 Jie Zhang
Jie Zhang Caixia Guo*
Caixia Guo*
- Cardiovascular Center, Beijing Tongren Hospital, Capital Medical University, Beijing, China
Ferroptosis, a newly recognized form of nonapoptotic regulated cell death, is characterized by iron-dependent lipid peroxidation. Biological processes, such as iron metabolism, lipid peroxidation, and amino acid metabolism, are involved in the process of ferroptosis. However, the related molecular mechanism of ferroptosis has not yet been completely clarified, and specific and sensitive biomarkers for ferroptosis need to be explored. Recently, studies have revealed that ferroptosis probably causes or exacerbates the progress of cardiovascular diseases, and could be the potential therapeutic target for cardiovascular diseases. In this review, we summarize the molecular mechanisms regulating ferroptosis, inducers or inhibitors of ferroptosis, and the current progresses of ferroptosis in cardiovascular diseases. Furthermore, we discuss the emerging challenges and future perspectives, which may provide novel insights into the treatment of cardiovascular diseases.
1. Introduction
Cardiovascular diseases (CVDs), a group of diseases, are currently the leading causes of death globally, imposing a significant economic burden on families and communities (1, 2). Therefore, understanding the pathophysiological progression of CVDs and related molecular mechanisms has become the main focus of medirelated cal studies.
Multiple types of cell death, including apoptosis, necrosis, and pyroptosis, are associated with the pathogenesis of CVDs (3). Accumulating evidence has demonstrated that ferroptosis, a newly recognized type of nonapoptotic regulated cell death (RCD), plays a central role in CVDs, which is triggered by iron-dependent lipid peroxidation (3, 4). Ferroptosis was first reported in 2012, with features of loss of mitochondria crista, cell swelling, and outer membrane rupture, without the change of nucleus (5).
In this review, we focus on effects of ferroptosis and related mechanisms in the occurrence and development of CVDs, and highlight the effective strategy for targeting ferroptosis in CVDs.
2. Cellular mechanisms of ferroptosis
Ferroptosis is a complicated process that is moderated by a series of signaling and molecules. The occurrence and execution of ferroptosis contain three main events, iron dysregulation, GSH depletion, and lipid membrane oxidation (6). The regulatory mechanisms of these events and their roles in cardiovascular diseases are summarized in Figure 1.
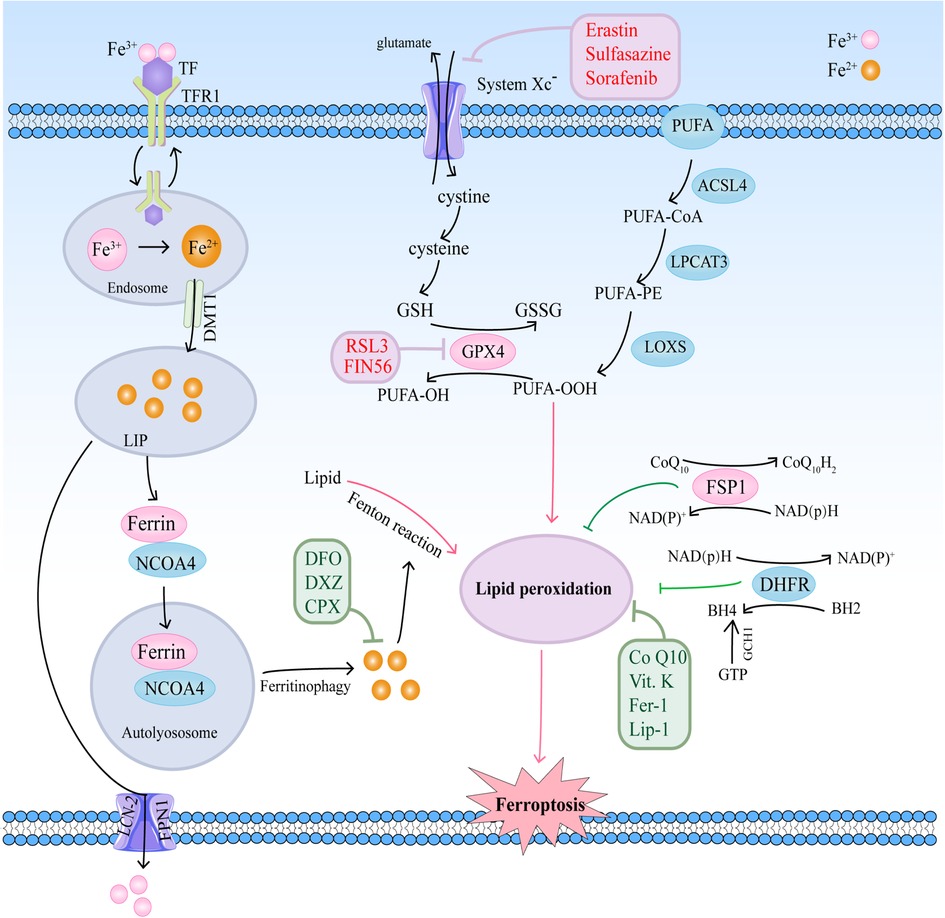
Figure 1. The regulatory mechanisms of ferroptosis in cardiovascular diseases.
2.1. Iron dysregulation
Iron homeostasis disorders are involved in the pathological processes of cardiovascular diseases. Iron as a necessary cofactor for several enzymes is essential for normal physiological activities, including cell oxygen transport, oxidation–reduction reactions, and energy metabolism (7). Circulating iron is present in two ionic forms, ferrous (Fe2+) and ferric (Fe3+) iron (8). In general, iron levels in different tissues are tightly regulated, which are accompanied by precise regulation of iron storage protein ferritin and other proteins. Extracellular iron binds to ferritin transferrin (TF) in the form of ferric iron, and is transported into cells by a cell-surface receptor, transferrin receptor 1 (TFR1) (8, 9). Six-transmembrane epithelial antigens of the prostate 3 (STEAP3) catalyzes the reduction of Fe3+ to Fe2+, and the transporter divalent metal transporter (DMT1) transports Fe2+ into the iron pool (LIP) through SLC11A2 (10, 11). The surplus Fe2+ is temporarily kept in the ferritin protein, composed of the ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1), or enters into the blood by ferroportin-1 (FPN) and lipocalin-2 (LCN-2) (12, 13).
FPN, the sole exporter in charge of the efflux of surplus iron, is degraded after being bound to hepcidin (14). Accordingly, hepcidin-FPN axis is essential for maintaining iron homeostasis. Altered FPN expression could cause iron deficiency or overload. Knockdown of FPN promotes ferroptosis with an accumulation of intracellular iron and the production of reactive oxygen species (ROS) by the Fenton reaction (13). High level of hepcidin is induced via iron overload and chronic inflammation. Hepcidin binds to FPN and leads to degradation of FPN, thereby causing intracellular iron restriction (15). In contrast, low level of hepcidin in response to iron deficiency or hypoxia stabilizes FPN, leading to more liberation of the iron (15).
Under pathological conditions, intracellular iron homeostasis is disrupted, including increased iron intake or decreased iron stores, which leads to increased Fe2+ concentration in extracellular space, and catalyzes the Fenton reaction to participate in the production of ROS, inducing lipid peroxidation and eventually ferroptosis (16). Recent studies have suggested that iron-bound ferritin could bind to nuclear receptor coactivator 4 (NCOA4), promoting the NCOA4-mediated autophagy pathway (namely ferrotinophagy) and resulting in lysosomal degradation of ferritin and iron release (17, 18). Upregulation of NCOA4 increases ferritin degradation and promotes ferroptosis (19). Therefore, both iron overload and ferritin deficiency could trigger ferroptosis.
