 Zeyu Zhao
Zeyu Zhao Xuemei Cui2
Xuemei Cui2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 21 June 2023
Sec. Cardiovascular Metabolism
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1202730
This article is part of the Research Topic Examining Genetic and Epigenetic Regulation in Cardiovascular Development, Regeneration and Disease View all 10 articles
Cardiac remodeling is a basic pathological process that enables the progression of multiple cardiac diseases to heart failure. Fibroblast growth factor 21 is considered a regulator in maintaining energy homeostasis and shows a positive role in preventing damage caused by cardiac diseases. This review mainly summarizes the effects and related mechanisms of fibroblast growth factor 21 on pathological processes associated with cardiac remodeling, based on a variety of cells of myocardial tissue. The possibility of Fibroblast growth factor 21 as a promising treatment for the cardiac remodeling process will also be discussed.
Cardiac remodeling refers to a series of changes in the heart that lead to increased ventricular dilation, cardiac dysfunction, and molecular changes which can be caused by a diminished variety of factors (1), such as myocardial infarction (MI), hypertension, obesity, and valvular heart diseases (2). Numerous kinds of cells, including endothelial cells, immune cells, and fibroblasts, are involved in the remodeling process, which aims to cope with risk factors, reduce myocardial damage and initiate repair processes to fill the damaged area (3). However, cardiac remodeling can also lead to diminishing cardiac contractility and a restricted supply of energy substrates and oxygen, resulting in elevated levels of cardiac fibrosis and hypertrophy (4, 5). Although cardiac remodeling is initially considered an adaptation to restore cardiac function after damage. The unlimited pathological change results in cardiomyocyte disarrangement and dysfunction (6), eventually leading to heart failure (HF), the outcome of cardiac adverse events (7). Accordingly, the use of interventions in the early stages to preserve the structure and cardiac function is necessary to delay the progression and improve patients' quality of life (8).
One potential intervention for cardiac remodeling is the use of fibroblast growth factor 21 (FGF21). FGF21 is initially identified in the liver and subsequently is also found to be expressed in brown adipose tissue, skeletal muscle, and pancreas, where it shows its effects mainly through a paracrine manner (9). While the heart is traditionally viewed as an effector organ of FGF21, studies have also shown that it can produce FGF21 in response to damage. By binding to the receptor fibroblast growth factor receptor (FGFR)1 and cofactors β-klotho, FGF21 activated the downstream genes to play a crucial role in cardioprotection (10). However, a recent study has pointed out that FGF21-FGFR4 signaling promoted cardiac hypertrophy (11).
After damage occurs, cardiomyocytes secrete FGF21 in an autocrine manner and reduced myocardial injury (12). Besides, FGF21 originating from other tissues has been shown to contribute to the limitation of cardiac damage and the reversal of cardiac remodeling. Following MI, the liver played a significant role as a major source of circulating FGF21 levels (13). Additionally, FGF21 derived from brown adipose tissue was found to attenuate adverse cardiac remodeling in mice with hypertension (14). Studies have shown that FGF21 resists cardiac remodeling and the following damage through ways such as promoting autophagy, alleviating inflammation, oxidative stress, and regulating energy metabolism (12, 15–17).
FGF21 has been shown to play a protective role in various cardiovascular diseases including MI, atherosclerosis, and diabetic cardiomyopathy in different studies (18–20), and FGF21 knockout mice tend to present pathological phenotypes (21). Moreover, FGF21 levels predict the prognosis of many heart diseases such as hypertension, dilated cardiomyopathy, and MI (22–24).
Recent studies report that FGF21 is involved in the prevention of cardiac damage. The review examines the mechanisms of FGF21 against cardiac remodeling, providing potential survival benefits through a range of pathways including autophagy, metabolic stability, oxidation, inflammation, and fibrosis. The mechanism by which FGF21 exerts protective effects on the heart are listed in Figure 1. Additionally, this review discusses the potential implications for clinical research on FGF21-related drugs.
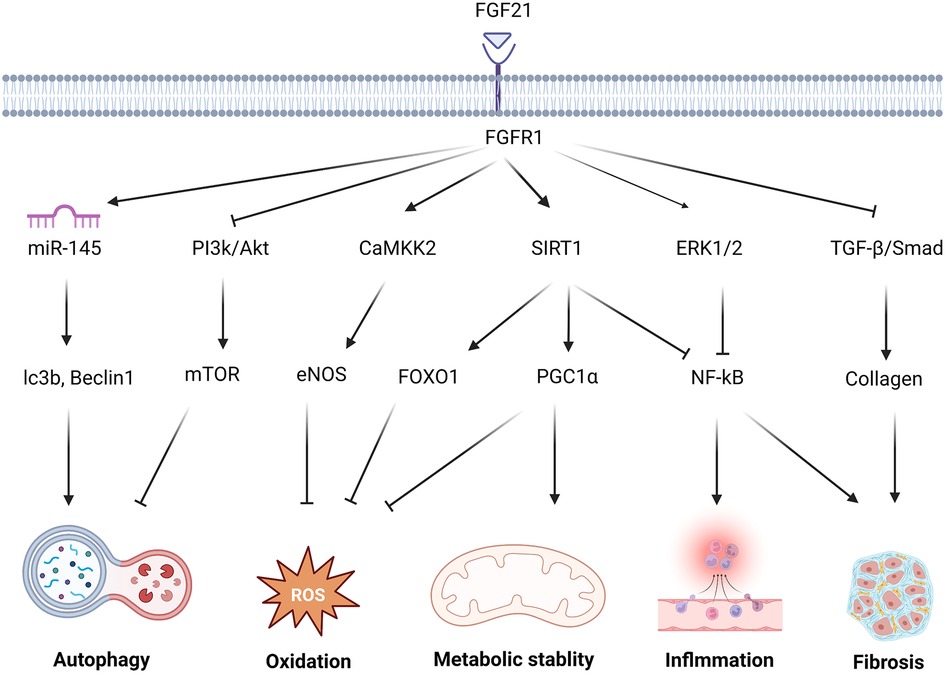
Figure 1. The molecular mechanism and effect of FGF21 in protecting against cardiac remodeling. FGF21 regulates multiple signaling pathways to protect against cardiac remodeling. FGF21 inhibited PI3K/Akt/mTOR pathway and upgraded the miR-145 level to enhance the autophagy level. FGF21 promotes CAMKK2-eNOS to elevate the vasomotor capacity and alleviate oxidative stress injury. via activation of FOXO1 and PGC1 α, SIRT1 results in multiple antioxidant proteins and transcription factors associated with oxidative stress levels. NF-kB, as a key transcription factor related to inflammation, under the action of FGF21, is inhibited by SIRT1, ERK1/2. FGF21 attenuates this adverse alteration. Reducing TGF-β/Smad signaling also helps avoid the secretion of large amounts of collagen by fibroblasts which may cause collagen deposition and fibrosis.
Cellular autophagy is a process that makes cellular energy reuse by degrading autophagic vesicles containing cytoplasmic material. It is a positive implication for cardiomyocytes in resisting stress (25, 26). Recycling misfolded proteins back as energy substrates reduces the demand of cells for external nutrients, removing mitochondria residuals, toxic metabolic substrates, and reactive oxygen species (ROS) in mitochondrial dysfunction (27).
