 Sarah M. Nwia
Sarah M. Nwia Ana Paula O. Leite
Ana Paula O. Leite Xiao Chun Li
Xiao Chun Li Jia Long Zhuo
Jia Long Zhuo- 1Tulane Hypertension and Renal Center of Excellence, Tulane University School of Medicine, New Orleans, LA, United States
- 2Department of Physiology, Tulane University School of Medicine, New Orleans, LA, United States
- 3Department of Pharmacology, Tulane University School of Medicine, New Orleans, LA, United States
Cardiovascular disease is a pathology that exhibits well-researched biological sex differences, making it possible for physicians to tailor preventative and therapeutic approaches for various diseases. Hypertension, which is defined as blood pressure greater than 130/80 mmHg, is the primary risk factor for developing coronary artery disease, stroke, and renal failure. Approximately 48% of American men and 43% of American women suffer from hypertension. Epidemiological data suggests that during reproductive years, women have much lower rates of hypertension than men. However, this protective effect disappears after the onset of menopause. Treatment-resistant hypertension affects approximately 10.3 million US adults and is unable to be controlled even after implementing ≥3 antihypertensives with complementary mechanisms. This indicates that other mechanisms responsible for modulating blood pressure are still unclear. Understanding the differences in genetic and hormonal mechanisms that lead to hypertension would allow for sex-specific treatment and an opportunity to improve patient outcomes. Therefore, this invited review will review and discuss recent advances in studying the sex-specific physiological mechanisms that affect the renin-angiotensin system and contribute to blood pressure control. It will also discuss research on sex differences in hypertension management, treatment, and outcomes.
Introduction
Hypertension, defined as blood pressure greater than 130/80 mmHg, has been firmly established as a primary risk factor associated with cardiovascular disease, stroke, and kidney diseases (1–4). In the United States alone, nearly 48% of American men and 43% of American women suffer from hypertension (2). Currently, most if not all available data from clinical studies in humans have consistently shown that premenopausal women are generally protected from the development of hypertension compared with age-matched men, but the prevalence of hypertension increases drastically in women during postmenopausal years. The mechanisms underlying these sex differences or sex dimorphism in the pathogenesis of hypertension in men vs. women remain incompletely understood. Historically, however, biological, physiological, and clinical research were conducted primarily on male cells, male animal models, and male human subjects, largely based upon the assumption that they are genetically, molecularly, and physiologically identical to their female counterparts (5–7). To further promote biomedical research in sex differences in all physiological and diseased models, the National Institute of Health (NIH) in 2014 began to mandate that all recipients of NIH funding are required to consider sex as biological variables in their experimental approaches to test their hypotheses. This policy has led to an explosion of the research on sex differences or sex dimorphism and the mechanisms involved across the board on the disease development and health outcomes (8).
Although hypertension is a multifactorial medical disorder, the renin-angiotensin-aldosterone system (RAAS) is recognized as one of the most important regulators of basal blood pressure homeostasis and a major contributor in the development of hypertension. This recognition is not only supported by extensive biomedical research in animal models of hypertension, but also by numerous clinical trials using the inhibitors of renin, angiotensin-converting enzyme (ACE), or type 1 angiotensin II (Ang II) receptor (AT1) or aldosterone receptor blockers to treat hypertension in human subjects (1–4). However, the RAAS is not only the targets for the development and treatment of hypertension, as many hypertensive patients require dual or multidrug therapy with a diuretic, calcium channel blocker, and an α or β blocker to control their blood pressure. Even then, appropriate >10 million Americans still suffer from resistant hypertension even treated with ≥3 antihypertensive medications with blood pressures persisting above the treatment threshold (1–4). The mechanisms underlying the development of resistant hypertension and the difficulty in treating resistant hypertension remain poorly understood. One of major problems may involve sex differences in the pathogenesis, mechanisms, and treatment of resistant hypertension between aging men and postmenopausal women. Thus, there is an urgent need for further studies of the sex differences in the mechanisms of hypertension and the contributions of the RAAS, which may offer more tailored or precision hypertensive treatments and achieve better therapeutic outcomes.
