 Osama Soliman1,2*†
Osama Soliman1,2*† Yogesh Acharya2,3,†
Yogesh Acharya2,3,† Martine Gilard2,4Garry Duffy2,5William Wijns1,2Venkatesh Kannan2,6
Martine Gilard2,4Garry Duffy2,5William Wijns1,2Venkatesh Kannan2,6 Sherif Sultan2,3,7,†
Sherif Sultan2,3,7,†
- 1Department of Cardiology, Galway University Hospital, Galway, Ireland
- 2CORRIB-CURAM-Vascular Group Collaborators, University of Galway, Galway, Ireland
- 3Western Vascular Institute, Department of Vascular and Endovascular Surgery, University Hospital Galway, University of Galway, Galway, Ireland
- 4Department of Cardiology, La Cavale Blanche Hospital, Brest, France
- 5Anatomy and Regenerative Medicine Institute (REMEDI), School of Medicine, University of Galway, Galway, Ireland
- 6Irish Centre for High-End Computing (ICHEC), University of Galway, Galway, Ireland
- 7Department of Vascular Surgery and Endovascular Surgery, Galway Clinic, Doughiska, Royal College of Surgeons in Ireland and University of Galway Affiliated Hospital, Galway, Ireland
Introduction: Neural crest cells (NCCs) are multipotent and are attributed to the combination of complex multimodal gene regulatory mechanisms. Cardiac neural crest (CNC) cells, originating from the dorsal neural tube, are pivotal architects of the cardio-neuro-vascular domain, which orchestrates the embryogenesis of critical cardiac and vascular structures. Remarkably, while the scientific community compiled a comprehensive inventory of neural crest derivatives by the early 1980s, our understanding of the CNC's role in various cardiovascular disease processes still needs to be explored. This review delves into the differentiation of NCC, specifically the CNC cells, and explores the diverse facets of non-syndromic cardiovascular neurocristopathies.
Methods: A systematic review was conducted as per the PRISMA Statement. Three prominent databases, PubMed, Scopus, and Embase, were searched, which yielded 1,840 studies. We excluded 1,796 studies, and the final selection of 44 studies formed the basis of this comprehensive review.
Results: Neurocristopathies are a group of genetic disorders that affect the development of cells derived from the NC. Cardiovascular neurocristopathy, i.e., cardiopathy and vasculopathy, associated with the NCC could occur in the form of (1) cardiac septation disorders, mainly the aortico-pulmonary septum; (2) great vessels and vascular disorders; (3) myocardial dysfunction; and (4) a combination of all three phenotypes. This could result from abnormalities in NCC migration, differentiation, or proliferation leading to structural abnormalities and are attributed to genetic, familial, sporadic or acquired causes.
Discussion: Phenotypic characteristics of cardiovascular neurocristopathies, such as bicuspid aortic valve and thoracic aortic aneurysm, share a common embryonic origin and are surprisingly prevalent in the general population, necessitating further research to identify the underlying pathogenic and genetic factors responsible for these cardiac anomalies. Such discoveries are essential for enhancing diagnostic screening and refining therapeutic interventions, ultimately improving the lives of individuals affected by these conditions.
1 Introduction
The cellular events during embryonic development unveils the remarkable complexity of life within which a particular group of cells, known as the neural crest cells (NCC), holds a pivotal role (1–3). Neurocristopathy a term that encompasses disorders within these NCC (1, 2, 4).
The cardiac neural crest (CNC) cells are specialized entities that assume a commanding role in the cardiovascular development of aortic arch arteries and the cardiac outflow tract (5, 6). The two interrelated conditions—cardiovascular neurocristopathy and NC aortopathy, usually manifest as a bicuspid aortic valve (BAV) (7, 8). BAV is marked by aortic dilation and/or acute aortic dissection (8–10).
The true magnitude of this aortopathy remains veiled, but recent revelations hint at its profound impact (11). While the scientific community once crafted an inventory of NC derivatives, it was not until the early 1980s that the heart revealed its secret—within the vagal NC lies a distinct and dynamic entity, the CNC (12). This population of cells not only shapes the cardiovascular system but also orchestrates the birth of the thymus, the thyroid glands, and the cardiac ganglia (3).
This systematic review is to unravel the intricate differentiation within NCC, with a specific focus on CNC cells. Our quest takes us into non-syndromic cardiovascular neurocristopathies, particularly within the young population, a vulnerable and often overlooked cohort.
2 We illuminate three crucial facets
1. The non-syndromic cardiovascular neurocristopathy, explores a myriad of phenotypes, from NC aortopathy to NC BAV and congenital heart disease (CHD).
2. We inspect the cardiovascular pathologies arising from disruptions in regulatory factors, and we spotlight the profound importance of various signalling pathways that thread through embryonic development.
3. We examine its connections to cerebro-vascular phenomena, offering insights into the mechanisms underpinning associated abnormalities. In doing so, we also explore potential therapeutic advancements.
3 Materials and methods
3.1 Selection strategy
To conduct a comprehensive review, we executed a systematic literature search with a sharp focus on our study objectives. Three prominent databases, PubMed, Scopus, and Embase, served as our sources of information. Our search revolved around the overarching theme of cardiovascular neurocristopathy and its various manifestations, using specific keywords such as “cardiovascular neurocristopathy,” “neural crest aortopathy,” “neural crest bicuspid aortic valve,” “neural crest heart disease,” and “non-syndromic aortopathy or neurocristopathy.” We restricted our search to articles published in the English language from the inception of each database until March 2023.
It is crucial to note that cardiovascular neurocristopathy encompasses both syndromic and non-syndromic cases. However, for this manuscript, our focus remained solely on non-syndromic neurocristopathy. This decision led to the exclusion of articles that discussed acquired causes and multi-organ involvement associated with syndromic neurocristopathy.
4 Results
4.1 Selection criteria
Our selection criteria adhered to the guidelines set by the PRISMA Statement (13). The initial search sought to map the existing literature concerning cardiovascular neurocristopathy, NC aortopathy, BAV and CHD. Subsequently, our search was fine-tuned to target the non-syndromic subset and the young-age population, defined as individuals between the ages of 18 and 65 as per WHO guidelines. The specific focus remained on non-syndromic NC aortic conditions and associated myocardial dysfunction, prompting the exclusion of articles dealing with syndromes, acquired causes, or multiorgan involvement.
Our diligent search yielded a total of 1,840 articles. Following a meticulous review process, 1,796 research articles were excluded, taking into account duplications and alignment with our inclusion criteria. This stringent process culminated in a final selection of 44 articles that form the basis of this comprehensive review.
4.2 Quality assessment and data collection
This review is anchored in original full-text research articles and review papers. The thoroughness of our approach was maintained by vigilant scrutiny to eliminate duplications. The abstracts of these articles underwent rigorous analysis to ensure their quality and relevance to our study objectives. Subsequently, each research paper was meticulously evaluated to gauge its suitability for inclusion in this review.
Following the removal of duplicate records, an additional four articles were excluded due to a lack of full-text availability. Ultimately, 40 articles were included in this final review paper, aligning with our objectives, which is outlined in the given PRISMA flow diagram (Figure 1).
