated innovative developments across diverse biological domains, with potential applications in many more non-syndromic aortic diseases.
5 Genetic abnormalities in thoracic aortic disease
In heritable TAD, irrespective of penetrance, 25% of patients have at least one first degree member affected (141, 142). Patients with a family history of TAD demonstrate more rapid aneurysmal growth than sporadic TAD or MFS aneurysm (31). Interestingly, there is significant variation in aortic event risk, even within functionally related genes, such as those regulating VSMC contraction (6). Such genetic information has been used to predict those at risk of severe, early onset TAD and those with late onset low penetrant TAD (6, 143).
Initially highly penetrant monogenic variants were identified with a candidate gene approach and linkage analysis. More recently, next generation sequencing has accelerated novel variant identification but poses some challenges. Firstly, it can be unclear whether the variant is causal, particularly if it is within a gene not previously associated with the condition. Judging causality of a genetic variant based on its presence within a known disease related gene is an inherently biased approach and could hinder discovery of new pathways. Identifying variants of unknown significance (VUS) poses a challenge to clinicians (144) and can make genetic counselling challenging and raise uncertainty in need for cascade screening. Additionally, penetrance classification and determining pathogenesis are critical in design of pharmacological therapy or determining prophylactic treatment. Despite this, the correlation between genotype and phenotype in most TAD cases remains poor and additional techniques to predict clinical course in patients are urgently required.
5.1 Novel variants with lower penetrance identified by next generation sequencing can be assessed for causality using iPSC-based disease modelling
iPSCs were first used to model genetic variants which result in substantial modification of a protein product, but this technology can be used in more advanced models where complex traits are investigated. Many genetic variants identified are within intronic or intergenic regions and can regulate gene function (145). Variable expressivity of monogenic disorders may be due to modulation by additional genetic variants and incomplete penetrance. A genome-wide association study (GWAS) of TAD in the Million Veteran Program revealed 21 risk loci with suggestion that TAD is a distinct vascular disease with complex traits. Importantly, this work argues that TAD is not inherited exclusively through protein altering variants of large effect size but are due to a combination of multiple at-risk genetic variants (146). Similarly, GWAS of the UK Biobank revealed 14 overlapping loci in ascending and descending thoracic aortic aneurysm (147), adding to findings from earlier GWAS (148–150). Given these interesting findings, the need for functional validation and exploration of biology underpinning disease is clear. Diseases which encompass a combination of numerous low effect risk loci, often identified by GWAS, pose a challenge in an iPSC model. Each individual variant may be too subtle to demonstrate a phenotype in vitro, which is in part due to gene-gene interactions not present outside of the patient's genetic context (151). However, patient derived iPSCs, which carry all potential risk variants, can be used to demonstrate which regions are more relevant in phenotypic manifestation. Multiple patient derived iPSCs can used in a pooled culture creating a “cell village” where the impact of variation can be studied at a population scale, providing in depth information of the effect of expression quantitative trait loci (eQLT) (152). The impact of eQLT on phenotypic manifestation of a genetic variant can be addressed using patient specific iPSCs and then CRISPR correcting these regions to create isogenic controls (153). As such, risk loci can be evaluated to assess their functional impact on common genetic variants which in turn can feed into individualised risk scoring.
5.2 Use of iPSCs for determining the pathogenicity of novel genetic variants
Heritable TAD is non syndromic and typically associated with a single mutation in a gene related to ECM, TGF-β signalling or VSMC contraction (154). Over time an increasing number of variants are being reported and genes classified as pathogenic (155). Substantial evidence exists for ACTA2, MYH11, MYLK, LOX and PRKG1 but other genes are implicated with varying levels of certainty (155). Most of the VUS are not causal but may be low penetrant risk variants contributing to TAD (143). Even in large, national whole genome sequencing studies the diagnosis rate in heritable TAD remains low (7). There is consequently great potential for using iPSC models from heritable TAD patients or by generating CRISPR knock ins in the hope of determining causality and providing clinicians with more biological information to inform future management plans.
Mutations in FBN1 are associated with MFS which is the commonest single gene aortopathy. Currently there are over 3,000 FBN1 pathogenic variants identified with most unique to a family (156). Genetic studies have demonstrated that cardiovascular risk is determined by the genetic variant possessed. Those with haploinsufficiency FBN1 variants have increased risk of major cardiovascular events than dominant negative mutations (157). Within the dominant negative group, variants which effect or create Cysteine residues and in frame deletions in exons 25–36 and 43–49 have over a 6-fold increase aortic event risk (156). Despite this, delineating the genotype-phenotype relationship, particularly in MFS, remains a major challenge. Genetic background is important as presence of another TAD associated variant can predispose to a more severe aortic disease phenotype (158). Genetic background can also partly explain the difference in phenotype severity within a family who carry the same FBN1 mutation (22) but there are other factors at play which remain uncovered. An iPSC model could be used to provide an in vitro prediction of severity once appropriate parameters of assays have been defined. Such an approach would be particularly useful in a young patient with a novel FBN1 variant where the impact of intensive lifelong surveillance is significant. Similarly, each LDS subtype carries different severity and therefore the relationship between genotype and phenotype is important. iPSC models of LDS have demonstrated abnormal contraction and enhanced proliferation of VSMCs (88) and can be utilised to explore genotype-phenotype correlation. As new variants are discovered in LDS related genes, there is an emerging need for gene-based classification and personalised risk scoring for each patient (159).
6 Complex three-dimensional disease models
The vasculature is a complex organ that involves crosstalk between the cells of the various layers and their extra-cellular micro-environment. In 2D systems, even in co-cultures of multiple cell types, the organisation of a vessel wall or physiological cell-cell contact cannot be recapitulated. In 2D systems, cells are cultured on hard plastic surfaces whose stiffness far surpasses that of a physiological matrix which can promote a more synthetic VSMC phenotype. Further, iPSC derived VSMCs are not as mature as those derived from tissue which is partly due to a lack of pulsatile flow and mechanical force in a monolayer culture. Hence, it is necessary to model the three-dimensional, multicellular vessel wall anatomy using cells embedded in an appropriate extra-cellular scaffold to capture the full phenotypic manifestation of a healthy or a diseased vessel. When comparing uniaxial stretch in 2D culture to a 3D construct, there is significantly higher increase in VSMC specific markers ACTA2 and calponin (160) suggesting that a 3D environment allows for more robust cell-cell and cell-matrix interactions. For this purpose, several 3D models (161) have been in development over the last several years that incorporate multiple cell types in a bio-degradable matrix. Some key formats include vascular organoids (VOs) (138), vessel on a chip microfluidic device (VoC) and bioink-based extrusion or bioprinting. Use of these 3D models is on the rise in conjunction with iPSC-derived vascular cells to understand how disease affects vasculature including formation or structure or integrity.
6.1 Vascular organoids mimic tissue niches with functional cell-cell and cell-ECM interactions
VOs can be made as simple, self-assembling structures with cells cultured in a hanging drop or in low adherence culture substrata. These are often spherical and in co-cultures of VSMCs and ECs, there is a level of self-organisation between the cell types. Co-culturing VSMCs and endothelial cells has been shown to improve SMC differentiation (162). However, these models tend to have high variability. To obtain a more controlled structure, scaffolds using extra-cellular matrix like collagen, Matrigel or other biomaterials are used and depending on the design, these can result in self-assembly of intricate branched vessel networks with a perfusable lumen lined by ECs and VSMCs or mural cells. VOs can be transplanted and engraft with host tissue to form vascular trees (163). A recent study used human iPSC-derived VOs to study basement membrane thickening seen in the skin microvasculature of diabetic patients which demonstrated vasculopathy. With exposure to high-glucose media, the VOs showed increased collagen IV production and captured the basement membrane phenotype (137). However, similar treatments of 2D iPSC-derived ECs did not show a similar result suggesting the usefulness of 3D models in capturing disease specific phenotypes.
6.2 Vessel on a chip enables shear stress, flow and associated mechanotransduction responses
VoCs on the other hand are specifically designed microfluidic devices that can then be lined with ECs, VSMCs and other cells to induce network and vessel formation (164). This model has been well characterised recently with robust vascular network formation comparing primary and iPSC-derived vascular cells, namely ECs, VSMCs and mural cells. The group also established the functionality of these vascular networks—agonist mediated stimulation of VSMCs—and crosstalk between VSMCs and ECs mimicking the environment in a vessel wall (165, 166) validating the usefulness of this system to model vascular diseases where such architecture and cell-cell interactions might be disrupted. Adding flow into a 3D culture system allows endothelial cells to detect shear stress and with this cue they can modulate SMC differentiation through signalling pathways such as nitric oxide (167).
Bio-printing uses cells mixed into collagen or other matrix materials as a bio-ink to be extruded into moulds where vessel formation would be observed (168–170). Numerous assays can be performed including transcriptomic and proteomic analysis, alongside classical ELISA, immunostaining, and Western blotting (164). Multiple chips can be connected in parallel to create complex models more reminiscent of human physiology (171). iPSC derived VSMCs from a patient with Hutchinson-Gilford Progeria syndrome in a chip system showed elevated DNA damage and inflammation, exacerbated by mechanical strain (124). An “aorta on a chip” approach in congenital aortic valve disease demonstrated impaired mitochondrial function due to suppression of NOTCH1, reducing VSMC contractility which was partially rescued with drug treatment (172). Liu et al. (173) developed a VoC model with VSMCs of all three aortic lineages in series and determined the differential response to Ciprofloxacin and identified the PI3K-Akt signalling pathway as key in response to stretch (173). It should be noted that these “aorta on a chip” models do not reflect the structure, scale and stress in the aortic wall and so their predictive power over simpler systems is debatable. Nevertheless, VoCs will continue to evolve, and are increasingly useful for their strength of potential downstream processing of cells for further investigation of mechanistic details especially with a systemic-like drug dosage procedure.