Against this background, the objective of this invited article is to review and discuss recent advances in studying sex differences or dimorphism in the RAAS and its contributions to the physiological regulation of blood pressure and in the development of hypertension, cardiovascular and kidney diseases. Our emphases will include sex differences in the RAAS and the mechanisms by which sex hormones and the RAAS contribute to normal blood pressure control and the development of hypertension, sex differences in the hypertension treatment and outcomes, as well as potential strategies for sex-specific treatment of resistant hypertension in humans.
Overview of the localization and roles of the RAAS in cardiovascular and kidney tissues
To help better understand the sex differences in the RAAS and its contributions to the regulation of cardiovascular and renal physiology and the development of hypertension and cardiovascular and kidney diseases, it is important to first review the localization and roles of the RAAS briefly. The RAAS has been delineated as a primary effector of the development of hypertension and two main axes responsible for blood pressure control have been established. The angiotensinogen (AGT)/renin/angiotensin-converting enzyme (ACE)/angiotensin II (Ang II)/AT1 receptor (AGT/renin/ACE/Ang II/AT1R) axis is the predominant pathway for Ang II formation and responsible for most if not all classic effects of Ang II in the development of hypertension and cardiovascular and kidney diseases (9) (Figure 1). The juxtaglomerular apparatus of the kidney tightly regulates renin release from the kidney via two important mechanisms—a baroreceptor mechanism that senses decreased blood pressure or blood volume loss within the renal vasculature and an osmoreceptor mechanism that senses NaCl delivery from the proximal nephron to the macula densa (10–14). Renin comprises the rate-limiting step in the activation of the RAAS, converting AGT to Ang I, so its expression levels are in constant balance via a variety of biological mechanisms (15). Ang I is then converted to the biologically active peptide Ang II by ACE. In addition to renin- and ACE-dependent pathways, non-renin/ACE independent pathways may also contribute to the formation and metabolism of Ang II in cardiovascular and kidney tissues (Figure 1). Chymase, a serine endopeptidase, is highly expressed in the heart of patients with cardiovascular diseases compared to ACE (16, 17), and reportedly ∼75% of Ang II is estimated to be generated from Ang (1–8, 10–13) in cardiac tissues by chymase rather than ACE (18, 19). The catalytic activity of chymase is reportedly about 20-fold higher compared to ACE (19, 20). In rats with pressure-overload, the expression of chymase was significantly increased in female than male rats (21). In the kidney, neprilysin (NEP), an endopeptidase, is highly expressed that directly cleaves Ang I into Ang (1-7) and shows much higher catalytic activity for Ang I compared ACE2 (22, 23). The expression of NEP in kidney is reportedly higher in female than male hypertensive mRen (2). Lewis rats (24). Thus, both renin/ACE-dependent and non-renin/ACE-dependent pathways may contribute to Ang II formation or metabolism in cardiovascular and kidney tissues in health and diseases (25, 26) (Figure 1).
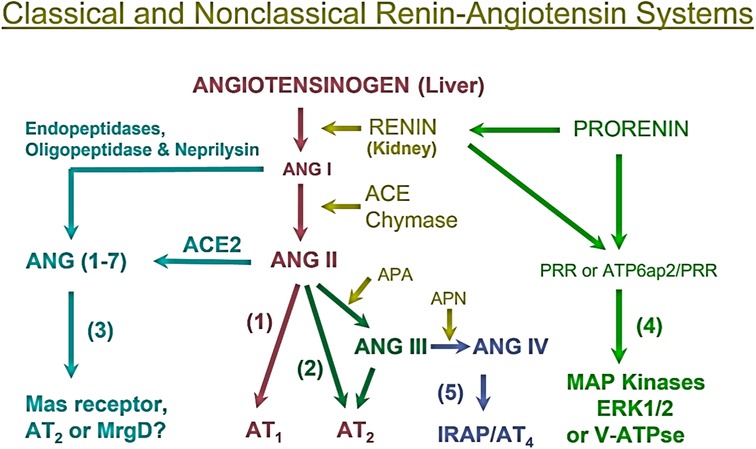
Figure 1. Classical renin/ACE-dependent and non-renin/ACE-dependent pathways for Ang II formation, metabolism, and actions in cardiovascular and kidney tissues. (1) The classical angiotensinogen/renin/ACE/ANG II/AT1 receptor axis. (2) The ANG II/APA/ANG III/AT2 receptor/NO/cGMP axis. (3) The ANG I/ANG II/ACE2-Neprilysin/ANG (1–7)/Mas receptor axis. (4) The prorenin/renin/prorenin receptor (PRR or ATP6ap2)/MAP kinases ERK1/2/V-ATPase axis. (5) The ANG III/APN/ANG IV/AT4 receptor/IRAP axis. Note that not only ACE but also chymase generate ANG II from ANG I, whereas neprilysin also cleaves ANG I to generate ANG (1-7). ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; APA, aminopeptidase A; APN, aminopeptidase N; IRAP, insulin-regulated aminopeptidase; PRR, prorenin receptor. Modified from reference (9) with permission.
