 Xun Wang1,†
Xun Wang1,† Dan Wang
Dan Wang Qiming Liu
Qiming Liu Yunbin Xiao
Yunbin Xiao- 1Department of Cardiology, Hunan Children’s Hospital, Changsha, China
- 2Department of Cardiovascular Medicine, The Second Xiangya Hospital of Central South University, Changsha, China
- 3Xiangya School of Medicine, Central South University, Changsha, China
This study reports the first case of a patient with chromosomal 2p16.1p15 microduplication syndrome complicated by pulmonary arterial hypertension (PAH). A female infant was admitted to the hospital suffering from dyskinesia and developmental delay, and conventional echocardiography revealed an atrial septal defect (ASD), which was not taken seriously or treated at that time. Two years later, preoperative right heart catheterization for ASD closure revealed a mean pulmonary artery pressure (mPAP) of 45 mmHg. The mPAP was reduced, and the condition was stabilized after drug therapy. A genomic copy number duplication (3×) of at least 2.58 Mb in the 2p16.1p15 region on the paternal chromosome was revealed. Multiple Online Mendelian Inheritance in Man (OMIM) genes are involved in this genomic region, such as BCL11A, EHBP1, FAM161A, PEX13, and REL. EHBP1 promotes a molecular phenotypic transformation of pulmonary vascular endothelial cells and is thought to be involved in the rapidly developing PAH of this infant. Collectively, our findings contribute to the knowledge of the genes involved and the clinical manifestations of the 2p16.1p15 microduplication syndrome. Moreover, clinicians should be alert to the possibility of PAH and take early drug intervention when facing patients with 2p16.1p15 microduplications.
Introduction
Pulmonary hypertension (PH) is a group of diseases characterized by a progressive increase of mean pulmonary arterial pressure (mPAP), with pulmonary vascular remodeling as the main pathological mechanism, which leads to right heart failure and eventually death. Pulmonary arterial hypertension (PAH) belongs to group 1 PH, and congenital cardiovascular disease is the most common cause of PH in children. Despite early detection and surgical repair of congenital cardiovascular defects, there is no better treatment for this type of PH. Moreover, it is considered that the underlying gene mutations or chromosomal aberrations are the major contributors to PH associated with pathological congenital cardiovascular disease.
Three case reports have documented 2p16.1p15 microduplication syndrome, and these cases have similar clinical manifestations, namely, developmental delay, intellectual disability, and congenital defects, but the length of the chromosomal microduplication region, the genes involved, and the number of genes differ (1–3). In addition to the developmental delay, this case also showed PH associated with a congenital defect of the heart and ocular lesions. Chromosomal alterations, especially duplication of certain segments, cause great genomic instability. Whole exome sequencing, karyotype analysis, copy number analysis, and single nucleotide polymorphism (SNP) analysis were performed to study this infant who suffered from PAH, developmental delay, movement disorders, and ocular lesions. A microduplication chromosomal abnormality in the 2p16.1p15 region was found. It is crucial to enrich the understanding of the genes involved and clinical manifestations of 2p16.1p15 microduplication syndrome.
Case presentation
A 2-year-old female infant was admitted to the Department of Cardiology of the Hunan Children's Hospital to undergo atrial septal defect (ASD) closure due to multiple echocardiographic examinations indicating an ASD (Figure 1). A preoperative echocardiogram revealed an ASD with mild tricuspid regurgitation. The ASD was carefully measured as 10 mm during the operation. The mPAP and pulmonary vascular resistance (PVR) were measured at 45 mmHg and 9.1 Wood units via right heart catheterization under transesophageal echocardiography guidance, respectively. The closure operation was canceled due to the infant having significant PAH. By puncturing the right femoral artery and inserting a right coronary catheter, we measure the blood pressure measured at various sites. The left ventricular pressure, ascending aortic pressure, and descending aortic pressure were 31, 48, and 48 mmHg, respectively, which ruled out pulmonary hypertension due to left heart-related disease. The oxygen saturations of the aorta, pulmonary artery, and superior vena cava were 100%, 72.3%, and 69.2%, respectively. PH crisis was considered, and the infant was given pure oxygen to prevent the possible occurrence of a PH crisis. The blood oxygen saturation of the infant was maintained above 90%, and the hemodynamics improved. There was a decrease in hemoglobin and transfusion of concentrated red blood cells used to improve anemia (Table 1).
