 Martha M. S. Sim1,2
Martha M. S. Sim1,2 Jeremy P. Wood
Jeremy P. Wood
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 23 October 2023
Sec. Cardiovascular Pharmacology and Drug Discovery
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1272971
Antithrombotic therapy is a delicate balance between the benefits of preventing a thrombotic event and the risks of inducing a major bleed. Traditional approaches have included antiplatelet and anticoagulant medications, require careful dosing and monitoring, and all carry some risk of bleeding. In recent years, several new targets have been identified, both in the platelet and coagulation systems, which may mitigate this bleeding risk. In this review, we briefly describe the current state of antithrombotic therapy, and then present a detailed discussion of the new generation of drugs that are being developed to target more safely existing or newly identified pathways, alongside the strategies to reverse direct oral anticoagulants, showcasing the breadth of approaches. Combined, these exciting advances in antithrombotic therapy bring us closer than we have ever been to the “holy grail” of the field, a treatment that separates the hemostatic and thrombotic systems, preventing clots without any concurrent bleeding risk.
Thrombosis is the pathological formation of a clot within an intact vessel, which blocks blood flow, resulting in an ischemic injury. Depending on their location, thrombi can be the direct cause of life-threatening events, such as myocardial infarctions, pulmonary emboli, and strokes (1). Thus, safe and effective antithrombotic therapies are a critical component of our medical system. However, all existing antithrombotics carry a risk of bleeding, which can also be life-threatening. This is because the same components responsible for thrombus formation (blood platelets and coagulation) are also necessary for hemostasis, the healthy formation of a blood clot in response to vessel injury (2). Therefore, research is ongoing to identify new targets in this system, which may provide safer treatment. Here, we summarize the current therapeutic strategies, targeting platelets and coagulation factors, and discuss the new approaches that are in development, emphasizing the diversity of strategies and targets being evaluated.
Antiplatelet therapy has become a fundamental component in the treatment of cardiovascular disease. At the site of vascular injury or in response to vascular pathology, such as rupture of an atherosclerotic plaque, platelets can activate and aggregate intravascularly, typically as arterial thrombi (1). Similarly, platelet activation and aggregation are associated with inflammatory conditions, and platelet depletion with the development of disseminated intravascular coagulopathy, in conditions such as sepsis (3). Antiplatelet drugs are intended to prevent or limit platelet activation and aggregation and are generally used in acute coronary syndrome (ACS) and ischemic stroke patients for long-term control or secondary prevention (4–7). As the same mechanistic processes are responsible for both physiological and pathological platelet aggregation, striking a balance between the beneficial and harmful effects of antiplatelet therapy continues to be a challenge. In practice, this is clinically managed by careful consideration of optimal therapeutic regimens and duration of therapy, while prioritizing treatment for patients whose thrombotic risk clearly outweighs their risk of bleeding complications. In this section, we discuss the existing and recently developed antiplatelet therapeutics, along with novel strategies that have been proposed based on animal studies.
The most commonly used antiplatelet agents target thromboxane A2 (TXA2) and ADP, which are secondary agonists of platelet activation (Figure 1, Table 1). Since the discovery of its antithrombotic effects in 1956, aspirin, used as an anti-inflammatory agent, has been one of the most important antiplatelet agents (8). Aspirin acts by irreversibly inhibiting cyclooxygenase-1 (COX-1), limiting the access of arachidonic acid to its active site and preventing prostaglandin G2 and H2 synthesis, and subsequent TXA2 production (9). Aspirin also inhibits COX-2, which is expressed by ∼10% of circulating platelets, though at ∼170-fold lesser potency compared to COX-1 (10). Its full antiplatelet effect is reached at a low dose of 75–100 mg/day, whereas COX-2 inhibitory effects are mainly observed with higher doses of >500 mg/day, which may result in side effects such as gastrointestinal bleeding, without additional benefit to the antiplatelet effect (11). Apart from its effects on platelet activation, aspirin also acetylates lysine residues on fibrinogen, enhancing fibrin clot permeability and lysis, and therefore, results in less stable clots (12).
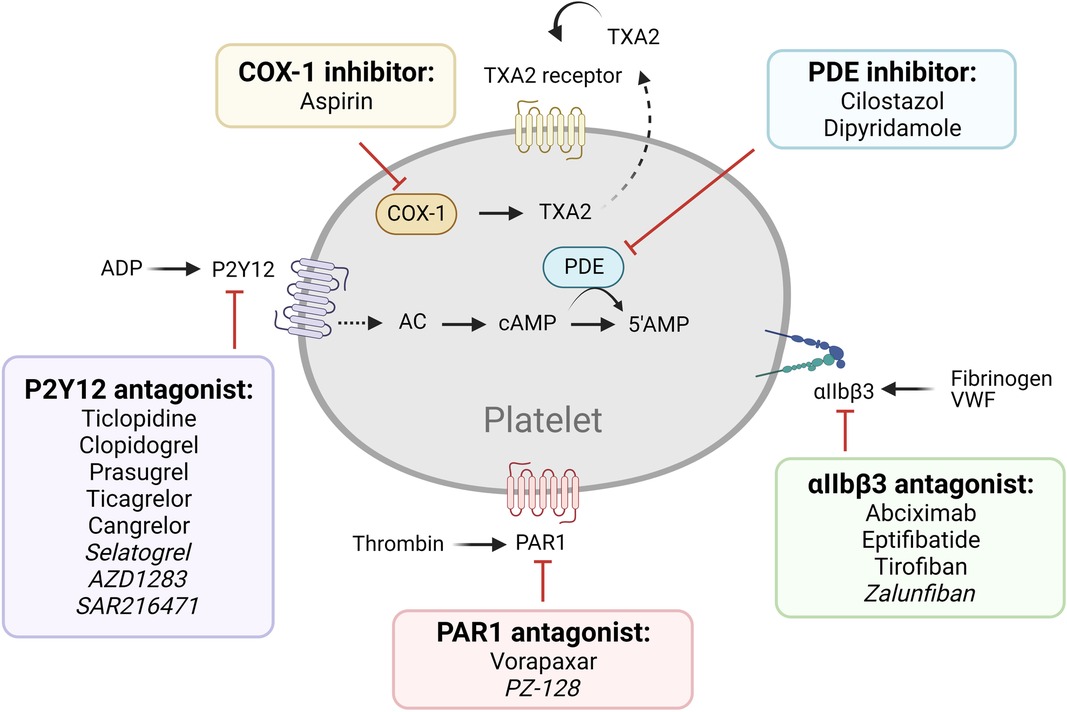
Figure 1. Older and newer generations of antiplatelet therapy targeting established pathways. Commonly used (regular font) and next generation (italicized) antiplatelet therapy of established pathways are listed.

Table 1. Current antithrombotic therapy.
P2Y12 antagonists are another category of main-line antiplatelet therapeutics. The thienopyridines class consists of ticlopidine, clopidogrel, and prasugrel; and the nucleoside-nucleotide derivatives include ticagrelor and cangrelor. Thienopyridines are prodrugs which require hepatic cytochrome P-450 (CYP450)-dependent metabolism (13). Clopidogrel blocks the P2Y12 receptor irreversibly by modifying a cysteine residue (14, 15). On the other hand, ticagrelor and cangrelor do not require liver-dependent metabolism and are reversible, competitive P2Y12 inhibitors (16). P2Y12 inhibitors are often used for patients with CAD after percutaneous coronary intervention (PCI) in combination with aspirin (dual antiplatelet therapy/ DAPT) and for patients with ACS, with or without PCI, with guidelines recommending varying durations for secondary prevention (17). Due to genetic factors involved in CYP450 metabolic pathways, clopidogrel shows widely variable inhibition of platelet activation, with ∼30% of treated individuals categorized as poor to intermediate responders to the drug (18). Genotype-guided strategies in clopidogrel therapy have been successful (19), suggesting value in individualized pharmacogenetics as a treatment strategy in clinical practice (18). Recently, the more potent and predictable ticagrelor and cangrelor have seen increasing use (20).
Other current antiplatelet therapy (Figure 1) includes phosphodiesterase (PDE) inhibitors (cilostazol, dipyridamole), αIIbβ3 antagonists (abciximab, eptifibatide, and tirofiban), and protease-activated receptor-1 (PAR1) antagonists (vorapaxar) (4–7). PDE inhibitors reduce platelet reactivity by increasing the cyclic nucleotides cAMP and/or cGMP, thereby dampening cytoskeletal rearrangement, integrin αIIbβ3 activation, and platelet secretion by interfering with activation signaling pathways (21). The combination of aspirin-dipyridamole is used for secondary prevention of cerebrovascular atherothrombotic events (22, 23). The αIIbβ3 antagonists prevent platelet aggregation by selectively blocking the fibrinogen receptor (24). Integrin αIIbβ3 can be targeted by a chimeric human-murine monoclonal antibody (abciximab), a synthetic cyclic heptapeptide based upon a sequence found in the snake venom platelet inhibitor disintegrin (eptifibatide), or an RGD-based peptidomimetic analog that specifically binds to αIIbβ3 on resting platelets (tirofiban) (24). The PAR1 antagonists block platelet activation by thrombin, the most potent agonist generated at vascular injury or plaque rupture sites via coagulation activation (25). Despite their great promise, however, several clinical trials have indicated safety concerns including elevated risk of major bleeding (26, 27).
Overall, antiplatelet therapy is still associated with non-negligible to high bleeding risk and thrombocytopenia, a meaningful variability in individual response due to genetic factors, and generally poor biological response in patients with comorbidities, such as diabetes and obesity (4–7, 18). In the next section, pharmacological approaches to platelet inhibition currently being considered, under development, or undergoing clinical testing, will be reviewed.
Several new antiplatelet agents, which target the pathways described above, are currently in clinical trials (Figure 1). These include:
(1) New P2Y12 antagonists: Selatogrel is a selective, potent, and reversible platelet P2Y12 antagonist, with the advantage of subcutaneous administration, allowing self-dosing and usage in the emergency setting of ACS or unconscious patients (28, 29). A Phase III clinical trial is ongoing. Other highly potent P2Y12 inhibitors are under development, including AZD1283 and SAR216471, both of which were associated with higher selectivity, less bleeding, and comparable antithrombotic efficacy compared to ticagrelor in animal models (30, 31). SAR216471 is currently in a Phase II study.
(2) New αIIbβ3 antagonists: Existing receptor antagonists are ligand-mimetics, which may cause a conformation change in αIIbβ3 to a high-affinity state, leading to either paradoxical platelet activation or exposure of ligand-induced binding sites that trigger antibody-mediated platelet clearance and thrombocytopenia in some patients (32, 33). They also are highly potent and associated with a significant increase in bleeding risk, and all require intravenous administration, limiting their utility for long-term therapy. Zalunfiban (RUC-4) is a small molecule inhibitor designed to alleviate this, as it binds to the metal ion-binding site on GPIIIa, maintaining the receptor in the low-affinity state incapable of fibrinogen binding, and therefore, does not induce a conformational change (34, 35). It can also be subcutaneously administered, which would favor its use in urgent settings (34). It showed efficacy in a Phase I study in healthy volunteers and stable CAD patients on aspirin, as it produced high-grade inhibition of ADP-induced platelet aggregation within 15 min of administration with rapid return to normal platelet function within the next 2 h (36). It is currently in a Phase IIb trial. Intracellular inhibitors of αIIbβ3 have also been developed which disrupt integrin activation, preventing the switch to the high-affinity state and subsequent outside-in signaling (37).
(3) New PAR1 antagonists: G protein-coupled receptors (GPCR) are cell surface receptors which upon ligand binding undergo conformational change and activate the associated cytosolic G protein, which further activates an intracellular signaling process. Pepducins are cell-penetrating lapidated peptides that are designed to selectively target the intracellular receptor-effector interface of a GPCR, by conjugating the intracellular loop portion of the receptor, including PAR1 (38). Existing PAR1 inhibitors interfere with both prothrombotic and cytoprotective downstream pathways, while the pepducin technology allows for selective control of downstream signaling pathways (39). PZ-128 is a pepducin inhibitor of PAR1 proposed for CAD treatment that has completed a Phase II clinical trial, showing that it was well tolerated in ACS patients undergoing PCI and did not cause bleeding even when administered on top of DAPT and heparin (40).
