 Yujian Wu
Yujian Wu Yun Zhu2,3,†
Yun Zhu2,3,† Xu Zhang
Xu Zhang Jinqing Feng
Jinqing Feng Huimin Xia
Huimin Xia Jia Li
Jia Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 11 August 2023
Sec. Pediatric Cardiology
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1215473
This article is part of the Research Topic Health Informatics in Pediatric Cardiology View all 6 articles
Objective: To examine the incidence and phenotypes of congenital heart disease (CHD) in a large cohort of patients with Hirschsprung's disease (HSCR).
Study design: Retrospective data review of children with HSCR between 2003 and 2020 was conducted at the Provincial Key Laboratory for Structural Birth Defects in Guangzhou, Guangdong, China. HSCR was confirmed by pathological diagnosis. CHD was defined as a gross structural abnormality of the heart or intrathoracic great vessels that is of functional significance.
Results: A total of 2,174 HSCR patients (84.7% males) were studied and 306 of them underwent echocardiography. Overall, 27 children (1.2%) had associated CHD. Among them, CHDs mostly presented as atrial and ventricular septal defects (n = 5 and 12 respectively) and patent ductus arteriosus (n = 4). Three patients (1.4‰) presented as a severe CHD including complete atrioventricular canal, congenitally corrected transposition of the great arteries and double-outlet of right ventricle. Among 14 patients carrying a chromosomal abnormality, CHD was detected in 4 infants (28.6%), all being mild forms of septal defects.
Conclusions: Some new and severe types of CHD were found in patients with HSCR. Patients with syndromic features had higher incidence of CHD.
Hirschsprung's disease (HSCR) is one of the most common congenital digestive tract disorders in neonates with an incidence of approximately 1 in 5,000 live births (1). In HSCR patients, the absence of ganglion cells in a variable length of the distal gut, beginning at the internal sphincter and extending proximally, results in absent peristalsis in the affected bowel. The final diagnosis depends on rectal suction biopsy by identifying the absence of ganglion cells (2).
HSCR is caused by deficient cranio-caudal migration, proliferation, differentiation and colonization of neural crest cells in the hindgut (3). Neural crest cells are pluripotential and then gives rise to crucial cell types ancestors, based on their migratory pathway, terminal location, and differential abilities (4). Cardiac neural crest cells, a smaller specified subgroup of neural crest cells, significantly contribute to proper cardiovascular formation, including development of the smooth muscle, septation of the cardiac outflow tract, and patterning of the septa, valves and arterial truncus (5). The developmental deficiencies in cardiac neural crest cells are considered to cause a variety of cardiac malformations, i.e., congenital heart defect (CHD) (6).
CHD is the most common congenital structural disorder in newborns affecting approximately 1% of live births (7). It is also the leading cause of mortality from birth defects (8). There is a view that both HSCR and CHD could be regarded as neurocristopathies. Several studies have reported the high incidence of CHD, being about 1.4%–17% in HSCR patients (9–14). Most of the cases were mild or moderate forms, including atrial or ventricular septal defects, mild aortic or tricuspid valve abnormalities and coarctation of the aorta. The sample size of those studies was small (ranging from 53 to 207 patients). One report described the association of hypoplastic left heart syndrome with HSCR (13). Other than this, there has been no report, to the best of our knowledge, about the occurrence of severe types of CHD in HSCR patients.
Our institution has established the Provincial Key Laboratory for Structural Birth Defects with a large number of HSCR patients. We therefore implemented a 20-year retrospective study on the patients with HSCR aiming to assess the incidence and phenotypes of associated CHDs based upon a fairly large cohort.
This study was approved by the Institutional Review Board of Guangzhou Women and Children's Medical Center (No. 2018052406). Informed consent was waived owing to the nature of this retrospective medical record review study. All HSCR patients admitted for further evaluation and/or surgical treatment to the Guangzhou Women and Children's Medical Center (Guangzhou, Guangdong province, China) between January 2003 and December 2020 were retrospectively reviewed. Inclusion criteria were patients with pathologically confirmed diagnosis of HSCR. Exclusion criteria were unavailable medical records for review.
