
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Cardiovasc. Med. , 22 October 2024
Sec. Heart Failure and Transplantation
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1332015
 Haoran Zheng1,2,†
Haoran Zheng1,2,† Xinxin Mao1,†
Xinxin Mao1,† Zhenyue Fu1,†
Zhenyue Fu1,† Chunmei Chen1Jiayu Lv1
Chunmei Chen1Jiayu Lv1 Yajiao Wang1Yuxin Wang1Huaqin Wu1Yvmeng Li1Yong Tan2Xiya Gao1Lu Zhao1
Yajiao Wang1Yuxin Wang1Huaqin Wu1Yvmeng Li1Yong Tan2Xiya Gao1Lu Zhao1 Xia Xu1*
Xia Xu1* Bingxuan Zhang1*
Bingxuan Zhang1* Qingqiao Song1*
Qingqiao Song1*
Background: Cytokines play a pivotal role in the progression of heart failure (HF) by modulating inflammatory responses, promoting vasoconstriction, and facilitating endothelial injury. However, it is now difficult to distinguish the causal relationship between HF and cytokines in observational studies. Mendelian randomization (MR) analyses of cytokines probably could enhance our comprehension to the underlying biological processes of HF.
Methods: This study was to explore the correlation between 41 cytokines with HF at the genetic level by MR analysis. We selected a HF dataset from the Heart Failure Molecular Epidemiology for Therapeutic Targets (HERMES) 2018 and a cytokine dataset from a meta-analysis of cytokine levels in Finns. Two-sample, bidirectional MR analyses were performed using Inverse Variance Weighted (IVW), Weighted Median and MR- egger, and the results were tested for heterogeneity and pleiotropy, followed by sensitivity analysis.
Results: Genetic prediction of high levels of circulating Macrophage inflammatory pro-tein-1β(MIP-1β) (P = 0.0389), Interferon gamma induced protein 10(IP-10) (P = 0.0029), and Regu-lated on activation, normal T cell expressed and secreted(RANTES) (P = 0.0120) expression was associated with an elevated risk of HF. HF was associated with the increased levels of circulating Interleukin-2 receptor, alpha subunit(IL-2ra) (P = 0.0296), Beta nerve growth fac-tor(β-NGF) (P = 0.0446), Interleukin-17(IL-17) (P = 0.0360), Basic fibroblast growth factor(FGF-basic) (P = 0.0220), Platelet derived growth factor BB(PDGF-BB) (P = 0.0466), and Interferon-gamma(IFN-γ) (P = 0.0222); and with decreased levels of Eotaxin (P = 0.0133). The heterogeneity and pleiotropy of the cytokines were acceptable, except for minor heterogeneity of FGF-basic and IL-17.
Conclusion: These findings provide compelling evidence for a genetically predictive relationship between cytokines and HF, emphasizing a great potential of targeted modulation of cytokines in slowing the progression of HF. This study draws further conclusions at the genetic level, providing a basis for future large-scale clinical trials.
Heart failure (HF) is a group of clinical syndromes characterized by the reduction of cardiac pumping and/or congestion function (1). On a global scale, approximately 64.3 million individuals grappled with HF in 2017 (2). Those afflicted with HF face a mortality rate exceeding 30% (3) and a re-hospitalization rate surpassing 50% (4) within the initial year of diagnosis. With the aging of the population, it is estimated that the prevalence and treatment expenses of HF are anticipated to surge, presenting both medical and economic challenges worldwide. Predictions indicate that by 2030, over 80,000 individuals in the United States will contend with HF, and the direct healthcare costs for HF treatment will escalate to $5.3 billion (5). So far, HF has turned out to be a systemic syndrome that afflicts all mankind, which means that its pathophysiology needs further exploration and must be accelerated.
Ever since Levine and his team reported the elevated levels of tumor necrosis factor (TNF) in the plasma of HF patients in 1990 (6), cytokines have played a crucial role in the pathophysiological research of HF. According to the cytokine hypothesis, cytokines are responsible for the progression of HF, and the cascade of cytokine activation following myocardial injury has a detrimental impact on the circulation (7). There are 2 classes of cytokines that have been identified to play a role in HF: vasoconstrictor cytokines and vasodepressor proinflammatory cytokines (7). Cytokines exert their effects through direct membrane action or by binding to specific receptors on the cell surface (8), and then modulating inflammatory responses, promoting vasoconstriction, and inducing endothelial damage to influence the progression of HF (9). In HF patients, inadequate tissue perfusion and hypoxia lead to excessive peripheral cytokine production, activating the inflammatory and immune systems (7, 10); elevated end-diastolic wall stress in the left ventricle results in excessive expression of myocardial cytokines, directly or indirectly affecting ventricular contractile function and remodeling, thereby exacerbating HF (11). A comprehensive study involving 29 cohorts revealed a close correlation between cytokines such as interleukin-6 (IL-6), IL-18, tumor necrosis factor-alpha (TNF-α) with the onset of HF, exhibiting an approximately logarithmic-linear relationship (12). Research suggests (13) that intravenous administration of TNFα can induce progressive left ventricular dilation in rats. P. Aukrust and colleagues (14) identified elevated levels of monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1 alpha (MIP-1α), and RANTES in patients with chronic HF, with MCP-1 and MIP-1α levels significantly negatively correlated with left ventricular ejection fraction (LVEF). However, the majority of studies investigating HF and cytokines have relied on observational research designs. Yet, observational study designs face challenges in deducing the directional flow of causality and distinguishing confounding factors. Additionally, clinical trials that intervene in cytokines are also challenging. Therefore, the causal sequence between cytokines and HF progression remains a topic for further consideration. Existing unidirectional MR studies on cytokines and HF (15, 16) have primarily focused on the upstream effects of cytokines, showing different directions from clinical practice and experimental research. Therefore, the hypothesis of a relationship between HF and cytokines urgently requires evidence at the genetic level, which means incorporating more types of cytokines and assessing their impact on the disease. Studies of the genetic prediction of HF and cytokines can help to shift clinical practice from treatment to prevention for at-risk populations primarily.
Compared to observational studies, Mendelian randomization (MR) is a natural randomized statistical method using genetic variations (single nucleotide polymorphisms, SNPs) as instrumental variables (IVs) (17). Since genetic variation occurs randomly, the random assignment of alleles from parent to offspring and the unidirectional flow from genotypes to phenotypes determine the randomness of genetic variation. This process is similar to the random assignment of control and treatment groups in a population (18). Under these random conditions, MR methods can be employed to estimate the directed effects of exposures on outcomes. MR has achieved significant success in exploring disease mechanisms, identifying potential biomarkers, and targeting treatments. Numerous studies have confirmed the reliability of MR in identifying genetic relationships between exposure factors and outcomes (19–21). In this study, we used a two-sample, bidirectional MR approach to investigate the relationship between inflammatory cytokines and HF, aiming to gain insight into the role of cytokines in the onset and progression of HF.
This study, based on a genome-wide association study (GWAS) dataset, employs a two-sample, bidirectional MR approach to systematically investigate the potential causal relationships between various cytokines and HF. The study is conducted in two parts. In the first part, cytokines are considered as exposures, and genetic variations are used as IVs to investigate their relationship, with HF as the outcome. In the second part, the exposure and outcome are reversed for analysis. The flowchart of the study is shown in Figure 1. Both parts of the study adhere to the three core assumptions of MR analysis (22):1. Relevance (23): genetic variations are related to the exposures. 2. Independence (24): no confounding factors are involved in the exposure-outcome pathway. 3. Exclusivity (23): the genetic variation does not affect the outcome except through its association with the exposure.