The most pertinent G protein-coupled receptors with which Ang II activates are AT1 and AT2 receptors. AT1 receptors can be classified further into two subtypes: AT1a and AT1b. In humans, there is only one AT1 receptor that is expressed, corresponding to the AT1a receptor found in rodents (27–29). The AT1 receptor is generally considered to have pro-hypertensive, pro-growth, and pro-proliferative downstream effects. Activation of the AT1 receptor promotes vasoconstriction, increased oxidative stress, aldosterone release, and renal sodium absorption which all contribute to the regulation of blood pressure and fluid homeostasis, as well as the development of hypertension and cardiovascular and kidney diseases (30, 31) (Figure 2). In the kidney, activation of AT1 receptors especially induces the sodium-hydrogen exchanger 3 (NHE3) expression in the proximal tubules and the ascending limp of loop of Henle, resulting in the impairment of the pressure-natriuresis response and an increase in blood pressure (32–36). Conversely, Ang II activation of AT2 receptors works against the pro-hypertensive, pro-growth, and proliferative effects of AT1 activation, causing vasodilation and increased natriuresis (Figure 2) (34, 37–40). However, Ang III, a biologically active metabolite of Ang II, also acts to increase the natriuresis response reportedly by regulating Na+/K+-ATPase activity and reducing NHE3 activity (41–44).
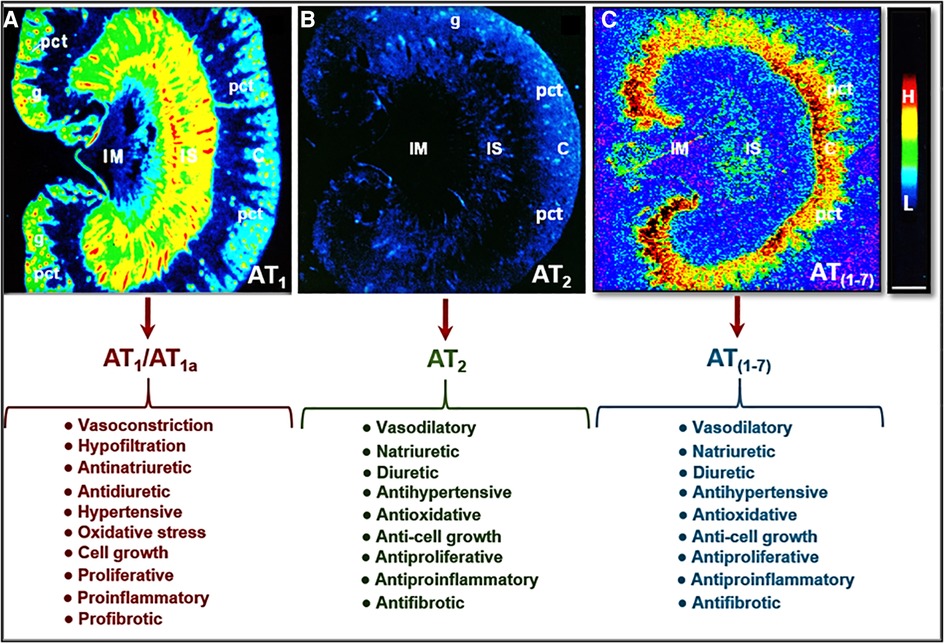
Figure 2. Localization of Ang II type 1 (AT1 or AT1a) and type 2 receptors (AT2) in the rat kidney using 125I-labeled Ang II receptor autoradiography and opposing actions of AT1 (AT1a), AT2, and/or AT (1-7) receptor activation in the kidney. (A) Shows the localization of AT1 or AT1a receptors with high levels in the glomerulus (g) and the inner stripe of the outer medulla corresponding to vasa recta bundles, and moderate levels in the proximal convoluted tubules (pct) in the cortex (C) and renomedullary interstitial cells (RMICs) in the inner stripe of the outer medulla between vasa recta bundles. The inner medulla (IM) expresses a very low level of AT1 or AT1a. (B) Shows the localization of AT2 receptors with low levels in the outer cortex, corresponding to the glomeruli and the proximal tubules, and the inner stripe of the outer medulla, corresponding to vasa recta bundles and RMICs. (C) Shows the localization of the receptor binding for Ang (1-7) in the kidney primarily in the inner cortex corresponding to the proximal tubules. Red represents high level (H), whereas dark blue represents background levels (L). Modified from reference (30) with permission.