2.2. Lipid peroxidation
Lipid peroxidation is a way that oxidants, including free radicals or non-radical species, attack lipids containing two or more double hydrocarbon chains on the carbon chain. Especially, polyunsaturated fatty acids (PUFAs) are more prone to lipid peroxidation under oxidative stress (20, 21). Cell membranes contain a large number of PUFAs that are primary targets of the attack by ROS (21). Lipid peroxidation can be non-enzymatically and enzymatically triggered. The former process is driven by free radicals, and the latter by enzymes, such as lipoxygenases (LOXs) (20).
PUFAs can be acylated by acyl-CoA synthetase long-chain family member 4 (ACSL4) to form PUFA acyl-coenzyme A (PUFA-CoA), and then reacts with lysophosphatidylcholine acyltransferase 3 (LPCAT3) to form PUFA-PE. PUFA-PE is subsequently oxidized by lipoxygenases (LOXs) to generates PUFAs-OOH (22, 23). Peroxidation of PUFA-PEs mainly depends on ACSL4 and LOXs. Thus, suppressed ACSL4 or LOXs could effectively inhibit ferroptosis. A study with human haploid cells has shown that downregulation of ACSL4 and LPCAT3 inhibits ferroptosis by limiting oxidation-sensitive fatty acids in cell membranes (24). Another study has also demonstrated that ACSL4 is a biomarker associated with ferroptosis sensitivity by regulating lipid metabolism (25).
Several reports have also shown that LOXs, the 15-LOX-1 isoform, especially are a key regulator of lipid peroxidation of ferroptosis (22, 26). Suppression of 15-LOX activity, but not cyclooxygenase (COX) and cytochrome P450 (CYP450), effectively inhibits ferroptosis (22). In addition, a study has indicated that 12-LOX is indirectly upregulated by p53, promoting its ability to oxygenate PUFAs upon ROS stress (27). Therefore, downregulation of certain enzymes (such as ACSL4, LPCAT3, and LOXs) inhibits ferroptosis by reducing lipid peroxidation.
2.3. Amino acid metabolism
Ferroptosis has been demonstrated to be controlled by amino acid metabolism. According to study reports, ferroptosis can be induced by erastin, a ferroptosis inducer. Erastin inhibits cystine uptake by the cystine/glutamate antiporter, therefore suppressing antioxidant defenses through GSH reduction (5, 28). The pathway that erastin inhibits cystine uptake is mainly dependent on cystine/glutamate antiporter (system Xc−), a membrane amino acid transporter, which consists of the catalytic subunit solute carrier family 7 member 11 (SLC7A11) and the solute carrier family 3 member 2 (SLC3A2) (29). System Xc− transports glutamate and cystine between the intracellular and extracellular at a 1:1 ratio, and reduces the cystine to cysteine, therefore providing the initial materials of GSH in the cell (30). Inhibition of System Xc− could directly decrease cellular cysteine, thus repressing the generation of GSH. Thus, System Xc− plays an important role in the process of ferroptosis-related amino-acid metabolism (31).
In addition, it is widely accepted that GPX4 is another key point in the amino acid metabolic pathway. GPX4 belongs to the selenoprotein family that uses GSH as a reducing agent and turns GSH into oxidized glutathione (GSSG) (32). In the process, PUFAs-OOH is reduced to PUFAs-OH by GPX4, suggesting that GPX4 plays a vital role in inhibiting lipid peroxidation (33). Restricted GPX4 decreases GSSG and increases ROS, thereby inducing ferroptosis (34).
3. Regulation of ferroptosis
Lipid peroxidation and iron homeostasis, as well as specific gene interventions, have been shown to regulate ferroptosis. Many molecules in cells, including system Xc− and GPX4, have been implicated in governing ferroptosis (28).
Transcription factors have been shown to impact on the determinants of ferroptosis through regulating the antioxidant system. Kelch-like ECH-associated protein 1 (Keap1)-Nuclear factor E2-related factor 2/antioxidant response element (Keap1-Nrf2-ARE) pathway is the main survival pathway in cells, which protect cells from oxidative and electrophilic insults (35). Nrf2 is considered as a master transcription factor for the antioxidant response. Under normal conditions, Nrf2 protein levels are kept basally low by binding to Keap1, and being then degraded through the ubiquitin-proteasome pathway (36). Under stress conditions, Nrf2 is released from Keap1 and translocated to the nucleus to initiate the transcription of the downstream gene (36). Notably, many proteins and enzymes responsible for the initiation of ferroptosis are the downstream of Nrf2, which are grouped into three broad categories according to their function: iron/metal metabolism, intermediate metabolism, and glutathione synthesis/metabolism (37). Therefore, the Keap1-Nrf2-ARE pathway is central to the ferroptosis.
Another cellular signaling pathway is the ferroptosis suppressor protein 1 (FSP1)–coenzyme Q10 (CoQ10)–nicotinamide adenine dinucleotide phosphate (NADPH) pathway, which is associated with GPX4 and glutathione (GSH) to prevent lipid peroxidation and ferroptosis independently (37). Together with NADPH, FSP1 functions as a CoQ oxidoreductase, and reduces CoQ10 to ubiquinol, a lipophilic radical-trapping antioxidant (RTA), which mediate lipid peroxidation, thereby suppressing ferroptosis (38, 39). These studies suggest that the NAD(P)H/FSP1/CoQ10 pathway is involved in the metabolic processes related to lipid peroxidation.
GTP cyclohydrolase-1 (GCH1)-tetrahydrobiopterin/dihydrobiopterin (BH4/BH2)-phospholipid axis has been shown to be a primary signaling pathway of ferroptosis resistance through upregulation of CoQ10, therefore suppressing oxidative damage and ferroptosis (40). GCH1 overexpression increases BH4/BH2, which could act as membrane-permeant antioxidant and preventing depletion of phospholipids with two PUFAs tails (40). Additionally, GCH1 overexpression does not affect GPX4 and GSH, suggesting that GCH1 inhibit ferroptosis independent of GPX4 (40).
4. Ferroptosis inducers and inhibitors
Ferroptosis plays a central role in CVDs, and regulating the levels of some key molecules related to ferroptosis may be helpful in treating CVDs (Table 1). Several compounds have been identified to induce or repress ferroptosis (Figure 1).
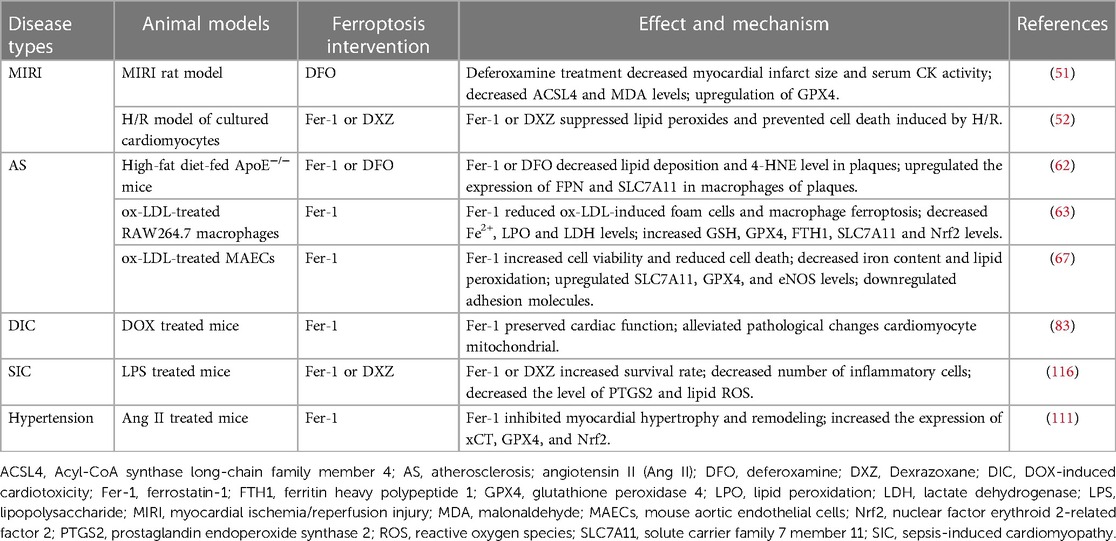
Table 1. Targeting ferroptosis in CVDs.