In addition to the development of a new generation of drugs targeting recognized pathways, new targets for antiplatelet therapy have also been identified, including platelet receptors and intracellular signaling pathways (Figure 2, Table 2). Many of these are thought to be non-essential for the hemostatic process, and so may provide safer alternatives to the current interventions.
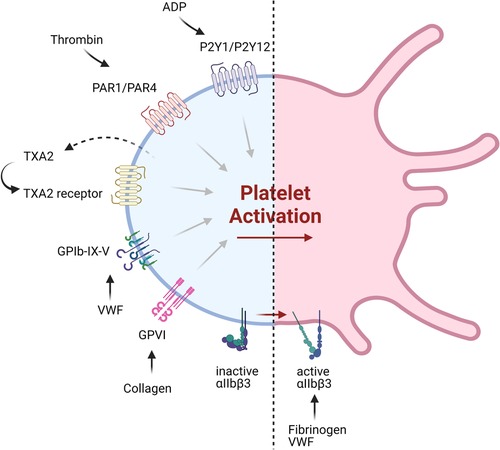
Figure 2. Current and emerging targets of antiplatelet therapeutics. Shown are various platelet receptors implicated in major pathways of platelet activation, which are being targeted by currently available antiplatelet therapy or those under development.

Table 2. Novel antithrombotic therapy.
A. Targeting platelet surface receptors (Figure 3).
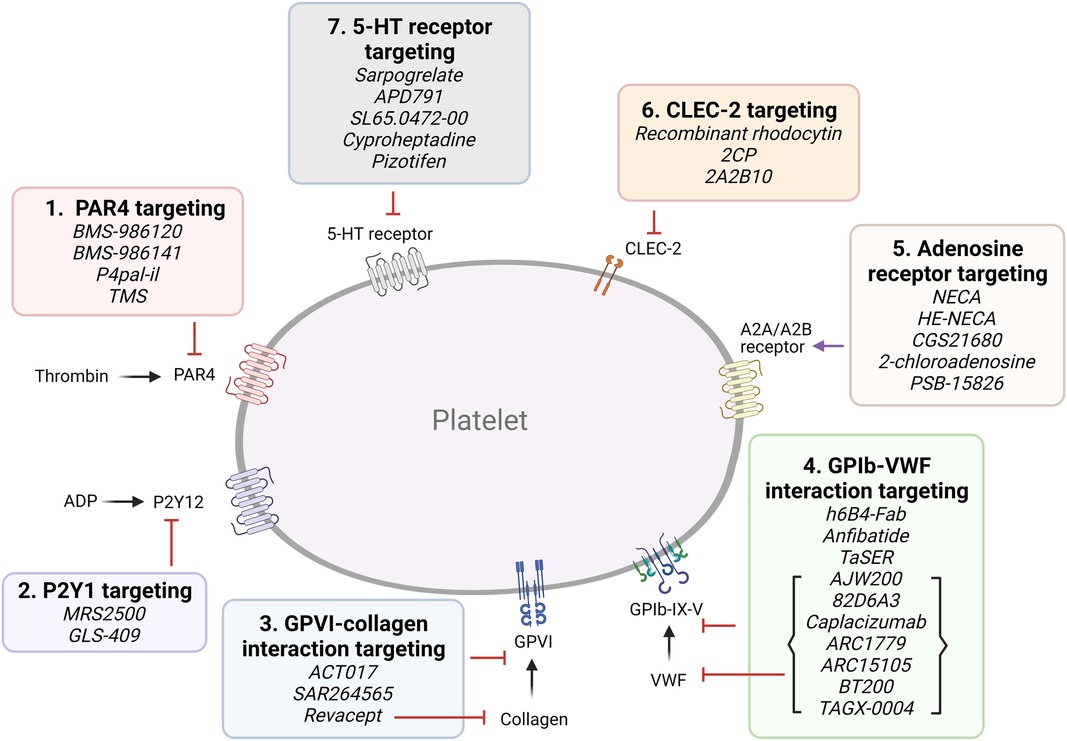
Figure 3. Targeting proposed pathways by modulating platelet receptors. Shown are novel approaches of currently emerging antiplatelet therapy by targeting platelet receptors.
(1) PAR4 targeting: Both PAR1 and PAR4 are expressed on platelets, and form heterodimers (41). PAR4 activation requires higher thrombin concentrations, and it was proposed to play a more important role in thrombosis than hemostasis (42). BMS-986120 and BMS-986141 are specific, small molecule inhibitors of PAR4, which show antithrombotic efficacy with very low bleeding effect in non-human primates and in healthy individuals, and the latter is showing promise in an ongoing clinical trial (42, 43). P4pal-i1 is a PAR4 pepducin inhibitor currently being investigated for antithrombotic properties, and has been shown to significantly decrease arterial occlusion in guinea pigs (44). And lastly, 3,5,2′,4′-tetramethoxystilbene (TMS) is a fully methylated analog of resveratrol, a phenol found in red wine. TMS binds PAR4 and has been shown to reduce thrombus formation in vitro (45).
(2) P2Y1 targeting: Platelets express P2Y1 and P2Y12 ADP-receptors (15, 46). MRS2500, an adenosine analog developed as a specific P2Y1 antagonist, has been shown to provide strong protection against systemic thromboembolism upon intravenous injection of collagen and adrenalin in mice, while only moderately prolonging bleeding time (47), and exhibited antithrombotic effects in cynomolgus monkeys (48). GLS-409, an analog of the naturally occurring compound adenosine tetraphosphate, inhibits ADP-induced platelet aggregation, and significantly inhibits thrombosis in animal models, with minimal increase in bleeding time (49). To date, there is no clinical trial assessing the P2Y1 antagonists.
(3) Glycoprotein VI (GPVI) targeting: GPVI is the major collagen receptor on platelets (50). Glenzocimab (ACT017) is a humanized monoclonal fragment antigen-binding (Fab) domain against platelet GPVI, which has been shown to inhibit aggregation and procoagulant activity on collagen-stimulated platelets, as well as adhesion and thrombus formation on collagen surfaces in vitro (51, 52). A Phase II/III trial is currently being conducted to evaluate its efficacy and safety in acute ischemic stroke and a Phase IIb in treating myocardial infarction is planned for the near future. SAR264565 is another humanized Fab directed against GPVI, which potently inhibits collagen-induced platelet aggregation (53). Revacept, on the other hand, is a soluble dimeric GPVI-Fc fusion protein containing the fragment crystallizable (Fc) portion of human IgG1, and was developed to specifically bind fibrillar collagen (54). Revacept binds collagen, preventing interaction with circulating platelets and von Willebrand factor (VWF), and resulting in antithrombotic effects. As it does not directly bind platelets, it does not interfere with platelet activity or cause thrombocytopenia (54, 55). It has recently completed a Phase II clinical trial (56, 57). Recently, new selective GPVI antagonists have been identified from large chemical database screening as reported by Olğaç et al. (58), which showed promising antithrombotic properties in vitro. Finally, nanobodies raised against the extracellular domain of GPVI have emerged as potential therapeutics (59).
(4) GPIb-VWF interaction targeting: GPIbα is a central component of the GPIb-IX-V complex expressed on platelets, which binds to VWF (60). VWF binding to collagen and its unfolding under higher shear rates exposes binding sites to platelet GPIb and mediates platelet adhesion and thrombus formation (61). Several GPIb-blocking antibodies have been developed and tested for their antiplatelet effects, such as the monoclonal antibody h6B4-Fab, which inhibits ristocetin-induced platelet aggregation in non-human primates, with only a mild prolongation of skin bleeding time (62, 63). Anfibatide is a C-type lectin (CLEC) purified from snake venom, which inhibits the binding of VWF and α-thrombin to GPIbα (64). It showed strong inhibition of murine and human platelet thrombus formation at low and high shears in ex vivo experiments (65), with a Phase I study in healthy volunteers showing good inhibitory effects without significant prolongation in bleeding time. Serine protease inhibitor (SERPIN)-based fusion proteins have also been proposed as antiplatelet therapy (66). A fusion protein called TaSER (targeted SERPIN), consisting of a variable heavy domain of heavy chain (VHH) with function-blocking activity against GPIbα and a thrombin-specific SERPIN, has been shown to block VWF binding and limit thrombus formation (67). Various monoclonal antibodies directed against VWF are also being pursued as antiplatelet therapy. AJW200 is a humanized IgG4 monoclonal antibody against the A1 domain of VWF, whereas 82D6A3 is directed against the A3 domain (68, 69). They both show promise in animal models and the former has completed a Phase I study, showing therapeutic promise without prolonging the skin bleeding time. ALX-0081 is a first-in-class, bivalent humanized nanobody directed against the A1 domain of VWF (70). The bivalency allows for high-affinity interaction with VWF-A1, leading to potent inhibition. Preclinical studies in cynomolgus monkeys showed potency in inhibiting ristocetin-induced platelet aggregation with 1.6- and 6-fold less prolongation of bleeding time compared to clopidogrel and abciximab, respectively (70), while studies in a guinea pigs thrombotic stroke model demonstrated reduction in brain infarct size (71). Caplacizumab, derived from ALX-0081, was approved in Europe in 2018 and in the USA in 2019 for use in acquired thrombotic thrombocytopenia purpura (TTP) (72). However, after an inconclusive Phase II trial, drug development for atherothrombotic indications was discontinued (73). Finally, nucleic acid aptamer technology has also been pursued. Aptamers are single-strand DNA or RNA molecules that form 3D structures which specifically bind to their target protein. They are potentially superior to antibodies due to their manufacturing cost, specificity, small size, lack of immunogenicity, and ease of reversal. ARC1779 is a nuclease-resistant aptamer designed to bind to VWF-A1, inhibiting VWF-dependent platelet aggregation (74). ARC15105 is a second generation VWF-A1 aptamer with improved potency and pharmacokinetics (75). BT200, derived from ARC15105, has been successfully tested in healthy volunteers and in a Phase IIa trial (76, 77). More recently, a novel DNA aptamer TAGX-0004, which targets VWF-A1 with very high affinity and specificity was generated (78). TAGX-0004 contains a unique mini-hairpin DNA structure which confers further resistance to nuclease degradation thus extending its half-life in vivo. It showed superior thrombus inhibition to ARC1779 and comparable to caplacizumab (78).
(5) Adenosine A2A and A2B receptor targeting: Adenosine is a purine metabolite in plasma resulting from ecto-50-nucleotidase activity (7). It has a very short (∼1 s) half-life due to enzymatic conversion and has been used widely as an antiarrhythmic agent (7). Platelets express adenosine GPCR receptors, and their activation leads to inhibition of platelet activation and aggregation by increasing the cellular cAMP level (79). Several adenosine receptor agonists are being evaluated as antiplatelet agents. Previously evaluated existing adenosine receptor agonists include 5′-N-ethylcarboxamidoadenosine (NECA), HE-NECA, CGS 21680, 2-chloroadenosine, and PSB-15826, which were shown to possess antiplatelet effects in vitro, and antithrombotic effects in vivo (80). However, relatively high doses were required for efficacy, leading to a strong vasodilatory activity and various off target effects.
(6) C-type lectin-like type II (CLEC-2) targeting: Platelets express CLEC-2, which activates various pathways including platelet activation and thromboinflammation via various endogenous ligands such as podoplanin and rhodocytin (81). A recombinant rhodocytin, derived from a snake venom protein, was developed and shown to inhibit CLEC-2 in in vitro and animal models (82). A small molecule, 2CP, was also reported to act as a chemical inhibitor of CLEC-2 (83). Anti-CLEC-2 mAb 2A2B10 has also been shown to suppress thrombus formation without significant bleeding effects in animal cancer model (84).