Demographic and clinical data were collected from the patient's records. During the hospitalization, all patients were assessed by clinical observation, physical examination and further specialist investigations based on clinical features. Echocardiographic examination was performed when a cardiovascular abnormality was suspected based on symptoms, depressed pulse oximetry, heart murmur, family history of congenital heart disease or abnormal prenatal ultrasonic findings. Detailed echo-scan was performed through a transthoracic approach with pulsed, continuous and color-Doppler using echocardiographic systems.
In our cohort, 4 types of HSCR were identified according to the length of aganglionosis: short segment HSCR (aganglionosis extending up to the left descending colon), long segment HSCR (aganglionosis extending up to the right transverse or ascending colon), total colonic aganglionosis (aganglionosis extending to the whole colon) and total intestinal aganglionosis (aganglionosis extending over the jejunum with less than 20 cm of normo-ganglionic bowel).
CHD was defined as a gross structural abnormality of the heart or intrathoracic great vessels that is actually or potentially of functional significance (15), diagnosed with echocardiography by the pediatric cardiologists. In according with many other epidemiologic studies, the definition of CHD excluded functionless abnormalities of the great veins (such as persistent left superior cava), bicuspid aortic valves, mitral valve prolapse, Marfan syndrome, cardiomyopathies, and congenital arrhythmias without an associated structural heart lesion (such as long Q-T syndrome). Atrial septal defects within oval fossa, patent foramen ovale and patent ductus arteriosus throughout the first 14 days of life were also excluded because they are considered normal findings. The CHD patients were grouped by disease severity. The category of severe CHD included those who were severely ill in early infancy (16).
SPSS for Windows, version 26.0 (SPSS Inc, Chicago, Illinois, USA) was used. Statistical analysis was performed using 2-tailed student t-test (for continuous variables) and Pearson's χ2 test or Fisher's exact test (for categorical variables). A P-value lower than 0.05 was considered significant.
A total of 2,174 patients were enrolled in this study (Figure 1). Male to female ratio was 5.5:1. Among them, 1,587 patients (72.9%) were affected by the short segment HSCR, 420 patients (19.3%) by the long segment HSCR, 129 patients (5.9%) by the total colonic aganglionosis, and 38 patients (1.7%) by the total intestinal aganglionosis. There were 306 patients undergoing transthoracic echocardiography during the study period. The lengthwise types of aganglionosis did not differ between patients with and without CHD findings (P = 0.051). Patients with HSCR associated with CHD had a higher proportion of syndromes compared to non-CHD patients (P < 0.001, Table 1).
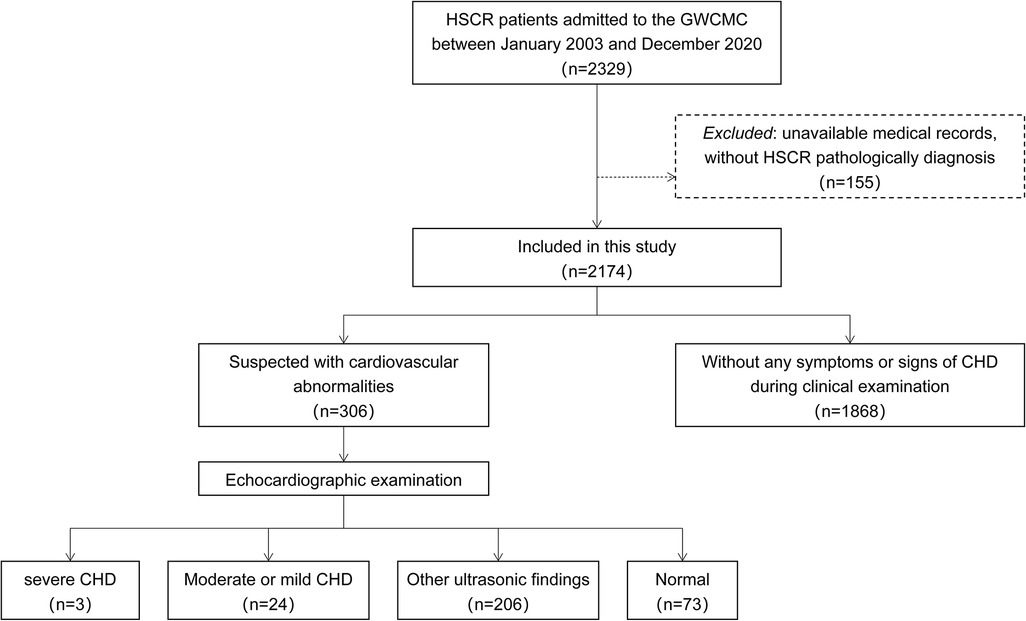
Figure 1. Flow chart of the study method. GWCMC, Guangzhou Women and Children's Medical Center; HSCR, Hirschsprung's disease; CHD, congenital heart disease.
