 Yuliang Liu
Yuliang Liu Yingying Guo2,†
Yingying Guo2,† Haipeng Guo
Haipeng Guo Man Xu
Man Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
EDITORIAL article
Front. Cardiovasc. Med. , 05 October 2023
Sec. Cardiovascular Metabolism
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1296935
This article is part of the Research Topic Crosstalk Between Lipid Metabolism and Ferroptosis in Cardiovascular Diseases View all 6 articles
Editorial on the Research Topic
Crosstalk between lipid metabolism and ferroptosis in cardiovascular diseases
Cardiovascular diseases (CVDs) pose a significant threat to human health, with their prevalence steadily increasing even among younger populations in developed countries (1). Moreover, CVD is still the primary cause of death in China, the world’s largest developing country (2). Therefore, devoting more attention to the pathogenesis and therapeutic measures of CVDs is crucial for maintaining health and reducing economic burden. The myocardium is not characterized by self-renewal, so the function and survival of cardiomyocytes are of particular concern in cardiovascular health. In recent years, ferroptosis has gradually been recognized as an essential type of death in cardiomyocytes and is usually initiated by lipid peroxidation. The metabolic pathways of ferroptosis mainly involve iron, lipid, and amino acid metabolism. Mitochondrial activity and intracellular redox homeostasis are also significantly associated with ferroptosis. Extensive previous studies have demonstrated a close association between different components of lipid metabolic pathways and CVDs. To identify the crosstalk between ferroptosis and lipid metabolism and explore how it affects cardiovascular health, we present new evidence regarding ferroptosis and lipid metabolism in the cardiovascular system, including clinically relevant drug applications, through this editorial. In addition, we summarize the new findings of the contributing articles on this research topic.
Recent studies indicated that iron deficiency may affect the prognosis of some patients with CVDs, and iron supplementation can benefit patients who suffer from heart failure with reduced ejection fraction or mildly reduced ejection fraction (HFrEF/HFmrEF) (3). However, there is growing evidence that cell death triggered by iron overload is deeply engaged in the emergence of many CVDs and a common pathophysiologic process in heart failure, cardiomyopathy, myocardial ischemia/reperfusion injury (I/RI), drug-associated myocardial damage, diabetic cardiomyopathy, and others (4). Teng and colleagues’ bibliometric analysis of ferroptosis in CVDs recently enriched our research topic. Their findings, presented through data visualization, indicate that research in this area has grown steadily in recent years. Specifically, studies have concentrated on I/RI, heart failure, and atherosclerosis. At the same time, they also provide scholars with the research direction of ferroptosis in the future, such as endothelial damage and gut microbiota-related cardiomyocyte ferroptosis. Potential inducers or inhibitors of ferroptosis are also promising research topics for CVDs.
Dixon et al. initially identified ferroptosis as a type of regulated cell death (RCD) that can be repressed by ferrostatin-1 and is activated by RAS-selective lethal (RSL) compounds, such as erastin and RSL3. Unlike other forms of RCD, electron microscopic views of mitochondrial outer membrane rupture, cristae reduction, and mitochondrial membrane condensation characterize ferroptosis (5). During amino acid metabolism, cystine is transported into the cell by the glutamate/cystine antiporter system Xc—, composed of the subunits SLC3A2 and SLC7A11. The intracellular cystine is then altered to cysteine and is involved in glutathione (GSH) synthesis. As an active peroxidase, glutathione peroxidase 4 (GPX4) rapidly oxidizes GSH to glutathione disulfide (GSSG) and inhibits lipid peroxidation or promotes the degradation of lipid peroxides. GSH depletion or reduced GPX4 activity can lead to impaired lipid peroxide metabolism and thus aggravate ferroptosis (6). Scavenging lipid peroxides reduces oxidative stress in cardiomyocytes and lessens ferroptosis severity (Figure 1).