(7) Serotonin/ 5-hydroxytryptamine (5-HT)-receptor interaction targeting: Platelets express 5-HT subtype 2A receptor (85). Serotonin, packaged in dense granules, is a mild platelet activator, and selective serotonin reuptake inhibitors (SSRIs) inhibit 5-HT reuptake by blocking the serotonin transporter (SERT), thereby acting as antiplatelet agents (86). Direct 5-HT2A receptor antagonists are also being investigated for antiplatelet activity. Currently tested SSRIs are MCI-9042 (sarpogrelate) is an SSRI currently being evaluated (87, 88), while several selective small molecule 5-HT2A receptor antagonists are being investigated, including APD791 (temanogrel), SL65.0472-00, and two existing antidepressant cyproheptadine and pizotifen (89–91). Sarpogrelate significantly reduced serotonin and collagen-induced acute pulmonary thromboembolic death in mice (87). In a trial with stroke patients, however, sarpogrelate failed to demonstrate noninferiority to aspirin, although it was associated with a reduced rate of bleeding complications (88).
B. Targeting platelet signaling components (Figure 4).
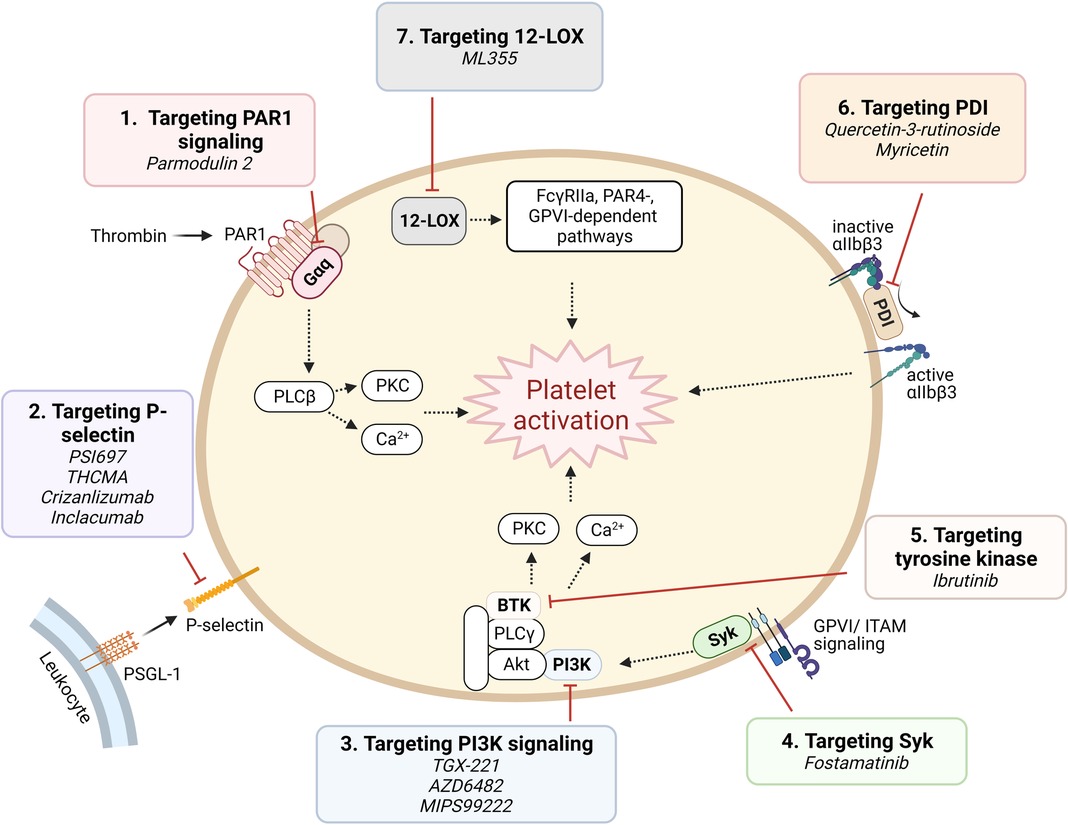
Figure 4. Targeting proposed pathways by modulating signaling component. Shown are novel approaches of currently emerging antiplatelet therapy by targeting platelet signaling component.
(1) Targeting signaling downstream of PAR1: Parmodulins are novel small molecule allosteric inhibitors of PAR1 which act on cytosolic Gαq subunit signaling downstream of the receptor, inhibiting integrin activation and platelet aggregation. Parmodulin 2, the most selective compound developed, further exhibits anti-inflammatory activity through PAR1 signaling inhibition in endothelial cells, in mouse models (92, 93). No parmodulin clinical trials have started yet.
(2) P-selectin targeting: Platelets store the adhesion molecule P-selectin in α granules and express it on the cell surface during activation (94). P-selectin plays a critical role in mediating platelet-leukocyte adhesion, and is therefore implicated in both thrombosis and inflammation (95). Antiplatelet effects of P-selectin inhibition have been recognized by investigations using small molecules or monoclonal antibodies. PSI697, a small molecule P-selectin antagonist, reduced both arterial and venous thrombosis in animals (96), while THCMA, a nanomolecule, was shown to reduce venous thrombosis without inducing bleeding in animal models (97). Crizanlizumab, a humanized IgG2 monoclonal antibody, was approved in 2019 for patients with sickle cell disease with vaso-occlusive symptoms (98). Inclacumab, a monoclonal antibody that reduces myocardial damage in NSTEMI patients treated with PCI without bleeding complications, is currently being investigated in a Phase III trial (99).
(3) Phosphoinositide 3-kinase β (PI3Kβ) targeting: PI3K signaling in platelets plays a major role in the formation of stable shear-dependent αIIbβ3 platelet adhesion (100). Platelets express class I and class II PI3Ks and among these, the class I PI3Kβ plays an important role in platelet activation responses triggered by the P2Y12 activation, GPVI ligation, and by αIIbβ3 outside-in signaling (100). Several PI3Kβ antagonists have been studied and shown to exhibit strong antithrombotic effects without significantly affecting hemostasis (101), including a peptide inhibitor TGX-221, which are consistent with the phenotype of PI3Kβ knockout mice (102). AZD6482, a selective inhibitor of PI3Kβ that blocks its interaction with ATP, is currently in a Phase IIa study (103). MIPS-9922, another selective inhibitor of PI3Kβ, showed promise in mouse studies, while berberine and 2v, its derivative, have been investigated and show antiplatelet aggregation effects (104, 105).
(4) Spleen tyrosine kinase (Syk)-targeting: Syk is a non-receptor tyrosine kinase important for immunoreceptor tyrosine-based activation motif (ITAM)-dependent platelet activation and in GPVI-induced platelet activation (106). Inhibition of Syk in a mouse model protects against arterial thrombosis without altering bleeding time (106). Fostamatinib is a first-in-class product and the only Syk inhibitor approved by the FDA, and is indicated for treating immune thrombocytopenia (ITP) (107). It provides additional, mild inhibition of platelet aggregation when combined with aspirin and/or ticagrelor (108).
(5) Tyrosine kinase targeting: Tyrosine kinase inhibitors (TKIs) are widely used as targeted strategies in cancer treatment, with many tyrosine kinases being highly expressed in platelets. Indeed, many TKIs are associated with suppression of platelet activation and mild bleeding. Ibrutinib, a Bruton tyrosine kinase irreversible inhibitor, was shown to reduce platelet adhesion and thrombus formation in vitro (109, 110). However, due to the ubiquitous expression of tyrosine kinases throughout the body, off-target effects might be inevitable.
(6) Protein disulfide isomerase (PDI)-targeting: PDI catalyzes disulfide bond formation and cleavage and acts as chaperone of protein folding (111). PDI is expressed on the surface of resting platelets and secreted upon activation (111). Platelet PDI was shown to regulate thrombus growth without affecting platelet adhesion or fibrin generation in an arterial injury mouse model. Anti-PDI antibodies and bacitracin, a nonselective PDI inhibitor, reduced thrombus formation and fibrin generation (112). As PDI is highly ubiquitous in tissue distribution, and furthermore, available PDI inhibitors are non-selective and often cytotoxic, off target effects remain a challenge in drug development. Recently, however, isoquercetin and myricetin, members of the flavonoid family, have gained interest and are being investigated as antiplatelet agents, with promising results in a Phase II clinical trial for the former (113, 114).
(7) 12-Lipoxygenase (12-LOX) targeting: 12-LOX is an oxygenase predominantly expressed in platelets, participates in arachidonic acid metabolism, producing downstream eicosanoids which are involved in platelet activation and proinflammatory response. In platelets, 12-LOX plays a role in αIIbβ3 activation and FcγRIIa-, PAR4-, and GPVI-mediated platelet activation (115, 116). ML355 is a potent and selective 12-LOX inhibitor, which has been investigated as an antiplatelet and shown promising results. ML355 inhibited platelet adhesion and thrombus formation under flow in vitro, and similarly reduced thrombus formation in mice without significantly affecting normal hemostasis function (117).
To summarize, several novel approaches to antiplatelet therapy have been eagerly pursued in the last few years, based on our increasing understanding of the mechanisms regulating platelet functions in hemostasis and thrombosis, with many offering promising preclinical results. Although the complete elimination of bleeding risk will probably remain elusive for a while, one or more agents directed against novel targets currently on clinical trial may accomplish this in the not-so-distant future.
Secondary hemostasis (blood coagulation) (Figure 5) is the process of generating thrombin to convert soluble fibrinogen into insoluble fibrin, in order to stabilize the platelet clot (118). Thrombin is produced through two pathways (termed the extrinsic and intrinsic, or contact, pathways), which culminate in the activation of the serine protease factor Xa (FXa), which then activates thrombin. Classic anticoagulation therapy (Table 1) has focused on inhibition of the final two enzymes in this pathway, factor Xa (FXa) and thrombin, or on a broader inhibition of this pathway. For example, warfarin reduces the availability of Vitamin K, Vitamin K is required to produce γ-carboxylated glutamic acid (Gla) residues, and Gla residues are necessary for the proper folding of multiple coagulation enzymes (119). As a result, factor VIIa (FVIIa), factor IXa (FIXa), FXa, and thrombin are all reduced in warfarin-treated individuals. Heparins have a similarly broad effect, and work by promoting the inhibition of coagulation serine proteases (primarily FXa and thrombin) by plasma antithrombin (120). Due to their potency, warfarin and heparins must be closely monitored. Heparins may be rapidly reversed by protamine, while warfarin is reversed by Vitamin K.
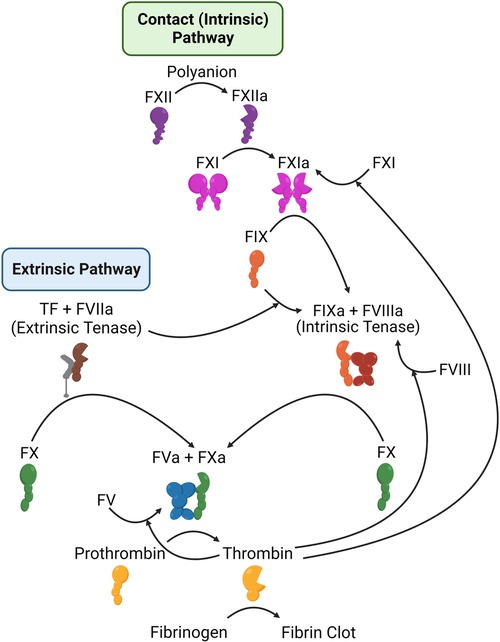
Figure 5. Schematic representation of the coagulation system. Shown are the components of the extrinsic (left) and intrinsic or contact (right) coagulation pathways, along with the mechanisms by which thrombin feeds back to amplify its own activation.
Direct oral anticoagulants (DOACs) have become popular alternatives due to their favorable pharmacological profiles (121). These differ from previous strategies in that they work by specifically inhibiting either FXa (rivaroxaban, apixaban, edoxaban, betrixaban) or thrombin (dabigatran), while leaving other parts of the coagulation system unimpaired.
Though they are safer than warfarin and heparin, direct FXa and thrombin inhibitors do carry risk of bleeding, including potentially fatal bleeds. Therefore, much of the research in this field has shifted towards the development of rapid reversal strategies. One advantage of the broader therapeutics (warfarin and heparins) has been the ease of reversal. Warfarin may be reversed in the short-term by replacement of the missing Vitamin K-dependent proteins, such as through administration of prothrombin complex concentrations or plasma transfusion (122–126) or in the long-term through administration of Vitamin K or by halting the warfarin (125, 127). Negatively-charged heparin is rapidly reversed by positively-charged protamine, though excess protamine itself has anticoagulant and other hazardous effects, so careful dosing is required (128, 129). Other broad anticoagulant reversal agents are also in development. For example, Meijers et al. (130) described OKL-1111, a cyclodextrin-based compound that broadly reverses antithrombotics, including every class of antiplatelet or anticoagulant medication that the authors tested, in a rat tail bleed model. The mechanism of action of OKL-1111 is unknown, though it has direct procoagulant activity in plasma.