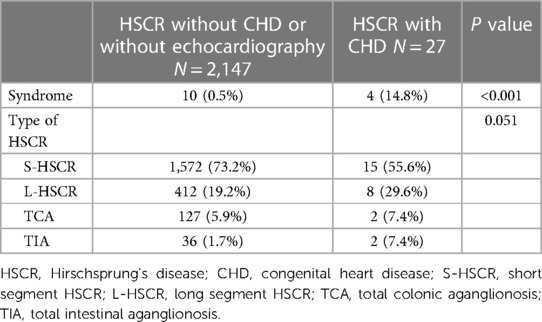
Table 1. Summary of syndromic and HSCR characteristics of overall patients grouped by CHD status.
There was no significant difference in sex or birthweight between individuals with and without CHD (P = 0.058 and 0.432 respectively, Table 2). The gestational age in patients with CHD was smaller (37.95 ± 2.07 vs. 38.86 ± 1.75, P = 0.041, Table 2).
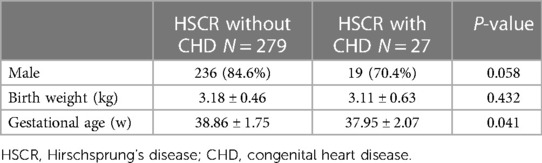
Table 2. Birth characteristics of overall HSCR patients with echocardiography.
The overall concomitant CHDs were detected in 1.2% (N = 27) of the HSCR patients (Table 3 for details). Echocardiography in the 306 patients showed that CHDs were mostly presented as septal defects, i.e., 12 patients had ventricular septal defect and 5 patients had atrial septal defect. Patent ductus arteriosus was detected in 4 patients, coarctation of the aorta in 2 and pulmonary stenosis in 1. Three patients (1.4‰) had a severe CHD, including complete atrioventricular canal, congenitally corrected transposition of the great arteries and double-outlet of right ventricle. Fourteen patients had chromosomal abnormalities, including Down's syndrome (N = 7), Waardenburg syndrome (N = 3), Cri-du-chat syndrome (N = 2), congenital central hypoventilation syndrome (N = 1) and Mowat–Wilson Syndrome (N = 1). Among these patients, CHD was detected in 4 patients (28.6%) being atrial (N = 1) and ventricular septal defects (N = 3). Out of 7 patients with Down's syndrome, 3 (42.9%) had atrial (N = 1) or ventricular septal defect (N = 2).