6.3 Larger vessel-like structures for assessment of structural integrity and tissue failure
Another interesting approach is to use 3D self-assembling tissue engineered vascular rings to provide a readout of aortic wall integrity. Dash et al. (174) fabricated vascular rings using iPSC derived VSMCs and were able to model supravalvular aortic stenosis with uniaxial mechanical testing. This approach is advantageous as it is relatively quick to fabricate the rings once VSMCs have been differentiated. Vascular rings can assess the effect of both mutant VSMCs and ECs in combination, or in isolation, and determine the impact on a clinically relevant readout of burst pressure. However, mechanical loading and integrity in the ring are dependent on the homogenous compaction of cells and deposition of collagen which can frequently be non-uniform leading to point weakness of the ring due to a technical issue (Sinha lab, unpublished). An engineered vascular tissue approach using murine VSMCs to study ECM remodelling allowed for tensile strength testing and assessment of responsive to contraction agonists (175). Here, both protein and proteoglycan accumulation changes over time with vascular calcification changes were demonstrated. While this model is still under development to be used for tensile strength testing of human iPSC VSMCs and its derivative ECM, it does provide a good basis for establishing the same. Use of TAD patient-derived iPSCs VSMCs or fibroblasts can help massively to study transient as well as long-term changes in ECM using 2D and 3D models. Recently, a tissue engineered blood vessel model of Hutchinson-Gilford Progeria Syndrome containing patient iPSC-derived VSMCs and ECs, was shown to serve as a useful drug testing platform where key disease-specific phenotypes such as vascular calcification, VSMC loss and extracellular matrix deposition were clearly captured (120–122).
7 Translational potential of iPSCs
Crucially, no drug treatment can halt aneurysmal degeneration of the aorta hence there is urgent need for new therapeutics. Promising animal data for Losartan, an angiotensin receptor blocker, in MFS were not replicated in human trials (176, 177). The AIMS trial with Irbesartan was successful but only showed a very small benefit (0.22 mm/year) in slowing of aortic wall aneurysmal dilatation (178). This was certainly less than seen in the dramatic mouse studies and may reflect differences between mouse and human disease as well as heterogeneity of human populations.
7.1 Use of functional genomics to model disease associated with predicted risk loci and severity in iPSC-based screens
Over the years, a multitude of screening methods have emerged as methodologies in functional genomics to offer profound insights into gene functionality and regulatory networks, through both loss-of-function and gain-of-function screens. These range from CRISPR based methods to RNA by employing siRNAs or shRNAs to post-transcriptionally mitigate gene expression. CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) screens, characterized by systematic overexpression and repression of target genes respectively, offer a platform for identifying genes fundamental to varied cellular activities, such as proliferation and drug resistance (179, 180). Moreover, coupling CRISPR screening with single-cell RNA sequencing delves into the cellular transcriptional outcomes consequent to genomic perturbations, providing an insight into cellular heterogeneity and the varying strategies cells deploy to manage therapeutic interventions (181). These methods have been instrumental in delineating genetic networks, understanding the heterogeneity in cellular responses to genetic perturbations, and unmasking the differential susceptibilities among individual cells within a population. Functional genomics also allows for modelling stroke and other cerebral small vessel diseases where predicted risk loci are validated using iPSC-derived vascular smooth muscle cells and testing for classes of drugs such as HDAC inhibitors and MMP inhibitors (182–184). Recent studies have highlighted the ability to employ iPSCs-derived cells, genomics, and machine learning-based analysis for predicting risk of occurrences of conditions like arrhythmia and heart failure susceptibility in cardiomyocytes (185, 186). Risk loci of coronary artery disease (CAD) (125) and schizophrenia severity (187) were also determined using iPSC-derived VSMCs and neurons respectively. In a recent move, single cell multi-omics that combines transcriptomic and epigenetic data has been used to determine the genetic determinants of arrhythmia (188). Large patient sample collection biobanks are being built across various countries thereby providing ample opportunity to apply functional genomics and build accurate severity prediction models employing the latest tools mentioned above.
7.2 Phenotypic screening and drug discovery and in iPSC models offers potential for disease severity prediction
iPSCs can be used to identify deregulated signalling pathways in vitro and assess response to compounds hypothesised to improve relevant readouts (113). As such, they serve as excellent drug testing platforms for development of disease-specific therapeutics. For example, pharmacological inhibition of PDGF signalling in iPSC-derived cardiomyocytes in LMNA-related dilated cardiomyopathy, ameliorated the arrhythmic phenotypes (189). Further, even disease severity modelling and prediction both have been successful in (a) Brugada syndrome where cardiomyocytes are affected by arrhythmogenesis of different severity (190) (b) Marfan syndrome where proteolytic degradation by vascular smooth muscle cells was used to scored disease severity and response to drugs inhibiting Gsk3β activity to restore proliferation, curtail apoptosis and MMP secretion (40) and (c) Alport syndrome where kidney organoids modelled severity based on extent of accumulation of Collagens and a marked reduction in the same was demonstrated by the use of chemical chaperones (191). Atherosclerotic plaque stability was demonstrated by KLF4 attenuation by ANGPTL4 treatment of iPSC-derived vascular smooth muscle cells and in mouse models (192). Currently, with very large drug and compound libraries being readily available through pharmaceutical companies, we are only limited by not having widespread use of robotic drug screening platforms and automated phenotype assessment modules (whether imaging or spectrophotometry based) to identify and validate disease modifying therapies.
7.3 Drug repurposing and use of omics approaches shorten therapeutics identification time and enhance efficiency
Current technologies of phenotypic drug screening have come far from relying on low throughput 2D cell culture assays along with time-consuming analysis, to high throughput or high content screening assisted by automated analysis (193). In high throughput drug screening, iPSCs can be used to find novel treatments, repurpose drugs or to predict effect (194). Pre-existing drugs for different pathologies are being repurposed for cardiovascular disease based on the phenotypic screening which assays for reducing disease-modifying effects. This kind of approach significantly reduces costs and time incurred in bringing a drug from benchside to clearing clinical trials. Re-engineering existing drugs to enhance efficacy using knowledge of medicinal chemistry, rational drug design to optimize design, delivery and pharmacokinetics in animal models is being done. Such re-engineered drugs are being screened and validated with iPSC models (195, 196). Going an extra mile to determine the complete extent of impact a given drug on modifying global gene expression in a diseased cell has been done in the latest studies by employing single cell transcriptomic profiling of drug response singularly or in combinations using Combi-seq (197). With such advancement we are no longer dependant on gene expression of 1–2 downstream targets to know the efficacy of drug treatment, rather an unbiased and wholistic assessment of direct and indirect target gene changes can be done.
7.4 “Clinical trials in a dish” and precision medicine
Drug screening with iPSC-derived cells can enable a precision medicine approach with individualised therapeutic plans based on response in iPSC models and with the knowledge about patient's lifestyle, genetic background, and environment (198). This platform can identify non-responders to drug therapy and in parallel be used to determine the molecular basis for this. Given that iPSCs can identify drug targets, predict response levels, screen novel compounds, and repurpose licensed therapeutics, they provide an excellent resource to bridge the gap between preclinical investigation and clinical trials. “Clinical trials in a dish” can fulfil the requirement of testing various drug therapy parameters in a high throughput fashion. Identifying those at higher risk of experiencing drug related complications can uncover the mechanisms leading to this and tailor therapy to mitigate the risk, such as reduced dosing regimens (199). Another important consideration is the vast resource and human impact of participating in a clinical trial. Using iPSC based “clinical trials in a dish” would complement animal studies and provide important biological information which could confirm the need for, or advise against, human trials. “Clinical trials in a dish” using iPSCs have been used for different cardiovascular cells such as endothelial cell function and crosstalk with cardiomyocytes improvement by Lovastatin in LMNA cardiomyopathy (200). Two hERG-blocking drugs, Dofetilide and Moxifloxacin, were also tested on iPSC derived cardiomyocytes for QT prolongation (201). With continued improvement in iPSC differentiations, assay development and 3D modelling, the utility of this approach will only increase.
7.5 Cell therapy in aortic disease
A stem cell therapy-based approach has been on keen interest within the cardiovascular field. In aortic context, work has focussed predominantly on AAA with use of mesenchymal stem cells (MSCs) (202), umbilical cord stem cells (203), and iPSC derived smooth muscle cell progenitors (IPSC-SMP) (204) with delivery methods including intravenous injection, direct injection and through collagen scaffolds.
Perivascular delivery of iPSC-SMP, integrated into a collagen scaffold, demonstrated feasibility, VSMC retention and reduced macrophage invasion in a murine model of AAA (204). However, in contrast to the primary VSMC collagen scaffolds, iPSC-SMP scaffolds did not result in reduced aneurysmal expansion. This could in part be due to using smooth muscle cell progenitors as the chosen cell type for delivery instead of more mature VSMCs which would be less likely to dedifferentiate and phenotype switch. An MSC intravenous infusion based clinical trial in patients with small AAA is underway with results awaited (205).
