
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med. , 24 August 2023
Sec. Cardiovascular Genetics and Systems Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1225667
 Chon-Hou Chan1,†
Chon-Hou Chan1,† Man-Fong Chu2,†
Man-Fong Chu2,† U-Po Lam2
U-Po Lam2 Toi-Meng Mok2
Toi-Meng Mok2 Weng-Chio Tam2*
Weng-Chio Tam2* Brian Tomlinson3Ricardo Coelho1Màrio Évora2
Brian Tomlinson3Ricardo Coelho1Màrio Évora2
Noonan syndrome with multiple lentigines (NSML, formerly known as LEOPARD syndrome) is a variant of Noonan syndrome which is an autosomal dominant disorder. Most cases of NSML are secondary to mutations of the protein-tyrosine phosphatase nonreceptor type 11 (PTPN11). Hypertrophic cardiomyopathy (HCM) remains the most frequent and serious cardiac abnormality in this inherited syndrome, and it may lead to sudden cardiac death related to HCM-associated outflow obstruction and fatal arrhythmia. Beyond cardiac involvement, NSML may present with multiple lentigines, ocular hypertelorism, genital anomalies, short stature and deafness. Herein, we report three patients with NSML among three generations in one family, all presenting with multiple lentigines, HCM and other distinctive clinical and molecular features, including facial dysmorphism, deafness, family history of sudden death and PTPN11 mutations. This case series highlights the importance of early echocardiography examinations for patients with NSML. Careful family screening and genetic counselling are also necessary, especially in patients with diffuse lentigines or a history of sudden death among family members. We also discuss the distinctive cardiac features and phenotypic characteristics at different stages of NSML, including childhood, adulthood and elderhood.
Noonan syndrome with multiple lentigines (NSML), formerly known as LEOPARD syndrome, is a rare multi-systemic disorder that is inherited in an autosomal-dominant manner. It was first described by Gorlin et al. in 1969, and is characterized by seven major features: lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, growth retardation, and deafness (1). About 200 cases have been reported in the English literature to date, but the incidence of NSML is not well documented. NSML is a “RASopathy” related to gene variants within the Ras/Mitogen-activated protein kinase (Ras/MAPK) pathway (2). Approximately 85% of patients with NSML are associated with missense mutations in exon 7, 12 or 13 of the PTPN11 on chromosome 12q24 (3, 4). PTPN11 mutations down-regulate the activity of Src homology-2 domain-containing protein-tyrosine phosphatase 2 (SHP-2), which enhances melanin synthesis in melanocytes causing diffuse lentigines (5). Hypertrophic cardiomyopathy (HCM), one of the diagnostic criteria of NSML, can be asymptomatic and easily missed at its early stage. Herein, we report three Han Chinese patients with NSML among three generations in one family, with an emphasis on their echocardiographic features and importance of family screening.
A 36-year-old male with a past history of bilateral congenital hearing loss presented to the dermatology clinic with diffuse black to brownish pigmented macules locating on his face, trunk and four limbs since early childhood, varying in shape and size, and with diameters ranging from 1 to 5 mm (Figures 1A–C). These skin lesions increased in number and darkened during his growing up. Café-au-lait macule was revealed (Figure 1C). Facial dysmorphic features including ocular hypertelorism, ptosis, thick lips (Figure 1A), retrognathism and bilateral low set ears (Figure 1D) were remarkable. He was referred to cardiology for cardiac assessment. The electrocardiogram showed sinus rhythm and left ventricular hypertrophy with strain pattern. Echocardiography demonstrated eccentric left ventricular hypertrophy (septal wall thickness: 22 mm) without outflow tract obstruction (Figures 1E,F, Supplementary Video S1). Cardiac magnetic resonance imaging revealed consistent findings with echocardiography (Figures 1G,H). NSML was confirmed based on the diagnostic criteria proposed by Voron et al. (6). Next generation sequencing revealed a heterozygous missense mutation in the PTPN11, exon 7, c.836 A > G, p.Tyr279Cys. This is a well-known genetic variant which has been published previously (3, 4, 7). After assessing his risk of sudden cardiac death (SCD), an implantable cardioverter defibrillator was not strongly indicated for primary prevention. We initiated beta-blockers and suggested him to avoid vigorous physical exercise. Detailed family screening was then performed, which found that his son (individual III-2), elder brother (individual II-2) and mother (individual I-8) also had multiple lentigines since childhood, and that his elder brother (individual II-2) had died at the age of 32 due to SCD (Pedigree, Figure 2).