The activation of autophagy helps to maintain cardiac homeostasis and prevent myocardium hypertrophy. The energy provided by autophagy supports cell survival, helping to maintain the number of cardiomyocytes and preserve cardiac function after stress (28). A study has reported that after MI, autophagy limited cardiac damage by reducing the scar size and holding cardiac function (29). The inhibition of autophagy would lead to pronounced signs of cellular hypertrophy in the myocardium (28). Angiogenesis is beneficial for improving postinfarction collateral perfusion, reducing infarct size, and attenuating the negative effect on contractility of the infarcted area, and autophagy plays a role in initiating angiogenesis (30).
Both mTORC1 and Beclin1 are autophagy-associated molecules, playing key roles in cardiac plasticity (31). As a serine/threonine kinase, mTORC1 acts to inhibit autophagy activity at various levels. By inhibiting the transport of nuclear transcription factor EB(TFEB), mTORC1 reduced the transcription of autophagy related (Atg) genes (32). The phosphorylation of DCP2 induced by mTORC1 made specific Atg mRNA degradation (33). The autophagosome formation discontinued after the phosphorylation of unc-51-like autophagy activating kinase 1 (Ulk1) at Ser757 (34). Beclin1 is another well-known protein in autophagy initiating, regarded as a marker of autophagy activity. By protein interaction, Beclin1 was one of the vital components of the Class III phosphatidylinositol 3-kinase (PI3K)-III complex, which acted as a platform to recruit multiple proteins to form mature autophagosomes (35).
FGF21 in cellular autophagy can be beneficial for attenuating cardiomyocyte damage after stress. As a peroxisome-proliferator-activated receptor agonist, the protective effect of Fenofibrate in the diabetes-induced cardiac remodeling was depended on FGF21- Sirtuin-1 (SIRT1) activated autophagy (17). The lack of FGF21 led to the impairment of autophagy and accumulation of fatty acid, making high-fat-diet mice cardiomyocytes hypertrophy (36).
FGF21 regulated autophagy through different pathways. By increasing TFEB transcriptional activity, FGF21 administration enhanced the autophagy effect and relieved the ischemia-reperfusion (I/R) injury in vascular endothelial cells (37). FGF21 protected against I/R injury, enhancing cell autophagy and reducing inflammation through regulating miR-145, with the upregulation of lc3b (lc3b I/II) and Beclin1 (38). Through KEGG pathway analysis, the PI3K-Akt-mTOR pathway was thought to be associated with FGF21. FGF21 promoted autophagy by inhibiting PI3K-Akt-mTOR transduction (39). Another study proved that FGF21 inhibited atherogenesis by up-regulating autophagy, enabling cholesterol efflux and reducing lipid accumulation in foam cells, this effect was related to the activated rack-1 pathway (18).
The alteration in energy supply is closely associated with pathological cardiac hypertrophy. They can even precede the appearance of a detectable cardiac hypertrophic result. The shift of substrate metabolism was viewed as an important feature of cardiac hypertrophy (40). Physiologically, cardiac energy was supplied by fatty acids, and approximately 70% ATP was provided by lipid substrates (41). However, cardiac tissue still preserved the ability to use various types of energy substrates including ketone bodies under different conditions, which was known as metabolic flexibility (42). Since deviating in metabolic pathways simultaneously led to cardiac hypofunction and maladaptive hypertrophy, the inhibition of related pathways might restore regular metabolic capacity while attenuating cardiac remodeling.
Cardiac diseases can exhibit diverse metabolic preference profiles. Heart failure relied on glycolysis and lactate for energy, whereas diabetes-induced cardiomyopathy was typically characterized by a dominance of lipid metabolism (43–45). For late-stage patients, the instability of the energy supply directly affected contractile function, and the limited metabolic pathways further interfered with ATP generation, ultimately causing HF (40, 46). Based on a proteomic study, FGF21 was found to restore the levels of pyruvate kinase isozymes M1/M2, which improved the energy supply after I/R injury (47).
FGF21 acted on upstream signals to regulate AMPK(Adenosine 5′-monophosphate -activated protein kinase)/SIRT1/PGC(peroxisome proliferators-activated receptor γ coactivator)-1α pathways. AMPK/SIRT1/PGC-1a was considered a target pathway involved in the regulation of metabolic stability, with its effects including the coordination of ATP levels, modulation of mitochondria function, and lipid metabolism (48–51). CD36 was a fatty acid translocase that can activate the AMPK pathway to accelerate lipid metabolism under fatty acid mediation (52). In FGF21 knockout mice's hearts, there was severe lipid accumulation and CD36 levels upregulation, accompanied by a decrease in phosphorylation levels of AMPK protein and PGC-1α expression, showing that CD36 cannot activate AMPK/SIRT1/PGC-1α pathway without FGF21 presence and thus led to metabolic dysregulation (16, 53). LKB1 was essential for AMPK activation. The inhibition of LKB1 weakened FGF21 promoting effect of mitochondrial function, demonstrating that the benefits of FGF21 promoting mitochondrial synthesis and mitochondrial oxidative capacity required the AMPK/SIRT1/PGC-1α pathways (54).
A study using angiopoietin 2-induced cardiac hypertrophy mice noted that FGF21 treatment increased SIRT1 deacetylation activity, promoted AMPK phosphorylation, and reduced cardiac hypertrophy in a SIRT1-dependent pathway (55). In brown adipose tissue, upon adenosine binding to the surface receptor A2AR, released FGF21 can be upregulated by phosphorylating AMPK and PGC-1 α against cardiac hypertrophy (14). In MI model mice with the activated AMPK/SIRT1/PGC-1α pathway, circulating FGF21 levels were greatly higher even at the early onset and lasted up to a week (56). The ability of the FGF21-AMPK pathway to rapidly respond to early ischemic injury suggested that it may serve as a key to reducing MI injury.
ROS-mediated oxidative stress injury contributed to the progression of cardiac remodeling and HF. The balance between various oxidant and antioxidant enzymes, including catalases, glutathione peroxidases, xanthine oxidases, and NADPH oxidase (NOX), generally maintains ROS levels at low levels. In situations of overloading or insufficient blood supply, mitochondrial dysfunction made hearts generate insufficient ATP, increased oxidative stress (57), overproduction of hydrogen peroxide, which ultimately led to cardiomyocyte necrosis, left ventricular dysfunction, and eventually HF (58). The experiment proved that cardiomyocyte apoptosis induced by oxidative stress was one of the reasons for myocardial decompensation (59).
ROS promoted the imbalanced growth of cardiomyocytes through multiple pathways, ultimately causing cardiac hypertrophy and remodeling (60). By directly activating the transcription factor NF-kB, ROS induced the transcription of more pro-hypertrophic genes (61). NADPH oxidase was a major source of superoxide in the cardiovascular system (62). The reaction between excessive superoxide anion and Nitric Oxide (NO) formed the peroxynitrite (ONOO-), decreasing NO bioavailability, causing vasoconstriction, and leading to the early formation of atherosclerotic plaques (63). Animal studies have shown a positive relationship between increased levels of oxidative stress and disease progression in multiple animal models of HF (64–66). Similarly, FGF21 levels were also specifically elevated in patients with poor prognosis HF (67), a coincidence that has led to thoughts about a possible relationship between FGF21 and the effects of oxidative stress.