The final cascade of the RAAS is the release and function of aldosterone from the adrenal glands. Ang II and Ang III both contribute to the stimulation of aldosterone release from the adrenal glands via binding to and activation of AT1 and AT2 receptors (Figure 3) (45, 46). Aldosterone is a mineralocorticoid that increases blood pressure by inducing the expression and activity of the epithelial sodium channel (ENaC) (47, 48). Previous studies have shown that Ang II stimulates aldosterone secretion in the zona glomerulosa cells (ZG) of the adrenal cortex and catecholamine release from chromaffin cells of the adrenal medulla. The catecholamines may stimulate aldosterone secretion via a paracrine mechanism (49, 50). Most if not all Ang II-induced aldosterone biosynthesis and release from the adrenal glands are mediated by AT1 (AT1a) receptors. Ang III has been demonstrated to have significant, if not equivocal aldosterone stimulating effects, to Ang II, but is hypothesized to primarily work through AT2 receptor activation (46, 51–54). Aldosterone acts to stimulate ENaC expression to increase sodium reabsorption primarily in the distal nephron and collecting tubules, resulting in blood pressure elevation (55). Additionally, increased levels of circulating aldosterone have been found to contribute to the pathogenesis of hypertension by causing endothelial dysfunction via increased production of reactive oxygen species (56).
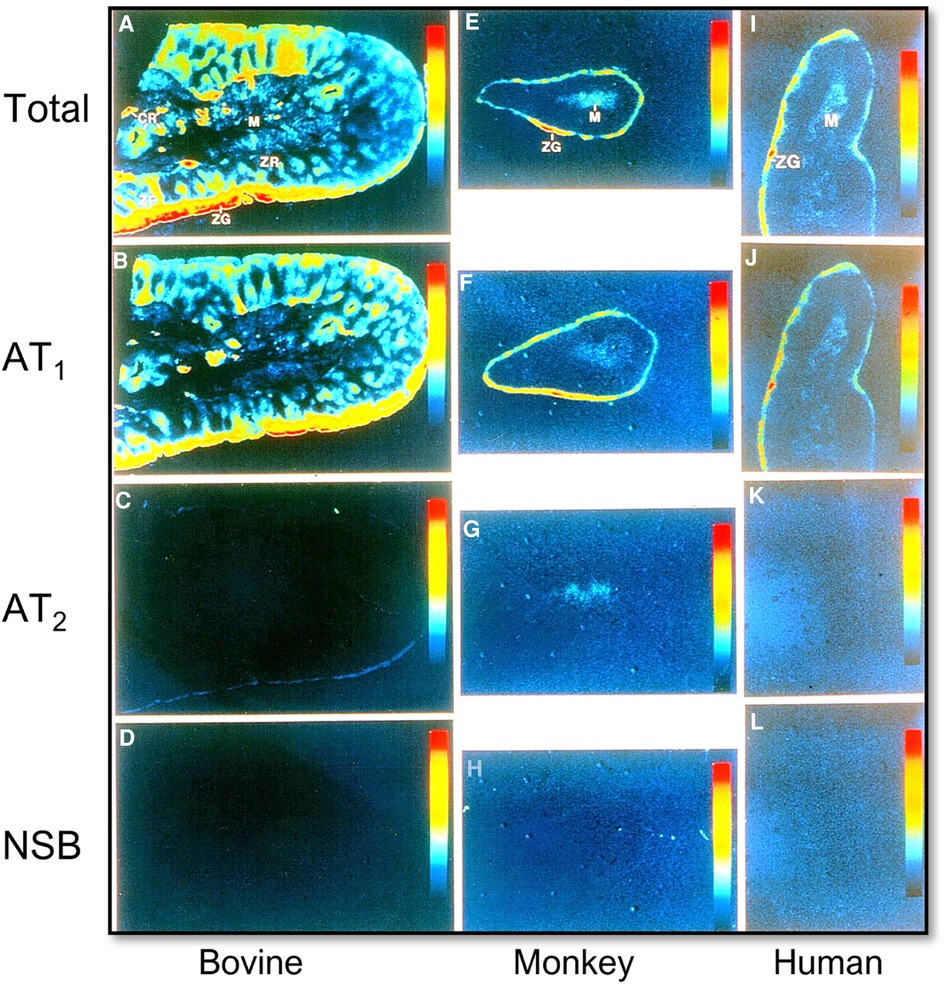
Figure 3. Localization of Ang II type 1 (AT1 or AT1a) and type 2 receptors (AT2) in the bovine, monkey, and human adrenal glands using quantitative 125I-labeled Ang II receptor autoradiography. (A,E,I) Represent total Ang II receptor binding; (B,F,J) represent AT1 receptor binding in the presence of an excess concentration of the AT2 receptor blocker PD123319 (10 µM); (C,G,K) represent AT2 receptor binding in the presence of an excess concentration of the AT1 receptor blocker losartan (10 µM); and (D,H,L) represent nonspecific binding in the presence of an excess concentration of unlabeled Ang II (10 µM), respectively. AT1 receptors predominate in the zona glomerulosa cells (ZG) of the adrenal cortex where aldosterone is synthesized and release into the circulation (B,F,J), and the adrenal medulla (M). AT2 receptors are low in the adrenal glands of bovine, monkey, and human adrenal glands (C,G,K). Red represents the highest level, whereas dark blue represents the background level of receptor binding. Modified from reference (32) with permission from the copyright holder.
