
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Cardiovasc. Med. , 14 September 2023
Sec. Cardio-Oncology
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1212174
This article is part of the Research Topic Far from Perfect: The long-lasting effects of cancer treatments View all 8 articles
 Ashita Jain1,†
Ashita Jain1,† Diego Casanova1,†
Diego Casanova1,† Alejandra Valdivia Padilla1,†
Alejandra Valdivia Padilla1,† Angelica Paniagua Bojorges1,†
Angelica Paniagua Bojorges1,† Sivareddy Kotla1Kyung Ae Ko1Venkata S. K. Samanthapudi1
Sivareddy Kotla1Kyung Ae Ko1Venkata S. K. Samanthapudi1 Khanh Chau2
Khanh Chau2 Minh T. H. Nguyen2Jake Wen1Selina L. Hernandez Gonzalez1Shaefali P. Rodgers2
Minh T. H. Nguyen2Jake Wen1Selina L. Hernandez Gonzalez1Shaefali P. Rodgers2 Elizabeth A. Olmsted-Davis2
Elizabeth A. Olmsted-Davis2 Dale J. Hamilton3
Dale J. Hamilton3 Cielito Reyes-Gibby4
Cielito Reyes-Gibby4 Sai-Ching J. Yeung4
Sai-Ching J. Yeung4 John P. Cooke2
John P. Cooke2 Joerg Herrmann5
Joerg Herrmann5 Eduardo N. Chini6
Eduardo N. Chini6 Xiaolei Xu7
Xiaolei Xu7 Syed Wamique Yusuf1
Syed Wamique Yusuf1 Momoko Yoshimoto8Philip L. Lorenzi9Brain Hobbs10Sunil Krishnan11
Momoko Yoshimoto8Philip L. Lorenzi9Brain Hobbs10Sunil Krishnan11 Efstratios Koutroumpakis1
Efstratios Koutroumpakis1 Nicolas L. Palaskas1
Nicolas L. Palaskas1 Guangyu Wang2
Guangyu Wang2 Anita Deswal1
Anita Deswal1 Steven H. Lin12
Steven H. Lin12 Jun-ichi Abe1*
Jun-ichi Abe1* Nhat-Tu Le2*†
Nhat-Tu Le2*†
Cardiovascular disease (CVD) is a leading cause of morbidity and mortality, especially among the aging population. The “response-to-injury” model proposed by Dr. Russell Ross in 1999 emphasizes inflammation as a critical factor in atherosclerosis development, with atherosclerotic plaques forming due to endothelial cell (EC) injury, followed by myeloid cell adhesion and invasion into the blood vessel walls. Recent evidence indicates that cancer and its treatments can lead to long-term complications, including CVD. Cellular senescence, a hallmark of aging, is implicated in CVD pathogenesis, particularly in cancer survivors. However, the precise mechanisms linking premature senescence to CVD in cancer survivors remain poorly understood. This article aims to provide mechanistic insights into this association and propose future directions to better comprehend this complex interplay.
The advent of advanced cancer treatments has revolutionized cancer management, offering new possibilities for individuals with various types of cancer. Screening, early detection, and treatment modalities such as targeted therapies, immunotherapy, radiation therapy (RT), and chemotherapy have significantly improved patient outcomes and survival rates. However, understanding the potential long-term effects and complications associated with these treatments is crucial. We need to explore the underlying mechanisms involved in cellular communication, treatment response, and systemic effects to develop targeted interventions and personalized approaches, optimizing the long-term health of cancer survivors (1–4).
Cancer management has witnessed remarkable advancements, with targeted therapies playing a crucial role in various types of cancer treatment. For example, sorafenib has been instrumental in hepatocellular carcinoma treatment (5), while drugs like sunitinib, sorafenib, temsirolimus, and bevacizumab have shown effectiveness in renal cell carcinoma (6). Bevacizumab, cetuximab, and panitumumab have shown benefit in colon cancer (7–9), and cetuximab has shown promise in head and neck cancers (10). In pancreatic cancer, erlotinib has yielded positive outcomes (11, 12). Undoubtedly, these targeted therapies have revolutionized treatment for their respective cancer types (13).
Novel targeted therapies has shown promise in overcoming treatment resistance. For example, lapatinib has proven effective in breast cancer cases resistant to other targeted therapies (14). Similarly, ponatinib and dasatinib have exhibited positive outcomes in chronic myeloid leukemia patients, addressing resistance concerns (15, 16). Monoclonal antibodies like rituximab, has been successful either as standalone treatments or in combination with chemotherapy, particularly in managing indolent lymphoma (17, 18). Furthermore, the incorporation of imatinib in the adjuvant setting for resected gastrointestinal stromal tumors has improved outcomes for these cases (19–25). Chemotherapeutic agents like oxaliplatin has enhanced survival rates for specific cancer types, such as colon cancer (26), while temozolomide has shown promise in treating malignant glioma (27, 28). Adjuvant chemotherapy has also yielded positive results in non-small cell lung cancer cases (29, 30).
Advancements in cancer screening and early detection, along with treatments such as targeted therapies, immunotherapy, RT, and chemotherapy (13, 29) have significantly improved cancer survival rates. However, it is important to recognize that cancer treatments can lead to long-term and latent effects (31, 32). Long-term effects emerge during treatment and persist over time, while latent effects may only appear years after treatment completion. A comprehensive review by Gegechkori et al. (2017) (33) summarizes common long-term and latent treatment effects associated with prevalent cancers. As the number of cancer survivors continues to increase, understanding and managing these treatment-related effects becomes more critical. In the United States alone, there are currently over 15 million cancer survivors, projected to exceed 20 million by 2026 (34). While many survivors may face other comorbidities overtime, it is crucial to acknowledge the lasting consequences of the treatments they underwent.
Certain cancer treatments, such as RT (35–39) and chemotherapy (40, 41), are linked to an increased risk of cardiovascular complications (42, 43), which may manifest years or decades after treatment completion (44). These complications encompass various cardiovascular conditions including hypertension, arrhythmias, coronary artery disease (CAD), heart failure (HF), valvular disease, thromboembolic disorders, peripheral vascular disease (PVD), stroke, pulmonary hypertension, and pericardial complications (35–39, 45, 46). Anthracycline-induced cardiotoxicity occurs dose-dependently, with the highest incidence typically observed within the first year after chemotherapy completion (47–49). However, there is significant variation in patients’ susceptibility to anthracyclines (47–50). Approximately 9% of adult patients experience this cardiotoxicity, with the risk of HF related to doxorubicin estimated to have a cumulative incidence of 26% (51–53). Cisplatin, another commonly used chemotherapy agent, is linked to an increased risk of cardiovascular events even years after completing treatment (54, 55). In breast cancer survivors treated with trastuzumab for HER2 + breast cancer, studies predict a high incidence of cardiotoxicity, reaching up to 30% (56). The combination of trastuzumab and doxorubicin raises the risk of HF by more than 7-fold (57). Cardiotoxicity related to trastuzumab progressively increases during the 3–5 years after treatment completion and can persist for many years (58). Additionally, adjuvant hormonal therapy with tamoxifen elevates the risk of venous thromboembolism (59, 60). RT has been associated with various cardiovascular complications, affecting different components of the cardiovascular system, including the pericardium, myocardium, valves, coronary arteries, and conduction system. Asymptomatic breast cancer survivors who received RT show regional perfusion defects, closely linked to the left ventricle’s volume within the radiation field (61–67). The estimated incidence of RT-induced cardiotoxicity ranges from 10% to 30% at 5 to 10 years after treatment. Breast cancer survivors face an increased risk of thrombus formation, attributed to imbalances in von Willebrand factor release, thrombomodulin, and adenosine diphosphatase production, leading to heightened platelet adherence and clot formation in irradiated capillaries and arteries (68–72). A cross-sectional study using the National Health and Nutrition Examination Survey assessed the 10-year risk of atherosclerotic CVD using Pooled Cohort Equations in cancer survivors and non-cancer patients, revealing higher risk for cancer survivors compared to non-cancer patients (73). This increased prevalence and mortality have been attributed to cancer therapies, including RT and chemotherapeutic agents (74).
As individual age, the prevalence of various disorders, including CVD, tends to increase within the population (75–79). Advanced age is a significant risk factor for doxorubicin-related cardiotoxicity, even at lower doses (80–82). CVD remains the leading cause of morbidity and mortality worldwide, particularly among individuals aged 65 and older. Scoring systems such as the Pooled Cohort Equations, Framingham Risk Score, and Reynolds Risk Score consistently highlight age as a significant risk factor for developing CVD, with both females and males facing an increased risk as they age (74, 83).
Cellular senescence, a hallmark of aging (84), plays a critical role in the development and progression of cardiac aging and CVD (74, 83, 85–87). Senescent cells accumulate in specific cardiovascular sites associated with CVD, such as atherosclerosis, HF, arterial stiffness, and hypertension (88, 89). Recent research has revealed that senescence is not limited to dividing cells due to replicative capacity loss (90–93). Instead, it is a broader cellular response to stress and damage, involving interconnected domains like DNA damage response (DDR), cell cycle arrest, senescence-associated secretory phenotype (SASP), senescence-associated mitochondrial dysfunction, autophagy/mitophagy dysfunction, nutrient and stress signaling, and epigenetic reprogramming. Activation of these domains occurs during senescence, with interactions between them. Cellular senescence significantly contributes to mammalian aging, playing a key role in age-related changes and dysfunction. Importantly, even post-mitotic cells, including cardiomyocytes, can undergo senescence (94, 95). Cardiac aging leads to various detrimental effects, including impaired angiogenesis (96), accelerated fibrosis (97), metabolic dysregulation (98), and the senescence and dysfunction of cardiomyocytes (99, 100). Senescent cardiomyocytes exhibit DNA damage, mitochondrial dysfunction, impaired contractile function, endoplasmic reticulum stress, hypertrophic growth, and secretion of SASP factors. Non-cardiomyocyte cells, such as fibroblasts, immune cells, and ECs, contribute to the regulation of cardiomyocyte senescence, further promoting cardiac aging and pathological remodeling (88, 101–104). Elevated activity of senescence-associated β-galactosidase (SA-β-gal), a widely used marker of cellular senescence, has been observed in ECs within atherosclerotic plaques in human coronary arteries (105).
The accumulation of senescent cells in the cardiovascular system during the aging process has been linked to age-associated disorders, including CVD (106–108). Senescent cells contribute to atherosclerotic plaque formation by secreting SASP factors, including proinflammatory cytokines and chemokines such as interleukin 1α, β (IL-1α, β), tumor necrosis factor α (TNFα), interferon γ (IFN γ), transforming growth factor β (TGFβ), and monocyte chemoattractant protein 1 (MCP-1). In early atherogenesis, oxidized low-density lipoprotein (oxLDL) accumulates in subendothelial spaces. SASP factors stimulates immune cell proliferation and activation, leading to monocyte recruitment into the subendothelial space. These monocytes differentiate into foam cell macrophages, contributing to plaque formation. Senescent ECs express vascular and intercellular cell-adhesion molecules (VCAM-1 and ICAM-1) and release cytokines, further attracting monocytes and promoting atherosclerotic plaque development (109–111). SASP factors also activate plasminogen activator inhibitor 1 (PAI-1), promoting thrombus formation (112). The persistent pro-inflammatory senescence phenotype induced by SASP factors leads to chronic inflammation, impairs cholesterol efflux from macrophages, and contributes to the accumulation of lipid-laden foam cells within the plaque (106–108).
Recent studies suggest cellular senescence’s involvement in plaque destabilization and rupture, leading to acute cardiovascular events. Senescent vascular smooth muscle cells (VSMCs) within the plaque show increased activity of matrix metalloproteinases (MMPs), contributing to plaque rupture and thrombosis (113). However, precise mechanisms connecting cellular senescence and CVD, such as chromatin structure remodeling, the local heart microenvironment, and the activation of DDR in cardiomyocytes leading to senescence, require further investigation.
Cellular senescence plays a dual role in cancer development and progression through the secretion of SASP factors (114). On one hand, it acts as a tumor-suppressor mechanism in various cancer treatments by inhibiting cell division and aiding in immune clearance of damaged cells, reducing the likelihood of tumorigenesis. However, cancer treatments inducing cellular senescence can have negative consequences, leading to persistent DNA damage, continued SASP factor secretion, and CVD development (115–122). SASP factors can induce inflammation, tissue remodeling, and angiogenesis, creating a microenvironment that supports cancer cell growth and spread (123, 124). The accumulation of senescent cells over time can lead to chronic inflammation, tissue dysfunction, and organ failure, increasing the risk of cancer development (125). Extensive research correlates senescent cell presence in tissues with an elevated cancer risk and poorer prognosis in cancer patients. Senescent cells can have both suppressive and promotive effects on cancer, depending on the context and specific cellular and molecular interactions involved.
Cellular senescence plays a crucial role linking cancer, cancer treatments, and CVD, offering promise for preventing various CVD conditions such as acute myocardial infarction, atherosclerosis, cardiac aging, pressure overload-induced hypertrophy, heart regeneration, hypertension, and abdominal aortic aneurysm. Targeting cellular senescence is especially relevant for CVD prevention in cancer survivors. However, the mechanisms underlying cellular senescence are complex, as it can have both beneficial and detrimental effects in cancer and CVD, depending on the circumstances (126, 127). The pioneering work of Hayflick and Moorhead (1961) revealed that human diploid fibroblasts undergo irreversible cell cycle arrest after a certain number of divisions, known as the Hayflick limit (90–93). This arrest occurs due to telomere shortening, hindering the formation of T-loop (90–93) and leading to RS (128–130).
Cellular senescence occurs through RS over repeated replication cycles, leading to the exponential accumulation of senescent cells in multiple tissues with increasing age (131, 132). Alternatively, cellular senescence can be triggered prematurely by stressors, known as SIPS (133). These stressors can originate from internal and external factors, including oncogenes, RT, cancer treatments, mitochondrial dysfunction, reactive oxygen species (ROS), etc. (Figure 1). SIPS is characterized by events such as upregulated cyclin-dependent kinase inhibitors p21Cip1/Waf1, p16INK4a, positive staining for SA-β-gal, and telomere shortening. Activation of the p53/p21Cip1/Waf1 and p16INK4a/pRB pathways plays a central role in regulating senescence (132). Unlike RS, SIPS can occur without significant telomere shortening, as we (106, 116, 117) and others (132, 136) have previously reviewed. Senescent cells secrete SASP factors, including various cytokines, growth factors, and proteases contributing to chronic inflammation and senescence-associated phenotypes (115–122). In cardio-oncology, SIPS has emerged as a critical mechanism contributing to CVD development after cancer treatments.
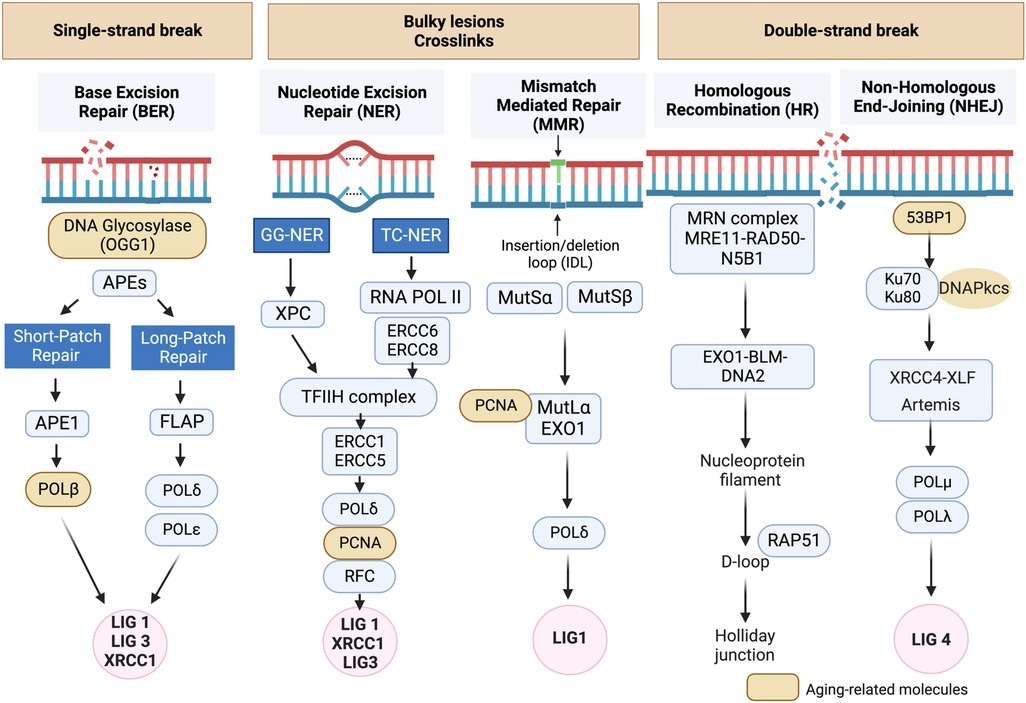
Figure 1. DNA damage response (DDR) mechanisms in aging-related disease: the DDR mechanisms depicted in this schematic are instrumental in preserving genomic stability and slowing down the aging process. However, as we grow older, the efficiency of these mechanisms may diminish, resulting in heightened accumulation of DNA damage and a decline in cellular function (134, 135). Schematic created using BioRender.com.
DNA damage can disrupt cellular homeostasis, leading to structural modifications that trigger cellular senescence during aging (137). Formation of γ-H2AX foci (γ-foci) indicates DNA damage is strongly associated with cellular senescence (11). DNA damage also induces mitochondrial ROS (mtROS) production and inflammation, further exacerbated by the mitochondria-nucleus feedback loop (115), as discussed in section (VI). Extrinsic factors such as free radicals, oxidizing agents, nitrosylating agents, alkylating agents, UV light, and certain cancer treatments like ionizing radiation (IR), can induce DNA damage (115, 137). In response to DNA damage, various DDR mechanisms are activated to recognize and repair different types of lesions, ensuring genomic integrity and DNA structure restoration. If the damage persists and cannot be effectively repaired, it can lead to RS (128–130), apoptosis, or SIPS (133) depending on the severity and nature of the damage and the cellular context (106, 116, 117, 132, 135–137). Cells utilize at least five distinct DDR mechanisms, including Based Excision Repair (BER) (138), Nucleotide excision repair (NER) (139, 140) and Mismatch repair (MMR) (141), each regulated differently to maintain genetic stability (Figure 1) (134, 135). Detailed mechanistic insights into these DDR mechanisms can be found elsewhere (134, 135, 137–140, 142–159).
Persistent activation of DDR pathways, crucial for cancer treatments, leads to the accumulation of inflammation, ultimately resulting in cellular senescence. DDR regulates cellular senescence and chronic sterile inflammation through various mechanisms. One mechanism involves the activation of DNA damage sensors that detect and respond to DNA damage signals. Another mechanism involves the alteration of the levels and activity of nicotinamide adenine dinucleotide (NAD+), an essential coenzyme involved in cellular metabolism and signaling pathways. DDR also influences mitochondrial function, linked to cellular senescence through ROS generation and metabolic changes. Furthermore, DDR modulates histone epigenetic changes and DNA modifications, leading to chromatin remodeling and alterations in gene expression patterns associated with cellular senescence (115, 137). Persistent activation of DDR also stimulates the secretion of SASP factors (115–122). For a comprehensive understanding of the mechanisms of cellular senescence regulation by DDR, each mechanism will be discussed in detail. As SASP factors play a critical role in CVD and cancer after cancer treatments, the discussion on SASP factors will be presented in a separate section (IV).
DDR activates major DDR pathways such as ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related (ATR) (Figures 2, 3). These sensors recognize DNA damage and initiate downstream phosphorylation events, involving checkpoint kinase 1/2 (CHK1/2) to coordinate the cellular response. Activated by ATM/ATR, p53 plays a critical role in responding to genotoxic stress, inducing apoptosis through targets like BAX and PUMA or promoting cell cycle arrest via p21Cip1/Waf1 and NOXA (115, 137, 163). p53 can inhibit NFκB activity, but its depletion may enhance tumor development by increasing inflammation (163–166). p53 also upregulates proinflammatory genes, such as IL6, CXCL1, and NFκB (167), with their secretion accelerated in senescent cells with high p53 activation (168, 169). In HF, inhibiting p53 can have cardioprotective effects by reducing cardiomyocyte apoptosis (170), but it may have atheroprone effects by increasing macrophage proliferation and/or inhibiting apoptosis, leading to larger plaques (171).
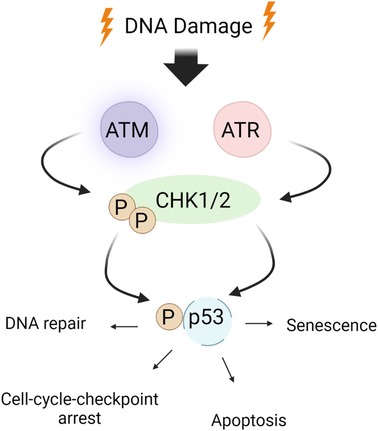
Figure 2. ATM/ATR signaling in the DDR: the DDR encompasses checkpoint kinases, downstream effector kinases, and a tumor suppressor protein, which collectively regulate apoptosis and cell cycle arrest. ATM/ATR, when activated by DNA damage, play critical roles in initiating the DDR. Upon activation, ATM/ATR phosphorylate various downstream targets, including CHK1/2. CHK1 phosphorylates p53 at S15 and Thr18 (160), thereby stabilizing p53 through prevention of p53 degradation and enhancing p53’s transcriptional activity. This ultimately leads to apoptosis and cell cycle arrest by inhibiting CDK activity. Schematic created using BioRender.com.
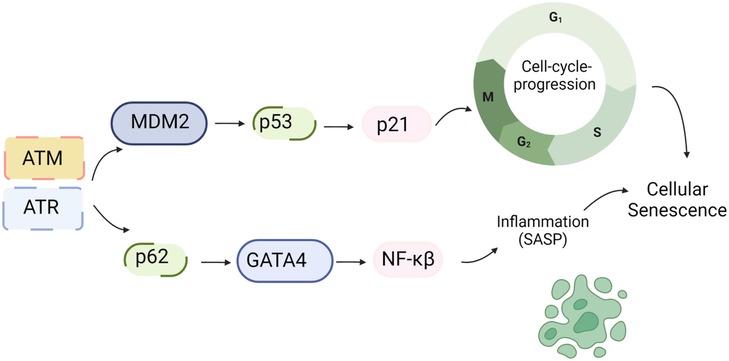
Figure 3. DDR and SASP: DNA damage triggers the activation of ATM/ATR, which subsequently phosphorylates CHK1/2. These phosphorylated kinases further activate p53. Upon activation, p53 transcriptionally upregulates p21, which inhibits CDK activity and leads to cell cycle arrest. Additionally, ATM/ATR can inhibit the interaction between p62 and GATA4, leading to the stabilization of GATA4. This stabilization event results in NF-kB activation and the subsequent induction of the SASP (161). Schematic created using BioRender.com.
Higo et al.’s research reveals the association between HF and unrepaired single-strand breaks (SSBs), leading to DDR activation and inflammation by upregulating NF-κB signaling. Depletion of ATM attenuates pressure overload-induced HF, highlighting DDR’s causal role in HF (172). ATM/ATR can hinder p62-GATA4 interaction, inhibiting autophagy and ultimately activating SASP through NF-κB (161). While ATM/ATR activation can induce SASP, its impact on CVD, especially atherosclerosis, can be intricate due to additional effects on inhibiting macrophages, VSMC, and fibroblast proliferation (173, 174).