4.1. Ferroptosis inducers
Ferroptosis inducers are compounds that act by inhibiting system Xc− or altering intracellular iron levels. Many ferroptosis inducers has been found, which can be divided into four classes, class I-IV. Class I ferroptosis inducers inhibit cystine uptake through the system Xc−, such as erastin and sorafenib. Erastin binds to SLC7A5, which is one of six light chains that bind SLC3A2 to import cystine, decreasing cellular glutathione, and inducing ferroptosis (5, 26, 33). Sorafenib and its analogs, such as SRS13-45, SRS13-60, SRS13-67, and SRS14-98, also inhibit cystine import via system Xc−, deplete glutathione and cause lipid ROS accumulation (41).
Class II inducers directly inhibit GPX4 activity, such as RAS-selective lethal 3 (RSL3). Class II inducers do not deplete GSH, but bind to and inactivate GPX4, resulting in the formation of ROS (33). Class III inducers (such as FIN56), newly identified inducers of ferroptosis via chemoproteomic analysis, induce ferroptosis by two different pathways. FIN56 induces GPX4 degradation through post-translational regulation and depletion of coenzyme Q10 and modulation of mevalonate pathway (42). Class IV inducers, including FINO2 (an endoperoxide-containing 1,2-dioxolane), induce ferroptosis via directly oxidizing labile iron or indirectly inhibiting GPX4 activity (43). The mechanisms by which FINO2 causes GPX4 inactivation remain unclear (44).
4.2. Ferroptosis inhibitors
A group of small molecules have been identified as ferroptosis inhibitors, which act by iron chelation, inhibiting lipid peroxidation, or eliminating lipid peroxides. Iron chelators, such as deferoxamine (DFO) and dexrazoxane (DXZ), could deplete the cellular iron pool, and promote the excretion of excess Fe2+, thereby preventing ferroptosis (20, 45). Ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1), two effective inhibitors of ferroptosis, prevent lipid hydroperoxides accumulation due to the reactivity as RTAs instead of the ability as inhibitors of LOXs (45). In addition, studies have shown that liproxstatin-1 and Vitamin E could regulate ferroptosis by binding to 15-LOX with high affinity, inhibiting the 15-LOX enzymatic activity and then suppressing the generation of oxygenated PE (22). Besides vitamin E, reduced vitamin K confer strong anti-ferroptotic function via FSP1-mediated reduction and act as an RTA (46). Reduced CoQ and its analogs, idebenone, also act as an RTA, suppressing lipid peroxidation and ferroptosis via potent free radical scavenging (47).
5. Ferroptosis and CVDs
Multiple cell death types are involved in the complicated mechanisms of CVDs. Recently, ferroptosis has been proven to play a central role in CVDs (3). The association between ferroptosis and various CVDs are summarized in the Table 2 and Table 3.
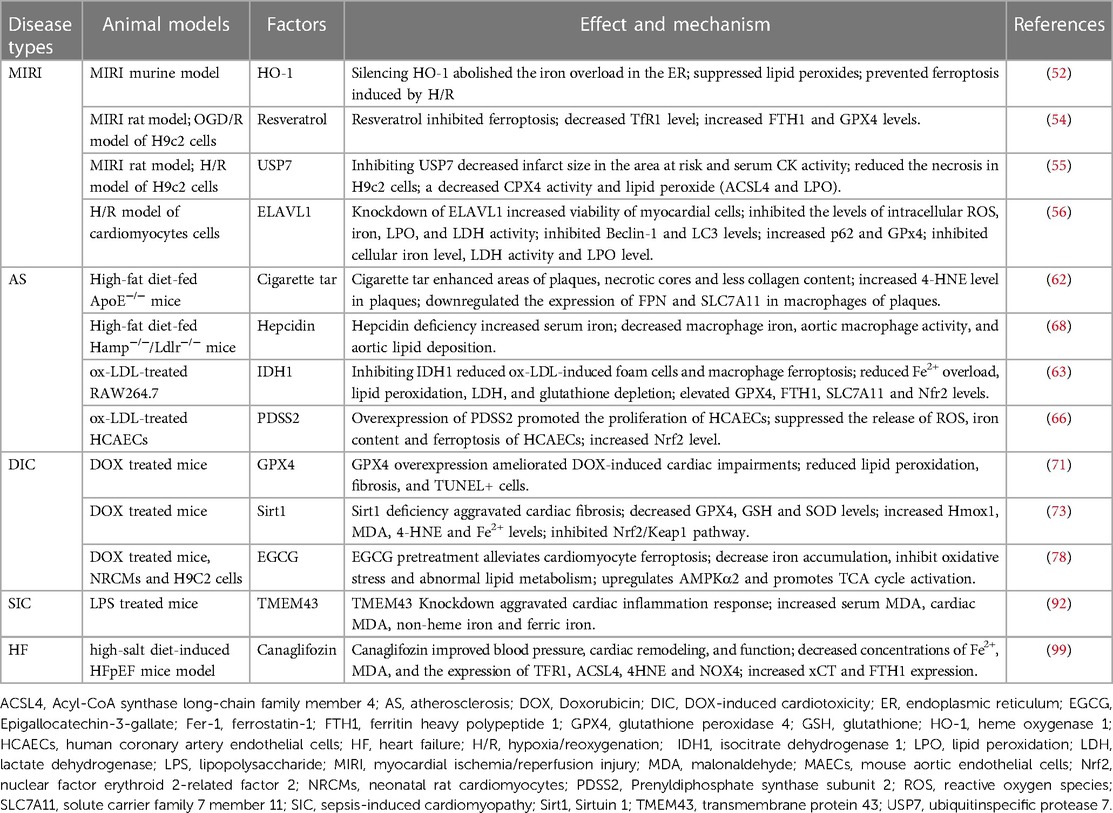
Table 2. Effect and mechanism of Ferroptosis in CVDs

Table 3. Animal models of ferroptosis related genes in CVDs.
5.1. Myocardial ischemia/reperfusion injury
Acute myocardial infarction (AMI) is one of the most common high-risk CVDs (48). Timely restoration of the blood supply remains the effective treatment for ischemic myocardium, called reperfusion (49). However, during this process, the myocardium generates a large number of free radicals, further aggravating myocardium injury, which is known as ischemia/reperfusion injury (MIRI) (49). Ferroptosis, characterized by excess iron accumulation and excessive lipid peroxides, participates in the pathogenesis of MIRI (50). Studies have suggested that ferroptosis happens mainly in myocardial reperfusion instead of ischemia, suggesting that ferroptosis intervention may have numerous beneficial effects on the reperfusion phase but not ischemic phase (51).