Cell based therapies for aortic disease have shown promise in animal models, but selection and optimisation of the delivered cell type is key. Further work is needed to elucidate whether injection or scaffold-based approaches are more efficacious.
8 Conclusion
Coupling the wealth of data from genomic discovery studies with functional validation tools such as iPSCs is key in uncovering further genetic landscape in TAD to aid diagnosis, personalised management and rationalise cascade screening. The versatility of iPSCs and the capability for genetic editing allows for a powerful humanised model for mechanistic insight and risk determination alongside evolving roles in drug discovery and validation. The increasing accuracy of genetic editing and advancement in 3D aortic disease modelling paves the way for patient and mutation specific management approaches and an exciting era of precision medicine in aortic and other cardiovascular diseases.
Author contributions
AS: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. DS: Writing – original draft, Writing – review & editing. AJ: Writing – original draft. SB: Writing – original draft, Writing – review & editing. SS: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by a BHF Senior Fellowship FS/18/46/33663 (SS and SB). SS was also supported by a BHF Programme Grant (RG/17/5/32936) and the British Heart Foundation Centre for Cardiovascular Research Excellence (RE/18/1/34212). AS was supported by the Wellcome Trust (227508/Z/23/Z). DS was supported by Cambridge Cardiovascular Research Bridge funding. AJ was supported by the Medical Research Council (G118531).
Acknowledgments
We also acknowledge core support from the Wellcome Trust and MRC to the Wellcome Trust—Medical Research Council Cambridge Stem Cell Institute. This research was funded in whole, or in part, by the Wellcome Trust [203151/Z/16/Z, 203151/A/16/Z] and the UKRI Medical Research Council [MC_PC_17230]. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology. Circ Res. (2015) 116(8):1448–61. doi: 10.1161/CIRCRESAHA.114.304936
2. Howard DP, Banerjee A, Fairhead JF, Perkins J, Silver LE, Rothwell PM. Population-based study of incidence and outcome of acute aortic dissection and premorbid risk factor control: 10-year results from the Oxford vascular study. Circulation. (2013) 127(20):2031–7. doi: 10.1161/CIRCULATIONAHA.112.000483
3. Obel LM, Lindholt JS, Lasota AN, Jensen HK, Benhassen LL, Mørkved AL, et al. Clinical characteristics, incidences, and mortality rates for type A and B aortic dissections: a nationwide Danish population-based cohort study from 1996 to 2016. Circulation. (2022) 146(25):1903–17. doi: 10.1161/CIRCULATIONAHA.122.061065
4. Meccanici F, Thijssen CGE, Dekker S, Bons LR, Gökalp AL, Rijke Y, et al. Circulating biomarkers associated with aortic diameter in male and female patients with thoracic aortic disease: a cross-sectional study. Open Heart. (2023) 10(1):e002317. doi: 10.1136/openhrt-2023-002317
5. Zhou W-Z, Zhang Y, Zhu G, Shen H, Zeng Q, Chen Q, et al. HTAADVar: aggregation and fully automated clinical interpretation of genetic variants in heritable thoracic aortic aneurysm and dissection. Genet Med. (2022) 24(12):2544–54. doi: 10.1016/j.gim.2022.08.024
6. Regalado ES, Morris SA, Braverman AC, Hostetler EM, De Backer J, Li R, et al. Comparative risks of initial aortic events associated with genetic thoracic aortic disease. J Am Coll Cardiol. (2022) 80(9):857–69. doi: 10.1016/j.jacc.2022.05.054
7. Catherine F, Yalda J, George G, Bernard K, Paul C, Tom F. 142 update on familial thoracic aortic aneurysm disease in the 100,000 genomes project: space for discovery. Heart. (2019) 105(Suppl 6):A117. doi: 10.1136/heartjnl-2019-BCS.139
8. Kuwabara JT, Tallquist MD. Tracking adventitial fibroblast contribution to disease: a review of current methods to identify resident fibroblasts. Arterioscler Thromb Vasc Biol. (2017) 37(9):1598–607. doi: 10.1161/ATVBAHA.117.308199
9. Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. (2009) 89(3):957–89. doi: 10.1152/physrev.00041.2008
10. Angoff R, Mosarla RC, Tsao CW. Aortic stiffness: epidemiology, risk factors, and relevant biomarkers. Front Cardiovasc Med. (2021) 8:1. doi: 10.3389/fcvm.2021.709396
11. Emerel L, Thunes J, Kickliter T, Billaud M, Phillippi JA, Vorp DA, et al. Predissection-derived geometric and distensibility indices reveal increased peak longitudinal stress and stiffness in patients sustaining acute type A aortic dissection: implications for predicting dissection. J Thorac Cardiovasc Surg. (2019) 158(2):355–63. doi: 10.1016/j.jtcvs.2018.10.116
12. Milewicz DM, Prakash SK, Ramirez F. Therapeutics targeting drivers of thoracic aortic aneurysms and acute aortic dissections: insights from predisposing genes and mouse models. Annu Rev Med. (2017) 68(1):51–67. doi: 10.1146/annurev-med-100415-022956
13. Oller J, Méndez-Barbero N, Ruiz EJ, Villahoz S, Renard M, Canelas LI, et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med. (2017) 23(2):200–12. doi: 10.1038/nm.4266
14. Rombouts KB, van Merrienboer TAR, Ket JCF, Bogunovic N, van der Velden J, Yeung KK. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur J Clin Investig. (2022) 52(4):e13697. doi: 10.1111/eci.13697
15. Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, et al. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. (2009) 138(6):1392–9. doi: 10.1016/j.jtcvs.2009.07.075
16. Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, Duraisamy S, et al. TGFBR2 Mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res. (2010) 88(3):520–9. doi: 10.1093/cvr/cvq230
17. Milewicz DM, Trybus KM, Guo DC, Sweeney HL, Regalado E, Kamm K, et al. Altered smooth muscle cell force generation as a driver of thoracic aortic aneurysms and dissections. Arterioscler Thromb Vasc Biol. (2017) 37(1):26–34. doi: 10.1161/ATVBAHA.116.303229
18. Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. (2007) 39(12):1488–93. doi: 10.1038/ng.2007.6
19. Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. (2006) 38(3):343–9. doi: 10.1038/ng1721
20. Wallace SE, Regalado ES, Gong L, Janda AL, Guo D-c, Russo CF, et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet Med. (2019) 21(1):144–51. doi: 10.1038/s41436-018-0038-0
21. Guo DC, Regalado E, Casteel DE, Santos-Cortez RL, Gong L, Kim JJ, et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. (2013) 93(2):398–404. doi: 10.1016/j.ajhg.2013.06.019
22. Davaapil H, Shetty DK, Sinha S. Aortic “disease-in-a-dish": mechanistic insights and drug development using iPSC-based disease modeling. Front Cell Dev Biol. (2020) 8:550504. doi: 10.3389/fcell.2020.550504
23. Jana S, Hu M, Shen M, Kassiri Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp Mol Med. (2019) 51(12):1–15. doi: 10.1038/s12276-019-0286-3
24. Pinard A, Jones GT, Milewicz DM. Genetics of thoracic and abdominal aortic diseases. Circ Res. (2019) 124(4):588–606. doi: 10.1161/CIRCRESAHA.118.312436
25. Rodrigues Bento J, Meester J, Luyckx I, Peeters S, Verstraeten A, Loeys B. The genetics and typical traits of thoracic aortic aneurysm and dissection. Annu Rev Genomics Hum Genet. (2022) 23:223–53. doi: 10.1146/annurev-genom-111521-104455
26. Uray IP, Uray K. Mechanotransduction at the plasma membrane-cytoskeleton interface. Int J Mol Sci. (2021) 22(21). doi: 10.3390/ijms222111566
27. Mohammed D, Versaevel M, Bruyère C, Alaimo L, Luciano M, Vercruysse E, et al. Innovative tools for mechanobiology: unraveling outside-in and inside-out mechanotransduction. Front Bioeng Biotechnol. (2019) 7:162. doi: 10.3389/fbioe.2019.00162
28. Ye GJ, Nesmith AP, Parker KK. The role of mechanotransduction on vascular smooth muscle myocytes’ [corrected] cytoskeleton and contractile function. Anat Rec (Hoboken). (2014) 297(9):1758–69. doi: 10.1002/ar.22983
29. Cook BL, Chao CJ, Alford PW. Architecture-dependent mechano-adaptation in single vascular smooth muscle cells. J Biomech Eng. (2021) 143(10). doi: 10.1115/1.4051117
30. Liu S, Lin Z. Vascular smooth muscle cells mechanosensitive regulators and vascular remodeling. J Vasc Res. (2022) 59(2):90–113. doi: 10.1159/000519845
31. Coady MA, Davies RR, Roberts M, Goldstein LJ, Rogalski MJ, Rizzo JA, et al. Familial patterns of thoracic aortic aneurysms. Arch Surg. (1999) 134(4):361–7. doi: 10.1001/archsurg.134.4.361
32. Arévalo Martínez M, Ritsvall O, Bastrup JA, Celik S, Jakobsson G, Daoud F, et al. Vascular smooth muscle-specific YAP/TAZ deletion triggers aneurysm development in mouse aorta. JCI Insight. (2023) 8(17). doi: 10.1172/jci.insight.170845
33. Swiatlowska P, Sit B, Feng Z, Marhuenda E, Xanthis I, Zingaro S, et al. Pressure and stiffness sensing together regulate vascular smooth muscle cell phenotype switching. Sci Adv. (2022) 8(15):eabm3471. doi: 10.1126/sciadv.abm3471
34. Karimi A, Milewicz DM. Structure of the elastin-contractile units in the thoracic aorta and how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Can J Cardiol. (2016) 32(1):26–34. doi: 10.1016/j.cjca.2015.11.004
35. Ahmed S, Johnson RT, Solanki R, Afewerki T, Wostear F, Warren DT. Using polyacrylamide hydrogels to model physiological aortic stiffness reveals that microtubules are critical regulators of isolated smooth muscle cell morphology and contractility. Front Pharmacol. (2022) 13:836710. doi: 10.3389/fphar.2022.836710