One area of recent research has been the creation of rapid reversal agents that are specific for the direct FXa and thrombin inhibitors, which have come in multiple forms:
(1) Monoclonal antibodies: Idarucizumab is a humanized monoclonal antibody Fab fragment targeted against dabigatran, and is able to reverse dabigatran anticoagulant activity within minutes of infusion (131). In addition, idarucizumab does not alter thrombin generation in the absence of dabigatran, suggesting that it does not pose a risk of over-correcting the hemostatic system and promoting thrombosis (132, 133).
(2) FXa mimetics: Andexanet alfa is a recombinant version of FXa in which the membrane-binding Gla domain has been removed and the active site serine residue mutated. Thus, andexanet alfa is catalytically inactive but still able to bind FXa inhibitors, either pharmacologic or endogenous (134). Unlike idarucizumab, andexanet alfa does appear to have some risk of overcorrection, as a recent safety study reported that ∼10% of patients experienced at least one thrombotic event within 30 days of receiving andexanet alfa, unless anticoagulation therapy was restarted (135). This is consistent with in vitro work, which indicated that andexanet alfa can increase plasma thrombin generation in the absence of FXa inhibition (136, 137). This effect may be due to the interaction of andexanet alfa with plasma FXa inhibitors, such as antithrombin and tissue factor pathway inhibitor (TFPI), and their neutralization.As an alternative approach, Thalji et al. described FXaI16l (138). This single amino acid substitution destabilizes the FXa active site enough that it is resistant to inactivation by plasma inhibitors, such as antithrombin and TFPI, and effectively reverses both rivaroxaban and dabigatran in murine models. FXaI16l has been safe in Phase I and Phase Ib clinical trials (139, 140). Similarly, Verhoef et al. described a FXa homolog found in the venom of Pseudonaja textilis (the eastern brown snake), which is resistant to direct FXa inhibitors (141). They utilized this homolog as the basis to design human FXa mutants, with alterations in the substrate binding pocket, which exhibit similar resistance properties. One of these compounds, termed VMX-C001, recently completed a Phase I clinical trial.
(3) Small molecules: Ciraparantag is a small peptide mimetic, which was originally developed to reverse heparins, but found to also reverse DOACs (both thrombin and FXa inhibitors) (142). Ciraparantag functions by directly binding the thrombin and FXa inhibitors and blocking interactions with their respective target proteases. It safely reversed the anticoagulant activities of edoxaban, apixaban, and rivaroxaban in healthy subjects (143, 144).
As summarized above, existing therapies, all of which carry a risk of major bleeding, all target FXa and/or thrombin, either by preventing their production (warfarin) or by blocking their activity. FXa and thrombin lie at the end of the coagulation pathway and are both critical for hemostasis. Deficiency in either is very rare and associated with bleeding. Similarly, homozygous deficiency of either is incompatible with life in mice (145–147), and no people have been described with total loss of either protein. To identify coagulation targets that would not adversely impact hemostasis, the focus has shifted instead to the beginning of coagulation. Coagulation can be initiated in two ways. First, the extrinsic pathway, consisting of tissue factor and FVIIa, is thought to be the primary initiator of hemostasis. Second, the intrinsic (or contact) pathway is initiated by contact with negatively charged surfaces, leading to factor XII (FXII) activation to FXIIa Meanwhile, high molecular weight kininogen (HK) acts as a bridge, bringing factor XI (FXI) and prekallikrein (PK) close to FXII. Within this cyclic system, FXIIa activates HK-bound PK, leading to the formation of kallikrein, which in turn activates additional FXII. Furthermore, FXIIa also activates FXI in a manner dependent on HK. The subsequent activation of FXIa contributes to the intrinsic coagulation pathway by activating factor IX (FIX), ultimately leading to the generation of thrombin (Figure 5). FXI can be activated independent of the contact system, through a feedback mechanism by thrombin (148, 149), and is thought to be necessary for the amplification of thrombin generation and stabilization of the clot. This may explain why FXII deficiency is not associated with bleeding risk, but FXI deficiency is.
In the laboratory, the contact pathway is activated by exposure of blood/plasma to negatively charged surfaces, such as glass or kaolin, a form of clay. For decades, the physiologic activator of the contact pathway has been unclear. Soil may be a physiologic activator, as the contact pathway may have evolved to recognize and respond to soil that enters the blood through an open wound (150). Many groups, though, have focused on biological polyanions, such as DNA and polyphosphates as potential activators of the coagulation system. DNA is released from activated inflammatory cells in the form of neutrophil extracellular traps (NETs) or the homologous monocyte extracellular traps (METs). NETs and METs are complexes of DNA, histones, and associated proteins, all of which can influence coagulation and platelet function. NETs promote thrombin activation in vitro. While there are likely many mechanisms involved, this activity is at least partially dependent on FXII and FXI. In addition, NETs have been shown to bind FXII and promote its activation, and to promote the thrombin-mediated activation of FXI. METs are less studied, compared to NETs, but likely have similar properties.
Polyphosphates are stored within the dense granules of circulating platelets and released upon platelet activation. Similar to NETs, polyphosphates promote FXI activation by thrombin and FXII activation, and promote thrombin generation in a FXII-dependent manner. Also similar to NETs, polyphosphates have contact pathway-independent effects on the coagulation system, such as inhibition of the anticoagulant tissue factor pathway inhibitor and promotion of factor V activation by thrombin. Here, we will discuss strategies that are in development or proposed to target components of the contact pathway, and its activators, as novel anticoagulant agents.
Current evidence favors FXI's role in thrombosis over FXII, with limited support for FXII's involvement in human thrombosis (151). Targeting FXII appears safer as it poses no significant bleeding risk. Solely targeting FXII may not be optimal, however, as thrombin from the extrinsic pathway can activate FXI independent of FXII (148, 149). In contrast, FXI inhibition may cause bleeding, especially in individuals with severe congenital FXI deficiency. FXI inhibition lacks bypass potential but carries off-target risks, like modulating inflammation via inhibiting bradykinin generation, as recently reviewed by Gigante and Ten Cate (152).
Multiple strategies have been developed to target FXII and FXI (Figure 6, Table 2):
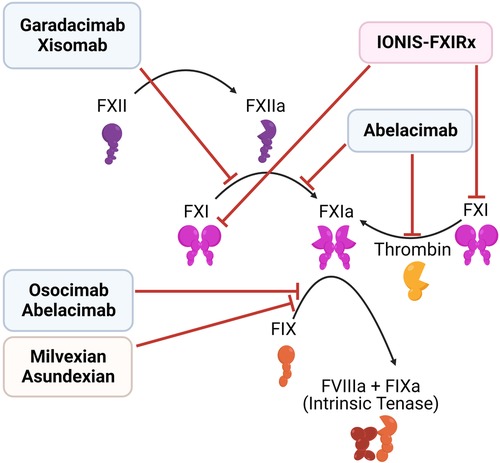
Figure 6. Inhibitors of the contact pathway. Shown are the targets of newly developed inhibitors of the contact system, including antisense oligonucleotides (red), monoclonal antibodies (blue), and small molecule inhibitors (orange).
(1) Antisense oligonucleotides (ASOs): Liver-directed ASOs can be used to selectively knockdown the expression of targeted proteins. ASOs which target components of the contact pathway were first reported in 2010, and have been shown to reduce thrombosis in multiple animal models, with low bleeding risk (153–156). IONIS-FXIRx (also called BAY-2306001, FXI ASO, ISIS 404071, ISIS-416858, and ISIS-FXIRX) was the first of these agents to be tested in humans, when it was tested in a cohort of patients undergoing total knee arthroplasty (157). This subcutaneous FXI-directed ASO reduced FXI levels effectively and lowered VTE risk in patients compared to enoxaparin, with no significant increase in bleeding.
(2) Monoclonal antibodies: Another approach involves parenteral administration of monoclonal antibodies that block clotting factor activation and/or activity. Several antibodies targeting FXI are in development, with differing modes of action: (a) Abelacimab binds the active site of FXI/XIa, locking it in an inactive zymogen-like state. As such, it both prevents the activation of FXI by thrombin or FXII and blocks the activity of FXIa (158, 159). (b) Osocimab binds an allosteric site on FXIa, blocking its activation of FIX (160, 161). Xisomab, inhibits the activation of FXI by FXIIa (162). Garadacimab (formerly CSL 312) is one of the few agents targeting FXII as a monoclonal antibody (163, 164). Unlike ASOs, monoclonal antibodies can be used both in acute and chronic settings, as they have a faster response. ASOs require uptake in the liver and clearance of the existing plasma protein, before their effect may be realized (151).
(3) Small molecule inhibitors: There are both orally and parenterally available small-molecule inhibitors of FXI, such as milvexian (BMS-986177/JNJ-70033093) and asundexian (BAY 2433334), in development. In a Phase II trial of 1242 knee arthroplasty patients, milvexian (25–200 mg) twice daily effectively prevented venous thromboembolism with lower bleeding risk than enoxaparin. Once daily milvexian also showed efficacy in VTE prevention (165). Milvexian is now being assessed in a Phase III trial. Similarly, asundexian at doses of 20 mg and 50 mg once daily demonstrated near-complete in vivo FXIa inhibition. These dosages resulted in reduced rates of bleeding, and similar rates of thrombosis, compared to the standard dosing of apixaban (166), suggesting that asundexian is a safer alternative. However, there are concerns about the efficacy of FXIa inhibition, based on these results (152).
(4) Targeting activators of the contact system: In addition to directly targeting the contact pathway, it may also be possible to target components that activate this pathway, such as NETs and polyphosphates. As recently reviewed elsewhere (167), multiple therapeutic strategies are in pre-clinical or clinical stages of development to target NETs, or the generation of NETs. These include approaches to directly or indirectly inhibit the release of nuclear content from neutrophils, the degradation of DNA by DNAse, and the targeting of NET-associated proteins, such as myeloperoxidase. In addition, Baron et al. recently reported that Selinexor, a first-in-class inhibitor of nuclear export approved for use in multiple myeloma patients, effectively prevents NET release in vitro (168). Selinexor may not be an effective antithrombotic, however, as it is associated with reduced platelet count, due to an off-target effect on megakaryocytes (169).Similarly, polyphosphates may be targeted to prevent thrombosis. La et al. developed a polycationic compounds (termed macromolecular polyanion inhibitors, MPIs) that bind and neutralizes polyphosphates (170). MPIs bind to platelet-released polyphosphates, and reduce thrombus formation in a cremaster muscle laser injury model in mice. In contrast, they did not impair hemostasis in a tail bleeding model.
To summarize, the contact pathway of coagulation is an appealing target for developing safer anticoagulants due to its potential to reduce thrombosis without increasing bleeding risk. Selective FXI or FXII inhibitors have shown promise in preclinical and early clinical studies, and agents that target activators of the contact pathway, such as NETs and polyphosphates are also in development.
For decades, the “holy grail” of antithrombotic treatment has been a drug that prevents thrombus formation but does not carry a concomitant bleeding risk. This has been a very difficult goal to achieve, as bleeding and thrombosis are closely related processes. Platelets and coagulation factors are required for both, and any compound that broadly inhibits either process has historically carried a bleeding risk. However, exciting progress has been made in recent years, and we may be closer than ever to obtaining the grail. Rapid reversal agents for DOACs have been developed, safer alternatives have been developed to existing antiplatelet and anticoagulant therapeutics, and new targets have been identified that appear to be more specifically involved in the thrombotic process. This is an exciting time for antithrombotic therapy, as we wait to see how effective these new targets and treatment strategies are in clinical trials.
MS: Writing – original draft, Writing – review & editing. SS: Writing – original draft, Writing – review & editing. JW: Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We thank Dlovan D Mahmood for careful proofreading of the manuscript.
