 Sha Su1
Sha Su1 Xiang Ji
Xiang Ji Yu Teng
Yu Teng Baofu Wang
Baofu Wang Mingjing Zhao
Mingjing Zhao- 1Key Laboratory of Chinese Internal Medicine of Ministry of Education and Beijing, Dongzhimen Hospital Affiliated to Beijing University of Chinese Medicine, Beijing, China
- 2Department of Cardiology, Dongzhimen Hospital Affiliated to Beijing University of Chinese Medicine, Beijing, China
Background/aims: To investigate the specific effects of sodium-glucose transporter 2 inhibitor (SGLT2i) on cardiac energy metabolism.
Methods: A systematic literature search was conducted in eight databases. The retrieved studies were screened according to the inclusion and exclusion criteria, and relevant information was extracted according to the purpose of the study. Two researchers independently screened the studies, extracted information, and assessed article quality.
Results: The results of the 34 included studies (including 10 clinical and 24 animal studies) showed that SGLT2i inhibited cardiac glucose uptake and glycolysis, but promoted fatty acid (FA) metabolism in most disease states. SGLT2i upregulated ketone metabolism, improved the structure and functions of myocardial mitochondria, alleviated oxidative stress of cardiomyocytes in all literatures. SGLT2i increased cardiac glucose oxidation in diabetes mellitus (DM) and cardiac FA metabolism in heart failure (HF). However, the regulatory effects of SGLT2i on cardiac FA metabolism in DM and cardiac glucose oxidation in HF varied with disease types, stages, and intervention duration of SGLT2i.
Conclusion: SGLT2i improved the efficiency of cardiac energy production by regulating FA, glucose and ketone metabolism, improving mitochondria structure and functions, and decreasing oxidative stress of cardiomyocytes under pathological conditions. Thus, SGLT2i is deemed to exert a benign regulatory effect on cardiac metabolic disorders in various diseases.
Systematic review registration: https://www.crd.york.ac.uk/, PROSPERO (CRD42023484295).
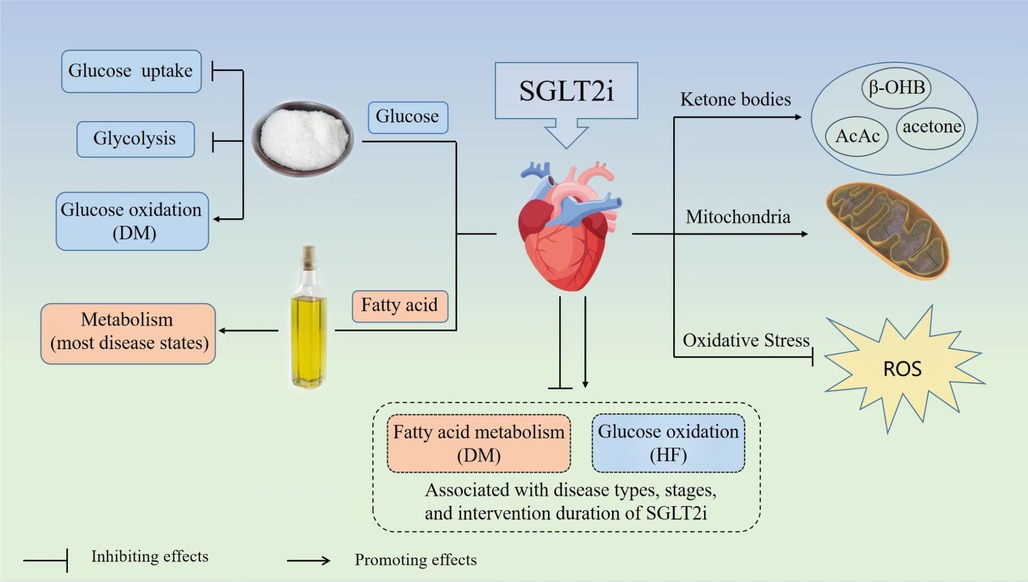
GRAPHICAL ABSTRACT. Graphical abstracts of this systematic review. SGLT2i, sodium-glucose transporter 2 inhibitor; DM, diabetes mellitus; HF, heart failure; ROS, reactive oxygen species; β-OHB, β-hydroxybutyric acid; AcAc, acetoacetate.
1. Introduction
The heart maintains blood circulation of the whole body through uninterrupted systolic and diastolic functions; this process requires a large supply of ATP. Therefore, normal energy metabolism is fundamental for the physiological functions of the heart. Several diseases lead to abnormal energy metabolism, which is one of the major pathological mechanisms of the disrupted physiological functions of the heart. Therefore, regulating energy metabolism is crucial to improving cardiac physiological functions.
As an oral hypoglycemic medication, sodium-glucose transporter 2 inhibitor (SGLT2i) blocks glucose reabsorption in the renal proximal tubules, improving urine glucose excretion and regulating plasma glucose levels (1). Clinically, SGLT2i is mainly used for patients with type 2 diabetes mellitus (T2DM), chronic kidney disease (CKD) and heart failure (HF). Moreover, SGLT2i has been included in the treatment guidelines for heart failure with reduced ejection fraction (HFrEF) (2, 3) and can improve the cardiovascular prognosis of high-risk patients (4), such as reducing the risk of cardiovascular mortality, HF hospitalizations and adverse kidney events (5–7). The benefits are noticeable early after SGLT2i treatment, and the absolute risk is reduced markedly. Thus, SGLT2i may exert a salutary role in the treatment of HFrEF by reducing hyperemia through diuretic/natriuretic properties.
Notably, SGLT2i regulates energy substrates metabolism in the heart and blood circulation (8–13), and also has effects on oxidative stress, mitochondria structure and functions of the heart (14, 15). Although it has been widely used in various diseases due to its cardiac benefits, the specific effects of SGLT2i on cardiac energy metabolism have not been discussed thoroughly. Some studies had shown that SGLT2i promoted the metabolism of cardiac energy substrates such as fatty acid (FA) and glucose in pathological conditions, while it played an inhibitory role in the metabolism of cardiac energy substrates in other diseases, so the results were inconsistent and confusing. Therefore, a systematic review to explore the specific effects of SGLT2i on cardiac energy metabolism is essential.
Based on the existing literatures, we analyzed the specific effects of SGLT2i on cardiac FA, glucose, ketone metabolism, mitochondria structure and functions, oxidative stress in clinical patients and experimental animals with different diseases to explicate the value of clinical application of SGLT2i in cardiac energy metabolism.
2. Methods
The report of this systematic review was prepared based on the PRISMA 2020 Statements.
2.1. Search strategy
Eight electronic databases (PubMed, Web of Science, Embase, Cochrane, China National Knowledge Infrastructure, WanFang Data, VIP Database, and SinoMed) were searched from inception up to April 20, 2023. The search strategy used the following general terms as MeSH terms or free terms: “Sodium Glucose Transporter 2 Inhibitors”, “SGLT-2 Inhibitors”, “SGLT 2 Inhibitors”, “SGLT2 Inhibitors”, “Sodium-Glucose Transporter 2 Inhibitor”, “Sodium Glucose Transporter 2 Inhibitor”, “SGLT2 Inhibitor”, “Inhibitor, SGLT2”, “Gliflozins”, “Gliflozin”, “SGLT-2 Inhibitor”, “Inhibitor, SGLT-2”, “SGLT 2 Inhibitor”, “Empagliflozin”, “Dapagliflozin”, “Canagliflozin”, “Ertugliflozin”, “heart, Myocardium”, “Cardiac Muscle”, “Muscle, Heart”, “Heart Muscle”, “Heart Muscles”, “Muscles, Heart”, “Myocardia”, “Muscle, Cardiac”, “Cardiac Muscles”, “Muscles, Cardiac”, “Energy Metabolism”, “Energy Metabolisms”, “Metabolism, Energy”, “Metabolisms, Energy”, “Energy Expenditure”, “Energy Expenditures”, “Expenditure, Energy”, “Expenditures”, “Energy”, “Bioenergetics”, “Bioenergetic”. For this search, no restrictions or filters were used. The comprehensive search strategy was found in supplement materials. A thorough search of the review articles and references for each identified study was done in order to find further pertinent studies.
2.2. Inclusion and exclusion criteria
The inclusion criteria of the clinical studies were as follows: (1) Participants: Patients were treated with SGLT2i, with or without the control group; (2) Method of research: randomized controlled trials (RCTs), cohort studies and cross-sectional studies were included; (3) Indicators related to cardiac energy metabolism were detected.
The inclusion criteria of the animal studies were as follows: (1) Animals had to be treated with SGLT2i; (2) The research measures were pertinent to cardiac energy metabolism; (3) There was no restriction on language usage and the literatures should be published in peer-reviewed journals.
The exclusion criteria were as follows: (1) Articles with incomplete information; (2) Duplicate publications; (3) Reviews, meta-analysis, and corresponding/conference abstracts.
2.3. Quality assessment
Two authors evaluated the publications' quality separately, and the Cochrane Collaboration tool was utilized to assess the risk of bias (ROB) of RCTs. There are six types of bias in it: selection, performance, detection, attrition, reporting and other sources of bias. “High risk”, “low risk”, or “unclear risk” was used to describe the concentration of each project. The selection of research population, compatibility of the study groups, and measurement of exposure factors were the three criteria used by the Newcastle–Ottawa scale (NOS) to evaluate the quality of cohort studies. Each study received a score of 0–9. Cross-sectional studies were evaluated using the National Institutes of Health (NIH) Quality Assessment Tool (16, 17). The tool comprises 14 questions, and studies with scores <7 were classified as poor, 7–9 as fair, and >9 as good.
SYRCLE's ROB tool was applied to assess the quality of animal studies. A total of ten items from six projects were used as evaluation criteria: sequence generation (i.e., selection bias), baseline characteristics (i.e., selection bias), allocation concealment (i.e., selection bias), random housing (i.e., performance bias), blinding (i.e., performance bias), random outcome assessment (i.e., detection bias), blinding (i.e., detection bias), incomplete outcomes data (i.e., attrition bias), selective outcome reporting (i.e., reporting bias), and other sources of bias. Each item was categorized as either “unclear”, “low risk”, or “high risk” (18). After discussing with the corresponding author, the differences were settled through consensus.
2.4. Data extraction
Data were extracted by two authors independently from the included literatures employing a standardized sheet created specifically for this systematic review. The basic characteristics of clinical studies were collected, consisting of first author's name, year of publication, country, diseases types, study design, group and sample size, SGLT2i names and dose, intervention time of SGLT2i and types of energy metabolism indicators. As for animal studies, the basic information of identified studies was extracted, including first author's name, year of publication, country, sex/animal strain, models (methods), group, SGLT2i names and dose, starting time/intervention time of SGLT2i and types of energy metabolism indicators. Any disagreement was discussed and settled in a consensus meeting with the corresponding author.
2.5. Data analysis
Due to the high heterogeneity of the included literatures, we only compared the general trends of changes in FA, glucose, ketone metabolism, mitochondria structure and functions, and oxidative stress related to cardiac energy metabolism in both clinical and animal studies. The outcome measures, which compared the SGLT2i groups with the model groups, were recorded as a significant uptrend marker “↑” or a significant downtrend marker “↓”. Therefore, a qualitative synthesis was adopted for this systematic review.
3. Results
3.1. Search results and study selection
A total of 618 records were identified from 8 electronic databases. After removing 233 duplicates, 385 potentially relevant literatures were assessed. Subsequently, 232 literatures were excluded after the evaluation of titles and abstracts. Of the 153 remaining literatures, we further excluded 119 after screening the full text. Finally, this systematic review contained 34 studies (10 clinical studies and 24 animal studies). The search procedure and study selection were depicted in the specific flow diagram (Figure 1).
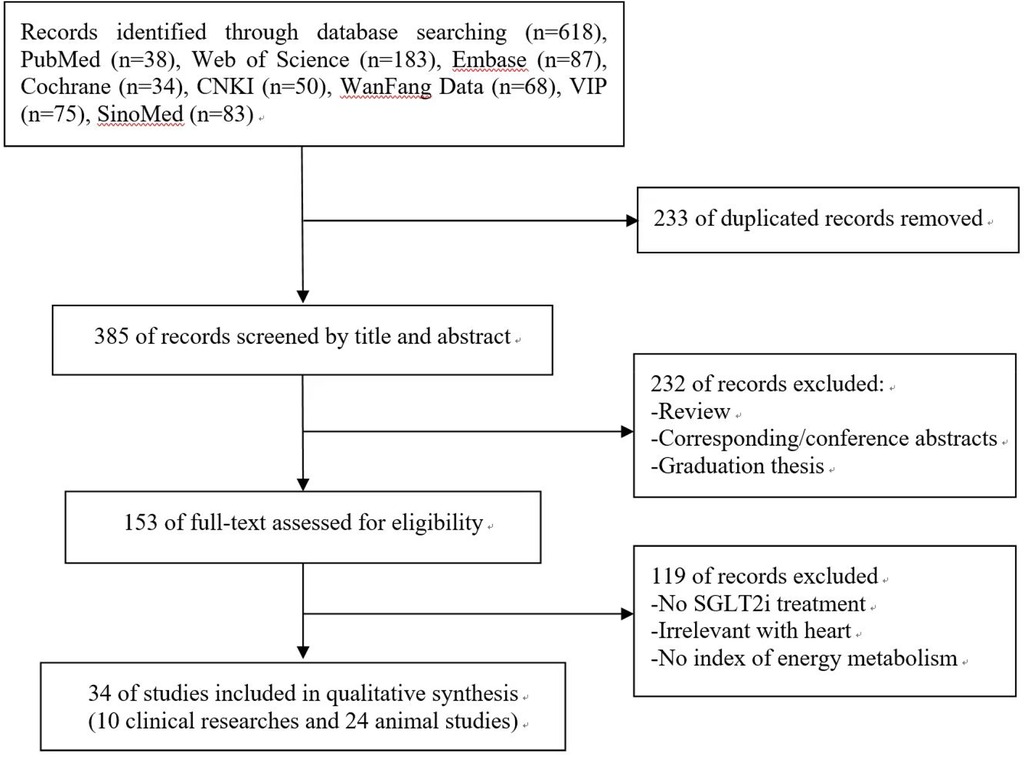
Figure 1. Flow chart of searching and screening studies.
3.2. Characteristics of included studies
3.2.1. Characteristics of clinical studies
As shown in Table 1, the included studies comprised 5 RCTs, 3 cohort studies, and 2 cross-sectional studies. The diseases types included pre-diabetic insulin resistance, T2DM, HFrEF, heart failure with preserved ejection fraction (HFpEF), and aortic or mitral valve replacement surgery. The 10 studies' sample sizes varied from 13 to 1,299, with the majority of them containing both male and female participants. The age range was 52–72.1 years. Among the included clinical studies, Empagliflozin was used in 5 studies, Dapagliflozin in 3 and Canagliflozin in 2. SGLT2i was administrated orally in 9 studies but was used to process human cardiomyocytes in 1 study. The intervention duration of SGLT2i ranged from 2 to 52 weeks. The samples were mainly collected from plasma, and indicators related to FA, glucose, ketone metabolism, and oxidative stress were assessed.
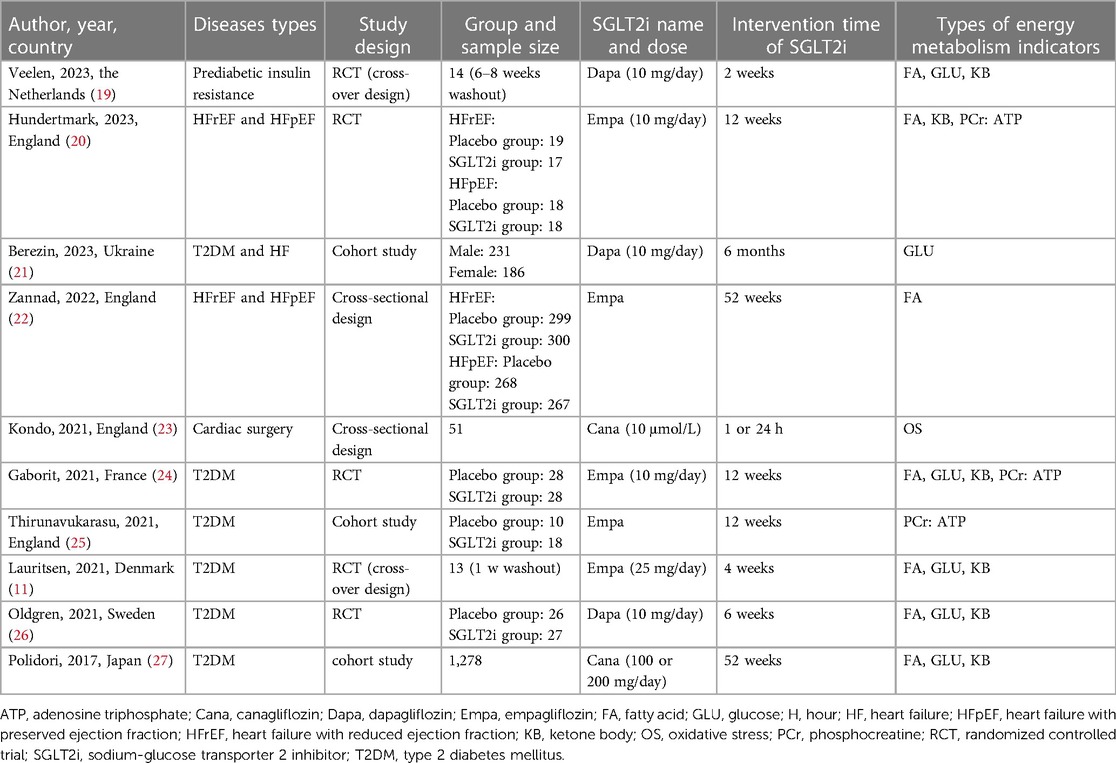
Table 1. Characteristics of the included clinical studies.