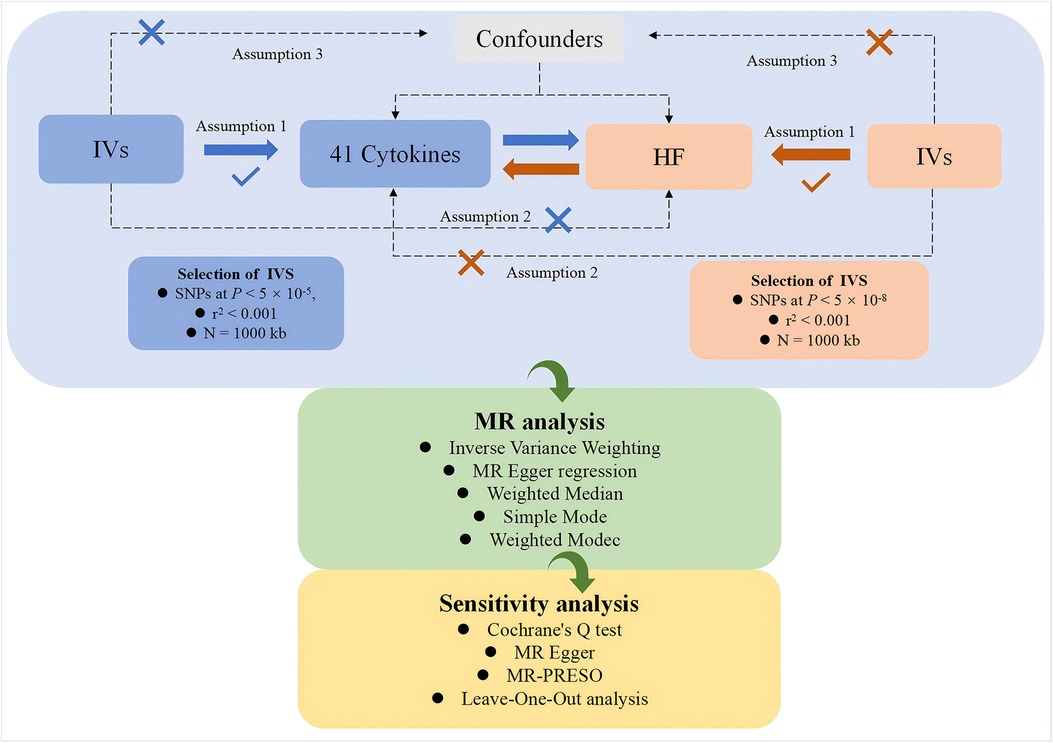
Figure 1. The study framework chart. IVs: instrumental variables; HF: heart failure.
Our study utilized publicly available GWAS datasets for MR analysis. Information regarding ethical committee approvals and participant informed consent can be found in the original research studies from each data source.
To mitigate potential differences in the distribution of genetic variations across different ethnic populations, we used the largest and most recent Genome-Wide Association Study (GWAS) in European populations as the data source. The definition of the European population is that at least 80% of the participants have European ancestry. We utilized the HF GWAS dataset provided by the Heart Failure Molecular Epidemiology for Therapeutic Targets (HERMES), which is a meta-analysis involving 26 studies. Diagnoses were based on criteria including the 9th/10th revision of the International Classification of Diseases codes or physician diagnoses. Adjustments for age and sex were made in the analysis of SNPs. The dataset comprises a total of 47,309 HF patients and 930,014 controls, with a total of 7,773,021 SNPs (25). Diagnostic information and baseline characteristics for each cohort are provided in Supplementary Table S1.
The cytokine GWAS data is derived from a meta-analysis of circulating cytokine levels in the Finnish population, which includes 8,293 samples from 3 cohorts: the Cardiovascular Risk in Young Finns Study (YFS), a multi-center follow-up study; FINRISK, a cross-sectional study monitoring chronic disease risk factors in the Finnish population, conducted every five years, our study extracts the results of FINRISK's 1997 and 2002 cross-sectional surveys. Final sample sizes for the three cohorts were 1980, 1,705, and 4,608 respectively. Cytokine levels were measured in participants' fasting plasma, half-fasting EDTA plasma, and half-fasting heparin plasma. For more details on cytokine classification and information, please refer to the [(Supplementary Table S2) and Baseline (Supplementary Table S3)] (26).
In accordance with the three fundamental assumptions of MR (22), IVs must satisfy the following three conditions:1. IVs are significantly associated with the exposure factor: In this study, to obtain enough SNPs as IVs (27, 28), we selected SNPs with a genome-wide significance level of P < 5 × 10−5 and an r2 < 0.001, withisn a genomic locus width of N = 1,000 kb. This ensures that the selected SNPs are in linkage equilibrium, guaranteeing their independence from each other. 2. IVs are unrelated to the outcome factor: SNPs should have a significance level of P > 5 × 10−8 in their association with the outcome. 3. Exposure factors are unrelated to confounding factors. Additionally, we computed the F-statistic for each IV, F = beta2/se2 (29–31), with beta representing the SNP-exposure effect estimate and se denoting its standard error. IVs with F-statistics less than 10 were classified as “weak IVs” (24) and were excluded from the MR analysis for the reliability of the analysis.
With five MR methods from the TwoSampleMR package, the ratios between the effects of SNPs on exposure and on outcome were calculated, then the results of each SNP calculation were combined to assess the potential causal relationship between exposure and outcome. The five MR methods include Inverse Variance Weighting (IVW), MR Egger regression, Weighted Median method, Simple Mode, and Weighted Mode (17). Among these methods,IVW is considered the most efficient, it could provide unbiased estimates with maximum statistical power after controlling for pleiotropy (21). Therefore, the results obtained from IVW analysis serve as the primary reference, with statistical significance denoted by P < 0.05. The Weighted Median method is suitable for cases with a higher proportion of invalid IVs and can generate effect estimates even when a substantial portion, possibly up to 50%, of IVs are invalid (18). The MR-Egger method exhibits higher tolerance for SNPs with horizontal pleiotropy, the MR Egger does not force the regression line to pass through the origin, allowing for targeted gene pleiotropy in the included IVs. The MR-PRESSO method allows for the exclusion of specific SNPs by excluding outliers to obtain estimates that are closer to the true values (32).
If the MR analysis results of the five methods are consistent, it is considered a stable and reliable result. To assess whether there was heterogeneity among the selected IVs, Cochrane's Q test was first performed, and if there was no significant heterogeneity at P > 0.05, the Wald estimates for each SNP were analyzed using the IVW fixed-effects model; otherwise, the random-effects model was used. MR-Egger regression assesses the horizontal pleiotropy of the analysis by the size of the intercept; and in the scatterplot, the correlation between the intercept and 0 is tested; if P > 0.05, it means that there is no horizontal pleiotropy, and vice versa (32). In addition, select exposure factors with SNPs ≥ 4 for MR-PRESO analysis, set K = 10,000, eliminate outlier SNPs, and estimate the corrected results (18). Finally, assess the bias of individual SNPs in MR by Leave-One-Out sensitivity analysis, exclude SNPs that potentially influence the results one by one; then re-run MR analysis to ensure the stability of the results.
In accordance with the filtering criteria mentioned earlier, relevant SNPs information was extracted, and the information for the SNPs used as IVs for exposures in the MR analysis can be found in Supplementary Tables S3 and S4, including the calculation of F-statistics. In the dataset for HF as the exposure, there are a total of 168 independent SNPs, and the F-statistics for individual SNPs range from 16.4398 to 83.0960. In the summary dataset for 41 cytokines, there are a total of 449 SNPs, with each cytokine having at least 3 SNPs. The F-statistics for individual cytokines range from 20.7962 to 782.4524, all of which are above 10. This suggests that, even after SNPs conditional cleaning in the HF and cytokine datasets, the genetic instruments remain robust and strong.
The results of the IVW method analysis for the impact of 41 cytokines on HF are presented in Figure 2. It shows that only two cytokines (MIP-1β and IP-10) exhibit a significant risk association with HF. Furthermore, RANTES demonstrates a stronger risk association with HF in the Weighted Median analysis, as detailed in Figure 3. For a comprehensive summary of the results of the 41 cytokines' impact on HF, refer to Supplementary Table S5.