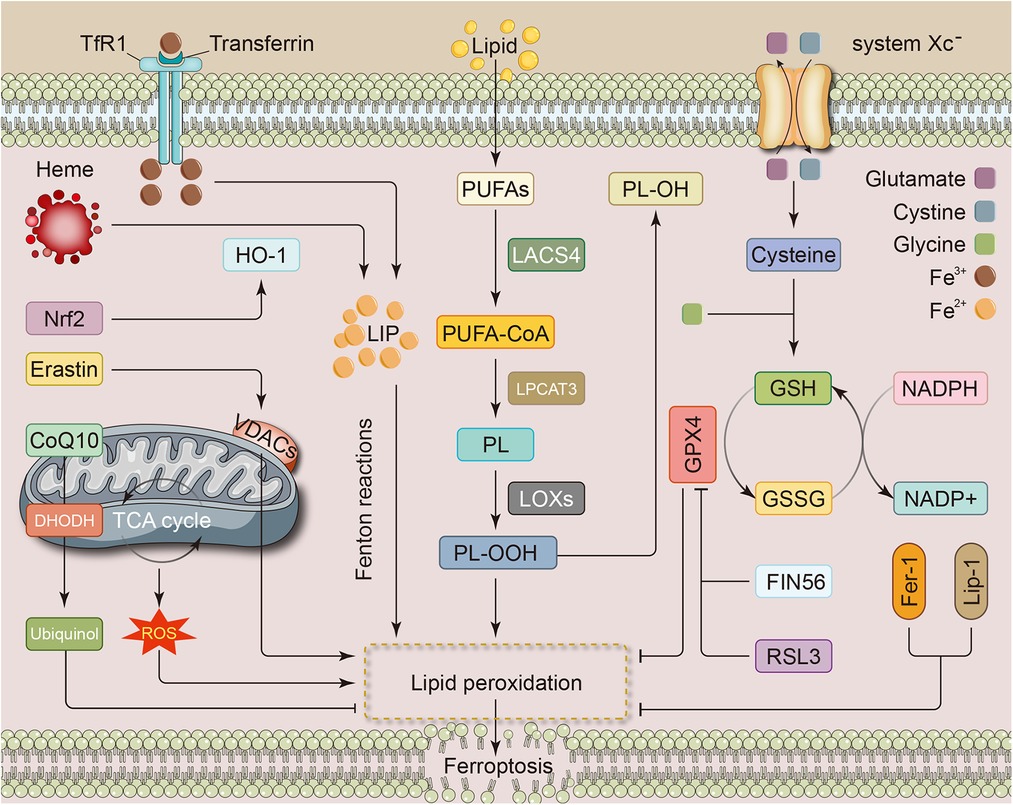
Figure 1. Metabolic pathways associated with ferroptosis in cardiovascular disease. The metabolic pathways of ferroptosis mainly involve iron, lipid, and amino acid metabolism. Iron overload from any cause can be a source of LIP and promote the development of ferroptosis, e.g., nrf2 promotes heme catabolism and thus increases free iron levels. Intracellular PUFAs are converted to toxic forms through a series of enzymatic reactions, which in turn lead to the accumulation of lipid peroxides. The system Xc—, consisting of the subunits SLC3A2 and SLC7A11 subunits, is responsible for transporting glutamate and cystine. Cystine undergoes conversion to cysteine and then participates in the synthesis of GSH. GPX4, which plays a role during the transformation of GSH to GSSG, inhibits lipid peroxidation and attenuates ferroptosis. In addition, several metabolic processes within the mitochondria and some substances, such as RSL3 and Fer-1, can also influence the development of ferroptosis through specific mechanisms. CoQ10, coenzyme Q10; DHODH, dihydroorotate dehydrogenase; Fer-1, ferrostatin-1; FIN56, ferroptosis inducing 56; GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, oxidized glutathione; HO-1, heme oxygenase-1; LACS4, long-chain fatty acid coenzyme A (CoA) ligase 4; LIP, labile iron pool; Lip-1, liproxstatin-1; LOX, lipoxygenase; LPCAT3, lysophosphatidylcholine acyltransferase3; NADPH, nicotinamide adenine dinucleotide phosphate; Nrf2, nuclear factor erythroid 2-related factor; PL, phospholipids; PUFA, polyunsaturated fatty acids; ROS, reactive oxygen species; RSL3, RAS-selective lethal 3; TCA, tricarboxylic acid; TfR1, transferrin receptor 1; VDAC, voltage-dependent anion channel.