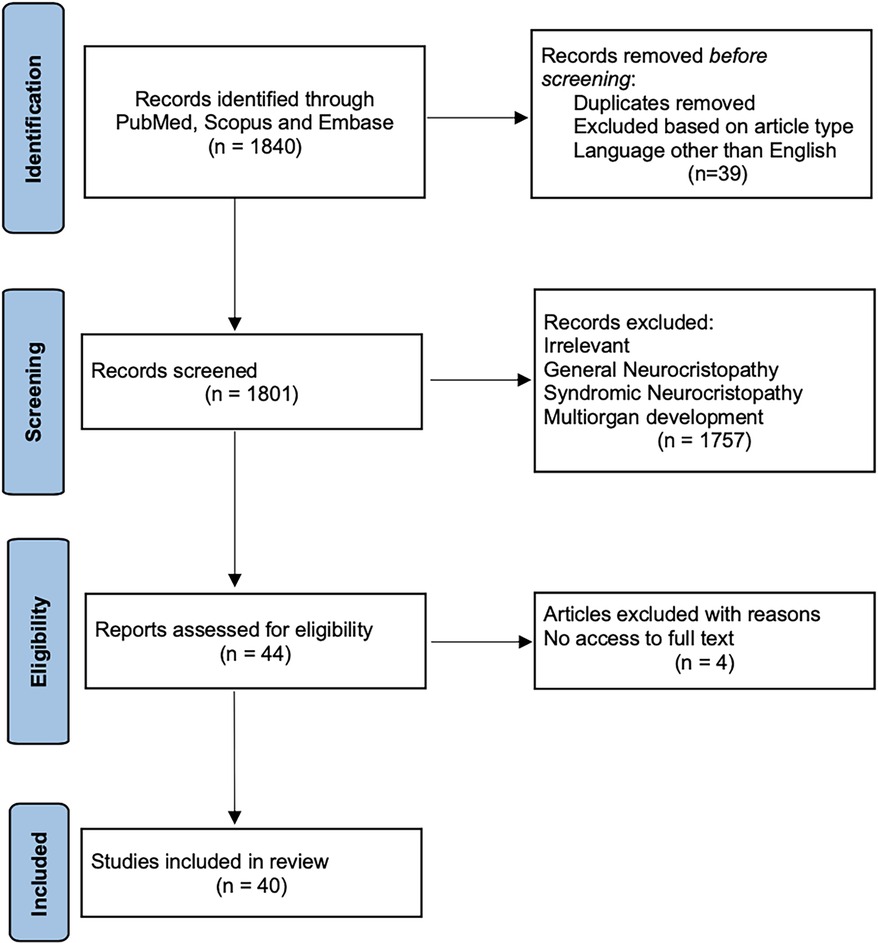
Figure 1. PRISMA flow diagram. This graph serves as a visual representation of the literature inclusion and exclusion at each stage, in accordance with the PRISMA Statement, guiding our systematic and methodical approach in conducting this comprehensive review.
4.3 Embryological origin of vascular tree
Vascular tree has a variable embryonic origin (Figure 2) (14–16). Embryonic fate-mapping studies have demonstrated that the aortic root, ascending aorta and aortic arch are populated by vascular smooth muscle cells (VSMCs) arising from NC, whereas VSMCs from the paraxial mesoderm or somites populate the descending aorta (17, 18). Studies have shown that CNS cells also contribute to the VSMCs found in some of the branchial arch arteries (19).
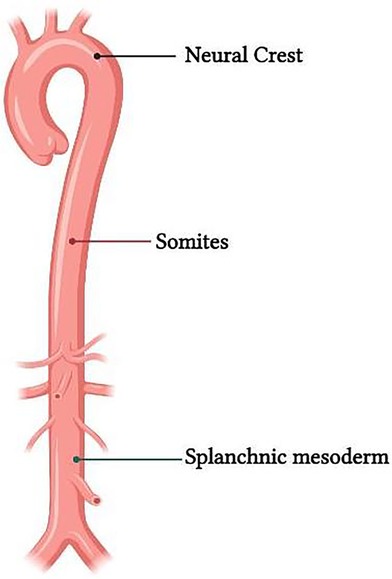
Figure 2. Representation of the difference in embryological origin of the thoracic and abdominal aorta.
4.4 Neural crest cells (NCC)
NCC originate from the neuroepithelium, i.e., the exterior layer or ectoderm, which is responsible for forming the neural tube that eventually becomes the central nervous system (3, 20). Gastrulation causes mesodermal structures to develop between the endoderm and ectoderm. The paraxial mesoderm divides into segmented groups of cells known as somites, which are found on either side of the neural tube. Although these somites are only temporary, they can produce a wide range of tissues and organs (21).
During the early stages of embryonic development, the NC exists as a narrow band of cells located between the neural and non-neural ectoderm (3, 20). The NCCs migrate dorsally and then delaminate in a rostrocaudal pattern as the neural tube shuts (Figure 3). The highly multipotent and temporarily migratory NC is a vertebrate-specific cell population divided into four main subpopulations based on their migratory path, terminal location, and differential abilities: cranial, vagal, trunk, and sacral (22).
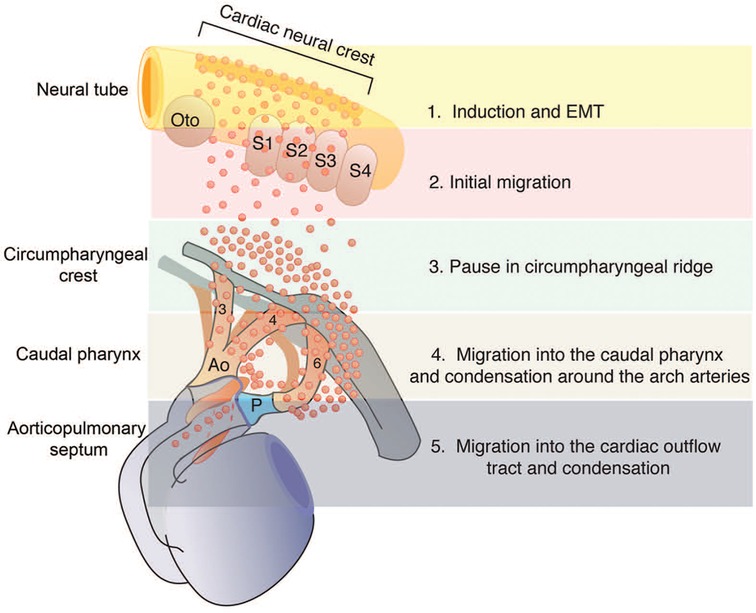
Figure 3. The migration and dispersion of cardiac neural crest cells (NCC). This graph displays the migration and dispersion of cardiac NCC from the neural tube to the caudal pharynx and then to the outflow tract (OFT). Reprinted with permission from Kirby et al. (22).
4.5 Cardiac neural crest (CNC) cells
Studies have indicated that the vagal NC consists of a smaller specified group of cells termed as the CNC, known to significantly contribute to cardiovascular development, along with aiding in the development of the thymus, thyroid glands, and cardiac ganglia (3, 23). CNC cells develop along the neural tube from the mid-diencephalon to the embryo's most caudal extremity. These crest cells are classified as cranial or trunk based on their ability to generate ecto-mesenchyme (22).
CNC cells have an impact on heart formation both directly and indirectly (6, 11) (Table 1). Alignment and outflow septation are two distinct phases that co-occur simultaneously during cardiac morphogenesis. In the alignment phase, the cardiac tube aligns with the developing vasculature, while in the outflow septation phase, the ventricular outflow tract is divided into the aorta and pulmonary artery. Also, CNC cells are necessary for correct looping and extension of the heart tube, including early myocardial function, and are crucial in the formation of the outflow septum and arch artery patterning (22, 24).
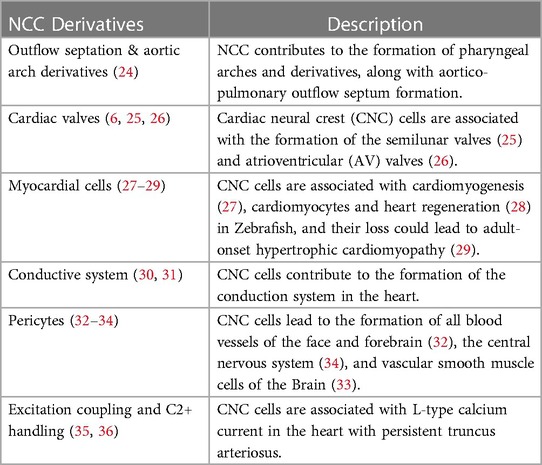
Table 1. Table showing the cardiovascular neural crest cells (NCC) derivatives.
4.6 Cellular mechanism of NC differentiation
During gastrulation, CNC cells are induced from the dorsal region of the neural plate border, orchestrated by various regulatory pathways such as Bmp, Wnt, Notch, and, more recently recognized, Hippo (Figure 4) (37).