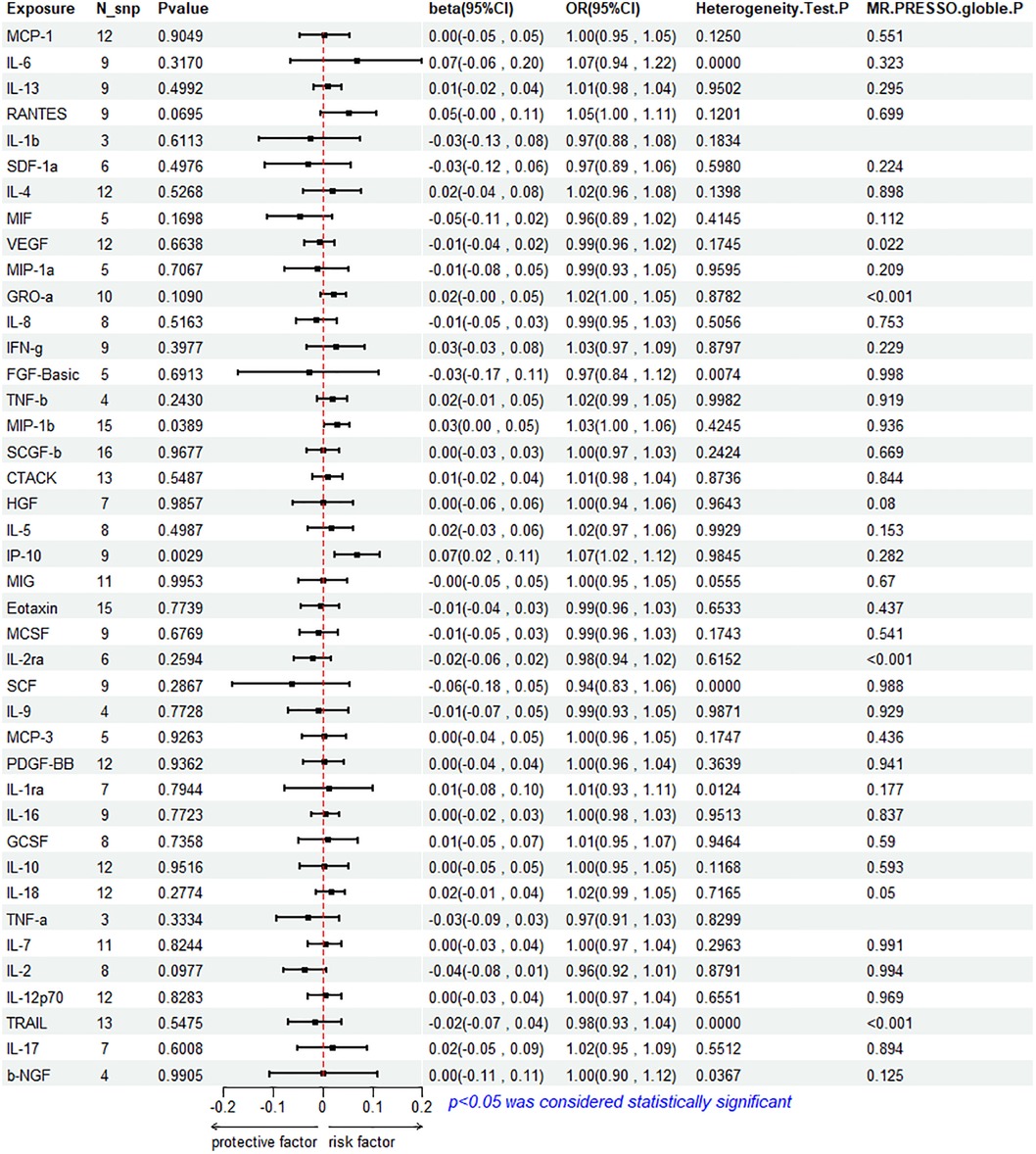
Figure 2. Mendelian randomization analysis of 41 cytokines and heart failure.
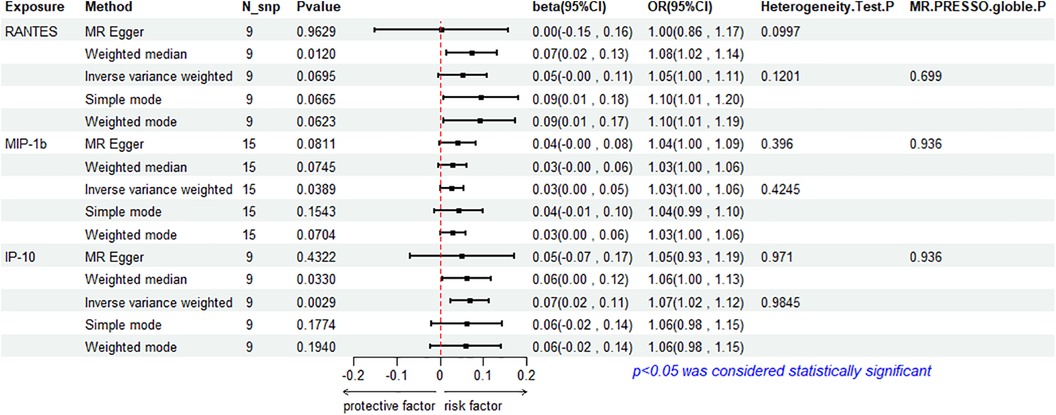
Figure 3. Mendelian randomization analysis of 3 cytokines and heart failure.
High genetic prediction of circulating MIP-1β is associated with an increased risk of HF (beta = 0.278, beta 95% CI = 0.0014–0.0541, OR = 1.0282, OR 95% CI = 1.0014–1.0056, P = 0.0389). Cochrane's Q test (P = 0.4245) did not reveal any heterogeneity (Supplementary Table S6). A fixed-effects model was used for the IVW method. All five MR analysis methods consistently show a positive association. The MR-Egger regression intercept is close to 0 (MR Egger-intercept = −0.0048, MR Egger-intercept P = 0.4455), suggesting no directional pleiotropy. The funnel plot is relatively symmetrical on both sides. Leave-one-out sensitivity analysis did not yield different results (Supplementary Figure S1). MR-PRESSO also found no evidence of horizontal pleiotropy (global P = 0.936), indicating the reliability of the MR analysis.
The IVW method reveals that an increase in plasma IP-10 levels is also associated with an increased risk of HF (beta = 0.0689, beta 95% CI = 0.0236–0.1141, OR = 1.0713, OR 95% CI = 1.0239–1.1209, P = 0.0029). All five MR methods show consistent risk relationships. Cochrane's Q test (P = 0.9845) did not reveal heterogeneity (Supplementary Table S6). The IVW method was conducted using a fixed-effects model. The MR-Egger regression intercept (MR Egger-intercept = 0.0032, MR Egger-intercept P = 0.7636) indicates no significant directional pleiotropy. MR-PRESSO testing yielded a global P-value of 0.282. Although the funnel plot is asymmetrical on both sides, leave-one-out sensitivity analysis did not produce different results (Supplementary Figure S1).
RANTES, on the other hand, demonstrates a stronger risk association with HF in the Weighted Median analysis (beta = 0.00738, beta 95% CI = 0.0162–0.1313, OR = 1.0765, OR 95% CI = 1.0164–1.1403, P = 0.0120). All five MR analysis results show consistent risk associations. The IVW method used a random-effects model, and the IVW analysis results remain non-significant (P = 0.0695) (Supplementary Table S5). The Cochrane's Q test (P = 0.1201) also did not reveal heterogeneity (Supplementary Table S6). MR-Egger regression intercept is close to 0 (MR Egger-intercept = 0.0097, MR Egger-intercept P = 0.5320), indicating no significant directional pleiotropy. MR-PRESSO testing found no significant horizontal pleiotropy (global P = 0.699). The funnel plot for the IVW method is relatively symmetric on both sides. Leave-one-out sensitivity analysis did not yield significantly different results (Supplementary Figure S1).