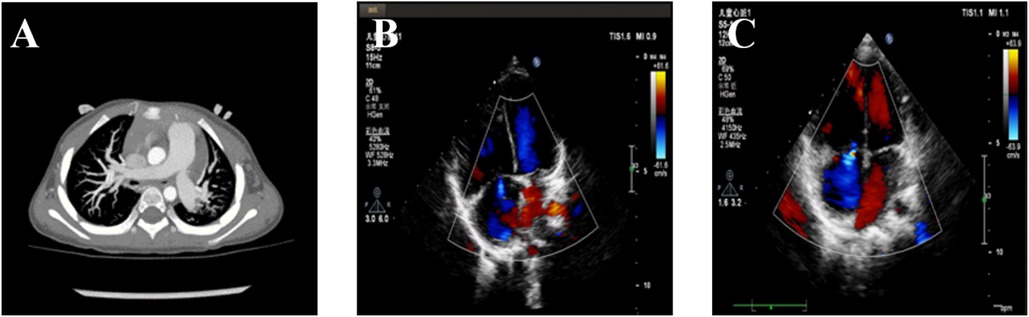
Figure 1. Pulmonary arterial hypertension of the patient. (A) The computerized tomography scan shows a dilated pulmonary artery. (B) The echocardiogram shows tricuspid regurgitation prior to treatment. (C) The echocardiogram shows tricuspid regurgitation after treatment.
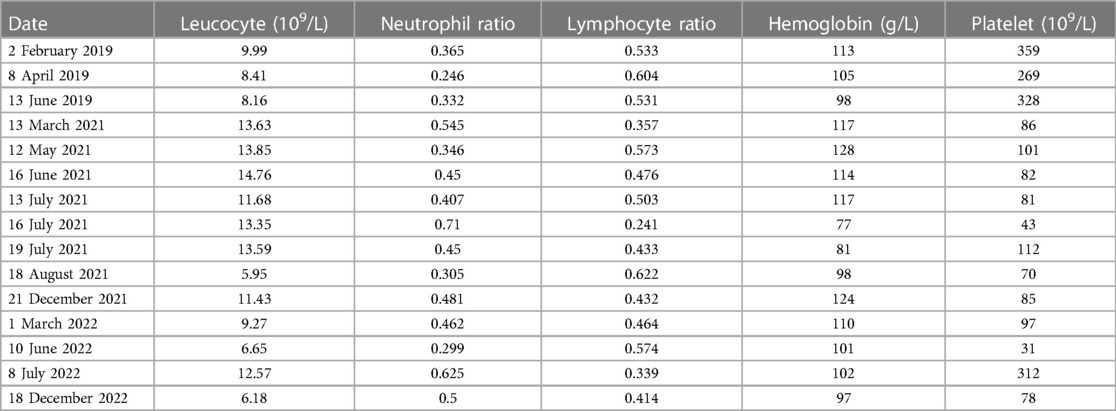
Table 1. Changes in blood cell count and ratio in the patient.
After her condition improved and her vital signs stabilized, bosentan (22 mg/time/12 h) and sildenafil (5 mg/time/8 h) were applied to lower the mPAP. After her condition improved and she was discharged from the hospital, she continued to take bosentan and sildenafil orally. Because the follow-up test of the blood routine showed that thrombocytopenia (Table 1) and pulmonary arterial pressure did not decrease significantly, she was rehospitalized in June 2022 in our hospital. Gamma globulin was used to increase the number of platelets for 3 days. Other therapeutic drugs included bosentan tablets (25 mg/time/12 h) and sildenafil (5 mg/time/8 h) to treat PAH. Because the mPAP was estimated to be increased by echocardiogram, sildenafil was changed to tadalafil (7 mg/time/day) and re-examination of echocardiogram showed that the pulmonary artery pressure decreased.
When the infant was 3 months old, she was admitted to the Hunan Children's Hospital for treatment due to poor head-up in the prone position. The doctor observed that her limbs moved slowly, and her pronunciation was less. Through physical examination, it was found that she could not turn over in the supine position while the upper limbs did not shift with the fulcrum on the face when trying to raise her head in the prone position. She had hypotonia in the limbs with grade II muscle tone in both upper limbs and grade I muscle tone in both lower limbs. Retinal examination revealed focal ridge-like changes and hemorrhages in the temporal periphery of both eyes. Brain MRI showed enlargement and deformation of the bilateral ventricle and third ventricle. After rehabilitation treatment, her motor ability has improved. Her parents and relatives did not have corresponding clinical manifestations or similar medical history.