In cardiomyocytes, FGF21 production was regulated by ROS level in an autocrine manner. High levels of ROS, which were stimulated by calcium ions released from endoplasmic reticulum, triggered a signal transduction pathway known as the unfolding protein response (UPR) which activated several transmembrane sensors, ultimately resulting in elevated FGF21 expression levels (68).
FGF21 treatment improved resistance to oxidative stress by elevating the expression levels of oxidative enzymes such as superoxide dismutase (SOD) and glutathione (69). It also activates nitric oxide synthase (eNOS) to increase NO production and reduce oxidation, guaranteeing the normal contractile function of blood vessels and reducing the adverse effects of peroxides on cells (70).
Studies have shown that FGF21 induced the expression of antioxidant genes. Through the Sirt1-FOXO1 pathway, FGF21 enhanced catalases, SOD2 levels, and the pro-apoptotic protein BIM lessened (55). In the absence of FGF21, SIRT1 effects that induced the levels of uncoupling protein (UCP)3, peroxiredoxin5, glutathione peroxidase1, catalase, and sequestosome1 were abolished (71).
Another study illustrated that recombinant FGF21 activated CaMKK2/AMPKα signaling, enhancing eNOS phosphorylation and reducing oxidative stress responses to ameliorate diabetes-induced aortic endothelial disorders (72).
Via AMPK/PGC1α/SIRT1 signaling, FGF21 led to a rise of nuclear factor erythroid 2–related factor 2 (Nrf2) and downstream antioxidative enzymes activity such as NOX4, UCP2 (73).
For atrial remodeling induced by oxidative stress, FGF21 functioned by regulating the degradation of myofibrils and levels of calpain, a calcium-dependent protease, which helped to maintain cardiac function, improved arrhythmia symptoms, and preserved electrophysiological function (74).
The main reason for acute inflammatory responses is cardiomyocyte necrosis in response to various adverse factors. After damage happened, innate immune cells localized in the heart were activated, recognizing endogenous damage-associated molecular patterns, clearing necrotic cells, and releasing proinflammatory factors. Circulating immune cells were then recruited, further infiltrating the damaged area (75). This early inflammatory response was necessary to initiate the repair process, protecting the heart from further damage. However, an uncontrolled chronic inflammatory response can further aggravate cardiac adaption (76).
Chronic inflammation was one of the most important stages in the progression process of cardiac remodeling. Patients with HF have shown significantly elevated levels of proinflammatory cells and chemokines. Inhibition a series of pro-inflammatory chemokines including IL-1 and TNF-α was beneficial for attenuating adverse cardiac remodeling, maintaining cardiac function, and reducing fibrotic area (77, 78).
Proinflammatory cell activation potentiated inflammatory responses through different pathways, and their phenotypic heterogeneity dictated that they played distinct roles in cardiac inflammation.
Despite comprising only 6%–8% of noncardiomyocytes in normal cardiac tissue, cardiac macrophages massively migrated and proliferated in the remote area during the chronic healing process of MI to mediate the onset of distant myocardial remodeling, and high levels of blood-derived macrophages had a strong link to heart failure with reduced ejection fraction (HFrEF), whereas heart confined macrophages might be more involved in post-MI repair (79).
Plasma FGF21 levels were significantly elevated in patients with HFrEF, regardless of the presence of cardiac cachexia. This elevation was independently associated with the inflammatory marker IL-6, suggesting that the inflammatory response was a contributing factor to elevated FGF21 levels (80).
Among peripheral blood leukocytes, FGF21 was highly expressed in monocytes and neutrophils (81). By increasing the expression of GLUT-1, FGF21 upregulated the ability to uptake glucose in activated monocytes, which can be beneficial in immune activation (82). FGF21 played a crucial role in aiding in clearing damaged cell remnants in phagosomes, resulting from the upregulation of NOX2 transcription and expression (81).
In addition to reducing infiltration of circulating immune cells, FGF21 shifted innate macrophages towards the anti-inflammatory M2 subtype rather than pro-inflammatory (83). FGF21 negatively regulated the early formation of atherosclerotic plaques by preventing the transformation of macrophages into foam cells and reducing macrophage migration by inhibiting the NF-kB signaling pathway (84).
CD4 + Th17 cells were thought to be directly involved in the progression of adverse remodeling from hypertrophy to HF in the situation of pressure overload (85). FGF21 regulated STAT3/RORγ phosphorylation and pro-T-cell expansion IL-23 levels, reducing splenic Th17 cell differentiation and proliferation, and ultimately downregulating the levels of pro-inflammatory factor IL-17, thereby alleviating arthritis in mice (86). This suggests that FGF21 can induce inflammation by downregulating T cells through the regulation of the Th17-IL-17 axis.
Multiple studies have demonstrated hemagglutination propensity, platelet activation, and levels of associated adhesion molecules all increase in HF patients (87). After activation and adhesion to the capillary wall, platelets secreted a variety of pro-inflammatory factors, promoted macrophage infiltration, and regulated precursor cell to foam cell transformation, ultimately leading to small vessel endothelial thickening and sclerotic plaque formation of large vessels. Agglutination of platelets following an acute cardiac event further enhanced microvascular obstruction and potentiated the inflammatory response (88).
To reduce reperfusion injury after MI, antiplatelet and antithrombotic drugs are routinely used in the clinic (89). FGF21 reduced factor VII expression which was a hallmark of the extrinsic coagulation pathway. Variations in the fluorescence intensity levels of platelet surface markers proved that FGF21 inhibited platelet activation, reducing hemagglutination. By regulating the ERK1/2 and TGF(Transforming Growth Factor)-β/Smad2 pathway, FGF21 promoted the expression of tPA and suppressed the expression of PAI, so that fibrinolysis was activated without bleeding risk (90).
FGF21 presented anti-inflammatory properties by modulating the balance between pro-inflammation and anti-inflammation factors. The administration of FGF21 led to a downregulation of several pro-inflammatory factors and increased the expression of anti-inflammatory cytokines over a long period (83). Another in vitro experiment pointed out that this kind of action of FGF21 was related to IL-10 in the ERK1/2 pathway (15). Additionally, FGF21 alleviated inflammation in vascular endothelial cells through SIRT1-mediated NF-kB deacetylation (91). Figure 2 summarizes the roles of FGF21 in different cells of myocardial tissue.