3.2.2. Characteristics of animal studies
A total of 24 animal studies were included in this systematic review. The detailed results were summarized in Table 2. The types of models included diabetes mellitus (DM) (4/24), myocardial infarction (MI) induced by left anterior descending coronary artery (LAD) ligation (2/24), cardiac pressure-overload by transverse aortic constriction (TAC) (4/24), ischemia-reperfusion (I/R) injury (6/24), gene knockout mouse (4/24), high-fat diet (HFD) (3/24), high-salt diet (1/24), high-carbohydrate diet (1/24), stress-induced cardiomyopathy (SCM) induced by isoproterenol (ISO) injected intraperitoneally (1/24), cardiotoxicity induced by doxorubicin (DOX) (1/24), and cardiac arrest (CA) induced by ventricular fibrillation (VF) (1/24), and 1 study (46) used normal animals without disease. The modeling time ranged from 10 min to 6 months. Among the 24 animal studies, 17 studies used Empagliflozin, 5 Dapagliflozin, 2 Canagliflozin, 2 Ertugliflozin, and 1 Sotagliflozin. The routes of SGLT2i administration included oral administration (n = 20), Langendorff heart infusion (n = 2), intraperitoneal injection (n = 1), and intravenous injection (n = 1). SGLT2i treatment duration ranged from 10 min to 12 weeks. The primary objective indicators in plasma samples were the same as in the included clinical studies, while animal studies included indicators related to energy metabolism in cardiac tissue.
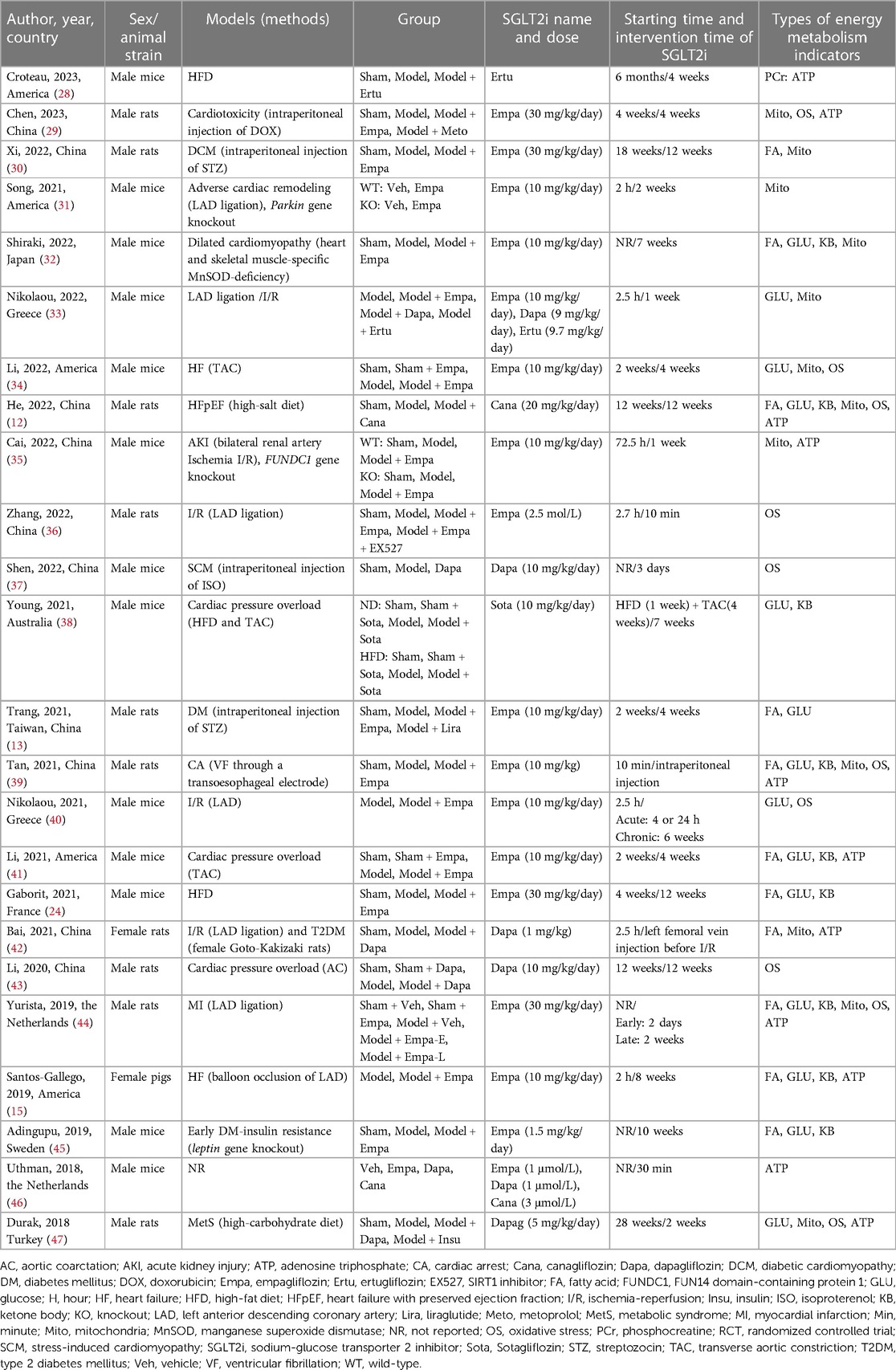
Table 2. Characteristics of the included animal studies.
3.3. Quality assessment of included studies
A total of 5 RCTs were evaluated using the Cochrane evaluation tool, and the results were summarized in Supplementary Table S1 (11, 19, 20, 24, 26). The evaluation results showed that all of 5 studies used random allocation; and attrition bias, reporting bias and other sources of bias were low risks and detection bias were unclear. 4 studies had a low risk about performance bias (11, 19, 20, 26). Allocation concealment was implemented in 2 studies (20, 24). The methodological quality results of the 3 cohort studies evaluated by NOS were shown in Supplementary Table S2. The included studies' mean NOS score was 7 and the overall scores ranged from 66 to 89%. The 2 cross-sectional studies were assessed using the NIH Risk bias tool with scores of 8 (22) and 9 (23), respectively. The detailed results were shown in Supplementary Table S3. According to the assessment of SYRCLE's ROB tool, 24 animal studies reported that the selective outcome was low risk, while the sources of biases, such as animal breeder, investigator blinding, random outcome assessment, and other biases, were unclear. In most studies, the sources of risk regarding sequence generation (22/24), baseline characteristics (19/24), allocation concealment (20/24), and implementation of blinding of outcome assessors (22/24), were unclear. Most studies had a low risk of random housing and incomplete outcome data (14/24). The details of the ROB of the experimental studies were provided in Supplementary Table S4.
3.4. Outcome analysis
This systematic review was comprised of 34 studies in total. These consisted of 10 clinical studies, of which 7 studies were related to DM and 3 studies were related to HF. The 24 animal studies included 4 DM, 2 LAD ligation, 4 TAC, 6 I/R, 4 gene knockout models, and other models types. The main sources of samples collection included plasma and hearts of patients and animals. The energy metabolism-related indicators mainly included FA, glucose, ketone metabolism, mitochondria structure and functions, and oxidative stress.
3.4.1. Effects of SGLT2i on cardiac FA metabolism
Of the 34 studies retrieved, 18 studies (7 clinical and 11 animal studies) addressed FA metabolism. The related indicators in plasma samples included the levels of free fatty acid (FFA), triglyceride (TG), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-c), and low-density lipoprotein cholesterol (LDL-c). The FA metabolism-related indicators in heart samples were as follows: the content of FA translocase (CD36), carnitine palmitoyl transferase-1 (CPT-1), peroxisome proliferator-activated receptor γ coactivator-1α (PGC1-α), peroxisome proliferator-activated receptor α (PPARα), the ratio of phosphorylated-adenosine monophosphate-activated protein kinase to adenosine monophosphate-activated protein kinase (p-AMPK/AMPK), the ratio of phosphorylated-acetyl CoA carboxylase to acetyl CoA carboxylase (p-ACC/ACC), myocardial TG content, myocardial FA uptake, epicardial fat volume, the total number of lipid droplets in cardiomyocytes, and proteomics and metabolomics of myocardial tissue (details were shown in Table 3). Among these, 6 studies showed that SGLT2i increased the FA metabolism of the heart (12, 15, 30, 41, 42, 44), including the expressions of FA uptake-related transporter protein CD36, FA from cytoplasm into mitochondria related protein CPT-1, FA oxidation-related proteins (PGC1-α, PPARα, AMPK, and ACC); notably, the EF values of 5/6 studies (12, 30, 41, 42, 44) were >50% which were similar to the EF values of HFpEF. Cardiac FA metabolism was reduced in 1 study (13) and remained unchanged in 2 studies (11, 32). This phenomenon might be related to the types and stages of diseases. In addition, we could not determine the specific effect of SGLT2i on cardiac FA metabolism due to limited detection indicators in 9 studies (19, 20, 22, 24, 26, 27, 39, 45). Besides, 12 studies (4 clinical and 8 animal studies) documented body weight alterations. 4 studies (12, 24, 27, 45) reported weight loss. 7 studies (11, 15, 19, 20, 30, 32, 41) showed no differences in changes to body weight and 1 study (13) reported increases in body weight. Table 3 displayed the specific results.
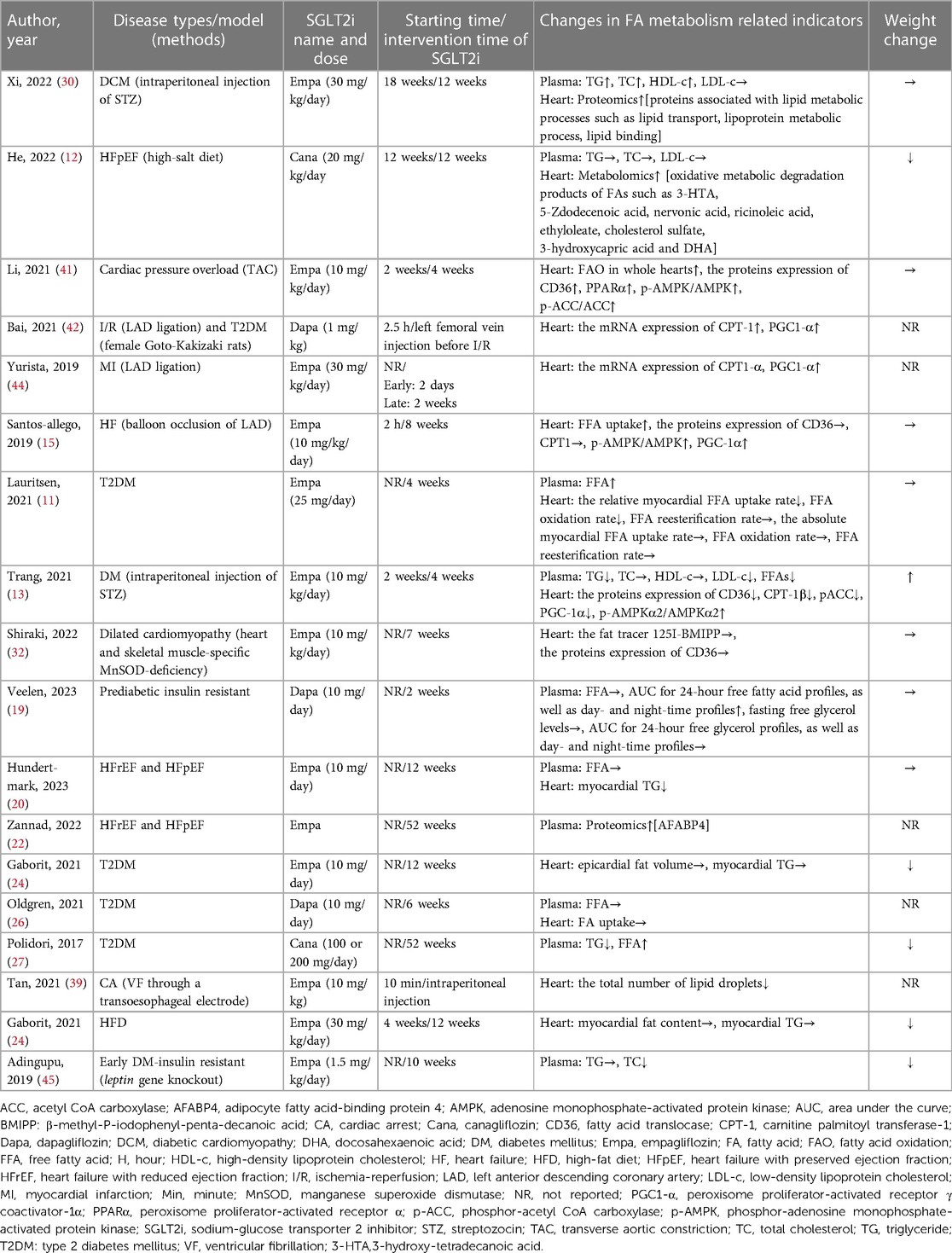
Table 3. Effects of SGLT2i on cardiac FA metabolism in different diseases.
3.4.2. Effects of SGLT2i on cardiac glucose metabolism
As shown in Table 4, 20 studies, including 6 clinical and 14 animal studies, were related to glucose metabolism. The indexes evaluated in plasma samples involved blood glucose and lactic acid (LA) levels. The indicators in myocardium tissue consisted of glucose uptake, glucose oxidation and glycolysis, specifically including the content of glucose transporter 4 (GLUT4) protein, pyruvate dehydrogenase (PDH), pyruvate dehydrogenase kinase 4 (PDK4), and lactate dehydrogenase (LDH). The results showed that myocardial glucose uptake ability decreased in 3 studies (11, 15, 41), while 2 studies (13, 44) demonstrated a significant increase in GLUT4 protein expression in the Empagliflozin group, regardless of T2DM. A total of 3 studies (13, 41, 44) reported that Empagliflozin upregulated glucose oxidation, while some studies (12, 15) put forth an opposite conclusion. Several animal studies (32, 39, 41) reported a decreased serum LA level. Another animal study suggested that Empagliflozin normalized myocardial LA consumption compared to higher net myocardial LA production in the control group (15). However, the level of serum LA remained unchanged in 2 clinical studies (11, 26). In addition, 12 studies could not decide the effect of SGLT2i on cardiac glucose metabolism due to incomplete indicators (19, 21, 24, 26, 27, 33, 34, 38, 40, 45, 47).