To assess the reverse effect, we used HF as the exposure factor and extracted seven cytokines that showed a significant correlation. The results of the IVW method analysis for the impact of HF on plasma cytokine levels are presented in Figure 4, which shows that the occurrence of HF affects the expression of two cytokines (IL-2ra and β-NGF). In addition, four cytokines (IL-17, FGF-basic, PDGF-BB, and IFN-γ) exhibit a stronger positive correlation in the Weighted Median analysis, while Eotaxin shows a significant negative correlation with HF in the MR Egger method. More details refer to Figure 5. (Supplementary Table S10).
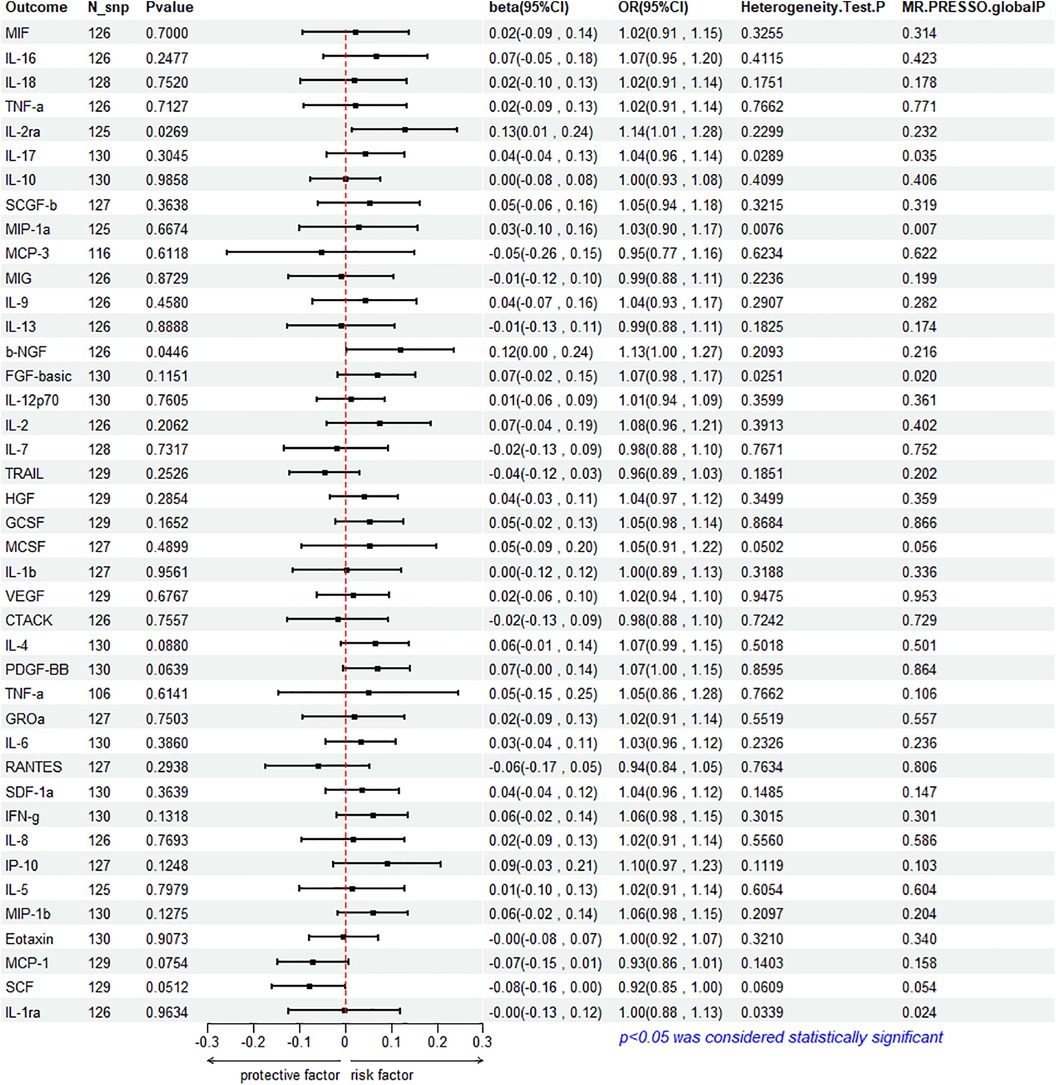
Figure 4. Mendelian randomization analysis of heart failure and 41 inflammatory factors.
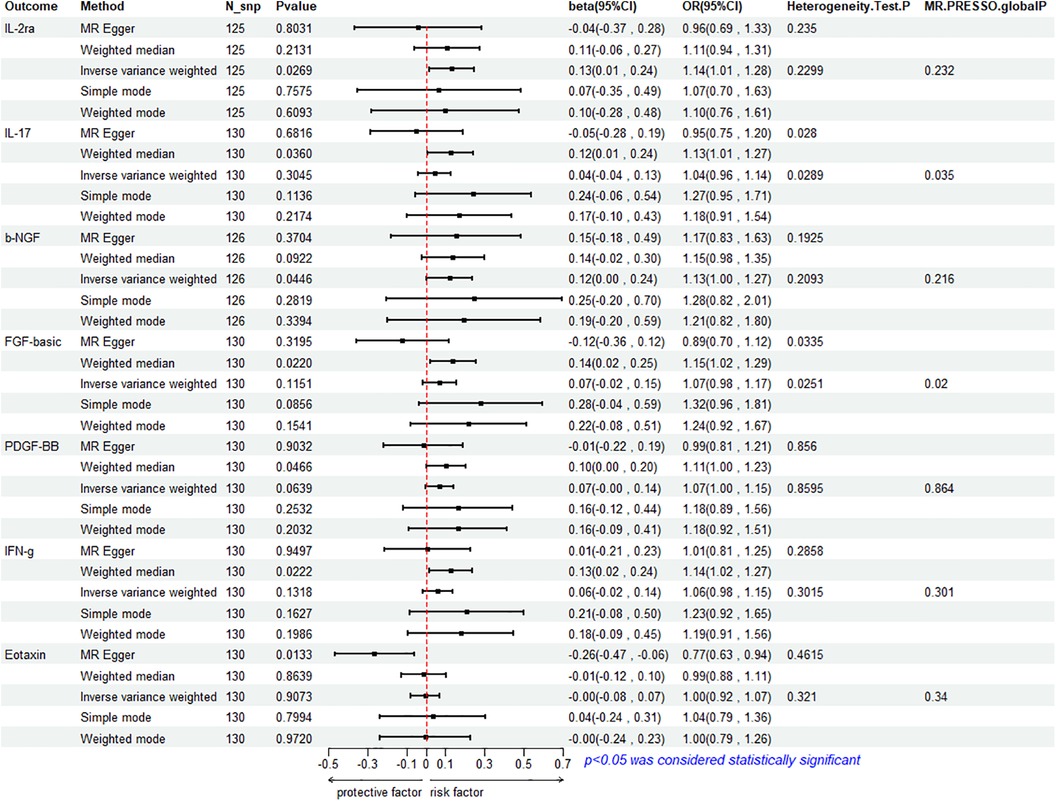
Figure 5. Mendelian analysis of heart failure and 7 cytokines.
The IVW method reveals a significant correlation between genetically predicted HF and increased circulating IL-2ra levels (beta = 0.1292, beta 95% CI = 0.0147–0.2436, OR = 1.1379, OR 95% CI = 1.0148–1.2759, P = 0.0296). Heterogeneity testing using Cochrane's Q test (P = 0.2299) did not reveal heterogeneity (Supplementary Table S11). The IVW method used a fixed-effects model. The funnel plot is generally symmetrical on both sides. Leave-one-out sensitivity analysis showed relatively robust results (Supplementary Figure S1). MR-PRESSO analysis did not detect any horizontal pleiotropy (global P = 0.232), indicating the robustness of the IVW results. When using the MR Egger method, beta = −0.0413, OR=0.9595, indicating a negative association with HF. Cochrane's Q test resulted in a P-value of 0.2299, and the MR-Egger regression intercept was close to 0 (MR Egger-intercept = 0.0089, MR Egger-intercept P = 0.2726), suggesting no significant directional pleiotropy.