JW has an investigator-initiated grant through Pfizer, Inc., which is unrelated to this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and platelets: an update. Eur Heart J. (2016) 38(11):785–91. doi: 10.1093/eurheartj/ehw550
2. Berndt MC, Metharom P, Andrews RK. Primary haemostasis: newer insights. Haemophilia. (2014) 20(Suppl 4):15–22. doi: 10.1111/hae.12427
3. Wagner DD, Burger PC. Platelets in inflammation and thrombosis. Arterioscl Thromb Vasc Biol. (2003) 23(12):2131–7. doi: 10.1161/01.ATV.0000095974.95122.EC
4. Majithia A, Bhatt DL. Novel antiplatelet therapies for atherothrombotic diseases. Arterioscler, Thromb, Vasc Biol. (2019) 39(4):546–57. doi: 10.1161/ATVBAHA.118.310955
5. Tscharre M, Michelson AD, Gremmel T. Novel antiplatelet agents in cardiovascular disease. J Cardiovasc Pharmacol Ther. (2020) 25(3):191–200. doi: 10.1177/1074248419899314
6. Jourdi G, Lordkipanidzé M, Philippe A, Bachelot-Loza C, Gaussem P. Current and novel antiplatelet therapies for the treatment of cardiovascular diseases. Int J Mol Sci. (2021) 22(23):13079. doi: 10.3390/ijms222313079
7. Gresele P, Momi S. Novel approaches to antiplatelet therapy. Biochem Pharmacol. (2022) 206:115297. doi: 10.1016/j.bcp.2022.115297
8. Miner J, Hoffhines A. The discovery of aspirin’s antithrombotic effects. Tex Heart Inst J. (2007) 34(2):179–86.17622365
9. Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J Clin Invest. (1975) 56(3):624–32. doi: 10.1172/JCI108132
10. Cipollone F, Rocca B, Patrono C. Cyclooxygenase-2 expression and inhibition in atherothrombosis. Arterioscler, Thromb, Vasc Biol. (2004) 24(2):246–55. doi: 10.1161/01.ATV.0000104005.92603.f2
11. Patrono C, García Rodríguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. (2005) 353(22):2373–83. doi: 10.1056/NEJMra052717
12. Undas A, Brummel-Ziedins KE, Mann KG. Antithrombotic properties of aspirin and resistance to aspirin: beyond strictly antiplatelet actions. Blood. (2007) 109(6):2285–92. doi: 10.1182/blood-2006-01-010645
13. Farid NA, Kurihara A, Wrighton SA. Metabolism and disposition of the thienopyridine antiplatelet drugs ticlopidine, clopidogrel, and prasugrel in humans. J Clin Pharmacol. (2010) 50(2):126–42. doi: 10.1177/0091270009343005
14. Sangkuhl K, Klein TE, Altman RB. Clopidogrel pathway. Pharmacogenet Genomics. (2010) 20(7):463–5. doi: 10.1097/FPC.0b013e3283385420
15. Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. (2001) 409(6817):202–7. doi: 10.1038/35051599
16. Wallentin L. P2y(12) inhibitors: differences in properties and mechanisms of action and potential consequences for clinical use. Eur Heart J. (2009) 30(16):1964–77. doi: 10.1093/eurheartj/ehp296
17. Sharma R, Kumar P, Prashanth SP, Belagali Y. Dual antiplatelet therapy in coronary artery disease. Cardiol Ther. (2020) 9(2):349–61. doi: 10.1007/s40119-020-00197-0
18. Castrichini M, Luzum JA, Pereira N. Pharmacogenetics of antiplatelet therapy. Annu Rev Pharmacol Toxicol. (2023) 63:211–29. doi: 10.1146/annurev-pharmtox-051921-092701
19. van den Broek WWA, Mani N, Azzahhafi J, Ten Berg JM. CYP2C9 Polymorphisms and the risk of cardiovascular events in patients treated with clopidogrel: combined data from the POPular genetics and POPular AGE trials. Am J Cardiovasc Drugs. (2023) 23(2):165–72. doi: 10.1007/s40256-022-00565-2
20. Dobesh PP, Varnado S, Doyle M. Antiplatelet agents in cardiology: a report on aspirin, clopidogrel, prasugrel, and ticagrelor. Curr Pharm Des. (2016) 22(13):1918–32. doi: 10.2174/1381612822666151208120106
21. Gresele P, Momi S, Falcinelli E. Anti-platelet therapy: phosphodiesterase inhibitors. Br J Clin Pharmacol. (2011) 72(4):634–46. doi: 10.1111/j.1365-2125.2011.04034.x
22. Halkes PH, van Gijn J, Kappelle LJ, Koudstaal PJ, Algra A. Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT): randomised controlled trial. Lancet (London, England). (2006) 367(9523):1665–73. doi: 10.1016/S0140-6736(06)68734-5
23. Halkes PH, Gray LJ, Bath PM, Diener HC, Guiraud-Chaumeil B, Yatsu FM, et al. Dipyridamole plus aspirin versus aspirin alone in secondary prevention after TIA or stroke: a meta-analysis by risk. J Neurol Neurosurg Psychiatr. (2008) 79(11):1218–23. doi: 10.1136/jnnp.2008.143875
24. De Luca G, Savonitto S, van't Hof AW, Suryapranata H. Platelet GP IIb-IIIa receptor antagonists in primary angioplasty: back to the future. Drugs. (2015) 75(11):1229–53. doi: 10.1007/s40265-015-0425-7
25. Franchi F, Rollini F, Park Y, Angiolillo DJ. Platelet thrombin receptor antagonism with vorapaxar: pharmacology and clinical trial development. Future Cardiol. (2015) 11(5):547–64. doi: 10.2217/fca.15.50
26. Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. (2012) 366(15):1404–13. doi: 10.1056/NEJMoa1200933
27. Bonaca MP, Scirica BM, Creager MA, Olin J, Bounameaux H, Dellborg M, et al. Vorapaxar in patients with peripheral artery disease: results from TRA2{degrees}P-TIMI 50. Circulation. (2013) 127(14):1522–9.e1–6. doi: 10.1161/CIRCULATIONAHA.112.000679
28. Storey RF, Gurbel PA, Ten Berg J, Bernaud C, Dangas GD, Frenoux JM, et al. Pharmacodynamics, pharmacokinetics, and safety of single-dose subcutaneous administration of selatogrel, a novel P2Y12 receptor antagonist, in patients with chronic coronary syndromes. Eur Heart J. (2020) 41(33):3132–40. doi: 10.1093/eurheartj/ehz807
29. Sinnaeve P, Fahrni G, Schelfaut D, Spirito A, Mueller C, Frenoux JM, et al. Subcutaneous selatogrel inhibits platelet aggregation in patients with acute myocardial infarction. J Am Coll Cardiol. (2020) 75(20):2588–97. doi: 10.1016/j.jacc.2020.03.059
30. Bach P, Antonsson T, Bylund R, Björkman JA, Österlund K, Giordanetto F, et al. Lead optimization of ethyl 6-aminonicotinate acyl sulfonamides as antagonists of the P2Y12 receptor. Separation of the antithrombotic effect and bleeding for candidate drug AZD1283. J Med Chem. (2013) 56(17):7015–24. doi: 10.1021/jm400820m
31. Delesque-Touchard N, Pflieger AM, Bonnet-Lignon S, Millet L, Salel V, Boldron C, et al. SAR216471, An alternative to the use of currently available P2Y₁₂ receptor inhibitors? Thromb Res. (2014) 134(3):693–703. doi: 10.1016/j.thromres.2014.06.034
32. Bougie DW, Wilker PR, Wuitschick ED, Curtis BR, Malik M, Levine S, et al. Acute thrombocytopenia after treatment with tirofiban or eptifibatide is associated with antibodies specific for ligand-occupied GPIIb/IIIa. Blood. (2002) 100(6):2071–6. doi: 10.1182/blood.V100.6.2071
33. Bassler N, Loeffler C, Mangin P, Yuan Y, Schwarz M, Hagemeyer CE, et al. A mechanistic model for paradoxical platelet activation by ligand-mimetic alphaIIb beta3 (GPIIb/IIIa) antagonists. Arterioscler, Thromb, Vasc Biol. (2007) 27(3):e9–15. doi: 10.1161/01.ATV.0000255307.65939.59
34. Li J, Vootukuri S, Shang Y, Negri A, Jiang JK, Nedelman M, et al. RUC-4: a novel αIIbβ3 antagonist for prehospital therapy of myocardial infarction. Arterioscler, Thromb, Vasc Biol. (2014) 34(10):2321–9. doi: 10.1161/ATVBAHA.114.303724
35. Zhu J, Choi WS, McCoy JG, Negri A, Zhu J, Naini S, et al. Structure-guided design of a high-affinity platelet integrin αIIbβ3 receptor antagonist that disrupts mg²+ binding to the MIDAS. Sci Transl Med. (2012) 4(125):125ra32. doi: 10.1126/scitranslmed.3003576
36. Kereiakes DJ, Henry TD, DeMaria AN, Bentur O, Carlson M, Seng Yue C, et al. First human use of RUC-4: a nonactivating second-generation small-molecule platelet glycoprotein IIb/IIIa (integrin αIIbβ3) inhibitor designed for subcutaneous point-of-care treatment of ST-segment-elevation myocardial infarction. J Am Heart Assoc. (2020) 9(17):e016552. doi: 10.1161/JAHA.120.016552
37. Gkourogianni AV, Kiouptsi K, Koloka V, Moussis V, Tsikaris V, Bachelot-Loza C, et al. Synergistic effect of peptide inhibitors derived from the extracellular and intracellular domain of α(IIb) subunit of integrin α(IIb)β(3) on platelet activation and aggregation. Platelets. (2018) 29(1):34–40. doi: 10.1080/09537104.2017.1293804
38. Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med. (2002) 8(10):1161–5. doi: 10.1038/nm760
39. Flaumenhaft R, De Ceunynck K. Targeting PAR1: now what? Trends Pharmacol Sci. (2017) 38(8):701–16. doi: 10.1016/j.tips.2017.05.001
40. Kuliopulos A, Gurbel PA, Rade JJ, Kimmelstiel CD, Turner SE, Bliden KP, et al. PAR1 (protease-activated receptor 1) pepducin therapy targeting myocardial necrosis in coronary artery disease and acute coronary syndrome patients undergoing cardiac catheterization: a randomized, placebo-controlled, phase 2 study. Arterioscler, Thromb, Vasc Biol. (2020) 40(12):2990–3003. doi: 10.1161/ATVBAHA.120.315168
41. Arachiche A, Mumaw MM, de la Fuente M, Nieman MT. Protease-activated receptor 1 (PAR1) and PAR4 heterodimers are required for PAR1-enhanced cleavage of PAR4 by alpha-thrombin. J Biol Chem. (2013) 288(45):32553–62. doi: 10.1074/jbc.M113.472373
42. Priestley ES, Banville J, Deon D, Dubé L, Gagnon M, Guy J, et al. Discovery of two novel antiplatelet clinical candidates (BMS-986120 and BMS-986141) that antagonize protease-activated receptor 4. J Med Chem. (2022) 65(13):8843–54. doi: 10.1021/acs.jmedchem.2c00359
43. Wilson SJ, Ismat FA, Wang Z, Cerra M, Narayan H, Raftis J, et al. PAR4 (protease-activated receptor 4) antagonism with BMS-986120 inhibits human ex vivo thrombus formation. Arterioscler, Thromb, Vasc Biol. (2018) 38(2):448–56. doi: 10.1161/ATVBAHA.117.310104
44. Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, et al. Blocking the protease-activated receptor 1-4 heterodimer in platelet-mediated thrombosis. Circulation. (2006) 113(9):1244–54. doi: 10.1161/CIRCULATIONAHA.105.587758
45. Chiang YC, Wu YS, Kang YF, Wang HC, Tsai MC, Wu CC. 3,5,2′,4′-Tetramethoxystilbene, a fully methylated resveratrol analog, prevents platelet aggregation and thrombus formation by targeting the protease-activated receptor 4 pathway. Chem-Biol Interact. (2022) 357:109889. doi: 10.1016/j.cbi.2022.109889