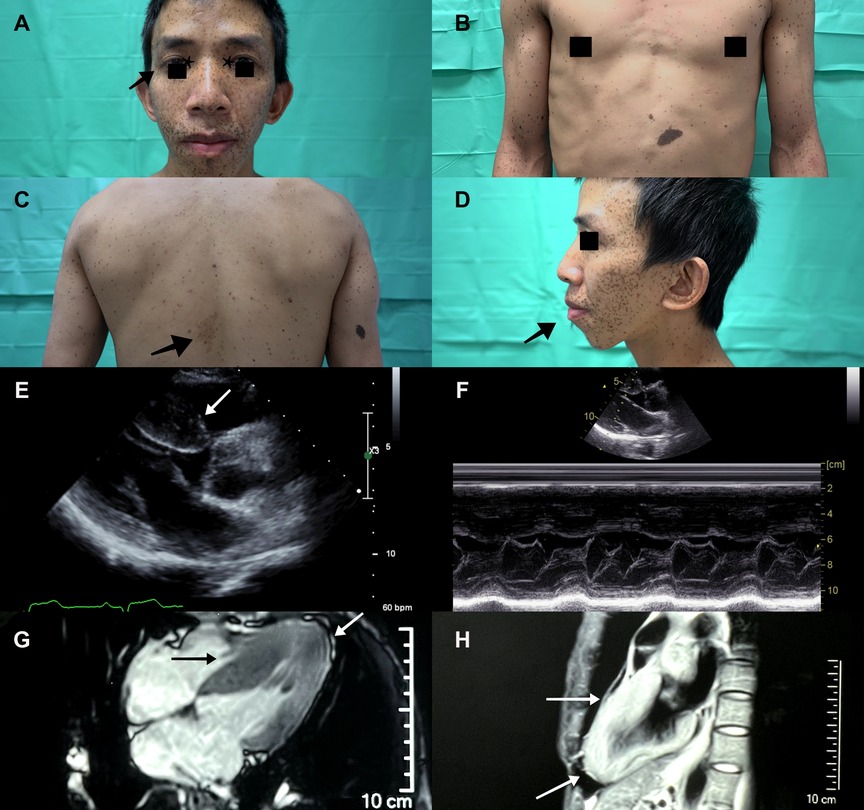
Figure 1. A Diffuse black to brownish lentigines seen mainly on the face of the proband (individual II-3). Ocular hypertelorism (asterisk), ptosis (arrowhead) and thick lips were remarkable. (B) Multiple lentigines over the anterior trunk and limbs varied in shape and size, and with diameters ranging from to 5 mm. (C) A café au lait macule (arrowhead) was seen on his mid-back. (D) Low set ears and retrognathism (arrowhead) were revealed. Echocardiography demonstrated (E) eccentric left ventricular hypertrophy (septal wall thickness: 22 mm, arrowhead), (F) without left ventricle outflow tract obstruction. (G,H) Cardiac magnetic resonance imaging showed hypertrophic segments over septal and apical walls (arrowhead).