Mitochondrial dysfunction is a hallmark of aging (84), characterized by decreased respiratory capacity, membrane potential, and increased free radical production. Doxorubicin administration exacerbates damage to myocardial mitochondrial DNA (mtDNA) and activates mast cells, leading to harm to cardiomyocytes and impaired self-repair mechanisms (175). The interplay between aging and mitochondrial dysfunction contributes to a senescent phenotype (175, 176). The mitochondrial electron transport chain (ETC) uses mobile electron carriers (ubiquinone and cytochrome c) and four enzyme complexes (I, II, III, and IV) in the inner mitochondrial membrane (IMM) (Figure 4) (177, 178) to generate adenosine triphosphate (ATP) (179–184) through oxidative phosphorylation (OXPHOS) (177, 178, 185–187). During ETC activity, mtROS are produced (178), playing a crucial role in signaling pathways and maintaining cellular function (188). mtROS is also involved in the persistent SASP associated with cancer treatments (189). With aging, reduced antioxidant production increases ROS levels, leading to oxidation of lipids, proteins, and DNA (190, 191). ROS exposure can modify DNA’s guanine (G) to form 8-oxo guanine (8-oxoG), causing base-pairing errors during replication, resulting in genomic instability (192, 193), particularly in mtDNA due to less efficient repair mechanisms (194). Thus, mitochondrial dysfunction-mediated mtDNA damage contributes significantly to pathological conditions, including aging-related CVD.
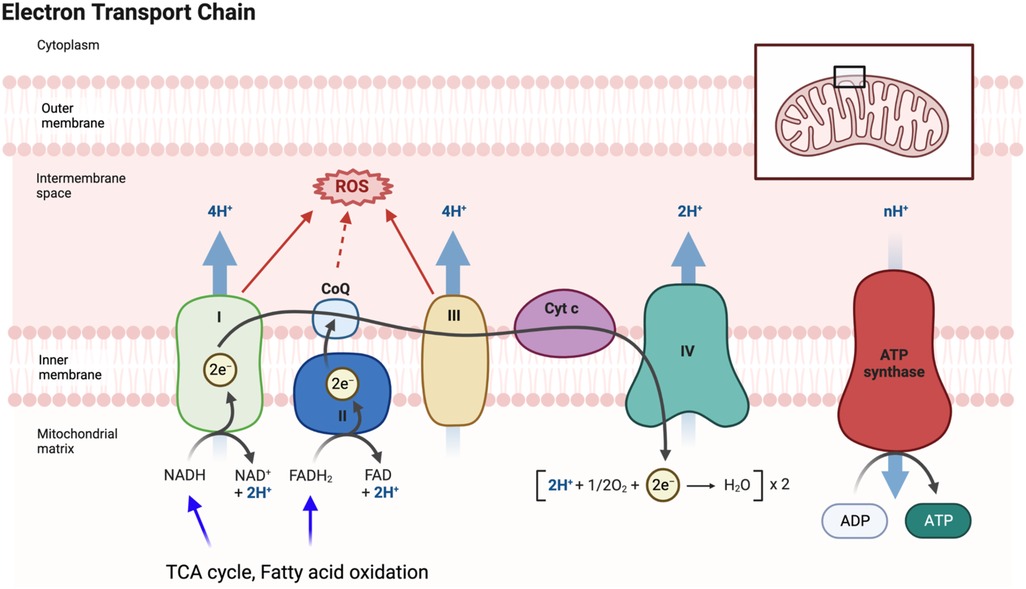
Figure 4. Electron transport chain (ETC) and mitochondrial ROS (mtROS) production: the ETC, located in the inner mitochondrial membrane, plays a vital role in the final steps of oxidative phosphorylation (OXPHOS). It facilitates the transfer of electrons from electron donors, such as NADH and FADH2, to oxygen, resulting in the production of ATP. As electrons flow through the ETC, protons are pumped across the membrane, creating a gradient that drives ATP synthesis by ATP synthase. During the electron transfer process in the ETC, there is also the generation of mtROS as a byproduct. These mtROS molecules can induce oxidative stress and cause damage to mitochondrial DNA, proteins, and lipids. FADH2 is a coenzyme involved in cellular respiration, and it donates electrons to the ETC. It is produced during the breakdown of fatty acids and certain amino acids. FADH2 specifically donates electrons to complex II of the ETC, bypassing complex I. This bypass leads to the production of fewer protons and consequently yields a lower amount of ATP compared to NADH. Figure generated using BioRender.com.
Mitochondrial dynamics, involving “fusion” (merging) and “fission” (separation) events, play a crucial role in maintaining mitochondrial health (Figures 5, 6). Mitofusin1/2 (MFN1/2) on the outer mitochondrial membrane (OMM) (197) facilitate fusion between adjacent mitochondria (198), regulated by dynamin-like GTPases (199). This process maintains inner membrane architecture through optic atrophy 1 (OPA1)-mediated fusion of the IMM (200). Conversely, mitochondrial fission is regulated by cytosolic GTPase dynamin-related protein 1 (DRP1) and mitochondria-bound proteins, such as mitochondrial fission factor (MFF), mitochondrial fission protein 1 (Fis1), and mitochondrial dynamics proteins of 51 and 49 kDa (MiD51 and MiD49). Defects in MFN1/2 or OPA1 can lead to mitochondrial dysfunction (201).
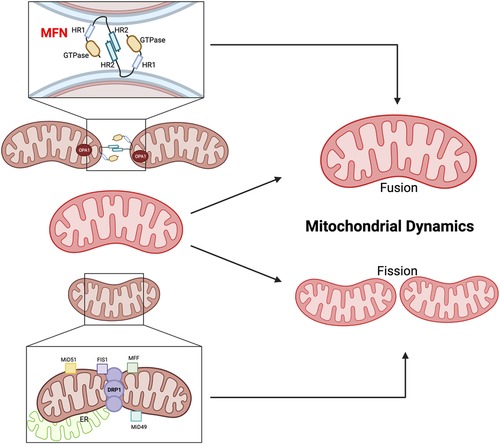
Figure 5. Mitochondrial fusion and fission. MFN1/2 and OPA1 are involved in OMM fusion, whereas OPA1 is involved in IMM fusion. The GTPase domain and Heptad repeat (HR) coiled-coil regions HR1/2 are shown. MFN2 interacts in transforming either homotypic or heterotypic (with MFN1) dimers to produce mitochondrial tethering and induces mitochondrial fusion. OPA1 binds the IMM. DRP1, MFF, Fis1, and the homologs MiD49 and MiD51 are involved in the fission process. MFF, Fis1, MiD49, and MiD51 act as receptors on the OMM to recruit and activate DRP1, which then oligomerizes and forms a ring-like structure around the mitochondrion at the site of division. The DRP1 ring generates tension, which causes constriction and eventual division of the mitochondrion into two daughter mitochondria (195). Figure created using BioRender.com.
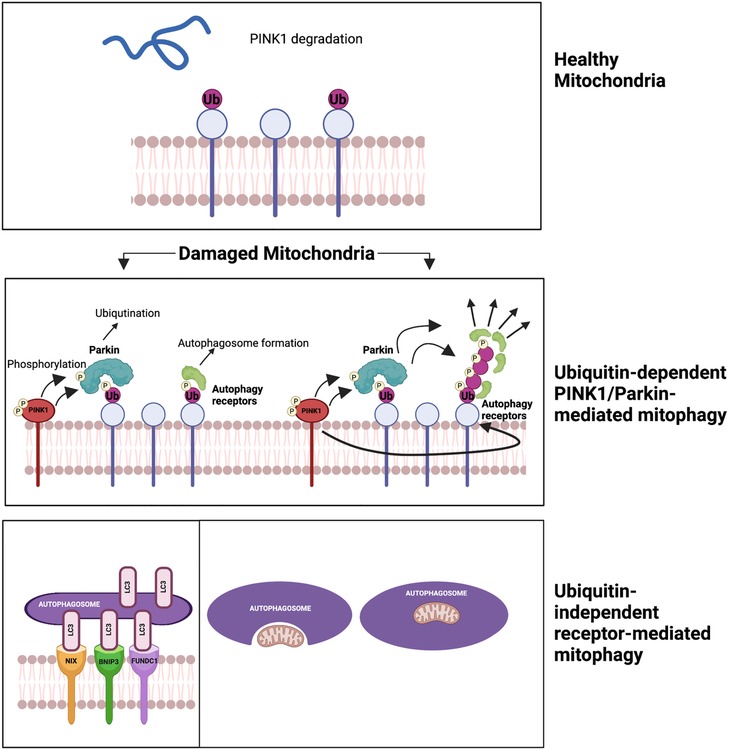
Figure 6. Two mechanisms of mitophagy (196) (please see the text) figure created using BioRender.com.
The balance between mitochondrial fusion and fission is critical for cellular homeostasis and preventing age-related pathologies. Perturbations in this balance can have significant consequences. Suppression of fission extends lifespan in Saccharomyces cerevisiae, while uncontrolled fission shortens lifespan (202). However, simultaneous suppression of both fission and fusion can lead to different effects on lifespan in yeast and worms (203). AMP-activated protein kinase (AMPK) and the target of rapamycin complex 1 (TORC1) are implicated in lifespan extension, with both fission and fusion playing roles in AMPK-induced longevity, while fusion appears particularly crucial for TORC1-mediated lifespan extension (204). These observations highlight the intricate involvement of mitochondrial dynamics in the aging process, but further research is needed to fully elucidate the precise roles of fission and fusion in aging and lifespan extension (120).
The dysregulation of fusion and fission-related molecules is associated with cardiac aging and diseases (205). DRP1 is crucial for mitochondrial fission and mitophagy (206). Cardiac-specific DRP1 knockout (KO) results in defective mitochondrial fission, increased mitophagy, mitochondrial loss, cardiomyocyte necrosis, and the development of dilated cardiomyopathy in mice (207–209). Conversely, cardiac-specific MFN1/2 KO leads to enhanced mitochondrial fragmentation, reduced mitophagy, mitochondrial accumulation, cardiomyocyte enlargement, progressive cardiac hypertrophy, and systolic dysfunction (210). Song et al. used cardiac-specific triple-KO mice lacking MFN1/2 and DRP1 to investigate the effects of eliminating both fission and fusion processes in the heart. The mice showed prolonged survival and developed pathological cardiac hypertrophy and HF resembling aged hearts. The researchers suggested that reduced fission and fusion processes leads to mitochondrial adynamism and senescence, impairing mitochondrial quantity control. These findings highlight how dysregulation of fission and fusion can contribute to cardiac dysfunction, hypertrophy, and aspects of aging in the heart (205).
In cancer, proteins promoting mitochondrial fission are often upregulated in tumor tissues compared to normal tissues (211), and their increased expression is associated with worse prognosis for cancer patients (211). The global fragmentation of mitochondria in cancer cells leads to mtROS generation and metabolic alterations, promoting cancer cell migration, invasion, and metastasis (211). For certain cancers like pancreatic ductal adenocarcinoma (PDAC), abnormally fragmented mitochondria are linked to the oncogenicity of the disease. Therapeutic approaches targeting mitochondrial fusion restoration and normalizing morphology have shown promising results in preclinical PDAC models, reducing OXPHOS, inhibiting tumor growth, and improving overall survival (212).
Low-dose IR can increase mitochondrial fusion, which may inhibit tumorigenesis. However, dysregulation of both fission and fusion, leading to mitochondrial adynamism, can be detrimental to the cardiovascular system (205). Therefore, it is crucial to cautiously evaluate cancer therapies targeting mitochondrial fragmentation and their potential impact on the circulatory system. A thorough understanding of the interplay between mitochondrial dynamics, cancer biology, and cardiovascular health is crucial for developing effective therapies while minimizing potential cardiovascular complications associated with cancer treatments. Further research is needed to archive this comprehensive understanding.
Mitochondrial autophagy, or “mitophagy,” was first described by John Lemasters (213). It selectively sequesters and encapsulates damaged or depolarized mitochondria into double-membraned autophagosomes, which then get degraded in lysosomes (214). There are two main mechanisms of mitophagy. The first mechanism is ubiquitin-dependent PINK1/Parkin-mediated mitophagy. It involves two key proteins, PTEN-induced putative kinase 1 (PINK1) and Parkin, recruited to damaged mitochondria. PINK1 phosphorylates ubiquitin, recruiting Parkin. Parkin then ubiquitinates mitochondrial proteins, forming ubiquitin chains signal the autophagic machinery. This triggers engulfment and degradation of the damaged mitochondria. PINK1’s phosphorylation of Parkin S65 is crucial for its activity and recruitment to the mitochondria, facilitating degradation through autophagy receptors (215, 216). The second mechanism is ubiquitin-independent receptor-mediated mitophagy, involving receptors on the OMM and not requiring ubiquitination of mitochondrial proteins. Autophagic receptors such as microtubule-associated protein light chain 3 (LC3), Bcl-2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3), NIP-3-like protein X (NIX or BNIP3l), and FUN14 domain-containing protein 1 (FUNDC1) are involved in this pathway. These receptors possess LC3-interacting regions (LIRs) that bind to damaged mitochondria, promoting their engulfment and degradation by the autophagic machinery (217). This pathway plays a critical role in maintaining mitochondrial quality control and preventing the accumulation of damaged mitochondria, and its dysregulation can contribute to diseases, including CVD (218, 219).
Mitophagy, the removal of damaged mitochondria, reduces mtROS production and prevents apoptosis (220, 221). Nuclear and mitochondrial genes coordinate mitochondrial biogenesis (222), ensuring mitochondrial function and cellular homeostasis. The balance between mitophagy and mitochondrial biogenesis determines mitochondrial turnover, regulated by the mTOR signaling pathway. Activation of mTORC1 inhibits mitophagy, while inhibiting mTORC1 or activating FoxO1 restores mitophagy. mTORC1 activation is associated with mitochondrial protein aging and p62/SQSTM1-positive mitochondria accumulation, possibly contributing to aging (223). Reduced mitophagy leads to the accumulation of damaged mitochondria in aged C. elegans (224), while upregulation extends lifespan of nematodes (225). Impaired mitophagy is linked to aging-related diseases such as Parkinson’s, Alzheimer’s, and CVD (226–228). Interventions such as caloric restriction and exercise promote mitophagy and enhance lifespan (229, 230).
Parkinson’s disease is linked to disruptions in calcium (Ca2+) homeostasis and increased mtROS production (226). Depletion of PINK1 inhibits mitophagy, leading to excessive mitochondrial Ca2+ buildup in response to dopamine. Elevated Ca2+ levels promote mtROS production and trigger neuronal cell death in midbrain neurons (231). Accumulation of damaged mitochondria results in overproduction of mtROS and release of pro-inflammatory cytokines, contributing to neurodegeneration (232). Mitophagy plays a critical role in regulating mitochondrial quantity and quality, controlling mtROS production, mitigating mtDNA damage, and preventing cellular apoptosis. Impairments in mitophagy can lead to persistent immune system activation and the development of aging-related neurological disorders (214). Rita Levi-Montalcini and Barbara Booker (1960) observed that mature neurons acquire resistance to apoptosis. The following year, Leonard Hayflick and Paul Moorhead (1961) described “senescence,” a stable replicative arrest of cells in vitro. Neuroscience and senescence in cell biology developed independently for six decades, each unraveling molecular mediators and defining phenotypes related to their observations. Neuroscientists noticed neuron’s remarkable survival ability despite chronic inflammation and degeneration in their environment. Similarly, SIPS leads to a change in cell fate instead of responding to cellular or DNA damage through entering a stable cell cycle arrest. These cells secrete SASP factors, negatively impact the cellular environment. These fields of neuroscience and senescence have now intersected, with neuroscientists applying the concept of senescence to the brain, including post-mitotic cells. This integration is advancing the understanding of brain aging, neurodegenerative diseases, and CVD (233).
Mitophagy is observed in aged hearts and vessels (234, 235). However, its role in CVD and tissue damage remains unclear. The outcome of mitophagy upregulation in CVD conditions may depend on specific mitophagy levels. Excessive mitophagy and mitochondrial clearance can potentially harm the compromised circulatory system, as seen in HF and ischemic heart diseases (236). In cancer treatments, mitophagy activation enhances cytotoxic effects on cancer cells (237–239), while autophagy and mitophagy inhibitors can promote antitumor effects (240). Mitophagy’s role in cancer treatments may vary depending on specific mitophagy levels. The involvement of autophagy and mitophagy in cancer therapy-related premature aging requires further investigation.
Mitochondrial accumulation of Ca2+ and ROS can trigger the opening of the mPTP, releasing of pro-apoptotic proteins and cytochrome c into the cytosol (232). The ETC at the IMM establishes an electrochemical gradient, driving the uptake of mitochondrial Ca2+ from the sarcoplasmic reticulum. The Na+/Ca2+ antiporter (NCLX) expels Ca2+ from the mitochondria. Cytosolic Ca2+ overload leads to sustained increase in mitochondrial Ca2+ levels, triggering PTP opening. This results in loss of mitochondrial membrane potential, decreased ATP synthesis, OMM, disruption, and cell death (241, 242). While voltage-dependent anion channels (VDACs), adenine nucleotide translocase (ANT), mitochondrial ATP synthase (F0F1), phosphate carrier (PiC), and cyclophilin D (CypD) contribute to the mPTP, its exact structural configuration remains incompletely understood.
The opening of the mPTP is closely associated with the aging process. Upregulation of cyclophilin D (CypD) and its interaction with p53 triggers mPTP opening, while downregulation of heat shock protein 90 (HSP90) in aged cells increases mPTP opening. SIRT3, a key player in aging, inhibits mPTP opening by modulating CypD activity (243). The decline of SIRT3 during aging contributes to mPTP opening and impacts the regulation of aging. Treatments like metformin and caloric restriction, known for promoting longevity, prevent mPTP opening, potentially contributing to their beneficial effects on extending lifespan (244–246).
Epigenetics refers to molecular modifications in chromatin (DNA and histone proteins) that regulate gene expression without altering the DNA sequence (247, 248). These modifications can be influenced by factors like the environment, diet, social conditions, and cancer treatments, potentially passing down to future generations (249). Epigenetics plays a crucial role in the development of heritable (248) and are implicated in various diseases, including cancer, autoimmune, degenerative, neuroendocrine, neuropsychiatric, and CVD (247, 249). Certain epigenetic alterations like histone modification and DNA methylation can contribute to age-related diseases such as cancer and CV (249–252).
One extensively studied chromatin modifications is the methylation of cytosine residues at carbon 5 (5 mC) within CpG dinucleotides. This modification plays a crucial role in gene expression regulation and is linked to carcinogenesis. Three DNA methyltransferases (DNMTs) have been identified: DNMT1, DNMT3a, and DNMT3b. DNMT1 maintains DNA methylation patterns by methylating hemimethylated DNA during replication. DNMT3a and DNMT3b act as de novo methyltransferases, establishing DNA methylation patterns, and coordinating chromatin templating processes. During embryonic development, DNMT3a and DNMT3b exhibit significant de novo methyltransferase activity (247).
Adverse alterations in DNA methylation can lead to genetic abnormalities, potentially causing cancer. These modifications may silence tumor suppressor genes or activate oncogenes. Different types of cancer show specific aberrations in DNA methylation, such as hypermethylation or hypomethylation in critical DNA regions, as well as acetylation or methylation of histone proteins, especially within CpG islands. The nature of these modifications depends on the repressed tumor suppressor genes, regulating cell malignant growth (249, 250).
Aging is associated with genome-wide DNA methylation alterations (253, 254). Epigenetic drift lads to gradual epigenetic changes over time, causing variations in gene expression even among genetically identical individuals. As twins age, differences in methylation conversion rates and global DNA methylation patterns may arise (255, 256), potentially contributing to age-related diseases. Age predictors based on DNA methylation levels have been developed (257), like the DNA methylation PhenoAge clock, which assesses health and lifespan by evaluating specific CpG sites (258). However, the precise mechanisms underlying age-related DNA methylation changes and their role in the aging process and age-related diseases remain unclear (254).
The nucleosome is a complex of four core histones (H2A, H2B, H3, and H4) wrapped around 147 base pairs of DNA, involved in DNA condensation and post-transcriptional modifications. Histone modifications include acetylation, methylation, ubiquitination, phosphorylation, and sumoylation (249).
Acetylation is a significant histone modification involving the addition of an acetyl group, affecting transcriptional pathways, DNA repair, and chromatin organization (247). Acetylation neutralizes the positive charge of lysine residues, weakening their interaction with histones and DNA. Two enzyme families, histone lysine acetyltransferases (HATs) and histone deacetylases (HDACs), dynamically and tightly regulate this process. HATs, including the CBP enzyme, have clinical relevance due to their role in neoplastic transformation and carcinogenesis (247, 250). Conversely, HDACs compact chromatin structure, repressing gene expression. HDAC inhibitors are used in cancer treatment to reactivate silenced tumor suppressor genes. HDACs are classified into four classes and can be involved in pathologies like leukemia through chimeric fusion proteins (247).
Aging is associated with alterations in histone modifications, particularly changes in acetylation and methylation patterns. Histone H4K16 acetylation, H4K20 trimethylation (H4K20me3), and H3K27 trimethylation (H3K27me3) are upregulated in aging (259). Nuclear SIRT deacetylation enzymes, such as SIRT1, SIRT6, and SIRT7 counteract aging effects. SIRT1 promotes chromatin silencing and transcriptional repression by deacetylating H1K26, H3K9, and H4K16 (260), while SIRT6 facilitates H3K9 and H3K56 deacetylation, contributing to the maintenance of genome stability and telomere function (261). Increased levels of H3K4 dimethylation (H3K4me2) are observed at stress response gene regions (262), and it plays a role in regulating hematopoietic stem cell identity and self-renewal abilities (263). Cells derived from progeria patients show an upregulation of repressive H4K29me3 (264, 265). The accumulation of repressive H3K27me3 in aged cells may lead to the permanent loss of transcriptional potential associated with aging.
HDAC inhibitors impact various hallmarks of aging, including epigenetic modifications, telomere attrition, genomic instability, loss of proteostasis, nutrient sensing dysregulation, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and intercellular communication (266). Trichostatin A, a selective inhibitor of HDAC class I and II, reduces pressure overload-induced cardiac hypertrophy by modulating relevant molecules (267). HDAC inhibitors like sodium butyrate has shown promise in inhibiting cardiac dysfunction in diabetic mice (268) and improving glucose metabolism in aged mice (269). These findings suggest that HDAC inhibition could be a potential therapeutic approach for cardiac diseases and may also have preventive effect against CVD by targeting the aging process.