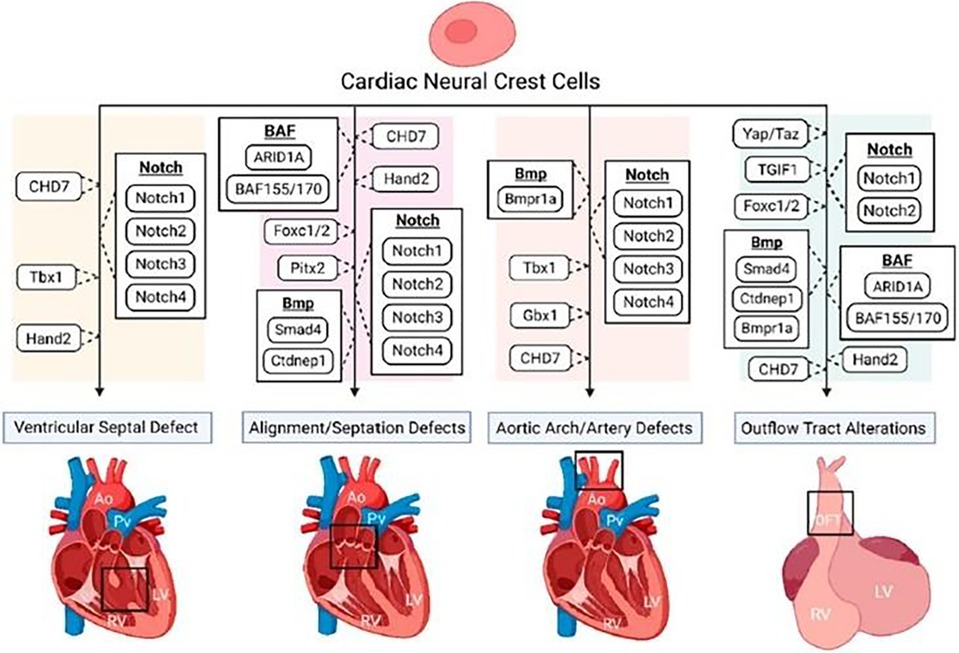
Figure 4. The disruption of various regulatory pathways of cardiac neural crest (CNC) cells. This figure shows the role of CNC cells in numerous congenital heart defect phenotypes. Reprinted with permission from Erhardt et al. (36).
NC progenitors obtain their migratory potential by undergoing epithelial-to-mesenchymal transition (EMT) (38). It has been demonstrated that different signaling pathways, such as BMP, WNT, and retinoic acid, play critical roles in regulating NC EMT and migration for proper cardiovascular development. However, more research into the crosstalk between signalling networks is required. Delamination from the neural tube is regulated via BMP-dependent Wnt1 activity, with Wnt1 expression turning off soon after the cells leave the neural tube (39). Many transcription factors and signalling molecules have been implicated in the later migration steps, proliferation, survival, and differentiation of the CNC.
Studies have also implicated the Notch signalling pathway for healthy NC development, including cellular proliferation and specification (40). Mutations in Notch-target genes such as Notch1 and Notch2, as well as Notch ligands such as Jagged1, have been demonstrated to produce a range of heart abnormalities in mice, including VSD and malformations of the cardiac OFT and great arteries (41–43).
Varadkar and colleagues (43) discovered that Notch2 is required for proper NC-derived aortic and pulmonary smooth muscle formation and disrupting Notch2 in post-migratory CNC cells with the Pax3-Cre driver results in narrowed OFT and OFT arteries (aorta and pulmonary) in E17.5 mouse embryos, indicating a cell-autonomous role for Notch2 in CNC cells. However, there is a lack of studies that look into how Notch manipulation contributes to CNC-derived heart development (37).
4.7 Multipotency of NCC
NCCs are multipotent, capable of differentiating into various cells in our bodies (18, 44). The multipotent capacity of the NCC are attributed to the combination of complex multimodal gene regulatory mechanism (44). Studies have shown that CNC could be differentiated into VSMCc, which could be employed for disease modelling and drug screening in relation to vasculopathies (17, 18).
Furthermore, second heart field (SHF) progenitors are multipotent cardiac progenitor cells that play an essential role with the cardiac NCCs in the development of the OFT and closely interact with each other (37). As such, SHF is responsible for forming the heart tube, myocardium, smooth muscle, and endothelial cells.
4.8 Cardiovascular neurocristopathy
Neurocristopathies are a group of genetic disorders that affect the development of cells derived from the NC. These could be from abnormalities in NCC migration, differentiation, or proliferation leading to structural abnormalities (45).
We have used the term cardiovascular neurocristopathy here to describe cardiopathy, aortopathy, and its related sequelae of the vascular and cardiac-specific anatomic region related to the CNC cell migration.
Cardiovascular neurocristopathy occurs in the form of (1) cardiac septation disorders, mainly the aortico-pulmonary septum; (2) great vessels and vascular disorders; (3) myocardial dysfunction; and (4) a combination of all three phenotypes. This could be attributed to genetic, familial, sporadic or acquired causes (Figure 5) (5). For ease of understanding, we have divided the cardiovascular neurocristopathy into cardiac and vascular neurocristopathy.
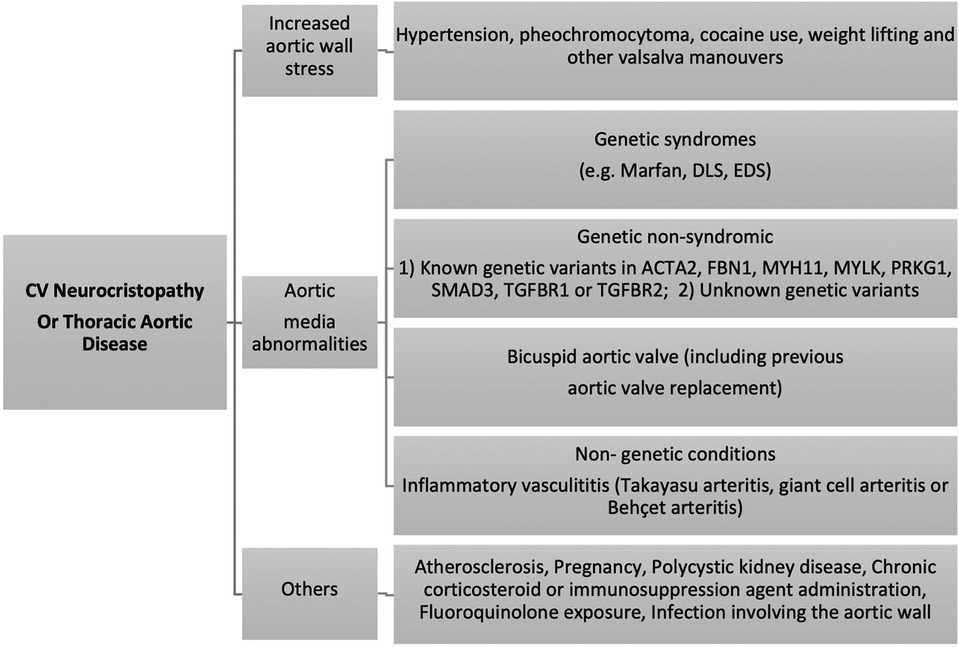
Figure 5. Cardiovascular neurocristopathy aetiologies and mechanisms.
Ablation studies have shown that NC abnormalities resulted in excitation-contraction (EC) coupling defects, especially contractility and L-type calcium current, causing abnormal myocardial function and death in-utero, resulting from cardiac failure (35, 36, 46). After CNC ablation, all the embryos showed abnormal myocardial function, mis-patterning of the arch arteries and glandular defects. In comparison, persistent truncus arteriosus was observed in 90%, while the remaining showed arterial pole misalignment defects, such as double-outlet right ventricles (5, 20, 35, 36, 46, 47).
4.8.1 Cardiac neurocristopathy
A failure of CNC cells to migrate to the developing heart can result in cardiac neurocristopathy, including abnormality of cardiac septation, outflow tract obstruction, myocardial dysfunction, and heart rhythm abnormality (6).