As a critical endogenous repressor of ferroptosis, GPX4 determines the severity of MIRI. The level of GPX4 is significantly down-regulated in MIRI-injured myocardium, and overexpression of GPX4 attenuates ferroptosis damage in MIRI (52). GPX4 has been suggested to be a critical regulator of ferroptosis in MIRI. Recent studies have reported that iron metabolism balance also plays a crucial role in MIRI. Inhibition of TfR1 reduces iron contents and ROS levels in H9C2 cells, thereby suppressing H/R-induced ferroptosis (53). Furthermore, downregulation of TfR1 and upregulation FTH1 and GPX4 could also attenuate ferroptosis and prevent MIRI in MIRI rat model (54). In addition, knockdown of ubiquitin-specific protease 7 (USP7) inhibits ferroptosis in p53/TfR1 pathway-dependent in H/R-treated H9c2 cells (55). A growing number of studies suggests that heme metabolism also exerts a significant role in ferroptosis. Heme oxygenase-1 (HO-1) level is increased both in the I/R-injured myocardium and cultured cardiomyocytes. HO-1 degrades heme and therefore causes excess Fe2+ in endoplasmic reticulum (ER), triggering ferroptosis after H/R in cardiomyocytes (56). Another study has indicated that overexpressed HO-1 inhibits ischemia-reperfusion-mediated ferroptosis via induction of anti-ferroptotic genes, such as Slc7a11 (57). In addition, it has been shown that ferroptosis partly relied on autophagy (58). Embryonic lethal-abnormal vision like protein 1 (ELAVL1), which is transcriptionally regulated by Forkhead box C1 (FOXC1), is reported to contribute to myocardial I/R-induced ferroptosis though increasing autophagy (59).
5.2. Atherosclerosis
Atherosclerosis (AS) characterized with arterial wall lipid deposition is the pathological basis for many vascular disorders. AS has been shown to be associated with inflammation, deposition of lipid peroxides and iron (60). Ferroptosis has been shown to regulate AS and plays a vital role in the development of early and late stage (61). Inhibition of macrophage ferroptosis may significantly improve the progression of AS. In ApoE−/− mice, hepcidin-knockdown or SLC7A11-overexpression inhibits plaque macrophage ferroptosis, therefore delaying the progression of atherosclerosis (62). Inhibiting ox-LDL-induced macrophage ferroptosis through reduced Fe2+ overload, lipid peroxidation, or elevated GPX4, FTH1, and SLC7A11 expression may alleviate atherosclerosis (63). Endothelial dysfunction is also critical in the onset and progression of AS (64). Iron overload in endothelial cells induces endothelial dysfunction by alleviating the oxidative stress (65). In human coronary artery endothelial cells (HCAECs), an enhanced PDSS2/Nrf2 axis inhibits ferroptosis by decreasing the release of ROS, providing a promising therapeutic target for AS (66). Moreover, in mouse aortic endothelial cells (MAECs), ferroptosis inhibitors could increase SLC7A11 and GPX4 levels, and reduce lipid peroxidation and iron content, therefore alleviating AS (67). Hepcidin, as a key regulator of iron homeostasis, could bind to and causes the degradation of ferroportin, thereby decreasing serum iron contents. Hepcidin deficiency reduces intracellular macrophage iron and proinflammatory phenotype of macrophages, and decreases atherosclerosis in the murine model (68).
5.3. Cardiomyopathy
5.3.1. Doxorubicin-induced cardiomyopathy
Doxorubicin (DOX), a chemotherapeutic drug, can cause cardiotoxicity termed doxorubicin-induced cardiomyopathy (DIC). Ferroptosis, a new type of cell death form, has been implicated in DIC mice. Mitochondria-dependent ferroptosis is involved in the pathogenesis of DIC (69). DOX downregulates glutathione peroxidase 4 (GPX4) and induces lipid peroxidation via DOX-Fe2+ complex in mitochondria, causing mitochondria-dependent ferroptosis (69). Both in vitro and in vivo, DOX downregulates GPX4 and NFS1 protein levels, and upregulates acyl-coenzyme A synthetase long-chain family member 4 (ACSL4) level, causing increased lipid peroxidation and ferroptosis (70, 71). In experimental animals, a novel SPATA2/CYLD pathway contributes to DIC by enhancing ferritinophagy through the deubiquitination of NCOA4 (72).
Many factors have been shown to participated in the process of ferroptosis in DIC. For example, Sirt1 exerts cardioprotective effects in DIC by inhibiting ferroptosis through the Nrf2/Keap1 pathway (73). Hydrogen sulfide (H2S) prevents DIC by inhibiting ferroptosis via targeting the OPA3-NFS1 axis (74). In H9c2 cells, resveratrol activates p62-Nrf2 axis to inhibit doxorubicin-induced ferroptosis in cardiomyocytes (75). In neonatal rat cardiomyocytes (NRCMs), exosomal thioredoxin-1 inhibits ferroptosis in DIC by targets mTORC1 signaling (76). In cultured rat aortic adventitial fibroblasts, Elabela blunts doxorubicin-induced ferroptosis by activating the KLF15/GPX4 signaling (77). Epigallocatechin-3-gallate alleviates doxorubicin-induced ferroptosis and cardiotoxicity by increasing AMPKα2 and activating adaptive autophagy both in in vitro and in vivo (78). In cultured neonatal rat primary ventricular cardiomyocytes (NRVMs), MITOL/MARCH5 inhibiting doxorubicin-induced ferroptosis by maintaining GSH homeostasis (79). In DIC cellular and animal models, disruption of histamine/H1R axis caused ferroptosis and exacerbates DIC likely by modulating STAT3-SLC7A11 pathway (80).
Ethoxyquin is a lipophilic antioxidant widely applied in food preservation, and has been shown to inhibit GPx4-deficient ferroptosis and prevent DIC both in vitro and in vivo (81). In a cellular and animal DIC model, melatonin alleviates doxorubicin caused mitochondrial damage and ferroptosis by upregulating YAP expression (82). Acyl-CoA thioesterase 1 in the biosynthesis of polyunsaturated fatty acid prevents cardiomyocytes from doxorubicin-induced ferroptosis by altering the lipid composition (83). In doxorubicin-treated mice, Empagliflozin (EMPA) reduces ferroptosis, fibrosis, apoptosis and inflammation through NLRP3 and MyD88-related pathways, causing significant improvements in cardiac functions (84). Therefore, ferroptosis in DIC could be manipulated with various approaches, which might be a therapeutic target.
5.3.2. Others chemical-induced cardiotoxicity
Ferroptosis has been reported to play a vital role in 5-Fluorouracil and other antitumor agents induced cardiotoxicity in cellular and animal levels. 5-fluorouracil (5-FU) is a potent antitumor drug, and its clinical use is limited for severe cardiotoxic effects. ROS and iron homeostasis dependent ferroptosis play a significant role in 5-Fluorouracil-induced cardiotoxicity in H9c2 cardiomyocytes and mice (85). Resveratrol reduces 5-FU-induced cardiotoxicity by attenuating ferroptosis in vitro and in vivo (86). Trastuzumab (TZM) is generally used for treat breast cancer. DNA damage and ferroptosis are the pivotal mechanisms in its anti-tumor drug cardiotoxicity. Empagliflozin attenuates trastuzumab-induced cardiotoxicity via suppressing DNA damage and ferroptosis (87). Mitophagy receptor protein FUNDC1 and JNK-mediated ferroptosis is involved in paraquat exposure-evoked cardiac and mitochondrial injury (88).
5.3.3. LPS-induced cardiomyopathy
Sepsis-induced cardiac injury may result in lethal consequences, and cardiomyocyte apoptosis, ferroptosis, and inflammation participate in the progress of sepsis-induced cardiomyopathy (SIC). Ferroptosis plays critical role in lipopolysaccharide (LPS)-induced cardiac injury. ICA69 upregulates of STING level, which further results intracellular lipid peroxidation, eventually aggravating ferroptosis and septic heart injury (89). Dex alleviates sepsis-induced myocardial cellular injury by attenuating sepsis-induced increased HO-1 and iron levels, and inhibits ferroptosis by enhancing GPX4 (90). Ferrostatin-1 (Fer-1), a ferroptosis inhibitor, ameliorates LPS-induced cardiac dysfunction partly by TLR4/NF-κB pathway (91). TMEM43 attenuates LPS-induced cardiac injury through ferroptosis inhibition in mice (92). In LPS-induced endotoxemia mouse and cell models, resveratrol regulates the miR-149/HMGB1 axis and inhibits ferroptosis to protect cardiomyocyte from injury (93). Puerarin inhibits the myocardial injury induced by LPS through AMPK-mediated ferroptosis signaling (94). Therefore, regulating ferroptosis is also a therapeutic target in sepsis-induced cardiomyopathy (SIC).