Autophagy is a conserved process involving degradation of macromolecules and organelles within lysosomes (270, 271). It has three forms: macroautophagy (autophagy), microautophagy, and chaperone-mediated autophagy (CMA) (272). Autophagy sequesters targets within autophagosomes, which fuse with lysosomes for degradation. Microautophagy involves lysosome membrane invagination, while CMA uses chaperones to translocate unfolded proteins into lysosomes (273). The ULK1/ATG13/ATG101/FIP200 complex initiates autophagy in response to low cellular nutrient levels. The Beclin1/ATG14/VPS15/VPS34 Class III PI3K complex is recruited to autophagic cargo sites, generating phosphatidylinositol 3-phosphate (PI3P) for phagophore formation. ATG proteins are then recruited to facilitate membrane elongation and closure, forming a complete autophagosome. The ATG5/ATG12/ATG16l complex enables ATG8 (LC3 and GABARAP) conjugation to the membrane. The ATG5/ATG12/ATG16l complex converts ATG4-cleaved LC3/GABARAP (LC3-I) to LC3-II by conjugating it to phosphatidylethanolamine (PE), essential for autophagosome formation. The autophagosome fuses with a lysosome, forming an autolysosome for cargo is degradation and recycling (274) (Figure 7).
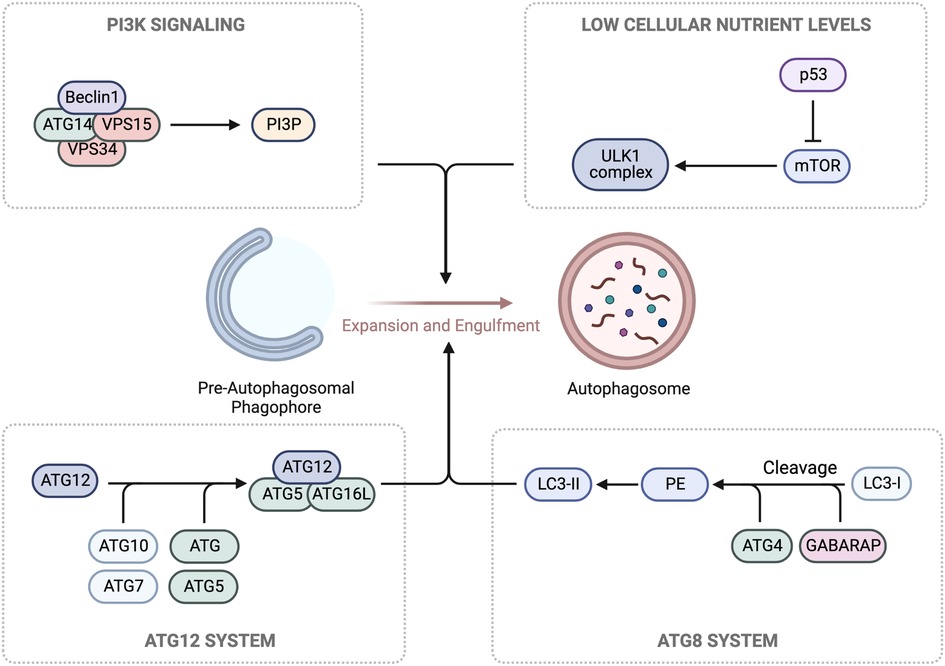
Figure 7. Autophagy machinery. In response to low glucose levels, AMPK pathways cause the phosphorylation and activation of the Unc-51-kinase 1 (ULK1) complex, which contributes to the growth of the pre-autophagosomal phagophore. Concurrently, ULK1 phosphorylation leads to the activation of the Class III PI3K complex, composed of Beclin1/ATG14/VPS15/VPS34, and the production of phosphatidylinositol 3-phosphate (PI3P) for further growth. For membrane binding and the final steps of autophagosome formation, conjugation of two ubiquitin-like complexes, ATG5/ATG12 and ATG10/ATG7, is necessary for the formation of the ATG5/ATG12/ATG16l complex. Hierarchically, this promotes the cleavage of LC3/GABARAP (LC3-I) by ATG4, generating phosphatidylethanolamine (PE), which forms LC3-II. Schematic created using BioRender.com.
Autophagy plays a vital role in various fields, including cancer and degenerative diseases (270), but it also becomes critical in aging. The aging process involves the accumulation of damaged proteins and organelles, particularly challenging for non-proliferating cardiomyocytes (275, 276). As organisms age, declining autophagy levels hinder efficient cellular clearance, impacting heart tissue homeostasis. This downregulation of autophagy in aging hearts increases the risk of developing senescence-associated diseases (277). Autophagy is regulated by transcription factors like FoxO, HIF-1, p53, E2F1, NFκB, KLF4, TFEB, and ZKSCAN3 (278), and in the aged heart, FoxO’s activity is inhibited, leading to reduced expression of autophagy-related molecules (278–280).
According to Levine and Klionsky, autophagy is a crucial component of the aging process in eukaryotic organisms, involved in the turnover of long-lived proteins and damaged organelles (281), Autophagy’s selective retention of components while eliminating senescent ones may slow down aging and age-related CVD. Autophagy has been observed in myocardial tissue cells, but its levels are relatively low. Conditions like congestive HF, CAD, hypertension, and aortic valvular disease can lead to the accumulation of autophagosome (282). CVD and stress can upregulate autophagy levels in the heart (283). Studies in ATG5-deficient mice have shown cardiomyopathy, hypertrophy, and contractile dysfunction, highlighting the significance of autophagy in maintaining cardiac function (284). However, it is important to note that increased autophagy levels have also been linked to HF and cardiomyocyte death through autophagy-induced degeneration (285). Thus, autophagy may have both beneficial and detrimental roles in the cardiovascular system.
In its oxidized state, NAD+ regulates metabolic networks, including glycolysis, the TCA cycle, FAO, and NADH production (185, 286). Age-related studies show decreased NAD+ levels in senescent cells (287–290), linked to premature senescence and age-related diseases (189, 291, 292). Treatments like doxorubicin and IR activate poly (ADP-ribose) polymerase (PARP), deplete NAD+, and induce premature senescence (189). NAD+ supplementation has shown potential in counteracting aging effects (293, 294). NAD+ depletion during aging is associated with changes in circadian oscillation. NAD+ is a promising therapeutic target for promoting proper autophagy, delaying senescence, and addressing age-related diseases (295), making it a focal point in therapeutic development.
NAD+ synthesis involves three pathways: the kynurenine (de novo) pathway, Preiss-Handler pathway, and NAD+ salvage pathway (296). In the kynurenine pathway, tryptophan is converted to NAD+ via multiple enzymatic steps. This includes the conversion of tryptophan to kynurenine, then to nicotinamide mononucleotide (NAMN), and finally to NAD+. The Preiss-Handler pathway utilizes nicotinic acid (NA), converted to NAMN by NA phosphoribosyltransferase (NAPRT). In the salvage pathway, extracellular nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN) can be converted to NAD+. NR is converted to NMN by nicotinamide riboside kinases (NRK) 1 and 2, and NMN is converted to NAD+ by NMNAT1-3. Nicotinamide (NAM) is recycled back to NAD+ via nicotinamide phosphoribosyltransferase (NAMPT) and CD73 (Figure 8).
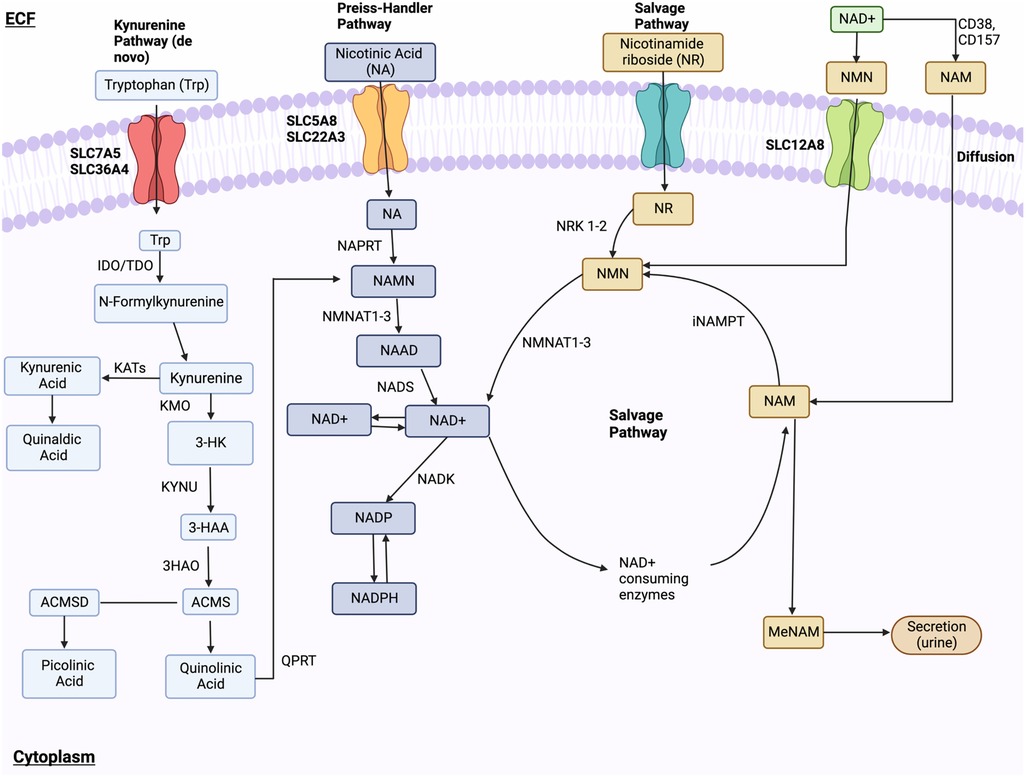
Figure 8. NAD+ metabolism. NAD+ synthesis happens through the kynurenine (de novo) pathway, Preiss-Handler pathway, and NAD+ salvage pathway. In the kynurenine pathway, Tryptophan enters through receptors SLC7A5 and SLC36A4 and is changed into formylkynurenine, then kynurenine, and finally, kynurenic acid or 3-HK. These final compounds are further modified to 3-HAA. Subsequent modifications of 3-HAA into ACMS, quinolinic acid, NAMN, and NAAD lead to final NAD+ synthesis via NADS. In the Preiss-Handler pathway, NA is transported via SLC5A8 and SLC22A13 and is converted to NAMN. NR is brought into the cell and is modified into NMN, which is then ultimately converted to NAD+ by NMNAT1-3. Lastly, the salvage pathway recycles NAM produced by the NAD+-consuming pathway into NMN, which is ultimately converted into NAD+. Ectoenzymes CD38 and CD157 convert NAD+into NAM, which enters the cell and is converted into NMN. Lastly, NMN is dephosphorylated by CD73 into NR. Figure created using BioRender.com.
The NAD+ salvage pathway is vital for recycling and converting NAM into NAD+. It replenishes NAD+ levels after its degradation by NAD+-consuming enzymes, such as glycohydrolases (CD38, CD157), sterile alpha and toll/IL-1 receptor (TIR) motif-containing 1 (SARM1), protein deacylases (such as sirtuins (SIRTs), and PARPs. NAM, a by-product of NAD+ degradation (Figure 8), can inhibit NAD+ biosynthesis by feedback inhibition of NAMPT, the rate-limiting enzyme in the pathway. NAMPT’s expression tends to decrease during aging (297–300), potentially contributing to age-related NAD+ depletion. The salvage pathway maintains cellular NAD+ levels and counteracts NAD+ depletion, ensuring a continuous supply for cellular processes.
NAD+-consuming and generating enzymes are localized in different subcellular compartments, regulating NAD+ homeostasis. Intracellular NAMPT (iNAMPT) and NMNAT2 in the cytoplasm produce NAD+, while NMNAT3 and SIRT3-SIRT5 in mitochondria consume NAD+. NAMAT1 salvage nuclear NAD+. Shuttlers and transporters, like SLC25A51 and the malate/aspartate shuttle, maintain NAD+ levels between compartments. NAD+ is predominantly recycled is recycled through salvage pathways rather than being generated de novo (292, 301, 302).
NAD+-consuming enzymes, such as CD38, CD157, SARM1, SIRTs, and PARPs contribute to NAD+ depletion. Targeting the NAD+ salvage pathway and these enzymes may offer potential therapeutic approach to address aging and age-related diseases.
CD38 is expressed in various cell types and plays a role in tumorigenesis, aging, and disease. CD157, a paralogue of CD38, is primarily expressed in lymphoid tissue and the gut (303). They act as NAD+ hydrolases, generating NAM, cyclic-ADPR (cADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP) from NAD+ degradation (304–306). Dysregulation of CD38 and CD157 is linked to diseases like Parkinson’s, ovarian cancer, and leukemia (307, 308). Increased CD38 and CD157 expression is observed during macrophage polarization and aging (288). CD38 KO mice show resistance to age-related NAD+ depletion and negative effects of a high-fat diet on NAD+ levels, liver health, and glucose metabolism (309, 310). CD38 overexpression reduces NAD+ levels and impairs mitochondrial function in mice (288, 310). CD38 activation can induce NAD+ depletion and EC dysfunction in the heart due to hypoxia-reoxygenation (311). CD38 inhibitors (thiazoloquin(az)olin(on)es and luteolinidin) prevent EC dysfunction and myocardial damage after ischemia (312, 313). Other CD38 inhibitors (apigenin and 78c) (314) show promise for treating aging-related diseases, including metabolic disorders and CVD (315).
SARM1 is a NAD+ cleavage enzyme found in neurons and other cell types (316–319) with two types of NADase activity. It hydrolyzes NAD+ to NAM and ADPR, and generates NAM and cADPR via ADP-ribosyl cyclase activity (317, 320). This depletion of cellular NAD+ levels and triggers axonal degeneration, which can be blocked by NAMPT or NMNAT overexpression and NR supplementation (321). SARM1 is crucial in regulating axonal degeneration after neural injury or disease (322). SARM1 KO rescues neuronal defects and embryonic lethality in NMNAT2 KO mice (323), indicating its NAD+-consuming role during embryonic development. SARM1 depletion also inhibits NMNAT2 deficiency-mediated axonopathy during aging (324). Notably, NAD+ inhibits SARM1 activity by competing with NMN in the presence of NMNAT2, and NMNAT2 depletion activates SARM1 (325, 326). SARM1 has been linked to age-dependent susceptibility to rotenone-induced neurotoxicity and may contribute to age-related neuronal loss (327). These findings provide insights into potential therapeutic targets for neurodegenerative diseases.
Mammalian cells contain seven different SIRTs, major NAD+-consuming enzymes, localized in the nucleus (SIRT 1,6, and 7), cytosol (SIRT 1,2, and 5), and mitochondria (SIRT 3,4, and 5) (328). SIRTs remove acetyl group from lysine residues of proteins, leading to deacetylated lysin, NAM, and 2′-O-acetyl-ADP-ribose (329). SIRT levels decline during cellular senescence and aging (330). SIRT1 downregulates SASP factors like IL6 and IL8 via increased histone H3 and H4 acetylation (331). Inhibiting SIRT1 and SIRT6 causes premature senescence in ECs (332, 333). SIRT-activating compounds (STACs), including resveratrol, SRT1720, SRT3025, and SRT2104, are potential intervention for cardiomyopathy, metabolic syndrome, EC function, and atherosclerosis (334).
Seventeen isoforms with poly-(ADP-ribosyl) or mono (ADP-ribosyl) transferase activity for PARPs have been found, which consume NAD+ by transferring ADP-ribose to proteins, generating NAM. PARP1 acts as a sensor for DNA damage and is activated when DNA is damaged, leading to NAD+ depletion. Anti-melanoma cancer treatments activate PARP1-NF-kb signaling, resulting in SASP (335). However, excessive DNA damage can lead to PARP1 overactivation, causing cell death known as “Parthanatos” (336). PARP inhibitors such as olaparib, INO-1001 (an isoindolinone-based PARP inhibitor), and veliparib are used to treat CVD, pulmonary arterial hypertension, and cardiac repolarization in cancer survivors. PARP1 activation also plays a role in telomere length and telomerase activity maintenance during aging (337, 338).
Premature aging diseases like Xeroderma Pigmentosum, Cockayne syndrome, and Ataxia-telangiectasia may connect ROS generation, NAD+ depletion, autophagy, and mitophagy. Mitophagy loss leads to NAD+ depletion and mitochondrial dysfunction (339–342). Persistent DNA damage in these diseases activates PARP, depleting NAD+, and inhibiting NAD+-dependent SIRT activity and autophagy. Hyperactivated PARP can cause ATP depletion and cell death (343–346). Jiang et al. demonstrated that low H2O2 exposure induces PARP1 activation-mediated parthanatos, involving mitochondrial membrane potential decline, apoptosis-inducing factor translocation, and cell death. PARP1 activation inhibits autophagy, crucial for cell survival (347). Muñoz-Gámez et al. showed that PARP inhibition prevents ATP and NAD+ depletion, triggers autophagy via mTOR activation, and has cytoprotective effects (348).
Various interventions, including caloric restriction, intermittent fasting, genetic alterations (e.g., cardiomyocyte-specific dnPI3K or global AKT2 KO), and transgenic mouse models (e.g., cardiomyocyte-specific Parkin transgenic mice, SIRT1 transgenic mice, and pharmacological interventions like spermidine, rapamycin, resveratrol, and SRT1720), extend lifespan and delay aging in model organisms. These approaches activate cellular stress response pathways, enhancing maintenance and repair mechanisms, resulting in improved healthspan and longevity (349).
Recent research emphasizes the therapeutic potential of supplementing exogenous NAD+ precursors. This supplementation increases NAD biosynthesis, offering therapeutic advantages for various conditions, including metabolic, cardiac, and neurodegenerative disorders (334, 350). Elevated NAD+ levels improve mitochondrial function, stimulate SIRT-dependent mitochondrial recycling, and enhance expression and activity of autophagy/mitophagy-related molecules (294, 295, 339, 351–353). This leads to better organelle and protein aggregate clearance, interrupting the cycle of damage and NAD+ depletion. Boosting NAD+ levels can counteract depletion and support the resolution of physiological stresses, promoting long-term cellular health. NAD+ supplementation shows promise for addressing aging-related diseases and disorders.
Senescent cells remain metabolically active, showing increased glycolytic activity (354, 355) and upregulated mtROS production and succinate induction, even with OXPHOS and glycolysis inhibition by low-dose IR, without undergoing necrosis or apoptosis (189). The SASP, consisting of soluble proteins and extracellular vesicles (EVs), includes pro-inflammatory cytokines, chemokines, growth factors, pro-angiogenic factors, small molecules, lipids (such as nitric oxide (NO) and prostaglandin E2 (PGE2)), ROS, and proteases (Figure 9) (356–358). Normally, SASP factors mediate communication between senescent cells and neighboring cells (106, 116, 117), facilitating immune cell recruitment for senescent cell removal. However, immunosenescence during aging (132, 136, 359) can compromise this clearance process, leading to the accumulation of senescent cells and their effects, contributing to CVD pathogenesis (360).
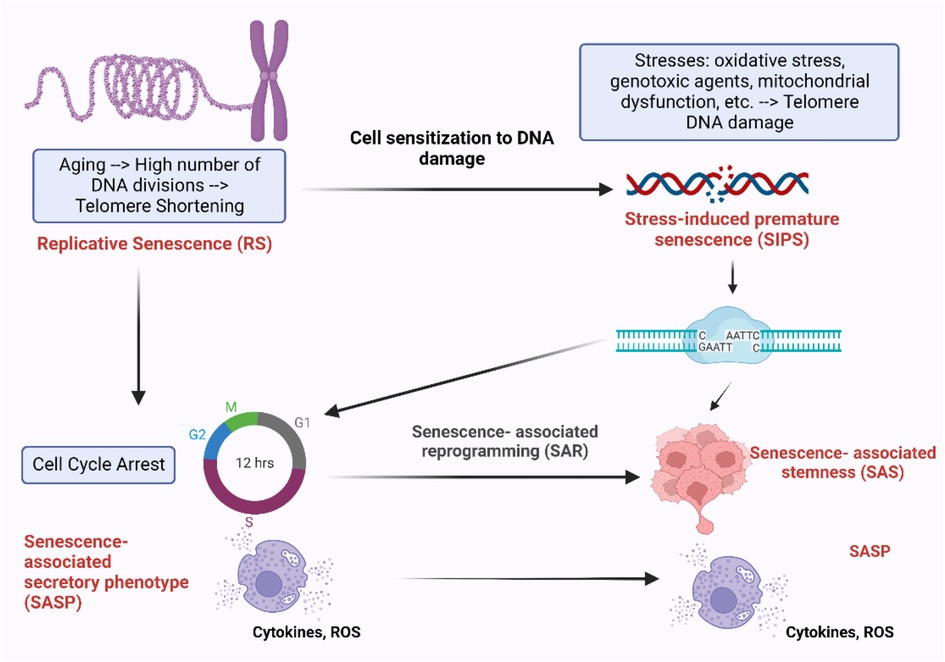
Figure 9. Different mechanisms of cellular senescence and their effects. During the process of aging, cells undergo irreversible cell cycle arrest after a certain number of divisions, RS. This process is accompanied by the progressive shortening of telomeres, the protective caps at the ends of chromosomes, which play a crucial role in cell division and survival (6–8). Cells experiencing RS enter cell cycle arrest and release pro-inflammatory cytokines, ROS, and other factors, known as SASP. Another form of senescence is SIPS, which is triggered by external factors such as oxidative stress, mitochondrial dysfunction, genotoxic agents, and others. Cells undergoing SIPS also enter cell cycle arrest and exhibit SASP. However, these senescent cells can bypass cell cycle arrest by undergoing senescence-associated reprogramming (SAR), leading to the development of SAS (68–70). Figure created using BioRender.com.
EVs, including exosomes and microvesicles, are small vesicles surrounded by a lipid bilayer membrane released by cells. They act as carriers of proteins, nucleic acids, and metabolites between cells, influencing the behavior of recipient cells (361, 362). Mesenchymal stem cell-derived EVs exhibit anti-inflammatory, anti-fibrotic, pro-proliferative, and pro-angiogenic properties through factors such as PGE2, tumor necrosis factor (TNF)-inducible gene 6 protein (TSG6), transforming growth factor β (TGFβ), interleukin 6 (IL6), interleukin-1 receptor antagonist (IL-1RA), NO produced by inducible NO synthase (iNOS), and kynurenine produced by indoleamine 2. 3-dioxygenase (IDO) (363–366). In contrast, activated senescent cells release more functional EVs than non-senescent cells due to upregulated p53 expression. These EVs can promote senescence in neighboring cells by enhancing ROS production (367, 368). EVs also contribute to cellular senescence by transporting senescence-inducing factors including miRNAs, long non-coding RNAs, and proteins that promote senescence in recipient cells (369).
RT administered to the chest region increases CVD risk in patients and may cause mitochondrial dysfunction and cellular senescence (117). To investigate this, we studied 16 thoracic cancer patients who received RT. We analyzed peripheral blood mononuclear cells (PBMCs) before and three months after RT using mass cytometry (CyTOF) to characterize immune cell lineages and examine senescence, DDR, efferocytosis, and determinants of clonal hematopoiesis of indeterminate potential (CHIP).
Our analysis showed reduced B cell subtypes after RT. Clustering of CyTOF data revealed 138 functional PBMC subsets. Post-RT samples displayed increased TBX21 (T-bet) expression in the largest subset of Ki67–/DNMT3a + naive B cells, with T-bet expression correlated with p90RSK phosphorylation. CD38 expression was elevated in naive B cells (CD27–) and CD8 + effector memory CD45RA T cells (TEMRA). In vitro experiments confirmed the crucial role of p90RSK activation in upregulating CD38+/T-bet + memory and naive B cells, myeloid cells, β-gal staining, and mitochondrial ROS following radiation exposure. These findings indicate the pivotal role of p90RSK activation in immunosenescence (370).