46. Leon C, Hechler B, Vial C, Leray C, Cazenave JP, Gachet C. The P2Y1 receptor is an ADP receptor antagonized by ATP and expressed in platelets and megakaryoblastic cells. FEBS Lett. (1997) 403(1):26–30. doi: 10.1016/S0014-5793(97)00022-7
47. Hechler B, Nonne C, Roh EJ, Cattaneo M, Cazenave JP, Lanza F, et al. MRS2500 [2-iodo-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate], a potent, selective, and stable antagonist of the platelet P2Y1 receptor with strong antithrombotic activity in mice. J Pharmacol Exp Ther. (2006) 316(2):556–63. doi: 10.1124/jpet.105.094037
48. Wong PC, Watson C, Crain EJ. The P2Y1 receptor antagonist MRS2500 prevents carotid artery thrombosis in cynomolgus monkeys. J Thromb Thrombolysis. (2016) 41(3):514–21. doi: 10.1007/s11239-015-1302-7
49. Gremmel T, Yanachkov IB, Yanachkova MI, Wright GE, Wider J, Undyala VV, et al. Synergistic inhibition of both P2Y1 and P2Y12 adenosine diphosphate receptors as novel approach to rapidly attenuate platelet-mediated thrombosis. Arterioscler, Thromb, Vasc Biol. (2016) 36(3):501–9. doi: 10.1161/ATVBAHA.115.306885
50. Moroi M, Jung SM, Okuma M, Shinmyozu K. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J Clin Invest. (1989) 84(5):1440–5. doi: 10.1172/JCI114318
51. Lecut C, Feeney LA, Kingsbury G, Hopkins J, Lanza F, Gachet C, et al. Human platelet glycoprotein VI function is antagonized by monoclonal antibody-derived fab fragments. J Thromb Haemost. (2003) 1(12):2653–62. doi: 10.1111/j.1538-7836.2003.00495.x
52. Lebozec K, Jandrot-Perrus M, Avenard G, Favre-Bulle O, Billiald P. Design, development and characterization of ACT017, a humanized fab that blocks platelet’s glycoprotein VI function without causing bleeding risks. mAbs. (2017) 9(6):945–58. doi: 10.1080/19420862.2017.1336592
53. Florian P, Wonerow P, Harder S, Kuczka K, Dubar M, Graff J. Anti-GPVI fab SAR264565 effectively blocks GPVI function in ex vivo human platelets under arterial shear in a perfusion chamber. Eur J Clin Pharmacol. (2017) 73(8):949–56. doi: 10.1007/s00228-017-2264-9
54. Grüner S, Prostredna M, Koch M, Miura Y, Schulte V, Jung SM, et al. Relative antithrombotic effect of soluble GPVI dimer compared with anti-GPVI antibodies in mice. Blood. (2005) 105(4):1492–9. doi: 10.1182/blood-2004-06-2391
55. Massberg S, Konrad I, Bültmann A, Schulz C, Münch G, Peluso M, et al. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. (2004) 18(2):397–9. doi: 10.1096/fj.03-0464fje
56. Mayer K, Hein-Rothweiler R, Schüpke S, Janisch M, Bernlochner I, Ndrepepa G, et al. Efficacy and safety of revacept, a novel lesion-directed competitive antagonist to platelet glycoprotein VI, in patients undergoing elective percutaneous coronary intervention for stable ischemic heart disease: the randomized, double-blind, placebo-controlled ISAR-PLASTER phase 2 trial. JAMA Cardiol. (2021) 6(7):753–61. doi: 10.1001/jamacardio.2021.0475
57. Uphaus T, Richards T, Weimar C, Neugebauer H, Poli S, Weissenborn K, et al. Revacept, an inhibitor of platelet adhesion in symptomatic carotid stenosis: a multicenter randomized phase II trial. Stroke. (2022) 53(9):2718–29. doi: 10.1161/STROKEAHA.121.037006
58. Olğaç S, Olğaç A, Yenicesu İ, Ozkan Y. Identification of novel antiplatelet agents by targeting glycoprotein VI: a combined virtual screening study. Bioorg Chem. (2022) 121:105661. doi: 10.1016/j.bioorg.2022.105661
59. Jooss NJ, Smith CW, Slater A, Montague SJ, Di Y, O’Shea C, et al. Anti-GPVI nanobody blocks collagen- and atherosclerotic plaque-induced GPVI clustering, signaling, and thrombus formation. J Thromb Haemost. (2022) 20(11):2617–31. doi: 10.1111/jth.15836
60. Shen Y, Romo GM, Dong JF, Schade A, McIntire LV, Kenny D, et al. Requirement of leucine-rich repeats of glycoprotein (GP) ibalpha for shear-dependent and static binding of von willebrand factor to the platelet membrane GP ib-IX-V complex. Blood. (2000) 95(3):903–10. doi: 10.1182/blood.V95.3.903.003k37_903_910
61. Gresele P, Momi S. Inhibitors of the interaction between von willebrand factor and platelet GPIb/IX/V. Handb Exp Pharmacol. (2012) 210:287–309. doi: 10.1007/978-3-642-29423-5_12
62. Cauwenberghs N, Meiring M, Vauterin S, van Wyk V, Lamprecht S, Roodt JP, et al. Antithrombotic effect of platelet glycoprotein ib-blocking monoclonal antibody fab fragments in nonhuman primates. Arterioscler, Thromb, Vasc Biol. (2000) 20(5):1347–53. doi: 10.1161/01.ATV.20.5.1347
63. Fontayne A, Meiring M, Lamprecht S, Roodt J, Demarsin E, Barbeaux P, et al. The humanized anti-glycoprotein ib monoclonal antibody h6B4-fab is a potent and safe antithrombotic in a high shear arterial thrombosis model in baboons. Thromb Haemostasis. (2008) 100(4):670–7. doi: 10.1160/TH08-02-0073
64. Lei X, Reheman A, Hou Y, Zhou H, Wang Y, Marshall AH, et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb Haemostasis. (2014) 111(2):279–89. doi: 10.1160/TH13-06-0490
65. Li TT, Fan ML, Hou SX, Li XY, Barry DM, Jin H, et al. A novel snake venom-derived GPIb antagonist, anfibatide, protects mice from acute experimental ischaemic stroke and reperfusion injury. Br J Pharmacol. (2015) 172(15):3904–16. doi: 10.1111/bph.13178
66. Li BX, Dai X, Xu XR, Adili R, Neves MAD, Lei X, et al. In vitro assessment and phase I randomized clinical trial of anfibatide a snake venom derived anti-thrombotic agent targeting human platelet GPIbα. Sci Rep. (2021) 11(1):11663. doi: 10.1038/s41598-021-91165-8
67. Sanrattana W, Smits S, Barendrecht AD, van Kleef ND, El Otmani H, Zivkovic M, et al. Targeted SERPIN (TaSER): a dual-action antithrombotic agent that targets platelets for SERPIN delivery. J Thromb Haemost. (2022) 20(2):353–65. doi: 10.1111/jth.15554
68. Kageyama S, Yamamoto H, Nakazawa H, Matsushita J, Kouyama T, Gonsho A, et al. Pharmacokinetics and pharmacodynamics of AJW200, a humanized monoclonal antibody to von willebrand factor, in monkeys. Arterioscler, Thromb, Vasc Biol. (2002) 22(1):187–92. doi: 10.1161/hq0102.101520
69. Wu D, Vanhoorelbeke K, Cauwenberghs N, Meiring M, Depraetere H, Kotze HF, et al. Inhibition of the von willebrand (VWF)-collagen interaction by an antihuman VWF monoclonal antibody results in abolition of in vivo arterial platelet thrombus formation in baboons. Blood. (2002) 99(10):3623–8. doi: 10.1182/blood.V99.10.3623
70. Ulrichts H, Silence K, Schoolmeester A, de Jaegere P, Rossenu S, Roodt J, et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood. (2011) 118(3):757–65. doi: 10.1182/blood-2010-11-317859
71. Momi S, Tantucci M, Van Roy M, Ulrichts H, Ricci G, Gresele P. Reperfusion of cerebral artery thrombosis by the GPIb-VWF blockade with the nanobody ALX-0081 reduces brain infarct size in Guinea pigs. Blood. (2013) 121(25):5088–97. doi: 10.1182/blood-2012-11-464545
72. Scully M, Cataland SR, Peyvandi F, Coppo P, Knöbl P, Kremer Hovinga JA, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic Purpura. N Engl J Med. (2019) 380(4):335–46. doi: 10.1056/NEJMoa1806311
73. Bartunek J, Barbato E, Heyndrickx G, Vanderheyden M, Wijns W, Holz JB. Novel antiplatelet agents: aLX-0081, a nanobody directed towards von willebrand factor. J Cardiovasc Transl Res. (2013) 6(3):355–63. doi: 10.1007/s12265-012-9435-y
74. Diener JL, Daniel Lagassé HA, Duerschmied D, Merhi Y, Tanguay JF, Hutabarat R, et al. Inhibition of von willebrand factor-mediated platelet activation and thrombosis by the anti-von willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost. (2009) 7(7):1155–62. doi: 10.1111/j.1538-7836.2009.03459.x
75. Siller-Matula JM, Merhi Y, Tanguay JF, Duerschmied D, Wagner DD, McGinness KE, et al. ARC15105 Is a potent antagonist of von willebrand factor mediated platelet activation and adhesion. Arterioscler, Thromb, Vasc Biol. (2012) 32(4):902–9. doi: 10.1161/ATVBAHA.111.237529
76. Kovacevic KD, Greisenegger S, Langer A, Gelbenegger G, Buchtele N, Pabinger I, et al. The aptamer BT200 blocks von willebrand factor and platelet function in blood of stroke patients. Sci Rep. (2021) 11(1):3092. doi: 10.1038/s41598-021-82747-7
77. Ay C, Pabinger I, Kovacevic KD, Gelbenegger G, Schörgenhofer C, Quehenberger P, et al. The VWF binding aptamer rondoraptivon pegol increases platelet counts and VWF/FVIII in type 2B von willebrand disease. Blood Adv. (2022) 6(18):5467–76. doi: 10.1182/bloodadvances.2022007805
78. Sakai K, Someya T, Harada K, Yagi H, Matsui T, Matsumoto M. Novel aptamer to von willebrand factor A1 domain (TAGX-0004) shows total inhibition of thrombus formation superior to ARC1779 and comparable to caplacizumab. Haematologica. (2020) 105(11):2631–8. doi: 10.3324/haematol.2019.235549
79. Poulsen SA, Quinn RJ. Adenosine receptors: new opportunities for future drugs. Bioorg Med Chem. (1998) 6(6):619–41. doi: 10.1016/S0968-0896(98)00038-8
80. Wolska N, Rozalski M. Blood platelet adenosine receptors as potential targets for anti-platelet therapy. Int J Mol Sci. (2019) 20(21):5475. doi: 10.3390/ijms20215475
81. Suzuki-Inoue K, Kato Y, Inoue O, Kaneko MK, Mishima K, Yatomi Y, et al. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J Biol Chem. (2007) 282(36):25993–6001. doi: 10.1074/jbc.M702327200
82. Sasaki T, Shirai T, Tsukiji N, Otake S, Tamura S, Ichikawa J, et al. Functional characterization of recombinant snake venom rhodocytin: rhodocytin mutant blocks CLEC-2/podoplanin-dependent platelet aggregation and lung metastasis. J Thromb Haemost. (2018) 16(5):960–72. doi: 10.1111/jth.13987
83. Chang YW, Hsieh PW, Chang YT, Lu MH, Huang TF, Chong KY, et al. Identification of a novel platelet antagonist that binds to CLEC-2 and suppresses podoplanin-induced platelet aggregation and cancer metastasis. Oncotarget. (2015) 6(40):42733–48. doi: 10.18632/oncotarget.5811
84. Shirai T, Inoue O, Tamura S, Tsukiji N, Sasaki T, Endo H, et al. C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J Thromb Haemost. (2017) 15(3):513–25. doi: 10.1111/jth.13604
85. Michal F. D-receptor for serotonin on blood platelets. Nature. (1969) 221(5187):1253–4. doi: 10.1038/2211253a0
86. Mammadova-Bach E, Mauler M, Braun A, Duerschmied D. Autocrine and paracrine regulatory functions of platelet serotonin. Platelets. (2018) 29(6):541–8. doi: 10.1080/09537104.2018.1478072