5.3.4. Diabetic cardiomyopathy
Diabetic cardiomyopathy (DCM), a major complication of type 2 diabetes, is a leading cause of heart failure and death in advanced diabetes. Ferroptosis also engages in the pathogenesis of DCM. A recent study has reported that ferroptosis is involved diabetes-induced endothelial dysfunction by activation of the p53-xCT-GSH axis (95). Astragaloside IV (AS-IV) attenuates cardiomyocyte injury through inhibiting CD36 expression, decreasing cellular lipid deposition, therefore suppressing ferroptosis in the models of DCM rats (96). Retinol dehydrogenase 10 (RDH10) metabolism disorder exists in the hearts of mice and the patients with type 2 diabetes mellitus (T2DM) (96). Reduction in RDH10 inhibits GPX4 and FSP1, promoting ferroptosis (97). In the model of T2D mice with DCM, sulforaphane (a Nrf2 activator) ameliorates cardiac ferroptosis in AMPK/Nrf2-dependent way (98). Canagliflozin (Cana), a hypoglycemic drug of SGLT2i, reduces ferroptosis both in DCM mice and cardiomyocytes by regulating iron metabolism and system Xc−/GSH/GPX4 axis (99).
5.4. Heart failure
Heart failure (HF) is a complex clinical syndrome with high mortality and morbidity (100, 101). Lipid peroxidation and cardiomyocyte ferroptosis have been reported in a rat model of heart failure (102). Since iron homeostasis is essential for the maintenance of normal function of cardiomyocytes, disrupted iron homeostasis contributes to HF. Knockdown of FTH1, which alters cardiac iron homeostasis and increases lipid peroxidation, leads to severe left ventricular (LV) hypertrophy via ferroptosis (103). Both SLC7A11 overexpression in cardiomyocytes and ferroptosis regulator Fer-1 may inhibit high-iron diet-induced severe cardiac damage (103). In a rat model of high-salt diet-induced HF with preserved ejection fraction (HFpEF), iron overloading and lipid peroxidation have been found in the myocardial tissue, as well as the expression of ACSL4, 4-HNE and NOX4 are increased, while GSH content is decreased (102). These changes eventually lead to ferroptosis, which are reversed by sodium-glucose cotransporter 2 inhibitors (SGLT2i) (102). In a transverse aortic constriction (TAC) mouse model, the circular RNA circSnx12 directly regulates iron metabolism via miR-224-5p/FTH1 regulatory network in myocardial cells, suggesting that circular RNA could be a potential therapeutic target for HF (104). Furthermore, expression of Toll-like receptor 4 (TLR4) and NADPH oxidase 4 (NOX4) is significantly increased in HF, and either TLR4 or NOX4 knockdown inhibits ferroptosis by regulating ferroptosis-related proteins, suggesting that TLR4-NOX4 pathway might be a potential therapeutic target for HF (105).
5.5. Atrial fibrillation
Atrial fibrillation (AF) is the most prevalent type of cardiac arrhythmia with higher morbidity and mortality, due to stroke, heart failure and other complications (106). Ferroptosis is related to frequent excessive alcohol consumption induced AF (107, 108). Mechanistically, inhibition of atrial ferroptosis via SIRT1-Nrf2-HO-1 signaling to reducing atrial mitochondrial damage, ROS accumulation and iron overload, eventually decrease the AF susceptibility (107). FPN, an iron exporter, is involved in LPS-induced endotoxemia rat model. Knockdown FPN increases intracellular iron concentration, worsens the calcium handling proteins dysregulation and exaggerates the AF vulnerability (109).
5.6. Hypertension
Hypertension is a major risk factor for numerous CVDs, and leads to pathological cardiac hypertrophy and eventually heart failure (110). Elabela (ELA), endogenous ligand for the apelin receptor, prevents myocardial remodeling through inhibiting ferroptosis via regulating the IL-6/STAT3/GPX4 signaling in angiotensin II (Ang II)-induced hypertensive mice (111). Moreover, Slc7a11 gene-encoded plasma membrane cystine/glutamate antiporter xCT exerts protective effects in Ang II-medicated cardiac hypertrophy by blocking ferroptosis (112). Ferroptosis is also involved in hypertensive renal injury. Sirtuin 7 alleviates renal ferroptosis and lipid peroxidation via Nrf2/GPX4/xCT and HO-1/NQO1signaling pathways in hypertensive mice (113).
6. Limitations and perspectives
Ferroptosis is a newly-identified iron-dependent form of cell death. Recent studies have suggested that ferroptosis could be a feasible therapeutic target for several cardiovascular diseases. However, the molecular mechanism of ferroptosis in cardiovascular diseases has not been fully understood. Reliable and sensitive biomarkers for ferroptosis need to be explored. Most investigations are confined to in vitro cell cultures, ex vivo models, and animal studies. The clinical studies relevant to ferroptosis in cardiovascular diseases are urgently needed. Currently, many chemical compounds have been found to affect the process of ferroptosis, including dysregulation of iron metabolism, lipid peroxidation, and abnormal amino acid metabolism. However, selectivity and side-effects of these compounds remain to be examined. It still faces great challenges in translating these compounds as future pharmacological drugs. In addition to ferroptosis, other forms of cell death, such as apoptosis, regulated necrosis and pyroptosis, have also been involved in the pathogenesis of CVDs. For examples, apoptosis and necroptosis are detected in diabetic mouse hearts at different ages (114). Palmitic acid induces the cell death of H9c2 cardiomyoblasts in a dose- and time-dependent manner (115). However, the occurrence timing of those cellular death forms in different CVDs is difficult to determine. The cross-talks may exist between those types of cell death. Therefore, it is unable to provide reliable and accurate information to guide the time window for intervention.
In conclusion, further studies are needed to identify specific biomarkers and central regulators of ferroptosis, providing solid evidence and basis for further exploration in clinical trials. Furthermore, it is important to clarify the different temporal stages of ferroptosis and other cell death forms in CVDs.