36. Qian W, Hadi T, Silvestro M, Ma X, Rivera CF, Bajpai A, et al. Microskeletal stiffness promotes aortic aneurysm by sustaining pathological vascular smooth muscle cell mechanosensation via Piezo1. Nat Commun. (2022) 13(1):512. doi: 10.1038/s41467-021-27874-5
37. Liu L, Jouve C, Henry J, Berrandou TE, Hulot JS, Georges A, et al. Genomic, transcriptomic, and proteomic depiction of induced pluripotent stem cells-derived smooth muscle cells as emerging cellular models for arterial diseases. Hypertension. (2023) 80(4):740–53. doi: 10.1161/HYPERTENSIONAHA.122.19733
38. Ellis MW, Riaz M, Huang Y, Anderson CW, Luo J, Park J, et al. Epigallocatechin gallate facilitates extracellular elastin fiber formation in induced pluripotent stem cell derived vascular smooth muscle cells for tissue engineering. J Mol Cell Cardiol. (2022) 163:167–74. doi: 10.1016/j.yjmcc.2021.12.014
39. Kapustin A, Tsakali SS, Whitehead M, Chennell G, Wu MY, Molenaar C, et al. Extracellular vesicles stimulate smooth muscle cell migration by presenting collagen VI. bioRxiv. (2023):5–9. doi: 10.1101/2023.08.17.551257
40. Davaapil H, McNamara M, Granata A, Macrae RGC, Hirano M, Fitzek M, et al. A phenotypic screen of Marfan syndrome iPSC-derived vascular smooth muscle cells uncovers GSK3β as a new target. Stem Cell Rep. (2023) 18(2):555–69. doi: 10.1016/j.stemcr.2022.12.014
41. Angbohang A, Huang L, Li Y, Zhao Y, Gong Y, Fu Y, et al. X-box binding protein 1-mediated COL4A1s secretion regulates communication between vascular smooth muscle and stem/progenitor cells. J Biol Chem. (2021) 296:100541. doi: 10.1016/j.jbc.2021.100541
42. Burgess JD, Amerna D, Norton ES, Parsons TM, Perkerson RB 3rd, Faroqi AH, et al. A mutant methionyl-tRNA synthetase-based toolkit to assess induced-mesenchymal stromal cell secretome in mixed-culture disease models. Res Sq [Preprint]. (2023). doi: 10.21203/rs.3.rs-2838195/v1. Update in: Stem Cell Res Ther. (2023) 14(1):289. PMID: 37205579; PMCID: PMC10187403
43. Rickel AP, Sanyour HJ, Leyda NA, Hong Z. Extracellular matrix proteins and substrate stiffness synergistically regulate vascular smooth muscle cell migration and cortical cytoskeleton organization. ACS Appl Bio Mater. (2020) 3(4):2360–9. doi: 10.1021/acsabm.0c00100
44. Hartman CD, Isenberg BC, Chua SG, Wong JY. Vascular smooth muscle cell durotaxis depends on extracellular matrix composition. Proc Natl Acad Sci U S A. (2016) 113(40):11190–5. doi: 10.1073/pnas.1611324113
45. Xie SA, Zhang T, Wang J, Zhao F, Zhang YP, Yao WJ, et al. Matrix stiffness determines the phenotype of vascular smooth muscle cell in vitro and in vivo: role of DNA methyltransferase 1. Biomaterials. (2018) 155:203–16. doi: 10.1016/j.biomaterials.2017.11.033
46. Camargo LL, Touyz RM. Phenotype-specific induced pluripotent stem cell-derived vascular smooth muscle cells to model vascular disease: implications of differentiation protocols. Hypertension. (2023) 80(4):754–6. doi: 10.1161/HYPERTENSIONAHA.123.20871
47. Atkins SK, Sonawane AR, Brouwhuis R, Barrientos J, Ha A, Rogers M, et al. Induced pluripotent stem cell-derived smooth muscle cells to study cardiovascular calcification. Front Cardiovasc Med. (2022) 9:925777. doi: 10.3389/fcvm.2022.925777
48. Meijer EM, Giles R, van Dijk CGM, Maringanti R, Wissing TB, Appels Y, et al. Effect of mechanical stimuli on the phenotypic plasticity of induced pluripotent stem-cell-derived vascular smooth muscle cells in a 3D hydrogel. ACS Appl Bio Mater. (2023) 6(12):5716–29. doi: 10.1021/acsabm.3c00840
49. Sinha S, Iyer D, Granata A. Embryonic origins of human vascular smooth muscle cells: implications for in vitro modeling and clinical application. Cell Mol Life Sci. (2014) 71(12):2271–88. doi: 10.1007/s00018-013-1554-3
50. El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. (2009) 6(12):771–86. doi: 10.1038/nrcardio.2009.191
51. Chaudhry SS, Cain SA, Morgan A, Dallas SL, Shuttleworth CA, Kielty CM. Fibrillin-1 regulates the bioavailability of TGFbeta1. J Cell Biol. (2007) 176(3):355–67. doi: 10.1083/jcb.200608167
52. Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. (2003) 33(3):407–11. doi: 10.1038/ng1116
53. Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. (2006) 312(5770):117–21. doi: 10.1126/science.1124287
54. Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, et al. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for marfan syndrome. Arterioscler Thromb Vasc Biol. (2015) 35(4):911–7. doi: 10.1161/ATVBAHA.114.305150
55. Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. (2014) 124(2):755–67. doi: 10.1172/JCI69942
56. Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, et al. Postnatal deletion of the type II transforming growth factor-β receptor in smooth muscle cells causes severe aortopathy in mice. Arterioscler Thromb Vasc Biol. (2015) 35(12):2647–56. doi: 10.1161/ATVBAHA.115.306573
57. Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest. (2010) 120(2):422–32. doi: 10.1172/JCI38136
58. Hara H, Takeda N, Fujiwara T, Yagi H, Maemura S, Kanaya T, et al. Activation of TGF-β signaling in an aortic aneurysm in a patient with loeys-dietz syndrome caused by a novel loss-of-function variant of TGFBR1. Hum Genome Var. (2019) 6:6. doi: 10.1038/s41439-019-0038-x
59. Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, et al. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest. (2014) 124(1):448–60. doi: 10.1172/JCI69666
60. MacFarlane EG, Parker SJ, Shin JY, Kang BE, Ziegler SG, Creamer TJ, et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J Clin Invest. (2019) 129(2):659–75. doi: 10.1172/JCI123547
61. Gong J, Zhou D, Jiang L, Qiu P, Milewicz DM, Chen YE, et al. In vitro lineage-specific differentiation of vascular smooth muscle cells in response to SMAD3 deficiency: implications for SMAD3-related thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol. (2020) 40(7):1651–63. doi: 10.1161/ATVBAHA.120.313033
62. Gao J, Cao H, Hu G, Wu Y, Xu Y, Cui H, et al. The mechanism and therapy of aortic aneurysms. Signal Transduct Target Ther. (2023) 8(1):55. doi: 10.1038/s41392-023-01325-7
63. Isselbacher EM, Lino Cardenas CL, Lindsay ME. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation. (2016) 133(24):2516–28. doi: 10.1161/CIRCULATIONAHA.116.009762
64. Zhu J, Angelov S, Yildirim IA, Wei H, Hu JH, Majesky MW, et al. Loss of transforming growth factor Beta signaling in aortic smooth muscle cells causes endothelial dysfunction and aortic hypercontractility. Arterioscler, Thromb, Vasc Biol. (2021) 41(6):1956–71. doi: 10.1161/ATVBAHA.121.315878
65. Chung AW, Au Yeung K, Cortes SF, Sandor GG, Judge DP, Dietz HC, et al. Endothelial dysfunction and compromised eNOS/akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br J Pharmacol. (2007) 150(8):1075–83. doi: 10.1038/sj.bjp.0707181
66. Galatioto J, Caescu CI, Hansen J, Cook JR, Miramontes I, Iyengar R, et al. Cell type-specific contributions of the angiotensin II type 1a receptor to aorta homeostasis and aneurysmal disease-brief report. Arterioscler Thromb Vasc Biol. (2018) 38(3):588–91. doi: 10.1161/ATVBAHA.117.310609
67. Méndez-Barbero N, Gutiérrez-Muñoz C, Blanco-Colio LM. Cellular crosstalk between endothelial and smooth muscle cells in vascular wall remodeling. Int J Mol Sci. (2021) 22(14):2–3. doi: 10.3390/ijms22147284
68. Hu Y, Xu Q. Adventitial biology: differentiation and function. Arterioscler Thromb Vasc Biol. (2011) 31(7):1523–9. doi: 10.1161/ATVBAHA.110.221176
69. Mackay CDA, Jadli AS, Fedak PWM, Patel VB. Adventitial fibroblasts in aortic aneurysm: unraveling pathogenic contributions to vascular disease. Diagnostics (Basel). (2022) 12(4):871. doi: 10.3390/diagnostics12040871
70. Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. (2007) 27(6):1248–58. doi: 10.1161/ATVBAHA.107.141069
71. Wu H, Xie C, Wang R, Cheng J, Xu Q, Zhao H. Comparative analysis of thoracic and abdominal aortic aneurysms across the segment and species at the single-cell level. Front Pharmacol. (2023) 13:1095757. doi: 10.3389/fphar.2022.1095757
72. Yao D, Mei S, Tang W, Xu X, Lu Q, Shi Z. AAAKB: a manually curated database for tracking and predicting genes of abdominal aortic aneurysm (AAA). PLoS One. (2023) 18(12):e0289966. doi: 10.1371/journal.pone.0289966
73. Cheung C, Bernardo AS, Pedersen RA, Sinha S. Directed differentiation of embryonic origin-specific vascular smooth muscle subtypes from human pluripotent stem cells. Nat Protoc. (2014) 9(4):929–38. doi: 10.1038/nprot.2014.059
74. Nakamura K, Dalal AR, Yokoyama N, Pedroza AJ, Kusadokoro S, Mitchel O, et al. Lineage-specific induced pluripotent stem cell-derived smooth muscle cell modeling predicts integrin alpha-V antagonism reduces aortic root aneurysm formation in Marfan syndrome mice. Arterioscler Thromb Vasc Biol. (2023) 43(7):1134–53. doi: 10.1161/ATVBAHA.122.318448
75. Cheung C, Bernardo AS, Trotter MW, Pedersen RA, Sinha S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat Biotechnol. (2012) 30(2):165–73. doi: 10.1038/nbt.2107
76. Serrano F, Bernard WG, Granata A, Iyer D, Steventon B, Kim M, et al. A novel human pluripotent stem cell-derived neural crest model of treacher Collins syndrome shows defects in cell death and migration. Stem Cells Dev. (2019) 28(2):81–100. doi: 10.1089/scd.2017.0234
77. Box GEP. Robustness in the strategy of scientific model building. In: Launer RL, Wilkinson GN, editors. Robustness in Statistics. New York: Academic Press (1979). p. 201–36.