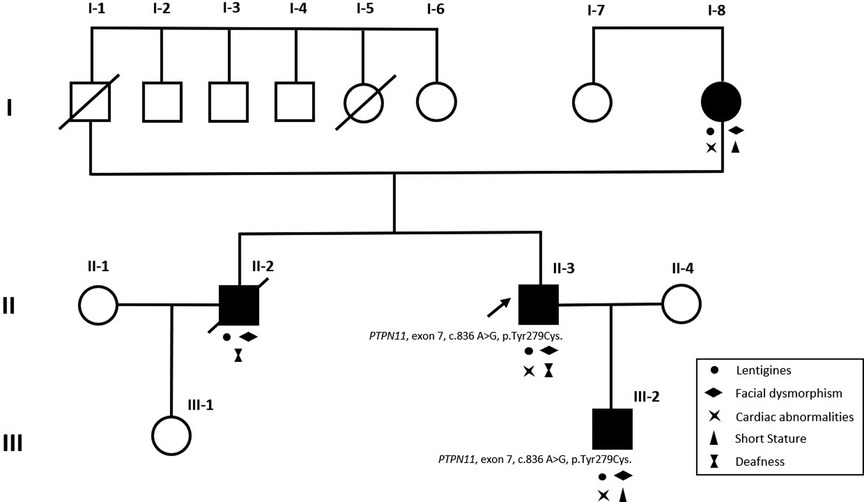
Figure 2. Pedigree of the proband (II-3, arrowhead). The black-filled squares and circle indicated the individuals with clinical evidence of NSML. Both proband (II-3), proband's mother (I-8), elder brother (II-2) and son (III-2) presented with multiple lentigines and facial dysmorphism. Clinical features of NSML were labelled with relevant symbols in the pedigree. Same heterozygous missense mutation of PTPN11 was confirmed in proband (II-3) and proband's son (III-2). Two unaffected family members (I-7 and III-1) showed no clinical evidence of NSML. Individual II-2 died at the age of 32 due to sudden cardiac death.
A 8-year-old boy with a 2-year history of multiple lentigines (Figures 3A,C) and two café-au-lait macules on his limbs since birth was found to have short stature (−3.0 standard deviation score from average), ocular hypertelorism (Figure 3A), low set ears (Figure 3B), pectus excavatum (Figure 3C) and systolic murmur upon physical examination. He denied any hearing loss. Echocardiography demonstrated normal left/right ventricle size and function, septal wall hypertrophy (maximal wall thickness: 17 mm) without left ventricle outflow tract obstruction (LVOTO) (Figure 3D and Supplementary Video S2), a secundum atrial septal defect (7 mm, shunting from left to right) (Figure 3E), and right pulmonary artery stenosis (peak pressure gradient around 14 mmHg) (Figure 3F). Next generation sequencing revealed the same heterozygous mutation in the PTPN11, exon 7, c.836 A > G, p.Tyr279Cys. He was treated with beta-blockers and serial follow-up at our cardiology department with periodic echocardiography.
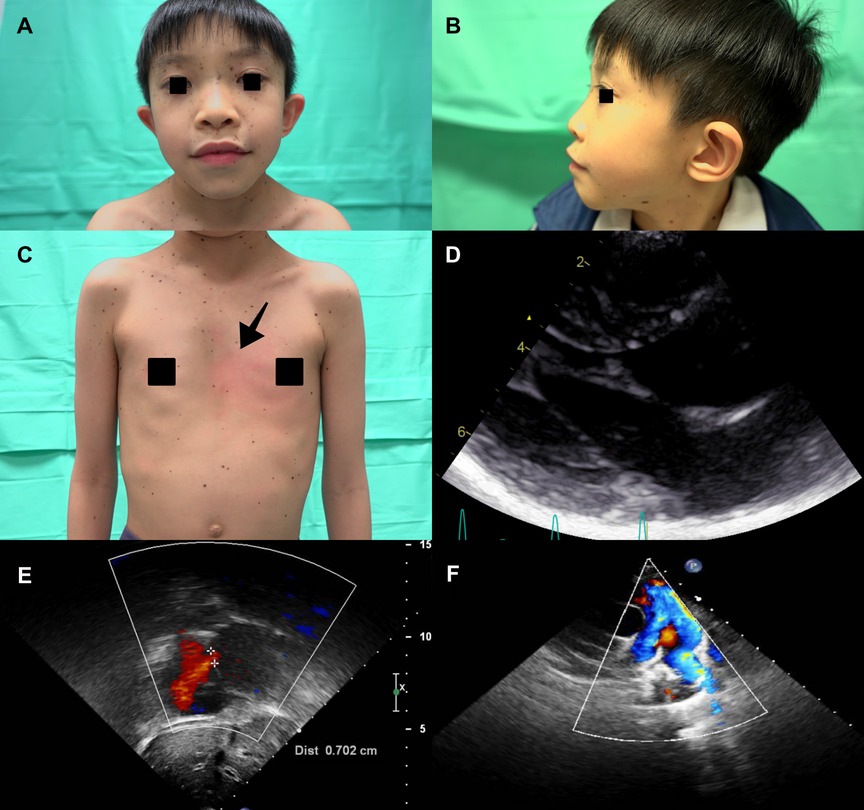