According to the IVW method, HF is closely related to an increase in circulating β-NGF levels (beta = 0.1203, beta 95% CI = 0.0029–0.2376, OR = 1.1272, OR 95% CI = 1.0029–1.2682, P = 0.0446). Heterogeneity testing using Cochrane's Q test (P = 0.2093) did not reveal heterogeneity (Supplementary Table S11). The IVW method used a fixed-effects model. The funnel plot is generally symmetrical on both sides. Leave-one-out sensitivity analysis showed relatively robust results. However, results from MR Egger, Weighted Median, Simple Mode, and Weighted Mode were consistent in direction but not statistically significant. The MR-Egger regression intercept was 0.0089, and the MR Egger-intercept P-value = 0.8379, suggesting no significant directional pleiotropy.
The results of Weighted Media suggest that IL-17 (beta = 0.1242, beta 95% CI = 0.0081∼0.2402, OR = 1.1322, OR 95% CI = 1.0082–1.2715, P = 0.0360), FGF basic (beta = 0.1369, beta 95% CI = 0.0197–0.2541, OR = 1.1467, OR 95% CI = 1.0199–1.2893, P = 0.0220), PDGF-BB (beta = 0.1027, beta 95%CI = 0.0015–0.2039, OR = 1.1082, OR 95% CI = 1.0015–1.2261, P = 0.0466), and IFN- γ (beta = 0.1283, beta 95% CI = 0.0183–0.2384, OR = 1.1369, OR 95% CI = 1.0185–1.2692, P = 0.0222) are significantly associated with the risk of HF. FGF basic and IL-17 exhibit heterogeneity, with Cochrane's Q test P of 0.0280 and 0.0335, respectively. The funnel plot is asymmetric and biased towards one side. Although the intercept of the MR Egger pleiotropy test for the four cytokines was close to 0 and there was no direct significant pleiotropy, MR-PRESSO analysis indicated potential horizontal pleiotropy for FGF-basic (global P = 0.02) and IL-17 (global P = 0.035), suggesting possible bias. However, after removing outliers, heterogeneity still persisted, indicating that the correlation could not be obtained for FGF-basic and IL-17 in all methods except Weighted Median, which compensates for the credibility of the results. Furthermore, leave-one-out forest plots showed that the effect of HF on the levels of these four cytokines remained relatively stable.
Eotaxin shows a significant protective correlation with HF in the MR Egger method. HF is associated with a decrease in plasma Eotaxin levels (beta = −0.2632, beta 95% CI = −0.4685 to −0.0578, OR = 0.7686, OR 95% CI = 0.6259–0.9438, P = 0.0133). Cochrane's Q test (P = 0.4615) did not reveal heterogeneity (Supplementary Table S11). The IVW method used a fixed-effects model. While the Simple Mode method showed a positive correlation, the results from the other four MR methods were all negative. The funnel plot is generally symmetrical on both sides. The MR-Egger regression intercept was 0.0137, and the MR Egger-intercept P-value was 0.0092, suggesting no directional pleiotropy. MR-PRESSO also found no horizontal pleiotropy (global P = 0.34). Leave-one-out sensitivity analysis did not yield significantly different results, after removing SNP (rs660240, rs10846742, rs10152199, rs117734706, rs35054810, rs17496249, rs78957967, rs146379019, rs1757223, rs34592354, rs77932705), the results are relatively robust.
In recent years, there has been extensive focus on the abnormal expression of biologically active factors in the development and progression of HF. Cytokines have received particular attention, following neurohormones, for their widespread mention in this context (33, 34). However, most of the previous studies of HF and cytokines have been observational, making it difficult to infer directed causation. There are also few studies of interventional cytokines. The natural randomized design of MR studies provides a higher level of clinical evidence. But the only MR studies that provide evidence at the gene level are unidirectional (15, 16), failed to identify inflammatory markers with a clear correlation with HF. The relationships between more cytokines, especially those displaying reverse effect, with HF are yet to be explored. To the best of our knowledge, this study is the first bidirectional, two-sample MR study of cytokines and HF, and utilizes the latest and largest genome-wide association data to evaluate the correlation between 41 cytokines and HF. Through this bidirectional analytical strategy, we could differentiate between upstream and downstream factors in the disease process, thus explore the pathophysiological mechanisms of HF. In the bidirectional MR analysis, we observed that elevated circulating levels of MIP-1β, IP-10, and RANTES, determined by genetics, were associated with an increased risk of HF. Conversely, the genetic susceptibility to HF was associated with elevated circulating levels of IL-2ra, β-NGF, IL-17, FGF-basic, PDGF-BB, IFN-γ, and a decrease in Eotaxin levels. Our study identified the responsible factors at various stages of HF, confirming the cytokine hypothesis in HF. Circulating cytokines play a crucial role in the formation and progression of HF, and HF may lead to worse outcomes through potential pro-inflammatory effects. Throughout the research, we conducted Cochrane's Q, leave-one-out heterogeneity tests, and MR-PRESSO and MR-Egger were applied for pleiotropy test to assess the reliability of MR analysis.
Previous results suggest that the elevated levels of MIP-1β are associated with an increased risk of HF. MIP-1β is a proinflammatory cytokine that can enhance the inflammatory response and promote inflammation progression. Observational studies (35) have found higher levels of MIP-1β in HF patients compared to the healthy group. Genetic testing (36) revealed that the mRNA expression of MIP-1β, which is regulated in HF patients, is eight times higher than that in the healthy control group. Furthermore, the corresponding receptor gene, CCR2, was also 2-fold higher than that of the control group and was negatively correlated with LVEF. IP-10 is a chemokine secreted by various cells, such as T cells, neutrophils, and endothelial cells (37), and it can recruit T cells, promote T cell differentiation, and regulate angiogenesis by binding to the chemokine receptor CXCR3 (38). We found that IP-10 is positively associated with the occurrence of HF, which aligns with previous unidirectional MR studies (39). In a mouse model of diastolic left ventricular dysfunction, the average increase in plasma IP-10 concentration is 140% of the NT-proBNP levels (40). The MONICA/KORA Augsburg 1984–2002 cohort study (41) discovered a positive association between IP-10 and several HF risk factors, including age, BMI, and coronary artery disease, and elevated plasma IP-10 levels precede the occurrence of coronary artery disease. Results from two prospective cohort studies also indicate that higher IP-10 concentrations are associated with an increased risk of HF (42). In our study, RANTES exhibited a strong positive association with HF in the Weighted Median analysis. RANTES, also known as CCL5, is primarily secreted by normal T cells and is involved in the transportation, homing, and proliferation of T cells (39). A decrease in serum RANTES levels with the severity of myocardial infarction was observed in patients with post-myocardial infarction HF (42), which contradicts our findings. The possible reasons for this discrepancy are mainly: first, the genetic information from genes to proteins has to go through the process of transcription and translation, and this process is regulated by many factors, so the discrepancy between the genetic information and the phenotype may occur; second, the level of RANTES in circulation could be influenced by different stages of the neuro-hormonal-immune pathway, so it may have pleiotropic effects at different stages of HF (43), leading to inconsistent observations. However, these hypotheses need to be verified by more animal experiments and clinical trials.