The karyotype analysis and blood metabolism screening of the infant showed no abnormalities. Pathogenic point variants that may be associated with clinical presentation were also not detected. Further whole exome sequencing revealed that the proband had a 2.58 Mb microduplication in the paternal chromosomal 2p16.1p15 region (Figures 2, 3). We concluded that this copy number variation (CNV) may be de novo with the fact that sequencing data showed that neither the father nor the mother of the infant had this copy number variation. This genomic region involves multiple Online Mendelian Inheritance in Man (OMIM) genes, among which BCL11A, EHBP1, FAM161A, PEX13, and REL genes are identified as OMIM pathogenic genes (Supplementary Material S1). Although there were no established triplosensitive genes in this region, its phenotype is highly consistent with that of the reported cases (1, 3) (Supplementary Material S1). Using the technical standards for the interpretation and reporting of constitutional copy number variants (4), we determined the pathogenicity of chromosomal microduplication as follows. First, chromosomal 2p16.1p15 microduplication regions contain protein-coding genes (BCL11A, EHBP1, FAM161A, PEX13, and REL) or other known functionally important elements (score 0). Second, one breakpoint is within an established haploinsufficient gene (BCL11A) and the phenotype of the patient is highly specific and consistent with what is expected for the loss of function (LOF) of that gene (5–7) (score 0.45). Third, the number of protein-coding genes wholly or partially included in the copy number gain is between 0 and 34 (score 0). Then, we performed detailed assessments of genomic content using cases from the published literature, public databases, and/or in-house laboratory data. We found that the reported phenotype of de novo occurrences in these cases is highly specific and relatively unique to the gene or genomic region (1, 3, 7) (score 0.45). Finally, the observed copy number gain in this case is de novo (score 0). Therefore, the chromosomal microduplication pathogenicity of this case is likely pathogenic (score 0.90).
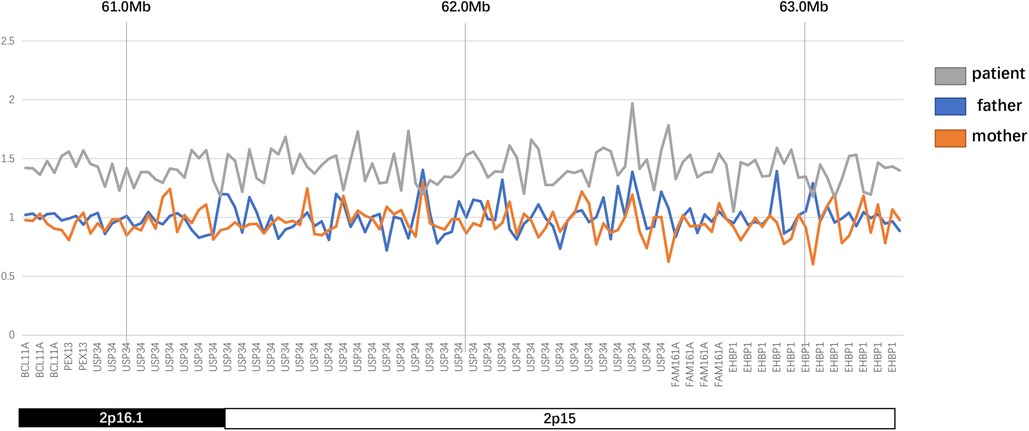
Figure 2. Results of CNV analysis in the patient.