Previous studies emphasized the critical role of p90RSK activation in immune cells and its involvement in the development of atherosclerosis through T-bet induction. T-bet directly binds to the CD38 promoter region, resulting in increased CD38 expression. Given the significant roles played by both T-bet and CD38 in immunosenescence, our data provide insights into the cellular and molecular mechanisms linking RT-induced p90RSK activation, immunosenescence, T-bet, and CD38 induction observed in thoracic cancer patients undergoing RT. Targeting the p90RSK/T-bet/CD38 pathway may prevent RT-associated CVD and improve cancer prognosis by inhibiting immunosenescence (370).
Senescence serves as a tumor-suppressor mechanism, but key regulators of senescence, such as p16INK4a, p21CIP1, p53, and H3K9me3, also impact stem cell functions, or “stemness.” Recent studies have shown that senescence can drive cancer cells towards stemness, promoting cancer stem cell regeneration (114, 371–376). Acquiring stemness properties in cancer cells can influence tumor aggressiveness and clinical outcomes. Milanovic et al. analyzed senescent and non-senescent B-cell lymphomas in mice (374), finding that senescent cells upregulated adult tissue stem cell signature, activated Wnt signaling, and distinct stem cell markers. Releasing cells from senescence led to enhanced clonogenic growth potential and higher tumor initiation potential in vivo, dependent on Wnt signaling. Inducing senescence in leukemia models reprogrammed non-stem bulk leukemia cells into self-renewing, leukemia-initiating stem cells. Human cancer cell lines and primary samples of hematological malignancies confirmed these findings. Milanovic et al. revealed that certain senescent cancer cells undergo reprogramming in response to chemotherapy, acquiring a proliferative phenotype known as SAS (374). In this state, they bypass senescence-induced cell cycle arrest and exhibit heightened clonogenic growth potential (374–376). SAS is regulated independently of cell cycle arrest (377, 378) and cell death (132, 335, 374, 375, 377–385). This adaptive mechanism can contribute to cancer cells developing resistance against chemotherapy and RT, leading to survival and continued proliferation despite treatment (379, 380, 382). These findings highlight the role of senescent cancer cells in treatment resistance and tumorigenesis, even in cancer survivors (371, 372). Additionally, SAS may also occur in vascular cells, including ECs and myeloid cells. Considering the contributions of both senescence and macrophage proliferation to CVD (386), the SAS phenotype in vascular cells may significantly impact the development and progression of CVD.
Extracellular signal-regulated kinase 5 (ERK5) is a protein with dual kinase-transcriptional activity (387–392). Inhibitors targeting ERK5's catalytic activity have been evaluated for cancer and inflammatory disease treatment (388, 393). However, recent evidence questions the significance of ERK5's catalytic activity in proliferation and inflammation. Our recent investigation aimed to understand how ERK5 drives the proinflammatory senescent phenotype in myeloid cells, contributing to atherosclerosis (394). We generated ERK5 S496A knock-in (KI) mice using CRISPR/Cas9 technology and induced hypercholesterolemia for atherosclerosis characterization. Plaque phenotyping was performed using imaging mass cytometry in both homozygous ERK5 S496A KI and wild-type (WT) mice. We also conducted RNA sequencing and in vitro assays, including senescence, mitochondrial ROS, inflammation assays, and metabolic extracellular flux analysis using bone marrow-derived macrophages from hypercholesterolemic mice. Our data analysis demonstrates that ERK5 S496A KI mice show inhibited atherosclerosis. We observed that ERK5 S496 phosphorylation mediates SASP and SAS through upregulation of the aryl hydrocarbon receptor (AHR) in plaques and macrophages from hypercholesterolemic mice. ERK5 S496 phosphorylation induces SUMOylation of NFE2-related factor 2 (NRF2) at K518, inhibiting NRF2’s transcriptional activity, without affecting ERK5's catalytic activity. Specific ERK5 kinase inhibitors (AX15836 and XMD8-92) can inhibit ERK5 S496 phosphorylation, suggesting that ERK5 S496 phosphorylation is involved in the anti-inflammatory effects of these inhibitors. Our study uncovers a novel mechanism involving the macrophage ERK5-NRF2 axis, significantly contributing to the SAS via AHR upregulation. This mechanism explains the paradoxical presence of senescence in proliferative plaques, allowing myeloid cells to bypass senescence-induced cell cycle arrest during atherosclerosis formation. Furthermore, the SAS phenotype provides valuable insights into the dynamics of cellular senescence within atherosclerotic lesions. These findings advance our understanding of atherogenesis at the molecular level and offer potential therapeutic strategies targeting the SAS phenotype (394).
Nakamura et al. made an important discovery, revealing the presence of γ-foci at uncapped telomeres and non-telomeric DNA damage sites in both humans and mice, suggesting that both types of DNA damage contribute to cellular senescence (11).
Telomeres, composed of repetitive DNA hexanucleotide TTAGGG sequences, are situated at the ends of mammalian chromosomes (395). They play a crucial role in protecting the linear chromosome structure. When unprotected, chromosome ends can resemble damaged DNA, triggering a DDR that leads to cell cycle arrest through p53-mediated p21 upregulation, ultimately resulting in cellular senescence (396). Telomere shortening occurs over multiple cell divisions, reaching the Hayflick limit, leading to RS and limited cell replication (90–93). Thus, telomere shortening serves as a marker of cellular aging and limited replicative capacity.
Recent studies challenge the direct correlation between telomere shortening and cellular senescence in non-dividing, quiescent, and terminally differentiated cells like cardiomyocytes. The link between telomere length, cardiac hypertrophy, fibrosis, and age-related cardiac dysfunction remains uncertain (397). Both human and murine cardiomyocytes show a senescent-like phenotype characterized by persistent DNA damage at telomere regions during aging, independent of telomere length and cell division, driven by mitochondrial dysfunction. This telomere damage activates senescence-inducing pathways (p21Cip1/Waf1 and p16INK4a) leading to a non-canonical SASP, contributing to age-related cardiac fibrosis and myocardial hypertrophy (77). Interestingly, cellular senescence can still occur with telomerase expression, as observed in immortalized human foreskin fibroblasts expressing telomerase (hTERT-BJ1) exposed to stressors like hydrogen peroxide or Ultraviolet B radiation (91). Renal tubular cells exposed to urine from patients with calcium oxalate kidney stones can undergo SIPS due to oxidative stress from oxalate and calcium oxalate monohydrate (398). Fairlie and Harrington’s study revealed that cell clonogenic survival following IR was dosage-dependent and increased when telomere lengths exceeded 17 kbp, even with telomeric DNA present in all chromosome ends (399). Post-mitotic cells like cardiomyocytes and neurons, which have longer telomeres, can experience persistent telomere DNA damage, leading to telomere-associated DDR foci (TAFs) in aging (77, 111, 400). In models of vascular aging, human telomerase overexpression restored telomere length and the shelterin complex expression, reducing DNA damage and its effects (401–403). Telomerase has broader effects on aging beyond telomere maintenance, including decreased secretion of inflammatory cytokines, normalization of cell and nuclear morphology, restoration of replicative capacity and cell functions (e.g., angiogenic processes), modulation of epigenetic marks, and adjustments to the transcriptional profile. In a murine model of Hutchison-Gilford Progeria syndrome, murine telomerase expression reversed vascular senescence and extended lifespan (403). Studies show that senescence can be induced in mouse embryonic fibroblasts exposed to 20% oxygen density, independent of concurrent telomere shortening (18). These findings suggest that telomere length alone may not be directly linked to the aging process, particularly in non-dividing, quiescent, rarely dividing/post-mitotic cardiomyocytes, or terminally differentiated cells, and that telomere shortening-independent pathways of senescence exist. These findings emphasize the critical role of telomere DNA damage and the resulting persistent activation of DDR. Telomere dysfunction, rather than telomere length alone, may be a more reliable indicator of SIPS. necessitating the evaluation of both factors for a comprehensive telomere status assessment and cellular age (399, 404–408). Various methods are available for measuring telomere length (409). Terminal restriction fragment (TRF) analysis involves Southern blotting and estimation of intensity and size distribution (410), while quantitative PCR-based measurement assesses the telomere-to-gene signal ratio (411, 412). In situ hybridization (Q-FISH) uses quantitative fluorescence on dividing cells (413–415), and single telomere length analysis (STELA) allows high-resolution measurements (416). The telomere short length assay (TeSLA) offers a more sensitive, efficient, and specific approach for detecting telomere length compared to other techniques.
Telomere DNA, rich in guanine, is susceptible to oxidation due to guanine’s low redox potential (417). Repair mechanisms for telomere DNA damage are less efficient compared to genomic DNA repair (400), and the repair process is significantly slower, taking up to 24 h (418, 419). This results in persistent DNA damage at telomere, leading to the continuous activation of DDR (397, 420). Research by Benetos et al. suggests that telomere attrition, rather than inherently short leukocyte telomere length at birth, primary contributes to shorter telomere length in atherosclerotic CVD patients (421). Telomere-binding proteins, such as Shelterin components, play a crucial role in preventing DNA damage (422, 423), and their dysfunction leads to the persistent activation of DDR at telomeres and the formation of TAFs (92, 111, 420, 424).
Telomere dysfunction arises from disruption in the Shelterin complex, composed of TRF1, TRF2, POT1, TIN2, TPP1, and TERF2IP (or RAP1). Shelterin plays a vital role in maintaining telomere length and preventing shortening (425). TRF1 and TRF2 bind to double-stranded telomeric DNA as homodimers, facilitated by the TRFH domain for dimerization and DNA binding (426). TIN2 associates with TRF1 and TRF2, stabilizes TRF1 and TRF2 at telomeres (427) and interacts with TPP1, which is essential for proper telomeric localization of TPP1 and POT1, ensuring telomere integrity (396, 428, 429).
ATM/ATR kinases are key regulators of DDR signaling pathways. They phosphorylate proteins at DNA damage sites, such as histone H2A (H2AX), forming phosphorylated γ-foci. CHK2 and CHK1 are also activated by ATM/ATR (430), potentially leading to cellular senescence (425). TRF2 suppresses the ATM pathway during DSBs, and POT1 suppresses the ATR pathway during SSBs (428), suggesting Shelterin prevents improper DDR activation and telomere dysfunction without affecting telomere length (431).
Adult tissues typically exhibit minimal or absent telomerase activity, suggesting that a decrease in the levels of Shelterin components can result in telomere shortening and cellular senescence (432). Telomeric DNA is susceptible to oxidation, leading to telomeric DNA oxidative damage and reducing the amount of bound TRF1 and TRF2 in vascular cells (417). Conversely, TRF2 overexpression in human fibroblasts reduces oxidative damage to telomeric DNA (433). In patients with CAD, there is an increased presence of senescent ECs with lower TRF1 levels and greater telomeric DNA oxidative damage (434). Serial passaging of human umbilical vein ECs, a cell culture model of aging, leads to decreased TRF1 expression (435). However, TRF1 overexpression in this model decreases TAFs (435) and SASP (436). This decrease in bound TRF1 and TRF2 may explain vascular cell senescence under oxidative stress. Studies on ECs and VSMCs reveal a connection between oxidative stress-induced telomere dysfunction and Shelterin protein expression. Overexpressing TRF2 in VSMCs reduces DNA damage and enhances DNA repair, highlighting the regulatory role of Shelterin proteins in oxidative stress-induced telomere dysfunction (437). Telomere dysfunction and SASP precede RS, potentially contributing to vascular dysfunction and the development of CVD by promoting early changes that lead to senescence in ECs and VSMCs (436). Preserving the structure and function of Shelterin presents an appealing strategy to control or prevent cellular senescence-associated CVD.
TERF2IP associates with TRF2 to bind to telomeres, enhancing TRF2's binding to these structures (438, 439). This complex is essential for inhibiting telomeric recombination, fragility, and shortening (440). Disturbed flow triggers EC activation and senescence by promoting TERF2IP S205 phosphorylation (113, 441, 442). Once phosphorylated at S205, TERF2IP and TRF2 undergo nuclear export. In the cytoplasm, TERF2IP interacts with cytosolic IKK, impairing its ability to inhibit NFκB and leading to EC activation. Concurrently, the removal of TRF2 from the nucleus induces telomere shortening, senescence, and apoptosis (443). This nuclear export of the TERF2IP-TRF2 complex connects inflammatory signaling with telomere shortening, representing a common pathway linking SASP and telomere shortening (443). Taken together, the expression of Shelterin complex and PTMs of TERF2IP play a critical role in regulating EC activation, apoptosis, senescence, and the development of atherosclerotic CVD (Figure 10).
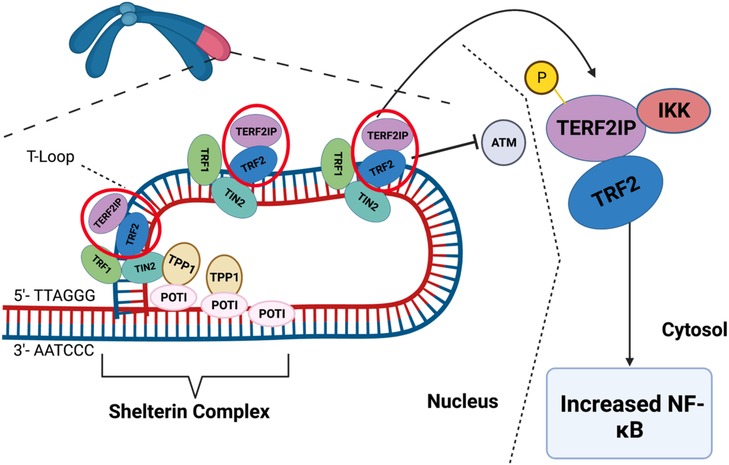
Figure 10. Scheme of Shelterin complex and t-loop. The ends of chromosomes have non-coding sequences called telomeres to protect the ends of DNA strands. The Shelterin complex is a group of proteins that maintain the T-loop, which protects the telomere ends from activating DDR. The Shelterin complex is made up of six proteins: TRF1, TRF2, POT1, TIN2, TERF2IP, and TPP1. These proteins can regulate telomere length, prevent telomere DNA damage and dysfunction, and maintain the structural integrity of telomeres. TERF2IP helps to regulate telomere length and prevent the activation of telomere DDR pathways. Note that TERF2IP-TRF2 phosphorylation induces telomere dysfunction and inflammation simultaneously. When TERF2IP is phosphorylated, the TERF2IP-TRF2 complex leaves the Shelterin complex and T-loop, reducing the protective action of the Shelterin complex. Furthermore, TERF2IP-TRF2 is exported into the cytoplasm, where TERF2IP binds to and represses IKK. Repressing this inhibitor of NF-κB induces inflammatory signaling. Therefore, TERF2IP phosphorylation can cause telomere dysfunction and inflammation simultaneously (443). Figure generated using BioRender.com.
The interplay between the nucleus and mitochondria is vital for forming the OXPHOS system, comprising protein complexes within the IMM. The nucleus supplies necessary components and regulatory factors for OXPHOS assembly, while mtDNA encodes crucial OXPHOS complex subunits. This coordinated process enables efficient synthesis, assembly, regulation, and maintenance of OXPHOS, facilitating cellular energy production and metabolic adaptation. The nuclear genome controls OXPHOS gene expression in response to cellular conditions, adjusting energy production based on demands and stresses. Nuclear transcription factors respond to nutrients, oxygen levels, and energy status, fine-tuning OXPHOS activity. The nuclear genome also maintain mtDNA integrity and stability, supporting proper mtDNA replication, repair, and protection. Nuclear-encoded subunits, synthesized in the cytoplasm, are imported into mitochondria to combine with mitochondria-encoded subunits. Critical nuclear-encoded factors facilitate import, assembly, and stability of mitochondria-encoded subunits, ensuring proper OXPHOS complexe formation and function (444).
Mice with a mutated mtDNA polymerase γ show a premature aging phenotype with increased mtDNA mutation rates (194). Surprisingly, despite the mutations, ROS production and antioxidant enzyme activity remain unchanged, suggesting mtDNA mutations may not significantly contribute to mtROS production or the premature aging phenotype. Other mechanisms, independent of mtDNA damage, may be responsible for the mtROS-mediated premature aging process.
Qian et al. reported that mtROS induce DNA damage in telomeres, activating DDR and peroxisome proliferator-activated receptor (PPAR) pathways (445). Chemoradiation-induced mtROS activates p90RSK, which phosphorylates ERK5 S496, inhibiting the activity of ERK5 and NRF2 transcription. This reduces NRF2 activity (189, 394) and downregulates antioxidant gene expression, including HO1 and TRX1. The decreased NRF2 activity establishes a persistent SASP state with senescence, inflammation, increased mtROS production, and impaired efferocytosis. Low-dose IR or doxorubicin-induced telomeric DNA damage enhances this SASP state.
Telomere DNA damage activates PARPs, depleting NAD+ and causing mitochondrial dysfunction (338). Chemoradiation treatment increases succinate levels and mtROS production via complex II. “Mitochondrial stunning” refers to a reversible mitochondrial dysfunction with severe ATP depletion without immediate cell death (189). This phenomenon activates the p90RSK-ERK5-NRF2 signaling pathway, establishing a positive feedback loop between mitochondrial dysfunction and nuclear DDR. This loop sustains mtROS production, contributing to premature aging through telomere erosion (189) (Figure 11).
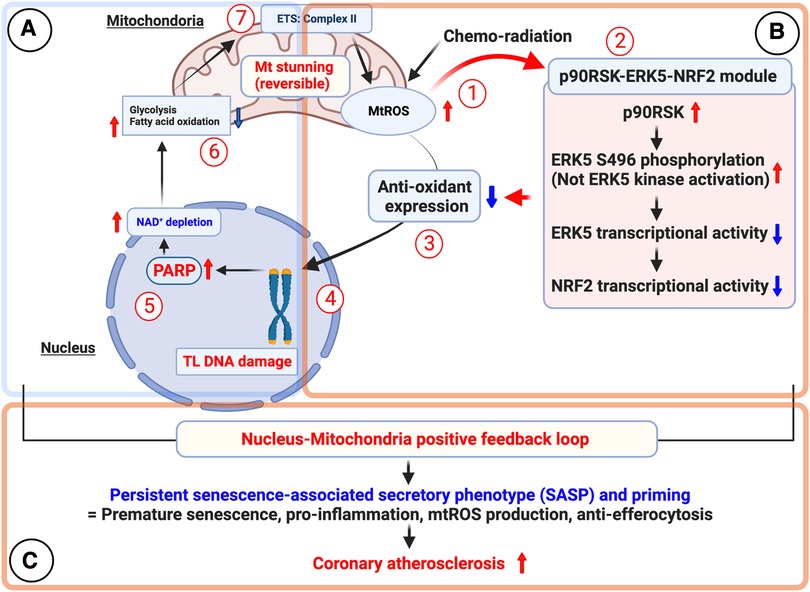
Figure 11. A nucleus-mitochondria positive feedback loop involving the p90RSK-ERK5-NRF2 module and PARP activation [modified from (189)]. (1) Chemo-radiation stimulates mtROS production and phosphorylates p90RSK-ERK5 S496. (2) Subsequently, chemo-radiation inhibits the activity of ERK5 and NRF2 transcription through mtROS-mediated ERK5 S496 phosphorylation. (3) Chemo-radiation decreases antioxidant expression (e.g., HO1 and Trx1), leading to a persistent SASP characterized by senescence, inflammation, mtROS production, and attenuation of efferocytosis. (4) Low dose IR and doxorubicin-induced mtROS production plays a critical role in telomere shortening. (5) Telomere DNA damage activates PARP. (6) Low dose IR and doxorubicin do not cause immediate cell death but lead to “mitochondrial stunning”, a metabolically active state with severe ATP depletion. Mitochondrial stunning induces persistent mtROS production and a late-lasting SASP, detectable long after cancer therapy completion. Schematic created using BioRender.com.
Transient inhibition of PARP during radiation exposure has been shown to reduce IR-induced coronary atherosclerosis, macrophage infiltration, and cardiac dysfunction (189, 446). However, long-term PARP inhibition may increase chromosomal instability and tumorigenesis risk, making it unsuitable for prolong clinical use as an anti-atherosclerosis drug (447). The data suggest that transient PARP inhibition specifically during RT effectively prevent cardiovascular damage without the potential tumorigenesis risk associated with prolonged PARP inhibition. This preclinical evidence supports using transient PARP inhibition as a therapeutic strategy to mitigate radiation-induced cardiovascular damage (189). These findings emohasize the crucial interplay between the nucleus and mitochondria in inducing SASP. Without this positive feedback loop, sustained SASP induction cannot occur, leading to premature aging-related CVD, especially following various cancer treatments.
The rising prevalence of CVD among older patients and cancer survivors has become a significant concern due to the expanding population of these individuals. Managing CVD in these groups pose challenges, given the complex molecular and intracellular events associated with cellular senescence (113). Senescence and premature senescence processes play both protective and detrimental roles in DNA damage-associated tumorigenesis and CVD. These events involve persistent DNA damage, chronic activation of DDR pathways, telomere dysfunction, mitochondrial dysfunction, metabolic and epigenetic alterations, mtROS production, SASP, and SAS (132, 335, 374–385). Therefore, it is crucial to carefully weigh the benefits and potential risks.
Chemotherapy (40) and RT, commonly cancer treatments, induce DNA damage and oxidative stress, leading to SIPS in various cardiovascular system cells (cardiomyocytes, ECs, myeloid cells). Both telomeric and non-telomeric DNA damage contribute to cellular senescence, but dysfunction, not telomere length, better reflects SIPS due to less efficient telomeric DDR repair compared to genomic DDR. Persistent telomeric DNA damage leads to the formation of TAFs, marking cellular senescence. Senescent cells (RS or SIPS) secrete pro-inflammatory factors known as SASP. Mitochondrial dysfunction plays a crucial role in SIPS and SASP induction through factors such as NAD+ depletion, increased mtROS production, impaired mitochondrial fission/fusion, and compromised mitophagy. Telomeric DDR activation and TAF formation, along with epigenetic changes, can create a nucleus-mitochondrial positive feedback loop, facilitated by PARP-mediated NAD+ depletion. Cancer therapies can activate this loop through a p90RSK-ERK5 module, resulting in chronic inflammation and tissue remodeling. Ultimately, these processes can contribute to CVD development after cancer treatments.
Our recent research has provided significant insights into the regulation of SASP and SAS in myeloid cells (392, 448). We found that p90RSK-mediated ERK5 S496 phosphorylation governs factors contributing to SASP (189), including mtROS induction, telomere shortening-triggered DNA damage, activation of p16INK4, p21Cip1/Waf1, p53, inflammation, and impaired efferocytosis in HIV patients on antiretroviral therapy (448). Cancer treatments can also induce SASP, linked to cancer cell proliferation treatment resistance. Our study demonstrated the important of p90RSK activation in immunosenescence (370) and atherosclerosis inhibition (394), indicating the potential significance of p90RSK-mediated ERK5 S496 phosphorylation in SASP and SAS regulation, and its implications for CVD and cancer treatment resistance. Further investigations are needed to fully understand the mechanisms of SASP modulation by p90RSK-mediated ERK5 S496 phosphorylation (392, 448). Additionally, research should explore the role of the p90RSK-ERK5-NRF2 axis in ECs. The interplay between SASP induced by cancer treatment and p90RSK-mediated ERK5 S496 phosphorylation may accelerate coronary atherosclerotic plaque formation.