78. Sakai LY, Keene DR, Renard M, De Backer J. FBN1: the disease-causing gene for Marfan syndrome and other genetic disorders. Gene. (2016) 591(1):279–91. doi: 10.1016/j.gene.2016.07.033
79. Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. (2004) 114(2):172–81. doi: 10.1172/JCI200420641
80. Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. (1999) 96(7):3819–23. doi: 10.1073/pnas.96.7.3819
81. van Andel MM, Indrakusuma R, Jalalzadeh H, Balm R, Timmermans J, Scholte AJ, et al. Long-term clinical outcomes of losartan in patients with Marfan syndrome: follow-up of the multicentre randomized controlled COMPARE trial. Eur Heart J. (2020) 41(43):4181–7. doi: 10.1093/eurheartj/ehaa377
82. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126(4):663–76. doi: 10.1016/j.cell.2006.07.024
83. Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. (2007) 448(7151):313–7. doi: 10.1038/nature05934
84. Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. (2008) 451(7175):141–6. doi: 10.1038/nature06534
85. Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. (2007) 448(7151):318–24. doi: 10.1038/nature05944
86. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. (2007) 318(5858):1917–20. doi: 10.1126/science.1151526
87. Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, et al. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet. (2017) 49(1):97–109. doi: 10.1038/ng.3723
88. Zhou D, Feng H, Yang Y, Huang T, Qiu P, Zhang C, et al. hiPSC modeling of lineage-specific smooth muscle cell defects caused by TGFBR1(A230T) variant, and its therapeutic implications for Loeys-Dietz syndrome. Circulation. (2021) 144(14):1145–59. doi: 10.1161/CIRCULATIONAHA.121.054744
89. Perik M, Rabaut L, Buccioli L, Verstraeten A, Van Laer L, Loeys B. IPSC-derived vascular smooth muscle cell model recapitulates Loeys-Dietz-Syndrome type V aortic phenotype. Eur Heart J. (2023) 44(Supplement_2):ehad655.2025. doi: 10.1093/eurheartj/ehad655.2025
90. Jiao J, Xiong W, Wang L, Yang J, Qiu P, Hirai H, et al. Differentiation defect in neural crest-derived smooth muscle cells in patients with aortopathy associated with bicuspid aortic valves. EBioMedicine. (2016) 10:282–90. doi: 10.1016/j.ebiom.2016.06.045
91. Kinnear C, Chang WY, Khattak S, Hinek A, Thompson T, de Carvalho Rodrigues D, et al. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl Med. (2013) 2(1):2–15. doi: 10.5966/sctm.2012-0054
92. Ge X, Ren Y, Bartulos O, Lee MY, Yue Z, Kim KY, et al. Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation. (2012) 126(14):1695–704. doi: 10.1161/CIRCULATIONAHA.112.116996
93. Choi KD, Yu J, Smuga-Otto K, Salvagiotto G, Rehrauer W, Vodyanik M, et al. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells. (2009) 27(3):559–67. doi: 10.1634/stemcells.2008-0922
94. Taura D, Sone M, Homma K, Oyamada N, Takahashi K, Tamura N, et al. Induction and isolation of vascular cells from human induced pluripotent stem cells–brief report. Arterioscler Thromb Vasc Biol. (2009) 29(7):1100–3. doi: 10.1161/ATVBAHA.108.182162
95. Trillhaase A, Maertens M, Aherrahrou Z, Erdmann J. Induced pluripotent stem cells (iPSCs) in vascular research: from two- to three-dimensional organoids. Stem Cell Rev Rep. (2021) 17(5):1741–53. doi: 10.1007/s12015-021-10149-3
96. Oh JE, Jung C, Yoon YS. Human induced pluripotent stem cell-derived vascular cells: recent progress and future directions. J Cardiovasc Dev Dis. (2021) 8(11):148. doi: 10.3390/jcdd8110148
97. Shen M, Quertermous T, Fischbein MP, Wu JC. Generation of vascular smooth muscle cells from induced pluripotent stem cells: methods, applications, and considerations. Circ Res. (2021) 128(5):670–86. doi: 10.1161/CIRCRESAHA.120.318049
98. Yang L, Geng Z, Nickel T, Johnson C, Gao L, Dutton J, et al. Differentiation of human induced-pluripotent stem cells into smooth-muscle cells: two novel protocols. PLoS One. (2016) 11(1):e0147155. doi: 10.1371/journal.pone.0147155
99. Zhang J, McIntosh BE, Wang B, Brown ME, Probasco MD, Webster S, et al. A human pluripotent stem cell-based screen for smooth muscle cell differentiation and maturation identifies inhibitors of intimal hyperplasia. Stem Cell Rep. (2019) 12(6):1269–81. doi: 10.1016/j.stemcr.2019.04.013
100. Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. (1992) 89(12):5547–51. doi: 10.1073/pnas.89.12.5547
101. Bertero A, Pawlowski M, Ortmann D, Snijders K, Yiangou L, Cardoso de Brito M, et al. Optimized inducible shRNA and CRISPR/Cas9 platforms for in vitro studies of human development using hPSCs. Development. (2016) 143(23):4405–18. doi: 10.1242/dev.138081
102. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. (2010) 40(2):179–204. doi: 10.1016/j.molcel.2010.09.019
103. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. (2020) 38(7):824–44. doi: 10.1038/s41587-020-0561-9
104. Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. (2016) 529(7587):490–5. doi: 10.1038/nature16526
105. Casini A, Olivieri M, Petris G, Montagna C, Reginato G, Maule G, et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol. (2018) 36(3):265–71. doi: 10.1038/nbt.4066
106. Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, et al. Diversity and evolution of class 2 CRISPR-cas systems. Nat Rev Microbiol. (2017) 15(3):169–82. doi: 10.1038/nrmicro.2016.184
107. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. (2016) 533(7603):420–4. doi: 10.1038/nature17946
108. Oliveros W, Delfosse K, Lato DF, Kiriakopulos K, Mokhtaridoost M, Said A, et al. Systematic characterization of regulatory variants of blood pressure genes. Cell Genomics. (2023) 3(7):100330. doi: 10.1016/j.xgen.2023.100330
109. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) 576(7785):149–57. doi: 10.1038/s41586-019-1711-4
110. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. (2018) 9(1):1911. doi: 10.1038/s41467-018-04252-2
111. Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, et al. RNA editing with CRISPR-Cas13. Science. (2017) 358(6366):1019–27. doi: 10.1126/science.aaq0180
112. Sa S, Gu M, Chappell J, Shao NY, Ameen M, Elliott KA, et al. Induced pluripotent stem cell model of pulmonary arterial hypertension reveals novel gene expression and patient specificity. Am J Respir Crit Care Med. (2017) 195(7):930–41. doi: 10.1164/rccm.201606-1200OC
113. Biel NM, Santostefano KE, DiVita BB, El Rouby N, Carrasquilla SD, Simmons C, et al. Vascular smooth muscle cells from hypertensive patient-derived induced pluripotent stem cells to advance hypertension pharmacogenomics. Stem Cells Transl Med. (2015) 4(12):1380–90. doi: 10.5966/sctm.2015-0126
114. Chen G, Orozco L, Parmisano S, Jahng JWS, Vera CD, Zhuge Y, et al. Generation of two induced pluripotent stem cell lines from patients suffering from pulmonary hypertension. Stem Cell Res. (2023) 72:103218. doi: 10.1016/j.scr.2023.103218
115. Suginobe H, Ishida H, Ishii Y, Ueda K, Yoshihara C, Ueyama A, et al. Isogenic pairs of induced-pluripotent stem-derived endothelial cells identify DYRK1A/PPARG/EGR1 pathway is responsible for down syndrome-associated pulmonary hypertension. Hum Mol Genet. (2023) 33(1):78–90. doi: 10.1093/hmg/ddad162
116. Devendran A, Kar S, Bailey R, Trivieri MG. The role of bone morphogenetic protein receptor type 2 (BMPR2) and the prospects of utilizing induced pluripotent stem cells (iPSCs) in pulmonary arterial hypertension disease modeling. Cells. (2022) 11(23). doi: 10.3390/cells11233823