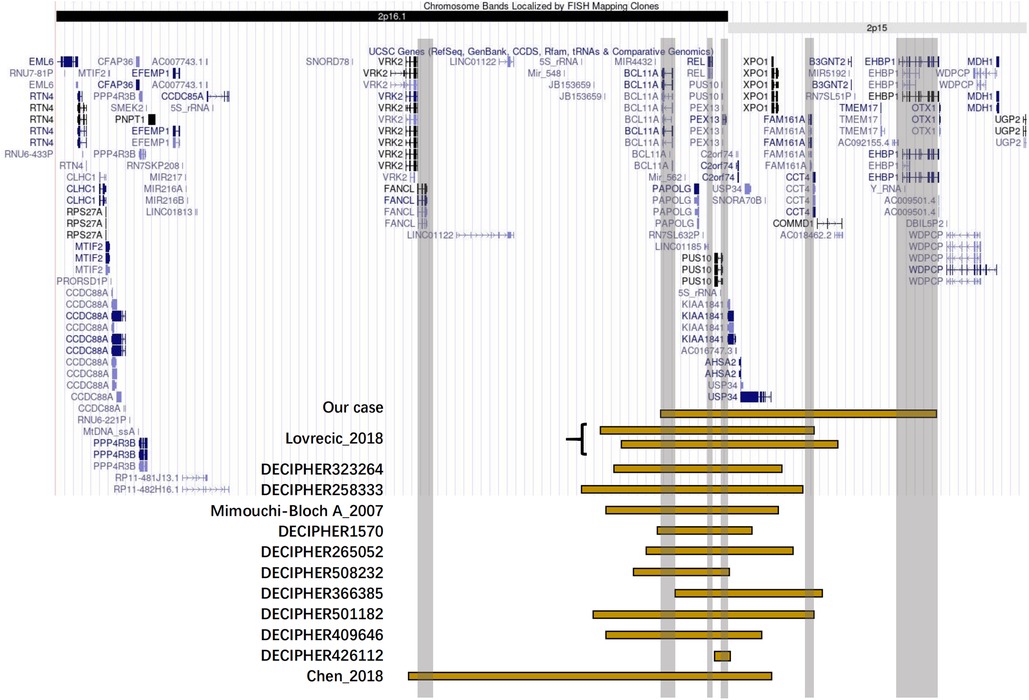
Figure 3. Positional plots of the microduplications on the 2p16.1p15 region.
Discussion
This study reports a female infant who suffered from PAH, developmental delay, ocular lesions, and bradykinesia. The PAH in this infant was discovered during right heart catheterization prior to the proposed repair of the ASD. ASD repair surgery was canceled, and routine treatment of PAH-targeted drugs, e.g., sildenafil and bosentan, was used leading to a decreased pulmonary arterial blood pressure. To clarify the possible genetic causes of PAH in the proband and promote awareness of the possibility of PAH in such children, we performed whole exome sequencing, and a 2.58 Mb microduplication in the chromosomal 2p16.1p15 region was revealed.
The chromosomal 2p16.1p15 region is prone to copy number variation. We aimed to identify the involved genes that may play key roles by collating the clinical characteristics of patients with similar copy number variants. Then, by searching the DECIPHER database (8) and the PubMed-indexed scientific literature, we included in the analysis a total of 13 cases for which phenotypes were described with four cases appearing as case reports (1–3) and another nine cases deposited in the DECIPHER database (Supplementary Table S1). The common clinical features of 14 patients with 2p16.1p15 microduplications including our report were delayed language development, developmental delay, intellectual disability, movement disorder, and special facial features. The patients reported by Mimouni-Bloch et al. (2) and DECIPHER 501182 had ADHD, whereas patients reported by DECIPHER 501182 and DECIPHER 508232 had autistic behaviors. Interestingly, three of the 14 patients with microduplications [Lovrecic et al. (3), DECIPHER 258333, and DECIPHER 508232] had macrocephaly, which was the opposite phenotype of microdeletion patients with 44% of patients showing microcephaly. Except for the patient in our case, none of the remaining 13 patients developed PH with two patients having atrial septal defect. Audiovisual impairment was present in some patients [DECIPHER426112, Mimouni-Bloch et al. (2), the patient in our case]. In terms of infections and the blood system, the patients in both our case and DECIPHER 258333 reported recurrent infections and abnormal blood counts.
Comparative analysis of OMIM genes involved in the symptoms of the patients and their microduplication regions provides an opportunity to find the key genes of specific phenotypes. The OMIM genes involved in microduplication regions in all reported cases are VRK2, FANCL, BCL11A, PAPOLG, REL, PUS10, PEX13, KIAA1841, C2orf74, ASHA2, USP34, XPO1, FAM161A, CCT4, COMMD1, B3GNT2, TMEM17, and EHBP1. Six of them, namely, BCL11A, PEX13, REL, FAM161A, EHBP1, and FANCL, are OMIM pathogenic genes. In this case report, chromosomal microduplication regions encompassed multiple genes, and five of them were identified as OMIM genes causing diseases, namely, BCL11A, PEX13, REL, FAM161A, and EHBP1. BCL11A and PEX13 are the two most shared OMIM pathogenic genes. REL and FAM161A were involved in 13 or five cases, respectively, while EHBP1 was only involved in our case.