Oncogene-induced senescence (OIS) occurs when non-tumor cells activate oncogenes, leading to stable cell cycle arrest (357). Recent research by Leon et al. discovered that OIS and overexpression of the oncogene HRASG12V in IMR90 cells increase active histone H3K79 di- and tri-methylation (H3K79me2/3) specifically at the IL1A gene locus (449). The histone methyltransferase disruptor of telomeric silencing 1-like (DOT1l) regulates this change, crucial for IL1A expression during OIS, but not directly involved in SASP. While epigenetic changes may contribute to irreversible SASP state in OIS cells (450, 451), their specific role in regulating SAS remains unknown. Further investigations are needed to understand epigenetic factors and their contribution to the SAS phenotype. Additionally, future research should explore other stressors inducing both SASP and SAS, broadening our knowledge of these cellular processes.
Senescence is linked to impaired angiogenesis (96), a critical process for forming new blood vessels. Inhibiting telomerase in ECs reduces angiogenesis in tumor and hind limb ischemia models. Targeting senescent cells offers a promising therapeutic approach for managing conditions with impaired angiogenesis (452, 453). Cancer treatment induces senescence in ECs (454), but the role of EC’s SASP in the delayed onset of CVD after cancer treatment remains unclear. Further research is needed to explore this aspect and understand the interplay between EC senescence, the SAS phenotype, and CVD in cancer survivors (113). These investigations will provide valuable insights into the cardiovascular consequences of cancer treatment and guide strategies for preventing and managing CVD in this population.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Write a manuscript: AJ, DC, JV, APB. Outline, Write a manuscript, and edit it: NTL, JA. Critical reading, edits, and suggestions: SK, KAK, VSKS, KC, MTN, JW, SLHG, SPR, EAOD, DJH, CRG, SCJY, JPC, JH, ENC, XX, SWY, MY, PLL, BH, SK, EK, NLP, GW, AD, and SHL. All authors contributed to the article and approved the submitted version
This work was supported by grants from the National Institutes of Health (NIH) to Drs. Abe (HL-149303, HL-163857, AI-156921), Cooke (HL-149303, 157790), Le (HL-134740, HL-149303, HL-163857, 157790), and Drs. Deswal, Lin, and Abe through M. D. Anderson’s Cancer Center Support Grant CA016672. It also received partial support from the University of Texas MD Anderson Cancer Center Institutional Research Grant (IRG) Program to KS.
SHL is an Advisory Board member of AstraZeneca, Beyond Spring Pharmaceuticals, STCube Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Partridge E, Teoh E, Nash C, Scott T, Charlwood C, Kostakis C. The increasing use and abuse of tapentadol and its incorporation into a validated quantitative method. J Anal Toxicol. (2018) 42:485–90. doi: 10.1093/jat/bky027
2. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
3. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. (1993) 362:801–9. doi: 10.1038/362801a0
4. Muhandiramge J, Zalcberg JR, van Londen GJ, Warner ET, Carr PR, Haydon A, et al. Cardiovascular disease in adult cancer survivors: a review of current evidence, strategies for prevention and management, and future directions for cardio-oncology. Curr Oncol Rep. (2022) 24:1579–92. doi: 10.1007/s11912-022-01309-w
5. Ben Mousa A. Sorafenib in the treatment of advanced hepatocellular carcinoma. Saudi J Gastroenterol. (2008) 14:40–2. doi: 10.4103/1319-3767.37808
6. Thompson Coon J, Hoyle M, Green C, Liu Z, Welch K, Moxham T, et al. Bevacizumab, sorafenib tosylate, sunitinib and temsirolimus for renal cell carcinoma: a systematic review and economic evaluation. Health Technol Assess. (2010) 14:1–184. iii-iv. doi: 10.3310/hta14020
7. Taniguchi H, Yamanaka T, Sakai D, Muro K, Yamazaki K, Nakata S, et al. Efficacy of panitumumab and cetuximab in patients with colorectal cancer previously treated with bevacizumab; a combined analysis of individual patient data from ASPECCT and WJOG6510G. Cancers (Basel). (2020) 12:1715. doi: 10.3390/cancers12071715
8. Garcia-Foncillas J, Sunakawa Y, Aderka D, Wainberg Z, Ronga P, Witzler P, et al. Distinguishing features of cetuximab and panitumumab in colorectal cancer and other solid tumors. Front Oncol. (2019) 9:849. doi: 10.3389/fonc.2019.00849
9. Hayashi K, Mitani S, Taniguchi H, Yasui H, Muro K, Mori K, et al. Panitumumab provides better survival outcomes compared to cetuximab for metastatic colorectal cancer patients treated with prior bevacizumab within 6 months. Oncology. (2019) 96:132–9. doi: 10.1159/000493321
10. Specenier P, Vermorken JB. Cetuximab: its unique place in head and neck cancer treatment. Biologics. (2013) 7:77–90. doi: 10.2147/BTT.S43628
11. Starling N, Neoptolemos J, Cunningham D. Role of erlotinib in the management of pancreatic cancer. Ther Clin Risk Manag. (2006) 2:435–45. doi: 10.2147/tcrm.2006.2.4.435
13. Debela DT, Muzazu SG, Heraro KD, Ndalama MT, Mesele BW, Haile DC, et al. New approaches and procedures for cancer treatment: current perspectives. SAGE Open Med. (2021) 9:20503121211034366. doi: 10.1177/20503121211034366
14. Wahdan-Alaswad R, Liu B, Thor AD. Targeted lapatinib anti-HER2/ErbB2 therapy resistance in breast cancer: opportunities to overcome a difficult problem. Cancer Drug Resist. (2020) 3:179–98. doi: 10.20517/cdr.2019.92
15. Aguilera DG, Tsimberidou AM. Dasatinib in chronic myeloid leukemia: a review. Ther Clin Risk Manag. (2009) 5:281–9. doi: 10.2147/tcrm.s3425
16. Muller MC, Cervantes F, Hjorth-Hansen H, Janssen J, Milojkovic D, Rea D, et al. Ponatinib in chronic myeloid leukemia (CML): consensus on patient treatment and management from a European expert panel. Crit Rev Oncol Hematol. (2017) 120:52–9. doi: 10.1016/j.critrevonc.2017.10.002
17. Kahl B. Chemotherapy combinations with monoclonal antibodies in non-Hodgkin’s lymphoma. Semin Hematol. (2008) 45:90–4. doi: 10.1053/j.seminhematol.2008.02.003
18. Sharkey RM, Goldenberg DM. Use of antibodies and immunoconjugates for the therapy of more accessible cancers. Adv Drug Deliv Rev. (2008) 60:1407–20. doi: 10.1016/j.addr.2008.04.011
19. Sugiyama Y, Sasaki M, Kouyama M, Tazaki T, Takahashi S, Nakamitsu A. Current treatment strategies and future perspectives for gastrointestinal stromal tumors. World J Gastrointest Pathophysiol. (2022) 13:15–33. doi: 10.4291/wjgp.v13.i1.15
20. Laurent M, Brahmi M, Dufresne A, Meeus P, Karanian M, Ray-Coquard I, et al. Adjuvant therapy with imatinib in gastrointestinal stromal tumors (GISTs)-review and perspectives. Transl Gastroenterol Hepatol. (2019) 4:24. doi: 10.21037/tgh.2019.03.07
21. Garlipp B, Bruns CJ. State of the art in the treatment of gastrointestinal stromal tumors. Gastrointest Tumors. (2014) 1:221–36. doi: 10.1159/000380788
22. Le Cesne A, Blay JY. Medical therapy of GIST; from palliative to curative treatment. Bull Acad Natl Med. (2012) 196:861–74. discussion 874-76.23550449
23. Zalcberg JR, Desai J. Dose optimization of tyrosine kinase inhibitors to improve outcomes in GIST. Asia Pac J Clin Oncol. (2012) 8:43–52. doi: 10.1111/j.1743-7563.2011.01491.x
24. Papaetis GS, Syrigos KN. Targeted therapy for gastrointestinal stromal tumors: current status and future perspectives. Cancer Metastasis Rev. (2010) 29:151–70. doi: 10.1007/s10555-010-9206-7
25. Apice G, Milano A, Bruni GS, Iaffaioli RV, Caponigro F. Medical treatment of gastrointestinal stromal tumors: state of the art and future perspectives. Rev Recent Clin Trials. (2006) 1:35–42. doi: 10.2174/157488706775246175
26. Comella P, Casaretti R, Sandomenico C, Avallone A, Franco L. Role of oxaliplatin in the treatment of colorectal cancer. Ther Clin Risk Manag. (2009) 5:229–38. doi: 10.2147/TCRM.S3583
27. Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. (2000) 6:2585–97.10914698
28. Friedman HS. Temozolomide in early stages of newly diagnosed malignant glioma and neoplastic meningitis. Semin Oncol. (2000) 27:35–40.10866348
29. Burney IA, Al-Moundhri MS. Major advances in the treatment of cancer: what does a non-oncologist need to know? Sultan Qaboos Univ Med J. (2008) 8:137–48.
30. Harada G, Neffa M, Bonadio RC, Mendoza EZ, Caparica R, Lauricella LL, et al. Effectiveness and toxicity of adjuvant chemotherapy in patients with non-small cell lung cancer. J Bras Pneumol. (2021) 47:e20200378. doi: 10.36416/1806-3756/e20200378
31. Jang RW, Le Maitre A, Ding K, Winton T, Bezjak A, Seymour L, et al. Quality-adjusted time without symptoms or toxicity analysis of adjuvant chemotherapy in non-small-cell lung cancer: an analysis of the national cancer institute of Canada clinical trials group JBR.10 trial. J Clin Oncol. (2009) 27:4268–73. doi: 10.1200/JCO.2008.20.5815
32. Bezjak A, Lee CW, Ding K, Brundage M, Winton T, Graham B, et al. Quality-of-life outcomes for adjuvant chemotherapy in early-stage non-small-cell lung cancer: results from a randomized trial, JBR.10. J Clin Oncol. (2008) 26:5052–9. doi: 10.1200/JCO.2007.12.6094
33. Gegechkori N, Haines L, Lin JJ. Long-term and latent Side effects of specific cancer types. Med Clin North Am. (2017) 101:1053–73. doi: 10.1016/j.mcna.2017.06.003
34. Miller KD, Nogueira L, Devasia T, Mariotto AB, Yabroff KR, Jemal A, et al. Cancer treatment and survivorship statistics, 2022. CA Cancer J Clin. (2022) 72:409–36. doi: 10.3322/caac.21731
35. Amatya B, Khan F, Lew TE, Dickinson M. Rehabilitation in patients with lymphoma: an overview of systematic reviews. J Rehabil Med. (2021) 53:jrm00163. doi: 10.2340/16501977-2810
36. Dash A, Knapp FF, Pillai MR. Targeted radionuclide therapy–an overview. Curr Radiopharm. (2013) 6:152–80. doi: 10.2174/18744710113066660023
37. Hassler MR, Schiefer AI, Egger G. Combating the epigenome: epigenetic drugs against non-Hodgkin’s lymphoma. Epigenomics. (2013) 5:397–415. doi: 10.2217/epi.13.39
38. Gustavsson A, Osterman B, Cavallin-Stahl E. A systematic overview of radiation therapy effects in non-Hodgkin’s lymphoma. Acta Oncol. (2003) 42:605–19. doi: 10.1080/02841860310014435
39. Lindley C. The lymphomas: Hodgkin’s disease and non-Hodgkin’s lymphomas. Am Pharm. (1991) NS31:46–51. doi: 10.1016/S0160-3450(16)33731-X
40. Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. (2017) 7:165–76. doi: 10.1158/2159-8290.CD-16-0241
41. Lerida-Viso A, Estepa-Fernandez A, Morella-Aucejo A, Lozano-Torres B, Alfonso M, Blandez JF, et al. Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol Res. (2022) 183:106356. doi: 10.1016/j.phrs.2022.106356
42. Chang HM, Okwuosa TM, Scarabelli T, Moudgil R, Yeh ETH. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 2. J Am Coll Cardiol. (2017) 70:2552–65. doi: 10.1016/j.jacc.2017.09.1095
43. Chang HM, Moudgil R, Scarabelli T, Okwuosa TM, Yeh ETH. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 1. J Am Coll Cardiol. (2017) 70:2536–51. doi: 10.1016/j.jacc.2017.09.1096
44. Nabialek-Trojanowska I, Lewicka E, Wrona A, Kaleta AM, Lewicka-Potocka Z, Raczak G, et al. Cardiovascular complications after radiotherapy. Cardiol J. (2020) 27:836–47. doi: 10.5603/CJ.a2018.0120
45. Zamorano JL, Lancellotti P, Rodriguez Munoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC committee for practice guidelines: the task force for cancer treatments and cardiovascular toxicity of the European society of cardiology (ESC). Eur Heart J. (2016) 37:2768–801. doi: 10.1093/eurheartj/ehw211
46. Zamorano JL, Lancellotti P, Rodriguez Munoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC committee for practice guidelines: the task force for cancer treatments and cardiovascular toxicity of the European society of cardiology (ESC). Eur J Heart Fail. (2017) 19:9–42. doi: 10.1002/ejhf.654
47. Meyersohn NM, Pursnani A, Neilan TG. Detection of cardiac toxicity due to cancer treatment: role of cardiac MRI. Curr Treat Options Cardiovasc Med. (2015) 17:396. doi: 10.1007/s11936-015-0396-8
48. Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. (2015) 131:1981–8. doi: 10.1161/CIRCULATIONAHA.114.013777
49. O'Hare M, Sharma A, Murphy K, Mookadam F, Lee H. Cardio-oncology part I: chemotherapy and cardiovascular toxicity. Expert Rev Cardiovasc Ther. (2015) 13:511–8. doi: 10.1586/14779072.2015.1032940
50. Volkova M, Russell R 3rd. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev. (2011) 7:214–20. doi: 10.2174/157340311799960645
51. Batist G, Harris L, Azarnia N, Lee LW, Daza-Ramirez P. Improved anti-tumor response rate with decreased cardiotoxicity of non-pegylated liposomal doxorubicin compared with conventional doxorubicin in first-line treatment of metastatic breast cancer in patients who had received prior adjuvant doxorubicin: results of a retrospective analysis. Anticancer Drugs. (2006) 17:587–95. doi: 10.1097/00001813-200606000-00014
52. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. (2003) 97:2869–79. doi: 10.1002/cncr.11407
53. Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. (2010) 115:155–62. doi: 10.1159/000265166
54. Hu Y, Sun B, Zhao B, Mei D, Gu Q, Tian Z. Cisplatin-induced cardiotoxicity with midrange ejection fraction: a case report and review of the literature. Medicine (Baltimore). (2018) 97:e13807. doi: 10.1097/MD.0000000000013807
55. Dugbartey GJ, Peppone LJ, de Graaf IA. An integrative view of cisplatin-induced renal and cardiac toxicities: molecular mechanisms, current treatment challenges and potential protective measures. Toxicology. (2016) 371:58–66. doi: 10.1016/j.tox.2016.10.001
56. Yang J, Ju J, Guo L, Ji B, Shi S, Yang Z, et al. Prediction of HER2-positive breast cancer recurrence and metastasis risk from histopathological images and clinical information via multimodal deep learning. Comput Struct Biotechnol J. (2022) 20:333–42. doi: 10.1016/j.csbj.2021.12.028
57. Beavers CJ, Rodgers JE, Bagnola AJ, Beckie TM, Campia U, Di Palo KE, et al. American heart association clinical pharmacology C, cardio-oncology committee of the council on clinical C, council on G, precision M and the council on peripheral vascular D. Cardio-oncology drug interactions: a scientific statement from the American heart association. Circulation. (2022) 145:e811–38. doi: 10.1161/CIR.0000000000001056
58. Riccio G, Coppola C, Piscopo G, Capasso I, Maurea C, Esposito E, et al. Trastuzumab and target-therapy side effects: is still valid to differentiate anthracycline type I from type II cardiomyopathies? Hum Vaccin Immunother. (2016) 12:1124–31. doi: 10.1080/21645515.2015.1125056
59. Peverill RE. Hormone therapy and venous thromboembolism. Best Pract Res Clin Endocrinol Metab. (2003) 17:149–64. doi: 10.1016/S1521-690X(02)00079-9
60. Meier CR, Jick H. Tamoxifen and risk of idiopathic venous thromboembolism. Br J Clin Pharmacol. (1998) 45:608–12. doi: 10.1046/j.1365-2125.1998.00733.x
61. Groarke JD, Nguyen PL, Nohria A, Ferrari R, Cheng S, Moslehi J. Cardiovascular complications of radiation therapy for thoracic malignancies: the role for non-invasive imaging for detection of cardiovascular disease. Eur Heart J. (2014) 35:612–23. doi: 10.1093/eurheartj/eht114
62. Correction to: radiation-induced cardiovascular disease: review of an underrecognized pathology. J Am Heart Assoc. (2023) 12:e027687. doi: 10.1161/JAHA.121.027687
63. Farzipour S, Jalali F, Alvandi M, Shaghaghi Z. Ferroptosis inhibitors as new therapeutic insights into radiation-induced heart disease. Cardiovasc Hematol Agents Med Chem. (2023) 21:2–9. doi: 10.2174/1871525720666220713101736
64. Vassileva J, Holmberg O. Radiation protection perspective to recurrent medical imaging: what is known and what more is needed? Br J Radiol. (2021) 94:20210477. doi: 10.1259/bjr.20210477
65. Amino M, Yoshioka K, Kamada T, Furusawa Y. The potential application of heavy ion beams in the treatment of arrhythmia: the role of radiation-induced modulation of connexin43 and the sympathetic nervous system. Int J Part Ther. (2018) 5:140–50. doi: 10.14338/IJPT-18-00022.1
66. Yusuf SW, Venkatesulu BP, Mahadevan LS, Krishnan S. Radiation-induced cardiovascular disease: a clinical perspective. Front Cardiovasc Med. (2017) 4:66. doi: 10.3389/fcvm.2017.00066
67. Cosset JM, Ferme C, Henry-Amar M, Carde P. The role of radiotherapy for limited stage Hodgkin’s disease in 1999: limitations and perspectives. Cancer Radiother. (1999) 3:112–8. doi: 10.1016/S1278-3218(99)80041-9
68. Lee C, Hahn RT. Valvular heart disease associated with radiation therapy: a contemporary review. Struct Heart. (2023) 7:100104. doi: 10.1016/j.shj.2022.100104
69. Ivashkevich A. The role of isoflavones in augmenting the effects of radiotherapy. Front Oncol. (2022) 12:800562. doi: 10.3389/fonc.2022.800562
70. Ferini G, Valenti V, Viola A, Umana GE, Martorana E. A critical overview of predictors of heart sparing by deep-inspiration-breath-hold irradiation in left-sided breast cancer patients. Cancers (Basel). (2022) 14:3477. doi: 10.3390/cancers14143477
71. Ellahham S, Khalouf A, Elkhazendar M, Dababo N, Manla Y. An overview of radiation-induced heart disease. Radiat Oncol J. (2022) 40:89–102. doi: 10.3857/roj.2021.00766
72. Gowda SN, Ali HJ, Hussain I. Overview of restrictive cardiomyopathies. Methodist Debakey Cardiovasc J. (2022) 18:4–16. doi: 10.14797/mdcvj.1078
73. Zhang X, Pawlikowski M, Olivo-Marston S, Williams KP, Bower JK, Felix AS. Ten-year cardiovascular risk among cancer survivors: the national health and nutrition examination survey. PLoS One. (2021) 16:e0247919. doi: 10.1371/journal.pone.0247919
74. Armenian SH, Armstrong GT, Aune G, Chow EJ, Ehrhardt MJ, Ky B, et al. Cardiovascular disease in survivors of childhood cancer: insights into epidemiology, pathophysiology, and prevention. J Clin Oncol. (2018) 36:2135–44. doi: 10.1200/JCO.2017.76.3920
75. Luu J, Palczewski K. Human aging and disease: lessons from age-related macular degeneration. Proc Natl Acad Sci U S A. (2018) 115:2866–72. doi: 10.1073/pnas.1721033115
76. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. (2016) 530:184–9. doi: 10.1038/nature16932
77. Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. (2019) 38:e100492. doi: 10.15252/embj.2018100492
78. Lewis-McDougall FC, Ruchaya PJ, Domenjo-Vila E, Shin Teoh T, Prata L, Cottle BJ, et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell. (2019) 18:e12931. doi: 10.1111/acel.12931
79. Walaszczyk A, Dookun E, Redgrave R, Tual-Chalot S, Victorelli S, Spyridopoulos I, et al. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell. (2019) 18:e12945. doi: 10.1111/acel.12945
80. Accordino MK, Neugut AI, Hershman DL. Cardiac effects of anticancer therapy in the elderly. J Clin Oncol. (2014) 32:2654–61. doi: 10.1200/JCO.2013.55.0459
81. Reddy P, Shenoy C, Blaes AH. Cardio-oncology in the older adult. J Geriatr Oncol. (2017) 8:308–14. doi: 10.1016/j.jgo.2017.04.001
82. Hershman DL, McBride RB, Eisenberger A, Tsai WY, Grann VR, Jacobson JS. Doxorubicin, cardiac risk factors, and cardiac toxicity in elderly patients with diffuse B-cell non-Hodgkin’s lymphoma. J Clin Oncol. (2008) 26:3159–65. doi: 10.1200/JCO.2007.14.1242
83. Kirkham AA, Davis MK. Exercise prevention of cardiovascular disease in breast cancer survivors. J Oncol. (2015) 2015:917606. doi: 10.1155/2015/917606
84. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
85. Huang Q, Gan Y, Yu Z, Wu H, Zhong Z. Endothelial to mesenchymal transition: an insight in atherosclerosis. Front Cardiovasc Med. (2021) 8:734550. doi: 10.3389/fcvm.2021.734550
86. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. (2016) 118:535–46. doi: 10.1161/CIRCRESAHA.115.307611
87. Wu CM, Zheng L, Wang Q, Hu YW. The emerging role of cell senescence in atherosclerosis. Clin Chem Lab Med. (2020) 59:27–38. doi: 10.1515/cclm-2020-0601
88. Childs BG, Li H, van Deursen JM. Senescent cells: a therapeutic target for cardiovascular disease. J Clin Invest. (2018) 128:1217–28. doi: 10.1172/JCI95146
89. Song P, Zhao Q, Zou MH. Targeting senescent cells to attenuate cardiovascular disease progression. Ageing Res Rev. (2020) 60:101072. doi: 10.1016/j.arr.2020.101072
90. Chuenwisad K, More-Krong P, Tubsaeng P, Chotechuang N, Srisa-Art M, Storer RJ, et al. Premature senescence and telomere shortening induced by oxidative stress from oxalate, calcium oxalate monohydrate, and urine from patients with calcium oxalate nephrolithiasis. Front Immunol. (2021) 12:696486. doi: 10.3389/fimmu.2021.696486
91. de Magalhaes JP, Chainiaux F, Remacle J, Toussaint O. Stress-induced premature senescence in BJ and hTERT-BJ1 human foreskin fibroblasts. FEBS Lett. (2002) 523:157–62. doi: 10.1016/S0014-5793(02)02973-3
92. Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. (2012) 3:708. doi: 10.1038/ncomms1708
93. Naka K, Tachibana A, Ikeda K, Motoyama N. Stress-induced premature senescence in hTERT-expressing ataxia telangiectasia fibroblasts. J Biol Chem. (2004) 279:2030–7. doi: 10.1074/jbc.M309457200
94. von Zglinicki T, Wan T, Miwa S. Senescence in post-mitotic cells: a driver of aging? Antioxid Redox Signal. (2021) 34:308–23. doi: 10.1089/ars.2020.8048