In addition to the AGT/renin/ACE/Ang II/AT1 receptor axis, there exits an alternative counteracting angiotensin-converting enzyme 2 (ACE2)/Ang (1-7)/Mas receptor/AT2 receptor (ACE2/Ang (1-7)/MasR/AT2R) axis in the cardiovascular and kidney tissues, which is responsible for inducing vasorelaxation, lower blood pressure, and natriuretic responses (9, 57, 58) (Figure 2). Ang (1-7) is a biologically active derivative of Ang I and Ang II that are enzymatically cleaved by ACE2 (57, 58). The primary effects of Ang (1-7) are to counter the effects of the AGT/renin/ACE/Ang II/AT1 receptor axis by binding to G-protein coupled Mas receptors (MasR) and inducing the release of nitric oxide (NO), prostaglandin E2, and bradykinin to promote vasodilation (59–64). Ang (1-7) infusion was also found to reduce plasma renin activity, which may contribute to its antihypertensive effect (65).
In the kidney, the (pro)renin receptor (PRR) is another receptor that has been established as an important RAAS modulator in the cardiovascular and kidney tissues. PRR is encoded by the ATP6AP2 gene on the X chromosome and has been localized to many tissues including adipose, heart, brain, vessel wall, placenta, and kidney (66–71). Three forms of the protein exist including PRR, soluble PRR (sPRR), and truncated PRR (tPRR). sPRR is released into the plasma, while tPRR remains within the cellular membrane. PRR binds to renin and prorenin resulting in approximately a 5-fold increase in angiotensinogen conversion to angiotensin I (72). PRR has been implicated in both water and sodium homeostasis, as well. During water deprivation trials, PRR and sPRR expression is markedly increased and animal models with principal cell specific PRR deletion have demonstrated significant reductions in AQP2 expression and urine osmolality (73–76). Ang II has also been found to increase AQP2 expression within the collecting duct through several intracellular signaling pathways (77). However, animal studies have demonstrated that chronic Ang II infusion augments sPRR expression which in turn augments water reabsorption via AQP2 demonstrating a positive feedback mechanism within the collecting duct (78). PRR in the collecting duct may cause a marked increase in blood pressure via increasing ENaC expression (76, 79, 80). The precise mechanisms and downstream effects of PRR and its derivatives on water, sodium, and blood pressure have been thoroughly reviewed elsewhere (81).