The abnormal aorto-left ventricular interaction could lead to altered aortic dilatation and loss of elasticity, which can induce left ventricular hypertrophy, reduce coronary blood flow, and eventually cause left ventricular failure (5, 6, 11). However, the most important clinical phenotype of cardiac neurocristopathy is the BAV.
BAV, the most common congenital cardiovascular abnormality, affects 0.9%–2% of the general population (7, 48). It causes greater morbidity and mortality than all other congenital cardiovascular abnormalities combined.
BAV patients' VSMCs are less differentiated due to a phenotypic switch failure, which results in considerably decreased expression of differentiated, contractile VSMC markers such as smoothelin, calponin, and SM22alpha. Furthermore, lamina A/C, which is important for VSMC differentiation, is considerably expressed lower in the BAV population compared to the tricuspid aortic valves (TAV) population (9).
Furthermore, although many CHDs are considered non-syndromic, certain hereditary syndromes, like DiGeorge and CHARGE, entail NC deficits with established cardiac phenotypes (37). Not only do NC deficits cause cardiac disorders, but they are also known to contribute to a variety of craniofacial malformations that are frequent in human cardio-craniofacial syndromes, including the most common multiple anomaly syndromes, DiGeorge, Noonan, and Velo-cardio-facial (49, 50).
4.8.2 Vascular neurocristopathy
Defects in NC migration and differentiation can also lead to abnormalities in the aortic arch and the great vessels of the head and neck, resulting in conditions such as coarctation of the aorta, interrupted aortic arch, and abnormalities in the origins of the brachiocephalic vessels (6, 51). This includes the thoracic aorta, innominate artery, its branches, left common carotid, left subclavian and OFT septae.
Aortic diseases involving the NC zone or vascular neurocristopathies could be categorized as (1) abnormal elasticity or increased aortic wall stress, (2) dilatation and aneurysm formation, and (3) dissection and rupture.
Vascular neurocristopathy constitutes aortic arch and carotid artery disorders, the most common phenotypes being the thoracic aortic aneurysm (TAA). As NC contributes to smooth muscle cells (SMCs) of the aortic arch, the innominate, and the right and left common carotid arteries, carotid artery dissection is other possible non-syndromic neurocristopathies. Additionally, defects in pericyte function can lead to weakened blood vessels, making them more prone to dissection. Pericytes in the aorta could have different origins, including SHF, neural crest, and somites (32, 33, 52, 53). Recent studies also suggest that gene mutations increase the risk of cervical carotid dissection. These mutations can affect the formation of the arterial wall, making it more susceptible to tearing (54).
Dilatation of the thoracic aorta could be due to intrinsic, hemodynamic abnormalities or both. Intrinsic here refers to the genetic theory, whereby the presence of aortic wall fragility is a consequence of a common developmental defect involving the aortic valve and the aortic wall (55, 56). TAA can be either hereditary or sporadic, the latter of which occurs without any known family history. It is estimated that genetic factors are responsible for about 25% of all TAA cases. Of this percentage, one-fifth are linked to a known genetic disorder, while the remaining patients with TAA have a family history of aneurysmal disease, but the specific gene causing it is yet unknown (54, 57).
One such condition is acute aortic syndrome (AAS), coined by Vilacosta et al. in 1998, which is a group of severe and potentially fatal aortic conditions, such as acute aortic dissection (AAD), traumatic aortic transection (TAT), intramural hematoma (IMH), penetrating aortic ulcers (PAU), and TAA post-SAD, all of which cause significant risk to life (58, 59).
Cervical carotid dissection and aberrant right subclavian artery are other such phenotypes linked with NCC, however, its exact cause is not yet fully understood (60). During embryonic development, the NCC migrate and differentiate into smooth muscle cells, which form the arterial wall, and pericytes, which support the developing blood vessels (32). Cardiac fibroblasts are derived from heterogeneous source, including the hematopoietic system, endothelium, epicardium, and neural crest (61).
Abnormalities in NCC migration, differentiation, or proliferation could lead to structural abnormalities in the arterial wall, making it more susceptible to tearing. Additionally, defects in pericyte function can lead to weakened blood vessels, making them more prone to dissection (32).
An aberrant right subclavian artery is also associated with abnormal NC migration. When the right fourth pharyngeal arch artery regresses improperly, it causes an aberrant right subclavian artery. In this situation, the right dorsal aorta remains abnormally cranial to the seventh intersegmental artery and creates the retroesophageal section of the right subclavian artery. This vascular abnormality is strongly linked to CHD affecting the outflow tract (OFT) (62).
4.8.3 Co-occurrence of cardiac & vascular neurocristopathy
The semilunar valves' embryonic genesis is linked to the formation of the ascending aorta (25). During late gestation, the NC plays a crucial role in developing the outflow endocardial cushions, the precursors of the semilunar valves. The NC is responsible for remodelling and positioning the cushions, mesenchymal apoptosis, and proper valve architecture (63). Moreover, it also contributes to forming the smooth muscle layer wall of the ascending aorta and aortic arch. Therefore, any defects in the CNC can result in semilunar valve functional abnormalities and/or aortic arch artery abnormalities. Abnormalities in the process of NCC condensation at the early stages of outflow cushion formation may provide a common mechanism underlying BAV and explain the link between arterial wall anomalies and outflow malalignment defects. As a result, a disruption in the early developing pathways might result not only in a BAV but also in the accompanying aortopathy. Given this, BAV patients are at a significantly elevated risk of developing a thoracic aortic aneurysm (Figure 6) (64).
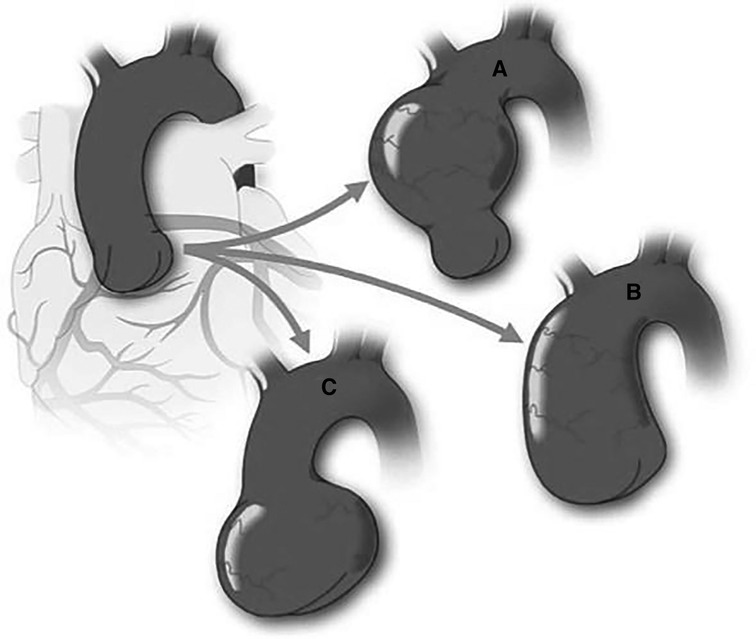
Figure 6. Schematic of variable aortic phenotypes encountered in bicuspid aortic valve (BAV). Figure demonstrates the different aortic dilatation patterns that may occur in BAV in comparison with a normal aorta (Top left). Although the most common portion to dilate is the tubular ascending aorta (A), the entire ascending aorta may be affected, including sinuses of Valsalva and tubular aorta with sinotubular junction effacement (B) There is a subgroup of BAV patients who exhibit dilatation of the sinuses of valsalva preferentially (C) This pattern is associated with type 1 (right-left fusion) BAV and male sex. Reprinted with permission from Michelena et al. (7). Copyrights 2023 Wolters Kluwer Health, Inc.
Patients with BAV might have medial abnormalities in the form of elastic fibre fragmentation and smooth muscle cell loss, previously termed “cystic medial necrosis”, which is like Marfan syndrome (65). However, the flow abnormalities associated with BAV may contribute to the aortopathy and aortic dilatation (9). TAAs form in roughly 50%–70% of BAV patients, much greater than the incidence of TAA generation in patients with tricuspid aortic valves (TAV) (66).