95. Sapieha P, Mallette FA. Cellular senescence in postmitotic cells: beyond growth arrest. Trends Cell Biol. (2018) 28:595–607. doi: 10.1016/j.tcb.2018.03.003
96. Moriya J, Minamino T. Angiogenesis, cancer, and vascular aging. Front Cardiovasc Med. (2017) 4:65. doi: 10.3389/fcvm.2017.00065
98. Costantino S, Paneni F, Cosentino F. Ageing, metabolism and cardiovascular disease. J Physiol. (2016) 594:2061–73. doi: 10.1113/JP270538
99. Shimizu I, Minamino T. Cellular senescence in cardiac diseases. J Cardiol. (2019) 74:313–9. doi: 10.1016/j.jjcc.2019.05.002
100. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
101. Tang X, Li PH, Chen HZ. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front Endocrinol (Lausanne). (2020) 11:280. doi: 10.3389/fendo.2020.00280
102. Cardoso AC, Lam NT, Savla JJ, Nakada Y, Pereira AHM, Elnwasany A, et al. Mitochondrial substrate utilization regulates cardiomyocyte cell cycle progression. Nat Metab. (2020) 2:167–78. doi: 10.1038/s42255-020-0169-x
103. Gevaert AB, Shakeri H, Leloup AJ, Van Hove CE, De Meyer GRY, Vrints CJ, et al. Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ Heart Fail. (2017) 10:e003806. doi: 10.1161/CIRCHEARTFAILURE.116.003806
104. Zhu F, Li Y, Zhang J, Piao C, Liu T, Li HH, et al. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PloS one. (2013) 8:e74535. doi: 10.1371/journal.pone.0074535
105. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. (2002) 105:1541–4. doi: 10.1161/01.CIR.0000013836.85741.17
106. Dominic A, Banerjee P, Hamilton DJ, Le NT, Abe JI. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. (2020) 37:101614. doi: 10.1016/j.redox.2020.101614
107. Morgan RG, Ives SJ, Lesniewski LA, Cawthon RM, Andtbacka RH, Noyes RD, et al. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol. (2013) 305:H251–8. doi: 10.1152/ajpheart.00197.2013
108. Rossman MJ, Kaplon RE, Hill SD, McNamara MN, Santos-Parker JR, Pierce GL, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. (2017) 313:H890–5. doi: 10.1152/ajpheart.00416.2017
109. Weber C, Soehnlein O. Apoe controls the interface linking lipids and inflammation in atherosclerosis. J Clin Invest. (2011) 121:3825–7. doi: 10.1172/JCI60457
110. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. (2016) 354:472–7. doi: 10.1126/science.aaf6659
111. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. (2016) 15:973–7. doi: 10.1111/acel.12458
112. McDonald AP, Meier TR, Hawley AE, Thibert JN, Farris DM, Wrobleski SK, et al. Aging is associated with impaired thrombus resolution in a mouse model of stasis induced thrombosis. Thromb Res. (2010) 125:72–8. doi: 10.1016/j.thromres.2009.06.005
113. Banerjee P, Rosales JE, Chau K, Nguyen MTH, Kotla S, Lin SH, et al. Possible molecular mechanisms underlying the development of atherosclerosis in cancer survivors. Front Cardiovasc Med. (2023) 10:1186679. doi: 10.3389/fcvm.2023.1186679
114. Schosserer M, Grillari J, Breitenbach M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front Oncol. (2017) 7:278. doi: 10.3389/fonc.2017.00278
115. Zhao Y, Simon M, Seluanov A, Gorbunova V. DNA damage and repair in age-related inflammation. Nat Rev Immunol. (2022) 28:75–89. doi: 10.1038/s41577-022-00751-y
116. Banerjee P, Olmsted-Davis EA, Deswal A, Nguyen MT, Koutroumpakis E, Palaskas NL, et al. Cancer treatment-induced NAD+ depletion in premature senescence and late cardiovascular complications. J Cardiovasc Aging. (2022) 2:28. doi: 10.20517/jca.2022.13
117. Banerjee P, Kotla S, Reddy Velatooru L, Abe RJ, Davis EA, Cooke JP, et al. Senescence-associated secretory phenotype as a hinge between cardiovascular diseases and cancer. Front Cardiovasc Med. (2021) 8:763930. doi: 10.3389/fcvm.2021.763930
118. Lagoumtzi SM, Chondrogianni N. Senolytics and senomorphics: natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic Biol Med. (2021) 171:169–90. doi: 10.1016/j.freeradbiomed.2021.05.003
119. Blokland KEC, Pouwels SD, Schuliga M, Knight DA, Burgess JK. Regulation of cellular senescence by extracellular matrix during chronic fibrotic diseases. Clin Sci (Lond). (2020) 134:2681–706. doi: 10.1042/CS20190893
120. Liu D, Richardson G, Benli FM, Park C, de Souza JV, Bronowska AK, et al. Inflammageing in the cardiovascular system: mechanisms, emerging targets, and novel therapeutic strategies. Clin Sci (Lond). (2020) 134:2243–62. doi: 10.1042/CS20191213
121. Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med. (2020) 288:518–36. doi: 10.1111/joim.13141
122. Sabbatinelli J, Prattichizzo F, Olivieri F, Procopio AD, Rippo MR, Giuliani A. Where metabolism meets senescence: focus on endothelial cells. Front Physiol. (2019) 10:1523. doi: 10.3389/fphys.2019.01523
123. Yang J, Liu M, Hong D, Zeng M, Zhang X. The paradoxical role of cellular senescence in cancer. Front Cell Dev Biol. (2021) 9:722205. doi: 10.3389/fcell.2021.722205
124. Chambers CR, Ritchie S, Pereira BA, Timpson P. Overcoming the senescence-associated secretory phenotype (SASP): a complex mechanism of resistance in the treatment of cancer. Mol Oncol. (2021) 15:3242–55. doi: 10.1002/1878-0261.13042
125. Machado-Oliveira G, Ramos C, Marques ARA, Vieira OV. Cell senescence, multiple organelle dysfunction and atherosclerosis. Cells. (2020) 9:2146. doi: 10.3390/cells9102146
126. Mehdizadeh M, Aguilar M, Thorin E, Ferbeyre G, Nattel S. The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nat Rev Cardiol. (2022) 19:250–64. doi: 10.1038/s41569-021-00624-2
127. Owens WA, Walaszczyk A, Spyridopoulos I, Dookun E, Richardson GD. Senescence and senolytics in cardiovascular disease: promise and potential pitfalls. Mech Ageing Dev. (2021) 198:111540. doi: 10.1016/j.mad.2021.111540
128. Campisi J. The biology of replicative senescence. Eur J Cancer. (1997) 33:703–9. doi: 10.1016/S0959-8049(96)00058-5
129. Campisi J. Aging and cancer: the double-edged sword of replicative senescence. J Am Geriatr Soc. (1997) 45:482–8. doi: 10.1111/j.1532-5415.1997.tb05175.x
130. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
131. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. (2014) 15:482–96. doi: 10.1038/nrm3823
132. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. (2021) 9:645593. doi: 10.3389/fcell.2021.645593
133. Raghuram GV, Mishra PK. Stress induced premature senescence: a new culprit in ovarian tumorigenesis? Indian J Med Res. (2014) 140(Suppl):S120–9.25673532
134. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. (2017) 58:235–63. doi: 10.1002/em.22087
135. Mazin AV, Mazina OM, Bugreev DV, Rossi MJ. Rad54, the motor of homologous recombination. DNA repair. (2010) 9:286–302. doi: 10.1016/j.dnarep.2009.12.006
136. Alessio N, Aprile D, Cappabianca S, Peluso G, Di Bernardo G, Galderisi U. Different stages of quiescence, senescence, and cell stress identified by molecular algorithm based on the expression of Ki67, RPS6, and beta-galactosidase activity. Int J Mol Sci. (2021) 22:3102. doi: 10.3390/ijms22063102
137. Yousefzadeh M, Henpita C, Vyas R, Soto-Palma C, Robbins P, Niedernhofer L. DNA damage-how and why we age? eLife. (2021) 10:e62852. doi: 10.7554/eLife.62852
138. Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. (2008) 18:27–47. doi: 10.1038/cr.2008.8
139. Sugasawa K. Mechanism and regulation of DNA damage recognition in mammalian nucleotide excision repair. Enzymes. (2019) 45:99–138. doi: 10.1016/bs.enz.2019.06.004
140. Kusakabe M, Onishi Y, Tada H, Kurihara F, Kusao K, Furukawa M, et al. Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Environ. (2019) 41:2. doi: 10.1186/s41021-019-0119-6
141. Pecina-Slaus N, Kafka A, Salamon I, Bukovac A. Mismatch repair pathway, genome stability and cancer. Front Mol Biosci. (2020) 7:122. doi: 10.3389/fmolb.2020.00122
142. Alekseev S, Coin F. Orchestral maneuvers at the damaged sites in nucleotide excision repair. Cell Mol Life Sci. (2015) 72:2177–86. doi: 10.1007/s00018-015-1859-5
143. Goosen N. Scanning the DNA for damage by the nucleotide excision repair machinery. DNA repair. (2010) 9:593–6. doi: 10.1016/j.dnarep.2010.02.015
144. Ferri D, Orioli D, Botta E. Heterogeneity and overlaps in nucleotide excision repair disorders. Clin Genet. (2020) 97:12–24. doi: 10.1111/cge.13545
145. Schumacher SB, Stucki M, Hubscher U. The N-terminal region of DNA polymerase delta catalytic subunit is necessary for holoenzyme function. Nucleic Acids Res. (2000) 28:620–5. doi: 10.1093/nar/28.2.620
146. Hedglin M, Perumal SK, Hu Z, Benkovic S. Stepwise assembly of the human replicative polymerase holoenzyme. eLife. (2013) 2:e00278. doi: 10.7554/eLife.00278
147. Wei L, Ploss A. Hepatitis B virus cccDNA is formed through distinct repair processes of each strand. Nat Commun. (2021) 12:1591. doi: 10.1038/s41467-021-21850-9
148. London RE. XRCC1—strategies For coordinating and assembling a versatile DNA damage response. DNA repair. (2020) 93:102917. doi: 10.1016/j.dnarep.2020.102917
149. Arakawa H, Iliakis G. Alternative okazaki fragment ligation pathway by DNA ligase III. Genes (Basel). (2015) 6:385–98. doi: 10.3390/genes6020385
150. Wright G, Gassman NR. Transcriptional dysregulation of base excision repair proteins in breast cancer. DNA repair. (2020) 93:102922. doi: 10.1016/j.dnarep.2020.102922
151. Ba X, Aguilera-Aguirre L, Rashid QT, Bacsi A, Radak Z, Sur S, et al. The role of 8-oxoguanine DNA glycosylase-1 in inflammation. Int J Mol Sci. (2014) 15:16975–97. doi: 10.3390/ijms150916975
152. Boiteux S, Coste F, Castaing B. Repair of 8-oxo-7,8-dihydroguanine in prokaryotic and eukaryotic cells: properties and biological roles of the fpg and OGG1 DNA N-glycosylases. Free Radic Biol Med. (2017) 107:179–201. doi: 10.1016/j.freeradbiomed.2016.11.042
153. Fleming AM, Burrows CJ. 8-Oxo-7,8-dihydroguanine, Friend and foe: epigenetic-like regulator versus initiator of mutagenesis. DNA repair. (2017) 56:75–83. doi: 10.1016/j.dnarep.2017.06.009
154. Markkanen E. Not breathing is not an option: how to deal with oxidative DNA damage. DNA repair. (2017) 59:82–105. doi: 10.1016/j.dnarep.2017.09.007
155. Sung JS, Demple B. Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. FEBS J. (2006) 273:1620–9. doi: 10.1111/j.1742-4658.2006.05192.x
156. Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. XRCC1 And DNA polymerase beta in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. (2008) 18:48–63. doi: 10.1038/cr.2008.7
157. Narayan S, Jaiswal AS, Law BK, Kamal MA, Sharma AK, Hromas RA. Interaction between APC and Fen1 during breast carcinogenesis. DNA repair. (2016) 41:54–62. doi: 10.1016/j.dnarep.2016.04.003
158. Boiteux S, Guillet M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA repair. (2004) 3:1–12. doi: 10.1016/j.dnarep.2003.10.002
159. Mohapatra P, Satapathy SR, Das D, Siddharth S, Choudhuri T, Kundu CN. Resveratrol mediated cell death in cigarette smoke transformed breast epithelial cells is through induction of p21Waf1/Cip1 and inhibition of long patch base excision repair pathway. Toxicol Appl Pharmacol. (2014) 275:221–31. doi: 10.1016/j.taap.2014.01.011
160. Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: chk1, Chk2, and MK2. Curr Opin Cell Biol. (2009) 21:245–55. doi: 10.1016/j.ceb.2009.01.018
161. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. (2015) 349:aaa5612. doi: 10.1126/science.aaa5612
162. Ou HL, Schumacher B. DNA Damage responses and p53 in the aging process. Blood. (2018) 131:488–95. doi: 10.1182/blood-2017-07-746396
163. Kawauchi K, Araki K, Tobiume K, Tanaka N. P53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. (2008) 10:611–8. doi: 10.1038/ncb1724
164. Son DS, Kabir SM, Dong YL, Lee E, Adunyah SE. Inhibitory effect of tumor suppressor p53 on proinflammatory chemokine expression in ovarian cancer cells by reducing proteasomal degradation of IkappaB. PLoS One. (2012) 7:e51116. doi: 10.1371/journal.pone.0051116
165. Gudkov AV, Komarova EA. P53 and the carcinogenicity of chronic inflammation. Cold Spring Harb Perspect Med. (2016) 6:a026161. doi: 10.1101/cshperspect.a026161
166. Uehara I, Tanaka N. Role of p53 in the regulation of the inflammatory tumor microenvironment and tumor suppression. Cancers (Basel). (2018) 10:219. doi: 10.3390/cancers10070219
167. Lowe JM, Menendez D, Bushel PR, Shatz M, Kirk EL, Troester MA, et al. P53 and NF-kappaB coregulate proinflammatory gene responses in human macrophages. Cancer Res. (2014) 74:2182–92. doi: 10.1158/0008-5472.CAN-13-1070
168. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. (2011) 192:547–56. doi: 10.1083/jcb.201009094
169. Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, et al. p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. (2013) 201:613–29. doi: 10.1083/jcb.201206006
170. Men H, Cai H, Cheng Q, Zhou W, Wang X, Huang S, et al. The regulatory roles of p53 in cardiovascular health and disease. Cell Mol Life Sci. (2021) 78:2001–18. doi: 10.1007/s00018-020-03694-6
172. Higo T, Naito AT, Sumida T, Shibamoto M, Okada K, Nomura S, et al. DNA single-strand break-induced DNA damage response causes heart failure. Nat Commun. (2017) 8:15104. doi: 10.1038/ncomms15104
173. Razani B, Raines EW. Can the DNA damage response be harnessed to modulate atherosclerotic plaque phenotype? Circ Res. (2015) 116:770–3. doi: 10.1161/CIRCRESAHA.115.305922
174. Gray K, Kumar S, Figg N, Harrison J, Baker L, Mercer J, et al. Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ Res. (2015) 116:816–26. doi: 10.1161/CIRCRESAHA.116.304921
175. Mitry MA, Laurent D, Keith BL, Sira E, Eisenberg CA, Eisenberg LM, et al. Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am J Physiol Cell Physiol. (2020) 318:C380–91. doi: 10.1152/ajpcell.00073.2019
176. Miwa S, Kashyap S, Chini E, von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest. (2022) 132:e158447. doi: 10.1172/JCI158447
177. Vercellino I, Sazanov LA. Structure and assembly of the mammalian mitochondrial supercomplex CIII(2)CIV. Nature. (2021) 598:364–7. doi: 10.1038/s41586-021-03927-z
178. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders—a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1066–77. doi: 10.1016/j.bbadis.2016.11.010
179. Gupta A, Varma A, Storey KB. New insights to regulation of fructose-1,6-bisphosphatase during anoxia in red-eared slider, trachemys scripta elegans. Biomolecules. (2021) 11:1548. doi: 10.3390/biom11101548
180. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. (2014) 5:592–602. doi: 10.1007/s13238-014-0082-8
181. Zhang X, Yang S, Chen J, Su Z. Unraveling the regulation of hepatic gluconeogenesis. Front Endocrinol (Lausanne). (2018) 9:802. doi: 10.3389/fendo.2018.00802
182. Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. (2015) 90:927–63. doi: 10.1111/brv.12140
184. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. (2013) 12:152. doi: 10.1186/1476-4598-12-152
185. Xiao W, Wang RS, Handy DE, Loscalzo J. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid Redox Signal. (2018) 28:251–72. doi: 10.1089/ars.2017.7216
186. Goodman RP, Calvo SE, Mootha VK. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J Biol Chem. (2018) 293:7508–16. doi: 10.1074/jbc.TM117.000258
187. Hassinen IE. Signaling and regulation through the NAD(+) and NADP(+) networks. Antioxid Redox Signal. (2019) 30:857–74. doi: 10.1089/ars.2017.7479
189. Kotla S, Zhang A, Imanishi M, Ko KA, Lin SH, Gi YJ, et al. Nucleus-mitochondria positive feedback loop formed by ERK5 S496 phosphorylation-mediated poly (ADP-ribose) polymerase activation provokes persistent pro-inflammatory senescent phenotype and accelerates coronary atherosclerosis after chemo-radiation. Redox Biol. (2021) 47:102132. doi: 10.1016/j.redox.2021.102132
190. Byrne NJ, Rajasekaran NS, Abel ED, Bugger H. Therapeutic potential of targeting oxidative stress in diabetic cardiomyopathy. Free Radic Biol Med. (2021) 169:317–42. doi: 10.1016/j.freeradbiomed.2021.03.046
191. Iakovou E, Kourti M. A comprehensive overview of the complex role of oxidative stress in aging, the contributing environmental stressors and emerging antioxidant therapeutic interventions. Front Aging Neurosci. (2022) 14:827900. doi: 10.3389/fnagi.2022.827900
192. van den Boogaard ML, Oka R, Hakkert A, Schild L, Ebus ME, van Gerven MR, et al. Defects in 8-oxo-guanine repair pathway cause high frequency of C > A substitutions in neuroblastoma. Proc Natl Acad Sci U S A. (2021) 118:e2007898118. doi: 10.1073/pnas.2007898118
193. Shields HJ, Traa A, Van Raamsdonk JM. Beneficial and detrimental effects of reactive oxygen Species on lifespan: a comprehensive review of comparative and experimental studies. Front Cell Dev Biol. (2021) 9:628157. doi: 10.3389/fcell.2021.628157
194. Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int. (2014) 2014:238463. doi: 10.1155/2014/238463
195. Chandhok G, Lazarou M, Neumann B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol Rev Camb Philos Soc. (2018) 93:933–49. doi: 10.1111/brv.12378
196. Sekine S, Youle RJ. PINK1 Import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. (2018) 16:2. doi: 10.1186/s12915-017-0470-7
197. Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. (2016) 27:105–17. doi: 10.1016/j.tem.2015.12.001
198. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. (2004) 305:858–62. doi: 10.1126/science.1099793
199. Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu B, et al. MFN1 Structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature. (2017) 542:372–6. doi: 10.1038/nature21077
200. Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. (2000) 26:207–10. doi: 10.1038/79936
201. Ichishita R, Tanaka K, Sugiura Y, Sayano T, Mihara K, Oka T. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in caenorhabditis elegans. J Biochem. (2008) 143:449–54. doi: 10.1093/jb/mvm245
202. Scheckhuber CQ, Erjavec N, Tinazli A, Hamann A, Nystrom T, Osiewacz HD. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat Cell Biol. (2007) 9:99–105. doi: 10.1038/ncb1524
203. Bernhardt D, Muller M, Reichert AS, Osiewacz HD. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci Rep. (2015) 5:7885. doi: 10.1038/srep07885
204. Zhang Y, Lanjuin A, Chowdhury SR, Mistry M, Silva-Garcia CG, Weir HJ, et al. Neuronal TORC1 modulates longevity via AMPK and cell nonautonomous regulation of mitochondrial dynamics in C. elegans. eLife. (2019) 8:e49158. doi: 10.7554/eLife.49158
205. Song M, Franco A, Fleischer JA, Zhang L, Dorn GW 2nd. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. (2017) 26:872–83. e5. doi: 10.1016/j.cmet.2017.09.023
206. Shirihai OS, Song M, Dorn GW 2nd. How mitochondrial dynamism orchestrates mitophagy. Circ Res. (2015) 116:1835–49. doi: 10.1161/CIRCRESAHA.116.306374
207. Song M, Dorn GW 2nd. Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. (2015) 21:195–205. doi: 10.1016/j.cmet.2014.12.019
208. Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. (2015) 21:273–86. doi: 10.1016/j.cmet.2014.12.011
209. Dorn GW 2nd, Song M, Walsh K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J Mol Cell Cardiol. (2015) 78:123–8. doi: 10.1016/j.yjmcc.2014.09.015
210. Chen Y, Liu Y, Dorn GW 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. (2011) 109:1327–31. doi: 10.1161/CIRCRESAHA.111.258723
211. Boulton DP, Caino MC. Mitochondrial fission and fusion in tumor progression to metastasis. Front Cell Dev Biol. (2022) 10:849962. doi: 10.3389/fcell.2022.849962
212. Yu M, Nguyen ND, Huang Y, Lin D, Fujimoto TN, Molkentine JM, et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight. (2019) 5:e126915. doi: 10.1172/jci.insight.126915
213. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. (2005) 8:3–5. doi: 10.1089/rej.2005.8.3
214. Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell Dev Biol. (2020) 8:467. doi: 10.3389/fcell.2020.00467
215. McWilliams TG, Muqit MM. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol. (2017) 45:83–91. doi: 10.1016/j.ceb.2017.03.013
216. Yamano K, Matsuda N, Tanaka K. The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep. (2016) 17:300–16. doi: 10.15252/embr.201541486
217. Padman BS, Nguyen TN, Uoselis L, Skulsuppaisarn M, Nguyen LK, Lazarou M. LC3/GABARAPs Drive ubiquitin-independent recruitment of optineurin and NDP52 to amplify mitophagy. Nat Commun. (2019) 10:408. doi: 10.1038/s41467-019-08335-6
218. Sulkshane P, Ram J, Thakur A, Reis N, Kleifeld O, Glickman MH. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. (2021) 45:102047. doi: 10.1016/j.redox.2021.102047
219. Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. (2014) 24:787–95. doi: 10.1038/cr.2014.75
220. Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem. (2007) 282:2355–62. doi: 10.1074/jbc.M608009200
221. Hickson-Bick DL, Jones C, Buja LM. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J Mol Cell Cardiol. (2008) 44:411–8. doi: 10.1016/j.yjmcc.2007.10.013
222. Zhang Y, Xu H. Translational regulation of mitochondrial biogenesis. Biochem Soc Trans. (2016) 44:1717–24. doi: 10.1042/BST20160071C
223. Bartolome A, Garcia-Aguilar A, Asahara SI, Kido Y, Guillen C, Pajvani UB, et al. MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol Cell Biol. (2017) 37:e00441–17. doi: 10.1128/MCB.00441-17
224. Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. (2015) 521:525–8. doi: 10.1038/nature14300
225. Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Felix AA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. (2016) 22:879–88. doi: 10.1038/nm.4132
226. Ludtmann MHR, Abramov AY. Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci Lett. (2018) 663:86–90. doi: 10.1016/j.neulet.2017.08.044
227. Poznyak AV, Nikiforov NG, Markin AM, Kashirskikh DA, Myasoedova VA, Gerasimova EV, et al. Overview of OxLDL and its impact on cardiovascular health: focus on atherosclerosis. Front Pharmacol. (2020) 11:613780. doi: 10.3389/fphar.2020.613780
228. Bernardini JP, Brouwer JM, Tan IK, Sandow JJ, Huang S, Stafford CA, et al. Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. (2019) 38:e99916. doi: 10.15252/embj.201899916
229. Chen G, Kroemer G, Kepp O. Mitophagy: an emerging role in aging and age-associated diseases. Front Cell Dev Biol. (2020) 8:200. doi: 10.3389/fcell.2020.00200
230. Shally A, McDonagh B. The redox environment and mitochondrial dysfunction in age-related skeletal muscle atrophy. Biogerontology. (2020) 21:461–73. doi: 10.1007/s10522-020-09879-7
231. Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. (2009) 33:627–38. doi: 10.1016/j.molcel.2009.02.013
232. Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. (1979) 195:453–9. doi: 10.1016/0003-9861(79)90371-0
233. Sah E, Krishnamurthy S, Ahmidouch MY, Gillispie GJ, Milligan C, Orr ME. The cellular senescence stress response in post-mitotic brain cells: cell survival at the expense of tissue degeneration. Life (Basel). (2021) 11:229. doi: 10.3390/life11030229
234. Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. (2013) 4:2308. doi: 10.1038/ncomms3308
235. Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. (2016) 61:654–66. doi: 10.1016/j.molcel.2016.01.028
236. Li A, Gao M, Liu B, Qin Y, Chen L, Liu H, et al. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. (2022) 13:444. doi: 10.1038/s41419-022-04906-6
237. Chang JY, Yi HS, Kim HW, Shong M. Dysregulation of mitophagy in carcinogenesis and tumor progression. Biochim Biophys Acta Bioenerg. (2017) 1858:633–40. doi: 10.1016/j.bbabio.2016.12.008
238. Zhang C, Liu Z, Bunker E, Ramirez A, Lee S, Peng Y, et al. Sorafenib targets the mitochondrial electron transport chain complexes and ATP synthase to activate the PINK1-Parkin pathway and modulate cellular drug response. J Biol Chem. (2017) 292:15105–20. doi: 10.1074/jbc.M117.783175
239. Cao S, Shen Z, Wang C, Zhang Q, Hong Q, He Y, et al. Resveratrol improves intestinal barrier function, alleviates mitochondrial dysfunction and induces mitophagy in diquat challenged piglets(1). Food Funct. (2019) 10:344–54. doi: 10.1039/C8FO02091D
240. Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming QL, et al. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1l-mediated mitochondrial fission. Autophagy. (2015) 11:1259–79. doi: 10.1080/15548627.2015.1056970
241. Angeli S, Foulger A, Chamoli M, Peiris TH, Gerencser A, Shahmirzadi AA, et al. The mitochondrial permeability transition pore activates the mitochondrial unfolded protein response and promotes aging. Elife. (2021) 10:e63453. doi: 10.7554/eLife.63453
242. Bauer TM, Murphy E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ Res. (2020) 126:280–93. doi: 10.1161/CIRCRESAHA.119.316306
243. Yang Y, Wang W, Tian Y, Shi J. Sirtuin 3 and mitochondrial permeability transition pore (mPTP): a systematic review. Mitochondrion. (2022) 64:103–11. doi: 10.1016/j.mito.2022.03.004
244. Yang J, Guo Q, Feng X, Liu Y, Zhou Y. Mitochondrial dysfunction in cardiovascular diseases: potential targets for treatment. Front Cell Dev Biol. (2022) 10:841523. doi: 10.3389/fcell.2022.841523
245. Kent AC, El Baradie KBY, Hamrick MW. Targeting the mitochondrial permeability transition pore to prevent age-associated cell damage and neurodegeneration. Oxid Med Cell Longev. (2021) 2021:6626484. doi: 10.1155/2021/6626484
246. Waseem M, Wang BD. Promising strategy of mPTP modulation in cancer therapy: an emerging progress and future insight. Int J Mol Sci. (2023) 24:5564. doi: 10.3390/ijms24065564
247. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. (2012) 150:12–27. doi: 10.1016/j.cell.2012.06.013
248. Harvey ZH, Chen Y, Jarosz DF. Protein-based inheritance: epigenetics beyond the chromosome. Mol Cell. (2018) 69:195–202. doi: 10.1016/j.molcel.2017.10.030
249. Zhang L, Lu Q, Chang C. Epigenetics in health and disease. Adv Exp Med Biol. (2020) 1253:3–55. doi: 10.1007/978-981-15-3449-2_1
250. Villanueva L, Alvarez-Errico D, Esteller M. The contribution of epigenetics to cancer immunotherapy. Trends Immunol. (2020) 41:676–91. doi: 10.1016/j.it.2020.06.002
251. Lin Z, Ding Q, Li X, Feng Y, He H, Huang C, et al. Targeting epigenetic mechanisms in vascular aging. Front Cardiovasc Med. (2021) 8:806988. doi: 10.3389/fcvm.2021.806988
252. Zhu D, Li X, Tian Y. Mitochondrial-to-nuclear communication in aging: an epigenetic perspective. Trends Biochem Sci. (2022) 47:645–59. doi: 10.1016/j.tibs.2022.03.008
253. Poulsen P, Esteller M, Vaag A, Fraga MF. The epigenetic basis of twin discordance in age-related diseases. Pediatr Res. (2007) 61:38R–42R. doi: 10.1203/pdr.0b013e31803c7b98
254. Salameh Y, Bejaoui Y, El Hajj N. DNA Methylation biomarkers in aging and age-related diseases. Front Genet. (2020) 11:171. doi: 10.3389/fgene.2020.00171
255. Lind GE, Skotheim RI, Fraga MF, Abeler VM, Henrique R, Saatcioglu F, et al. The loss of NKX3.1 expression in testicular–and prostate–cancers is not caused by promoter hypermethylation. Mol Cancer. (2005) 4:8. doi: 10.1186/1476-4598-4-8
256. Levesque ML, Casey KF, Szyf M, Ismaylova E, Ly V, Verner MP, et al. Genome-wide DNA methylation variability in adolescent monozygotic twins followed since birth. Epigenetics. (2014) 9:1410–21. doi: 10.4161/15592294.2014.970060
257. Lowe D, Horvath S, Raj K. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget. (2016) 7:8524–31. doi: 10.18632/oncotarget.7383
258. Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). (2018) 10:573–91. doi: 10.18632/aging.101414
259. Dube CT, Jahan FRS, Lim CY. Key changes in chromatin mark mammalian epidermal differentiation and ageing. Epigenetics. (2022) 17:444–59. doi: 10.1080/15592294.2021.1917812
260. Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. (2004) 16:93–105. doi: 10.1016/j.molcel.2004.08.031
261. Tennen RI, Chua KF. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem Sci. (2011) 36:39–46. doi: 10.1016/j.tibs.2010.07.009
262. Han Y, Han D, Yan Z, Boyd-Kirkup JD, Green CD, Khaitovich P, et al. Stress-associated H3K4 methylation accumulates during postnatal development and aging of rhesus macaque brain. Aging Cell. (2012) 11:1055–64. doi: 10.1111/acel.12007
263. Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, et al. ZMYND11 Links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. (2014) 508:263–8. doi: 10.1038/nature13045
264. Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. (2006) 103:8703–8. doi: 10.1073/pnas.0602569103
265. Sarg B, Koutzamani E, Helliger W, Rundquist I, Lindner HH. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J Biol Chem. (2002) 277:39195–201. doi: 10.1074/jbc.M205166200
266. McIntyre RL, Daniels EG, Molenaars M, Houtkooper RH, Janssens GE. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol Med. (2019) 11:e9854. doi: 10.15252/emmm.201809854
267. Ooi JY, Tuano NK, Rafehi H, Gao XM, Ziemann M, Du XJ, et al. HDAC Inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics. (2015) 10:418–30. doi: 10.1080/15592294.2015.1024406
268. Chen Y, Du J, Zhao YT, Zhang L, Lv G, Zhuang S, et al. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol. (2015) 14:99. doi: 10.1186/s12933-015-0262-8
269. Walsh ME, Bhattacharya A, Sataranatarajan K, Qaisar R, Sloane L, Rahman MM, et al. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell. (2015) 14:957–70. doi: 10.1111/acel.12387
270. Khandia R, Dadar M, Munjal A, Dhama K, Karthik K, Tiwari R, et al. A comprehensive review of autophagy and its Various roles in infectious, non-infectious, and lifestyle diseases: current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells. (2019) 8:674. doi: 10.3390/cells8070674
271. Sharma V, Verma S, Seranova E, Sarkar S, Kumar D. Selective autophagy and xenophagy in infection and disease. Front Cell Dev Biol. (2018) 6:147. doi: 10.3389/fcell.2018.00147
272. Wong SQ, Kumar AV, Mills J, Lapierre LR. Autophagy in aging and longevity. Hum Genet. (2020) 139:277–90. doi: 10.1007/s00439-019-02031-7
273. Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. (2009) 335:1–32. doi: 10.1007/978-3-642-00302-8_1
274. Deng Z, Purtell K, Lachance V, Wold MS, Chen S, Yue Z. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol. (2017) 27:491–504. doi: 10.1016/j.tcb.2017.01.001
275. Abdellatif M, Ljubojevic-Holzer S, Madeo F, Sedej S. Autophagy in cardiovascular health and disease. Prog Mol Biol Transl Sci. (2020) 172:87–106. doi: 10.1016/bs.pmbts.2020.04.022
276. Abdellatif M, Sedej S, Carmona-Gutierrez D, Madeo F, Kroemer G. Autophagy in cardiovascular aging. Circ Res. (2018) 123:803–24. doi: 10.1161/CIRCRESAHA.118.312208
277. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. (2008) 132:27–42. doi: 10.1016/j.cell.2007.12.018
278. Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. (2010) 107:1470–82. doi: 10.1161/CIRCRESAHA.110.227371
279. Liao X, Zhang R, Lu Y, Prosdocimo DA, Sangwung P, Zhang L, et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest. (2015) 125:3461–76. doi: 10.1172/JCI79964
280. Chauhan S, Goodwin JG, Chauhan S, Manyam G, Wang J, Kamat AM, et al. ZKSCAN3 Is a master transcriptional repressor of autophagy. Mol Cell. (2013) 50:16–28. doi: 10.1016/j.molcel.2013.01.024
281. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. (2004) 6:463–77. doi: 10.1016/S1534-5807(04)00099-1
282. Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res. (2005) 68:355–65. doi: 10.1016/j.cardiores.2005.08.014
283. Nakai K, Hayashi T, Nagaya S, Toyoda H, Yamamoto M, Shiku H, et al. Shear stress-induced myosin association with cytoskeleton and phosphorylation in human platelets. Life Sci. (1997) 60:L181–91.
284. Nishinaka Y, Masutani H, Oka S, Matsuo Y, Yamaguchi Y, Nishio K, et al. Importin alpha1 (Rch1) mediates nuclear translocation of thioredoxin-binding protein-2/vitamin D(3)-up-regulated protein 1. J Biol Chem. (2004) 279:37559–65. doi: 10.1074/jbc.M405473200
285. Takemura G, Miyata S, Kawase Y, Okada H, Maruyama R, Fujiwara H. Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy. (2006) 2:212–4. doi: 10.4161/auto.2608
286. Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, et al. NAD(+) metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther. (2020) 5:227. doi: 10.1038/s41392-020-00311-7
287. Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PloS one. (2011) 6:e19194. doi: 10.1371/journal.pone.0019194
288. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. (2016) 23:1127–39. doi: 10.1016/j.cmet.2016.05.006
289. Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. (2013) 154:430–41. doi: 10.1016/j.cell.2013.06.016
290. Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A. (2015) 112:2876–81. doi: 10.1073/pnas.1417921112
291. Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S, et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol. (2019) 21:397–407. doi: 10.1038/s41556-019-0287-4
292. Covarrubias AJ, Perrone R, Grozio A, Verdin E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol. (2021) 22:119–41. doi: 10.1038/s41580-020-00313-x
293. Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. (2015) 350:1208–13. doi: 10.1126/science.aac4854
294. Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. (2016) 352:1436–43. doi: 10.1126/science.aaf2693
295. Fang EF, Hou Y, Lautrup S, Jensen MB, Yang B, SenGupta T, et al. NAD(+) augmentation restores mitophagy and limits accelerated aging in werner syndrome. Nat Commun. (2019) 10:5284. doi: 10.1038/s41467-019-13172-8
296. Lin Q, Zuo W, Liu Y, Wu K, Liu Q. NAD(+) and cardiovascular diseases. Clin Chim Acta. (2021) 515:104–10. doi: 10.1016/j.cca.2021.01.012
297. Frederick DW, Loro E, Liu L, Davila A Jr., Chellappa K, Silverman IM, et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. (2016) 24:269–82. doi: 10.1016/j.cmet.2016.07.005
298. Stein LR, Imai S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. (2014) 33:1321–40. doi: 10.1002/embj.201386917
299. van der Veer E, Ho C, O'Neil C, Barbosa N, Scott R, Cregan SP, et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J Biol Chem. (2007) 282:10841–5. doi: 10.1074/jbc.C700018200
300. Chang HC, Guarente L. SIRT1 Mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. (2013) 153:1448–60. doi: 10.1016/j.cell.2013.05.027
301. Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. (2004) 61:19–34. doi: 10.1007/s00018-003-3161-1
302. Magni G, Amici A, Emanuelli M, Raffaelli N, Ruggieri S. Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol Biol. (1999) 73:135–82. doi: 10.1002/9780470123195.ch5
303. Hernandez-Campo PM, Almeida J, Sanchez ML, Malvezzi M, Orfao A. Normal patterns of expression of glycosylphosphatidylinositol-anchored proteins on different subsets of peripheral blood cells: a frame of reference for the diagnosis of paroxysmal nocturnal hemoglobinuria. Cytometry B Clin Cytom. (2006) 70:71–81. doi: 10.1002/cyto.b.20087
304. Berthelier V, Tixier JM, Muller-Steffner H, Schuber F, Deterre P. Human CD38 is an authentic NAD(P)+ glycohydrolase. Biochem J. (1998) 330(Pt 3):1383–90. doi: 10.1042/bj3301383
305. Kirchberger T, Guse AH. Measuring CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) activity by reverse-phase HPLC. Cold Spring Harb Protoc. (2013) 2013:569–73. doi: 10.1101/pdb.prot073007
306. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. (2008) 88:841–86. doi: 10.1152/physrev.00035.2007
307. Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, et al. CD38 And CD157: a long journey from activation markers to multifunctional molecules. Cytometry B Clin Cytom. (2013) 84:207–17. doi: 10.1002/cyto.b.21092
308. Preugschat F, Carter LH, Boros EE, Porter DJ, Stewart EL, Shewchuk LM. A pre-steady state and steady state kinetic analysis of the N-ribosyl hydrolase activity of hCD157. Arch Biochem Biophys. (2014) 564:156–63. doi: 10.1016/j.abb.2014.09.008
309. Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, et al. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. (2013) 62:1084–93. doi: 10.2337/db12-1139
310. Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, et al. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. (2007) 21:3629–39. doi: 10.1096/fj.07-8290com
311. Boslett J, Helal M, Chini E, Zweier JL. Genetic deletion of CD38 confers post-ischemic myocardial protection through preserved pyridine nucleotides. J Mol Cell Cardiol. (2018) 118:81–94. doi: 10.1016/j.yjmcc.2018.02.015
312. Boslett J, Hemann C, Zhao YJ, Lee HC, Zweier JL. Luteolinidin protects the postischemic heart through CD38 inhibition with preservation of NAD(P)(H). J Pharmacol Exp Ther. (2017) 361:99–108. doi: 10.1124/jpet.116.239459
313. Boslett J, Reddy N, Alzarie YA, Zweier JL. Inhibition of CD38 with the thiazoloquin(az)olin(on)e 78c protects the heart against postischemic injury. J Pharmacol Exp Ther. (2019) 369:55–64. doi: 10.1124/jpet.118.254557
314. Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, et al. Discovery, synthesis, and biological evaluation of thiazoloquin(az)olin(on)es as potent CD38 inhibitors. J Med Chem. (2015) 58:3548–71. doi: 10.1021/jm502009h
315. Chillemi A, Zaccarello G, Quarona V, Ferracin M, Ghimenti C, Massaia M, et al. Anti-CD38 antibody therapy: windows of opportunity yielded by the functional characteristics of the target molecule. Mol Med. (2013) 19:99–108. doi: 10.2119/molmed.2013.00009
316. Conforti L, Gilley J, Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. (2014) 15:394–409. doi: 10.1038/nrn3680
317. Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron. (2017) 93:1334–43. e5. doi: 10.1016/j.neuron.2017.02.022
318. Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J Neurosci. (2013) 33:13569–80. doi: 10.1523/JNEUROSCI.1197-13.2013
319. Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, et al. Dsarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. (2012) 337:481–4. doi: 10.1126/science.1223899
320. Hopkins EL, Gu W, Kobe B, Coleman MP. A novel NAD signaling mechanism in axon degeneration and its relationship to innate immunity. Front Mol Biosci. (2021) 8:703532. doi: 10.3389/fmolb.2021.703532
321. Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 Activation triggers axon degeneration locally via NAD(+) destruction. Science. (2015) 348:453–7. doi: 10.1126/science.1258366
322. Gerdts J, Summers DW, Milbrandt J, DiAntonio A. Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron. (2016) 89:449–60. doi: 10.1016/j.neuron.2015.12.023
323. Gilley J, Orsomando G, Nascimento-Ferreira I, Coleman MP. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. (2015) 10:1974–81. doi: 10.1016/j.celrep.2015.02.060
324. Gilley J, Ribchester RR, Coleman MP. Sarm1 deletion, but Not Wld(S), confers lifelong rescue in a mouse model of severe axonopathy. Cell Rep. (2017) 21:10–6. doi: 10.1016/j.celrep.2017.09.027
325. Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD(+) ratio to trigger axon degeneration. Neuron. (2021) 109:1118–36. e11. doi: 10.1016/j.neuron.2021.02.009
326. Waller TJ, Collins CA. An NAD+/NMN balancing act by SARM1 and NMNAT2 controls axonal degeneration. Neuron. (2021) 109:1067–9. doi: 10.1016/j.neuron.2021.03.021
327. Sur M, Dey P, Sarkar A, Bar S, Banerjee D, Bhat S, et al. Sarm1 induction and accompanying inflammatory response mediates age-dependent susceptibility to rotenone-induced neurotoxicity. Cell Death Discov. (2018) 4:114. doi: 10.1038/s41420-018-0119-5
328. Covarrubias AJ, Kale A, Perrone R, Lopez-Dominguez JA, Pisco AO, Kasler HG, et al. Author correction: senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat Metab. (2021) 3:120–1. doi: 10.1038/s42255-020-00328-w
329. Feldman JL, Dittenhafer-Reed KE, Denu JM. Sirtuin catalysis and regulation. J Biol Chem. (2012) 287:42419–27. doi: 10.1074/jbc.R112.378877
330. Lee HC, Zhao YJ. Resolving the topological enigma in Ca(2+) signaling by cyclic ADP-ribose and NAADP. J Biol Chem. (2019) 294:19831–43. doi: 10.1074/jbc.REV119.009635
331. Hayakawa T, Iwai M, Aoki S, Takimoto K, Maruyama M, Maruyama W, et al. SIRT1 Suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PloS one. (2015) 10:e0116480. doi: 10.1371/journal.pone.0116480
332. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. (2006) 124:315–29. doi: 10.1016/j.cell.2005.11.044
333. Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. (2007) 43:571–9. doi: 10.1016/j.yjmcc.2007.08.008
334. Kane AE, Sinclair DA. Sirtuins and NAD(+) in the development and treatment of metabolic and cardiovascular diseases. Circ Res. (2018) 123:868–85. doi: 10.1161/CIRCRESAHA.118.312498
335. Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. (2011) 25:1245–61. doi: 10.1101/gad.625811
336. Lv ZC, Li F, Wang L, Zhao QH, Gang GS, Wu Y, et al. Impact of parthanatos on the increased risk of onset and mortality in male patients with pulmonary hypertension. Am J Mens Health. (2021) 15:15579883211029458. doi: 10.1177/15579883211029458
337. Savelyev NV, Shepelev NM, Lavrik OI, Rubtsova MP, Dontsova OA. PARP1 Regulates the biogenesis and activity of telomerase Complex through modification of H/ACA-proteins. Front Cell Dev Biol. (2021) 9:621134. doi: 10.3389/fcell.2021.621134
338. Gomez M, Wu J, Schreiber V, Dunlap J, Dantzer F, Wang Y, et al. PARP1 Is a TRF2-associated poly(ADP-ribose)polymerase and protects eroded telomeres. Mol Biol Cell. (2006) 17:1686–96. doi: 10.1091/mbc.e05-07-0672
339. Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, et al. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. (2016) 24:566–81. doi: 10.1016/j.cmet.2016.09.004
340. Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. (2014) 157:882–96. doi: 10.1016/j.cell.2014.03.026
341. Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. (2014) 20:840–55. doi: 10.1016/j.cmet.2014.10.005
342. Scheibye-Knudsen M, Ramamoorthy M, Sykora P, Maynard S, Lin PC, Minor RK, et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med. (2012) 209:855–69. doi: 10.1084/jem.20111721
343. Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagne JP, et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. (2014) 111:10209–14. doi: 10.1073/pnas.1405158111
344. Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, et al. PARP-1 Inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. (2011) 13:461–8. doi: 10.1016/j.cmet.2011.03.004
345. Fouquerel E, Goellner EM, Yu Z, Gagne JP, Barbi de Moura M, Feinstein T, et al. ARTD1/PARP1 Negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. (2014) 8:1819–31. doi: 10.1016/j.celrep.2014.08.036
346. Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. (2005) 280:43121–30. doi: 10.1074/jbc.M506162200
347. Jiang HY, Yang Y, Zhang YY, Xie Z, Zhao XY, Sun Y, et al. The dual role of poly(ADP-ribose) polymerase-1 in modulating parthanatos and autophagy under oxidative stress in rat cochlear marginal cells of the stria vascularis. Redox Biol. (2018) 14:361–70. doi: 10.1016/j.redox.2017.10.002
348. Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G, et al. PARP-1 Is involved in autophagy induced by DNA damage. Autophagy. (2009) 5:61–74. doi: 10.4161/auto.5.1.7272
349. Sedlackova L, Korolchuk VI. The crosstalk of NAD, ROS and autophagy in cellular health and ageing. Biogerontology. (2020) 21:381–97. doi: 10.1007/s10522-020-09864-0
350. Lautrup S, Sinclair DA, Mattson MP, Fang EF. NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. (2019) 30:630–55. doi: 10.1016/j.cmet.2019.09.001
351. Hou Y, Lautrup S, Cordonnier S, Wang Y, Croteau DL, Zavala E, et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci U S A. (2018) 115:E1876–85. doi: 10.1073/pnas.1718819115
352. Schondorf DC, Ivanyuk D, Baden P, Sanchez-Martinez A, De Cicco S, Yu C, et al. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep. (2018) 23:2976–88. doi: 10.1016/j.celrep.2018.05.009
353. Vannini N, Campos V, Girotra M, Trachsel V, Rojas-Sutterlin S, Tratwal J, et al. The NAD-booster nicotinamide riboside potently stimulates hematopoiesis through increased mitochondrial clearance. Cell Stem Cell. (2019) 24:405–18. e7. doi: 10.1016/j.stem.2019.02.012
354. Sohn JY, Kwak HJ, Rhim JH, Yeo EJ. AMP-Activated protein kinase-dependent nuclear localization of glyceraldehyde 3-phosphate dehydrogenase in senescent human diploid fibroblasts. Aging (Albany NY). (2022) 14:4–27. doi: 10.18632/aging.203825
355. Bittles AH, Harper N. Increased glycolysis in ageing cultured human diploid fibroblasts. Biosci Rep. (1984) 4:751–6. doi: 10.1007/BF01128816
356. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
357. Coppe JP, Kauser K, Campisi J, Beausejour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. (2006) 281:29568–74. doi: 10.1074/jbc.M603307200
358. Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. (2009) 11:973–9. doi: 10.1038/ncb1909
359. Salminen A. Feed-forward regulation between cellular senescence and immunosuppression promotes the aging process and age-related diseases. Ageing Res Rev. (2021) 67:101280. doi: 10.1016/j.arr.2021.101280
360. Akboga MK, Canpolat U, Yayla C, Ozcan F, Ozeke O, Topaloglu S, et al. Association of platelet to lymphocyte ratio with inflammation and severity of coronary atherosclerosis in patients with stable coronary artery disease. Angiology. (2016) 67:89–95. doi: 10.1177/0003319715583186
361. Zhang X, Liu D, Gao Y, Lin C, An Q, Feng Y, et al. The biology and function of extracellular vesicles in cancer development. Front Cell Dev Biol. (2021) 9:777441. doi: 10.3389/fcell.2021.777441
362. Fuloria S, Subramaniyan V, Dahiya R, Dahiya S, Sudhakar K, Kumari U, et al. Mesenchymal stem cell-derived extracellular vesicles: regenerative potential and challenges. Biology (Basel). (2021) 10:172. doi: 10.3390/biology10030172
363. Jeske R, Chen X, Ma S, Zeng EZ, Driscoll T, Li Y. Bioreactor expansion reconfigures metabolism and extracellular vesicle biogenesis of human adipose-derived stem cells in vitro. Biochem Eng J. (2022) 188:108711. doi: 10.1016/j.bej.2022.108711
364. Torres Crigna A, Uhlig S, Elvers-Hornung S, Kluter H, Bieback K. Human adipose tissue-derived stromal cells suppress human, but not murine lymphocyte proliferation, via indoleamine 2,3-dioxygenase activity. Cells. (2020) 9:2419. doi: 10.3390/cells9112419
365. Antunes MA, Braga CL, Oliveira TB, Kitoko JZ, Castro LL, Xisto DG, et al. Mesenchymal stromal cells from emphysematous donors and their extracellular vesicles are unable to reverse cardiorespiratory dysfunction in experimental severe emphysema. Front Cell Dev Biol. (2021) 9:661385. doi: 10.3389/fcell.2021.661385
366. Femmino S, Penna C, Margarita S, Comita S, Brizzi MF, Pagliaro P. Extracellular vesicles and cardiovascular system: biomarkers and cardioprotective effectors. Vascul Pharmacol. (2020) 135:106790. doi: 10.1016/j.vph.2020.106790
367. Burger D, Kwart DG, Montezano AC, Read NC, Kennedy CR, Thompson CS, et al. Microparticles induce cell cycle arrest through redox-sensitive processes in endothelial cells: implications in vascular senescence. J Am Heart Assoc. (2012) 1:e001842. doi: 10.1161/JAHA.112.001842
368. Riquelme JA, Takov K, Santiago-Fernandez C, Rossello X, Lavandero S, Yellon DM, et al. Increased production of functional small extracellular vesicles in senescent endothelial cells. J Cell Mol Med. (2020) 24:4871–6. doi: 10.1111/jcmm.15047
369. Simoncini S, Chateau AL, Robert S, Todorova D, Yzydorzick C, Lacroix R, et al. Biogenesis of pro-senescent microparticles by endothelial colony forming cells from premature neonates is driven by SIRT1-dependent epigenetic regulation of MKK6. Sci Rep. (2017) 7:8277. doi: 10.1038/s41598-017-08883-1
370. Imanishi M, Cheng H, Kotla S, Deswal A, Le NT, Chini E, et al. Radiation therapy induces immunosenescence mediated by p90RSK. Front Cardiovasc Med. (2022) 9:988713. doi: 10.3389/fcvm.2022.988713
371. Triana-Martinez F, Loza MI, Dominguez E. Beyond tumor suppression: senescence in cancer stemness and tumor dormancy. Cells. (2020) 9:346. doi: 10.3390/cells9020346
372. Zhang DY, Monteiro MJ, Liu JP, Gu WY. Mechanisms of cancer stem cell senescence: current understanding and future perspectives. Clin Exp Pharmacol Physiol. (2021) 48:1185–202. doi: 10.1111/1440-1681.13528
373. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. (2019) 19:439–53. doi: 10.1038/s41568-019-0156-2
374. Milanovic M, Yu Y, Schmitt CA. The senescence-stemness alliance—a cancer-hijacked regeneration principle. Trends Cell Biol. (2018) 28:1049–61. doi: 10.1016/j.tcb.2018.09.001
375. Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. (2018) 553:96–100. doi: 10.1038/nature25167
376. Dou Z, Berger SL. Senescence elicits stemness: a surprising mechanism for cancer relapse. Cell Metab. (2018) 27:710–1. doi: 10.1016/j.cmet.2018.03.009
377. Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. (2011) 286:36396–403. doi: 10.1074/jbc.M111.257071
378. Ferrand M, Kirsh O, Griveau A, Vindrieux D, Martin N, Defossez PA, et al. Screening of a kinase library reveals novel pro-senescence kinases and their common NF-kappaB-dependent transcriptional program. Aging (Albany NY). (2015) 7:986–1003. doi: 10.18632/aging.100845
379. Saleh T, Tyutynuk-Massey L, Cudjoe EK Jr., Idowu MO, Landry JW, Gewirtz DA. Non-Cell autonomous effects of the senescence-associated secretory phenotype in cancer therapy. Front Oncol. (2018) 8:164. doi: 10.3389/fonc.2018.00164
380. Saleh T, Tyutyunyk-Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, et al. Tumor cell escape from therapy-induced senescence. Biochem Pharmacol. (2019) 162:202–12. doi: 10.1016/j.bcp.2018.12.013
381. Salminen A, Kaarniranta K, Kauppinen A. Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci. (2019) 76:1901–18. doi: 10.1007/s00018-019-03048-x
382. Munoz DP, Yannone SM, Daemen A, Sun Y, Vakar-Lopez F, Kawahara M, et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight. (2019) 5:e124716. doi: 10.1172/jci.insight.124716
383. Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. (2012) 23:2240–52. doi: 10.1091/mbc.e11-11-0926
384. Gong YN, Crawford JC, Heckmann BL, Green DR. To the edge of cell death and back. FEBS J. (2019) 286:430–40. doi: 10.1111/febs.14714
385. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
386. Tang J, Lobatto ME, Hassing L, van der Staay S, van Rijs SM, Calcagno C, et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. (2015) 1:e1400223. doi: 10.1126/sciadv.1400223
387. Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. (2006) 18:753–60. doi: 10.1016/j.cellsig.2005.11.003
388. Drew BA, Burow ME, Beckman BS. MEK5/ERK5 Pathway: the first fifteen years. Biochim Biophys Acta. (2012) 1825:37–48. doi: 10.1016/j.bbcan.2011.10.002
389. Lochhead PA, Gilley R, Cook SJ. ERK5 And its role in tumour development. Biochem Soc Trans. (2012) 40:251–6. doi: 10.1042/BST20110663
390. Nithianandarajah-Jones GN, Wilm B, Goldring CE, Muller J, Cross MJ. ERK5: structure, regulation and function. Cell Signal. (2012) 24:2187–96. doi: 10.1016/j.cellsig.2012.07.007
391. Le NT, Takei Y, Izawa-Ishizawa Y, Heo KS, Lee H, Smrcka AV, et al. Identification of activators of ERK5 transcriptional activity by high-throughput screening and the role of endothelial ERK5 in vasoprotective effects induced by statins and antimalarial agents. J Immunol. (2014) 193:3803–15. doi: 10.4049/jimmunol.1400571
392. Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, et al. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation. (2013) 127:486–99. doi: 10.1161/CIRCULATIONAHA.112.116988
393. Cook SJ, Tucker JA, Lochhead PA. Small molecule ERK5 kinase inhibitors paradoxically activate ERK5 signalling: be careful what you wish for. Biochem Soc Trans. (2020) 48:1859–75. doi: 10.1042/BST20190338
394. Abe JI, Imanishi M, Li S, Zhang A, Ko KA, Samanthapudi VSK, et al. An ERK5-NRF2 axis mediates senescence-associated stemness and atherosclerosis. Circ Res. (2023) 133:25–44. doi: 10.1161/CIRCRESAHA.122.322017
395. Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A. (1988) 85:6622–6. doi: 10.1073/pnas.85.18.6622
396. de Lange T. How telomeres solve the end-protection problem. Science. (2009) 326:948–52. doi: 10.1126/science.1170633
397. Rossiello F, Jurk D, Passos JF, d'Adda di Fagagna F. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. (2022) 24:135–47. doi: 10.1038/s41556-022-00842-x
398. Nakamura AJ, Chiang YJ, Hathcock KS, Horikawa I, Sedelnikova OA, Hodes RJ, et al. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin. (2008) 1:6. doi: 10.1186/1756-8935-1-6
399. Fairlie J, Harrington L. Enforced telomere elongation increases the sensitivity of human tumour cells to ionizing radiation. DNA Repair (Amst). (2015) 25:54–9. doi: 10.1016/j.dnarep.2014.11.005
400. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
401. Li Y, Zhou G, Bruno IG, Zhang N, Sho S, Tedone E, et al. Transient introduction of human telomerase mRNA improves hallmarks of progeria cells. Aging Cell. (2019) 18:e12979. doi: 10.1111/acel.12979
402. Li Y, Zhou G, Bruno IG, Cooke JP. Telomerase mRNA reverses senescence in progeria cells. J Am Coll Cardiol. (2017) 70:804–5. doi: 10.1016/j.jacc.2017.06.017
403. Mojiri A, Walther BK, Jiang C, Matrone G, Holgate R, Xu Q, et al. Telomerase therapy reverses vascular senescence and extends lifespan in progeria mice. Eur Heart J. (2021) 42:4352–69. doi: 10.1093/eurheartj/ehab547
404. Sishc BJ, Nelson CB, McKenna MJ, Battaglia CL, Herndon A, Idate R, et al. Telomeres and telomerase in the radiation response: implications for instability, reprograming, and carcinogenesis. Front Oncol. (2015) 5:257. doi: 10.3389/fonc.2015.00257
405. Smith S. Telomerase can't handle the stress. Genes Dev. (2018) 32:597–9. doi: 10.1101/gad.316042.118
406. Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P. Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. Br Med J. (2014) 349:g4227. doi: 10.1136/bmj.g4227
407. Aviv A. Genetics of leukocyte telomere length and its role in atherosclerosis. Mutat Res. (2012) 730:68–74. doi: 10.1016/j.mrfmmm.2011.05.001
408. Aviv A, Levy D. Telomeres, atherosclerosis, and the hemothelium: the longer view. Annu Rev Med. (2012) 63:293–301. doi: 10.1146/annurev-med-050311-104846
409. Lai TP, Zhang N, Noh J, Mender I, Tedone E, Huang E, et al. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat Commun. (2017) 8:1356. doi: 10.1038/s41467-017-01291-z
410. Kimura M, Stone RC, Hunt SC, Skurnick J, Lu X, Cao X, et al. Measurement of telomere length by the southern blot analysis of terminal restriction fragment lengths. Nat Protoc. (2010) 5:1596–607. doi: 10.1038/nprot.2010.124
411. Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. (2009) 10:478–87. doi: 10.1038/nrm2718
412. Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. (2002) 30:e47. doi: 10.1093/nar/30.10.e47
413. Montpetit AJ, Alhareeri AA, Montpetit M, Starkweather AR, Elmore LW, Filler K, et al. Telomere length: a review of methods for measurement. Nurs Res. (2014) 63:289–99. doi: 10.1097/NNR.0000000000000037
414. Nussey DH, Baird D, Barrett E, Boner W, Fairlie J, Gemmell N, et al. Measuring telomere length and telomere dynamics in evolutionary biology and ecology. Methods Ecol Evol. (2014) 5:299–310. doi: 10.1111/2041-210X.12161
415. Vera E, Blasco MA. Beyond average: potential for measurement of short telomeres. Aging (Albany NY). (2012) 4:379–92. doi: 10.18632/aging.100462
416. Baird DM, Rowson J, Wynford-Thomas D, Kipling D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat Genet. (2003) 33:203–7. doi: 10.1038/ng1084
417. Fleming AM, Burrows CJ. G-quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem Res Toxicol. (2013) 26:593–607. doi: 10.1021/tx400028y
418. Gioia U, Francia S, Cabrini M, Brambillasca S, Michelini F, Jones-Weinert CW, et al. Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Sci Rep. (2019) 9:6460. doi: 10.1038/s41598-019-42892-6
419. Schumann S, Scherthan H, Pfestroff K, Schoof S, Pfestroff A, Hartrampf P, et al. DNA damage and repair in peripheral blood mononuclear cells after internal ex vivo irradiation of patient blood with (131)I. Eur J Nucl Med Mol Imaging. (2021) 49:1447–55. doi: 10.1007/s00259-021-05605-8
420. Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol. (2012) 14:355–65. doi: 10.1038/ncb2466
421. Benetos A, Toupance S, Gautier S, Labat C, Kimura M, Rossi PM, et al. Short leukocyte telomere length precedes clinical expression of atherosclerosis: the blood-and-muscle model. Circ Res. (2018) 122:616–23. doi: 10.1161/CIRCRESAHA.117.311751
422. Sarthy J, Bae NS, Scrafford J, Baumann P. Human RAP1 inhibits non-homologous end joining at telomeres. EMBO J. (2009) 28:3390–9. doi: 10.1038/emboj.2009.275
423. Bae NS, Baumann P. A RAP1/TRF2 complex inhibits nonhomologous end-joining at human telomeric DNA ends. Mol Cell. (2007) 26:323–34. doi: 10.1016/j.molcel.2007.03.023
424. Fumagalli M, Rossiello F, Mondello C, d'Adda di Fagagna F. Stable cellular senescence is associated with persistent DDR activation. PLoS One. (2014) 9:e110969. doi: 10.1371/journal.pone.0110969
425. de Lange T. Shelterin-Mediated telomere protection. Annu Rev Genet. (2018) 52:223–47. doi: 10.1146/annurev-genet-032918-021921
426. Fairall L, Chapman L, Moss H, de Lange T, Rhodes D. Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol Cell. (2001) 8:351–61. doi: 10.1016/S1097-2765(01)00321-5
427. Martinez P, Blasco MA. Role of shelterin in cancer and aging. Aging Cell. (2010) 9:653–66. doi: 10.1111/j.1474-9726.2010.00596.x
428. Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. (2012) 336:593–7. doi: 10.1126/science.1218498
429. Mir SM, Samavarchi Tehrani S, Goodarzi G, Jamalpoor Z, Asadi J, Khelghati N, et al. Shelterin complex at telomeres: implications in ageing. Clin Interv Aging. (2020) 15:827–39. doi: 10.2147/CIA.S256425
430. Uryga A, Gray K, Bennett M. DNA damage and repair in vascular disease. Annu Rev Physiol. (2016) 78:45–66. doi: 10.1146/annurev-physiol-021115-105127
431. Liu Y, Bloom SI, Donato AJ. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation. (2019) 26:e12487. doi: 10.1111/micc.12487
432. Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. (2011) 11:161–76. doi: 10.1038/nrc3025
433. Richter T, Saretzki G, Nelson G, Melcher M, Olijslagers S, von Zglinicki T. TRF2 overexpression diminishes repair of telomeric single-strand breaks and accelerates telomere shortening in human fibroblasts. Mech Ageing Dev. (2007) 128:340–5. doi: 10.1016/j.mad.2007.02.003
434. Voghel G, Thorin-Trescases N, Farhat N, Nguyen A, Villeneuve L, Mamarbachi AM, et al. Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech Ageing Dev. (2007) 128:662–71. doi: 10.1016/j.mad.2007.09.006
435. Rothkamm K, Barnard S, Moquet J, Ellender M, Rana Z, Burdak-Rothkamm S. DNA Damage foci: meaning and significance. Environ Mol Mutagen. (2015) 56:491–504. doi: 10.1002/em.21944
436. Hohensinner PJ, Kaun C, Buchberger E, Ebenbauer B, Demyanets S, Huk I, et al. Age intrinsic loss of telomere protection via TRF1 reduction in endothelial cells. Biochim Biophys Acta. (2016) 1863:360–7. doi: 10.1016/j.bbamcr.2015.11.034
437. Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. (2015) 132:1909–19. doi: 10.1161/CIRCULATIONAHA.115.016457
438. Rai R, Chen Y, Lei M, Chang S. TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat Commun. (2016) 7:10881. doi: 10.1038/ncomms10881
439. Janouskova E, Necasova I, Pavlouskova J, Zimmermann M, Hluchy M, Marini V, et al. Human Rap1 modulates TRF2 attraction to telomeric DNA. Nucleic Acids Res. (2015) 43:2691–700. doi: 10.1093/nar/gkv097
440. Sfeir A, Kabir S, van Overbeek M, Celli GB, de Lange T. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science. (2010) 327:1657–61. doi: 10.1126/science.1185100
441. Collins C, Tzima E. Hemodynamic forces in endothelial dysfunction and vascular aging. Exp Gerontol. (2011) 46:185–8. doi: 10.1016/j.exger.2010.09.010
442. Mortuza R, Chen S, Feng B, Sen S, Chakrabarti S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS One. (2013) 8:e54514. doi: 10.1371/journal.pone.0054514
443. Kotla S, Vu HT, Ko KA, Wang Y, Imanishi M, Heo KS, et al. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2-interacting protein. JCI Insight. (2019) 4:e124867. doi: 10.1172/jci.insight.124867
444. Shoubridge EA. Nuclear genetic defects of oxidative phosphorylation. Hum Mol Genet. (2001) 10:2277–84. doi: 10.1093/hmg/10.20.2277
445. Qian W, Kumar N, Roginskaya V, Fouquerel E, Opresko PL, Shiva S, et al. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc Natl Acad Sci U S A. (2019) 116:18435–44. doi: 10.1073/pnas.1910574116
446. Pacher P, Szabo C. Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: the therapeutic potential of PARP inhibitors. Cardiovasc Drug Rev. (2007) 25:235–60. doi: 10.1111/j.1527-3466.2007.00018.x
447. Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. (2011) 108:3406–11. doi: 10.1073/pnas.1013715108
448. Singh MV, Kotla S, Le NT, Ae Ko K, Heo KS, Wang Y, et al. Senescent phenotype induced by p90RSK-NRF2 signaling sensitizes monocytes and macrophages to oxidative stress in HIV-positive individuals. Circulation. (2019) 139:1199–216. doi: 10.1161/CIRCULATIONAHA.118.036232
449. Leon KE, Buj R, Lesko E, Dahl ES, Chen CW, Tangudu NK, et al. DOT1l modulates the senescence-associated secretory phenotype through epigenetic regulation of IL1A. J Cell Biol. (2021) 220:e202008101. doi: 10.1083/jcb.202008101
450. Zhu W, Zhang X, Yu M, Lin B, Yu C. Radiation-induced liver injury and hepatocyte senescence. Cell Death Discov. (2021) 7:244. doi: 10.1038/s41420-021-00634-6
451. Nacarelli T, Liu P, Zhang R. Epigenetic basis of cellular senescence and its implications in aging. Genes (Basel). (2017) 8:343. doi: 10.3390/genes8120343
452. Falchetti ML, Mongiardi MP, Fiorenzo P, Petrucci G, Pierconti F, D'Agnano I, et al. Inhibition of telomerase in the endothelial cells disrupts tumor angiogenesis in glioblastoma xenografts. Int J Cancer. (2008) 122:1236–42. doi: 10.1002/ijc.23193
453. Zaccagnini G, Gaetano C, Della Pietra L, Nanni S, Grasselli A, Mangoni A, et al. Telomerase mediates vascular endothelial growth factor-dependent responsiveness in a rat model of hind limb ischemia. J Biol Chem. (2005) 280:14790–8. doi: 10.1074/jbc.M414644200
Keywords: premature senescence, cardio-oncology, DNA damage, telomere dysfunction, mitochondrial dysfunction, fission and fusion, autophagy, NAD+
Citation: Jain A, Casanova D, Padilla AV, Paniagua Bojorges A, Kotla S, Ko KA, Samanthapudi VSK, Chau K, Nguyen MTH, Wen J, Hernandez Gonzalez SL, Rodgers SP, Olmsted-Davis EA, Hamilton DJ, Reyes-Gibby C, Yeung S-CJ, Cooke JP, Herrmann J, Chini EN, Xu X, Yusuf SW, Yoshimoto M, Lorenzi PL, Hobbs B, Krishnan S, Koutroumpakis E, Palaskas NL, Wang G, Deswal A, Lin SH, Abe J-i and Le N-T (2023) Premature senescence and cardiovascular disease following cancer treatments: mechanistic insights. Front. Cardiovasc. Med. 10:1212174. doi: 10.3389/fcvm.2023.1212174
Received: 25 April 2023; Accepted: 3 August 2023;
Published: 14 September 2023.
Edited by:
Goo Taeg Oh, Ewha Womans University, Republic of KoreaReviewed by:
Gavin Richardson, Newcastle University, United Kingdom© 2023 Jain, Casanova, Padilla, Paniagua Bojorges, Kotla, Ko, Samanthapudi, Chau, Nguyen, Wen, Hernandez Gonzalez, Rodgers, Olmsted-Davis, Hamilton, Reyes-Gibby, Yeung, Cooke, Herrmann, Chini, Xu, Yusuf, Yoshimoto, Lorenzi, Hobbs, Krishnan, Koutroumpakis, Palaskas, Wang, Deswal, Lin, Abe and Le. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nhat-Tu Le TmhsZUBob3VzdG9ubWV0aG9kaXN0Lm9yZw== Jun-ichi Abe amFiZUBtZGFuZGVyc29uLm9yZw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.