Figure 3. (A) The proband's son (individual III-2) presented with diffuse lentigines over the face, thick lips and ocular hypertelorism. (B) Low set ears and (C) Diffuse lentigines over the trunk and pectus excavatum (arrowhead) were also revealed. Echocardiography showed (D) septal wall hypertrophy without intra-cavity or left ventricle outflow tract obstruction. (E) Secundum atrial septal defect (cross-mark) (defect size about 7 mm, shunting from left to right). (F) Narrowing color-flow at the right pulmonary artery, indicating right pulmonary artery stenosis.
A 66-year-old woman with a history of diffuse lentigines since childhood (Figures 4A–C) came to the cardiology clinic with intermittent exertional dyspnea and exercise intolerance in the recent few months. Ocular hypertelorism was revealed (Figure 4A). She had no hearing loss. Electrocardiography showed sinus rhythm with left ventricular hypertrophy using voltage criteria. Echocardiography revealed eccentric septal hypertrophy (septal wall thickness: 26 mm) (Figure 4D), apical aneurysm and mid-cavity obstruction with pressure gradient (17 mmHg) (Figures 4E,F and Supplementary Video S3). Implantation of an implantable cardioverter defibrillator followed by septal wall myomectomy was proposed based on SCD risk stratification (8). Table 1 summarizes the clinical manifestations and cardiac phenotypes of these three patients (individuals II-3, III-2, I-8). Supplementary Table S1 also summarizes clinical phenotypic features of the unaffected family members (individuals I-7 & III-1).
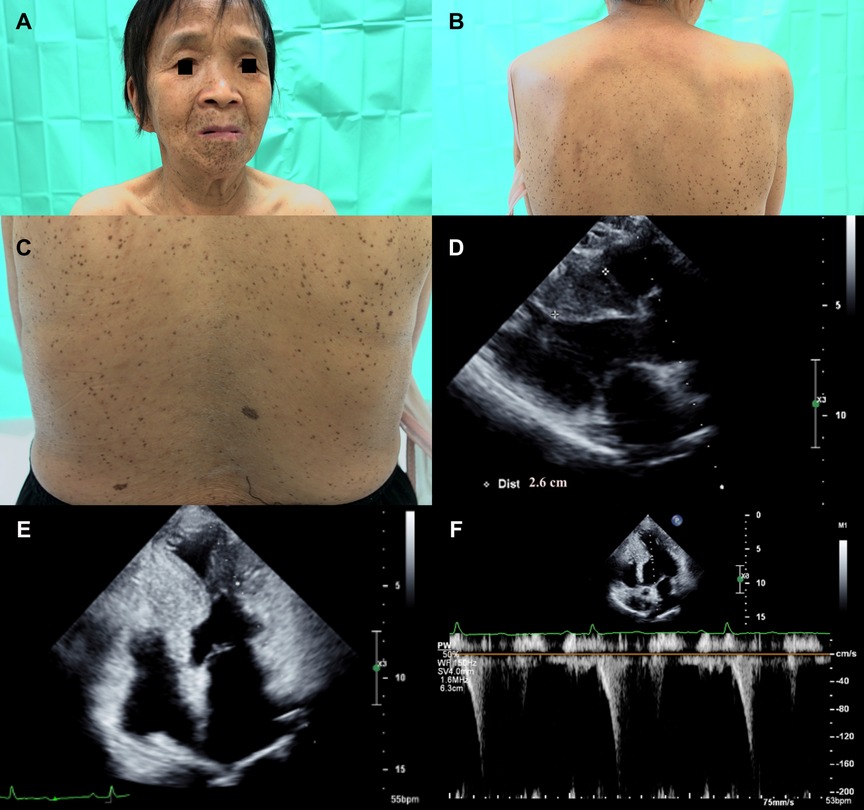
Figure 4. (A) The proband's mother (individual I-8) presented with ocular hypertelorism and diffuse black to brownish facial lentigines. (B) Numerous lentigines on her back, varying in size and shape. (C) Close-up of back lentigines with diameters ranging from 1 mm to 5 mm. Echocardiography revealed (D) significant septal wall hypertrophy (septal wall thickness: 26 mm, cross-mark). (E) Apical aneurysm formation was revealed with midventricular obstruction. (F) Pulsed wave Doppler showed mid-cavity pressure gradient was around 17 mmHg.