Aortic dilatation occurs early in the BAV person and is often characterized by mid-ascending dilation. It is still uncertain whether aneurysms in BAV patients are caused by changed hemodynamic forces caused by the defective valve or by an underlying genetic flaw that causes both BAV and TAA (48). The evaluation of hemodynamic flow in BAV patients reveals major changes in flow patterns compared to TAV patients (67).
As BAV is a heterogeneous disease with different aortic phenotypes, any decision to extend a proximal aortic repair into the arch while repairing the BAV valve must be individualized. Small subsets of patients may benefit from prophylactic hemiarch resection during proximal repair. However, it is challenging to identify those patients who could potentially develop proximal arch disease.
Furthermore, people with BAV have an eight-fold increased risk of aortic dissection (68). Despite aortic valve replacement to improve hemodynamics, some BAV patients develop an ascending aortic aneurysm (69). Using aorta tissue from BAV patients, researchers discovered possible indications of aortopathy of the aortic wall, including immature SMCs (9). Aneurysms frequently include the aortic root, ascending aorta, and aortic arch, although the descending aorta is rarely compromised in BAV/TAA (10).
Congenital BAV is the first nonsyndromic CHD where aortic dissection and dilation are documented (21). Aortic dissection is nine times more common in BAV patients than in tricuspid aortic valve patients and presents at a relatively younger age (55 vs. 63 years) (57). In BAV patients, aortic dilatation starts in childhood, independent of aortic regurgitation or aortic stenosis (70).
Histopathological studies of non- and dilated ascending aortic wall samples from BAV and TAV patients have shown several structural discrepancies in recent years. Larson et al. reported one of the first studies that revealed variations in the ascending aorta of BAV and TAV in 1984 (71). Vascular smooth muscle cells (VSMCs) play an important role in distinguishing BAV from TAV histology. VSMCs are not only detected in apoptosis, but they are also physically different in the BAV population (72).
4.9 CNC ablation phenotypes
The significance of CNC cells in heart development and function has come from research on chick embryos after the pre-migratory CNC was removed (11). This NC ablation paradigm was the first reliable animal experimental model of congenital heart abnormalities and has served as the “gold” standard for characterizing the aetiology of heart problems in later experimental models, including transgenic mice (24).
The major component of the CNC ablation phenotype is myocardial function abnormalities. Myocardial dysfunction, like the looping defect, is initially noticed in NC-ablated embryos around the time NCC should migrate into the caudal pharyngeal arches, many days before the typical entry of NCC into the outflow canal (73). CNC ablation also results in altered SHF proliferation and abnormal myocardial function as secondary effects (25). Following CNC ablation, abnormal looping is the earliest visible defect observed, even before CNC cells reach the OFT. Furthermore, defective looping could also be seen if the OFT myocardium fails to add to the heart tube from the anterior subpopulation of SHF cells. Thus, the looping defects observed after CNC ablation suggest that CNC cells are required for the normal deployment of SHF cells. Altered looping could also lead to hemodynamic changes, potentially causing flow related pathologies (74).
These embryos adjust for reduced contractility by dilatation of the ventricles, allowing them to retain enough cardiac output for survival (75). These functional compensations during very early cardiac development are hypothesized to play an etiologic role in the eventual development of structural heart abnormalities. At older ages, when septation would ordinarily be complete, these embryos have a lower ejection percentage and poor contractility in the myocardium (49). Reduced embryo weight and oedema also suggest poor cardiac function (76).
CNC ablation also results in altered SHF proliferation and abnormal myocardial function as secondary effects (77). Abnormal looping is the earliest defect seen after CNC ablation and can be observed before the CNC cells reach the OFT. Defective looping may be caused by the failure of the addition of the OFT myocardium to the heart tube from the anterior subpopulation of SHF cells. Thus, the looping defects observed after CNC ablation suggest that CNC cells are required for the normal deployment of SHF cells (78).
4.10 Scrutiny of the systematic review for 40 articles
While this systematic review has endeavoured to shed light on the complex realm of neurocristopathy, it is important to acknowledge its inherent limitations. The vast heterogeneity observed in the 40 manuscripts analyzed underscores the intricate and multifaceted nature of this field, revealing that the science surrounding neurocristopathy is still in its infancy. Here, we outline the key limitations of this review:
4.11 Heterogeneity of data
The foremost limitation arises from the remarkable heterogeneity observed within the selected manuscripts. The diversity in study design, patient populations, and research methodologies across the included articles made it challenging to conduct a uniform analysis. This heterogeneity emphasizes the need for standardized approaches in future investigations in neurocristopathies.
4.12 Limited sample size
Many of the manuscripts under review presented data from relatively small sample sizes. This limitation raises questions about the generalizability of the findings and underscores the need for larger and more comprehensive studies to draw definitive conclusions.
4.13 Heterogeneity in definitions and diagnostic criteria
The lack of universally accepted definitions and diagnostic criteria for neurocristopathy-related conditions posed a significant challenge. This variability in terminology and classification across studies hinders efforts to consolidate and compare data accurately.
4.14 Data quality and reporting
The overall quality of data and reporting in some of the included manuscripts was variable. Incomplete data, vague reporting of methods, and limited statistical analyses in certain articles hindered our ability to perform a robust and consistent review.
4.15 Publication bias
Systematic reviews are inherently susceptible to publication bias, where positive findings are more likely to be published than negative or inconclusive ones. This bias may have influenced the selection of studies included in this review, potentially skewing the overall findings.
4.16 Inherent challenges of a nascent field
The review's limitations are compounded by the fact that the field of neurocristopathy is still in its infancy. The limited body of research and the evolving understanding of the subject matter pose inherent challenges in conducting a comprehensive review.
4.17 Lack of longitudinal data
Many of the manuscripts presented cross-sectional data, lacking longitudinal follow-up of patients. Long-term outcomes and the natural history of neurocristopathy-related conditions remain areas of uncertainty.
4.18 Language and geographic bias
The restriction to articles published in the English language may introduce language bias, potentially omitting relevant research published in other languages.
As the science surrounding neurocristopathy is still evolving and much work remains to be done, addressing these limitations requires collaborative efforts from the scientific community to standardize definitions, conduct larger and more comprehensive studies, and foster a global and multidisciplinary approach to advancing our understanding. Despite the challenges, this systematic review represents a critical step in the ongoing journey to unravel the mysteries of neurocristopathy and pave the way for more effective diagnosis and treatment in the future.
5 Discussion
The traditional perspective on atherosclerosis as a leading player in the development of aortic aneurysms and dissections has been challenged by emerging research. While the well-established link between atherosclerosis and these conditions cannot be denied, recent studies have introduced complexity to this narrative. These studies raise questions about the multifactorial nature of aortopathy, suggesting that the causes might extend beyond atherosclerosis (79).
Herein, we contemplate the role of CNC cells and other factors, such as the gut microbiome and intrinsic inflammatory processes, in shaping the landscape of aortopathy. This evolving understanding urges us to explore the intricate web of interconnected factors that contribute to the pathogenesis of aortopathy (80).
Of paramount importance is the role played by CNC cells in the genesis of CHDs. As our ability to screen and detect CHDs in childhood has improved, we have witnessed a rise in the recognition of CNC deficit phenotypes associated with CHDs. This observation underscores the interplay between aortopathy and cardiac abnormalities and prompts the need for further research to identify the underlying pathogenic factors and genetic deficits responsible for these cardiac anomalies. Such discoveries are essential for enhancing diagnostic screening and refining therapeutic interventions, ultimately improving the lives of individuals affected by these conditions (57, 81).
Phenotypes characteristic of cardiovascular neurocristopathy, such as BAV and TAA, share a common embryonic origin in CNC cells and are surprisingly prevalent in the general population. BAV, with or without associated aortopathy, affects around 2% of the population, while TAA is observed in 1% of individuals. Within this realm, TAA encompasses hereditary and sporadic cases, with an estimated 25% of all TAA cases having a genetic component (7, 48).