BCL11A (OMIM 606557) encodes B-cell chronic lymphocytic leukemia (CLL)/lymphoma 11A which is a zinc finger protein and regulates gene transcription by interacting with COUP-TF protein (5). BCL11A is highly expressed in the human cerebral cortex, hippocampus, and cerebellum. Mutation or deletion of BCL11A has been shown to cause various diseases or dysfunctions. Heterozygous mutant BCL11A is associated with autosomal dominant Dias–Logan syndrome or intellectual developmental disability with persistent fetal hemoglobin (OMIM 617101). Soblet et al. (6) reported that BCL11A frameshift mutations lead to dyskinesia and hypotonia. Cai et al. (9) found BCL11A mutations in patients with autism and intellectual disability. Basak et al. (10) found that loss of BCL11A causes schizophrenia and attention deficit hyperactivity disorder. Dias et al. (7) found that BCL11A haploinsufficiency can lead to neurodevelopmental defects, developmental delay, and intellectual disability. Peter et al. (11) reported the association of BCL11A deletion with severe speech impairment. The reason for the above phenotypes may be that BCL11A is related to the development and growth of nerve cells. Wiegreffe et al. (5) found that BCL11A controls the migration of cortical projection neurons through Sema3c. Fox et al. (12) found that BCL11A affects the number and types of neurons by affecting the differentiation time program of neural stem cells. Kuo et al. (13) found that BCL11A controls axon branching and dendrite growth by regulating the expression of DCC and MAP1B. Kuo et al. (14) also found that the known X-linked intellectual disability gene CASK interacts with BCL11A to regulate axon branching and growth. All of the above are the result of BCL11A copy number deficiency or mutation, and the significance of BCL11A copy number gain remains unclear. However, the BCL11A knockdown model of zebra fish developed microcephaly with reduced size (15), which was in contrast to macrocephaly in three cases. Therefore, the phenotypes caused by increased or decreased BCL11A copy number may be opposite in some respects. Three cases reported macrocephaly, and according to the fact that the genes shared by the microduplication region in these three cases included BCL11A, the copy number increase of BCL11A may be able to explain to some extent why patients with 2p16.1p15 microduplication syndrome have intellectual disability and developmental disability and special facial features.
PEX13 (OMIM 601789) encodes peroxisome biogenesis factor 13, which is a peroxisome membrane protein. Deletion of PEX13 is associated with autosomal recessive disorders of peroxisome biogenesis 11A and 11B (16, 17). REL (OMIM 164910) encodes c-Rel, a transcription factor of the REL/NFKB family. REL is required for long-term synaptic plasticity and memory function in the hippocampus (18). REL promotes survival and apoptosis resistance in hippocampal neurons (19) and its knockout promotes a Parkinson's disease-like phenotype (20). DNA binding of NF-κB/c-Rel is reduced in the substantia nigra and peripheral blood mononuclear cells of patients with Parkinson's disease (21). FAM161A (OMIM613596) is a microtubule-binding protein expressed throughout the photoreceptor inner segment, enriched in the photoreceptor base connecting cilia and basal body (22, 23), which interacts with C8orf37 to promote the survival of photoreceptors (24). Deletion of FAM161A is associated with autosomal recessive retinitis pigmentosa 28 (RP28) (23). After knocking out Fam161a in mice, Beryozkin et al. (25) found that the outer segmental discs of photoreceptors were disorganized in the vertical direction and that the base of the outer segments was wider and shorter than that of those in the WT mice and eventually resulted in retinal denaturation. This may be related to retinal damage in our case.