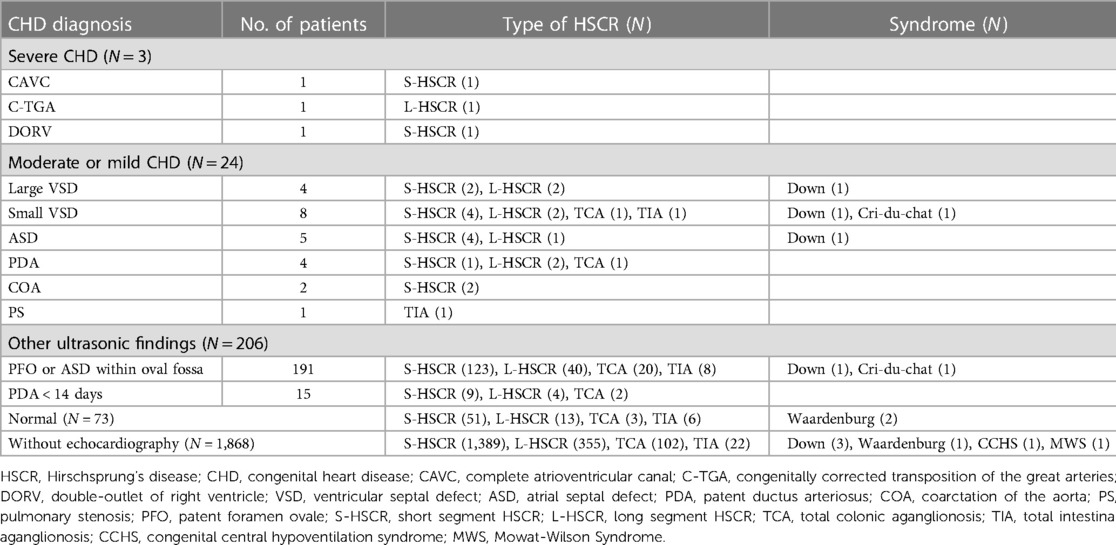
Table 3. Details of overall HSCR patients.
HSCR was first described in 1,886 by a Danish physician, Harald Hirschsprung, and the chief culprit was revealed as aganglionosis in all or distal part of the colon. Colonic lesions fail to relax, causing functional colonic obstruction over time (17). HSCR occurs as an isolated phenotype in most cases, while a number of HSCR-associated anomalies such as gastrointestinal, neurologic, and genitourinary anomalies have been reported (10). In particular, concomitant CHDs have been drawn attention on account of its influence on the long-term prognosis.
Several studies over the last few decades have reported CHD in fairly small populations of HSCR. A prospective observational study published in 2013 detected a prevalence rate of 4.7% out of 106 children with HSCR, with coarctation of the aorta in 1 and septal defects in 4 cases (9). In another prospective study, 133 consecutive HSCR patients underwent cardiac screening and 11 patients (8.3%) presented CHDs which were mostly septal defects or small patent ductus arteriosus (14). Two epidemiological studies reported the incidence of 4/207 (1.4%, 3 with ventricular septal defect and 1 with patent ductus arteriosus) and 2/126 (1.6%, no details given) respectively (10, 18). One report showed higher incidence of HSCR in patients with hypoplastic left heart syndrome (13). In our largest cohort of patients with HSCR ever reported, we found that 1.2% of children had concomitant CHD, which were mostly presented by atrial or ventricular septal defects. The incidence of severe CHD was 1.4‰ (N = 3), including complete atrioventricular canal, congenitally corrected transposition of the great arteries and double-outlet of right ventricle. None of them has ever been reported. In patients with syndromic disorders, the prevalence of associated CHD was 28.6%. Among 7 patients with Down's syndrome, 42.9% had associated CHD, in concordance with previous studies (9, 14, 19).
There are several reasons which could lead to a wide range in estimates of incidence. It may be attributed to the dissimilarities in definition of CHD and methods among the studies (16). High incidences were found in the prospective studies, as they detected mild or moderate CHDs, which are often asymptomatic and do not have significant murmurs. By contrast, estimates of prevalence of severe CHD are more reliable, since that few patients in the severe group will be misdiagnosed in the children's medical centers. Most epidemiological studies reported an estimated number of 1.0–1.7 cases per 1,000 live births for severe CHD (16, 20, 21). A similar incidence was found in our HSCR population.