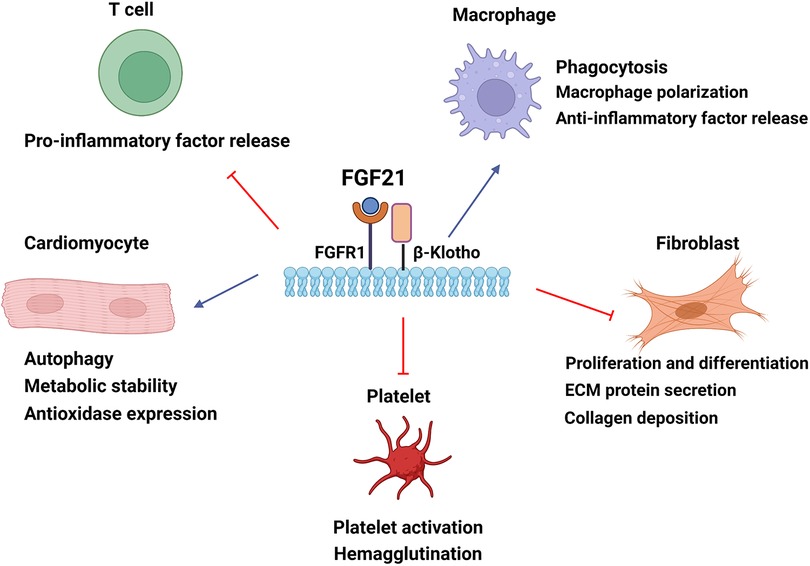
Figure 2. The cellular mechanisms by which FGF21 confers cardioprotection in cardiac tissue. FGF21 modulates cardiomyocyte energy metabolism and autophagy and improves oxidative stress damage. FGF21 can mediate macrophage polarization to the anti-inflammatory subtype and regulate cytokine levels to alleviate inflammatory responses. FGF21 upregulates phagocytosis to eliminate damaged remnants. The inhibition of fibroblast reduces collagen deposition of the extracellular matrix and ameliorates cardiac fibrosis by FGF21. FGF21 also downregulates hemagglutination by reducing platelet activation. Blue arrow, upregulation; red arrow, downregulation. ECM, extracellular matrix.
Fibrosis is considered as a crucial process in heart remodeling. After encountering pressure, fibroblasts secreted a large number of collagen fibers to the interstitium, and this excessive fiber deposition triggered the disarrangement of cardiomyocytes (92). The hypertension phenotype presented an increased wall/lumen ratio in resistance arteries, which was related to the activation and decomposition of collagens and other extracellular matrix components (ECM) components (93). After MI, fibroblast activation resulted in the deposition of collagen to form a scar, which helped to ensure the intactness of residual myocardial tissue and avoided serious consequences such as cardiac rupture (4, 94). During the post-infarction repair period, the distal area of the myocardium without infarction had massive fibrin deposition, leading to interstitial remodeling, reducing cardiac compliance, and promoting ventricular diastolic dysfunction development (95).
By using an efficient carrier, FGF21 administration attenuated cardiac fibrosis, resulting in decreased mRNA levels of cardiac hypertrophy and fibrosis markers in diabetic cardiomyopathy mice (96). FGF21 released from brown adipose tissue ameliorated cardiac fibrosis and hypertension-induced myocardial remodeling (14). In vitro experiments showed that FGF21 significantly decreased myofibroblast markers α-SMA and ACTA2 mRNA levels, suggesting that FGF21 was related to attenuating cardiac fibrosis (14, 97). The administration of the homolog of FGF21, LY2405319 in mice with liver fibrosis, reduced the levels of type 1 collagen and α-SMA in tissue, resulting from inhibiting the succinate- G-protein coupled receptor 91 pathway (98).
TGFβ is one of the key factors leading to cardiac remodeling, accelerating fibroblast proliferation, and inducing accumulation of ECM components, which led to impaired cardiac function (99). FGF21 treatment attenuated the fibrotic phenotype by upregulating the transcription factor EGR1 (100). FGF21 also decreased collagen synthesis in MI mice through TGF-β1/Smad2/3-MMP (metalloproteinase)2/9 signaling (101). FGF21 downregulated MMP9 levels through NF-kB [Z.-C (102)]. Through the FGFR1/Syk/NLRP3 inflammasome pathway, FGF21 restrained the proliferation and migration of vascular smooth muscle cells and neointimal hyperplasia, marked by increasing in PCNA, cyclin D1 and MMP9 (103).
Galectin(Gal)-3 is mainly secreted by macrophages in the heart, released into the cytoplasmic matrix in association with collagen receptor action. It can promote macrophage migration, cardiac fibroblast proliferation, and collagen deposition in the matrix, leading to reduced ejection fraction (104). FGF21 regulated Gal-3 levels in H9c2 cells in a dose-dependent manner and ECM-related proteins (fibronectin, collagen I) levels were also decreased under FGF21 treatment (105). This suggested that FGF21 may play a protective role in the heart by reducing the level of cardiac fibrosis through downregulating Gal-3 levels. However, due to the inadequacy of the available evidence, more studies are still needed to further determine the relationship between FGF21 and Gal-3.
FGF21, as a metabolically associated protective factor, has been shown to have potential clinical applications in various diseases. Elevated serum levels of FGF21 have been observed in response to stress stimuli and have been associated with a higher risk of morbidity and poor prognosis in multiple diseases. One analysis indicated that FGF21 levels were useful in predicting the severity and prognostic risk in patients with community-acquired pneumonia (106). Clinical studies have also shown that FGF21 is useful in predicting HF (107–110). Serum FGF21 levels were higher under acute insufficient sleep conditions when adipose tissue FGF21 promoter region was methylated (111).
Due to the short half-life and susceptibility to plasma proteases, FGF21 was difficult to use in clinical practice. Therefore, diverse chemical modifications have been used to increase its stability and applicability (112). Current clinical studies targeting FGF21 mainly focus on its ameliorative effects on metabolic diseases. The use of fibroblast activation protein inhibitors, BR103354, successfully elevates FGF21 levels in cynomolgus monkeys (113). Treatment of nonalcoholic fatty liver disease using bms-986036, a PEGylated FGF21 analog, resulting in a considerable decrease in liver fat and fibrosis in patients after 16 weeks of treatment, with very limited adverse effects (114).
Preclinical pharmacological studies have extensively validated the protective effects of FGF21 in damaged hearts. AMPK-FGF21 helped regulate the survival of cardiomyocytes under ischemic conditions after MI (56). FGF21 ameliorated atrial fibrillation and tachycardia by reducing excessive fibrosis (74). FGF21 has been shown to improve post-infarction arrhythmias and preserve electrophysiological function through mediating cardiac sodium current and inward rectifier potassium current (115). By improving FGFR1 phosphorylation, long-term FGF21 administration helped to alleviate high-fat-diet-induced left heart dysfunction and restored FGF21 sensitivity (116). In alcoholic cardiomyopathy, FGF21 improved adverse effects related to dysfunctional mitochondria by mediating autophagic pathways (117). However, a recent study reported that FGF21-FGFR4 signaling upregulated the activity of ERK1/2, enhancing diabetes mouse concentric cardiac hypertrophy and adverse cardiac remodeling (11). The FGF21 analog shows promise as a candidate for the diagnosis and therapy of cardiac diseases, but further investigation is needed to support its use in the future.
The review mainly expounds on the possibility that FGF21, as a promising cardioprotective factor, improves cardiac function and ultimately retarding the progress of cardiac diseases. The actions of FGF21 for the heart are extensive. On the one hand, FGF21 has a regulatory effect targeted to different populations of cardiac tissue, including cardiomyocytes, immune cells, and fibroblasts. On the other hand, by acting on AMPK/SIRT1/PGC-1α, PI3K-Akt, ERK1/2, and TGF-β/Smad2 to mediate vital mechanisms, including autophagy, energy metabolism, oxidative stress, inflammation, and fibrosis, it is verified that FGF21 acts as a possible cardiac protective molecule. Therefore, the next steps in the development of relevant studies on targeted therapy of FGF21 in cardiac remodeling will be promising directions.