In this study from HF to circulating cytokines, we observed a significant increase in plasma IL-2ra levels with the occurrence of HF. IL-2ra is one of the heterotrimers that make up the high-affinity IL-2 receptor (IL-2r), which is involved in T cell activation and immune responses (44). Previous research has found increased serum IL-2R levels in patients with stable angina (45) and coronary artery disease (46). A case-control study at the genetic level (47) identified a significant association between a single SNP (rs12569923) in IL-2ra and the increased risk of diabetes in coronary artery disease patients. β-NGF is a neurotrophic factor involved in the differentiation of central and peripheral neurons and selective innervation of target tissues (48). NGF affects the strength and quantity of the development of synapses, enhances synaptic transmission between autonomic neurons and cardiomyocyte (49); in addition, overexpressed NGF binds to its high-affinity transmembrane receptor TrKA, leading to its dimerization and activation, which recruits and activates downstream signaling molecules through a signaling cascade reaction, triggering a variety of physiological responses (50), improving endothelial cell survival and promoting neovascularization (51). It was shown that NGF can support the survival of sympathetic neurons at low levels and synaptic transmission requires over ten times of the normal NGF levels (49). In patients with HF and in rats with post-myocardial infarction HF, NGF expression decreases due to the dysfunction of cardiac sympathetic nerve endings (52). However, in mice with acute myocardial infarction, delivering NGF-related genes around the infarcted myocardium can improve myocardial cell survival and cardiac function (51), suggesting that NGF can participate in cardiac repair and therapy. IL-17 is an interleukin, and consistent with MR results, studies have found increased plasma IL-17 levels in patients with congestive HF (53). In a study on calcium in HF patients (54), IL-17 levels in the circulation and cardiac tissue were significantly increased in HF patients and mouse models. Knockdown of IL-17-related genes in mice increased the magnitude of calcium transients and cell shortening, ultimately improving heart function. In patients with ischemic HF, anti-IL-17 neutralizing antibodies can inhibit myocardial cell fibrosis and apoptosis, providing cardiac protection (55).
IFN-γ is a proinflammatory cytokine with antiviral, antitumor, and immune-regulatory functions. Animal experiments have shown an increase in plasma IFN-γ levels in mice with post-myocardial infarction HF (56), consistent with our MR results. Interestingly, a recent clinical observational study found lower IFN-γ levels in HF patients compared to healthy patients, which aligns with the IVW results. However, this negative correlation trend did not extend to the correlation between disease duration and LVEF. Instead, IL-9 levels, which did not exhibit significant correlation in our MR results, were negatively correlated with LVEF and duration of illness (35). Eotaxin is a key chemokine in the inflammatory process and is involved in the recruitment of eosinophils at the site of injury. MR results revealed a gradual decrease in plasma Eotaxin levels with the onset of HF, which may be related to the rarity of eosinophils in atherosclerotic lesions (57). However, Eotaxin and its receptor CCR3 were found to be elevated in both atherosclerotic and healthy control populations (58). A cohort study found that a T/A mutation in amino acid 23 on the Eotaxin gene increased the risk of myocardial infarction (57), and provided evidence for the involvement of Eotaxin in the formation of atherosclerosis, also providing evidence for the involvement of Eotaxin in the formation of atherosclerosis.
Our findings emphasize the intricate relationship between cytokines and HF. Interestingly, there was no overlap between cytokines associated with increased risk of HF and those expressed as a result of HF. This suggests that the mechanisms underlying the onset and progression of HF may be different. Cohort studies have demonstrated a dose-dependent relationship between higher than baseline body mass index (BMI) levels and downstream risk of HF hospitalization events (59). In HF patients, the adverse outcomes were dominated by renal failure, infection, and multi-organ failure, while the prevalence of chronic kidney disease in HF patients was 63%, with an 11% increase in hospitalization and a 17% increase in mortality; meanwhile, there was a U-shaped relationship between eGFR and mortality in HF patients (60). Given these conflicting findings, further research, particularly focusing on underlying mechanisms, is essential to clarify the potential role of cytokines in the development of HF. Its mechanism may be related to increased oxidative stress, chronic inflammation, myocardial hypertrophy, perivascular and interstitial collagen deposition/cross-linking, abnormal intracellular calcium processing, endothelial and mitochondrial dysfunction, and cell apoptosis (61).
Cytokine therapy was one of the earliest forms of immunotherapy approved by the US Food and Drug Administration (FDA). Research has found that in patients with non ST segment elevation myocardial infarction, administering a single dose of tocilizumab before coronary angiography can reduce the release of troponin T and systemic inflammation (62). The efficacy of TNF-α antagonism in patients with HF has been further investigated through the administration of soluble TNFR, but no significant clinical benefit has been observed (63). To date, the clinical benefit of cytokine therapy has not surpassed that of current classic agents such as diuretics. Moreover, the utilization of cytokine therapy may impact the inflammatory state in patients with co-infection. The heterogeneity of HF patients in terms of disease complexity and severity poses challenges for cytokine-based therapy implementation. Long-term administration of cytokine therapy is deemed suboptimal. HF may not be an ideal candidate disease for cytokine therapy due to its complex nature. But that doesn't mean it's not worth pursuing, brief cytokine therapy in conditions such as myocardial infarction and myocarditis has shown potential in altering inflammatory responses and potentially preventing progression towards HF. Of all these cytokines, MIP-1β, IP-10, RANTES, IL-2r, β-NGF, PDGF-BB, FGF-basic, IL-17, and IFN-γ are especially desirable for immediate study.
The novelty of this study lies in the fact that it is the first time that a bidirectional, two-sample MR approach was employed to explore the association between circulating cytokines and HF. The database of this study is the largest GWAS dataset on circulating cytokines and HF ever published. Due to the natural randomization of the MR and the strict screening criteria for SNPs, the results of the study are less susceptible to confounding factors and reverse causal associations. However, our study also has some limitations. First, the data sources for our exposure and outcome are based on studies conducted on populations of European ancestry, and the exposures and outcomes did not originate from the same population, so it is unclear whether these findings are applicable to other ethnicities or regions. Second, the relationship between cytokines and different phenotypes of HF still needs to be further explored because of the lack of datasets stratified by cardiac function class and ejection fraction. Third, to assess the relationship between as many cytokines as possible and HF, we used SNPs for exposure factors with a significance of P < 5 × 10−5, and the process of IVs screening may still carry some risk of confounders, such as the possibility that different cytokines may be confounders for each other. Finally, the dataset of circulating cytokines we used did not provide effector allele frequencies (EAFs), and there may be differences in the direction of alignment of the SNPs for exposure and outcome, but we also applied MR-Egger and MR-PRESSO tests for horizontal pleiotropy to reduce bias. In the future MR studies, in order to minimize all these bias, larger gene-wide databases of cytokines and diseases should be established, which should contain multi-ethnic populations and disease phenotypes. And animal experiments and clinical trials should also be used to validate the results of MR analyses and to provide more scientific evidence for the conclusions.