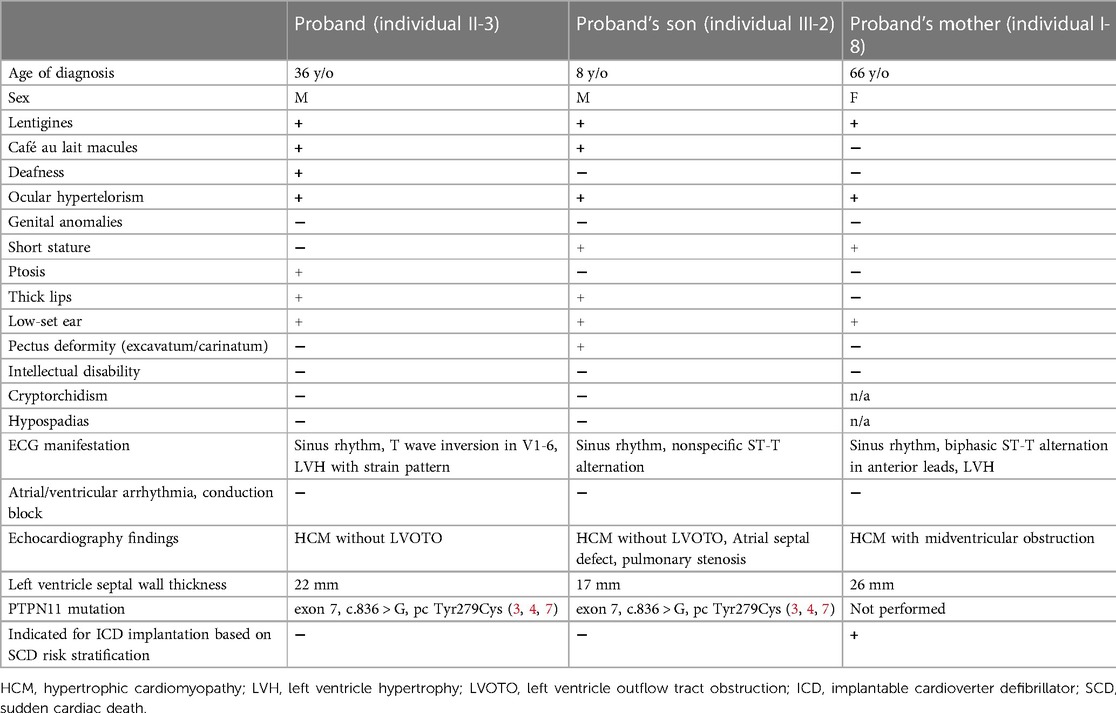
Table 1. Clinical characteristics and cardiac phenotypes of the three patients with NSML.
In this case report, we document the clinical manifestations of NSML among three different generations in one family, with an emphasis on the cardiac and cutaneous presentations at different stages of the same disease in childhood, adulthood and elderhood. Of note, none of the three patients received any treatment or evaluations until the diagnosis of NSML was confirmed. Our report demonstrates that NSML-related HCM without treatment can cause left ventricle outflow/midventricular obstruction, leading to sudden cardiac death. We also identified the distinctive clinical manifestations and evolution of the disease, especially the cardiac phenotypes at different stages.