EHBP1 (OMIM609922) may play a role in actin reorganization which links clathrin-mediated endocytosis to the actin cytoskeleton. EHBP1 regulates vesicle trafficking by recruiting Rab8 family members and Eps15 homology domain-containing protein 1/2 (EHD1/2) (26–28). In all 14 cases, EHBP1 was the only OMIM causative gene that was not shared and was found in children suffering from sudden PAH. Intersectin-1s (ITSN) deficiency and expression of bioactive ITSN fragments are characteristic of PAH human and animal model lung tissue (29). Bioactive ITSN fragments promote EC proliferation in PAH lungs (30). Predescu et al. (31) found that EHBP1 was involved in the proliferative effect of EHIRSN on endothelial cells by changing the subcellular localization of EHBP1. The unexpected occurrence of PAH based on an atrial septal defect in our case may be due to a copy number gain of the EHBP1 gene that promotes EHBP1 expression in endothelial cells and diverts physiological vesicle trafficking to an alternative endocytic pathway, ultimately leading to molecular phenotypes of dysfunctional pulmonary vascular endothelial cells. This requires further evidence.
Although this case report involves only one patient, it is unique in several ways. First, this patient had a longer chromosomal microduplication region length and a greater portion of the 2p15 region, in contrast to most reported 2p16.1p15 microduplication copy number changes, which mainly occurred in the 2p16.1 region. Despite this discrepancy, this patient had clinical features, such as developmental delay and movement disturbance, similar to those of patients with 2p16.1p15 microduplication carriers. A second unique feature of this case is that the father and mother of the proband had no similar copy number variation and all chromosome copy numbers were normal, suggesting that the microduplication was a de novo mutation. Finally, the 2p16.1p15 microduplication in the proband involved an OMIM causative gene, EHBP1, which was not involved in all reported case reports in the PubMed and DECIPHER databases. EHBP1 may be related to the rapid onset of PAH in this case.
This report has limitations. First, the cause for the 2p16.1p15 microduplication of the chromosome of the patient was not explained in this report. Whole genome sequencing and further bioinformatics should be performed to explore any possible causes. Second, the reasons for PAH in this infant have not been thoroughly explored. Although EHBP1 was suggested as the most likely cause, quantitative polymerase chain reaction and Western blot experiments on tissue or blood samples from the infant were not performed to determine the high expression of EHBP1. Third, subsequent in vitro cell experiments to verify the effect of EHBP1 on endothelial cell proliferation phenotype should also be performed.
In conclusion, we report in detail a case of rare 2p16.1p15 microduplication syndrome involving EHBP1, which suggests that the occurrence of PAH should be alerted if microduplication mutations occur in the 2p16.1p15 region and involve EHBP1.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Hunan Children's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the legal guardians/next of kin of the participant. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YX, XW, and ZZ conceptualized and designed the study, drafted the initial manuscript, and reviewed and revised the manuscript. WZ, DW, and QL collected data, carried out the initial analyses, and reviewed and revised the manuscript. FY designed the data collection instruments, coordinated and supervised data collection, and critically reviewed the manuscript. All authors agreed to be accountable for all aspects of the work. All authors contributed to the article and approved the submitted version.
Funding
All phases of this study were supported by grants from the Hunan Province Major Special Project (No. 2020SK1013), Hunan Provincial Health Commission Project (No. 20200483), National Natural Sciences Foundation of China (No. 81500041), and Changsha Natural Science Foundation (No. KQ220239).