In conclusion, our study indicates that, among people of European ancestry, genetically increased plasma levels of MIP-1β, IP-10, and RANTES are positively associated with an increased risk of HF, while genetically increased risk of HF is associated with elevated plasma levels of IL-17, FGF-basic, PDGF-BB, IFN-γ, and decreased levels of Eotaxin. These findings provide an epidemiologic basis for cytokine-targeted drugs to prevent and treat HF, however, more animal experiments, clinical trials based on large samples is still needed in the future to support the conclusions at the gene level.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
HZ: Conceptualization, Writing – original draft. XM: Writing – original draft. ZF: Writing – original draft. CC: Data curation, Writing – review & editing. JL: Formal Analysis, Writing – review & editing. YaW: Formal Analysis, Writing – review & editing. YuW: Validation, Writing – review & editing. HW: Methodology, Writing – review & editing. YL: Methodology, Writing – review & editing. YT: Writing – review & editing. XG: Investigation, Writing – review & editing. LZ: Investigation, Writing – review & editing. XX: Supervision, Writing – review & editing. BZ: Writing – review & editing. QS: Project administration, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by the Clinical Medical Research Center Construction Special Project, Guang ‘anmen Hospital, China Academy of Chinese Medical Sciences (No. 2022LYJSZX05).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1332015/full#supplementary-material
1. Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC, Coats AJS. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res. (2023) 118(17):3272–87. doi: 10.1093/cvr/cvac013 Erratum in: Cardiovasc Res. 2023 Jun 13;119(6):1453.35150240
2. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. (2018) 392(10159):1789–858. doi: 10.1016/S0140-6736(18)32279-7 Erratum in: Lancet. 2019 Jun 22;393(10190):e44.30496104
3. Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. (2016) 13(6):368–78. doi: 10.1038/nrcardio.2016.25
4. Lawson CA, Zaccardi F, Squire I, Ling S, Davies MJ, Lam CSP, et al. 20-year Trends in cause-specific heart failure outcomes by sex, socioeconomic status, and place of diagnosis: a population-based study. Lancet Public Health. (2019) 4(8):e406–20. doi: 10.1016/S2468-2667(19)30108-2
5. Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, et al. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. (2013) 6(3):606–19. doi: 10.1161/HHF.0b013e318291329a
6. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. (1990) 323(4):236–41. doi: 10.1056/NEJM199007263230405
7. Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail. (1996) 2(3):243–9. doi: 10.1016/s1071-9164(96)80047-9
8. Kagan BL, Baldwin RL, Munoz D, Wisnieski BJ. Formation of ion-permeable channels by tumor necrosis factor-alpha. Science. (1992) 255(5050):1427–30. doi: 10.1126/science.1371890
9. Amin MN, Siddiqui SA, Ibrahim M, Hakim ML, Ahammed MS, Kabir A, et al. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. (2020) 8:2050312120965752. doi: 10.1177/2050312120965752
10. Kapadia S, Dibbs Z, Kurrelmeyer K, Kalra D, Seta Y, Wang F, et al. The role of cytokines in the failing human heart. Cardiol Clin. (1998) 16(4):645–56. viii. doi: 10.1016/s0733-8651(05)70041-2
11. Adamopoulos S, Parissis JT, Kremastinos DT. A glossary of circulating cytokines in chronic heart failure. Eur J Heart Fail. (2001) 3(5):517–26. doi: 10.1016/s1388-9842(01)00156-8
12. Kaptoge S, Seshasai SR, Gao P, Freitag DF, Butterworth AS, Borglykke A, et al. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Eur Heart J. (2014) 35(9):578–89. doi: 10.1093/eurheartj/eht367
13. Bozkurt B, Kribbs SB, Clubb FJ Jr, Michael LH, Didenko VV, Hornsby PJ, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. (1998) 97(14):1382–91. doi: 10.1161/01.cir.97.14.1382
14. Aukrust P, Ueland T, Müller F, Andreassen AK, Nordøy I, Aas H, et al. Elevated circulating levels of C-C chemokines in patients with congestive heart failure. Circulation. (1998) 97(12):1136–43. doi: 10.1161/01.cir.97.12.1136
15. Li X, Peng S, Guan B, Chen S, Zhou G, Wei Y, et al. Genetically determined inflammatory biomarkers and the risk of heart failure: a Mendelian randomization study. Front Cardiovasc Med. (2021) 8:734400. doi: 10.3389/fcvm.2021.734400
16. Remmelzwaal S, van Oort S, Handoko ML, van Empel V, Heymans SRB, Beulens JWJ. Inflammation and heart failure: a two-sample Mendelian randomization study. J Cardiovasc Med. (2022) 23(11):728–35. doi: 10.2459/JCM.0000000000001373
17. Emdin CA, Khera AV, Kathiresan S. Mendelian Randomization. JAMA. (2017) 318(19):1925–6. doi: 10.1001/jama.2017.17219
18. Davey Smith G, Hemani G. Mendelian Randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. (2014) 23(R1):R89–98. doi: 10.1093/hmg/ddu328
19. Liu K, Zou J, Fan H, Hu H, You Z. Causal effects of gut microbiota on diabetic retinopathy: a Mendelian randomization study. Front Immunol. (2022) 13:930318. doi: 10.3389/fimmu.2022.930318
20. Wu S, Ye Z, Yan Y, Zhan X, Ren L, Zhou C, et al. The causal relationship between autoimmune diseases and osteoporosis: a study based on Mendelian randomization. Front Endocrinol (Lausanne). (2023) 14:1196269. doi: 10.3389/fendo.2023.1196269
21. Yang J, Liu P, Wang S, Jiang T, Zhang Y, Liu W. Causal relationship between sarcopenia and osteoarthritis: a bi-directional two-sample Mendelian randomized study. Eur J Med Res. (2023) 28(1):327. doi: 10.1186/s40001-023-01322-0
22. Skrivankova VW, Richmond RC, Woolf BAR, Yarmolinsky J, Davies NM, Swanson SA, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: the STROBE-MR statement. JAMA. (2021) 326(16):1614–21. doi: 10.1001/jama.2021.18236
23. Larsson SC, Butterworth AS, Burgess S. Mendelian randomization for cardiovascular diseases: principles and applications. Eur Heart J. (2023) 44(47):4913–24. doi: 10.1093/eurheartj/ehad736
24. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Br Med J. (2018) 362:k601. doi: 10.1136/bmj.k601
25. Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. (2020) 1(1):163. doi: 10.1038/s41467-019-13690-5
26. Ahola-Olli AV, Würtz P, Havulinna AS, Aalto K, Pitkänen N, Lehtimäki T, et al. Genome-wide association study identifies 27 loci influencing concentrations of circulating cytokines and growth factors. Am J Hum Genet. (2017) 100(1):40–50. doi: 10.1016/j.ajhg.2016.11.007
27. Liu D, Qin Z, Yi B, Xie H, Liang Y, Zhu L, et al. Inflammatory cytokine profiles in erectile dysfunction: a bidirectional Mendelian randomization. Front Immunol. (2024) 15:1342658. doi: 10.3389/fimmu.2024.1342658
28. Jiang Z, Cai X, Yao X, Zhang S, Lan W, Jin Z, et al. The causal effect of cytokine cycling levels on osteoarthritis: a bidirectional Mendelian randomized study. Front Immunol. (2024) 14:1334361. doi: 10.3389/fimmu.2023.1334361
29. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-egger regression: the role of the I2 statistic. Int J Epidemiol. (2016) 45(6):1961–74. doi: 10.1093/ije/dyw220
30. Chen H, Peng L, Wang Z, He Y, Zhang X. Exploring the causal relationship between periodontitis and gut microbiome: unveiling the oral-gut and gut-oral axes through bidirectional Mendelian randomization. J Clin Periodontol. (2024) 51(4):417–30. doi: 10.1111/jcpe.13906
31. Huang YF, Zhang WM, Wei ZS, Huang H, Mo QY, Shi DL, et al. Causal relationships between gut microbiota and programmed cell death protein 1/programmed cell death-ligand 1: a bidirectional Mendelian randomization study. Front Immunol. (2023) 14:1136169. doi: 10.3389/fimmu.2023.1136169
32. Birney E. Mendelian Randomization. Cold Spring Harb Perspect Med. (2022) 12(4):a041302. doi: 10.1101/cshperspect.a041302
33. Nagai T, Komuro I. Gene and cytokine therapy for heart failure: molecular mechanisms in the improvement of cardiac function. Am J Physiol Heart Circ Physiol. (2012) 303(5):H501–12. doi: 10.1152/ajpheart.00130.2012