ZZ conceptualized and wrote the manuscript. XC drew the figures and revised the manuscipt. ZL revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (82060657). Figures were created with Biorender.com
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Diez J, Ertl G. A translational approach to myocardial remodelling. Cardiovasc Res. (2008) 81(3):409–11. doi: 10.1093/cvr/cvn352
2. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol. (2000) 35(3):569–82. doi: 10.1016/S0735-1097(99)00630-0
3. Frantz S, Bauersachs J, Ertl G. Post-Infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res. (2008) 81(3):474–81. doi: 10.1093/cvr/cvn292
4. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. (1990) 81(4):1161–72. doi: 10.1161/01.CIR.81.4.1161
5. Miura T, Miki T. Limitation of myocardial infarct size in the clinical setting: current Status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol. (2008) 103(6):501–13. doi: 10.1007/s00395-008-0743-y
6. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. (2009) 123(2):255–78. doi: 10.1016/j.pharmthera.2009.05.002
7. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2016) 37(27):2129–200. doi: 10.1093/eurheartj/ehw128
8. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. (2008) 358(13):1370–80. doi: 10.1056/NEJMra072139
9. Zhang Y, Liu D, Long X-X, Fang Q-C, Jia W-P, Li H-T. The role of FGF21 in the pathogenesis of cardiovascular disease. Chin Med J. (2021) 134(24):2931–43. doi: 10.1097/CM9.0000000000001890
10. Lü Y, Liu J-H, Zhang L-K, Jie DU, Zeng X-J, Hao G, et al. Fibroblast growth factor 21 as a possible endogenous factor inhibits apoptosis in cardiac endothelial cells. Chin Med J. (2010) 123(23):3417–21. doi: 10.3760/cma.j.issn.0366-6999.2010.23.008
11. Yanucil C, Kentrup D, Li X, Grabner A, Schramm K, Martinez EC, et al. FGF21-FGFR4 Signaling in cardiac myocytes promotes concentric cardiac hypertrophy in mouse models of diabetes. Sci Rep. (2022) 12(1):7326. doi: 10.1038/s41598-022-11033-x
12. Planavila A, Redondo I, Hondares E, Vinciguerra M, Munts C, Iglesias R, et al. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat Commun. (2013) 4(1):2019. doi: 10.1038/ncomms3019
13. Sun J-Y, Du L-J, Shi X-R, Zhang Y-Y, Liu Y, Wang Y-L, et al. An IL-6/STAT3/MR/FGF21 axis mediates heart-liver cross-talk after myocardial infarction. Sci Adv. (2023) 9(14):eade4110. doi: 10.1126/sciadv.ade4110
14. Ruan C-C, Kong L-R, Chen X-H, Ma Y, Pan X-X, Zhang Z-B, et al. A2a receptor activation attenuates hypertensive cardiac remodeling via promoting brown adipose tissue-derived FGF21. Cell Metab. (2018) 28(3):476–89. e5. doi: 10.1016/j.cmet.2018.06.013
15. Li J-y, Wang N, Khoso MH, Shen C-b, Guo M-z, Pang X-x, et al. FGF-21 elevated IL-10 production to correct LPS-induced inflammation. Inflammation. (2018a) 41(3):751–59. doi: 10.1007/s10753-018-0729-3
16. Yan X, Chen J, Zhang C, Zhou S, Zhang Z, Chen J, et al. FGF21 deletion exacerbates diabetic cardiomyopathy by aggravating cardiac lipid accumulation. J Cell Mol Med. (2015) 19(7):1557–68. doi: 10.1111/jcmm.12530
17. Zhang J, Cheng Y, Gu J, Wang S, Zhou S, Wang Y, et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of type 1 diabetic mice. Clin Sci. (2016) 130(8):625–41. doi: 10.1042/CS20150623
18. Xiaolong L, Dongmin G, Liu M, Zuo W, Huijun H, Qiufen T, et al. FGF21 Induces autophagy-mediated cholesterol efflux to inhibit atherogenesis via RACK1 up-regulation. J Cell Mol Med. (2020) 24(9):4992–5006. doi: 10.1111/jcmm.15118
19. Xu Z, Sun J, Tong Q, Lin Q, Qian L, Park Y, et al. The role of ERK1/2 in the development of diabetic cardiomyopathy. Int J Mol Sci. (2016) 17(12):2001. doi: 10.3390/ijms17122001
20. Liu SQ, Roberts D, Kharitonenkov A, Zhang B, Hanson SM, Li YC, et al. Endocrine protection of ischemic myocardium by FGF21 from the liver and adipose tissue. Sci Rep. (2013) 3(1):2767. doi: 10.1038/srep02767
21. Pan X, Shao Y, Wu F, Wang Y, Xiong R, Zheng J, et al. FGF21 prevents angiotensin II-induced hypertension and vascular dysfunction by activation of ACE2/angiotensin-(1–7) axis in mice. Cell Metab. (2018) 27(6):1323–37. e5. doi: 10.1016/j.cmet.2018.04.002
22. Zhang W, Chu S, Ding W, Wang F. Serum level of fibroblast growth factor 21 is independently associated with acute myocardial infarction. PLOS ONE. (2015) 10(6):e0129791. doi: 10.1371/journal.pone.0129791
23. Semba RD, Crasto C, Strait J, Sun K, Schaumberg DA, Ferrucci L. Elevated serum fibroblast growth factor 21 is associated with hypertension in community-dwelling adults. J Hum Hypertens. (2013) 27(6):397–99. doi: 10.1038/jhh.2012.52
24. Gu L, Jiang W, Zheng R, Yao Y, Ma G. Fibroblast growth factor 21 correlates with the prognosis of dilated cardiomyopathy. Cardiology. (2021) 146(1):27–33. doi: 10.1159/000509239
25. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. (2018) 80(1):1–26. doi: 10.1146/annurev-physiol-021317-121427
26. Levine B, Klionsky DJ. Development by self-digestion. Dev Cell. (2004) 6(4):463–77. doi: 10.1016/S1534-5807(04)00099-1
27. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science. (2011) 333(6046):1109–12. doi: 10.1126/science.1201940
28. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. (2007a) 13(5):619–24. doi: 10.1038/nm1574
29. Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and bcl-2. Nat Med. (2013) 19(11):1478–88. doi: 10.1038/nm.3322
30. Lu Q, Yao Y, Hu Z, Hu C, Song Q, Ye J, et al. Angiogenic factor AGGF1 activates autophagy with an essential role in therapeutic angiogenesis for heart disease. PLoS Biol. (2016) 14(8):e1002529. doi: 10.1371/journal.pbio.1002529
31. Shi B, Ma M, Zheng Y, Pan Y, Lin X. MTOR and Beclin1: two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J Cell Physiol. (2019) 234(8):12562–68. doi: 10.1002/jcp.28125
32. Noda T. Regulation of autophagy through TORC1 and MTORC1. Biomolecules. (2017) 7(4):52. doi: 10.3390/biom7030052
33. Hu G, McQuiston T, Bernard A, Park Y-D, Qiu J, Vural A, et al. A conserved mechanism of TOR-dependent RCK-mediated MRNA degradation regulates autophagy. Nat Cell Biol. (2015) 17(7):930–42. doi: 10.1038/ncb3189