Approximately 95% of cases of NSML are caused by mutations in 3 loci: PTPN11 on chromosome 12q24, RAF1 on chromosome 3p25.2, and BRAF on chromosome 7q34. About 85% of all cases of NSML are related to PTPN11 mutations, which are mostly detected on exons 7, 12, and 13 (2). The cardiac phenotypes may be related to specific exon mutations. For example, mutations on exon 7 and 12 are associated with HCM, while mutations on exon 13 are associated with SCD (9, 10). Regarding multiple lentigines formation, plexin B1 can be associated with c-MET, which inhibits the HGF/c-MET pathway by blocking SHP-2 activity, followed by the abrogation of MAPK/ERK and PI3K/AKT activation in melanocytes. This may explain why mutations in SHP-2 cause generalized lentigines (11). Also, the lentigines usually appear first in one's early childhood with main involvement of face and trunk, and then gradually increase in size and numbers with darkening as one grows up. We demonstrated these findings in our case report. The proband's son (individual III-2) presented with fewer lentigines while proband (individual II-3) and proband's mother (individual I-8) were evident with numerous and increased size of lentigines, predominantly distributed on the face and upper part of the trunk.
The PTPN11, located in chromosome 12q24, encodes a protein with the Src homology-2 (SH2) domain and tyrosine phosphatase domain containing the active site (12). NS and NSML are two disorders that are categorized as RASopathies. Pathogenic SHP-2 variants are found in NS, NSML and other RASopathies, and large variations among phenotypes have also been observed (13, 14). Several PTPN11 missense mutations that cause NSML have been identified and p.Tyr279Cys in exon 7 is one of the most frequent mutations reported in the literature including in one Han Chinese subject (4, 7). Germline mutations in SHP-2 are known to cause both NS and NSML, two clinically similar autosomal dominant developmental disorders (15). Although NS and NSML patients display multiple overlapping phenotypic traits in early childhood, including short stature and facial dysmorphic features, NSML is difficult to differentiate from NS based on clinical manifestation at the early stage of the disease until the presentation of typical multiple lentigines and genetic confirmation. However, some specific features reflect the unique mechanisms of NS and NSML. NS and NSML SHP-2 variants cause opposite effects on the phosphatase activity of SHP-2 (15). Recent studies demonstrated that NS was associated with gain-of-function mutations of PTPN11 encoding SHP-2, which were mainly located in the interface between amino-terminal src-homology 2 (N-SH2) and protein-tyrosine phosphatase (PTP) domains. In contrast, NSML was associated with dominant-negative or loss-of-function mutations of SHP-2, which were located only in the PTP domain and are closed to the substrate binding or catalytic sites of SHP-2 (16, 17). The NSML associated SHP-2 mutant Tyr279Cys impaired phosphatase activity and weakened the intramolecular interaction between N-SH2 and PTP domains. The weakened interaction resulted in the N-SH2 domain being more readily activated by competing phospho-tyrosine ligands. Simultaneously, the NSML associated SHP-2 mutant Tyr279Cys increased the propensity to undergo the transition from a closed, autoinhibited conformation to an open, activated state (15, 18). The open conformation of NSML associated SHP2 mutations enhance SH2 domain protein-protein interactions that likely lead to propagating aberrant signaling and relate to the pathogenesis of NSML (15). Genotype-related phenotype heterogeneity relies on the developmental time. NSML patients may have the higher risk of hearing loss. While both NS and NSML patients develop congenital heart defects, the proportion of patients are different: the majority (85%) of NSML patients present with HCM, whereas pulmonary stenosis is predominant in NS patients (19).