More interestingly, 2 of the severe CHDs were conotruncal developmental anomalies, i.e., congenitally corrected transposition of the great arteries and double-outlet of right ventricle. This may be expected as the critical embryologic role of neural crest cells in the development of both enteric nervous system and cardiac outflow tract and septation. After undergoing epithelial-mesenchymal transition, multipotent cells of the neural crest migrate and subsequently differentiate into a wide variety of cell type ancestors that are responsible for the development of various organs (22, 23). Various animal models have repeatedly confirmed that vagal neural crest cells are the main source of the enteric nervous system constituents (3, 24). The crucial role of neural crest cells in conotruncal separation during proper heart formation have been demonstrated (5, 6, 25). Although progress has been made in revealing possible embryological connection between CHD and HSCR, the developmental processes of neural crest cells are extraordinary complexities. Future experimental work in this area is needed to determine the regulatory pathways and factors during the development of cardiovascular and enteric nervous system.
In our cohort, 14 patients (0.6%) had Down's syndrome or other syndromes. Several particular reasons probably account for the lower than reported percentage (4.3%–6.6%) of Down's syndrome in our live-born series (9, 10, 14, 19). In China, prenatal screening for Down's syndrome is offered as part of routine antenatal care, and termination of pregnancy for Down's syndrome is legal (26, 27). From the viewpoint of Down's syndrome, the most frequent associated anomalies are cardiac disorders, which has been reported to occur in roughly 44%–60% (28, 29). Besides, the clinical association between Down's syndrome and HSCR has been well-established (30, 31). However, the general incidence of CHD associated with HSCR-Down's syndrome has not substantially changed compared with all Down's syndrome patients – 3 out of 7 HSCR-Down's syndrome patients (42.9%) presented also an associated CHD and similar proportions in other studies (36.1%–62.5%) (9, 14, 19). The past three nationwide surveys in Japan found that cardiovascular anomalies were increased over time among HSCR-Down's syndrome patients (19). The incremental trends were consistent with the increasing diagnosed incidence in general population over the years, which primarily due to the rising frequency of echocardiographic ultrasound screening and the growing ability to detect mild lesions. Nonetheless, the associations between CHD, HSCR and genetic syndromes are still of significance in revealing the possible abnormal genetic background and pathogenesis and in evaluating the prognosis.
Our study has several limitations. The observational retrospective study had intrinsic risks of information bias and temporal bias, and the specific biological mechanisms are speculative. Besides, our echocardiographic evaluation was conducted in only 306 patients depending on referral, which tended to underestimate the number of mild lesions, particular atrial septal defect. But it is unlikely that major cardiac defects were missed. In addition, it represents a single-institutional experience for a referral center for HSCR and should extrapolate with care. A meta-analysis combining numerous studies may help to identify further associations. Furthermore, missing information about long-term follow-up resulted in inability to assess the effect of associated CHDs on the outcomes of patients with HSCR.
Our retrospective study during the last twenty years in China demonstrated CHDs in 1.2% of 2,174 HSCR patients. Echocardiography evaluation in 306 patients helped to identify 3 patients (1.4‰) with severe CHD including complete atrioventricular canal, congenitally corrected transposition of the great arteries and double-outlet of right ventricle, which have never been reported.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Guangzhou Women and Children's Medical Center. Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
YW and JL: conceptualized and designed the study. YW and YZ: performed formal analysis and wrote the article. JF: participated in the acquisition of data. XZ and HX: contributed to the interpretation of data. YZ: and JL: supervised the project and contributed to critical revision of the article. All authors contributed to the article and approved the submitted version.