117. Gu M, Donato M, Guo M, Wary N, Miao Y, Mao S, et al. iPSC-endothelial cell phenotypic drug screening and in silico analyses identify tyrphostin-AG1296 for pulmonary arterial hypertension. Sci Transl Med. (2021) 13(592):eaba6480. doi: 10.1126/scitranslmed.aba6480
118. Kiskin FN, Chang CH, Huang CJZ, Kwieder B, Cheung C, Dunmore BJ, et al. Contributions of BMPR2 mutations and extrinsic factors to cellular phenotypes of pulmonary arterial hypertension revealed by induced pluripotent stem cell modeling. Am J Respir Crit Care Med. (2018) 198(2):271–5. doi: 10.1164/rccm.201801-0049LE
119. Gu M, Shao NY, Sa S, Li D, Termglinchan V, Ameen M, et al. Patient-specific iPSC-derived endothelial cells uncover pathways that protect against pulmonary hypertension in BMPR2 mutation carriers. Cell Stem Cell. (2017) 20(4):490–504.e5. doi: 10.1016/j.stem.2016.08.019
120. Abutaleb NO, Atchison L, Choi L, Bedapudi A, Shores K, Gete Y, et al. Lonafarnib and everolimus reduce pathology in iPSC-derived tissue engineered blood vessel model of Hutchinson-Gilford progeria syndrome. Sci Rep. (2023) 13(1):5032. doi: 10.1038/s41598-023-32035-3
121. Atchison L, Abutaleb NO, Snyder-Mounts E, Gete Y, Ladha A, Ribar T, et al. iPSC-derived endothelial cells affect vascular function in a tissue-engineered blood vessel model of Hutchinson-Gilford progeria syndrome. Stem Cell Rep. (2020) 14(2):325–37. doi: 10.1016/j.stemcr.2020.01.005
122. Atchison L, Zhang H, Cao K, Truskey GA. A tissue engineered blood vessel model of Hutchinson-Gilford progeria syndrome using human iPSC-derived smooth muscle cells. Sci Rep. (2017) 7(1):8168. doi: 10.1038/s41598-017-08632-4
123. Matrone G, Thandavarayan RA, Walther BK, Meng S, Mojiri A, Cooke JP. Dysfunction of iPSC-derived endothelial cells in human Hutchinson-Gilford progeria syndrome. Cell Cycle. (2019) 18(19):2495–508. doi: 10.1080/15384101.2019.1651587
124. Ribas J, Zhang YS, Pitrez PR, Leijten J, Miscuglio M, Rouwkema J, et al. Biomechanical strain exacerbates inflammation on a progeria-on-a-chip model. Small. (2017) 13(15). doi: 10.1002/smll.201603737
125. Trillhaase A, Schmidt B, Märtens M, Haferkamp U, Erdmann J, Aherrahrou Z. The CAD risk locus 9p21 increases the risk of vascular calcification in an iPSC-derived VSMC model. Stem Cell Res Ther. (2021) 12(1):166. doi: 10.1186/s13287-021-02229-5
126. Mayner JM, Masutani EM, Demeester E, Kumar A, Macapugay G, Beri P, et al. Heterogeneous expression of alternatively spliced lncRNA mediates vascular smooth cell plasticity. Proc Natl Acad Sci U S A. (2023) 120(24):e2217122120. doi: 10.1073/pnas.2217122120
127. Tokairin K, Hamauchi S, Ito M, Kazumata K, Sugiyama T, Nakayama N, et al. Vascular smooth muscle cell derived from IPS cell of moyamoya disease—comparative characterization with endothelial cell transcriptome. J Stroke Cerebrovasc Dis. (2020) 29(12):105305. doi: 10.1016/j.jstrokecerebrovasdis.2020.105305
128. Xue Y, Zhang Q, Wang LJ, Tu WJ, Zhao J. Application of induced pluripotent stem cells in moyamoya disease: progress and promises. Curr Stem Cell Res Ther. (2023) 18(6):733–9. doi: 10.2174/1574888X17666220607121027
129. Matsuo M, Nadanaka S, Soga M, Sugiyama T, Serigano S, Shimano K, et al. Vulnerability to shear stress caused by altered peri-endothelial matrix is a key feature of moyamoya disease. Sci Rep. (2021) 11(1):1552. doi: 10.1038/s41598-021-81282-9
130. Hamauchi S, Shichinohe H, Uchino H, Yamaguchi S, Nakayama N, Kazumata K, et al. Cellular functions and gene and protein expression profiles in endothelial cells derived from moyamoya disease-specific iPS cells. PLoS One. (2016) 11(9):e0163561. doi: 10.1371/journal.pone.0163561
131. Hitomi T, Habu T, Kobayashi H, Okuda H, Harada KH, Osafune K, et al. Downregulation of securin by the variant RNF213 R4810K (rs112735431, G > A) reduces angiogenic activity of induced pluripotent stem cell-derived vascular endothelial cells from moyamoya patients. Biochem Biophys Res Commun. (2013) 438(1):13–9. doi: 10.1016/j.bbrc.2013.07.004
132. Hayes G, Pinto J, Sparks SN, Wang C, Suri S, Bulte DP. Vascular smooth muscle cell dysfunction in neurodegeneration. Front Neurosci. (2022) 16:1010164. doi: 10.3389/fnins.2022.1010164
133. Zhang W, Zhao X, Qi X, Kimber SJ, Hooper NM, Wang T. Induced pluripotent stem cell model revealed impaired neurovascular interaction in genetic small vessel disease cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Front Cell Neurosci. (2023) 17:1195470. doi: 10.3389/fncel.2023.1195470
134. Wang J, Zhang L, Wu G, Wu J, Zhou X, Chen X, et al. Correction of a CADASIL point mutation using adenine base editors in hiPSCs and blood vessel organoids. J Genet Genomics. (2023). doi: 10.1016/j.jgg.2023.04.013
135. Yamamoto Y, Kojima K, Taura D, Sone M, Washida K, Egawa N, et al. Human iPS cell-derived mural cells as an in vitro model of hereditary cerebral small vessel disease. Mol Brain. (2020) 13(1):38. doi: 10.1186/s13041-020-00573-w
136. Toyohara T, Roudnicky F, Florido MHC, Nakano T, Yu H, Katsuki S, et al. Patient hiPSCs identify vascular smooth muscle arylacetamide deacetylase as protective against atherosclerosis. Cell Stem Cell. (2020) 27(1):147–57.e7. doi: 10.1016/j.stem.2020.04.018
137. Wimmer RA, Leopoldi A, Aichinger M, Wick N, Hantusch B, Novatchkova M, et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature. (2019) 565(7740):505–10. doi: 10.1038/s41586-018-0858-8
138. Naderi-Meshkin H, Cornelius VA, Eleftheriadou M, Potel KN, Setyaningsih WAW, Margariti A. Vascular organoids: unveiling advantages, applications, challenges, and disease modelling strategies. Stem Cell Res Ther. (2023) 14(1):292. doi: 10.1186/s13287-023-03521-2
139. Iosef C, Pedroza AJ, Cui JZ, Dalal AR, Arakawa M, Tashima Y, et al. Quantitative proteomics reveal lineage-specific protein profiles in iPSC-derived Marfan syndrome smooth muscle cells. Sci Rep. (2020) 10(1):20392. doi: 10.1038/s41598-020-77274-w
140. Kelleher J, Dickinson A, Cain S, Hu Y, Bates N, Harvey A, et al. Patient-specific iPSC model of a genetic vascular dementia syndrome reveals failure of mural cells to stabilize capillary structures. Stem Cell Rep. (2019) 13(5):817–31. doi: 10.1016/j.stemcr.2019.10.004
141. Albornoz G, Coady MA, Roberts M, Davies RR, Tranquilli M, Rizzo JA, et al. Familial thoracic aortic aneurysms and dissections—incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. (2006) 82(4):1400–5. doi: 10.1016/j.athoracsur.2006.04.098
142. Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg. (1997) 25(3):506–11. doi: 10.1016/S0741-5214(97)70261-1
143. Kwartler CS, Gong L, Chen J, Wang S, Kulmacz R, Duan XY, et al. Variants of unknown significance in genes associated with heritable thoracic aortic disease can be low penetrant “risk variants”. Am J Hum Genet. (2018) 103(1):138–43. doi: 10.1016/j.ajhg.2018.05.012
144. Ziganshin BA, Bailey AE, Coons C, Dykas D, Charilaou P, Tanriverdi LH, et al. Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Ann Thorac Surg. (2015) 100(5):1604–11. doi: 10.1016/j.athoracsur.2015.04.106