34. Hartman MHT, Groot HE, Leach IM, Karper JC, van der Harst P. Translational overview of cytokine inhibition in acute myocardial infarction and chronic heart failure. Trends Cardiovasc Med. (2018) 28(6):369–79. doi: 10.1016/j.tcm.2018.02.003
35. Cappuzzello C, Di Vito L, Melchionna R, Melillo G, Silvestri L, Cesareo E, et al. Increase of plasma IL-9 and decrease of plasma IL-5, IL-7, and IFN-γ in patients with chronic heart failure. J Transl Med. (2011) 9:28. doi: 10.1186/1479-5876-9-28
36. Damås JK, Gullestad L, Aass H, Simonsen S, Fjeld JG, Wikeby L, et al. Enhanced gene expression of chemokines and their corresponding receptors in mononuclear blood cells in chronic heart failure–modulatory effect of intravenous immunoglobulin. J Am Coll Cardiol. (2001) 38(1):187–93. doi: 10.1016/s0735-1097(01)01335-3
37. Sopova K, Tual-Chalot S, Mueller-Hennessen M, Vlachogiannis NI, Georgiopoulos G, Biener M, et al. Effector T cell chemokine IP-10 predicts cardiac recovery and clinical outcomes post-myocardial infarction. Front Immunol. (2023) 14:1177467. doi: 10.3389/fimmu.2023.1177467
38. Cheng C, Tempel D, van Haperen R, de Boer HC, Segers D, Huisman M, et al. Shear stress-induced changes in atherosclerotic plaque composition are modulated by chemokines. J Clin Invest. (2007) 117(3):616–26. doi: 10.1172/JCI28180
39. Wei T, Zhu Z, Liu L, Liu B, Wu M, Zhang W, et al. Circulating levels of cytokines and risk of cardiovascular disease: a Mendelian randomization study. Front Immunol. (2023) 14:1175421. doi: 10.3389/fimmu.2023.1175421
40. Altara R, Gu YM, Struijker-Boudier HA, Thijs L, Staessen JA, Blankesteijn WM. Left ventricular dysfunction and CXCR3 ligands in hypertension: from animal experiments to a population-based pilot study. PLoS One. (2015) 10(10):e0141394. doi: 10.1371/journal.pone.0141394
41. Herder C, Baumert J, Thorand B, Martin S, Löwel H, Kolb H, et al. Chemokines and incident coronary heart disease: results from the MONICA/KORA augsburg case-cohort study, 1984–2002. Arterioscler Thromb Vasc Biol. (2006) 26(9):2147–52. doi: 10.1161/01.ATV.0000235691.84430.86
42. Leavitt C, Zakai NA, Auer P, Cushman M, Lange EM, Levitan EB, et al. Interferon gamma-induced protein 10 (IP-10) and cardiovascular disease in African Americans. PLoS One. (2020) 15(4):e0231013. doi: 10.1371/journal.pone.0231013
43. Need AC, Goldstein DB. Next generation disparities in human genomics: concerns and remedies. Trends Genet. (2009) 25(11):489–94. doi: 10.1016/j.tig.2009.09.012
44. Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc Natl Acad Sci U S A. (1997) 94(7):3168–71. doi: 10.1073/pnas.94.7.3168
45. Simon AD, Yazdani S, Wang W, Schwartz A, Rabbani LE. Elevated plasma levels of interleukin-2 and soluble IL-2 receptor in ischemic heart disease. Clin Cardiol. (2001) 24(3):253–6. doi: 10.1002/clc.4960240315
46. Sakamoto A, Ishizaka N, Saito K, Imai Y, Morita H, Koike K, et al. Serum levels of IgG4 and soluble interleukin-2 receptor in patients with coronary artery disease. Clin Chim Acta. (2012) 413(5-6):577–81. doi: 10.1016/j.cca.2011.11.023
47. Chen X, Wang X, Zhang Z, Chen Y, Wang C. Role of IL-9, IL-2RA, and IL-2RB genetic polymorphisms in coronary heart disease. Herz. (2021) 46(6):558–66. (in English). doi: 10.1007/s00059-020-05004-z
48. Edwards RH, Rutter WJ, Hanahan D. Directed expression of NGF to pancreatic beta cells in transgenic mice leads to selective hyperinnervation of the islets. Cell. (1989) 58(1):161–70. doi: 10.1016/0092-8674(89)90412-1
49. Lockhart ST, Turrigiano GG, Birren SJ. Nerve growth factor modulates synaptic transmission between sympathetic neurons and cardiac myocytes. J Neurosci. (1997) 17(24):9573–82. doi: 10.1523/JNEUROSCI.17-24-09573.1997
50. Wang Q, Zhao Y, Dong X, Li C, Zhou L, Zou C, et al. The occurrence of valvular atrial fibrillation: involvement of NGF/TrKA signaling pathway. J Invest Surg. (2021) 34(12):1379–86. doi: 10.1080/08941939.2020.1798570 Erratum in: J Invest Surg. 2020 August 17;:1-2.32781864
51. Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van Linthout S, et al. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. (2010) 106(7):1275–84. doi: 10.1161/CIRCRESAHA.109.210088
52. Qin F, Vulapalli RS, Stevens SY, Liang CS. Loss of cardiac sympathetic neurotransmitters in heart failure and NE infusion is associated with reduced NGF. Am J Physiol Heart Circ Physiol. (2002) 282(1):H363–71. doi: 10.1152/ajpheart.00319.2001
53. Rahmati Z, Amirzargar AA, Saadati S, Rahmani F, Mahmoudi MJ, Rahnemoon Z, et al. Association of levels of interleukin 17 and T-helper 17 count with symptom severity and etiology of chronic heart failure: a case-control study. Croat Med J. (2018) 59(4):139–48. doi: 10.3325/cmj.2018.59.139
54. Xue GL, Li DS, Wang ZY, Liu Y, Yang JM, Li CZ, et al. Interleukin-17 upregulation participates in the pathogenesis of heart failure in mice via NF-κB-dependent suppression of SERCA2a and Cav1.2 expression. Acta Pharmacol Sin. (2021) 42(11):1780–9. doi: 10.1038/s41401-020-00580-6
55. Kaye DM, Vaddadi G, Gruskin SL, Du XJ, Esler MD. Reduced myocardial nerve growth factor expression in human and experimental heart failure. Circ Res. (2000) 86(7):E80–4. doi: 10.1161/01.res.86.7.e80
56. Vistnes M, Waehre A, Nygård S, Sjaastad I, Andersson KB, Husberg C, et al. Circulating cytokine levels in mice with heart failure are etiology dependent. J Appl Physiol (1985). (2010) 108(5):1357–64. doi: 10.1152/japplphysiol.01084.2009
57. Zee RY, Cook NR, Cheng S, Erlich HA, Lindpaintner K, Lee RT, et al. Threonine for alanine substitution in the eotaxin (CCL11) gene and the risk of incident myocardial infarction. Atherosclerosis. (2004) 175(1):91–4. doi: 10.1016/j.atherosclerosis.2004.01.042
58. Mosedale DE, Smith DJ, Aitken S, Schofield PM, Clarke SC, McNab D, et al. Circulating levels of MCP-1 and eotaxin are not associated with presence of atherosclerosis or previous myocardial infarction. Atherosclerosis. (2005) 183(2):268–74. doi: 10.1016/j.atherosclerosis.2004.11.028
59. Nakamura M, Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol (Lond). (2020) 598:2977–93. doi: 10.1113/JP276747
60. Wanner C, Schuchhardt J, Bauer C, Lindemann S, Brinker M, Kong SX, et al. Clinical characteristics and disease outcomes in non-diabetic chronic kidney disease: retrospective analysis of a US healthcare claims database. J Nephrol. (2023) 36(1):45–54. doi: 10.1007/s40620-022-01340-x
61. Putnam K, Shoemaker R, Yiannikouris F, Cassis LA. The renin-angiotensin system: a target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am J Physiol Heart Circ Physiol. (2012) 302:H1219–30. doi: 10.1152/ajpheart.00796.2011
62. Kleveland O, Kunszt G, Bratlie M, Ueland T, Broch K, Holte E, et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur Heart J. (2016) 37:2406–13. doi: 10.1093/eurheartj/ehw171
Keywords: cytokines, heart failure, Mendelian randomization, genetics, bidirectional, two-sample
Citation: Zheng H, Mao X, Fu Z, Chen C, Lv J, Wang Y, Wang Y, Wu H, Li Y, Tan Y, Gao X, Zhao L, Xu X, Zhang B and Song Q (2024) The role of circulating cytokines in heart failure: a bidirectional, two-sample Mendelian randomization study. Front. Cardiovasc. Med. 11:1332015. doi: 10.3389/fcvm.2024.1332015
Received: 2 November 2023; Accepted: 19 July 2024;
Published: 22 October 2024.
Edited by:
Syamal K. Bhattacharya, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Thomas E. Sharp III, University of South Florida, United States© 2024 Zheng, Mao, Fu, Chen, Lv, Wang, Wang, Wu, Li, Tan, Gao, Zhao, Xu, Zhang and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Xu, MTc2MDMyMTkwMkBxcS5jb20=; Bingxuan Zhang, emJ4MTExOEAxMjYuY29t; Qingqiao Song, U29uZ3FxYmpAMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.