This research was supported by the Basic Research Program of Guangzhou Municipal Science and Technology Bureau (grant no. 2023A04J1874) and start-up fund of Guangzhou Women and Children's Medical Center.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Arshad A, Powell C, Tighe MP. Hirschsprung’s disease. Br Med J. (2012) 345:e5521. doi: 10.1136/bmj.e5521
2. Das K, Mohanty S. Hirschsprung disease - current diagnosis and management. Indian J Pediatr. (2017) 84:618–23. doi: 10.1007/s12098-017-2371-8
3. Klein M, Varga I. Hirschsprung’s disease-recent understanding of embryonic aspects, etiopathogenesis and future treatment avenues. Medicina (Kaunas). (2020) 56:611. doi: 10.3390/medicina56110611
4. Takahashi Y, Sipp D, Enomoto H. Tissue interactions in neural crest cell development and disease. Science. (2013) 341:860–3. doi: 10.1126/science.1230717
5. George RM, Maldonado-Velez G, Firulli AB. The heart of the neural crest: cardiac neural crest cells in development and regeneration. Development. (2020) 147:dev.188706. doi: 10.1242/dev.188706
6. Erhardt S, Zheng M, Zhao X, Le TP, Findley TO, Wang J. The cardiac neural crest cells in heart development and congenital heart defects. J Cardiovasc Dev Dis. (2021) 8:89. doi: 10.3390/jcdd8080089
7. Bouma BJ, Mulder BJ. Changing landscape of congenital heart disease. Circ Res. (2017) 120:908–22. doi: 10.1161/CIRCRESAHA.116.309302
8. Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics–2012 update: a report from the American heart association. Circulation. (2012) 125:e2–220. doi: 10.1161/CIR.0b013e31823ac046
9. Pini Prato A, Rossi V, Mosconi M, Holm C, Lantieri F, Griseri P, et al. A prospective observational study of associated anomalies in Hirschsprung’s disease. Orphanet J Rare Dis. (2013) 8:184. doi: 10.1186/1750-1172-8-184
10. Russell MB, Russell CA, Niebuhr E. An epidemiological study of Hirschsprung’s disease and additional anomalies. Acta Paediatr. (1994) 83:68–71. doi: 10.1111/j.1651-2227.1994.tb12955.x
11. Mery CM, De Leon LE, Rodriguez JR, Nieto RM, Zhang W, Adachi I, et al. Effect of gastrointestinal malformations on the outcomes of patients with congenital heart disease. Ann Thorac Surg. (2017) 104:1590–6. doi: 10.1016/j.athoracsur.2017.04.042
12. Hasserius J, Hedbys J, Graneli C, Hagelsteen K, Stenstrom P. Treatment and patient reported outcome in children with Hirschsprung disease and concomitant congenital heart disease. Biomed Res Int. (2017) 2017:1703483. doi: 10.1155/2017/1703483
13. Ahola JA, Koivusalo A, Sairanen H, Jokinen E, Rintala RJ, Pakarinen MP. Increased incidence of Hirschsprung’s disease in patients with hypoplastic left heart syndrome–a common neural crest-derived etiology? J Pediatr Surg. (2009) 44:1396–400. doi: 10.1016/j.jpedsurg.2008.11.002
14. Tuo G, Pini Prato A, Derchi M, Mosconi M, Mattioli G, Marasini M. Hirschsprung’s disease and associated congenital heart defects: a prospective observational study from a single institution. Front Pediatr. (2014) 2:99. doi: 10.3389/fped.2014.00099
15. Mitchell SC, Korones SB, Berendes HW. Congenital heart disease in 56,109 births. Incidence and natural history. Circulation. (1971) 43:323–32. doi: 10.1161/01.cir.43.3.323
16. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. (2002) 39:1890–900. doi: 10.1016/s0735-1097(02)01886-7
17. Tam PK. Hirschsprung’s disease: a bridge for science and surgery. J Pediatr Surg. (2016) 51:18–22. doi: 10.1016/j.jpedsurg.2015.10.021