145. Villar D, Frost S, Deloukas P, Tinker A. The contribution of non-coding regulatory elements to cardiovascular disease. Open Biol. (2020) 10(7):200088. doi: 10.1098/rsob.200088
146. Klarin D, Devineni P, Sendamarai AK, Angueira AR, Graham SE, Shen YH, et al. Genome-wide association study of thoracic aortic aneurysm and dissection in the million veteran program. Nat Genet. (2023) 55(7):1106–15. doi: 10.1038/s41588-023-01420-z
147. Pirruccello JP, Chaffin MD, Chou EL, Fleming SJ, Lin H, Nekoui M, et al. Deep learning enables genetic analysis of the human thoracic aorta. Nat Genet. (2022) 54(1):40–51. doi: 10.1038/s41588-021-00962-4
148. LeMaire SA, McDonald ML, Guo DC, Russell L, Miller CC 3rd, Johnson RJ, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. (2011) 43(10):996–1000. doi: 10.1038/ng.934
149. Roychowdhury T, Lu H, Hornsby WE, Crone B, Wang GT, Guo DC, et al. Regulatory variants in TCF7L2 are associated with thoracic aortic aneurysm. Am J Hum Genet. (2021) 108(9):1578–89. doi: 10.1016/j.ajhg.2021.06.016
150. Guo DC, Grove ML, Prakash SK, Eriksson P, Hostetler EM, LeMaire SA, et al. Genetic variants in LRP1 and ULK4 are associated with acute aortic dissections. Am J Hum Genet. (2016) 99(3):762–9. doi: 10.1016/j.ajhg.2016.06.034
151. Badano JL, Katsanis N. Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet. (2002) 3(10):779–89. doi: 10.1038/nrg910
152. Neavin DR, Steinmann AM, Farbehi N, Chiu HS, Daniszewski MS, Arora H, et al. A village in a dish model system for population-scale hiPSC studies. Nat Commun. (2023) 14(1):3240. doi: 10.1038/s41467-023-38704-1
153. Brooks IR, Garrone CM, Kerins C, Kiar CS, Syntaka S, Xu JZ, et al. Functional genomics and the future of iPSCs in disease modeling. Stem Cell Rep. (2022) 17(5):1033–47. doi: 10.1016/j.stemcr.2022.03.019
154. Ostberg NP, Zafar MA, Ziganshin BA, Elefteriades JA. The genetics of thoracic aortic aneurysms and dissection: a clinical perspective. Biomolecules. (2020) 10(2):182. doi: 10.3390/biom10020182
155. Faggion Vinholo T, Brownstein AJ, Ziganshin BA, Zafar MA, Kuivaniemi H, Body SC, et al. Genes associated with thoracic aortic aneurysm and dissection: 2019 update and clinical implications. Aorta (Stamford). (2019) 7(4):99–107. doi: 10.1055/s-0039-3400233
156. Takeda N, Inuzuka R, Maemura S, Morita H, Nawata K, Fujita D, et al. Impact of pathogenic FBN1 variant types on the progression of aortic disease in patients with Marfan syndrome. Circ Genom Precis Med. (2018) 11(6):e002058. doi: 10.1161/CIRCGEN.117.002058
157. Franken R, Groenink M, de Waard V, Feenstra HM, Scholte AJ, van den Berg MP, et al. Genotype impacts survival in Marfan syndrome. Eur Heart J. (2016) 37(43):3285–90. doi: 10.1093/eurheartj/ehv739
158. Aubart M, Gazal S, Arnaud P, Benarroch L, Gross MS, Buratti J, et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur J Hum Genet. (2018) 26(12):1759–72. doi: 10.1038/s41431-018-0164-9
159. Camerota L, Ritelli M, Wischmeijer A, Majore S, Cinquina V, Fortugno P, et al. Genotypic categorization of Loeys-Dietz syndrome based on 24 novel families and literature data. Genes (Basel). (2019) 10(10):9–10. doi: 10.3390/genes10100764
160. Bono N, Pezzoli D, Levesque L, Loy C, Candiani G, Fiore GB, et al. Unraveling the role of mechanical stimulation on smooth muscle cells: a comparative study between 2D and 3D models. Biotechnol Bioeng. (2016) 113(10):2254–63. doi: 10.1002/bit.25979
161. Sellahewa SG, Li JY, Xiao Q. Updated perspectives on direct vascular cellular reprogramming and their potential applications in tissue engineered vascular grafts. J Funct Biomater. (2022) 14(1):17–25. doi: 10.3390/jfb14010021
162. Miyagawa K, Shi M, Chen PI, Hennigs JK, Zhao Z, Wang M, et al. Smooth muscle contact drives endothelial regeneration by BMPR2-Notch1-mediated metabolic and epigenetic changes. Circ Res. (2019) 124(2):211–24. doi: 10.1161/CIRCRESAHA.118.313374
163. Wimmer RA, Leopoldi A, Aichinger M, Kerjaschki D, Penninger JM. Generation of blood vessel organoids from human pluripotent stem cells. Nat Protoc. (2019) 14(11):3082–100. doi: 10.1038/s41596-019-0213-z
164. Palasantzas V, Tamargo-Rubio I, Le K, Slager J, Wijmenga C, Jonkers IH, et al. iPSC-derived organ-on-a-chip models for personalized human genetics and pharmacogenomics studies. Trends Genet. (2023) 39(4):268–84. doi: 10.1016/j.tig.2023.01.002
165. Vila Cuenca M, Cochrane A, van den Hil FE, de Vries AAF, Lesnik Oberstein SAJ, Mummery CL, et al. Engineered 3D vessel-on-chip using hiPSC-derived endothelial- and vascular smooth muscle cells. Stem Cell Rep. (2021) 16(9):2159–68. doi: 10.1016/j.stemcr.2021.08.003
166. Bulut M, Vila Cuenca M, de Graaf M, van den Hil FE, Mummery CL, Orlova VV. Three-dimensional vessels-on-a-chip based on hiPSC-derived vascular endothelial and smooth muscle cells. Curr Protoc. (2022) 2(10):e564. doi: 10.1002/cpz1.564
167. Collado MS, Cole BK, Figler RA, Lawson M, Manka D, Simmers MB, et al. Exposure of induced pluripotent stem cell-derived vascular endothelial and smooth muscle cells in coculture to hemodynamics induces primary vascular cell-like phenotypes. Stem Cells Transl Med. (2017) 6(8):1673–83. doi: 10.1002/sctm.17-0004
168. Dogan L, Scheuring R, Wagner N, Ueda Y, Schmidt S, Worsdorfer P, et al. Human iPSC-derived mesodermal progenitor cells preserve their vasculogenesis potential after extrusion and form hierarchically organized blood vessels. Biofabrication. (2021) 13(4). doi: 10.1088/1758-5090/ac26ac
169. Fazal F, Raghav S, Callanan A, Koutsos V, Radacsi N. Recent advancements in the bioprinting of vascular grafts. Biofabrication. (2021) 13(3). doi: 10.1088/1758-5090/ac0963
170. Xu Y, Hu Y, Liu C, Yao H, Liu B, Mi S. A novel strategy for creating tissue-engineered biomimetic blood vessels using 3D bioprinting technology. Materials (Basel). (2018) 11(9):1581. doi: 10.3390/ma11091581
171. Low LA, Mummery C, Berridge BR, Austin CP, Tagle DA. Organs-on-chips: into the next decade. Nat Rev Drug Discov. (2021) 20(5):345–61. doi: 10.1038/s41573-020-0079-3
172. Abudupataer M, Zhu S, Yan S, Xu K, Zhang J, Luo S, et al. Aorta smooth muscle-on-a-chip reveals impaired mitochondrial dynamics as a therapeutic target for aortic aneurysm in bicuspid aortic valve disease. Elife. (2021) 10:8–17. doi: 10.7554/eLife.69310
173. Liu G, Li J, Ming Y, Xiang B, Zhou X, Chen Y, et al. A hiPSC-derived lineage-specific vascular smooth muscle cell-on-a-chip identifies aortic heterogeneity across segments. Lab Chip. (2023) 23(7):1835–51. doi: 10.1039/D2LC01158A
174. Dash BC, Levi K, Schwan J, Luo J, Bartulos O, Wu H, et al. Tissue-engineered vascular rings from human iPSC-derived smooth muscle cells. Stem Cell Rep. (2016) 7(1):19–28. doi: 10.1016/j.stemcr.2016.05.004
175. Reed E, Fellows A, Lu R, Rienks M, Schmidt L, Yin X, et al. Extracellular matrix profiling and disease modelling in engineered vascular smooth muscle cell tissues. Matrix Biol Plus. (2022) 16:100122. doi: 10.1016/j.mbplus.2022.100122
176. Teixido-Tura G, Forteza A, Rodríguez-Palomares J, González Mirelis J, Gutiérrez L, Sánchez V, et al. Losartan versus atenolol for prevention of aortic dilation in patients with Marfan syndrome. J Am Coll Cardiol. (2018) 72(14):1613–8. doi: 10.1016/j.jacc.2018.07.052