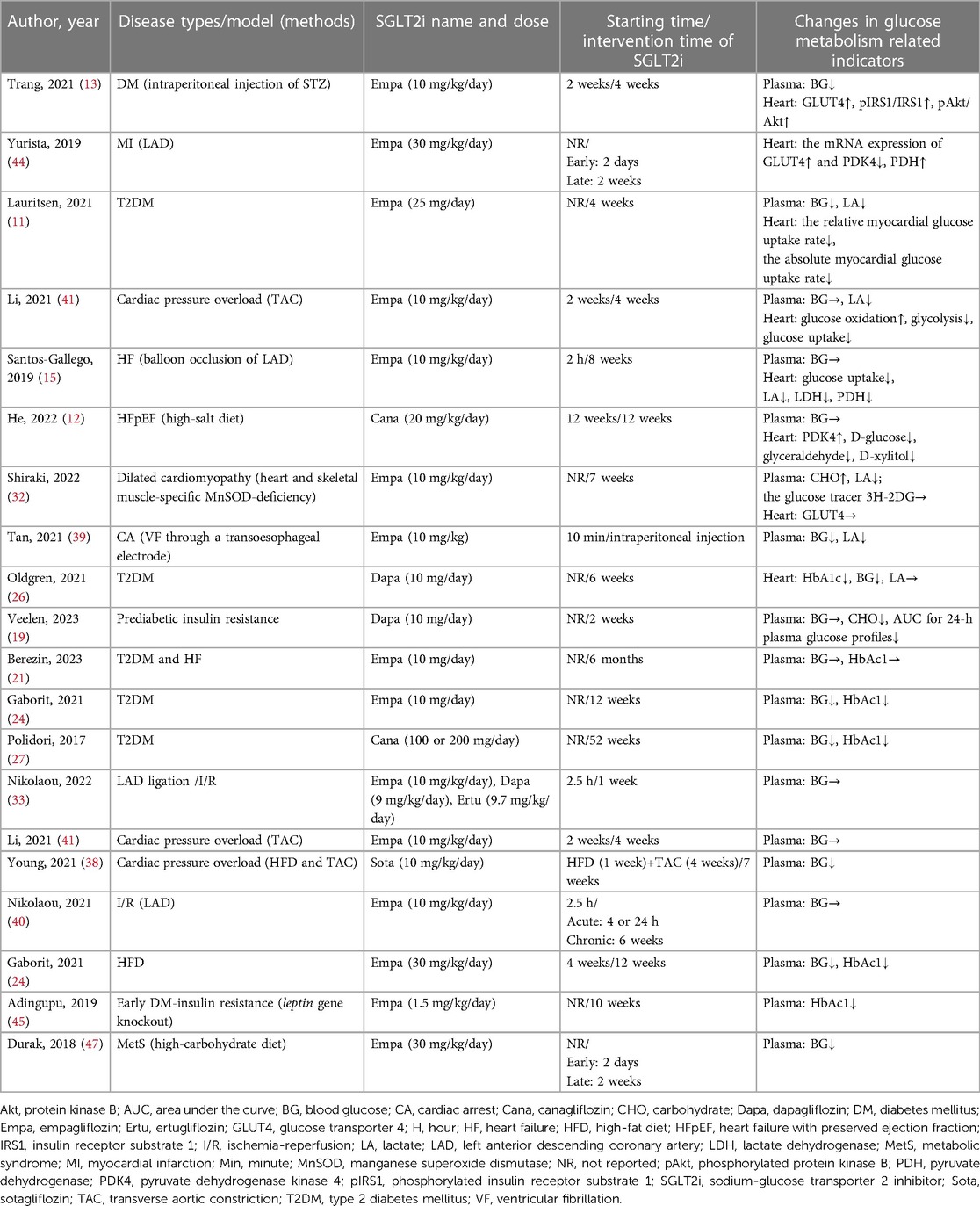
Table 4. Effects of SGLT2i on cardiac glucose metabolism in different diseases.
3.4.3. Effects of SGLT2i on cardiac ketone metabolism
Of the 34 studies included in this systematic review, 15 studies (6 clinical and 9 animal studies) addressed ketone metabolism. The related indexes of ketone metabolism in plasma samples included the levels of ketone body (KB), total ketone bodies (TKB), β-hydroxybutyrate (β-OHB), and acetoacetate (AcAc). The ketone metabolic indicators in heart samples included the content of KB, TKB, β-OHB, KB transporter monocarboxylate transporter 1 (MCT1), monocarboxylate transporter 2 (MCT2), β-hydroxybutyrate dehydrogenase 1 (BDH1), β-hydroxybutyrate dehydrogenase 2 (BDH2), and succinyl-CoA: 3-ketoacid CoA transferase (SCOT). As illustrated in Table 5, 10 studies showed that SGLT2i increased the levels of ketone bodies (KBs) in plasma (11, 12, 15, 19, 24, 27, 39, 44, 45); 4 studies showed that SGLT2i increased the cardiac ketone metabolism (12, 15, 39, 44); 2 studies did not show any significant change, which might be related to the types of disease models; 4 studies showed no change in plasma ketone levels after SGLT2i treatment (20, 26, 32, 41). The detailed results were shown in Table 5.
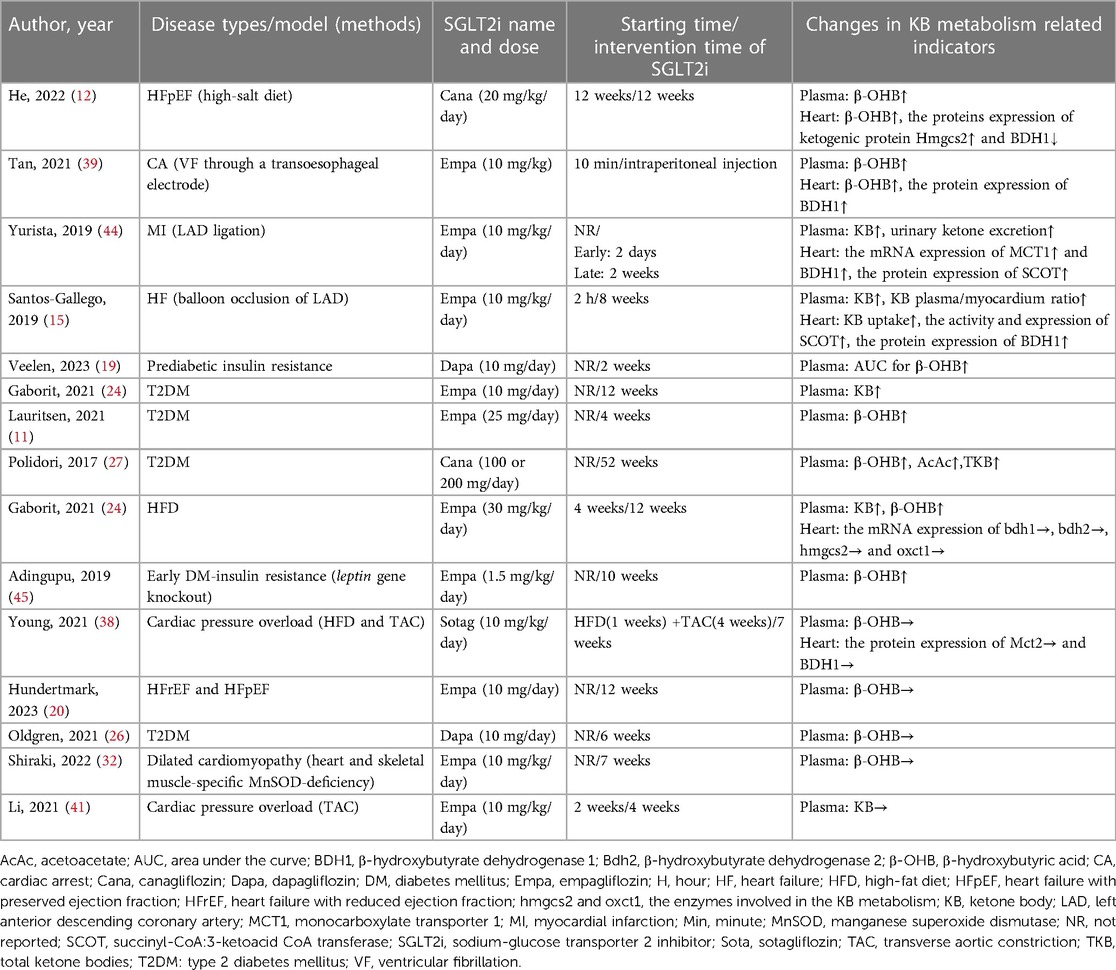
Table 5. Effects of SGLT2i on cardiac ketone metabolism in different diseases.
3.4.4. Effects of SGLT2i on mitochondria structure and functions of the heart
This systematic review consisted of 12 animal studies related to cardiac mitochondria structure and functions. The main detection indicators included mitochondria number, morphology, the average area, mitochondria membrane potential (MMP), the baseline oxygen consumption rate (OCR), the activity and oxidative phosphorylation (OXPHOS) capacity of mitochondria respiratory complexes I–III (COX I–III), the mitochondria DNA (mtDNA), the content of pro-fission dynein-related protein 1 (DRP1), mitochondria fission 1 protein (FIS1), mitochondria pro-fusion proteins mitofusin 1 (MFN-1), mitofusin 2 (MFN-2), optic nerve atrophy 1 (OPA1), p-AMPK, sirtuin-1 (SIRT1), and PGC-1α. These studies confirmed the advantages of SGLT2i treatment on cardiac mitochondria structure and functions. Table 6 provided a full description of the outcomes.
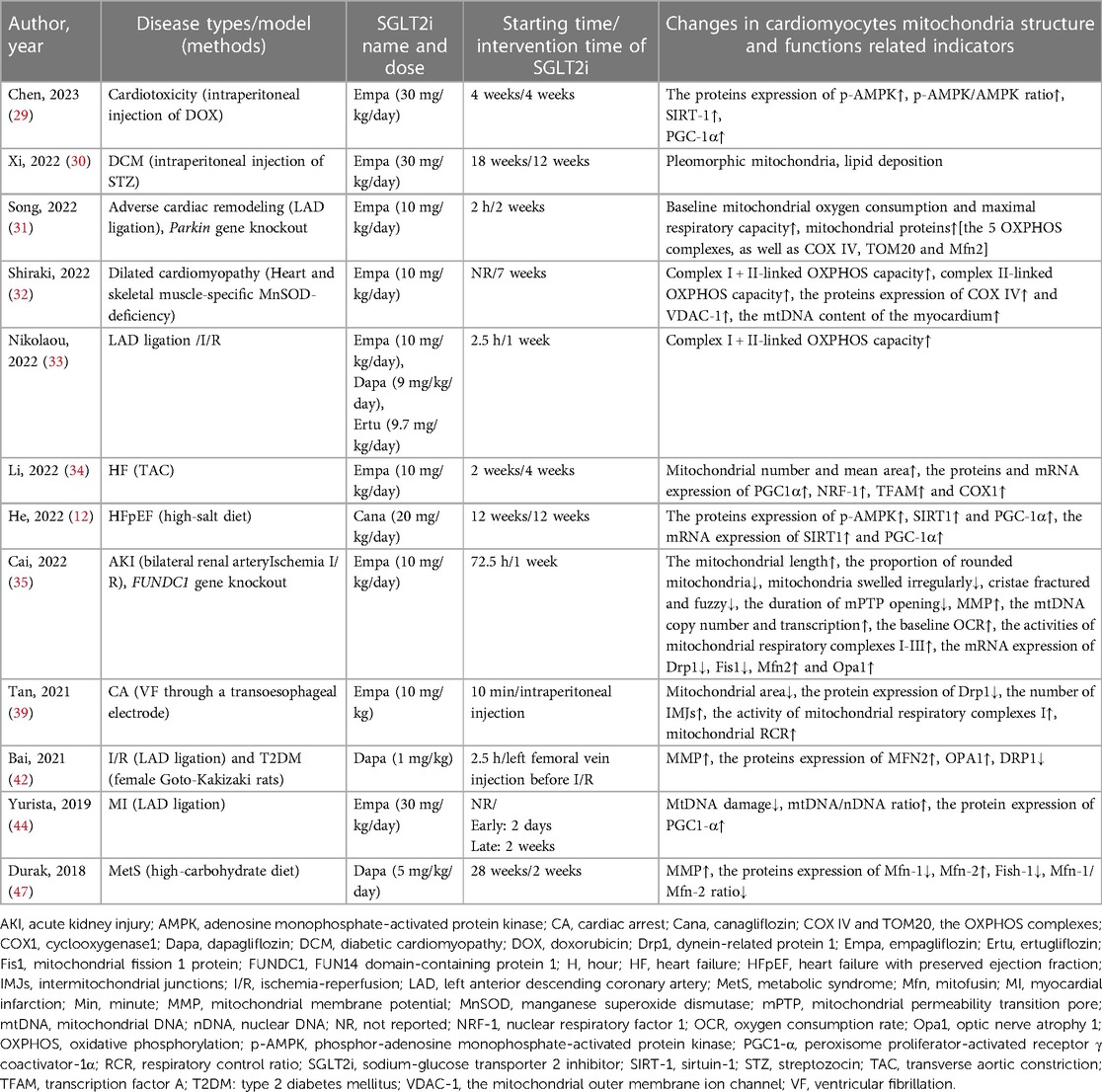
Table 6. Effects of SGLT2i on mitochondria structure and functions of the heart in different diseases.
3.4.5. Effects of SGLT2i on oxidative stress of the heart
As shown in Table 7, 11 studies (1 clinical study and 10 animal studies) were connected with cardiac oxidative stress. The indicators included reactive oxygen species (ROS), superoxide dismutase (SOD), malondialdehyde acid (MDA), NADPH oxidase (NOX), and advanced oxidation protein product (AOPP). The disease types included HF, I/R, SCM, metabolic syndrome (MetS), and so on. 1 study showed that Canagliflozin improved oxidative stress in myocardial tissue (23). Reportedly, the treatment with Empagliflozin or Dapagliflozin greatly attenuated MDA and elevated SOD levels in myocardial tissue (29, 36, 37, 40, 43). 4 studies (34 36, 39, 47), revealed markedly inhibited cardiac ROS production. The expression of AOPP and NOX4 proteins was reduced in 2 studies (12, 44).
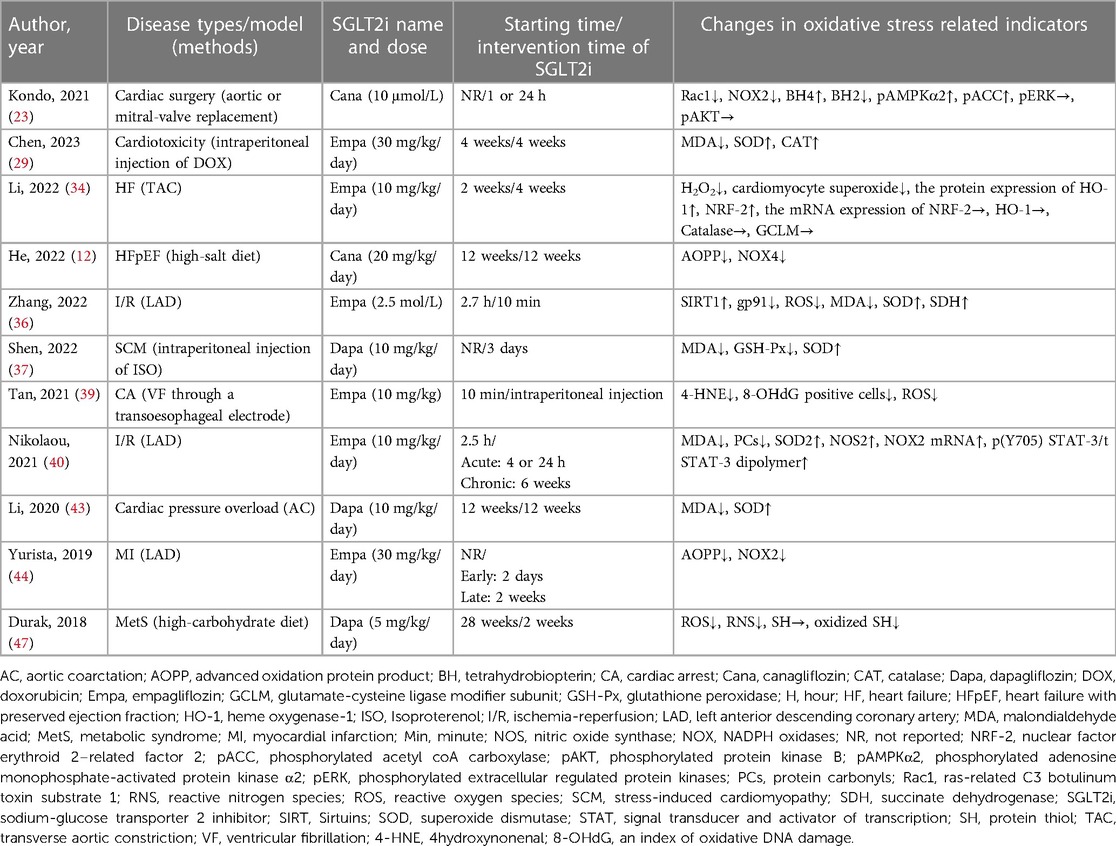
Table 7. Effects of SGLT2i on oxidative stress of the heart in different diseases.