34. Rabanal-Ruiz Y, Otten EG, Korolchuk VI. MTORC1 as the main gateway to autophagy. Essays Biochem. (2017) 61(6):565–84. doi: 10.1042/EBC20170027
35. McKnight NC, Yue Z. Beclin 1, an essential component and master regulator of PI3K-III in health and disease. Curr Pathobiol Rep. (2013) 1(4):231–38. doi: 10.1007/s40139-013-0028-5
36. Rupérez C, Lerin C, Ferrer-Curriu G, Cairo M, Mas-Stachurska A, Sitges M, et al. Autophagic control of cardiac steatosis through FGF21 in obesity-associated cardiomyopathy. Int J Cardiol. (2018) 260:163–70. doi: 10.1016/j.ijcard.2018.02.109
37. Chen F, Zhan J, Yan X, al Mamun A, Zhang Y, Xu Y, et al. FGF21 alleviates microvascular damage following limb ischemia/reperfusion injury by TFEB-mediated autophagy enhancement and anti-oxidative response. Signal Transduct Targeted Ther. (2022) 7(1):349. doi: 10.1038/s41392-022-01172-y
38. Hu S, Cao S, Tong Z, Liu J. FGF21 Protects myocardial ischemia-reperfusion injury through reduction of MiR-145-mediated autophagy. Am J Transl Res. (2018) 10(11):3677–88. 30662618 6291727.30662618
39. Dai H, Hu W, Zhang L, Jiang F, Mao X, Yang G, et al. FGF21 Facilitates autophagy in prostate cancer cells by inhibiting the PI3K–Akt–MTOR signaling pathway. Cell Death Dis. (2021) 12(4):303. doi: 10.1038/s41419-021-03588-w
40. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. (2018) 15(8):457–70. doi: 10.1038/s41569-018-0044-6
41. Stanley W. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions potential for pharmacological interventions. Cardiovasc Res. (1997) 33(2):243–57. doi: 10.1016/S0008-6363(96)00245-3
42. Kim TT, Dyck JRB. Is AMPK the savior of the failing heart? Trends Endocrinol Metab. (2015) 26(1):40–8. doi: 10.1016/j.tem.2014.11.001
43. Rijzewijk LJ, van der Meer RW, Lamb HJ, de Jong HWAM, Lubberink M, Romijn JA, et al. Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy. J Am Coll Cardiol. (2009) 54(16):1524–32. doi: 10.1016/j.jacc.2009.04.074
44. Choi YS, de Mattos ABM, Shao D, Li T, Nabben M, Kim M, et al. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J Mol Cell Cardiol. (2016) 100:64–71. doi: 10.1016/j.yjmcc.2016.09.001
45. Kolwicz SC, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-Specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. (2012) 111(6):728–38. doi: 10.1161/CIRCRESAHA.112.268128
46. Neubauer S. The failing heart — an engine out of fuel. N Engl J Med. (2007) 356(11):1140–51. doi: 10.1056/NEJMra063052
47. Cong W-T, Ling J, Tian H-S, Ling R, Wang Y, Huang B-B, et al. Proteomic study on the protective mechanism of fibroblast growth factor 21 to ischemia–reperfusion injury. Can J Physiol Pharmacol. (2013) 91(11):973–84. doi: 10.1139/cjpp-2012-0441
48. Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK Regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. (2009) 458(7241):1056–60. doi: 10.1038/nature07813
49. Matsushima S, Sadoshima J. The role of sirtuins in cardiac disease. Am J PhysiolHeart Circ Physiol. (2015) 309(9):H1375–89. doi: 10.1152/ajpheart.00053.2015
50. Cantó C, Auwerx J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. (2009) 20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4
51. Hardie DG. AMP-Activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. (2007) 8(10):774–85. doi: 10.1038/nrm2249
52. Samovski D, Sun J, Pietka T, Gross RW, Eckel RH, Su X, et al. Regulation of AMPK activation by CD36 links fatty acid uptake to β-oxidation. Diabetes. (2015) 64(2):353–59. doi: 10.2337/db14-0582
53. Chen C, Meng Z, Zheng Y, Hu B, Shen E. Fibroblast growth factor 21 inhibition aggravates cardiac dysfunction in diabetic cardiomyopathy by improving lipid accumulation. Exp Ther Med. (2018) 15(1):75–84. doi: 10.3892/etm.2017.5375
54. Chau MDL, Gao J, Yang Q, Wu Z, Gromada J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK–SIRT1–PGC-1α pathway. Proc Natl Acad Sci USA. (2010) 107(28):12553–58. doi: 10.1073/pnas.1006962107
55. Li S, Zhu Z, Xue M, Yi X, Liang J, Niu C, et al. Fibroblast growth factor 21 protects the heart from angiotensin II-induced cardiac hypertrophy and dysfunction via SIRT1. Biochim Biophys Acta (BBA). (2019) 1865(6):1241–52. doi: 10.1016/j.bbadis.2019.01.019
56. Sunaga H, Koitabashi N, Iso T, Matsui H, Obokata M, Kawakami R, et al. Activation of cardiac AMPK-FGF21 feed-forward loop in acute myocardial infarction: role of adrenergic overdrive and lipolysis byproducts. Sci Rep. (2019) 9(1):11841. doi: 10.1038/s41598-019-48356-1
57. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko Av, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. (2018) 50(2):121–27. doi: 10.1080/07853890.2017.1417631
58. Nickel AG, von Hardenberg A, Hohl M, Löffler JR, Kohlhaas M, Becker J, et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. (2015) 22(3):472–84. doi: 10.1016/j.cmet.2015.07.008
59. Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, et al. Oxidative stress–mediated cardiac cell death is a Major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. (2001) 89(3):279–86. doi: 10.1161/hh1501.094115
60. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301(6):H2181–90. doi: 10.1152/ajpheart.00554.2011
61. Kumar S, Seqqat R, Chigurupati S, Kumar R, Baker KM, Young D, et al. Inhibition of nuclear factor ΚB regresses cardiac hypertrophy by modulating the expression of extracellular matrix and adhesion molecules. Free Radic BiolMed. (2011) 50(1):206–15. doi: 10.1016/j.freeradbiomed.2010.10.711
62. Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. (2010) 106(7):1253–64. doi: 10.1161/CIRCRESAHA.109.213116
63. Violi F, Basili S, Nigro C, Pignatelli P. Role of NADPH oxidase in atherosclerosis. Future Cardiol. (2009) 5(1):83–92. doi: 10.2217/14796678.5.1.83
64. Dai D-F, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintrón M, Chen T, et al. Mitochondrial oxidative stress mediates angiotensin II–induced cardiac hypertrophy and gαq overexpression–induced heart failure. Circ Res. (2011) 108(7):837–46. doi: 10.1161/CIRCRESAHA.110.232306