Author contributions
JZ: Conceptualization, Writing – original draft. CG: Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by the Beijing Natural Science Foundation (7232022), National Natural Science Foundation of China (82171808) and the Young Outstanding Talent Foundation (2021-YJJ-ZZL-001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: executive summary: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation. (2019) 140(11):e563–95. doi: 10.1161/CIR.0000000000000677
2. Lüscher TF. Prevention: some important steps forward, but many unmet needs in a world with cardiovascular disease as the leading cause of death. Eur Heart J. (2016) 37(42):3179–81. doi: 10.1093/eurheartj/ehw566
3. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99(4):1765–817. doi: 10.1152/physrev.00022.2018
4. Huang T, Wang K, Li Y, Ye Y, Chen Y, Wang J, et al. Construction of a novel ferroptosis-related gene signature of atherosclerosis. Front Cell Dev Biol. (2022) 9:800833. doi: 10.3389/fcell.2021.800833
5. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
6. Bertrand RL. Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events. Med Hypotheses. (2017) 101:69–74. doi: 10.1016/j.mehy.2017.02.017
7. Ward DM, Cloonan SM. Mitochondrial iron in human health and disease. Annu Rev Physiol. (2019) 81:453–82. doi: 10.1146/annurev-physiol-020518-114742
8. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. (2016) 23(3):369–79. doi: 10.1038/cdd.2015.158
9. Chen X, Kang R, Kroemer G, Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med. (2021) 218(6):e20210518. doi: 10.1084/jem.20210518
10. Lakhal-Littleton S, Wolna M, Chung YJ, Christian HC, Heather LC, Brescia M, et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. Elife. (2016) 5:e19804. doi: 10.7554/eLife.19804
11. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
12. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. (2022) 82(12):2215–27. doi: 10.1016/j.molcel.2022.03.022
13. Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. (2018) 22(12):3826–36. doi: 10.26355/eurrev_201806_15267
14. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. (2004) 306(5704):2090–3. doi: 10.1126/science.1104742
15. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell. (2017) 168(3):344–61. doi: 10.1016/j.cell.2016.12.034
16. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. (2015) 59(2):298–308. doi: 10.1016/j.molcel.2015.06.011
17. Santana-Codina N, Gikandi A, Mancias JD. The role of NCOA4-mediated ferritinophagy in ferroptosis. Adv Exp Med Biol. (2021) 1301:41–57. doi: 10.1007/978-3-030-62026-4_4
18. Santana-Codina N, Mancias JD. The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals (Basel). (2018) 11(4):114. doi: 10.3390/ph11040114
19. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12(8):1425–8. doi: 10.1080/15548627.2016.1187366
20. Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Lipid peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci. (2022) 24(1):449. doi: 10.3390/ijms24010449
21. Wu ZF, Liu XY, Deng NH, Ren Z, Jiang ZS. Outlook of ferroptosis-targeted lipid peroxidation in cardiovascular disease. Curr Med Chem. (2023) 30(31):3550–61. doi: 10.2174/0929867330666221111162905
22. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
23. Golej DL, Askari B, Kramer F, Barnhart S, Vivekanandan-Giri A, Pennathur S, et al. Long-chain acyl-CoA synthetase 4 modulates prostaglandin E₂ release from human arterial smooth muscle cells. J Lipid Res. (2011) 52(4):782–93. doi: 10.1194/jlr.M013292
24. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. (2015) 10(7):1604–9. doi: 10.1021/acschembio.5b00245
25. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 Dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
26. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
27. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 Is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. (2019) 21(5):579–91. doi: 10.1038/s41556-019-0305-6
28. Huang F, Yang R, Xiao Z, Xie Y, Lin X, Zhu P, et al. Targeting ferroptosis to treat cardiovascular diseases: a new continent to be explored. Front Cell Dev Biol. (2021) 9:737971. doi: 10.3389/fcell.2021.737971
29. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 Promotes erastin-induced ferroptosis by suppressing system Xc. Cell Death Differ. (2020) 27(2):662–75. doi: 10.1038/s41418-019-0380-z
30. Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. (2018) 16(5):e2006203. doi: 10.1371/journal.pbio.2006203
31. Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids. (2012) 42(1):231–46. doi: 10.1007/s00726-011-0867-5
32. Xie Y, Kang R, Klionsky DJ, Tang D. GPX4 In cell death, autophagy, and disease. Autophagy. (2023) 19(10):2621–38. doi: 10.1080/15548627.2023.2218764
33. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. (2014) 156(1–2):317–31. doi: 10.1016/j.cell.2013.12.010
34. Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. (2008) 8(3):237–48. doi: 10.1016/j.cmet.2008.07.005
35. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. (2017) 6(8):e371. doi: 10.1038/oncsis.2017.65
36. Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. (2015) 88(Pt B):101–7. doi: 10.1016/j.freeradbiomed.2015.05.034
37. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 Plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
38. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 Is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
39. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
40. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6(1):41–53. doi: 10.1021/acscentsci.9b01063
41. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. (2014) 3:e02523. doi: 10.7554/eLife.02523
42. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. (2016) 12(7):497–503. doi: 10.1038/nchembio.2079
43. Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO2 Initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. (2018) 14(5):507–15. doi: 10.1038/s41589-018-0031-6
44. Xie LH, Fefelova N, Pamarthi SH, Gwathmey JK. Molecular mechanisms of ferroptosis and relevance to cardiovascular disease. Cells. (2022) 11(17):2726. doi: 10.3390/cells11172726
45. Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. (2017) 3(3):232–43. doi: 10.1021/acscentsci.7b00028
46. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. (2022) 608(7924):778–83. doi: 10.1038/s41586-022-05022-3
47. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. (2019) 12(7):497–503. doi: 10.1038/nchembio.2079
48. Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. Heart disease and stroke statistics-2023 update: a report from the American heart association. Circulation. (2023) 147(8):e93–e621. doi: 10.1161/CIR.0000000000001123
49. Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest. (1985) 76(5):1713–9. doi: 10.1172/JCI112160
50. Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. (2018) 314(3):H659–68. doi: 10.1152/ajpheart.00452.2017
51. Tang LJ, Luo XJ, Tu H, Chen H, Xiong XM, Li NS, et al. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol. (2021) 394(2):401–10. doi: 10.1007/s00210-020-01932-z
52. Miyamoto HD, Ikeda M, Ide T, Tadokoro T, Furusawa S, Abe K, et al. Iron overload via heme degradation in the endoplasmic Reticulum triggers ferroptosis in myocardial ischemia-reperfusion injury. JACC Basic Transl Sci. (2022) 7(8):800–19. doi: 10.1016/j.jacbts.2022.03.012