3.4.6. Effects of SGLT2i on cardiac energy metabolism in DM and HF
This systematic review covered a total of 12 studies pertaining to HF (3 clinical and 9 animal studies) and 11 studies pertaining to DM (7 clinical and 4 animal studies). Subsequently, we summarized the indicators related to energy metabolism as class indicators, including: FA metabolism (lipid levels in plasma and the content of proteins related to FA metabolism in the heart), glucose metabolism (cardiac glucose uptake, glucose oxidation, and glycolysis), KB metabolism (the level of KB in the circulation and the heart), myocardial mitochondria structure and functions, and oxidative stress in cardiomyocytes. The outcomes demonstrated that SGLT2i decreased the absorption of glucose and glycolysis, increased the circulating and cardiac level of KB in DM and HF, improved mitochondria structure and functions, and reduced oxidative stress in cardiomyocytes. In addition, SGLT2i increased the cardiac glucose oxidation in DM and FA metabolism in HF. However, SGLT2i had inconsistent regulatory effects on FA metabolism in DM and glucose oxidation in HF. Finally, because of the differences in the research purpose of each study, indicators related to energy metabolism were not mentioned in some literatures. The detailed results were shown in Tables 8, 9.
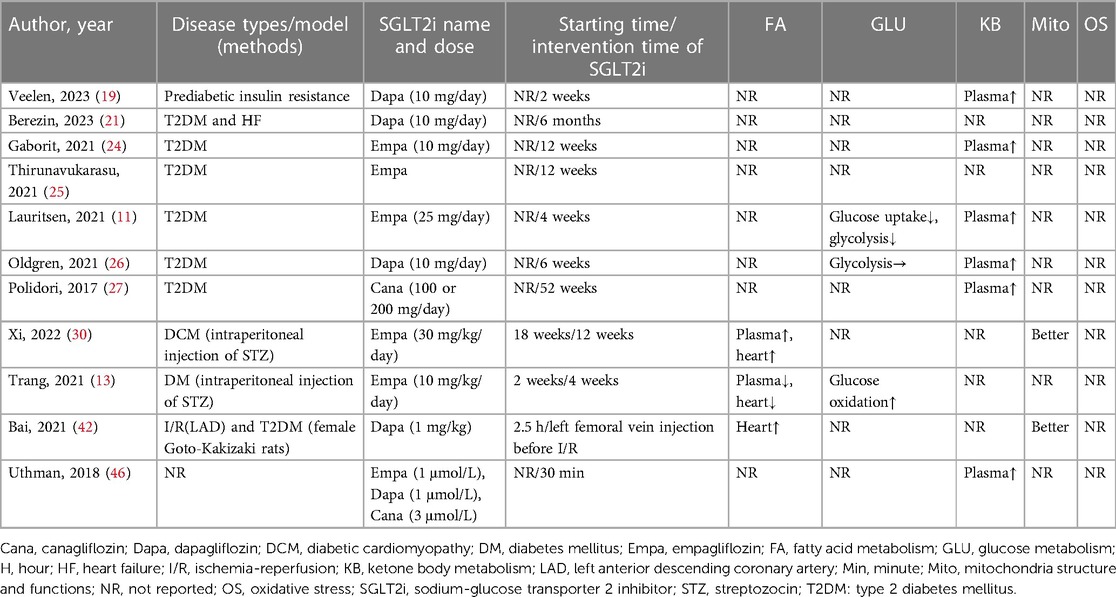
Table 8. Effects of SGLT2i on cardiac energy metabolism in DM.
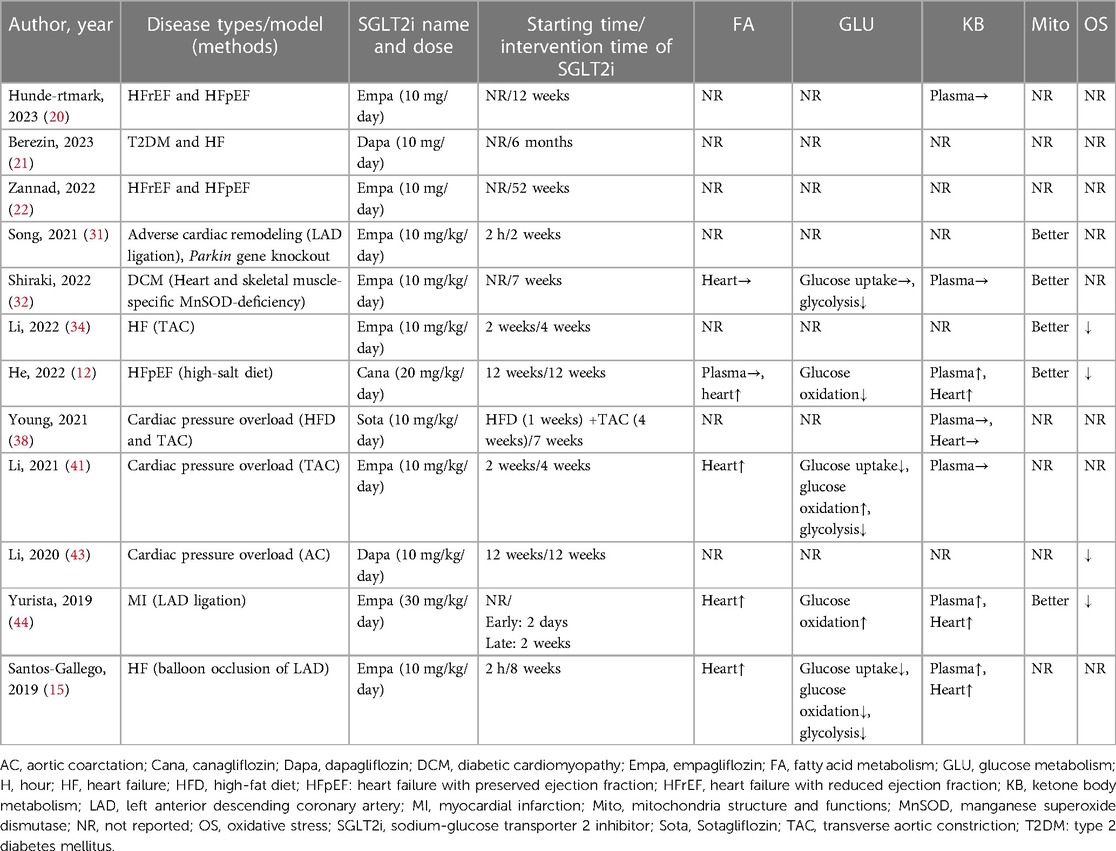
Table 9. Effects of SGLT2i on cardiac energy metabolism in HF.
4. Discussion
Many clinical diseases lead to cardiac involvement and cause cardiac energy metabolism disorders. The effects of various diseases on myocardial energy metabolism might vary due to different pathogenesis. SGLT2i has been widely used to treat heart diseases in clinical practice. Although existing studies had shown its cardiovascular benefits, the specific pharmacological effects of SGLT2i on cardiac energy metabolism were not yet clarified. After analyzing 10 clinical and 24 animal studies, we found that SGLT2i optimized the cardiac energy production environment, improved cardiac energy metabolism, and increased cardiac energy efficiency through the pharmacological effects on cardiac FA, glucose, ketone metabolism, mitochondria structure and functions, and oxidative stress. Moreover, we also explored the effects of SGLT2i on cardiac energy metabolism in DM and HF from a disease perspective.
4.1. Effects of SGLT2i on cardiac FA metabolism
Under physiological conditions, the heart utilizes FAs (40%–60%) and glucose (20%–40%) for ATP production, while LA, KBs, and amino acids contribute minimally, reflecting the diversity of cardiac metabolic substrates (48–51). FAs are taken up from the circulation into the cardiomyocytes cytoplasm by CD36 and transported into the mitochondria for β-oxidation via CPT-1, from where they enter into the tricarboxylic acid cycle and undergo aerobic oxidation through a series of metabolic enzymes to produce ATP. PGC1-α, PPARα, AMPK and ACC are proteins related to FA metabolism. The myocardial FA oxidation (FAO) rate changes with the progression of diseases. Cardiac FA metabolism decreased in various diseases. Many studies had showed that FAO was downregulated in patients with idiopathic dilated cardiomyopathy (IDCM), Dahl salt-sensitive rats fed a high-salt diet, Wister rats post-MI, and canine model of cardiac pacing (52–59). The decrease in FAO was not a consistent finding. According to several researches, individuals with congestive HF had higher FA uptake, while patients with IDCM showed no differences in FA uptake (60–62). Other studies showed that HFpEF, especially when HFpEF was associated with DM and obesity, upregulated FAO (63–65). T2DM men with cardiomyopathy and obese women with left ventricular hypertrophy exhibited increased myocardial FA uptake and oxidation (49, 64, 65). Moreover, FAO was also increased in murine HFpEF model involving HFD and aging (66). In addition to diseases types, different stages of diseases also caused changes in cardiac FA metabolism. Interestingly, FA metabolism showed temporal dynamic changes in ischemic heart disease-induced HF. In the acute ischemic stage, cardiac FAO was basically normal (67), and FA metabolism gradually decreased with HF progression from the early stage to the decompensated stage (52, 59, 67, 68). While there was a notable rise in cardiac FA uptake and oxidation, during the early stage of DM (65, 69–74). However, with the progression of DM, high rates of cardiac FA β-oxidation negatively affect cardiac efficiency (myocardial oxygen consumption/cardiac work) in humans (74, 75) and animals (76–78). Additionally, the increased FA dependence of the diabetic heart impaired the antioxidant capacity of the mitochondria, cellular ATP shuttle (79, 80), and caused cardiac lipotoxicity initiated by a mismatch between FA uptake and β-oxidation (66, 81–86). Therefore, cardiac FA metabolism might decrease in the end-stage of diabetic cardiomyopathy (DCM) due to the above adverse consequences, leading to a deficiency in cardiac energy metabolism. In conclusion, patterns of FA utilization might be connected with the types and stages of diseases. SGLT2i was closely related to FA metabolism and Empagliflozin had been identified as a regulator of adiposity and energy metabolism in adipose tissue. According to certain researches, SGLT2i prevented tubular epithelial cells from undergoing the metabolic transition from FAO to glycolysis, which is mediated by the hypoxia-inducible factor 1-α (HIF-1α) (87). When glucose is substituted for FAs in podocytes as an energy substrate in experimental Alport syndrome (AS), lipotoxicity is mitigated and renal function is enhanced by Empagliflozin (88).
Importantly, SGLT2i was beneficial for cardiac FA metabolism (11, 15, 41, 44, 89, 90). According to the literatures, 6 animal studies showed that SGLT2i promoted cardiac FA metabolism and the disease models included high-salt diet-induced HFpEF (12), TAC-induced cardiac pressure overload (41), LAD ligation-induced MI (44), HF induced by balloon occlusion of LAD (15), DCM established by intraperitoneal injection of STZ (30), and composite models of DM and I/R (42). Cardiac FA metabolism was decreased in the pathological states, while SGLT2i treatment increased the FA metabolism in 5 animal studies (12, 15, 41, 42, 44). Xi et al. demonstrated that SGLT2i promoted FA metabolism despite its upregulation in the diabetic group (30). The proteomics data suggested that the proteins related to the mechanism of lipid metabolism, such as monocarboxylic acid, cellular lipid and lipoprotein metabolism, as well as FA β-oxidation and lipid transport, were increased in the diabetic group, which was consistent with our descriptions. Also, Empagliflozin group exhibited upregulated levels of proteins related to lipid metabolic processes compared to the diabetic group (30). Hence, the role of SGLT2i in promoting cardiac FA metabolism needed to be investigated further. Among the above 6 animal studies on SGLT2i-promoted cardiac FA metabolism, 4 belonged to HFpEF, and the disease models were DCM created by intraperitoneal injection of STZ (30), HFpEF brought on by high-salt diet (12), TAC-induced cardiac pressure overload (41), and composite models of DM and I/R (42). Although the LAD ligation-induced MI model was generally used to establish HFrEF, the EF value of was >50% with this model in Yurista et al.'s study (44). In the study by Santos-Gallego et al. (15), although the EF value reached the level of HFrEF, the results showed that SGLT2i improved cardiac function and cardiac hypoxia. HFpEF is a multisystem syndrome, with metabolic stress/obesity, hypertension, and aging being the most common comorbidities. Some studies confirmed that the heart of HFpEF was in a state where hypoxia was not severe, while energy deprivation was existing, and resting ATP levels in the heart had been reduced to 20%–40% of the normal (53, 91–94). In the 6 animal studies wherein SGLT2i promoted cardiac FA metabolism (12, 15, 30, 41, 42, 44), the hearts were in a state of energy metabolism: ATP was scarce, but hypoxia was not yet serious, thereby, increasing the total amount of ATP production was crucial. When compared to other substrates, FAO generated the most ATP molecules per mole (129 ATP molecules for complete oxidation of one molecule of palmitate). Therefore, when the hypoxia of the heart was not severe, SGLT2i increased the total ATP production of the whole heart by promoting cardiac FA metabolism, hence improving the adverse situation of cardiac energy deficiency.
However, 3 studies showed that myocardial FA metabolism did not increase with SGLT2i treatment. Trang et al. showed that the levels of proteins related to FAs metabolism were lower in the Empagliflozin group compared to the DM group (13), which might be due to the shorter induction time (2 weeks) of the DM model than the general time (4–8 weeks); therefore, we speculated that SGLT2i inhibited FA uptake and metabolism in the early stage of DM. Lauritsen et al. showed that the relative uptake rate of myocardial FFA was reduced, and the absolute uptake rate was unchanged after Empagliflozin treatment (11). This phenomenon could be attributed to the milder severity of the disease in DM patients (diabetes duration only >1 year and metformin as the only antidiabetic pharmacological treatment), as well as the high FFA and low insulin state during SGLT2i treatment, which increased CD36 degradation and thus decreased myocardial FFA uptake capacity (95, 96). Shiraki et al. used the heart and skeletal muscle-specific manganese superoxide dismutase (MnSOD) deficiency dilated cardiomyopathy mouse model and demonstrated that the amount of cardiac FA uptake and the CD36 content in the Empagliflozin group did not alter significantly compared to the model group (32). Hence, we suspected that this disease model did not affect cardiac FA metabolism, but influenced the oxidative stress of the heart.
Numerous studies had shown that SGLT2i reduce body weight, with the initial weight loss being attributed to osmotic diuresis and urinary glucose (97). By lowering calories and improving hypothalamic insulin response, SGLT2i treatment might cause adipocytes in perivascular adipose tissues to become smaller and promote them to spend more energy. Significant weight loss would follow, along with reductions in visceral fat, subcutaneous fat, and overall adiposity (98–100). But SGLT2i treatment-induced weight loss was limited. The rise in FFA levels was observed to peak at week 24 and then to fall from week 24 to week 52, indicating that the improvement of anti-lipolysis effects might have mitigated the weight loss resulting from long-term SGLT2i medication (101). This review found that SGLT2i could cause weight loss through prompting lipid mobilization and lipolysis in DM patients and animals with metabolic diseases caused by high salt diet, high fat diet or leptin gene knockout. However, in patients or animals with HF, dilated cardiomyopathy and other heart diseases or DM patients with short duration of medication (2–4 weeks), the body weight remained unchanged, which might be related to the absence of an increase in body fat mass under the disease state. However, in the STZ-induced DM animal models, STZ only destroyed islet cells without causing an increase of body fat mass, resulting in a significant weight loss in the pathological state. Thus, animal body weight remained unchanged or increased after SGLT2i application.
In conclusion, the changes in cardiac FA metabolism might be connected with the types and stages of diseases; however, SGLT2i enhanced cardiac ATP production by promoting FA metabolism under the condition of poor ATP source but relatively sufficient oxygen, thereby improving the efficiency of cardiac energy production. Moreover, SGLT2i also affect body weight in different diseases.
4.2. Effects of SGLT2i on cardiac glucose metabolism
First-choice myocardial fuel, FA, is primarily oxidized by the mitochondria to meet the high energy requirements of a healthy heart. Nonetheless, the contribution of glucose consumption for cardiac ATP production is increased by the elevated levels of insulin and plasma glucose during the feeding state. During moderate-intensity exercise, the glucose uptake and oxidation in cardiomyocytes were also found increased (102–104). Because of its metabolic flexibility, the heart can utilize various substrates based on workload and substrate availability. As another important cardiac energy source, glucose has higher oxygen efficiency, indicating that glucose produces more ATP while consuming an equivalent amount of oxygen. Glucose is mainly absorbed into cardiomyocytes from the circulation by GLUT4 in physiological state, but less by GLUT1. Nevertheless, in pathological conditions, such as cardiac hypertrophy and HF, the heart returned to the fetal phenotype characterized by downregulated GLUT4 and upregulated GLUT1 (105). Following intake, glucose is quickly phosphorylated to glucose 6-phosphate and undergoes a series of metabolic processes. In the cytoplasm, glucose is converted into pyruvate by glycolytic pathway. Under aerobic environment, pyruvate is carried into the mitochondria where PDH decarboxylates it to generate acetyl CoA. Acetyl CoA then proceeds through the tricarboxylic acid cycle to produce ATP. PDH is a key regulator promoting glucose oxidation but is inactivated by PDK4 after phosphorylation (106). When oxygen is insufficient or myocardium hypoxia is severe, pyruvate in the cytoplasm could be glycolysed into LA by LDH, and excessive accumulation of LA in cardiomyocytes could lead to acidosis.