65. Dai D-F, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, et al. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res. (2012) 93(1):79–88. doi: 10.1093/cvr/cvr274
66. Mollnau H, Oelze M, August M, Wendt M, Daiber A, Schulz E, et al. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler, Thromb, Vasc Biol. (2005) 25(12):2554–59. doi: 10.1161/01.ATV.0000190673.41925.9B
67. Fan L, Gu L, Yao Y, Ma G. Elevated Serum fibroblast growth factor 21 is relevant to heart failure patients with reduced ejection fraction. Comput Math Methods Med. (2022) 2022:1–6. doi: 10.1155/2022/7138776
68. Tabari FS, Karimian A, Parsian H, Rameshknia V, Mahmoodpour A, Majidinia M, et al. The roles of FGF21 in atherosclerosis pathogenesis. Rev Endocr Metab Disord. (2019) 20(1):103–14. doi: 10.1007/s11154-019-09488-x
69. Zhang S, Yu D, Wang M, Huang T, Wu H, Zhang Y, et al. FGF21 Attenuates pulmonary fibrogenesis through ameliorating oxidative stress in vivo and in vitro. Biomed Pharmacother. (2018) 103:1516–25. doi: 10.1016/j.biopha.2018.03.100
70. Wang X-M, Song S-S, Xiao H, Gao P, Li X-J, Si L-Y. Fibroblast growth factor 21 protects against high glucose induced cellular damage and dysfunction of endothelial nitric-oxide synthase in endothelial cells. Cell Physiol Biochem. (2014) 34(3):658–71. doi: 10.1159/000363031
71. Planavila A, Redondo-Angulo I, Ribas F, Garrabou G, Casademont J, Giralt M, et al. Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc Res. (2015) 106(1):19–31. doi: 10.1093/cvr/cvu263
72. Ying L, Li N, He Z, Zeng X, Nan Y, Chen J, et al. Fibroblast growth factor 21 ameliorates diabetes-induced endothelial dysfunction in mouse aorta via activation of the CaMKK2/AMPKα signaling pathway. Cell Death Dis. (2019) 10(9):665. doi: 10.1038/s41419-019-1893-6
73. Kawakami R, Sunaga H, Iso T, Kaneko R, Koitabashi N, Obokata M, et al. Ketone body and FGF21 coordinately regulate fasting-induced oxidative stress response in the heart. Sci Rep. (2022) 12(1):7338. doi: 10.1038/s41598-022-10993-4
74. Chen M, Zhong J, Wang Z, Xu H, Chen H, Sun X, et al. Fibroblast growth factor 21 protects against atrial remodeling via reducing oxidative stress. Front Cardiovasc Med. (2021) 8:720581. doi: 10.3389/fcvm.2021.720581
75. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. (2020) 116(6):1101–12. doi: 10.1093/cvr/cvz336
76. Dick SA, Epelman S. Chronic heart failure and inflammation. Circ Res. (2016) 119(1):159–76. doi: 10.1161/CIRCRESAHA.116.308030
77. Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, et al. Tumor necrosis factor-α mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. (2007) 115(11):1398–407. doi: 10.1161/CIRCULATIONAHA.106.643585
78. Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. (2008) 173(1):57–67. doi: 10.2353/ajpath.2008.070974
79. Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res. (2016) 119(7):853–64. doi: 10.1161/CIRCRESAHA.116.309001
80. Refsgaard Holm M, Christensen H, Rasmussen J, Johansen ML, Schou M, Faber J, et al. Fibroblast growth factor 21 in patients with cardiac cachexia: a possible role of chronic inflammation. ESC Heart Fail. (2019) 6(5):983–91. doi: 10.1002/ehf2.12502
81. Wang W-f, Ma L, Liu M-y, Zhao T-t, Zhang T, Yang Y-b, et al. A novel function for fibroblast growth factor 21: stimulation of NADPH oxidase-dependent ROS generation. Endocrine. (2015) 49(2):385–95. doi: 10.1007/s12020-014-0502-9
82. Wang N, Li J-y, Li S, Guo X-c, Wu T, Wang W-f, et al. Fibroblast growth factor 21 regulates foam cells formation and inflammatory response in ox-LDL-induced THP-1 macrophages. Biomed Pharmacother. (2018a) 108:1825–34. doi: 10.1016/j.biopha.2018.09.143
83. Wang D, Liu F, Zhu L, Lin P, Han F, Wang X, et al. FGF21 Alleviates neuroinflammation following ischemic stroke by modulating the temporal and spatial dynamics of microglia/macrophages. J Neuroinflammation. (2020) 17(1):257. doi: 10.1186/s12974-020-01921-2
84. Wang N, Li J-Y, Zhao T-t, Li S-m, Shen C-B, Li D-S, et al. FGF-21 Plays a crucial role in the glucose uptake of activated monocytes. Inflammation. (2018b) 41(1):73–80. doi: 10.1007/s10753-017-0665-7
85. Gröschel C, Sasse A, Röhrborn C, Monecke S, Didié M, Elsner L, et al. T helper cells with specificity for an antigen in cardiomyocytes promote pressure overload-induced progression from hypertrophy to heart failure. Sci Rep. (2017) 7(1):15998. doi: 10.1038/s41598-017-16147-1
86. Li S-m, Yu Y-h, Li L, Wang W-f, Li D-s. Treatment of CIA mice with FGF21 down-regulates TH17-IL-17 axis. Inflammation. (2016) 39(1):309–19. doi: 10.1007/s10753-015-0251-9
87. Chung I, Lip GYH. Platelets and heart failure. Eur Heart J. (2006) 27(22):2623–31. doi: 10.1093/eurheartj/ehl305
88. Badimon L, Padró T, Vilahur G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur Heart J. (2012) 1(1):60–74. doi: 10.1177/2048872612441582
89. Bienvenu LA, Maluenda A, McFadyen JD, Searle AK, Yu E, Haller C, et al. Combined antiplatelet/anticoagulant drug for cardiac ischemia/reperfusion injury. Circ Res. (2020) 127(9):1211–13. doi: 10.1161/CIRCRESAHA.120.317450
90. Li S, Jia H, Liu Z, Wang N, Guo X, Cao M, et al. Fibroblast growth factor-21 as a novel metabolic factor for regulating thrombotic homeostasis. Sci Rep. (2022) 12(1):400. doi: 10.1038/s41598-021-00906-2
91. Zhou X, Wang X, Lu L, Deng M, Shi X. Fibroblast growth factor 21 improves lipopolysaccharide-induced pulmonary microvascular endothelial cell dysfunction and inflammatory response through SIRT1-mediated NF-ΚB deacetylation. Can J Physiol Pharmacol. (2022) 100(6):492–99. doi: 10.1139/cjpp-2021-0454
92. Volders PGA, Willems IEMG, Cleutjens JPM, Aren J-W, Havenith MG, Daemen MJAP. Interstitial collagen is increased in the non-infarcted human myocardium after myocardial infarction. J Mol Cell Cardiol. (1993) 25(11):1317–23. doi: 10.1006/jmcc.1993.1144
93. Intengan HD, Schiffrin EL. Vascular remodeling in hypertension. Hypertension. (2001) 38(3):581–87. doi: 10.1161/hy09t1.096249
94. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction. Circ Res. (2016) 119(1):91–112. doi: 10.1161/CIRCRESAHA.116.303577
95. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. (2016) 365(3):563–81. doi: 10.1007/s00441-016-2431-9
96. Gao J, Liu J, Meng Z, Li Y, Hong Y, Wang L, et al. Ultrasound-Assisted C3F8-filled PLGA nanobubbles for enhanced FGF21 delivery and improved prophylactic treatment of diabetic cardiomyopathy. Acta Biomater. (2021) 130:395–408. doi: 10.1016/j.actbio.2021.06.015
97. Ferrer-Curriu G, Redondo-Angulo I, Guitart-Mampel M, Ruperez C, Mas-Stachurska A, Sitges M, et al. Fibroblast growth factor-21 protects against fibrosis in hypertensive heart disease. J Pathol. (2019) 248(1):30–40. doi: 10.1002/path.5226
98. Le CT, Nguyen G, Park SY, Choi DH, Cho E-H. LY2405319, An analog of fibroblast growth factor 21 ameliorates α-smooth muscle actin production through inhibition of the succinate—g-protein couple receptor 91 (GPR91) pathway in mice. PLOS ONE. (2018) 13(2):e0192146. doi: 10.1371/journal.pone.0192146
99. Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol. (2011) 51(4):600–6. doi: 10.1016/j.yjmcc.2010.10.033
100. Li J, Gong L, Zhang R, Li S, Yu H, Liu Y, et al. Fibroblast growth factor 21 inhibited inflammation and fibrosis after myocardial infarction via EGR1. Eur J Pharmacol. (2021a) 910:174470. doi: 10.1016/j.ejphar.2021.174470
101. Ma Y, Kuang Y, Bo W, Liang Q, Zhu W, Cai M, et al. Exercise training alleviates cardiac fibrosis through increasing fibroblast growth factor 21 and regulating TGF-Β1-Smad2/3-MMP2/9 signaling in mice with myocardial infarction. Int J Mol Sci. (2021) 22(22):12341. doi: 10.3390/ijms222212341
102. Pan Z-C, Wang S-P, Ou T-T, Liu H, Ma J-W, Wang W-X, et al. A study on the expression of FGF-21 and NF-ΚB pathway in the tissues of atherosclerotic mice. Eur Rev Med Pharmacol Sci. (2017) 21(3 Suppl):102–7. 28745781.28745781
103. Wei W, Li X-X, Xu M. Inhibition of vascular neointima hyperplasia by FGF21 associated with FGFR1/syk/NLRP3 inflammasome pathway in diabetic mice. Atherosclerosis. (2019) 289:132–42. doi: 10.1016/j.atherosclerosis.2019.08.017
104. Lin Y-H, Lin L-Y, Wu Y-W, Chien K-L, Lee C-M, Hsu R-B, et al. The relationship between Serum galectin-3 and Serum markers of cardiac extracellular matrix turnover in heart failure patients. Clin Chim Acta. (2009) 409(1–2):96–9. doi: 10.1016/j.cca.2009.09.001
105. Sun M, Jin L, Bai Y, Wang L, Zhao S, Ma C, et al. Fibroblast growth factor 21 protects against pathological cardiac remodeling by modulating galectin-3 expression. J Cell Biochem. (2019) 120(12):19529–40. doi: 10.1002/jcb.29260
106. Ebrahimi F, Wolffenbuttel C, Blum CA, Baumgartner C, Mueller B, Schuetz P, et al. Fibroblast growth factor 21 predicts outcome in community-acquired pneumonia: secondary analysis of two randomised controlled trials. Eur Respir J. (2019) 53(2):1800973. doi: 10.1183/13993003.00973-2018
107. Gan F, Huang J, Dai T, Li M, Liu J. Serum level of fibroblast growth factor 21 predicts long-term prognosis in patients with both diabetes Mellitus and coronary artery calcification. Ann Palliat Med. (2020) 9(2):368–74. doi: 10.21037/apm.2020.03.28
108. Sommakia S, Almaw NH, Lee SH, Ramadurai DKA, Taleb I, Kyriakopoulos CP, et al. FGF21 (fibroblast growth factor 21) defines a potential cardiohepatic signaling circuit in End-stage heart failure. Circulation. (2022) 15(3):e008910. doi: 10.1161/CIRCHEARTFAILURE.121.008910
109. Ianoș RD, Pop C, Iancu M, Rahaian R, Cozma A, Procopciuc LM. Diagnostic performance of Serum biomarkers fibroblast growth factor 21, galectin-3 and copeptin for heart failure with preserved ejection fraction in a sample of patients with type 2 diabetes Mellitus. Diagnostics. (2021) 11(9):1577. doi: 10.3390/diagnostics11091577
110. Chou R-H, Huang P-H, Hsu C-Y, Chang C-C, Leu H-B, Huang C-C, et al. Circulating fibroblast growth factor 21 is associated with diastolic dysfunction in heart failure patients with preserved ejection fraction. Sci Rep. (2016) 6(1):33953. doi: 10.1038/srep33953
111. Brandão M, Eduardo L, Espes D, Westholm JO, Martikainen T, Westerlund N, et al. Acute sleep loss alters circulating fibroblast growth factor 21 levels in humans: a randomised crossover trial. J Sleep Res. (2022) 31(2):e13472. doi: 10.1111/jsr.13472
112. Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol. (2020) 16(11):654–67. doi: 10.1038/s41574-020-0386-0
113. Cho JM, Yang EH, Quan W, Nam EH, Cheon HG. Discovery of a novel fibroblast activation protein (FAP) inhibitor, BR103354, with anti-diabetic and anti-steatotic effects. Sci Rep. (2020) 10(1):21280. doi: 10.1038/s41598-020-77978-z
114. Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. (2018) 392(10165):2705–17. doi: 10.1016/S0140-6736(18)31785-9
115. Li J, Li Y, Liu Y, Yu H, Xu N, Huang D, et al. Fibroblast growth factor 21 ameliorates NaV1.5 and Kir2.1 channel dysregulation in human AC16 cardiomyocytes. Front Pharmacol. (2021b) 12:715466. doi: 10.3389/fphar.2021.715466
116. Tanajak P, Sa-nguanmoo P, Wang X, Liang G, Li X, Jiang C, et al. Fibroblast growth factor 21 (FGF21) therapy attenuates left ventricular dysfunction and metabolic disturbance by improving FGF21 sensitivity, cardiac mitochondrial redox homoeostasis and structural changes in Pre-diabetic rats. Acta Physiol. (2016) 217(4):287–99. doi: 10.1111/apha.12698
Keywords: fibroblast growth factor 21 (FGF21), heart remodeling, energy metabolism, oxidation, inflammation, autophagy
Citation: Zhao Z, Cui X and Liao Z (2023) Mechanism of fibroblast growth factor 21 in cardiac remodeling. Front. Cardiovasc. Med. 10:1202730. doi: 10.3389/fcvm.2023.1202730
Received: 9 April 2023; Accepted: 7 June 2023;
Published: 21 June 2023.
Edited by:
Yoshiyuki Ikeda, Kagoshima University, JapanReviewed by:
Hiroya Ohta, Tokushima Bunri University, Japan© 2023 Zhao, Cui and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhangping Liao bGlhb3pwMTk4MEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.