87. Hara H, Kitajima A, Shimada H, Tamao Y. Antithrombotic effect of MCI-9042, a new antiplatelet agent on experimental thrombosis models. Thromb Haemost. (1991) 66(4):484–8. doi: 10.1055/s-0038-1646443
88. Shinohara Y, Nishimaru K, Sawada T, Terashi A, Handa S, Hirai S, et al. Sarpogrelate-aspirin comparative clinical study for efficacy and safety in secondary prevention of cerebral infarction (S-ACCESS): a randomized, double-blind, aspirin-controlled trial. Stroke. (2008) 39(6):1827–33. doi: 10.1161/STROKEAHA.107.505131
89. Adams JW, Ramirez J, Shi Y, Thomsen W, Frazer J, Morgan M, et al. APD791, 3-methoxy-n-(3-(1-methyl-1h-pyrazol-5-yl)-4-(2-morpholinoethoxy)phenyl)benzamide, a novel 5-hydroxytryptamine 2A receptor antagonist: pharmacological profile, pharmacokinetics, platelet activity and vascular biology. J Pharmacol Exp Ther. (2009) 331(1):96–103. doi: 10.1124/jpet.109.153189
90. Lin OA, Karim ZA, Vemana HP, Espinosa EV, Khasawneh FT. The antidepressant 5-HT2A receptor antagonists pizotifen and cyproheptadine inhibit serotonin-enhanced platelet function. PloS One. (2014) 9(1):e87026. doi: 10.1371/journal.pone.0087026
91. Berry CN, Lorrain J, Lochot S, Delahaye M, Lalé A, Savi P, et al. Antiplatelet and antithrombotic activity of SL65.0472, a mixed 5-HT1B/5-HT2A receptor antagonist. Thromb Haemost. (2001) 85(3):521–8. doi: 10.1055/s-0037-1615615
92. De Ceunynck K, Peters CG, Jain A, Higgins SJ, Aisiku O, Fitch-Tewfik JL, et al. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc Natl Acad Sci U S A. (2018) 115(5):E982–91. doi: 10.1073/pnas.1718600115
93. Aisiku O, Peters CG, De Ceunynck K, Ghosh CC, Dilks JR, Fustolo-Gunnink SF, et al. Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood. (2015) 125(12):1976–85. doi: 10.1182/blood-2014-09-599910
94. Blann AD, Nadar SK, Lip GY. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J. (2003) 24(24):2166–79. doi: 10.1016/j.ehj.2003.08.021
95. Pircher J, Engelmann B, Massberg S, Schulz C. Platelet-Neutrophil crosstalk in atherothrombosis. Thromb Haemost. (2019) 119(8):1274–82. doi: 10.1055/s-0039-1692983
96. Bedard PW, Clerin V, Sushkova N, Tchernychev B, Antrilli T, Resmini C, et al. Characterization of the novel P-selectin inhibitor PSI-697 [2-(4-chlorobenzyl)-3-hydroxy-7,8,9,10-tetrahydrobenzo[h] quinoline-4-carboxylic acid] in vitro and in rodent models of vascular inflammation and thrombosis. J Pharmacol Exp Ther. (2008) 324(2):497–506. doi: 10.1124/jpet.107.128124
97. Feng Q, Wang M, Muhtar E, Wang Y, Zhu H. Nanoparticles of a new small-molecule P-selectin inhibitor attenuate thrombosis, inflammation, and tumor growth in two animal models. Int J Nanomed. (2021) 16:5777–95. doi: 10.2147/IJN.S316863
98. Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. (2017) 376(5):429–39. doi: 10.1056/NEJMoa1611770
99. Stähli BE, Gebhard C, Duchatelle V, Cournoyer D, Petroni T, Tanguay JF, et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention according to timing of infusion: insights from the SELECT-ACS trial. J Am Heart Assoc. (2016) 5(11):e004255. doi: 10.1161/JAHA.116.004255
100. Ribes A, Oprescu A, Viaud J, Hnia K, Chicanne G, Xuereb JM, et al. Phosphoinositide 3-kinases in platelets, thrombosis and therapeutics. Biochem J. (2020) 477(22):4327–42. doi: 10.1042/BCJ20190402
101. Martin V, Guillermet-Guibert J, Chicanne G, Cabou C, Jandrot-Perrus M, Plantavid M, et al. Deletion of the p110beta isoform of phosphoinositide 3-kinase in platelets reveals its central role in akt activation and thrombus formation in vitro and in vivo. Blood. (2010) 115(10):2008–13. doi: 10.1182/blood-2009-04-217224
102. Canobbio I, Stefanini L, Cipolla L, Ciraolo E, Gruppi C, Balduini C, et al. Genetic evidence for a predominant role of PI3Kbeta catalytic activity in ITAM- and integrin-mediated signaling in platelets. Blood. (2009) 114(10):2193–6. doi: 10.1182/blood-2009-03-208074
103. Nylander S, Kull B, Björkman JA, Ulvinge JC, Oakes N, Emanuelsson BM, et al. Human target validation of phosphoinositide 3-kinase (PI3K)β: effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J Thromb Haemost. (2012) 10(10):2127–36. doi: 10.1111/j.1538-7836.2012.04898.x
104. Wang C, Cheng Y, Zhang Y, Jin H, Zuo Z, Wang A, et al. Berberine and its main metabolite berberrubine inhibit platelet activation through suppressing the class I PI3Kβ/Rasa3/Rap1 pathway. Front Pharmacol. (2021) 12:734603. doi: 10.3389/fphar.2021.734603
105. Fan T, Cheng Y, Wei W, Zeng Q, Guo X, Guo Z, et al. Palmatine derivatives as potential antiplatelet aggregation agents via protein kinase G/vasodilator-stimulated phosphoprotein and phosphatidylinositol 3-kinase/akt phosphorylation. J Med Chem. (2022) 65(10):7399–413. doi: 10.1021/acs.jmedchem.2c00592
106. Andre P, Morooka T, Sim D, Abe K, Lowell C, Nanda N, et al. Critical role for syk in responses to vascular injury. Blood. (2011) 118(18):5000–10. doi: 10.1182/blood-2011-06-360743
107. Bussel J, Arnold DM, Grossbard E, Mayer J, Treliński J, Homenda W, et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: results of two phase 3, randomized, placebo-controlled trials. Am J Hematol. (2018) 93(7):921–30. doi: 10.1002/ajh.25125
108. Newland A, McDonald V. Fostamatinib: a review of its clinical efficacy and safety in the management of chronic adult immune thrombocytopenia. Immunotherapy. (2020) 12(18):1325–40. doi: 10.2217/imt-2020-0215
109. Karel MFA, Tullemans BME, D'Italia G, Lemmens TP, Claushuis TAM, Kuijpers MJE, et al. The effect of bruton’s tyrosine kinase inhibitor ibrutinib on atherothrombus formation under stenotic flow conditions. Thromb Res. (2022) 212:72–80. doi: 10.1016/j.thromres.2022.02.020
110. Kohs TCL, Olson SR, Pang J, Jordan KR, Zheng TJ, Xie A, et al. Ibrutinib inhibits BMX-dependent endothelial VCAM-1 expression in vitro and pro-atherosclerotic endothelial activation and platelet adhesion in vivo. Cell Mol Bioeng. (2022) 15(3):231–43. doi: 10.1007/s12195-022-00723-1
111. Essex DW, Chen K, Swiatkowska M. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood. (1995) 86(6):2168–73. doi: 10.1182/blood.V86.6.2168.bloodjournal8662168
112. Jasuja R, Furie B, Furie BC. Endothelium-derived but not platelet-derived protein disulfide isomerase is required for thrombus formation in vivo. Blood. (2010) 116(22):4665–74. doi: 10.1182/blood-2010-04-278184
113. Lin L, Gopal S, Sharda A, Passam F, Bowley SR, Stopa J, et al. Quercetin-3-rutinoside inhibits protein disulfide isomerase by binding to its b'x domain. J Biol Chem. (2015) 290(39):23543–52. doi: 10.1074/jbc.M115.666180
114. Gaspar RS, da Silva SA, Stapleton J, Fontelles JLL, Sousa HR, Chagas VT, et al. Myricetin, the main flavonoid in syzygium cumini leaf, is a novel inhibitor of platelet thiol isomerases PDI and ERp5. Front Pharmacol. (2019) 10:1678. doi: 10.3389/fphar.2019.01678
115. Yeung J, Tourdot BE, Fernandez-Perez P, Vesci J, Ren J, Smyrniotis CJ, et al. Platelet 12-LOX is essential for FcγRIIa-mediated platelet activation. Blood. (2014) 124(14):2271–9. doi: 10.1182/blood-2014-05-575878
116. Yeung J, Apopa PL, Vesci J, Stolla M, Rai G, Simeonov A, et al. 12-lipoxygenase Activity plays an important role in PAR4 and GPVI-mediated platelet reactivity. Thromb Haemost. (2013) 110(3):569–81. doi: 10.1160/TH13-01-0014
117. Adili R, Tourdot BE, Mast K, Yeung J, Freedman JC, Green A, et al. First selective 12-LOX inhibitor, ML355, impairs thrombus formation and vessel occlusion in vivo with minimal effects on hemostasis. Arterioscler, Thromb, Vasc Biol. (2017) 37(10):1828–39. doi: 10.1161/ATVBAHA.117.309868
118. Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. (1990) 76(1):1–16. doi: 10.1182/blood.V76.1.1.1
119. Bovill EG, Malhotra OP, Mann KG. Mechanisms of vitamin K antagonism. Baillieres Clin Haematol. (1990) 3(3):555–81. doi: 10.1016/S0950-3536(05)80019-8
120. Damus PS, Hicks M, Rosenberg RD. Anticoagulant action of heparin. Nature. (1973) 246(5432):355–7. doi: 10.1038/246355a0
121. Hoffman R, Brenner B. The promise of novel direct oral anticoagulants. Best Pract Res Clin Haematol. (2012) 25(3):351–60. doi: 10.1016/j.beha.2012.06.004
122. Frumkin K. Rapid reversal of warfarin-associated hemorrhage in the emergency department by prothrombin complex concentrates. Ann Emerg Med. (2013) 62(6):616–26.e8. doi: 10.1016/j.annemergmed.2013.05.026
123. Chai-Adisaksopha C, Hillis C, Siegal DM, Movilla R, Heddle N, Iorio A, et al. Prothrombin complex concentrates versus fresh frozen plasma for warfarin reversal. A systematic review and meta-analysis. Thromb Haemostasis. (2016) 116(5):879–90. doi: 10.1160/TH16-04-0266
124. Ostermann H, von Heymann C. Prothrombin complex concentrate for vitamin K antagonist reversal in acute bleeding settings: efficacy and safety. Expert Rev Hematol. (2019) 12(7):525–40. doi: 10.1080/17474086.2019.1624520
125. Pagano MB, Foroutan F, Goel R, Allen ES, Cushing MM, Garcia DA, et al. Vitamin K antagonist reversal strategies: systematic review and network meta-analysis from the AABB. Transfusion. (2022) 62(8):1652–61. doi: 10.1111/trf.17010
126. Jansma B, Montgomery J, Dietrich S, Mixon MA, Peksa GD, Faine B. Emergent warfarin reversal with fixed-dose 4-factor prothrombin Complex concentrate. Ann Pharmacother. (2020) 54(11):1090–5. doi: 10.1177/1060028020920855
127. Farrow GS, Delate T, McNeil K, Jones AE, Witt DM, Crowther MA, et al. Vitamin K versus warfarin interruption alone in patients without bleeding and an international normalized ratio > 10. J Thromb Haemost. (2020) 18(5):1133–40. doi: 10.1111/jth.14772
128. Boer C, Meesters MI, Veerhoek D, Vonk ABA. Anticoagulant and side-effects of protamine in cardiac surgery: a narrative review. Br J Anaesth. (2018) 120(5):914–27. doi: 10.1016/j.bja.2018.01.023
129. Sokolowska E, Kalaska B, Miklosz J, Mogielnicki A. The toxicology of heparin reversal with protamine: past, present and future. Expert Opin Drug Metab Toxicol. (2016) 12(8):897–909. doi: 10.1080/17425255.2016.1194395