No known SGLT2 receptor was found in the heart (107), however, several studies had revealed a direct protective effect of SGLT2i in HF, with several factors influencing this effect. It had been discovered that SGLT2i might directly inhibit GLUT1 (108). In this systematic review, 3 studies indicated that Empagliflozin reduced cardiac glucose uptake, inducing a shift in the utilization of myocardium substrates from glucose toward other sources (11, 15, 41). The analysis of metabolites revealed a decline in cardiac glucose intake, while the contents of GLUT1 and GLUT4 were not detected. Li et al. discovered that GLUT1 and GLUT4 were highly affinified for Empagliflozin in the heart as assessed by molecular docking. These findings indicated that SGLT2i blocked the direct combination of glucose and GLUT1/4 in the heart, such that the cardiac glucose intake decreased (41). However, we also found that SGLT2i improved the gene and protein levels of GLUT4 (13, 44), although glucose uptake and GLUT1 content were not detected. Lauritsen et al. suggested that SGLT2i stimulated the expression of p-AMPKα2 protein, which significantly enhanced the cardiac energy metabolism (109), and increased AMPK activation, which enhanced the transcription and translocation of GLUT4, thus augmenting GLUT4 gene and protein expression in the heart. Moreover, whether the alterations in glucose uptake and glucose transporters were related to the types of diseases and different stages of the model remained to be further explored and confirmed.
3 animal studies showed that SGLT2i treatment enhanced glucose oxidation in T2DM (13), cardiac pressure overload (41) and MI (44). When mitochondria oxidative metabolism was damaged due to hypoxia, oxidative stress and other pathological factors, such as high glycolysis rate and low glucose oxidation rate could lead to increased decoupling of glycolysis and glucose oxidation, as well as changes in ion homeostasis, thus reducing cardiac energy generation efficiency (110). The current results showed that Empagliflozin increased cardiac glucose oxidation and restored the coupling between glycolysis and glucose oxidation (41, 102). Some studies revealed that after aortic constriction in rats, cardiac glucose oxidation increased in initial stage, while in the later stage of HF, glucose uptake and utilization were reduced. Other studies demonstrated that Empagliflozin restored myocardial glucose oxidation to non-diabetic level in diabetic rats, promoted the normalization of cardiac glucose oxidative metabolism and ATP generation, enhanced cardiac function, and optimized cardiac hypertrophy and fibrosis in non-diabetic rats after MI (44, 76). Nevertheless, 2 studies (12, 15) also concluded that the glucose oxidation decreased after the SGLT2i intervention. One study used Dahl salt-sensitive (DSS) rats fed with 8% high-salt diet for 12 weeks (12), and the other study used balloon occlusion of LAD for 2 h to establish HF model. The main reason for decreased glucose oxidation might be the increase in FA and ketone metabolism after the long-term intervention of SGLT2i (12, 15). Whether this phenomenon was related to the disease types and intervention time needed further investigation. 4 studies confirmed that SGLT2i reduced anaerobic or cardiac glycolytic metabolism, and decreased intracellular LA accumulation and acidosis in cardiomyocytes (15, 32, 39, 41). The lower blood LA level produced by anaerobic metabolism in Empagliflozin group suggested that the cardiac mitochondria was capable of supplying adequate energy for the heart to beat, thus reducing the dependence on the compensatory energy supply of anaerobic metabolism (32). 2 clinical studies found that circulating LA levels were not affected (11, 26), which might be due to the circulating LA level of T2DM patients fluctuate slightly little, and drugs were not sensitive to this index.
Taken together, SGLT2i inhibited cardiac glucose uptake but upregulated the expression of GLUT4. It also increased the proportion of glucose oxidation under most circumstances. In addition, SGLT2i inhibited glycolysis and reduced the occurrence of intracellular LA accumulation and acidosis in cardiomyocytes.
4.3. Effects of SGLT2i on cardiac ketone metabolism
Typically, ketone metabolism accounts for approximately 10% of the substrates of cardiac energy metabolism (48, 49, 51, 111). However, KBs are recently recognized as crucial energy substrates for the heart (112, 113). When the heart developed energy metabolism disorders due to various diseases, the normal oxidative metabolism of FAs and glucose was disrupted, making the heart depend on KBs as fuels for metabolism (114–116). Under physiological conditions, KBs are mainly produced by FAs in the liver when supply of carbohydrates is restricted, such as ketogenic diets, fasting, and extended exercise. The human body produces three distinct ketones: β-OHB, acetoacetate (AcAc), and acetone. 80% of the circulating ketone concentration is made up of β-OHB, which is the primary KBs oxidized in the heart (117). MCT1 promotes cardiomyocytes uptake β-OHB, and then, β-OHB passes through a series of metabolic enzymes, such as BDH1 and SCOT, to produce acetyl CoA, which then enters the tricarboxylic acid cycle. KBs had a beneficial effect on hemodynamics and metabolism in individuals with various illnesses, such as overweight, T2DM, ischemic HF and cognitive impairment (118–122). A modest increase in cardiac ketone metabolism was salutary for overall cardiac function. Some studies showed that the infusion of β-OHB or ketone ester improved contractile performance in patients with HFrEF and decreased adverse remodeling in animals with HF (120, 123, 124). The cardiac consumption of β-OHB resulted in a considerable improvement in heart function and membrane potential stability, which in turn strengthened the cardiomyocytes' antiarrhythmic potential (125). Nevertheless, it was challenging to keep the high level of circulating ketone with either ketone or ketone ester infusions. This issue could be resolved by SGLT2i. Although originally developed as an anti-hyperglycemic agents for the treatment of DM, SGLT2i had recently been shown to exert significant cardioprotective effects in HF patients, potentially by increasing circulating KBs and affecting cardiac ketone metabolism (6, 126–128).
Our review showed that SGLT2i enhanced cardiac ketone metabolism and levels of circulating ketone with respect to the already increased ketone level and ketone metabolism under the diseases states. 4 clinical (11, 19, 24, 27) and 6 animal studies (12, 15, 24, 39, 44, 45) demonstrated that SGLT2i increased the circulating level of ketone. The result could be attributed to the following: in one sense, SGLT2i stimulated hepatic ketogenesis by reducing insulin levels, thereby increasing lipolysis and circulating FFA levels. But in another, it might be linked to a lower renal clearance of KBs (29, 129, 130). The elevated levels of circulating KBs promoted cardiac ketone utilization (112, 113), which was confirmed by 4 studies (12, 15, 39, 44) in our systematic review. KB produces ATP in a higher oxygen-efficient manner than FA (P/O 2.5 for KB vs. 2.33 for palmitate), and KB oxidation liberates more energy than glucose (244 kcal/mol for KB vs. 224 kcal/mol for glucose) (114, 115). Thus, KBs are energetically efficient fuels for cardiac energy production which are beneficial to increase cardiac ATP production and improve overall cardiac work efficiency. In addition, KBs, as substrates for mitochondria energy metabolism, do not produce LA, and reduced the risk of intracellular LA accumulation and acidosis in cardiomyocytes. However, no significant change was detected in cardiac ketone metabolism after SGLT2i treatment in 2 studies (24, 38). Both these studies used HFD, which might be the major cause for the lack of significant effect of SGLT2i on cardiac ketone metabolism. SGLT2i acts on the renal proximal tubules, but after HFD, fat is deposited on renal proximal tubules. Increased excretion of KIM-1 in urine, sign of damage to the proximal tubule, had been reported in HFD mice (1). Some studies also reported disrupted sodium handling in HFD mice, suggesting impaired renal proximal function (2). Therefore, renal proximal tubular injury caused by HFD resulted in a decline in the diuretic and glycosuria effects of SGLT2i, and cardiac ketone metabolism remains unaltered ultimately. In addition, 2 clinical (20, 26) and 2 animal studies (32, 41) showed that the plasma ketone levels did not alter significantly after SGLT2i treatment, which might be related to low SGLT2i dose and short treatment time.
Therefore, without affecting the activity site of SGLT2i—the renal proximal tubules, SGLT2i could improve cardiac ketone metabolism, thus increasing the efficiency of ATP production with reducing the occurrence of adverse metabolic consequences such as acidosis, which could reverse the cardiac energy metabolism disorder in the pathological state.
4.4. Effects of SGLT2i on mitochondria structure and functions
As an energy-processing plant, the heart generates enormous ATP to sustain systolic and diastolic function from two main sources: mitochondria oxidative phosphorylation and glycolysis. In the physiological state, 95% of myocardial ATP requirements are typically met by mitochondria OXPHOS, with glycolysis providing the remaining 5% (131–134). Cardiomyocytes have more mitochondria than other cell types, therefore, structural damage and dysfunction of the mitochondria inevitably result in decreased cardiac energy production (135). Mitochondria undergo continuous cycles of fusion and fission that regulate mitochondrial homeostasis, recycling, and biogenesis to sustain energy production, hence, the structure and functions of mitochondria depend on the balance between mitochondrial fusion and fission (136). The outer membrane protein mitofusin (MFN) and the inner membrane protein OPA1 are associated with mitochondrial fusion (49, 137, 138). DRP1 and FIS1 are involved in mitochondrial division. In our systematic review, 3 studies (35, 42, 47) confirmed that SGLT2i regulated the fusion and fission of the mitochondria. The disease models used in these 3 studies include acute kidney injury (AKI) induced by bilateral renal artery ischemia (35), I/R in GK rats (42), and MetS induced by high-carbohydrate diet (47). The results demonstrated that the pro-fusion proteins MFN1/2 and OPA1 were upregulated, and the pro-fission proteins DRP1 and FIS1 were downregulated after SGLT2i treatment (35, 42, 47). According to Koizumi et al., Empagliflozin brought the altered atrial protein expression associated with fission and fusion of mitochondria back to levels comparable to those observed in the control group (139). Additionally, 6 studies showed that SGLT2i increased the mitochondria number (34), length (35), pleomorphism (30, 35), mean area (34, 39) and MMP (42, 47) while alleviating mitochondria irregular swelling, fractured cristae and fuzziness (35), implying an improvement in mitochondria structure. Consistent with our results, Mizuno et al. similarly showed that Empagliflozin normalized mitochondria size and quantity in DM hearts following MI (140).
Biogenesis and OXPHOS are two important functions of cardiac mitochondria. Mitochondrial biogenesis is characterized by generating new mitochondria and increasing mitochondrial content and number. Alterations in the mitochondria fusion and fission processes as well as mitophagy suppression might lead to a decrease in mitochondrial biogenesis (141). PGC1-α is a vital transcriptional regulator of mitochondrial biogenesis and can activate the transcriptional expression of nuclear respiratory factor-1/2 (NRF-1/2). These are target genes encoding the proteins that mediate mitochondria replication, maintenance, and the generation of electron transport chain (ETC) components (142). Previous studies had demonstrated that AMPK/SIRT-1/PGC-1α signaling pathway was an energy-sensing network that significantly regulated mitochondrial biogenesis, energy metabolism, and oxidative stress (143). In our review, 4 studies elucidated that SGLT2i increased the proteins or mRNA expression of AMPK, SIRT-1, and PGC-1α in cardiomyocytes, exerting a cardioprotective role by upregulating mitochondrial biogenesis and enhancing OXPHOS (12, 29, 34, 44). The decline in the PGC1-α level might contribute to mitochondria dysfunction observed in HF (144). Acting as an upstream factor, SIRT1 directly stimulated PGC1-α expression (145, 146). Zarei et al. observed that stimulating the SIRT1/PGC1-α pathway reduced the cardiomyocytes toxicity of D-galactose-induced by improving mitochondrial biogenesis (141). According to Santos-Gallego et al., Empagliflozin therapy enhanced heart function and mitochondria energetics in porcine model of HF, which was achieved by upregulating the expression of AMPK and PGC-1α (15). These studies supported the conclusions of our review. SGLT2i was also advantageous for OXPHOS of cardiac mitochondria. In mitochondria, energy is generated via OXPHOS, wherein the electrons are transferred from negative to positive redox potentials along the ETC (147) which consists of 4 proteins: mitochondria respiratory complexes I–IV (COX I–IV), localized on the inner mitochondria membrane. In our systematic review, 3 studies showed that SGLT2i increased the OXPHOS capacity of cardiac mitochondria COX I, II and III (31–33). Upregulated OXPHOS capacity made mitochondria less reliant on anaerobic metabolism's compensatory energy source, allowing cardiac mitochondria to obtain sufficient energy from aerobic metabolism for a continuously beating heart. According to Nambu et al., Empagliflozin ameliorated the reduced skeletal muscle capacity for exercise by restoring the mitochondrial OXPHOS in murine HF (148).
In summary, SGLT2i protected mitochondria structure by maintaining the balance between mitochondrial fusion and fission and restoring mitochondria number, mean area, and MMP. Moreover, SGLT2i protected mitochondria functions by increasing its biogenesis and the ability of OXPHOS, thereby exerting beneficial effects on cardiac energy metabolism.
4.5. Effects of SGLT2i on oxidative stress
Sustained oxidative stress and related inflammation play a crucial role in myocardial injury, forming the core pathological link of HF and other cardiovascular diseases (149, 150). The re-oxidation of myocardial tissue led to excessive ROS and increased oxidative stress, which induced the break of metabolic homeostasis. It has long been recognized that the primary source of ROS generated by NOX is the mitochondria. Among the members of the NOX family that transport electrons across the plasma membrane and produce superoxide (151), cardiomyocytes primarily express NOX2 and NOX4 (152, 153). Under physiological conditions, ROS advocates the basic signaling function of regulating mitochondria activity and cell adaptation to stressors. However, when cardiac mitochondria were damaged, ROS production was enhanced. Long-term exposure to oxidative stress and ROS accumulation had been linked to reduced antioxidant capacity, imbalanced redox signals, and macromolecular damage, finally, ROS-mediated myocardial injury and cardiomyocytes death was effectuated (154, 155). Also, excessive ROS led to defective myocardial remodeling and impaired myocardial contractility, that in turn aggravated HF (156). Some studies showed that hyperglycemia stimulated ROS production, and the excessive ROS might lead to the long-term development of diabetic complications (157, 158).
The oxidative stress produced by the diseases reported in 11 studies was shown to be mitigated in cardiomyocytes by the application of SGLT2i. By activating the AMPK/SIRT-1/PGC-1α pathway, Empagliflozin protected mitochondria functions and reduced oxidative stress (29). The enhanced protein expression of SIRT1 inhibited ROS accumulation, NOX activity and MDA content, and upregulated the activities of antioxidant enzymes like SOD and SDH, increasing endogenous heart antioxidant capacity and alleviating oxidative stress (29, 36, 43). In addition, the expression of AOPP and NOX2 was suppressed by Empagliflozin in the rat model of non-diabetic MI (44). Canagliflozin directly inhibited oxidative stress through SGLT1/AMPK/Rac1 signaling and played a crucial role in suppressing the pro-inflammatory and pro-apoptotic effects of TNF-α, IL-1, and NF-κb pathways (23). Shen et al. showed that Dapagliflozin reduced oxidative stress and regulated the survival, apoptosis, growth of cardiomyocytes through PI3K/Akt signaling pathway (37). Also, chronic administration of Empagliflozin induced dimerization of p(Y705) STAT-3, upregulated the downstream proteins SOD2 and VEGF, and reduced the cardiac oxidative stress response after I/R injury in non-diabetic mice (40). Altogether, SGLT2i played an antioxidant role by reducing cardiac oxidative stress for a direct cardioprotective effect.