It is now well-recognized that multiple RAAS axes are working concomitantly to regulate blood pressure and tissue perfusion (32, 34, 43, 82–86). The circulating or classical RAAS including all major components that have well-recognized endocrine effects (15, 32, 34). By contrast, the RAAS in the kidney may represent an important paracrine/autocrine/intracrine system, eliciting a more local and intracellular effect within the kidney tissue, especially within the proximal tubules (32, 34, 43, 82–86). Notably, the intrarenal RAAS has been found to have markedly higher concentrations of Ang II when compared to circulating plasma concentrations (87–93). Chronic Ang II exposure typically causes a down-regulation of AT1 receptors in different cardiac and vascular tissues; however, within the intrarenal RAAS, AT1 receptor expression is either constant or upregulated during the development of hypertension, cardiovascular and kidney diseases (94, 95).
Recently, there is evidence supporting a functional role for an intracellular and mitochondrial RAS as well. Initial animal studies demonstrated the presence of Ang II binding sites within hepatic cells (33, 92, 96–99). Since then, significant progress has been made in characterizing intracellular RAS within other tissue types. Within the kidney, high-density specific receptors for Ang II and Ang (1-7) were localized to cortical nuclei in sheep and rats (100–103). A fully functional RAS has also been demonstrated within the mitochondria (33, 104, 105). The exact origin of the intracellular RAS and its role in blood pressure homeostasis is yet to be determined, but there is evidence suggesting that they both serve physiological functions in the context of Ang II-induced hypertension (106, 107).
Clearly, recent studies in delineating the vasoconstrictive properties of the AGT/renin/ACE/Ang II/AT1 receptor and the vasodilatory properties of the counteracting ACE2/Ang (1-7)/Mas receptor/AT2 receptor axes have greatly expanded the therapeutic targets available to treat hypertension and cardiovascular and kidney diseases. Currently, first-line pharmacological treatments for hypertension include monotherapy or combination therapy using ACE inhibitors and angiotensin AT1 receptor blockers (ARBs), thiazide diuretics, and long-acting dihydropyridine calcium channel blockers (108, 109). Alpha- and β-blockers have also been identified as adjunctive treatments for hypertension, but they have additional side effects that may make them intolerable to patients including asthma exacerbations, insomnia, worsening glucose intolerance, bradycardia, and sick sinus syndrome (110, 111). Treatment-resistant hypertension is defined as hypertension that is unable to be controlled after the implementation of three antihypertensives with complementary mechanisms (1–4). Now affecting nearly 10.3 million Americans, it has become increasingly prevalent in the United States, indicating a need for alternative or additional therapies (2). Since the classical RAAS has been expanded in recent years, various new drugs have been developed to target these new substrates and receptors. Preclinical data has supported Ang (1-7) and AT2 agonists as viable treatment targets, but whether they are effective therapeutic targets in hypertension, cardiovascular and kidney diseases remains to be confirmed in clinical trials (112–114).
Sex differences in the RAAS and their roles in cardiovascular and renal physiology and hypertension
Sex differences in vascular dysfunction
Evidence has repeatedly demonstrated that there is an age-dependent difference in the prevalence of hypertension between men and women. Until age 45, women are less likely to develop hypertension than men, while this difference is not present between ages 46 and 64 (2, 115, 116). After age 65, the prevalence among women increases significantly. It is estimated that 85% of women over 75 have hypertension compared with 79% of men within the same age group (2, 115, 116). Recent studies are ongoing to further characterize these differences and underlying mechanisms in the RAAS between males and females, which may contribute to this age-dependent difference in the prevalence of hypertension between men and women.