18. Singh SJ, Croaker GD, Manglick P, Wong CL, Athanasakos H, Elliott E, et al. Hirschsprung’s disease: the Australian paediatric surveillance unit’s experience. Pediatr Surg Int. (2003) 19:247–50. doi: 10.1007/s00383-002-0842-z
19. Ieiri S, Higashi M, Teshiba R, Saeki I, Esumi G, Akiyoshi J, et al. Clinical features of Hirschsprung’s disease associated with down syndrome: a 30-year retrospective nationwide survey in Japan. J Pediatr Surg. (2009) 44:2347–51. doi: 10.1016/j.jpedsurg.2009.07.055
20. van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. (2011) 58:2241–7. doi: 10.1016/j.jacc.2011.08.025
21. van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJ. The changing epidemiology of congenital heart disease. Nat Rev Cardiol. (2011) 8:50–60. doi: 10.1038/nrcardio.2010.166
22. York JR, McCauley DW. The origin and evolution of vertebrate neural crest cells. Open Biol. (2020) 10:190285. doi: 10.1098/rsob.190285
23. Brandon AA, Almeida D, Powder KE. Neural crest cells as a source of microevolutionary variation. Semin Cell Dev Biol. (2022) 145:42–51. doi: 10.1016/j.semcdb.2022.06.001
24. Tjaden NEB, Trainor PA. The developmental etiology and pathogenesis of Hirschsprung disease. Transl Res. (2013) 162:1–15. doi: 10.1016/j.trsl.2013.03.001
25. Yamagishi H. Cardiac neural crest. Cold Spring Harb Perspect Biol. (2021) 13:a036715. doi: 10.1101/cshperspect.a036715
26. Shang W, Wan Y, Chen J, Du Y, Huang J. Introducing the non-invasive prenatal testing for detection of down syndrome in China: a cost-effectiveness analysis. BMJ Open. (2021) 11:e046582. doi: 10.1136/bmjopen-2020-046582
27. Li B, Sahota DS, Lao TT, Xu J, Hu SQ, Zhang L, et al. Applicability of first-trimester combined screening for fetal trisomy 21 in a resource-limited setting in mainland China. BJOG. (2016) 123(Suppl 3):23–9. doi: 10.1111/1471-0528.14004
28. Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, et al. Down syndrome. Nat Rev Dis Primers. (2020) 6:9. doi: 10.1038/s41572-019-0143-7
29. Asim A, Kumar A, Muthuswamy S, Jain S, Agarwal S. Down syndrome: an insight of the disease. J Biomed Sci. (2015) 22:41. doi: 10.1186/s12929-015-0138-y
30. Prato AP, Arnoldi R, Sgro A, Felici E, Racca F, Nozza P, et al. Hirschsprung disease and down syndrome: from the reappraisal of risk factors to the impact of surgery. J Pediatr Surg. (2019) 54:1838–42. doi: 10.1016/j.jpedsurg.2019.01.053
Keywords: Hirschsprung’s disease, congenital heart disease, chromosomal abnormality, complete atrioventricular canal, congenitally corrected transposition of the great arteries, double-outlet of right ventricle
Citation: Wu Y, Zhu Y, Zhang X, Feng J, Xia H, Zhang Y and Li J (2023) Associated congenital heart disease with Hirschsprung's disease: a retrospective cohort study on 2,174 children. Front. Cardiovasc. Med. 10:1215473. doi: 10.3389/fcvm.2023.1215473
Received: 2 May 2023; Accepted: 31 July 2023;
Published: 11 August 2023.
Edited by:
Arpit Kumar Agarwal, Baylor College of Medicine, United StatesReviewed by:
Siqi Hu, Seventh Medical Center of PLA General Hospital, China© 2023 Wu, Zhu, Zhang, Feng, Xia, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Zhang eWFubml6eUBnd2NtYy5vcmc= Jia Li amlhbGlfYmVpamluZ0AxMjYuY29t
†These authors have contributed equally to this work
‡These authors have jointly supervised this work
Abbreviations CHD, congenital heart disease; HSCR, Hirschsprung's disease.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.