As illustrated in this case report, the same PTPN11 mutation in exon 7, c.836 A > G, p.Tyr279Cys was detected in the proband (individual II-3) and proband's son (individual III-2). Interestingly, other than HCM, the proband's son (individual III-2) presented with pulmonary stenosis and atrial septal defect, which are usually associated with NS resulting from exon 3 and 8 mutations of the PTPN11. To our best knowledge, these echocardiography findings have rarely been reported as the cardiac phenotypes of exon 7 mutations of the PTPN11. Further research on genotype and phenotype correlations is warranted.
In general, 55% of HCM cases are familial and 45% are sporadic (20). Approximately 75% of familial form of HCM is autosomal dominance inheritance (8). The natural history of HCM, particularly patterns of left ventricular remodeling may be related to clinical outcomes and risk of SCD (21). In NSML, if one parent is affected, a 50% recurrence risk is appropriate (22). Thus, complete family screening and genetic counselling is needed. Clinicians may consider whole exome sequencing study to determine the deleterious and pathogenic variants, and simultaneously to clarify any potential disease contributing NSML variation, particularly some individuals with atypical phenotypic features and proband's offspring.
In summary, NSML is a rare autosomal dominant disease characterized by facial dysmorphism, specific cutaneous and cardiac manifestations. This case report highlights the clinical spectrum of this rare disease in childhood, adulthood and elderhood. As HCM-related SCD remains the leading cause of death, early and periodic cardiovascular assessments are of extreme importance for such patients. Complete family screening and genetic counselling of different generations should be performed. Additional genetic testing could shed light on genotype-phenotype correlations.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the 0041/MEC/N/2022 Centro Hospitalar Conde de São Januário Macau Medical Ethical Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Concept – WT; Design – MC; Supervision – ME; Materials – RC and CC; Data collection and/ or processing – CC; Analysis and/or interpretation – MC; Literature search – UL; Writing – CC; Critical review – TM and BT. All authors contributed to the article and approved the submitted version.
This study was supported by association of diagnosis and therapeutic intervention of Macau.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1225667/full#supplementary-material
1. Gelb BD, Tartaglia M. Noonan syndrome with multiple lentigines. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al., editors. Genereviews((R)). Seattle, WA: University of Washington, Seattle (1993). Available from: https://www.ncbi.nlm.nih.gov/books/NBK1383/
2. Limongelli G, Sarkozy A, Pacileo G, Calabro P, Digilio MC, Maddaloni V, et al. Genotype-phenotype analysis and natural history of left ventricular hypertrophy in leopard syndrome. Am J Med Genet A. (2008) 146A(5):620–8. doi: 10.1002/ajmg.a.32206
3. Martinez-Quintana E, Rodriguez-Gonzalez F. Leopard syndrome caused by Tyr279cys mutation in the Ptpn11 gene. Mol Syndromol. (2012) 2(6):251–3. doi: 10.1159/000335995
4. Wang Y, Chen C, Wang DW. Leopard syndrome caused by heterozygous missense mutation of Tyr 279 Cys in the Ptpn11 gene in a sporadic case of Chinese han. Int J Cardiol. (2014) 174(3):e101–4. doi: 10.1016/j.ijcard.2014.04.161
5. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in Ptpn11, encoding the protein tyrosine phosphatase shp-2, cause Noonan syndrome. Nat Genet. (2001) 29(4):465–8. doi: 10.1038/ng772
6. Voron DA, Hatfield HH, Kalkhoff RK. Multiple lentigines syndrome. Case report and review of the literature. Am J Med. (1976) 60(3):447–56. doi: 10.1016/0002-9343(76)90764-6
7. National Center for Biotechnology Information. Clinvar (2013). Available at: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000013328.61 (Accessed July 1, 2023).
8. Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 Aha/Acc guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. (2020) 142(25):e558–631. doi: 10.1161/CIR.0000000000000937
9. Kauffman H, Ahrens-Nicklas RC, Calderon-Anyosa RJC, Ritter AL, Lin KY, Rossano JW, et al. Genotype-phenotype association by echocardiography offers incremental value in patients with Noonan syndrome with multiple lentigines. Pediatr Res. (2021) 90(2):444–51. doi: 10.1038/s41390-020-01292-7
10. Sarkozy A, Conti E, Seripa D, Digilio MC, Grifone N, Tandoi C, et al. Correlation between Ptpn11 gene mutations and congenital heart defects in Noonan and leopard syndromes. J Med Genet. (2003) 40(9):704–8. doi: 10.1136/jmg.40.9.704
11. Soong J, Scott G. Plexin B1 inhibits met through direct association and regulates Shp2 expression in melanocytes. J Cell Sci. (2013) 126(Pt 2):688–95. doi: 10.1242/jcs.119487
12. Pandey R, Saxena M, Kapur R. Role of Shp2 in hematopoiesis and leukemogenesis. Curr Opin Hematol. (2017) 24(4):307–13. doi: 10.1097/MOH.0000000000000345
13. Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, et al. Ptpn11 mutations in Noonan syndrome: molecular Spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet. (2002) 70(6):1555–63. doi: 10.1086/340847
14. Shoji Y, Ida S, Niihori T, Aoki Y, Okamoto N, Etani Y, et al. Genotype-phenotype correlation analysis in Japanese patients with Noonan syndrome. Endocr J. (2019) 66(11):983–94. doi: 10.1507/endocrj.EJ18-0564
15. Yu ZH, Zhang RY, Walls CD, Chen L, Zhang S, Wu L, et al. Molecular basis of gain-of-function leopard syndrome-associated Shp2 mutations. Biochemistry. (2014) 53(25):4136–51. doi: 10.1021/bi5002695
16. Hijikata A, Tsuji T, Shionyu M, Shirai T. Decoding disease-causing mechanisms of missense mutations from supramolecular structures. Sci Rep. (2017) 7(1):8541. doi: 10.1038/s41598-017-08902-1
17. Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. Ptpn11 (Shp2) mutations in leopard syndrome have dominant negative, not activating, effects. J Biol Chem. (2006) 281(10):6785–92. doi: 10.1074/jbc.M513068200
18. Qiu W, Wang X, Romanov V, Hutchinson A, Lin A, Ruzanov M, et al. Structural insights into Noonan/leopard syndrome-related mutants of protein-tyrosine phosphatase Shp2 (Ptpn11). BMC Struct Biol. (2014) 14:10. doi: 10.1186/1472-6807-14-10
19. Grant AR, Cushman BJ, Cave H, Dillon MW, Gelb BD, Gripp KW, et al. Assessing the gene-disease association of 19 genes with the rasopathies using the clingen gene curation framework. Hum Mutat. (2018) 39(11):1485–93. doi: 10.1002/humu.23624
20. Cam FS, Guray M. Hypertrophic cardiomyopathy: pathological features and molecular pathogenesis. Anadolu Kardiyol Derg. (2004) 4(4):327–30.15590362
21. Monda E, Prosnitz A, Aiello R, Lioncino M, Norrish G, Caiazza M, et al. Natural history of hypertrophic cardiomyopathy in Noonan syndrome with multiple lentigines. Circ Genom Precis Med. (2023) 3:e003861. doi: 10.1161/CIRCGEN.122.003861
Keywords: hypertrophic cardiomyopathy, Noonan syndrome with multiple lentigines, inherited disorder, sudden cardiac death, echocardiography
Citation: Chan C-H, Chu M-F, Lam U-P, Mok T-M, Tam W-C, Tomlinson B, Coelho R and Évora M (2023) Case report: Distinctive cardiac features and phenotypic characteristics of Noonan syndrome with multiple lentigines among three generations in one family. Front. Cardiovasc. Med. 10:1225667. doi: 10.3389/fcvm.2023.1225667
Received: 19 May 2023; Accepted: 8 August 2023;
Published: 24 August 2023.
Edited by:
Georges Michel Nemer, Hamad bin Khalifa University, QatarReviewed by:
Jirko Kühnisch, Charité University Medicine Berlin, Germany© 2023 Chan, Chu, Lam, Mok, Tam, Tomlinson, Coelho and Évora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weng-Chio Tam Y2hpbzIwMDEyQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.