Lipid metabolism affects phospholipid peroxidation and various other cellular processes associated with it, so lipid metabolism may regulate ferroptosis through various signaling pathways, e.g., by regulating the levels of lipid peroxides and related lipid metabolizing enzymes. Indeed, excess lipid peroxides are considered to be key drivers in ferroptosis (7). The acylation of polyunsaturated fatty acids (PUFA) to toxic forms by the enzyme long-chain fatty acid CoA ligase 4 (LACS4) is a significant process of lipid peroxidation in ferroptosis, makes cells more sensitive to ferroptosis and promotes the initiation of iron-dependent cell death cascade responses (8). In contrast, LACS3 reduces the accumulation of reactive oxygen species (ROS) and lipid peroxides at the plasma membrane by activating exogenous monounsaturated fatty acids (MUFAs) (9). In addition, PUFAs are sequentially converted by lysophosphatidylcholine acyltransferase3 (LPCAT3) and lipoxygenase (LOX) to PUFA-containing membrane phospholipid and lipid ROS, which then mediate the membrane injury (10). As main substances produced by the process of lipid peroxidation, malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) (11) are also present in cardiac tissues subjected to oxidative stress injury and are down-regulated by the nuclear factor erythroid 2-related factor (Nrf2) activator resveratrol (12). Notably, during doxorubicin (DOX)-induced myocardial injury, increased heme oxygenase-1 (HO-1) induced by Nrf2 promotes heme degradation and free iron release, exacerbating ferroptosis and myocardial injury (13). However, numerous previous investigations have shown that the Nrf2/HO-1 system is essential for ROS scavenging and attenuating oxidative stress injury. Thus, ferroptosis may impair myocardial viability through its unique role, although it is involved in oxidative stress. As represented by acute infarction, ischemic heart disease is also characterized by a high mortality rate, and cell death exhibits key effects in I/RI. Feng and colleagues discovered that liproxstatin-1, a ferroptosis inhibitor, lessened I/RI in mice myocardium by restoring GPX4 concentration, decreasing ROS production and accumulation in mitochondria, preserving mitochondrial function, and reducing myocardial infarction area (14).
As previously described, SLC7A11, one of the subunits of the system Xc—, attenuates ferroptosis and cardiovascular injury by promoting GSH synthesis and ROS clearance. Interestingly, Hu et al. showed that in the animal model of pulmonary arterial hypertension, erastin could attenuate pulmonary vascular remodeling by inducing ferroptosis to attenuate the proliferation of pulmonary arterial smooth muscle and subsequently alleviate pulmonary vascular remodeling. The mechanism of action was that erastin administration decreased the levels of SLC7A11 and GPX4 (15). Conversely, Xie et al. concluded that ferrostatin-1, a ferroptosis inhibitor, helped attenuate MCT-induced pulmonary hypertension, restored right ventricular function, and attenuated inflammatory damage to pulmonary artery endothelial cells (16). Recently, Xu and Bu provided new perspectives on Hmox1 and SLC7A11 in CVDs to our research topic. They analyzed two gene expression datasets, GSE185754 and GSE171546, and then identified Hmox1 (HO-1) and SLC7A11 as ferroptosis-related targets. Subsequently, the authors used siRNA to knock down these two genes in the model of LPS-induced myocardial injury, demonstrating that downregulation of Hmox1 and SLC7A11 contributes to restoring cardiomyocyte viability and morphology, potentially attenuating septic cardiomyopathy. Disturbed lipid metabolism is a characteristic metabolic abnormality of atherosclerosis. In the high-fat diet-induced atherosclerosis in ApoE-/- mice model, ferrostatin-1 restored the expression of SLC7A11 and GPX4 while also reducing iron content. In addition, inhibition of ferroptosis represented attenuated lipid peroxidation, which was closely associated with improved arterial endothelial function (17).