Acknowledgments
We formally acknowledge the patient and her family for entrusting our team with the clinical care.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1219480/full#supplementary-material
References
1. Chen C-P, Chern S-R, Wu P-S, Chen S-W, Lai S-T, Chuang T-Y, et al. Prenatal diagnosis of a 3.2-Mb 2p16.1-p15 duplication associated with familial intellectual disability. Taiwan J Obstet Gynecol. (2018) 57(4):578–82. doi: 10.1016/j.tjog.2018.06.018
2. Mimouni-Bloch A, Yeshaya J, Kahana S, Maya I, Basel-Vanagaite L. A de-novo interstitial microduplication involving 2p16.1-p15 and mirroring 2p16.1-p15 microdeletion syndrome: clinical and molecular analysis. Eur J Paediatr Neurol. (2015) 19(6):711–5. doi: 10.1016/j.ejpn.2015.07.013
3. Lovrecic L, Gnan C, Baldan F, Franzoni A, Bertok S, Damante G, et al. Microduplication in the 2p16.1p15 chromosomal region linked to developmental delay and intellectual disability. Mol Cytogenet. (2018) 11:39. doi: 10.1186/s13039-018-0388-y
4. Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. (2020) 22(2):245–57. doi: 10.1038/s41436-019-0686-8
5. Wiegreffe C, Simon R, Peschkes K, Kling C, Strehle M, Cheng J, et al. Bcl11a (Ctip1) controls migration of cortical projection neurons through regulation of Sema3c. Neuron. (2015) 87(2):311–25. doi: 10.1016/j.neuron.2015.06.023
6. Soblet J, Dimov I, von Kalckreuth CG, Cano-Chervel J, Baijot S, Pelc K, et al. BCL11A frameshift mutation associated with dyspraxia and hypotonia affecting the fine, gross, oral, and speech motor systems. Am J Med Genet Part A. (2018) 176(1):201–8. doi: 10.1002/ajmg.a.38479
7. Dias C, Estruch SB, Graham SA, McRae J, Sawiak SJ, Hurst JA, et al. BCL11A haploinsufficiency causes an intellectual disability syndrome and dysregulates transcription. Am J Hum Genet. (2016) 99(2):253–74. doi: 10.1016/j.ajhg.2016.05.030
8. Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet. (2009) 84:524–33. doi: 10.1016/j.ajhg.2009.03.010
9. Cai T, Chen X, Li J, Xiang B, Yang L, Liu Y, et al. Identification of novel mutations in the HbF repressor gene BCL11A in patients with autism and intelligence disabilities. Am J Hematol. (2017) 92(12):E653–6. doi: 10.1002/ajh.24902
10. Basak A, Hancarova M, Ulirsch JC, Balci TB, Trkova M, Pelisek M, et al. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J Clin Invest. (2015) 125(6):2363–8. doi: 10.1172/JCI81163
11. Peter B, Matsushita M, Oda K, Raskind W. De novo microdeletion of BCL11A is associated with severe speech sound disorder. Am J Med Genet Part A. (2014) 164a(8):2091–6. doi: 10.1002/ajmg.a.36599
12. Fox PM, Tang JLY, Brand AH. The Drosophila homologue of CTIP1 (Bcl11a) and CTIP2 (Bcl11b) regulates neural stem cell temporal patterning. Development. (2022) 149(17):dev200677. doi: 10.1242/dev.200677
13. Kuo TY, Hong CJ, Hsueh YP. Bcl11A/CTIP1 regulates expression of DCC and MAP1b in control of axon branching and dendrite outgrowth. Mol Cell Neurosci. (2009) 42(3):195–207. doi: 10.1016/j.mcn.2009.07.006
14. Kuo TY, Chen CY, Hsueh YP. Bcl11A/CTIP1 mediates the effect of the glutamate receptor on axon branching and dendrite outgrowth. J Neurochem. (2010) 114(5):1381–92. doi: 10.1111/j.1471-4159.2010.06852.x
15. Bagheri H, Badduke C, Qiao Y, Colnaghi R, Abramowicz I, Alcantara D, et al. Identifying candidate genes for 2p15p16.1 microdeletion syndrome using clinical, genomic, and functional analysis. JCI Insight. (2016) 1(3):e85461. doi: 10.1172/jci.insight.85461
16. Catharina Müller C, Nguyen TH, Ahlemeyer B, Meshram M, Santrampurwala N, Cao S, et al. Pex13 inactivation in the mouse disrupts peroxisome biogenesis and leads to a Zellweger syndrome phenotype. Mol Cell Biol. (2003) 23(16):5947–57. doi: 10.1128/mcb.23.16.5947-5957.2003
17. Müller CC, Nguyen TH, Ahlemeyer B, Meshram M, Santrampurwala N, Cao S, et al. PEX13 deficiency in mouse brain as a model of Zellweger syndrome: abnormal cerebellum formation, reactive gliosis and oxidative stress. Dis Model Mech. (2011) 4(1):104–19. doi: 10.1242/dmm.004622