130. Meijers JCM, Bakhtiari K, Zwiers A, Peters SLM. OKL-1111, a modified cyclodextrin as a potential universal reversal agent for anticoagulants. Thromb Res. (2023) 227:17–24. doi: 10.1016/j.thromres.2023.05.003
131. Pollack CV Jr, Reilly PA, Eikelboom J, Glund S, Verhamme P, Bernstein RA, et al. Idarucizumab for dabigatran reversal. N Engl J Med. (2015) 373(6):511–20. doi: 10.1056/NEJMoa1502000
132. Honickel M, Treutler S, van Ryn J, Tillmann S, Rossaint R, Grottke O. Reversal of dabigatran anticoagulation ex vivo: porcine study comparing prothrombin complex concentrates and idarucizumab. Thromb Haemost. (2015) 113(4):728–40. doi: 10.1160/TH14-08-0712
133. Glund S, Moschetti V, Norris S, Stangier J, Schmohl M, van Ryn J, et al. A randomised study in healthy volunteers to investigate the safety, tolerability and pharmacokinetics of idarucizumab, a specific antidote to dabigatran. Thromb Haemost. (2015) 113(5):943–51. doi: 10.1160/TH14-12-1080
134. Lu G, DeGuzman FR, Hollenbach SJ, Karbarz MJ, Abe K, Lee G, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med. (2013) 19(4):446–51. doi: 10.1038/nm.3102
135. Milling TJ Jr, Middeldorp S, Xu L, Koch B, Demchuk A, Eikelboom JW, et al. Final study report of andexanet alfa for Major bleeding with factor Xa inhibitors. Circulation. (2023) 147(13):1026–38. doi: 10.1161/CIRCULATIONAHA.121.057844
136. Siddiqui F, Tafur A, Ramacciotti LS, Jeske W, Hoppensteadt D, Ramacciotti E, et al. Reversal of factor Xa inhibitors by andexanet alfa may increase thrombogenesis compared to pretreatment values. Clin Appl Thromb Hemost. (2019) 25:1076029619863493. doi: 10.1177/1076029619863493
137. Lu G, Lin J, Bui K, Curnutte JT, Conley PB. Andexanet versus prothrombin complex concentrates: differences in reversal of factor Xa inhibitors in in vitro thrombin generation. Res Pract Thromb Haemost. (2020) 4(8):1282–94. doi: 10.1002/rth2.12418
138. Thalji NK, Ivanciu L, Davidson R, Gimotty PA, Krishnaswamy S, Camire RM. A rapid pro-hemostatic approach to overcome direct oral anticoagulants. Nat Med. (2016) 22(8):924–32. doi: 10.1038/nm.4149
139. Parsons-Rich D, Hua F, Li G, Kantaridis C, Pittman DD, Arkin S. Phase 1 dose-escalating study to evaluate the safety, pharmacokinetics, and pharmacodynamics of a recombinant factor Xa variant (FXa(I16l)). J Thromb Haemost. (2017) 15(5):931–7. doi: 10.1111/jth.13673
140. Silva Blas Y, Diringer MN, Lo B, Masjuan J, de la Ossa N P, Cardinal M, et al. Phase 1b study to evaluate safety, tolerability, and Maximum tolerated dose of PF-05230907 for intracerebral hemorrhage. Stroke. (2021) 52(1):294–8. doi: 10.1161/STROKEAHA.120.029789
141. Verhoef D, Visscher KM, Vosmeer CR, Cheung KL, Reitsma PH, Geerke DP, et al. Engineered factor Xa variants retain procoagulant activity independent of direct factor Xa inhibitors. Nat Commun. (2017) 8(1):528. doi: 10.1038/s41467-017-00647-9
142. Ansell J, Laulicht BE, Bakhru SH, Burnett A, Jiang X, Chen L, et al. Ciraparantag, an anticoagulant reversal drug: mechanism of action, pharmacokinetics, and reversal of anticoagulants. Blood. (2021) 137(1):115–25. doi: 10.1182/blood.2020007116
143. Ansell J, Bakhru S, Laulicht BE, Tracey G, Villano S, Freedman D. Ciraparantag reverses the anticoagulant activity of apixaban and rivaroxaban in healthy elderly subjects. Eur Heart J. (2022) 43(10):985–92. doi: 10.1093/eurheartj/ehab637
144. Ansell JE, Bakhru SH, Laulicht BE, Steiner SS, Grosso MA, Brown K, et al. Single-dose ciraparantag safely and completely reverses anticoagulant effects of edoxaban. Thromb Haemost. (2017) 117(2):238–45. doi: 10.1160/TH16-03-0224
145. Dewerchin M, Liang Z, Moons L, Carmeliet P, Castellino FJ, Collen D, et al. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb Haemost. (2000) 83(2):185–90. doi: 10.1055/s-0037-1613783
146. Mullins ES, Kombrinck KW, Talmage KE, Shaw MA, Witte DP, Ullman JM, et al. Genetic elimination of prothrombin in adult mice is not compatible with survival and results in spontaneous hemorrhagic events in both heart and brain. Blood. (2009) 113(3):696–704. doi: 10.1182/blood-2008-07-169003
147. Sun WY, Witte DP, Degen JL, Colbert MC, Burkart MC, Holmback K, et al. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci U S A. (1998) 95(13):7597–602. doi: 10.1073/pnas.95.13.7597
148. Gailani D, Broze GJ Jr. Factor XI activation in a revised model of blood coagulation. Science. (1991) 253(5022):909–12. doi: 10.1126/science.1652157
149. Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. (1991) 266(12):7353–8. doi: 10.1016/S0021-9258(20)89453-8
150. Juang LJ, Mazinani N, Novakowski SK, Prowse ENP, Haulena M, Gailani D, et al. Coagulation factor XII contributes to hemostasis when activated by soil in wounds. Blood Adv. (2020) 4(8):1737–45. doi: 10.1182/bloodadvances.2019000425
151. Weitz JI, Fredenburgh JC. Factors XI and XII as targets for new anticoagulants. Front Med (Lausanne). (2017) 4:19. doi: 10.3389/fmed.2017.00019
152. Gigante B, Ten Cate H. Factor XI inhibitors in patients with cardiovascular disease and a high risk of bleeding: a cautionary tale. Nat Rev Cardiol. (2023) 20(8):511–2. doi: 10.1038/s41569-023-00872-4
153. Revenko AS, Gao D, Crosby JR, Bhattacharjee G, Zhao C, May C, et al. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood. (2011) 118(19):5302–11. doi: 10.1182/blood-2011-05-355248
154. Tweddell JS, Kharnaf M, Zafar F, Riggs KW, Reagor JA, Monia BP, et al. Targeting the contact system in a rabbit model of extracorporeal membrane oxygenation. Blood Adv. (2023) 7(8):1404–17. doi: 10.1182/bloodadvances.2022007586
155. Yau JW, Liao P, Fredenburgh JC, Stafford AR, Revenko AS, Monia BP, et al. Selective depletion of factor XI or factor XII with antisense oligonucleotides attenuates catheter thrombosis in rabbits. Blood. (2014) 123(13):2102–7. doi: 10.1182/blood-2013-12-540872
156. Zhang H, Lowenberg EC, Crosby JR, MacLeod AR, Zhao C, Gao D, et al. Inhibition of the intrinsic coagulation pathway factor XI by antisense oligonucleotides: a novel antithrombotic strategy with lowered bleeding risk. Blood. (2010) 116(22):4684–92. doi: 10.1182/blood-2010-04-277798
157. Buller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. (2015) 372(3):232–40. doi: 10.1056/NEJMoa1405760
158. Verhamme P, Yi BA, Segers A, Salter J, Bloomfield D, Buller HR, et al. Abelacimab for prevention of venous thromboembolism. N Engl J Med. (2021) 385(7):609–17. doi: 10.1056/NEJMoa2105872
159. Yi BA, Freedholm D, Widener N, Wang X, Simard E, Cullen C, et al. Pharmacokinetics and pharmacodynamics of abelacimab (MAA868), a novel dual inhibitor of factor XI and factor XIa. J Thromb Haemost. (2022) 20(2):307–15. doi: 10.1111/jth.15577
160. Schaefer M, Buchmueller A, Dittmer F, Strassburger J, Wilmen A. Allosteric inhibition as a new mode of action for BAY 1213790, a neutralizing antibody targeting the activated form of coagulation factor XI. J Mol Biol. (2019) 431(24):4817–33. doi: 10.1016/j.jmb.2019.09.008
161. Weitz JI, Bauersachs R, Becker B, Berkowitz SD, Freitas MCS, Lassen MR, et al. Effect of osocimab in preventing venous thromboembolism among patients undergoing knee arthroplasty: the FOXTROT randomized clinical trial. JAMA. (2020) 323(2):130–9. doi: 10.1001/jama.2019.20687
162. Lorentz CU, Verbout NG, Wallisch M, Hagen MW, Shatzel JJ, Olson SR, et al. Contact activation inhibitor and factor XI antibody, AB023, produces safe, dose-dependent anticoagulation in a phase 1 first-in-human trial. Arterioscler, Thromb, Vasc Biol. (2019) 39(4):799–809. doi: 10.1161/ATVBAHA.118.312328
163. Cao H, Biondo M, Lioe H, Busfield S, Rayzman V, Nieswandt B, et al. Antibody-mediated inhibition of FXIIa blocks downstream bradykinin generation. J Allergy Clin Immunol. (2018) 142(4):1355–8. doi: 10.1016/j.jaci.2018.06.014
164. McKenzie A, Roberts A, Malandkar S, Feuersenger H, Panousis C, Pawaskar D. A phase I, first-in-human, randomized dose-escalation study of anti-activated factor XII monoclonal antibody garadacimab. Clin Transl Sci. (2022) 15(3):626–37. doi: 10.1111/cts.13180
165. Weitz JI, Strony J, Ageno W, Gailani D, Hylek EM, Lassen MR, et al. Milvexian for the prevention of venous thromboembolism. N Engl J Med. (2021) 385(23):2161–72. doi: 10.1056/NEJMoa2113194
166. Piccini JP, Caso V, Connolly SJ, Fox KAA, Oldgren J, Jones WS, et al. Safety of the oral factor XIa inhibitor asundexian compared with apixaban in patients with atrial fibrillation (PACIFIC-AF): a multicentre, randomised, double-blind, double-dummy, dose-finding phase 2 study. Lancet (London, England). (2022) 399(10333):1383–90. doi: 10.1016/S0140-6736(22)00456-1
167. Chamardani TM, Amiritavassoli S. Inhibition of NETosis for treatment purposes: friend or foe? Mol Cell Biochem. (2022) 477(3):673–88. doi: 10.1007/s11010-021-04315-x
168. Baron S, Rashal T, Vaisman D, Elhasid R, Shukrun R. Selinexor, a selective inhibitor of nuclear export, inhibits human neutrophil extracellular trap formation in vitro. Front Pharmacol. (2022) 13:1030991. doi: 10.3389/fphar.2022.1030991
169. Machlus KR, Wu SK, Vijey P, Soussou TS, Liu ZJ, Shacham E, et al. Selinexor-induced thrombocytopenia results from inhibition of thrombopoietin signaling in early megakaryopoiesis. Blood. (2017) 130(9):1132–43. doi: 10.1182/blood-2016-11-752840
Keywords: thrombosis, hemostasis, platelet, anticoagulation, thrombin
Citation: Sim MMS, Shiferawe S and Wood JP (2023) Novel strategies in antithrombotic therapy: targeting thrombosis while preserving hemostasis. Front. Cardiovasc. Med. 10:1272971. doi: 10.3389/fcvm.2023.1272971
Received: 4 August 2023; Accepted: 6 October 2023;
Published: 23 October 2023.
Edited by:
Joon-Young Park, Baylor University, United StatesReviewed by:
Heiko Rühl, University Hospital Bonn, Germany© 2023 Sim, Shiferawe and Wood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy P. Wood SmVyZW15Lldvb2RAdWt5LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.