Researchers have also progressively begun to understand ferroptosis at the genetic level in recent years. Wu et al. discovered several ferroptosis-associated genes, such as CA9, CBS, CEBPG, and HSPB1, which are considered to be strongly associated with coronary artery disease (18). In addition, several studies on this topic have identified ferroptosis-related genes, offering new targets for ferroptosis. Li et al. identified genes associated with ferroptosis in calcific aortic valve disease for the first time. In addition, they conducted GSEA and KEGG pathway analyses, followed by validation in vitro experiments. Finally, they indicated that IL-6, BID, PRKAA2, HIF-1, and HMOX1 genes are relevant targets for ferroptosis; accordingly, the NAFLD and the HIF-1 pathway act as ferroptosis-related signaling pathways that influence the development of calcific aortic valve disease. Similarly, Huang et al. analyzed gene expression datasets in ischemic cardiomyopathy (ICM) and identified dozens of differentially expressed genes associated with ferroptosis. They also considered IL-6, JUN, STAT3, MAP3K5, and ATM as the top five hub genes, which were also confirmed to be significantly altered in expression by qRT-PCR in clinical blood samples from ICM. Subsequently, functional enrichment analysis revealed that ferroptosis in ICM may involve oxidative stress, immune-related, and inflammation-related signaling pathways. In summary, further studies are being conducted to identify numerous ferroptosis-related genes, and the mechanisms that affect CVD deserve further exploration.
Based on various preclinical studies, certain substances like ferrostatin-1 have shown effective anti-ferroptosis properties through mechanisms like preventing iron overload, scavenging ROS, activating GPX4, and reducing lipid peroxidation (4). Wang et al. showed that tanshinoneIIA, similar to ferrostatin-1, restored GPX4 and SLC7A11 expression in high-power microwave (HPM)-damaged cardiomyocytes while decreasing the levels of Hmox1 and ACSL4, which ultimately attenuated the severity of HPM-induced ferroptosis. Future studies should focus more on collecting evidence for the role of ferroptosis in human specimens or in vivo, sorting out the complex relationship between ferroptosis and lipid metabolism, ultimately providing a basis for the discovery of new markers and the development of novel ferroptosis inducers and/or inhibitors. In addition, the risk factors of ferroptosis in cardiomyocytes, the predictive indicators of ferroptosis, the predictive value of ferroptosis and its degree in different CVDs, such as what degree of ferroptosis can predict the possible recurrence of inflammation and oxidative stress, and the timing or degree of intervention for ferroptosis targets, are all topics that may need to be explored in the future.
Overall, this research topic addresses the role of ferroptosis in CVDs and highlights the diverse mechanisms by which it affects disease progression. Based on the current evidence, we firmly believe that ferroptosis and lipid metabolism have the potential to be effective therapeutic targets. However, more in-depth research is required to clarify the crosstalk between ferroptosis and lipid metabolism, ultimately determining the reliability of its transformation into clinical application. We hope this research topic will not only provide readers with new insights into ferroptosis and cardiovascular health but also inspire new advances in this research field and even other research topics.
YL: Project administration, Writing – original draft. YG: Project administration, Writing – review and editing. PL: Investigation, Writing – review and editing. HG: Writing – original draft, Writing – review and editing. MX: Writing – original draft, Writing – review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by grants from the National Natural Science Foundation of China (82000229 and 82172165), the Taishan Young Scholar Program of Shandong Province (tsqn202103171) and the Clinical Research Center of Shandong University (2020SDUCRCC007).