About one-fifth of TAA cases are linked to known genetic disorders, while the remaining cases have a positive family history of the condition, albeit with an unknown causative gene (70). This complex landscape emphasizes the need for a deeper understanding of thoracic aorta development and both normal and defective aortic valves (6). In particular, it is critical, given the lack of available medications to prevent TAA in BAV patients.
The development of aortopathies, including aneurysms and dissections, is a potentially life-threatening process, fraught with the lifelong risk of catastrophic aortic events. Detecting thoracic aortic aneurysms early and preventing acute aortic syndromes is a formidable challenge.
Historically, formal screening of aortic diameter has been the primary method for identifying these conditions and preventing life-threatening complications. However, the increasing incidence of potentially fatal aortopathies, such as acute aortic syndromes, demands a more profound understanding of aortic pathologies at the cellular level and their genetic underpinnings. This knowledge is essential for anticipating the likelihood of aortic complications in specific patients and tailoring early preventive and therapeutic interventions to reduce cardiovascular morbidity and mortality (57).
In essence, a better grasp of cardiovascular neurocristopathies paves the way for a deeper understanding of disease pathophysiology, enabling the formulation of precision management strategies for these potentially fatal cardiovascular conditions. This journey towards precision medicine is one that holds the promise of better outcomes for those grappling with aortopathies and related cardiac anomalies.
5.1 Future perspective
It is imperative to gain a precise understanding of the ultimate phenotype of central cardiovascular structures. This understanding will pave the way for the separation of processes contributing to this final phenotype. By defining novel models of cardiovascular dysmorphogenesis, we can explore interactions of structure and function, a dynamic relationship that ultimately shapes the phenotype. This journey will enable us to comprehend the involvement of specific genes in the early stages of dysmorphogenesis and the factors influencing cardiovascular patterning.
Molecular research in cardiovascular neurocristopathy represents a pivotal moment in our understanding of cardiovascular health, offering the promise of insights into the intricate mechanisms guiding the formation of the heart.
Genetic abnormalities associated with aortopathies underscore the critical role played by smooth muscle cells and the extracellular matrix in shaping the destiny of cardiovascular neurocristopathies. These revelations provide a glimpse into cardiovascular system, shedding light on the genetic underpinnings of these conditions.
Furthermore, the highly heritable nature of aortic size and elasticity points to the potential role of multiomics and polygenic risk scores in the early detection of sporadic cases of cardiovascular neurocristopathies. This promising avenue could revolutionize our approach to screening and diagnosis, enabling early intervention and personalized management strategies.
Molecular insights, genetic revelations, and advanced screening techniques converge to offer a brighter and healthier future for those who contend with these intricate cardiovascular conditions. This exciting period in heart development promises to reshape the landscape of cardiovascular medicine and enhance our ability to prevent, diagnose, and treat these challenging disorders.
6 Conclusion
Cardiovascular neurocristopathy emerges as a possible instigator of a spectrum of cardiac and vascular aortopathies, calling into question the very foundations of our understanding of these conditions. To navigate this uncharted territory and pave the way for a brighter, healthier future for those who grapple with these intricate cardiovascular anomalies, we advocate for a concerted effort in preclinical and clinical research.
In the realm of cardiovascular health, the enigmatic world of neurocristopathies unfolds as a potential harbinger of thoracic aneurysms, dissections, and the catastrophic rupture of life's vital conduits. It beckons us to explore the uncharted territories of the heart and vascular system, revealing a panorama of pathologies that remain largely unaddressed by conventional disease paradigms. By delving into the differentiation of CNC cells, we can unlock the mysteries of cardiac neurocristopathies. This knowledge holds the potential to reshape our approach to diagnosis, treatment, and prevention, offering a lifeline to those at risk of these devastating cardiovascular events.
The cardiovascular community is poised to rewrite the narrative of aortic pathologies and cardiac dysmorphogenesis. The quest for a deeper understanding of CNC cells and their role in cardiac neurocristopathies represents a beacon of hope, promising to illuminate the path toward better cardiovascular health and a future where these potentially life-threatening conditions are conquered.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
OS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. YA: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing, Validation. MG: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. GD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. WW: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. VK: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. SS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bolande RP. The neurocristopathies: a unifying concept of disease arising in neural crest maldevelopment. Hum Pathol. (1974) 5(4):409–29. doi: 10.1016/S0046-8177(74)80021-3
2. Bolande RP. Neurocristopathy: its growth and development in 20 years. Pediatr Pathol Lab Med. (1997) 17(1):1–25. doi: 10.1080/15513819709168343
3. Mayor R, Theveneau E. The neural crest. Development. (2013) 140(11):2247–51. doi: 10.1242/dev.091751
4. Komiyama M. Cardio-cephalic neural crest syndrome: a novel hypothesis of vascular neurocristopathy. Interv Neuroradiol. (2017) 23(6):572–6. doi: 10.1177/1591019917726093
5. Keyte A, Hutson MR. The neural crest in cardiac congenital anomalies. Differntiation. (2012) 84(1):25–40. doi: 10.1016/j.diff.2012.04.005
6. Plein A, Fantin A, Ruhrberg C. Neural crest cells in cardiovascular development. Curr Top Dev Biol. (2015) 111:183–200. doi: 10.1016/bs.ctdb.2014.11.006
7. Michelena HI, Prakash SK, Della Corte A, Bissell MM, Anavekar N, Mathieu P, et al. Bicuspid aortic valve: identifying knowledge gaps and rising to the challenge from the international bicuspid aortic valve consortium (BAVCon). Circulation. (2014) 129(25):2691–704. doi: 10.1161/CIRCULATIONAHA.113.007851
8. Jiao J, Xiong W, Wang L, Yang J, Qiu P, Hirai H, et al. Differentiation defect in neural crest-derived smooth muscle cells in patients with aortopathy associated with bicuspid aortic valves. EBioMedicine. (2016) 10:282–90. doi: 10.1016/j.ebiom.2016.06.045
9. Grewal N, Gittenberger-de Groot AC, Poelmann RE, Klautz RJ, Lindeman JH, Goumans M-J, et al. Ascending aorta dilation in association with bicuspid aortic valve: a maturation defect of the aortic wall. J Thorac Cardiovasc Surg. (2014) 148(4):1583–90. doi: 10.1016/j.jtcvs.2014.01.027