4.6. Effects of SGLT2i on cardiac energy metabolism in DM and HF
In DM patients, myocardial metabolism flexibility was impaired, FA β-oxidation became the main source of ATP generation, and glucose oxidation was significantly reduced. These changes had been reported in preclinical models, T1DM and T2DM patients (65, 70, 72–74, 159). SGLT2i promotes urine glucose excretion, which is not affected by islet function and insulin resistance (160). Several clinical studies illustrated that patients with T2DM and atherosclerotic diseases reflected a low HF hospitalization rate and mortality under SGLT2i treatment (161–163). In the 34 included studies, 11 studies were associated with DM (7 clinical and 4 animal studies). The diseases in the clinical studies involved T2DM, pre-diabetic insulin resistance, and T2DM complicated with HF, and the disease models in animal studies included DM, DCM and insulin resistance. These clinical and animal studies related to DM showed that the cardiac glucose intake remained unchanged or decreased but the cardiac glucose oxidation and the KBs levels in circulation were elevated with treatment of SGLT2i. The reason might be attributed to the fact that SGLT2i reduced the dependence on FA metabolism and avoided lipotoxicity-mediated myocardial damage by improving the oxidative utilization of glucose (13, 164). Some studies demonstrated that SGLT2i reduced the blood glucose concentration through reduced reabsorption of glucose by the kidney, thus treating T2DM (45, 165). However, we found the FA metabolism in the myocardium was inhibited after SGLT2i treatment for 4 weeks in DM, but was upregulated after treatment for 12 weeks in DCM and after a single intravenous administration in a composite model of DM and I/R, which might be related to models types, stages of DM development, and the intervention time of SGLT2i. Thereby, we discovered that the application of SGLT2i generally promoted the conversion of cardiac metabolism substrates, increased cardiac glucose oxidation and ketone metabolism, regulated FA metabolism to the direction of benefit to the whole body according to the disease states, and finally restored the balance between myocardial energy demand and supply.
The alteration of cardiac energy metabolism is one of the core pathological mechanisms of HF. The energy metabolism alterations in HF are complex and depend on the severity and types of HF as well as the coexistence of common comorbidities, such as obesity and T2DM. An energy deficit is present in the failing heart, and the relative contribution of various metabolism substrates is also changed compared to the physiological state. Of the 34 included studies, 12 studies were associated with HF (3 clinical and 9 animal studies). The analysis showed that SGLT2i increased cardiac FA (12, 15, 41, 44) and ketone metabolism (12, 15, 44), with increasing (41, 44) or decreasing (12, 15) glucose oxidation and decreasing glycolysis (15, 32, 41). 2 studies showed that SGLT2i reduced the rate of cardiac glucose oxidation (12, 15), which could be due to increased metabolism of FAs and KBs produced more abundant ATP. Typically, cardiac glucose uptake and glycolysis were increased in HF, which was linked to the elevated GLUT1 protein expression (166, 167). Some studies showed that cardiac glucose oxidation and FAO in HF influenced each other, and the changes were uncertain, which were dependent on the types of HF. For example, myocardial glucose oxidation was reduced and FAO was increased in HF associated with DM and obesity (63, 64, 168–170); in some researches using mouse models of HF with lower FAO rates, the proportional contribution of glucose oxidation to tricarboxylic acid cycle flux might be increased (171). Recent studies demonstrated that myocardial KB oxidation was increased in HF (172–174), which might be related to the inhibition of FAO, reflecting the mutual regulation of increased KB oxidation and FAO. These metabolic changes ultimately reduced the heart's efficiency of producing energy, while SGLT2i exerted a benign regulatory effect on cardiac energy metabolism in HF. In a word, the results indicated that SGLT2i increased cardiac utilization and metabolism of FAs and KBs in HF, regulated glucose oxidation, and increased cardiac ATP production, exerting beneficial effects on cardiac energy metabolism.
In clinical researches, patients had a disadvantage due to the diversity of their pathological conditions and confounding factors compared to the advantages of controllable research conditions of experimental animals in basic researches. However, clinical patients could reflect the real situations of the diseases. Therefore, it was necessary to combine the two for comprehensive analysis. Moreover, the clinical patients’ indicators were limited to blood lipids, blood glucose, blood ketone, and blood LA, and lack heart-specific metabolic markers, so the changes of indicators were not sensitive enough. Animal studies could detect the levels of proteins closely associated to the cardiac metabolism of FA, glucose, and KB, which directly and pertinently represented the alterations in cardiac metabolism of energy. Overall, SGLT2i's effects on cardiac metabolic indicators in clinical patients and experimental animals were basically consistent, however, the circulating LA level of clinical patients was generally unchanged, while that in the animal heart was significantly decreased.
It was uncertain whether the role of SGLT2i differed in gender. Previous studies had shown that SGLT2i reduced adverse cardiovascular events in both sexes (175), but there had been some reports showed that sex differences might contribute to different responsiveness to SGLT2i (176, 177). In 10 clinical studies of this review, the ratio of male to female was balanced. However, male animals were used in all but one of the literature which used female pigs as research subjects. Moreover, the effect of SGLT2i on gender was not addressed in 34 studies we included, thus more researches is still needed.
4.7. SGLT2i adverse effects
SGLT2 inhibitors are generally well-tolerated, but like any medication, they can have adverse effects. Common side effects include increased urination (due to their diuretic effect), urinary tract infections, genital mycotic infections, and a slightly increased risk of hypotension and dehydration, especially in certain populations. Of the 10 clinical studies in this systematic review, only 1 study reported adverse effects after taking SGLT2i, which were not different from those in the placebo group. It showed that the treatment with SGLT2i was well tolerated and remains positive of the overall risk-benefit profile.
5. Limitations
Nevertheless, the present study had some limitations. Firstly, the disease models included in the literatures were diverse and not limited to cardiovascular diseases, resulting in the heart might not be the main lesion site and the energy metabolism of the heart might not be affected. Secondly, the main research purpose of some studies was whole-body energy metabolism; thus, not every study contained indicators closely related to cardiac energy metabolism. Finally, we were unable to draw accurate conclusions from the meta-analysis because we employed qualitative synthesis rather than quantitative methods due to the diversity of models and the inconsistency of interventions.
6. Conclusions
Among the 34 included studies (including 10 clinical and 24 animal studies), the effects of SGLT2i on cardiac energy metabolism were described in different diseases, such as HF, T2DM, I/R injury, MI, cardiac pressure overload, dilated cardiomyopathy, SCM, DCM, MetS, CA, and other diseases. The findings of the current systematic review demonstrated that SGLT2i reduced cardiac glucose uptake and glycolysis, increased FA metabolism of the heart in most disease states, upregulated ketone metabolism, improved the structure and functions of myocardial mitochondria, and alleviated oxidative stress of cardiomyocytes in all the studies. However, the effects of SGLT2i on FA and glucose metabolism were not consistent due to the different diseases types, stages, and the time of SGLT2i intervention. SGLT2i increased cardiac glucose oxidation in DM and FA metabolism in HF. In addition, it could adjust cardiac FA metabolism in DM and cardiac glucose oxidation of in HF according to the different diseases stages to optimize the heart’ metabolic condition. All of the SGLT2i effects listed above on the regulation of energy metabolism could increase cardiac ATP production and improving cardiac work efficiency. This systematic review discussed the specific effects of SGLT2i on cardiac energy metabolism in detail, providing clinical values for the application of SGLT2i against diseases related to cardiac energy metabolism disorders.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
SS: Formal Analysis, Methodology, Project administration, Writing – original draft, Writing – review & editing, Data curation, Software, Supervision, Validation. XJ: Methodology, Supervision, Writing – original draft, Writing – review & editing, Data curation, Formal Analysis, Validation. TL: Conceptualization, Investigation, Validation, Writing – review & editing. YT: Data curation, Formal Analysis, Project administration, Software, Writing – review & editing. BW: Project administration, Resources, Validation, Writing – review & editing. XH: Resources, Supervision, Visualization, Writing – review & editing. MZ: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This study was supported by the National Natural Science Foundation Project of China (no. 82274461).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1291450/full#supplementary-material
References
1. Ferrannini E, Solini A. SGLT2 Inhibition in diabetes mellitus: rationale and clinical prospects. Nat Rev Endocrinol. (2012) 8:495–502. doi: 10.1038/nrendo.2011.243
2. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American college of cardiology foundation/American heart association task force on practice guidelines. J Am Coll Cardiol. (2013) 62:e147–239. doi: 10.1016/j.jacc.2013.05.019
3. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: developed by the task force for the diagnosis and treatment of acute and chronic heart failure of the European society of cardiology (ESC). With the special contribution of the heart failure association (HFA) of the ESC. Eur J Heart Fail. (2022) 24:4–131. doi: 10.1002/ejhf.2333
4. Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. (2019) 393:31–9. doi: 10.1016/S0140-6736(18)32590-X
5. Zannad F, Ferreira JP, Pocock SJ, Anker SD, Butler J, Filippatos G, et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-reduced and DAPA-HF trials. Lancet. (2020) 396:819–29. doi: 10.1016/S0140-6736(20)31824-9
6. McMurray J, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. (2019) 381:1995–2008. doi: 10.1056/NEJMoa1911303
7. Kosiborod MN, Jhund PS, Docherty KF, Diez M, Petrie MC, Verma S, et al. Effects of dapagliflozin on symptoms, function, and quality of life in patients with heart failure and reduced ejection fraction: results from the DAPA-HF trial. Circulation. (2020) 141:90–9. doi: 10.1161/CIRCULATIONAHA.119.044138
8. Mazidi M, Rezaie P, Gao HK, Kengne AP. Effect of sodium-glucose cotransport-2 inhibitors on blood pressure in people with type 2 diabetes Mellitus: a systematic review and meta-analysis of 43 randomized control trials with 22,528 patients. J Am Heart Assoc. (2017) 6:e004007. doi: 10.1161/JAHA.116.004007
9. Muzurović E, Mikhailidis DP. Impact of glucagon-like peptide 1 receptor agonists and sodium-glucose transport protein 2 inhibitors on blood pressure and lipid profile. Expert Opin Pharmacother. (2020) 21:2125–35. doi: 10.1080/14656566.2020.1795132
10. Op den Kamp Y, de Ligt M, Dautzenberg B, Kornips E, Esterline R, Hesselink M, et al. Effects of the SGLT2 inhibitor dapagliflozin on energy metabolism in patients with type 2 diabetes: a randomized, double-blind crossover trial. Diabetes Care. (2021) 44:1334–43. doi: 10.2337/dc20-2887
11. Lauritsen KM, Nielsen B, Tolbod LP, Johannsen M, Hansen J, Hansen TK, et al. SGLT2 inhibition does not affect myocardial fatty acid oxidation or uptake, but reduces myocardial glucose uptake and blood flow in individuals with type 2 diabetes: a randomized double-blind, placebo-controlled crossover trial. Diabetes. (2021) 70:800–8. doi: 10.2337/db20-0921
12. He L, Ma S, Zuo Q, Zhang G, Wang Z, Zhang T, et al. An effective sodium-dependent glucose transporter 2 inhibition, canagliflozin, prevents development of hypertensive heart failure in dahl salt-sensitive rats. Front Pharmacol. (2022) 13:856386. doi: 10.3389/fphar.2022.856386
13. Trang NN, Chung CC, Lee TW, Cheng WL, Kao YH, Huang SY, et al. Empagliflozin and liraglutide differentially modulate cardiac metabolism in diabetic cardiomyopathy in rats. Int J Mol Sci. (2021) 22:1177. doi: 10.3390/ijms22031177
14. Radlinger B, Hornsteiner F, Folie S, Salvenmoser W, Haubner BJ, Schuetz T, et al. Cardioprotective effects of short-term empagliflozin treatment in db/db mice. Sci Rep. (2020) 10:19686. doi: 10.1038/s41598-020-76698-8
15. Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. (2019) 73:1931–44. doi: 10.1016/j.jacc.2019.01.056
16. Aljadani AH, Alshammari TS, Sadaqir RI, Alrashede N, Aldajani BM, Almehmadi SA, et al. Prevalence and risk factors of attention deficit-hyperactivity disorder in the Saudi population: a systematic review and meta-analysis. Saudi J Med Med Sci. (2023) 11:126–34. doi: 10.4103/sjmms.sjmms_528_22
17. Ma LL, Wang YY, Yang ZH, Huang D, Weng H, Zeng XT. Methodological quality (risk of bias) assessment tools for primary and secondary medical studies: what are they and which is better. Mil Med Res. (2020) 7:7. doi: 10.1186/s40779-020-00238-8
18. Hooijmans CR, Rovers MM, de Vries RB, Leenaars M, Ritskes-Hoitinga M, Langendam MW. SYRCLE’s risk of bias tool for animal studies. BMC Med Res Methodol. (2014) 14(43):43–51. doi: 10.1186/1471-2288-14-43
19. Veelen A, Andriessen C, Op den Kamp Y, Erazo-Tapia E, de Ligt M, Mevenkamp J, et al. Effects of the sodium-glucose cotransporter 2 inhibitor dapagliflozin on substrate metabolism in prediabetic insulin resistant individuals: a randomized, double-blind crossover trial. Metab Clin Exp. (2023) 140:155396. doi: 10.1016/j.metabol.2022.155396
20. Hundertmark MJ, Adler A, Antoniades C, Coleman R, Griffin JL, Holman RR, et al. Assessment of cardiac energy metabolism, function, and physiology in patients with heart failure taking empagliflozin: the randomized, controlled EMPA-VISION trial. Circulation. (2023) 147:1654–69. doi: 10.1161/CIRCULATIONAHA.122.062021
21. Berezin AA, Obradovic Z, Fushtey IM, Berezina TA, Novikov EV, Schmidbauer L, et al. The impact of SGLT2 inhibitor dapagliflozin on adropin Serum levels in men and women with type 2 diabetes Mellitus and chronic heart failure. Biomedicines. (2023) 11:457. doi: 10.3390/biomedicines11020457
22. Zannad F, Ferreira JP, Butler J, Filippatos G, Januzzi JL, Sumin M, et al. Effect of empagliflozin on circulating proteomics in heart failure: mechanistic insights into the EMPEROR programme. Eur Heart J. (2022) 43:4991–5002. doi: 10.1093/eurheartj/ehac495
23. Kondo H, Akoumianakis I, Badi I, Akawi N, Kotanidis CP, Polkinghorne M, et al. Effects of canagliflozin on human myocardial redox signalling: clinical implications. Eur Heart J. (2021) 42:4947–60. doi: 10.1093/eurheartj/ehab420
24. Gaborit B, Ancel P, Abdullah AE, Maurice F, Abdesselam I, Calen A, et al. Effect of empagliflozin on ectopic fat stores and myocardial energetics in type 2 diabetes: the EMPACEF study. Cardiovasc Diabetol. (2021) 20:57. doi: 10.1186/s12933-021-01237-2
25. Thirunavukarasu S, Jex N, Chowdhary A, Hassan IU, Straw S, Craven TP, et al. Empagliflozin treatment is associated with improvements in cardiac energetics and function and reductions in myocardial cellular volume in patients with type 2 diabetes. Diabetes. (2021) 70:2810–22. doi: 10.2337/db21-0270
26. Oldgren J, Laurila S, Åkerblom A, Latva-Rasku A, Rebelos E, Isackson H, et al. Effects of 6 weeks of treatment with dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on myocardial function and metabolism in patients with type 2 diabetes: a randomized, placebo-controlled, exploratory study. Diabetes Obes Metab. (2021) 23:1505–17. doi: 10.1111/dom.14363
27. Polidori D, Iijima H, Goda M, Maruyama N, Inagaki N, Crawford PA. Intra- and inter-subject variability for increases in serum ketone bodies in patients with type 2 diabetes treated with the sodium glucose co-transporter 2 inhibitor canagliflozin. Diabetes Obes Metab. (2018) 20:1321–6. doi: 10.1111/dom.13224
28. Croteau D, Baka T, Young S, He H, Chambers JM, Qin F, et al. SGLT2 Inhibitor ertugliflozin decreases elevated intracellular sodium, and improves energetics and contractile function in diabetic cardiomyopathy. Biomed Pharmacother. (2023) 160:114310. doi: 10.1016/j.biopha.2023.114310
29. Chen M. Empagliflozin attenuates doxorubicin-induced cardiotoxicity by activating AMPK/SIRT-1/PGC-1α-mediated mitochondrial biogenesis. Toxicol Res (Camb). (2023) 12:216–23. doi: 10.1093/toxres/tfad007
30. Xi Y, Chen D, Dong Z, Zhang J, Lam H, He J, et al. Multi-omics insights into potential mechanism of SGLT2 inhibitors cardiovascular benefit in diabetic cardiomyopathy. Front Cardiovasc Med. (2022) 9:999254. doi: 10.3389/fcvm.2022.999254
31. Song Y, Huang C, Sin J, Germano JF, Taylor D, Thakur R, et al. Attenuation of adverse postinfarction left ventricular remodeling with empagliflozin enhances mitochondria-linked cellular energetics and mitochondrial biogenesis. Int J Mol Sci. (2021) 23:437. doi: 10.3390/ijms23010437
32. Shiraki A, Oyama JI, Shimizu T, Nakajima T, Yokota T, Node K. Empagliflozin improves cardiac mitochondrial function and survival through energy regulation in a murine model of heart failure. Eur J Pharmacol. (2022) 931:175194. doi: 10.1016/j.ejphar.2022.175194
33. Nikolaou PE, Mylonas N, Makridakis M, Makrecka-Kuka M, Iliou A, Zerikiotis S, et al. Cardioprotection by selective SGLT-2 inhibitors in a non-diabetic mouse model of myocardial ischemia/reperfusion injury: a class or a drug effect. Basic Res Cardiol. (2022) 117:27. doi: 10.1007/s00395-022-00934-7
34. Li X, Flynn ER, do Carmo JM, Wang Z, da Silva AA, Mouton AJ, et al. Direct cardiac actions of sodium-glucose cotransporter 2 inhibition improve mitochondrial function and attenuate oxidative stress in pressure overload-induced heart failure. Front Cardiovasc Med. (2022) 9:859253. doi: 10.3389/fcvm.2022.859253
35. Cai C, Wu F, Zhuang B, Ou Q, Peng X, Shi N, et al. Empagliflozin activates Wnt/β-catenin to stimulate FUNDC1-dependent mitochondrial quality surveillance against type-3 cardiorenal syndrome. Mol Metab. (2022) 64:101553. doi: 10.1016/j.molmet.2022.101553
36. Fan Z, Li C, Xu DC, Chun HZ, Xiao LC. Empagliflozin alleviates myocardial ischemia-reperfusion injury by up-regulating silent information regulator 1. J Anhui Med Univ. (2022) 57:1195–200. doi: 10.19405/j.cnki.issn1000-1492.2022.08.004
37. Xiu FS, Wen JL, Hui YZ, Guo HZ, Fu YL. Effects of dapagliflozin on inflammation and oxidative stress in mice with stress-induced cardiomyopathy. Jiangsu Medicine. (2022) 48:441–5+541. doi: 10.19460/j.cnki.0253-3685.2022.05.003
38. Young SL, Ryan L, Mullins TP, Flint M, Steane SE, Walton SL, et al. Sotagliflozin, a dual SGLT1/2 inhibitor, improves cardiac outcomes in a normoglycemic mouse model of cardiac pressure overload. Front Physiol. (2021) 12:738594. doi: 10.3389/fphys.2021.738594
39. Tan Y, Yu K, Liang L, Liu Y, Song F, Ge Q, et al. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function after cardiac arrest in rats by enhancing mitochondrial energy metabolism. Front Pharmacol. (2021) 12:758080. doi: 10.3389/fphar.2021.758080
40. Nikolaou PE, Efentakis P, Abu Qourah F, Femminò S, Makridakis M, Kanaki Z, et al. Chronic empagliflozin treatment reduces myocardial infarct size in nondiabetic mice through STAT-3-mediated protection on microvascular endothelial cells and reduction of oxidative stress. Antioxid Redox Signal. (2021) 34:551–71. doi: 10.1089/ars.2019.7923
41. Li X, Lu Q, Qiu Y, do Carmo JM, Wang Z, da Silva AA, et al. Direct cardiac actions of the sodium glucose co-transporter 2 inhibitor empagliflozin improve myocardial oxidative phosphorylation and attenuate pressure-overload heart failure. J Am Heart Assoc. (2021) 10:e018298. doi: 10.1161/JAHA.120.018298
42. Jing B, Jing X, Yu JY, Ya PW, Wen BL. Net of Glenn on diabetic myocardial ischemia rat myocardial cell energy metabolism and blood rheology of action research. Mod Biomed Adv. (2021) 21:4223–8. doi: 10.13241/j.cnki.pmb.2021.22.005
43. Gang L, Da CY, Tian HL, Chao PY, Qiang W. Effects of dapagliflozin on cardiac hypertrophy and myocardial fibrosis. Med Chongqing. (2020) 49:13–7.