177. Milleron O, Arnoult F, Ropers J, Aegerter P, Detaint D, Delorme G, et al. Marfan Sartan: a randomized, double-blind, placebo-controlled trial. Eur Heart J. (2015) 36(32):2160–6. doi: 10.1093/eurheartj/ehv151
178. Mullen M, Jin XY, Child A, Stuart AG, Dodd M, Aragon-Martin JA, et al. Irbesartan in marfan syndrome (AIMS): a double-blind, placebo-controlled randomised trial. Lancet. (2019) 394(10216):2263–70. doi: 10.1016/S0140-6736(19)32518-8
179. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. (2014) 159(3):647–61. doi: 10.1016/j.cell.2014.09.029
180. Bock C, Datlinger P, Chardon F, Coelho MA, Dong MB, Lawson KA, et al. High-content CRISPR screening. Nat Rev Methods Primers. (2022) 2(1). doi: 10.1038/s43586-021-00093-4
181. Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. (2016) 167(7):1853–66.e17. doi: 10.1016/j.cell.2016.11.038
182. Granata A, Kasioulis I, Serrano F, Cooper JD, Traylor M, Sinha S, et al. The histone deacetylase 9 stroke-risk variant promotes apoptosis and inflammation in a human iPSC-derived smooth muscle cells model. Front Cardiovasc Med. (2022) 9:849664. doi: 10.3389/fcvm.2022.849664
183. Granata A. Functional genomics in stroke: current and future applications of iPSCs and gene editing to dissect the function of risk variants. BMC Cardiovasc Disord. (2023) 23(1):223. doi: 10.1186/s12872-023-03227-6
184. Al-Thani M, Goodwin-Trotman M, Bell S, Patel K, Fleming LK, Vilain C, et al. A novel human iPSC model of COL4A1/A2 small vessel disease unveils a key pathogenic role of matrix metalloproteinases. Stem Cell Reports. (2023) 18(12):2386–99. doi: 10.1016/j.stemcr.2023.10.014
185. Serrano R, Feyen DAM, Bruyneel AAN, Hnatiuk AP, Vu MM, Amatya PL, et al. A deep learning platform to assess drug proarrhythmia risk. Cell Stem Cell. (2023) 30(1):86–95.e4. doi: 10.1016/j.stem.2022.12.002
186. Patel KK, Venkatesan C, Abdelhalim H, Zeeshan S, Arima Y, Linna-Kuosmanen S, et al. Genomic approaches to identify and investigate genes associated with atrial fibrillation and heart failure susceptibility. Hum Genomics. (2023) 17(1):47. doi: 10.1186/s40246-023-00498-0
187. Page SC, Sripathy SR, Farinelli F, Ye Z, Wang Y, Hiler DJ, et al. Electrophysiological measures from human iPSC-derived neurons are associated with schizophrenia clinical status and predict individual cognitive performance. Proc Natl Acad Sci U S A. (2022) 119(3):e2109395119. doi: 10.1073/pnas.2109395119
188. Engel JL, Zhang X, Lu DR, Vila OF, Arias V, Lee J, et al. Single cell multi-omics of an iPSC model of human sinoatrial node development reveals genetic determinants of heart rate and arrhythmia susceptibility. bioRxiv [Preprint]. (2023). doi: 10.1101/2023.07.01.547335
189. Lee J, Termglinchan V, Diecke S, Itzhaki I, Lam CK, Garg P, et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature. (2019) 572(7769):335–40. doi: 10.1038/s41586-019-1406-x
190. Sun Y, Su J, Wang X, Wang J, Guo F, Qiu H, et al. Patient-specific iPSC-derived cardiomyocytes reveal variable phenotypic severity of Brugada syndrome. EBioMedicine. (2023) 95:104741. doi: 10.1016/j.ebiom.2023.104741
191. Hirayama R, Toyohara K, Watanabe K, Otsuki T, Araoka T, Mae SI, et al. iPSC-derived type IV collagen α5-expressing kidney organoids model Alport syndrome. Commun Biol. (2023) 6(1):854. doi: 10.1038/s42003-023-05203-4
192. Cho DI, Ahn MJ, Cho HH, Cho M, Jun JH, Kang BG, et al. ANGPTL4 Stabilizes atherosclerotic plaques and modulates the phenotypic transition of vascular smooth muscle cells through KLF4 downregulation. Exp Mol Med. (2023) 55(2):426–42. doi: 10.1038/s12276-023-00937-x
193. Grafton F, Ho J, Ranjbarvaziri S, Farshidfar F, Budan A, Steltzer S, et al. Deep learning detects cardiotoxicity in a high-content screen with induced pluripotent stem cell-derived cardiomyocytes. Elife. (2021) 10:10–21. doi: 10.7554/eLife.68714
194. Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. (2017) 9(377). doi: 10.1126/scitranslmed.aaf2584
195. Cashman JR, Ryan D, McKeithan WL, Okolotowicz K, Gomez-Galeno J, Johnson M, et al. Antiarrhythmic hit to lead refinement in a dish using patient-derived iPSC cardiomyocytes. J Med Chem. (2021) 64(9):5384–403. doi: 10.1021/acs.jmedchem.0c01545
196. McKeithan WL, Feyen DAM, Bruyneel AAN, Okolotowicz KJ, Ryan DA, Sampson KJ, et al. Reengineering an antiarrhythmic drug using patient hiPSC cardiomyocytes to improve therapeutic potential and reduce toxicity. Cell Stem Cell. (2020) 27(5):813–21.e6. doi: 10.1016/j.stem.2020.08.003
197. Mathur L, Szalai B, Du NH, Utharala R, Ballinger M, Landry JJM, et al. Combi-seq for multiplexed transcriptome-based profiling of drug combinations using deterministic barcoding in single-cell droplets. Nat Commun. (2022) 13(1):4450. doi: 10.1038/s41467-022-32197-0
198. Turnbull IC, Bajpai A, Jankowski KB, Gaitas A. Single-cell analysis of Contractile forces in iPSC-derived cardiomyocytes: paving the way for precision medicine in cardiovascular disease. Int J Mol Sci. (2023) 24(17):13416. doi: 10.3390/ijms241713416
199. Fermini B, Coyne ST, Coyne KP. Clinical trials in a dish: a perspective on the coming revolution in drug development. SLAS Discov. (2018) 23(8):765–76. doi: 10.1177/2472555218775028
200. Sayed N, Liu C, Ameen M, Himmati F, Zhang JZ, Khanamiri S, et al. Clinical trial in a dish using iPSCs shows lovastatin improves endothelial dysfunction and cellular cross-talk in LMNA cardiomyopathy. Sci Transl Med. (2020) 12(554). doi: 10.1126/scitranslmed.aax9276
201. Blinova K, Schocken D, Patel D, Daluwatte C, Vicente J, Wu JC, et al. Clinical trial in a dish: personalized stem cell-derived cardiomyocyte assay compared with clinical trial results for two QT-prolonging drugs. Clin Transl Sci. (2019) 12(6):687–97. doi: 10.1111/cts.12674
202. Hashizume R, Yamawaki-Ogata A, Ueda Y, Wagner WR, Narita Y. Mesenchymal stem cells attenuate angiotensin II-induced aortic aneurysm growth in apolipoprotein E-deficient mice. J Vasc Surg. (2011) 54(6):1743–52. doi: 10.1016/j.jvs.2011.06.109
203. Wen H, Wang M, Gong S, Li X, Meng J, Wen J, et al. Human umbilical cord mesenchymal stem cells attenuate abdominal aortic aneurysm progression in sprague-dawley rats: implication of vascular smooth muscle cell phenotypic modulation. Stem Cells Dev. (2020) 29(15):981–93. doi: 10.1089/scd.2020.0058
204. Mulorz J, Shayan M, Hu C, Alcazar C, Chan AHP, Briggs M, et al. Peri-adventitial delivery of smooth muscle cells in porous collagen scaffolds for treatment of experimental abdominal aortic aneurysm. Biomater Sci. (2021) 9(20):6903–14. doi: 10.1039/D1BM00685A
Keywords: thoracic aortic disease, genetic variants, induced pluripotent stem cells, disease modelling, aortic aneurysm, aortic dissection, 3D models
Citation: Singh AA, Shetty DK, Jacob AG, Bayraktar S and Sinha S (2024) Understanding genomic medicine for thoracic aortic disease through the lens of induced pluripotent stem cells. Front. Cardiovasc. Med. 11:1349548. doi: 10.3389/fcvm.2024.1349548
Received: 4 December 2023; Accepted: 31 January 2024;
Published: 19 February 2024.
Edited by:
Daniel Greif, Yale University, United StatesReviewed by:
Lingfeng Luo, Stanford University, United StatesYanming Li, Baylor College of Medicine, United States
© 2024 Singh, Shetty, Jacob, Bayraktar and Sinha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanjay Sinha c3M2NjFAY2FtLmFjLnVr