53. Lei D, Li B, Isa Z, Ma X, Zhang B. Hypoxia-elicited cardiac microvascular endothelial cell-derived exosomal miR-210-3p alleviate hypoxia/reoxygenation-induced myocardial cell injury through inhibiting transferrin receptor 1-mediated ferroptosis. Tissue Cell. (2022) 79:101956. doi: 10.1016/j.tice.2022.101956
54. Li T, Tan Y, Ouyang S, He J, Liu L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene. (2022) 808:145968. doi: 10.1016/j.gene.2021.145968
55. Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang JJ, Luo XJ, et al. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic Biol Med. (2021) 162:339–52. doi: 10.1016/j.freeradbiomed
56. Machado SE, Spangler D, Stacks DA, Darley-Usmar V, Benavides GA, Xie M, et al. Counteraction of myocardial ferritin heavy chain deficiency by heme oxygenase-1. Int J Mol Sci. (2022) 23(15):8300. doi: 10.3390/ijms23158300
57. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
58. Zhou Y, Shen Y, Chen C, Sui X, Yang J, Wang L, et al. The crosstalk between autophagy and ferroptosis: what can we learn to target drug resistance in cancer? Cancer Biol Med. (2019) 16(4):630–46. doi: 10.20892/j.issn.2095-3941.2019.0158
59. Chen HY, Xiao ZZ, Ling X, Xu RN, Zhu P, Zheng SY. ELAVL1 Is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating autophagy. Mol Med. (2021) 27(1):14. doi: 10.1186/s10020-021-00271-w
60. Lin L, Zhang MX, Zhang L, Zhang D, Li C, Li YL. Autophagy, pyroptosis, and ferroptosis: new regulatory mechanisms for atherosclerosis. Front Cell Dev Biol. (2022) 9:809955. doi: 10.3389/fcell.2021.809955
61. Ouyang S, You J, Zhi C, Li P, Lin X, Tan X, et al. Ferroptosis: the potential value target in atherosclerosis. Cell Death Dis. (2021) 12(8):782. doi: 10.1038/s41419-021-04054-3
62. Bao X, Luo X, Bai X, Lv Y, Weng X, Zhang S, et al. Cigarette tar mediates macrophage ferroptosis in atherosclerosis through the hepcidin/FPN/SLC7A11 signaling pathway. Free Radic Biol Med. (2023) 201:76–88. doi: 10.1016/j.freeradbiomed.2023.03.006
63. Li B, Wang C, Lu P, Ji Y, Wang X, Liu C, et al. IDH1 Promotes foam cell formation by aggravating macrophage ferroptosis. Biology (Basel). (2022) 11(10):1392. doi: 10.3390/biology11101392
64. Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118(4):620–36. doi: 10.1161/CIRCRESAHA.115.306301
65. Xu S. Iron and atherosclerosis: the link revisited. Trends Mol Med. (2019) 25(8):659–61. doi: 10.1016/j.molmed.2019.05.012
66. Yang K, Song H, Yin D. PDSS2 Inhibits the ferroptosis of vascular endothelial cells in atherosclerosis by activating Nrf2. J Cardiovasc Pharmacol. (2021) 77(6):767–76. doi: 10.1097/FJC.0000000000001030
67. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med. (2020) 160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026
68. Malhotra R, Wunderer F, Barnes HJ, Bagchi A, Buswell MD, O'Rourke CD, et al. Hepcidin deficiency protects against atherosclerosis. Arterioscler Thromb Vasc Biol. (2019) 39(2):178–87. doi: 10.1161/ATVBAHA.118.312215
69. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. (2019) 116(7):2672–80. doi: 10.1073/pnas.1821022116
70. Li X, Liang J, Qu L, Liu S, Qin A, Liu H, et al. Exploring the role of ferroptosis in the doxorubicin-induced chronic cardiotoxicity using a murine model. Chem Biol Interact. (2022) 363:110008. doi: 10.1016/j.cbi.2022.110008
71. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. (2023) 8(6):e169756. doi: 10.1172/jci.insight.169756
72. Zhou YJ, Duan DQ, Lu LQ, Tang LJ, Zhang XJ, Luo XJ, et al. The SPATA2/CYLD pathway contributes to doxorubicin-induced cardiomyocyte ferroptosis via enhancing ferritinophagy. Chem Biol Interact. (2022) 368:110205. doi: 10.1016/j.cbi.2022.110205
73. Wang W, Zhong X, Fang Z, Li J, Li H, Liu X, et al. Cardiac sirtuin1 deficiency exacerbates ferroptosis in doxorubicin-induced cardiac injury through the Nrf2/Keap1 pathway. Chem Biol Interact. (2023) 377:110469. doi: 10.1016/j.cbi.2023.110469
74. Wang Y, Ying X, Wang Y, Zou Z, Yuan A, Xiao Z, et al. Hydrogen sulfide alleviates mitochondrial damage and ferroptosis by regulating OPA3-NFS1 axis in doxorubicin-induced cardiotoxicity. Cell Signal. (2023) 107:110655. doi: 10.1016/j.cellsig.2023.110655
75. Yu W, Chen C, Xu C, Xie D, Wang Q, Liu W, et al. Activation of p62-NRF2 axis protects against doxorubicin-induced ferroptosis in cardiomyocytes: a novel role and molecular mechanism of resveratrol. Am J Chin Med. (2022) 50(8):2103–23. doi: 10.1142/S0192415X22500902
76. Yu Y, Wu T, Lu Y, Zhao W, Zhang J, Chen Q, et al. Exosomal thioredoxin-1 from hypoxic human umbilical cord mesenchymal stem cells inhibits ferroptosis in doxorubicin-induced cardiotoxicity via mTORC1 signaling. Free Radic Biol Med. (2022) 193(Pt 1):108–21. doi: 10.1016/j.freeradbiomed.2022.10.268
77. Zhang MW, Li XT, Zhang ZZ, Liu Y, Song JW, Liu XM, et al. Elabela blunts doxorubicin-induced oxidative stress and ferroptosis in rat aortic adventitial fibroblasts by activating the KLF15/GPX4 signaling. Cell Stress Chaperones. (2023) 28(1):91–103. doi: 10.1007/s12192-022-01317-6
78. He H, Wang L, Qiao Y, Yang B, Yin D, He M. Epigallocatechin-3-gallate pretreatment alleviates doxorubicin-induced ferroptosis and cardiotoxicity by upregulating AMPKα2 and activating adaptive autophagy. Redox Biol. (2021) 48:102185. doi: 10.1016/j.redox.2021.102185
79. Kitakata H, Endo J, Matsushima H, Yamamoto S, Ikura H, Hirai A, et al. MITOL/MARCH5 determines the susceptibility of cardiomyocytes to doxorubicin-induced ferroptosis by regulating GSH homeostasis. J Mol Cell Cardiol. (2021) 161:116–29. doi: 10.1016/j.yjmcc.2021.08.006
80. Zhu X, Wang X, Zhu B, Ding S, Shi H, Yang X. Disruption of histamine/H1R-STAT3-SLC7A11 axis exacerbates doxorubicin-induced cardiac ferroptosis. Free Radic Biol Med. (2022) 192:98–114. doi: 10.1016/j.freeradbiomed.2022.09.012
81. Tadokoro T, Ikeda M, Abe K, Ide T, Miyamoto HD, Furusawa S, et al. Ethoxyquin is a competent radical-trapping antioxidant for preventing ferroptosis in doxorubicin cardiotoxicity. J Cardiovasc Pharmacol. (2022) 80(5):690–9. doi: 10.1097/FJC.0000000000001328
82. Sun X, Sun P, Zhen D, Xu X, Yang L, Fu D, et al. Melatonin alleviates doxorubicin-induced mitochondrial oxidative damage and ferroptosis in cardiomyocytes by regulating YAP expression. Toxicol Appl Pharmacol. (2022) 437:115902. doi: 10.1016/j.taap.2022.115902
83. Liu Y, Zeng L, Yang Y, Chen C, Wang D, Wang H. Acyl-CoA thioesterase 1 prevents cardiomyocytes from doxorubicin-induced ferroptosis via shaping the lipid composition. Cell Death Dis. (2020) 11(9):756. doi: 10.1038/s41419-020-02948-2
84. Quagliariello V, De Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A, et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. (2021) 20(1):150. doi: 10.1186/s12933-021-01346-y
85. Li D, Song C, Zhang J, Zhao X. ROS And iron homeostasis dependent ferroptosis play a vital role in 5-fluorouracil induced cardiotoxicity in vitro and in vivo. Toxicology. (2022) 468:153113. doi: 10.1016/j.tox.2022.153113
86. Li D, Song C, Zhang J, Zhao X. Resveratrol alleviated 5-FU-induced cardiotoxicity by attenuating GPX4 dependent ferroptosis. J Nutr Biochem. (2023) 112:109241. doi: 10.1016/j.jnutbio.2022.109241