44. Yurista SR, Silljé H, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, et al. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. (2019) 21:862–73. doi: 10.1002/ejhf.1473
45. Adingupu DD, Göpel SO, Grönros J, Behrendt M, Sotak M, Miliotis T, et al. SGLT2 Inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob(-/-) mice. Cardiovasc Diabetol. (2019) 18:16. doi: 10.1186/s12933-019-0820-6
46. Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet J, Koeman A, et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia. (2018) 61:722–6. doi: 10.1007/s00125-017-4509-7
47. Durak A, Olgar Y, Degirmenci S, Akkus E, Tuncay E, Turan B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc Diabetol. (2018) 17:144. doi: 10.1186/s12933-018-0790-0
48. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of metabolic flexibility in the failing heart. Front Cardiovasc Med. (2018) 5:68. doi: 10.3389/fcvm.2018.00068
49. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. (2021) 128:1487–513. doi: 10.1161/CIRCRESAHA.121.318241
50. Glatz J, Nabben M, Young ME, Schulze PC, Taegtmeyer H, Luiken J. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165579. doi: 10.1016/j.bbadis.2019.165579
51. Karwi QG, Jörg AR, Lopaschuk GD. Allosteric, transcriptional and post-translational control of mitochondrial energy metabolism. Biochem J. (2019) 476:1695–712. doi: 10.1042/BCJ20180617
52. Rosenblatt-Velin N, Montessuit C, Papageorgiou I, Terrand J, Lerch R. Postinfarction heart failure in rats is associated with upregulation of GLUT-1 and downregulation of genes of fatty acid metabolism. Cardiovasc Res. (2001) 52:407–16. doi: 10.1016/s0008-6363(01)00393-5
53. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. (2010) 3:420–30. doi: 10.1161/CIRCHEARTFAILURE.109.888479
54. Dávila-Román VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. (2002) 40:271–7. doi: 10.1016/s0735-1097(02)01967-8
55. Tuunanen H, Engblom E, Naum A, Någren K, Hesse B, Airaksinen KE, et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation. (2006) 114:2130–7. doi: 10.1161/CIRCULATIONAHA.106.645184
56. Neglia D, De Caterina A, Marraccini P, Natali A, Ciardetti M, Vecoli C, et al. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. (2007) 293:H3270–8. doi: 10.1152/ajpheart.00887.2007
57. Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. (1996) 94:2837–42. doi: 10.1161/01.cir.94.11.2837
58. Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, et al. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation. (2002) 106:606–12. doi: 10.1161/01.cir.0000023531.22727.c1
59. Heather LC, Cole MA, Lygate CA, Evans RD, Stuckey DJ, Murray AJ, et al. Fatty acid transporter levels and palmitate oxidation rate correlate with ejection fraction in the infarcted rat heart. Cardiovasc Res. (2006) 72:430–7. doi: 10.1016/j.cardiores.2006.08.020
60. Taylor M, Wallhaus TR, Degrado TR, Russell DC, Stanko P, Nickles RJ, et al. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F]fluoro-6-thia-heptadecanoic acid and [18F]FDG in patients with congestive heart failure. J Nucl Med. (2001) 42:55–62.11197981
61. Funada J, Betts TR, Hodson L, Humphreys SM, Timperley J, Frayn KN, et al. Substrate utilization by the failing human heart by direct quantification using arterio-venous blood sampling. PLoS One. (2009) 4:e7533. doi: 10.1371/journal.pone.0007533
62. Voros G, Ector J, Garweg C, Droogne W, Van Cleemput J, Peersman N, et al. Increased cardiac uptake of ketone bodies and free fatty acids in human heart failure and hypertrophic left ventricular remodeling. Circ Heart Fail. (2018) 11:e004953. doi: 10.1161/CIRCHEARTFAILURE.118.004953
63. Kenny HC, Abel ED. Heart failure in type 2 diabetes Mellitus. Circ Res. (2019) 124:121–41. doi: 10.1161/CIRCRESAHA.118.311371
64. Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation. (2004) 109:2191–6. doi: 10.1161/01.CIR.0000127959.28627.F8
65. Rijzewijk LJ, van der Meer RW, Lamb HJ, de Jong HW, Lubberink M, Romijn JA, et al. Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: studies with cardiac positron emission tomography and magnetic resonance imaging. J Am Coll Cardiol. (2009) 54:1524–32. doi: 10.1016/j.jacc.2009.04.074
66. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, et al. Targeting mitochondria-inflammation circuit by β-hydroxybutyrate mitigates HFpEF. Circ Res. (2021) 128:232–45. doi: 10.1161/CIRCRESAHA.120.317933
67. Remondino A, Rosenblatt-Velin N, Montessuit C, Tardy I, Papageorgiou I, Dorsaz PA, et al. Altered expression of proteins of metabolic regulation during remodeling of the left ventricle after myocardial infarction. J Mol Cell Cardiol. (2000) 32:2025–34. doi: 10.1006/jmcc.2000.1234
68. Amorim PA, Nguyen TD, Shingu Y, Schwarzer M, Mohr FW, Schrepper A, et al. Myocardial infarction in rats causes partial impairment in insulin response associated with reduced fatty acid oxidation and mitochondrial gene expression. J Thorac Cardiovasc Surg. (2010) 140:1160–7. doi: 10.1016/j.jtcvs.2010.08.003
69. Doria A, Nosadini R, Avogaro A, Fioretto P, Crepaldi G. Myocardial metabolism in type 1 diabetic patients without coronary artery disease. Diabet Med. (1991) 8(Spec No):S104–7. doi: 10.1111/j.1464-5491.1991.tb02168.x
70. Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C, et al. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. (2006) 47:598–604. doi: 10.1016/j.jacc.2005.09.030
71. Monti LD, Lucignani G, Landoni C, Moresco RM, Piatti P, Stefani I, et al. Myocardial glucose uptake evaluated by positron emission tomography and fluorodeoxyglucose during hyperglycemic clamp in IDDM patients. Role of free fatty acid and insulin levels. Diabetes. (1995) 44:537–42. doi: 10.2337/diab.44.5.537
72. Hällsten K, Virtanen KA, Lönnqvist F, Janatuinen T, Turiceanu M, Rönnemaa T, et al. Enhancement of insulin-stimulated myocardial glucose uptake in patients with type 2 diabetes treated with rosiglitazone. Diabet Med. (2004) 21:1280–7. doi: 10.1111/j.1464-5491.2004.01332.x
73. Lautamäki R, Airaksinen KE, Seppänen M, Toikka J, Luotolahti M, Ball E, et al. Rosiglitazone improves myocardial glucose uptake in patients with type 2 diabetes and coronary artery disease: a 16-week randomized, double-blind, placebo-controlled study. Diabetes. (2005) 54:2787–94. doi: 10.2337/diabetes.54.9.2787
74. Mather KJ, Hutchins GD, Perry K, Territo W, Chisholm R, Acton A, et al. Assessment of myocardial metabolic flexibility and work efficiency in human type 2 diabetes using 16-[18F]fluoro-4-thiapalmitate, a novel PET fatty acid tracer. Am J Physiol Endocrinol Metab. (2016) 310:E452–60. doi: 10.1152/ajpendo.00437.2015
76. Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, Teoh H, et al. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl Sci. (2018) 3:575–87. doi: 10.1016/j.jacbts.2018.07.006
77. Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. (2004) 53:2366–74. doi: 10.2337/diabetes.53.9.2366
78. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. (2007) 56:2457–66. doi: 10.2337/db07-0481
79. Shug AL, Shrago E, Bittar N, Folts JD, Koke JR. Acyl-CoA inhibition of adenine nucleotide translocation in ischemic myocardium. Am J Physiol. (1975) 228:689–92. doi: 10.1152/ajplegacy.1975.228.3.689
80. Woldegiorgis G, Yousufzai SY, Shrago E. Studies on the interaction of palmitoyl coenzyme A with the adenine nucleotide translocase. J Biol Chem. (1982) 257:14783–7. doi: 10.1016/S0021-9258(18)33348-9
81. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. (2002) 109:121–30. doi: 10.1172/JCI14080
82. Rask-Madsen C, Li Q, Freund B, Feather D, Abramov R, Wu IH, et al. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. (2010) 11:379–89. doi: 10.1016/j.cmet.2010.03.013
83. Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. (2000) 97:1784–9. doi: 10.1073/pnas.97.4.1784
84. Chun L, Junlin Z, Aimin W, Niansheng L, Benmei C, Minxiang L. Inhibition of ceramide synthesis reverses endothelial dysfunction and atherosclerosis in streptozotocin-induced diabetic rats. Diabetes Res Clin Pract. (2011) 93:77–85. doi: 10.1016/j.diabres.2011.03.017
85. Sharma V, Dhillon P, Wambolt R, Parsons H, Brownsey R, Allard MF, et al. Metoprolol improves cardiac function and modulates cardiac metabolism in the streptozotocin-diabetic rat. Am J Physiol Heart Circ Physiol. (2008) 294:H1609–20. doi: 10.1152/ajpheart.00949.2007
86. Onay-Besikci A, Guner S, Arioglu E, Ozakca I, Ozcelikay AT, Altan VM. The effects of chronic trimetazidine treatment on mechanical function and fatty acid oxidation in diabetic rat hearts. Can J Physiol Pharmacol. (2007) 85:527–35. doi: 10.1139/y07-036
87. Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. (2020) 11:390. doi: 10.1038/s41419-020-2544-7
88. Ge M, Molina J, Kim JJ, Mallela SK, Ahmad A, Varona Santos J, et al. Empagliflozin reduces podocyte lipotoxicity in experimental alport syndrome. Elife. (2023) 12:e83353. doi: 10.7554/eLife.83353
89. Koyani CN, Plastira I, Sourij H, Hallström S, Schmidt A, Rainer PP, et al. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol Res. (2020) 158:104870. doi: 10.1016/j.phrs.2020.104870
90. Lee HC, Shiou YL, Jhuo SJ, Chang CY, Liu PL, Jhuang WJ, et al. The sodium-glucose co-transporter 2 inhibitor empagliflozin attenuates cardiac fibrosis and improves ventricular hemodynamics in hypertensive heart failure rats. Cardiovasc Diabetol. (2019) 18:45. doi: 10.1186/s12933-019-0849-6
91. Capone F, Vettor R, Schiattarella GG. Cardiometabolic HFpEF: nASH of the heart. Circulation. (2023) 147:451–3. doi: 10.1161/CIRCULATIONAHA.122.062874
92. Phan TT, Abozguia K, Nallur Shivu G, Mahadevan G, Ahmed I, Williams L, et al. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J Am Coll Cardiol. (2009) 54:402–9. doi: 10.1016/j.jacc.2009.05.012
93. Beer M, Seyfarth T, Sandstede J, Landschütz W, Lipke C, Köstler H, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. (2002) 40:1267–74. doi: 10.1016/s0735-1097(02)02160-5
94. Conway MA, Allis J, Ouwerkerk R, Niioka T, Rajagopalan B, Radda GK. Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet. (1991) 338:973–6. doi: 10.1016/0140-6736(91)91838-l
95. Smith J, Su X, El-Maghrabi R, Stahl DP, Abumrad NA. Opposite regulation of CD36 ubiquitination by fatty acids and insulin: effects on fatty acid uptake. J Biol Chem. (2008) 283:13578–85. doi: 10.1074/jbc.M800008200
96. Coburn CT, Knapp FF Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem. (2000) 275:32523–9. doi: 10.1074/jbc.M003826200
97. Fonseca-Correa JI, Correa-Rotter R. Sodium-glucose cotransporter 2 inhibitors mechanisms of action: a review. Front Med (Lausanne). (2021) 8:777861. doi: 10.3389/fmed.2021.777861
98. Li J, Zhou L, Gong H. New insights and advances of sodium-glucose cotransporter 2 inhibitors in heart failure. Front Cardiovasc Med. (2022) 9:903902. doi: 10.3389/fcvm.2022.903902
99. Kabir M, Bergman RN, Porter J, Stefanovski D, Paszkiewicz RL, Piccinini F, et al. Dapagliflozin prevents abdominal visceral and subcutaneous adipose tissue dysfunction in the insulin-resistant canine model. Obesity (Silver Spring). (2023) 31:1798–811. doi: 10.1002/oby.23771
100. Dong M, Chen H, Wen S, Yuan Y, Yang L, Li Y, et al. The neuronal and non-neuronal pathways of sodium-glucose cotransporter-2 inhibitor on body weight-loss and insulin resistance. Diabetes Metab Syndr Obes. (2023) 16:425–35. doi: 10.2147/DMSO.S399367
101. Yoshida A, Matsubayashi Y, Nojima T, Suganami H, Abe T, Ishizawa M, et al. Attenuation of weight loss through improved antilipolytic effect in adipose tissue via the SGLT2 inhibitor tofogliflozin. J Clin Endocrinol Metab. (2019) 104:3647–60. doi: 10.1210/jc.2018-02254
102. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24. doi: 10.1161/CIRCRESAHA.113.300376
103. Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J Clin Invest. (1988) 82:2017–25. doi: 10.1172/JCI113822
104. Jiang H, Jia D, Zhang B, Yang W, Dong Z, Sun X, et al. Exercise improves cardiac function and glucose metabolism in mice with experimental myocardial infarction through inhibiting HDAC4 and upregulating GLUT1 expression. Basic Res Cardiol. (2020) 115:28. doi: 10.1007/s00395-020-0787-1
105. Bertrand L, Auquier J, Renguet E, Angé M, Cumps J, Horman S, et al. Glucose transporters in cardiovascular system in health and disease. Pflugers Arch. (2020) 472:1385–99. doi: 10.1007/s00424-020-02444-8
106. Capone F, Sotomayor-Flores C, Bode D, Wang R, Rodolico D, Strocchi S, et al. Cardiac metabolism in HFpEF: from fuel to signalling. Cardiovasc Res. (2023) 118:3556–75. doi: 10.1093/cvr/cvac166
107. Riggs K, Ali H, Taegtmeyer H, Gutierrez AD. The use of SGLT-2 inhibitors in type 2 diabetes and heart failure. Metab Syndr Relat Disord. (2015) 13:292–7. doi: 10.1089/met.2015.0038
108. Packer M. SGLT2 Inhibitors: role in protective reprogramming of cardiac nutrient transport and metabolism. Nat Rev Cardiol. (2023) 20:443–62. doi: 10.1038/s41569-022-00824-4
109. Lu Q, Li X, Liu J, Sun X, Rousselle T, Ren D, et al. AMPK Is associated with the beneficial effects of antidiabetic agents on cardiovascular diseases. Biosci Rep. (2019) 39:BSR20181995. doi: 10.1042/BSR20181995
110. Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol. (2014) 171:2080–90. doi: 10.1111/bph.12475
111. Lopaschuk GD. Metabolic modulators in heart disease: past, present, and future. Can J Cardiol. (2017) 33:838–49. doi: 10.1016/j.cjca.2016.12.013
112. Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. (2013) 304:H1060–76. doi: 10.1152/ajpheart.00646.2012
113. Karwi QG, Biswas D, Pulinilkunnil T, Lopaschuk GD. Myocardial ketones metabolism in heart failure. J Card Fail. (2020) 26:998–1005. doi: 10.1016/j.cardfail.2020.04.005
114. Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis. Diabetes Care. (2016) 39:1115–22. doi: 10.2337/dc16-0542
115. Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care. (2016) 39:1108–14. doi: 10.2337/dc16-0330
116. Mizuno Y, Harada E, Nakagawa H, Morikawa Y, Shono M, Kugimiya F, et al. The diabetic heart utilizes ketone bodies as an energy source. Metab Clin Exp. (2017) 77:65–72. doi: 10.1016/j.metabol.2017.08.005
117. Abdul Kadir A, Clarke K, Evans RD. Cardiac ketone body metabolism. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165739. doi: 10.1016/j.bbadis.2020.165739
118. Rusek M, Pluta R, Ułamek-Kozioł M, Czuczwar SJ. Ketogenic diet in Alzheimer’s disease. Int J Mol Sci. (2019) 20:3892. doi: 10.3390/ijms20163892
119. Kosinski C, Jornayvaz FR. Effects of ketogenic diets on cardiovascular risk factors: evidence from animal and human studies. Nutrients. (2017) 9:517. doi: 10.3390/nu9050517
120. Nielsen R, Møller N, Gormsen LC, Tolbod LP, Hansson NH, Sorensen J, et al. Cardiovascular effects of treatment with the ketone body 3-hydroxybutyrate in chronic heart failure patients. Circulation. (2019) 139:2129–41. doi: 10.1161/CIRCULATIONAHA.118.036459
121. Mayer SB, Jeffreys AS, Olsen MK, McDuffie JR, Feinglos MN, Yancy WS Jr. Two diets with different haemoglobin A1c and antiglycaemic medication effects despite similar weight loss in type 2 diabetes. Diabetes Obes Metab. (2014) 16:90–3. doi: 10.1111/dom.12191
122. Luong TV, Nielsen EN, Falborg L, Kjærulff M, Tolbod LP, Søndergaard E, et al. Intravenous and oral whole body ketone dosimetry, biodistribution, metabolite correction and kinetics studied by (R)-[1-(11)C]β-hydroxybutyrate ([(11)C]OHB) PET in healthy humans. EJNMMI Radiopharm Chem. (2023) 8:12. doi: 10.1186/s41181-023-00198-z
123. Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight. (2019) 4:e124079. doi: 10.1172/jci.insight.124079
124. Yurista SR, Matsuura TR, Silljé H, Nijholt KT, McDaid KS, Shewale SV, et al. Ketone ester treatment improves cardiac function and reduces pathologic remodeling in preclinical models of heart failure. Circ Heart Fail. (2021) 14:e007684. doi: 10.1161/CIRCHEARTFAILURE.120.007684
125. Papazafiropoulou AK, Georgopoulos MM, Katsilambros NL. Ketone bodies and the heart. Arch Med Sci Atheroscler Dis. (2021) 6:e209–14. doi: 10.5114/amsad.2021.112475
126. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
127. Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. (2020) 383:1413–24. doi: 10.1056/NEJMoa2022190
128. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) inhibitors: a state-of-the-art review. JACC Basic Transl Sci. (2020) 5:632–44. doi: 10.1016/j.jacbts.2020.02.004
129. MacIsaac RJ, Jerums G, Ekinci EI. Cardio-renal protection with empagliflozin. Ann Transl Med. (2016) 4:409. doi: 10.21037/atm.2016.10.36
130. Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, et al. Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes. (2016) 65:1190–5. doi: 10.2337/db15-1356
131. Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. (2010) 106:360–8. doi: 10.1016/j.amjcard.2010.03.032
132. Oshima H, Miki T, Kuno A, Mizuno M, Sato T, Tanno M, et al. Empagliflozin, an SGLT2 inhibitor, reduced the mortality rate after acute myocardial infarction with modification of cardiac metabolomes and antioxidants in diabetic rats. J Pharmacol Exp Ther. (2019) 368:524–34. doi: 10.1124/jpet.118.253666
133. Saddik M, Lopaschuk GD. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J Biol Chem. (1991) 266:8162–70. doi: 10.1016/S0021-9258(18)92956-X
134. Wisneski JA, Stanley WC, Neese RA, Gertz EW. Effects of acute hyperglycemia on myocardial glycolytic activity in humans. J Clin Invest. (1990) 85:1648–56. doi: 10.1172/JCI114616
135. Vásquez-Trincado C, García-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. (2016) 594:509–25. doi: 10.1113/JP271301
136. Ketelhuth D. The immunometabolic role of indoleamine 2,3-dioxygenase in atherosclerotic cardiovascular disease: immune homeostatic mechanisms in the artery wall. Cardiovasc Res. (2019) 115:1408–15. doi: 10.1093/cvr/cvz067
137. Zhang XL, Shen Y, Ma XJ, Lu ZG, Xu YT, Xiong Q, et al. Low serum osteocalcin levels are correlated with left ventricular systolic dysfunction and cardiac death in Chinese men. Acta Pharmacol Sin. (2019) 40:486–91. doi: 10.1038/s41401-018-0080-0
138. Wang A, Zhang D, Liu J, Yan H, Zhang P, Yuan H, et al. Guanxinning injection combined with ischemic postconditioning attenuate myocardial ischemic reperfusion injury in chronic renal failure rats by modulating mitochondrial dynamics. Front Cardiovasc Med. (2022) 9:905254. doi: 10.3389/fcvm.2022.905254
139. Koizumi T, Watanabe M, Yokota T, Tsuda M, Handa H, Koya J, et al. Empagliflozin suppresses mitochondrial reactive oxygen species generation and mitigates the inducibility of atrial fibrillation in diabetic rats. Front Cardiovasc Med. (2023) 10:1005408. doi: 10.3389/fcvm.2023.1005408
140. Mizuno M, Kuno A, Yano T, Miki T, Oshima H, Sato T, et al. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol Rep. (2018) 6:e13741. doi: 10.14814/phy2.13741
141. Zarei M, Sarihi A, Zamani A, Raoufi S, Karimi SA, Ramezani-Aliakbari F. Mitochondrial biogenesis and apoptosis as underlying mechanisms involved in the cardioprotective effects of gallic acid against D-galactose-induced aging. Mol Biol Rep. (2023) 50:8005–14. doi: 10.1007/s11033-023-08670-4
142. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. (2006) 116:615–22. doi: 10.1172/JCI27794
143. Cantó C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. (2009) 20:98–105. doi: 10.1097/MOL.0b013e328328d0a4
144. Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. (2018) 15:335–46. doi: 10.1016/j.redox.2017.12.019
145. Zhang WX, He BM, Wu Y, Qiao JF, Peng ZY. Melatonin protects against sepsis-induced cardiac dysfunction by regulating apoptosis and autophagy via activation of SIRT1 in mice. Life Sci. (2019) 217:8–15. doi: 10.1016/j.lfs.2018.11.055
146. Ma T, Huang X, Zheng H, Huang G, Li W, Liu X, et al. SFRP2 Improves mitochondrial dynamics and mitochondrial biogenesis, oxidative stress, and apoptosis in diabetic cardiomyopathy. Oxid Med Cell Longev. (2021) 2021:9265016. doi: 10.1155/2021/9265016
147. Müller M, Donhauser E, Maske T, Bischof C, Dumitrescu D, Rudolph V, et al. Mitochondrial integrity is critical in right heart failure development. Int J Mol Sci. (2023) 24:11108. doi: 10.3390/ijms241311108
148. Nambu H, Takada S, Fukushima A, Matsumoto J, Kakutani N, Maekawa S, et al. Empagliflozin restores lowered exercise endurance capacity via the activation of skeletal muscle fatty acid oxidation in a murine model of heart failure. Eur J Pharmacol. (2020) 866:172810. doi: 10.1016/j.ejphar.2019.172810
149. Trachtenberg BH, Hare JM. Inflammatory cardiomyopathic syndromes. Circ Res. (2017) 121:803–18. doi: 10.1161/CIRCRESAHA.117.310221
150. Dhalla NS, Elimban V, Bartekova M, Adameova A. Involvement of oxidative stress in the development of subcellular defects and heart disease. Biomedicines. (2022) 10:393. doi: 10.3390/biomedicines10020393
151. Vermot A, Petit-Härtlein I, Smith S, Fieschi F. NADPH Oxidases (NOX): an overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants (Basel). (2021) 10:890. doi: 10.3390/antiox10060890
152. Nabeebaccus A, Zhang M, Shah AM. NADPH Oxidases and cardiac remodelling. Heart Fail Rev. (2011) 16:5–12. doi: 10.1007/s10741-010-9186-2
153. Joseph LC, Barca E, Subramanyam P, Komrowski M, Pajvani U, Colecraft HM, et al. Inhibition of NAPDH oxidase 2 (NOX2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes. PLoS One. (2016) 11:e0145750. doi: 10.1371/journal.pone.0145750
154. Patil KD, Halperin HR, Becker LB. Cardiac arrest: resuscitation and reperfusion. Circ Res. (2015) 116:2041–9. doi: 10.1161/CIRCRESAHA.116.304495
155. Hoehlschen J, Hofreither D, Tomin T, Birner-Gruenberger R. Redox-driven cardioprotective effects of sodium-glucose co-transporter-2 inhibitors: comparative review. Cardiovasc Diabetol. (2023) 22:101. doi: 10.1186/s12933-023-01822-7
156. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301:H2181–90. doi: 10.1152/ajpheart.00554.2011
157. Zhang M, Kho AL, Anilkumar N, Chibber R, Pagano PJ, Shah AM, et al. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. (2006) 113:1235–43. doi: 10.1161/CIRCULATIONAHA.105.581397
158. Kaludercic N, Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med. (2020) 7:12. doi: 10.3389/fcvm.2020.00012
159. Herrero P, McGill J, Lesniak DS, Dence CS, Scott SW, Kisrieva-Ware Z, et al. PET Detection of the impact of dobutamine on myocardial glucose metabolism in women with type 1 diabetes mellitus. J Nucl Cardiol. (2008) 15:791–9. doi: 10.1007/BF03007360
160. Latva-Rasku A, Honka MJ, Kullberg J, Mononen N, Lehtimäki T, Saltevo J, et al. The SGLT2 inhibitor dapagliflozin reduces liver fat but does not affect tissue insulin sensitivity: a randomized, double-blind, placebo-controlled study with 8-week treatment in type 2 diabetes patients. Diabetes Care. (2019) 42:931–7. doi: 10.2337/dc18-1569
161. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2019) 380:347–57. doi: 10.1056/NEJMoa1812389
162. Fitchett D, Inzucchi SE, Cannon CP, McGuire DK, Scirica BM, Johansen OE, et al. Empagliflozin reduced mortality and hospitalization for heart failure across the Spectrum of cardiovascular risk in the EMPA-REG OUTCOME trial. Circulation. (2019) 139:1384–95. doi: 10.1161/CIRCULATIONAHA.118.037778
163. Perkovic V, de Zeeuw D, Mahaffey KW, Fulcher G, Erondu N, Shaw W, et al. Canagliflozin and renal outcomes in type 2 diabetes: results from the CANVAS program randomised clinical trials. Lancet Diabetes Endocrinol. (2018) 6:691–704. doi: 10.1016/S2213-8587(18)30141-4
164. Garg V, Verma S, Connelly K. Mechanistic insights regarding the role of SGLT2 inhibitors and GLP1 agonist drugs on cardiovascular disease in diabetes. Prog Cardiovasc Dis. (2019) 62:349–57. doi: 10.1016/j.pcad.2019.07.005
165. Møller N. Ketone body, 3-hydroxybutyrate: minor metabolite—major medical manifestations. J Clin Endocrinol Metab. (2020) 105:dgaa370. doi: 10.1210/clinem/dgaa370
166. Lei B, Lionetti V, Young ME, Chandler MP, d'Agostino C, Kang E, et al. Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J Mol Cell Cardiol. (2004) 36:567–76. doi: 10.1016/j.yjmcc.2004.02.004
167. Diakos NA, Navankasattusas S, Abel ED, Rutter J, McCreath L, Ferrin P, et al. Evidence of glycolysis up-regulation and pyruvate mitochondrial oxidation mismatch during mechanical unloading of the failing human heart: implications for cardiac reloading and conditioning. JACC Basic Transl Sci. (2016) 1:432–44. doi: 10.1016/j.jacbts.2016.06.009
168. Christe ME, Rodgers RL. Cardiac glucose and fatty acid oxidation in the streptozotocin-induced diabetic spontaneously hypertensive rat. Hypertension. (1995) 25:235–41. doi: 10.1161/01.hyp.25.2.235
169. Sankaralingam S, Abo Alrob O, Zhang L, Jaswal JS, Wagg CS, Fukushima A, et al. Lowering body weight in obese mice with diastolic heart failure improves cardiac insulin sensitivity and function: implications for the obesity paradox. Diabetes. (2015) 64:1643–57. doi: 10.2337/db14-1050
170. Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res. (2007) 101:335–47. doi: 10.1161/CIRCRESAHA.107.150417
171. Lahey R, Wang X, Carley AN, Lewandowski ED. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation. (2014) 130:1790–9. doi: 10.1161/CIRCULATIONAHA.114.011687
172. Ho KL, Zhang L, Wagg C, Al Batran R, Gopal K, Levasseur J, et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc Res. (2019) 115:1606–16. doi: 10.1093/cvr/cvz045
173. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The failing heart relies on ketone bodies as a fuel. Circulation. (2016) 133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
174. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. (2016) 133:706–16. doi: 10.1161/CIRCULATIONAHA.115.017545
175. Rådholm K, Zhou Z, Clemens K, Neal B, Woodward M. Effects of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes in women versus men. Diabetes Obes Metab. (2020) 22:263–6. doi: 10.1111/dom.13876
176. Pahud de Mortanges A, Salvador D Jr, Laimer M, Muka T, Wilhelm M, Bano A. The role of SGLT2 inhibitors in atherosclerosis: a narrative Mini-review. Front Pharmacol. (2021) 12:751214. doi: 10.3389/fphar.2021.751214
Keywords: sodium-glucose transporter 2 inhibitor, heart, energy metabolism, fatty acid, glucose, ketone body
Citation: Su S, Ji X, Li T, Teng Y, Wang B, Han X and Zhao M (2023) The changes of cardiac energy metabolism with sodium-glucose transporter 2 inhibitor therapy. Front. Cardiovasc. Med. 10:1291450. doi: 10.3389/fcvm.2023.1291450
Received: 9 September 2023; Accepted: 13 November 2023;
Published: 6 December 2023.
Edited by:
David Della-Morte, University of Miami, United StatesReviewed by:
Ana Carolina Mieko Omoto, University of Mississippi Medical Center, United StatesElena N. Dedkova, University of California, Davis, United States
© 2023 Su, Ji, Li, Teng, Wang, Han and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingjing Zhao bWpneDIwMDRAMTYzLmNvbQ==