18. Ahn HJ, Hernandez CM, Levenson JM, Lubin FD, Liou HC, Sweatt JD. c-Rel, an NF-kappaB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn Mem. (2008) 15(7):539–49. doi: 10.1101/lm.866408
19. Wang Z, Dong H, Wang J, Huang Y, Zhang X, Tang Y, et al. Pro-survival and anti-inflammatory roles of NF-κB c-Rel in the Parkinson’s disease models. Redox Biol. (2020) 30:101427. doi: 10.1016/j.redox.2020.101427
20. Parrella E, Bellucci A, Porrini V, Benarese M, Lanzillotta A, Faustini G, et al. NF-κB/c-Rel deficiency causes Parkinson’s disease-like prodromal symptoms and progressive pathology in mice. Transl Neurodegener. (2019) 8:16. doi: 10.1186/s40035-019-0154-z
21. Porrini V, Pilotto A, Vezzoli M, Lanzillotta A, Gennari MM, Bonacina S, et al. NF-κB/c-Rel DNA-binding is reduced in substantia nigra and peripheral blood mononuclear cells of Parkinson’s disease patients. Neurobiol Dis. (2023) 180:106067. doi: 10.1016/j.nbd.2023.106067
22. Di Gioia SA, Letteboer SJF, Kostic C, Bandah-Rozenfeld D, Hetterschijt L, Sharon D, et al. FAM161A, associated with retinitis pigmentosa, is a component of the cilia-basal body complex and interacts with proteins involved in ciliopathies. Hum Mol Genet. (2012) 21(23):5174–84. doi: 10.1093/hmg/dds368
23. Zach F, Grassmann F, Langmann T, Sorusch N, Wolfrum U, Stöhr H. The retinitis pigmentosa 28 protein FAM161A is a novel ciliary protein involved in intermolecular protein interaction and microtubule association. Hum Mol Genet. (2012) 21(21):4573–86. doi: 10.1093/hmg/dds268
24. Liu Y, Chen J, Sager R, Sasaki E, Hu H. Interactions between C8orf37 and FAM161A, two ciliary proteins essential for photoreceptor survival. Int J Mol Sci. (2022) 23(19):10233. doi: 10.3390/ijms231912033
25. Beryozkin A, Matsevich C, Obolensky A, Kostic C, Arsenijevic Y, Wolfrum U, et al. A new mouse model for retinal degeneration due to Fam161a deficiency. Sci Rep. (2021) 11(1):2030. doi: 10.1038/s41598-021-81414-1
26. Guilherme A, Soriano NA, Bose S, Holik J, Bose A, Pomerleau DP, et al. EHD2 and the novel EH domain binding protein EHBP1 couple endocytosis to the actin cytoskeleton. J Biol Chem. (2004) 279(11):10593–605. doi: 10.1074/jbc.M307702200
27. Rai A, Bleimling N, Vetter IR, Goody RS. The mechanism of activation of the actin binding protein EHBP1 by Rab8 family members. Nat Commun. (2020) 11(1):4187. doi: 10.1038/s41467-020-17792-3
28. Rai A, Oprisko A, Campos J, Fu Y, Friese T, Itzen A, et al. bMERB domains are bivalent Rab8 family effectors evolved by gene duplication. eLife. (2016) 5:e18675. doi: 10.7554/eLife.18675
29. Patel M, Predescu D, Tandon R, Bardita C, Pogoriler J, Bhorade S, et al. A novel p38 mitogen-activated protein kinase/Elk-1 transcription factor-dependent molecular mechanism underlying abnormal endothelial cell proliferation in plexogenic pulmonary arterial hypertension. J Biol Chem. (2013) 288(36):25701–16. doi: 10.1074/jbc.M113.502674
30. Predescu DN, Neamu R, Bardita C, Wang M, Predescu SA. Impaired caveolae function and upregulation of alternative endocytic pathways induced by experimental modulation of intersectin-1s expression in mouse lung endothelium. Biochem Res Int. (2012) 2012:672705. doi: 10.1155/2012/672705
Keywords: pulmonary arterial hypertension, developmental delay, microduplication, copy number mutation, 2p16.1p15
Citation: Wang X, Zhang Z, Zuo W, Wang D, Yang F, Liu Q and Xiao Y (2023) Case Report: Identification of microduplication in the chromosomal 2p16.1p15 region in an infant suffering from pulmonary arterial hypertension. Front. Cardiovasc. Med. 10:1219480. doi: 10.3389/fcvm.2023.1219480
Received: 9 May 2023; Accepted: 3 October 2023;
Published: 23 October 2023.
Edited by:
Kostas Dimopoulos, Royal Brompton Hospital, United KingdomReviewed by:
Raj Sewduth, VIB-KU Leuven Center for Cancer Biology, BelgiumVenkatesh Katari, University of Toledo, United States
© 2023 Wang, Zhang, Zuo, Wang, Yang, Liu, and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunbin Xiao eGlhb3l1bmJpbnJ1aUAxMjYuY29t
†These authors have contributed equally to this work and share first authorship