There are several baseline physiological differences that contribute to the development of hypertension that have been observed in male and female subjects. Nitric oxide (NO), which has vasodilatory effects, has been established as a key mechanism of blood pressure homeostasis (117, 118). NO plays a protective role in the development of hypertension because of its vasodilatory effects and ability to quickly react with superoxide to counteract the latter's effects (119). Animal studies have shown that females have greater NO bioavailability compared with males due to higher NO-generating capacity in females and increased oxidative stress levels in males (120–125). Oxidative stress causes endothelial dysfunction due to vasoconstriction and the activation of the RAAS in blood vessels. In vivo studies have shown that Ang II causes mesangial cells in the kidney to produce superoxide, while the inhibition of the RAAS has been shown to reduce oxidative stress (126, 127). More recent data has demonstrated that mice treated with buthionine sulfoximine (BSO), a substance that induces oxidative stress, had higher levels of AT1 receptors within the proximal tubules. Additionally, they demonstrated a more dramatic downstream signaling effect, indicating that oxidative stress sensitizes kidney cells to produce an amplified RAS response (128). An inflammatory response to oxidative stress is also activated by Ang II via AT1 receptors, leading to nuclear factor-κB (NF-κB) transcription factor expression (128, 129).
Sex differences and the cardioprotective roles of estrogen
In view of the age differences well-recognized in hypertension prevalence between males and females, the interactions between estrogen and the RAAS have become an important research focus (130). Estrogen is a steroid hormone that binds to two nuclear receptors, estrogen receptor-α (ER-α) and estrogen receptor-β (ER-β), and G protein-coupled estrogen receptor 1 (GPER1) (130–133). ER-α is abundantly expressed in the vascular endothelium and helps promote vasodilation, endothelial repair, and NO production (134). ER-β activation primarily results in NO production (134, 135). Together, the binding of estrogen to these two receptors increases vasodilation and has a protective effect against hypertension. Esqueda et al. demonstrated that after ovariectomy, estrogen-supplemented, salt-sensitive rats had restored ER-β expression levels. The same was not demonstrated for ER-α, implying that the imbalance between ER-α and ER-β might contribute to the development of hypertension after menopause (136).
In animal studies, estradiol has been found to have a role in protecting against hypertension. In spontaneously hypertensive rats (SHRs), young male rats have demonstrated higher mean blood pressures than young female rats (137–140). This difference was eliminated through pharmacological RAS inhibition and the cessation of estrous cycling, implicating estrogen as the cardioprotective factor and accounting for the sex and age-related differences (139, 141, 142). Aging SHRs have been established as a model for postmenopausal hypertension due to their non-cycling, low serum estradiol and the ensuing increase in blood pressure (142, 143).
In human studies, 17β-estradiol (E2) has been determined to regulate the RAS via the changes in this enzyme expression. For example, Proudler et al. investigated the effect of estrogen/progesterone combined hormone replacement therapy (HRT) on ACE activity in postmenopausal women. They determined that ACE activity was reduced by 20% in treated women when compared to their untreated controls; however, this study was limited by sample size, including only 28 women in the treatment group and 16 in the untreated group (144). Soon after, Schunkert et al. measured and compared renin and angiotensinogen levels between women treated with estrogen replacement therapy (ERT) and those who were not. Renin levels were found to be significantly increased in women without ERT, measuring 16.6 ± 0.9 mU/L compared to 12.0 ± 0.7 mU/L in the treated group. Angiotensinogen levels were found to be higher in women with ERT, compared to those without, indicating a reduced rate of conversion by renin (145). Thus, these studies provide the evidence for estrogen's cardioprotective effects in part by regulating the expression or activity of the RAS.