10. Fazel SS, Mallidi HR, Lee RS, Sheehan MP, Liang D, Fleischman D, et al. The aortopathy of bicuspid aortic valve disease has distinctive patterns and usually involves the transverse aortic arch. J Thorac Cardiovasc Surg. (2008) 135(4):901–7.2. doi: 10.1016/j.jtcvs.2008.01.022
11. Hutson MR, Kirby ML. Neural crest and cardiovascular development: a 20-year perspective. Birth defects res. Part C Embryo Today. (2003) 69(1):2–13. doi: 10.1002/bdrc.10002
12. Tang W, Bronner ME. Neural crest lineage analysis: from past to future trajectory. Development. (2020) 147(20):dev193193. doi: 10.1242/dev.193193
13. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Ann Intern Med. (2009) 151(4):264–9. doi: 10.7326/0003-4819-151-4-200908180-00135
14. Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. (2007) 27(6):1248–58. doi: 10.1161/ATVBAHA.107.141069
15. Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. (2000) 127(8):1607–16. doi: 10.1242/dev.127.8.1607
16. Wasteson P, Johansson BR, Jukkola T, Breuer S, Akyürek LM, Partanen J, et al. Developmental origin of smooth muscle cells in the descending aorta in mice. Development. (2008) 135(10):1823–32. doi: 10.1242/dev.020958
17. Shen M, Quertermous T, Fischbein MP, Wu JC. Generation of vascular smooth muscle cells from induced pluripotent stem cells: methods, applications, and considerations. Circulation Res. (2021) 128(5):670–86. doi: 10.1161/CIRCRESAHA.120.318049
18. Shen M, Liu C, Wu JC. Generation of embryonic origin-specific vascular smooth muscle cells from human induced pluripotent stem cells. Methods Mol Biol. (2022) 2429:233–46. doi: 10.1007/978-1-0716-1979-7_15
19. Oh J, Richardson JA, Olson EN. Requirement of myocardin-related transcription factor-B for remodeling of branchial arch arteries and smooth muscle differentiation. Proc Natl Acad Sci U S A. (2005) 102(42):15122–7. doi: 10.1073/pnas.0507346102
20. Noisa P, Raivio T. Neural crest cells: from developmental biology to clinical interventions. Birth Defects Res C Embryo Today. (2014) 102(3):263–74. doi: 10.1002/bdrc.21074
21. Sato Y. Dorsal aorta formation: separate origins, lateral-to-medial migration, and remodeling. Dev Growth Diff. (2013) 55(1):113–29. doi: 10.1111/dgd.12010
22. Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. (1983) 220(4601):1059–61. doi: 10.1126/science.6844926
23. Gilbert SF. An introduction to early developmental processes. In: Developmental Biology, 6th ed. Sunderland, MA: Sinauer Associates (2000). https://www.ncbi.nlm.nih.gov/books/NBK9992/
24. Kirby ML, Waldo KL. Neural crest and cardiovascular patterning. Circ Res. (1995) 77(2):211–5. doi: 10.1161/01.RES.77.2.211
25. Jain R, Engleka KA, Rentschler SL, Manderfield LJ, Li L, Yuan L, et al. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J Clin Invest. (2011) 121(1):422–30. doi: 10.1172/JCI44244
26. Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. (2006) 98(12):1547–54. doi: 10.1161/01.RES.0000227505.19472.69
27. Sato M, Yost HJ. Cardiac neural crest contributes to cardiomyogenesis in zebrafish. Dev Biol. (2003) 257(1):127–39. doi: 10.1016/S0012-1606(03)00037-X
28. Tang W, Martik ML, Li Y, Bronner ME. Cardiac neural crest contributes to cardiomyocytes in amniotes and heart regeneration in zebrafish. Elife. (2019) 8:e47929. doi: 10.7554/eLife.47929
29. Abdul-Wajid S, Demarest BL, Yost HJ. Loss of embryonic neural crest derived cardiomyocytes causes adult onset hypertrophic cardiomyopathy in zebrafish. Nat Commun. (2018) 9(1):4603. doi: 10.1038/s41467-018-07054-8
30. Gorza L, Schiaffino S, Vitadello M. Heart conduction system: a neural crest derivative? Brain Res. (1988) 457(2):360–6. doi: 10.1016/0006-8993(88)90707-X
31. van den Hoff MJ, Moorman AF. Cardiac neural crest: the holy grail of cardiac abnormalities? Cardiovasc Res. (2000) 47(2):212–6. doi: 10.1016/S0008-6363(00)00127-9
32. Etchevers HC, Vincent C, Le Douarin NM, Couly GF. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development. (2001) 128(7):1059–68. doi: 10.1242/dev.128.7.1059
33. Korn J, Christ B, Kurz H. Neuroectodermal origin of brain pericytes and vascular smooth muscle cells. J Comp Neurol. (2002) 442(1):78–88. doi: 10.1002/cne.1423
34. Girolamo F, Errede M, Bizzoca A, Virgintino D, Ribatti D. Central nervous system pericytes contribute to health and disease. Cells. (2022) 11(10):1707. doi: 10.3390/cells11101707
35. Creazzo TL. Reduced L-type calcium current in the embryonic chick heart with persistent truncus arteriosus. Circ Res. (1990) 66(6):1491–8. doi: 10.1161/01.RES.66.6.1491
36. Creazzo TL, Godt RE, Leatherbury L, Conway SJ, Kirby ML. Role of cardiac neural crest cells in cardiovascular development. Annu Rev Physiol. (1998) 60:267–86. doi: 10.1146/annurev.physiol.60.1.267
37. Erhardt S, Zheng M, Zhao X, Le TP, Findley TO, Wang J. The cardiac neural crest cells in heart development and congenital heart defects. J Cardiovasc Dev Dis. (2021) 8(8):89. doi: 10.3390/jcdd8080089
38. Chen T, You Y, Jiang H, Wang ZZ. Epithelial-mesenchymal transition (EMT): a biological process in the development, stem cell differentiation, and tumorigenesis. J Cell Physiol. (2017) 232(12):3261–72. doi: 10.1002/jcp.25797
39. Basch ML, Bronner-Fraser M. Neural crest inducing signals. Adv Exp Med Biol. (2006) 589:24–31. doi: 10.1007/978-0-387-46954-6_2
40. Lai EC. Notch signaling: control of cell communication and cell fate. Development. (2004) 131(5):965–73. doi: 10.1242/dev.01074
41. McElhinney DB, Krantz ID, Bason L, Piccoli DA, Emerick KM, Spinner NB, et al. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or alagille syndrome. Circulation. (2002) 106(20):2567–74. doi: 10.1161/01.CIR.0000037221.45902.69
42. High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, et al. An essential role for notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Investig. (2007) 117(2):353–63. doi: 10.1172/JCI30070
43. Varadkar P, Kraman M, Despres D, Ma G, Lozier J, McCright B. Notch2 is required for the proliferation of cardiac neural crest-derived smooth muscle cells. Dev Dyn. (2008) 237(4):1144–52. doi: 10.1002/dvdy.21502
44. Sauka-Spengler T, Bronner-Fraser M. A gene regulatory network orchestrates neural crest formation. Nat Rev Mol Cell Biol. (2008) 9(7):557–68. doi: 10.1038/nrm2428
45. Huang X, Saint-Jeannet J-P. Induction of the neural crest and the opportunities of life on the edge. Dev Biol. (2004) 275(1):1–11. doi: 10.1016/j.ydbio.2004.07.033
46. Conway SJ, Godt RE, Hatcher CJ, Leatherbury L, Zolotouchnikov VV, Brotto MA, et al. Neural crest is involved in development of abnormal myocardial function. J Mol Cell Cardiol. (1997) 29(10):2675–85. doi: 10.1006/jmcc.1997.0499
47. Gopalan D, Thomas SM. Pharmacotherapy for patients undergoing carotid stenting. Eur J Radiol. (2006) 60(1):14–9. doi: 10.1016/j.ejrad.2006.05.024
48. Prakash SK, Bossé Y, Muehlschlegel JD, Michelena HI, Limongelli G, Della Corte A, et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: insights from the international BAVCon (bicuspid aortic valve consortium). J Am Coll Cardiol. (2014) 64(8):832–9. doi: 10.1016/j.jacc.2014.04.073
49. Hutson MR, Kirby ML. Model systems for the study of heart development and disease: cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol. (2007) 18(1):101–10. doi: 10.1016/j.semcdb.2006.12.004
50. Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pr Res Clin Endocrinol Metab. (2011) 25(1):161–79. doi: 10.1016/j.beem.2010.09.002