We thank all the authors and reviewers for contributing to this Research Topic.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Andersson C, Vasan RS. Epidemiology of cardiovascular disease in young individuals. Nat Rev Cardiol. (2018) 15(4):230–40. doi: 10.1038/nrcardio.2017.154
2. Liu S, Li Y, Zeng X, Wang H, Yin P, Wang L, et al. Burden of cardiovascular diseases in China, 1990–2016: findings from the 2016 global burden of disease study. JAMA Cardiol. (2019) 4(4):342–52. doi: 10.1001/jamacardio.2019.0295
3. Savarese G, von Haehling S, Butler J, Cleland JGF, Ponikowski P, Anker SD. Iron deficiency and cardiovascular disease. Eur Heart J. (2023) 44(1):14–27. doi: 10.1093/eurheartj/ehac569
4. Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. (2023) 20(1):7–23. doi: 10.1038/s41569-022-00735-4
5. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
6. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
7. Dixon SJ, Stockwell BR. The hallmarks of ferroptosis. In: Jacks T, Sawyers CL, editors. Annual review of cancer biology, vol 3. Annual Review of Cancer Biology-3. Palo Alto: Annual Reviews (2019). p. 35–54. doi: 10.1146/annurev-cancerbio-030518-055844
8. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
9. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. (2019) 26(3):420–32.e9. doi: 10.1016/j.chembiol.2018.11.016
10. Zhang Y, Xin L, Xiang M, Shang C, Wang Y, Wang Y, et al. The molecular mechanisms of ferroptosis and its role in cardiovascular disease. Biomed Pharmacother. (2022) 145:112423. doi: 10.1016/j.biopha.2021.112423
11. Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. (1991) 11(1):81–128. doi: 10.1016/0891-5849(91)90192-6
12. Bai T, Hu X, Zheng Y, Wang S, Kong J, Cai L. Resveratrol protects against lipopolysaccharide-induced cardiac dysfunction by enhancing SERCA2a activity through promoting the phospholamban oligomerization. Am J Physiol Heart Circ Physiol. (2016) 311(4):H1051–h62. doi: 10.1152/ajpheart.00296.2016
13. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. (2019) 116(7):2672–80. doi: 10.1073/pnas.1821022116
14. Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. (2019) 520(3):606–11. doi: 10.1016/j.bbrc.2019.10.006
15. Hu P, Xu Y, Jiang Y, Huang J, Liu Y, Wang D, et al. The mechanism of the imbalance between proliferation and ferroptosis in pulmonary artery smooth muscle cells based on the activation of SLC7A11. Eur J Pharmacol. (2022) 928:175093. doi: 10.1016/j.ejphar.2022.175093
16. Xie SS, Deng Y, Guo SL, Li JQ, Zhou YC, Liao J, et al. Endothelial cell ferroptosis mediates monocrotaline-induced pulmonary hypertension in rats by modulating NLRP3 inflammasome activation. Sci Rep. (2022) 12(1):3056. doi: 10.1038/s41598-022-06848-7
17. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med. (2020) 160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026
Keywords: ferroptosis, cell death, lipid metabolism, lipid peroxidation, cardiovascular disease
Citation: Liu Y, Guo Y, Li P, Guo H and Xu M (2023) Editorial: Crosstalk between lipid metabolism and ferroptosis in cardiovascular diseases. Front. Cardiovasc. Med. 10:1296935. doi: 10.3389/fcvm.2023.1296935
Received: 19 September 2023; Accepted: 20 September 2023;
Published: 5 October 2023.
Edited and Reviewed by: Ichiro Manabe, Chiba University, Japan
© 2023 Liu, Guo, Li, Guo and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Man Xu eHVtYW45ODdAd2h1LmVkdS5jbg== Haipeng Guo SGFpcGVuZzE5ODMzNEAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.