87. Min J, Wu L, Liu Y, Song G, Deng Q, Jin W, et al. Empagliflozin attenuates trastuzumab-induced cardiotoxicity through suppression of DNA damage and ferroptosis. Life Sci. (2023) 312:121207. doi: 10.1016/j.lfs.2022.121207
88. Peng H, Fu S, Wang S, Xu H, Dhanasekaran M, Chen H, et al. Ablation of FUNDC1-dependent mitophagy renders myocardium resistant to paraquat-induced ferroptosis and contractile dysfunction. Biochim Biophys Acta Mol Basis Dis. (2022) 1868(9):166448. doi: 10.1016/j.bbadis.2022.166448
89. Kong C, Ni X, Wang Y, Zhang A, Zhang Y, Lin F, et al. ICA69 Aggravates ferroptosis causing septic cardiac dysfunction via STING trafficking. Cell Death Discov. (2022) 8(1):187. doi: 10.1038/s41420-022-00957-y
90. Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang CF, et al. Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol Med Rep. (2020) 22(1):175–84. doi: 10.3892/mmr.2020.11114
91. Xiao Z, Kong B, Fang J, Qin T, Dai C, Shuai W, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered. (2021) 12(2):9367–76. doi: 10.1080/21655979.2021.2001913
92. Chen Z, Cao Z, Gui F, Zhang M, Wu X, Peng H, et al. TMEM43 Protects against sepsis-induced cardiac injury via inhibiting ferroptosis in mice. Cells. (2022) 11(19):2992. doi: 10.3390/cells11192992
93. Wang X, Simayi A, Fu J, Zhao X, Xu G. Resveratrol mediates the miR-149/HMGB1 axis and regulates the ferroptosis pathway to protect myocardium in endotoxemia mice. Am J Physiol Endocrinol Metab. (2022) 323(1):E21–32. doi: 10.1152/ajpendo.00227.2021
94. Zhou B, Zhang J, Chen Y, Liu Y, Tang X, Xia P, et al. Puerarin protects against sepsis-induced myocardial injury through AMPK-mediated ferroptosis signaling. Aging (Albany NY). (2022) 14(8):3617–32. doi: 10.18632/aging.204033
95. Luo EF, Li HX, Qin YH, Qiao Y, Yan GL, Yao YY, et al. Role of ferroptosis in the process of diabetes-induced endothelial dysfunction. World J Diabetes. (2021) 12(2):124–37. doi: 10.4239/wjd.v12.i2.124
96. Li X, Li Z, Dong X, Wu Y, Li B, Kuang B, et al. Astragaloside IV attenuates myocardial dysfunction in diabetic cardiomyopathy rats through downregulation of CD36-mediated ferroptosis. Phytother Res. (2023) 37(7):3042–56. doi: 10.1002/ptr.7798
97. Wu Y, Huang T, Li X, Shen C, Ren H, Wang H, et al. Retinol dehydrogenase 10 reduction mediated retinol metabolism disorder promotes diabetic cardiomyopathy in male mice. Nat Commun. (2023) 14(1):1181. doi: 10.1038/s41467-023-36837-x
98. Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. (2022) 12(2):708–22. doi: 10.1016/j.apsb.2021.10.005
99. Du S, Shi H, Xiong L, Wang P, Shi Y. Canagliflozin mitigates ferroptosis and improves myocardial oxidative stress in mice with diabetic cardiomyopathy. Front Endocrinol (Lausanne). (2022) 13:1011669. doi: 10.3389/fendo.2022.1011669
100. Kaspar M, Fette G, Güder G, Seidlmayer L, Ertl M, Dietrich G, et al. Underestimated prevalence of heart failure in hospital inpatients: a comparison of ICD codes and discharge letter information. Clin Res Cardiol. (2018) 107(9):778–87. doi: 10.1007/s00392-018-1245-z
101. Woulfe KC, Bruns DR. From pediatrics to geriatrics: mechanisms of heart failure across the life-course. J Mol Cell Cardiol. (2019) 126:70–6. doi: 10.1016/j.yjmcc.2018.11.009
102. Ma S, He LL, Zhang GR, Zuo QJ, Wang ZL, Zhai JL, et al. Canagliflozin mitigates ferroptosis and ameliorates heart failure in rats with preserved ejection fraction. Naunyn Schmiedebergs Arch Pharmacol. (2022) 395(8):945–62. doi: 10.1007/s00210-022-02243-1
103. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. (2020) 127(4):486–501. doi: 10.1161/CIRCRESAHA.120.316509
104. Zheng H, Shi L, Tong C, Liu Y, Hou M. Circsnx12 is involved in ferroptosis during heart failure by targeting miR-224-5p. Front Cardiovasc Med. (2021) 8:656093. doi: 10.3389/fcvm.2021.656093
105. Chen X, Xu S, Zhao C, Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun. (2019) 516(1):37–43. doi: 10.1016/j.bbrc.2019.06.015
106. Nattel S, Heijman J, Zhou L, Dobrev D. Molecular basis of atrial fibrillation pathophysiology and therapy: a translational perspective. Circ Res. (2020) 127(1):51–72. doi: 10.1161/CIRCRESAHA.120.316363
107. Yu LM, Dong X, Huang T, Zhao JK, Zhou ZJ, Huang YT, et al. Inhibition of ferroptosis by icariin treatment attenuates excessive ethanol consumption-induced atrial remodeling and susceptibility to atrial fibrillation, role of SIRT1. Apoptosis. (2023) 28(3–4):607–26. doi: 10.1007/s10495-023-01814-8
108. Dai C, Kong B, Qin T, Xiao Z, Fang J, Gong Y, et al. Inhibition of ferroptosis reduces susceptibility to frequent excessive alcohol consumption-induced atrial fibrillation. Toxicology. (2022) 465:153055. doi: 10.1016/j.tox.2021.153055
109. Fang J, Kong B, Shuai W, Xiao Z, Dai C, Qin T, et al. Ferroportin-mediated ferroptosis involved in new-onset atrial fibrillation with LPS-induced endotoxemia. Eur J Pharmacol. (2021) 913:174622. doi: 10.1016/j.ejphar.2021.174622
110. Di Palo KE, Barone NJ. Hypertension and heart failure: prevention, targets, and treatment. Heart Fail Clin. (2020) 16(1):99–106. doi: 10.1016/j.hfc.2019.09.001
111. Zhang Z, Tang J, Song J, Xie M, Liu Y, Dong Z, et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic Biol Med. (2022) 181:130–42. doi: 10.1016/j.freeradbiomed.2022.01.020
112. Zhang X, Zheng C, Gao Z, Chen H, Li K, Wang L, et al. SLC7A11/xCT Prevents cardiac hypertrophy by inhibiting ferroptosis. Cardiovasc Drugs Ther. (2022) 36(3):437–47. doi: 10.1007/s10557-021-07220-z
113. Li XT, Song JW, Zhang ZZ, Zhang MW, Liang LR, Miao R, et al. Sirtuin 7 mitigates renal ferroptosis, fibrosis and injury in hypertensive mice by facilitating the KLF15/Nrf2 signaling. Free Radic Biol Med. (2022) 193(Pt 1):459–73. doi: 10.1016/j.freeradbiomed.2022.10.320
114. Gao P, Cao M, Jiang X, Wang X, Zhang G, Tang X, et al. Cannabinoid receptor 2-centric molecular feedback loop drives necroptosis in diabetic heart injuries. Circulation. (2023) 147(2):158–74. doi: 10.1161/CIRCULATIONAHA.122.059304
115. Wang N, Ma H, Li J, Meng C, Zou J, Wang H, et al. HSF1 Functions as a key defender against palmitic acid-induced ferroptosis in cardiomyocytes. J Mol Cell Cardiol. (2021) 150:65–76. doi: 10.1016/j.yjmcc.2020.10.010
116. Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. (2020) 160:303–18. doi: 10.1016/j.freeradbiomed.2020.08.009
117. Liu L, Pang J, Qin D, Li R, Zou D, Chi K, et al. Deubiquitinase OTUD5 as a novel protector against 4-HNE-triggered ferroptosis in myocardial ischemia/reperfusion injury. Adv Sci (Weinh). (2023) 10(28):e2301852. doi: 10.1002/advs.202301852
Keywords: cardiovascular disease, ferroptosis, iron metabolism, lipid peroxidation, amino acid metabolism
Citation: Zhang J and Guo C (2023) Current progress of ferroptosis in cardiovascular diseases. Front. Cardiovasc. Med. 10:1259219. doi: 10.3389/fcvm.2023.1259219
Received: 15 July 2023; Accepted: 29 September 2023;
Published: 24 October 2023.
Edited by:
Michio Shimabukuro, Fukushima Medical University, JapanReviewed by:
Zhihua Wang, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaPan Gao, Fudan University, China
© 2023 Zhang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caixia Guo Y3hnYmJAMTYzLmNvbQ==