51. Kappetein A, Gittenberger-de Groot A, Zwinderman A, Rohmer J, Poelmann R, Huysmans H. The neural crest as a possible pathogenetic factor in coarctation of the aorta and bicuspid aortic valve. J Thorac Cardiovasc Surg. (1991) 102(6):830–6. doi: 10.1016/S0022-5223(19)33931-5
52. Yamazaki T, Mukouyama YS. Tissue specific origin, development, and pathological perspectives of pericytes. Front Cardiovasc Med. (2018) 5:78. doi: 10.3389/fcvm.2018.00078
53. Joseph NM, Mukouyama YS, Mosher JT, Jaegle M, Crone SA, Dormand EL, et al. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development. (2004) 131(22):5599–612. doi: 10.1242/dev.01429
54. Bax M, Romanov V, Junday K, Giannoulatou E, Martinac B, Kovacic JC, et al. Arterial dissections: common features and new perspectives. Front Cardiovasc Med. (2022) 9:1055862. doi: 10.3389/fcvm.2022.1055862
55. Francois K. Aortopathy associated with congenital heart disease: a current literature review. Ann Pediatr Cardiol. (2015) 8(1):25–36. doi: 10.4103/0974-2069.149515
56. Girdauskas E, Borger MA, Secknus MA, Girdauskas G, Kuntze T. Is aortopathy in bicuspid aortic valve disease a congenital defect or a result of abnormal hemodynamics? A critical reappraisal of a one-sided argument. Eur J Cardiothorac Surg. (2011) 39(6):809–14. doi: 10.1016/j.ejcts.2011.01.001
57. Milewicz DM, Regalado ES. Use of genetics for personalized management of heritable thoracic aortic disease: how do we get there? J Thorac Cardiovasc Surg. (2015) 149(2):S3–5. doi: 10.1016/j.jtcvs.2014.07.070
58. Vilacosta I, San Román JA, Aragoncillo P, Ferreirós J, Mendez R, Graupner C, et al. Penetrating atherosclerotic aortic ulcer: documentation by transesophageal echocardiography. J Am Col Cardiol. (1998) 32(1):83–9. doi: 10.1016/S0735-1097(98)00194-6
59. Sultan S, Acharya Y, Long KCV, Hatem M, Hezima M, Veerasingham D, et al. Management of acute aortic syndrome with evolving individualized precision medicine solutions: lessons learned over two decades and literature review. Front Surg. (2023) 10:1157457. doi: 10.3389/fsurg.2023.1157457
60. Tzourio C, Cohen A, Lamisse N, Biousse VR, Bousser M-G. Aortic root dilatation in patients with spontaneous cervical artery dissection. Circulation. (1997) 95(10):2351–3. doi: 10.1161/01.CIR.95.10.2351
61. Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res. (2014) 115(7):625–35. doi: 10.1161/CIRCRESAHA.115.303794
62. Yamagishi H. Cardiac neural crest. Cold Spring Harb Perspect Biol. (2021) 13(1):a036715. doi: 10.1101/cshperspect.a036715
63. Phillips HM, Mahendran P, Singh E, Anderson RH, Chaudhry B, Henderson DJ. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc Res. (2013) 99(3):452–60. doi: 10.1093/cvr/cvt132
64. Tzemos N, Therrien J, Yip J, Thanassoulis G, Tremblay S, Jamorski MT, et al. Outcomes in adults with bicuspid aortic valves. JAMA. (2008) 300(11):1317–25. doi: 10.1001/jama.300.11.1317
65. Mckusick VA, Logue RB, Bahnson HT. Association of aortic valvular disease and cystic medial necrosis of the ascending aorta: report of four instances. Circulation. (1957) 16(2):188–94. doi: 10.1161/01.CIR.16.2.188
66. Fedak PW, David TE, Borger M, Verma S, Butany J, Weisel RD. Bicuspid aortic valve disease: recent insights in pathophysiology and treatment. Expert Rev Cardiovasc Ther. (2005) 3(2):295–308. doi: 10.1586/14779072.3.2.295
67. Keane MG, Wiegers SE, Plappert T, Pochettino A, Bavaria JE, Sutton MGSJ. Bicuspid aortic valves are associated with aortic dilatation out of proportion to coexistent valvular lesions. Circulation. (2000) 102(suppl_3):Iii-35–9. doi: 10.1161/circ.102.suppl_3.III-35
68. Michelena HI, Khanna AD, Mahoney D, Margaryan E, Topilsky Y, Suri RM, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA. (2011) 306(10):1104–12. doi: 10.1001/jama.2011.1286
69. Itagaki S, Chikwe JP, Chiang YP, Egorova NN, Adams DH. Long-term risk for aortic complications after aortic valve replacement in patients with bicuspid aortic valve versus Marfan syndrome. J Am Coll Cardiol. (2015) 65(22):2363–9. doi: 10.1016/j.jacc.2015.03.575
70. Albornoz G, Coady MA, Roberts M, Davies RR, Tranquilli M, Rizzo JA, et al. Familial thoracic aortic aneurysms and dissections—incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. (2006) 82(4):1400–5. doi: 10.1016/j.athoracsur.2006.04.098
71. Larson EW, Edwards WD. Risk factors for aortic dissection: a necropsy study of 161 cases. Am J Cardiol. (1984) 53(6):849–55. doi: 10.1016/0002-9149(84)90418-1
72. Mohamed S, Misfeld M, Hanke T, Charitos E, Bullerdiek J, Belge G, et al. Inhibition of caspase-3 differentially affects vascular smooth muscle cell apoptosis in the concave versus convex aortic sites in ascending aneurysms with a bicuspid aortic valve. Ann Anat. (2010) 192(3):145–50. doi: 10.1016/j.aanat.2010.02.006
73. Leatherbury L, Gauldin HE, Waldo K, Kirby ML. Microcinephotography of the developing heart in neural crest-ablated chick embryos. Circulation. (1990) 81(3):1047–57. doi: 10.1161/01.CIR.81.3.1047
74. Courchaine K, Rykiel G, Rugonyi S. Influence of blood flow on cardiac development. Prog Biophys Mol Biol. (2018) 137:95–110. doi: 10.1016/j.pbiomolbio.2018.05.005
75. Tomita H, Connuck DM, Leatherbury L, Kirby ML. Relation of early hemodynamic changes to final cardiac phenotype and survival after neural crest ablation in chick embryos. Circulation. (1991) 84(3):1289–95. doi: 10.1161/01.CIR.84.3.1289
76. Creazzo TL, Burch J, Redmond S, Kumiski D. Myocardial enlargement in defective heart development. Anat Rec. (1994) 239(2):170–6. doi: 10.1002/ar.1092390207
77. Farrell MJ, Burch JL, Wallis K, Rowley L, Kumiski D, Stadt H, et al. FGF-8 in the ventral pharynx alters development of myocardial calcium transients after neural crest ablation. J Clin Invest. (2001) 107(12):1509–17. doi: 10.1172/JCI9317
78. Waldo KL, Hutson MR, Stadt HA, Zdanowicz M, Zdanowicz J, Kirby ML. Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol. (2005) 281(1):66–77. doi: 10.1016/j.ydbio.2005.02.011
79. Agmon Y, Khandheria BK, Meissner I, Schwartz GL, Sicks JD, Fought AJ, et al. Is aortic dilatation an atherosclerosis-related process? Clinical, laboratory, and transesophageal echocardiographiccorrelates of thoracic aortic dimensions in the populationwith implications for thoracic aortic aneurysm formation. J Am Coll Cardiol. (2003) 42(6):1076–83. doi: 10.1016/S0735-1097(03)00922-7
80. Wu W, He J, Shao X. Incidence and mortality trend of congenital heart disease at the global, regional, and national level, 1990–2017. Medicine (Baltimore). (2020) 99(23):e20593. doi: 10.1097/MD.0000000000020593
Keywords: neural crest cells, cardiac neural crest cells, cardiovascular neurocristopathy, cardiac neurocristopathy, vascular neurocristopathy, aortopathy, bicuspid aortic valve
Citation: Soliman O, Acharya Y, Gilard M, Duffy G, Wijns W, Kannan V and Sultan S (2024) Systematic review of cardiovascular neurocristopathy—contemporary insights and future perspectives. Front. Cardiovasc. Med. 11:1333265. doi: 10.3389/fcvm.2024.1333265
Received: 2 December 2023; Accepted: 22 March 2024;
Published: 9 April 2024.
Edited by:
Paolo Ciliberti, Bambino Gesù Children’s Hospital (IRCCS), ItalyReviewed by:
Elena Bennati, Meyer Children’s Hospital, ItalyRobert Poelmann, Leiden University, Netherlands
© 2024 Soliman, Acharya, Gilard, Duffy, Wijns, Kannan and Sultan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Osama Soliman by5pLnNvbGltYW5AZ21haWwuY29t
†ORCID Osama Soliman orcid.org/0000-0003-0758-3539 Yogesh Acharya orcid.org/0000-0003-1829-5911 Sherif Sultan orcid.org/0000-0001-8767-4929