Sex differences in the classical RAS and the role of estrogen
New data has recently built upon these previous studies to elucidate the mechanisms by which estrogen modulates the classical RAS. Essentially, estrogen can alter RAS activities by regulating the levels of key substrate, enzyme, and receptor expression, and protein production. Animal studies have shown that the expression of the RAS enzymes was significantly altered in the presence or absence of estrogen. In young male SHRs, ACE mRNA expression in the kidneys was significantly increased when compared to their female counterparts (146, 147). Similar results were found in two-kidney, one-clip (2K1C) renal hypertension animal models (147). This difference in intratubular enzyme concentrations is attenuated between aging SHR male and female rats (148). In aging SHRs, plasma renin activity (PRA) and concentrations of AGT and Ang II, which are measures of the circulatory RAS activation, were not significantly different between aging male and female SHRs. However, intratubular AGT expression was increased in males when compared to females, whereas aging females were found to have higher Ang II expression (148). These data suggest that in young rats, males have higher levels of intratubular RAS enzyme expression and cascade activation compared to females. In aging rats, when the protective effect of estrogen has diminished, females have increased intrarenal RAS activation and higher levels of Ang II. In addition to the regulation of renin and ACE, estrogen also regulates the renin- and ACE-independent enzymes in the RAS. Ahmad et al. and others compared the metabolic pathway for Ang II formation in cardiac tissues of gonadal-intact and ovariectomized (OVX) adult Wistar Kyoto (WKY) and SHR rats, and found that estrogen depletion significantly increased chymase activity, but not ACE activity (24, 25). Li et al. demonstrated that estrogen inhibits chymase release from cardiac mast cells to prevent pressure overload-induced adverse cardiac remodeling (20). The latter studies suggest that estrogen status may play an important role in the regulation of cardiac chymase expression and cardiovascular protection in adult female animals (20, 24, 25).
Estrogen also plays an important role in regulating the RAS through the modulation of AT1 and AT2 receptor expression (141). In animal studies comparing arterial AT1 expression in male rats, ovariectomized rats, and estrogen-supplemented ovariectomized rats, AT1 receptor density was found to be significantly increased in the males and ovariectomized rats when compared to those supplemented with estrogen (140, 149). In aging SHRs, this difference is eliminated and AT1 expression was found to be the same between male and female rats (148). Silva-Antonnialli et al. demonstrated that AT2 receptor expression was similar among male, female, oophorectomized females, and estrogen-replaced females, causing the AT1/AT2 ratio in estrogen-treated females to be higher (140). These studies suggest that estrogen's protective role can be partially attributed to its ability to downregulate AT1 receptor expression. Indeed, these differences are supported by the studies showing a significant difference in the response to AT1 blockers. For instance, aging male rats were observed to have 52% decrease in mean arterial blood pressure, while females only had a 37% drop (148). Increased Ang II or its AT1 receptor expression in the kidneys of postmenopausal female rats may explain why postmenopausal women are more susceptible to the development of hypertension and the roles of estrogen in sex differences in hypertension.
The third mechanism by which estrogen can influence blood pressure via the classical RAS is by regulating aldosterone secretion. Aldosterone is known to cause increased salt retention and blood pressure. In animal studies, estrogen was found to reduce AT1 receptor expression in the adrenal glands, which in part contribute to reduced aldosterone secretion (150). More recent clinical studies have shown that when consuming high salt diets, men had significantly higher plasma aldosterone, extracellular volume, and systolic blood pressure than women (151). These two studies further suggest that aldosterone secretion may be a key contributor to the sex differences in hypertension prevalence between men and women.
However, the sex differences or the sexual dimorphism of PRR and its role in the development of hypertension remain poorly understood. A study on type 2 diabetic men and women reported that plasma sPRR was significantly higher in women compared to men and that sPRR concentrations appeared to correlate with age, BMI, eGFR, and plasma renin activity in female subjects, though not statistically significant in the male subjects (152). The finding that increased age correlates with increased sPRR and systemic RAS activation suggests that the transition to an estrogen-deficient state of menopause causes increased sPRR expression and RAS activation. However, more work is necessary to characterize the mechanism by which estrogen and PRR interact in further studies.
Sex differences in the vasoprotective axis of the RAS and the role of estrogen
In addition to inhibitory effects on the classical RAS system, estrogen exerts antihypertensive effects via upregulation of the substrate and enzymes in the counterregulatory RAS pathways. Lee et al. studied ACE2 expression in control and 2K1C male and female rats and demonstrated that female rats showed increased intratubular ACE2 expression regardless of 2K1C treatment status, suggesting estrogen's protective role in increasing Ang II metabolism to Ang (1-7) (147). In studies using human umbilical vein endothelial cells (HUVEC), estrogen activation of ER-α receptors was shown to elevate intracellular ACE and ACE2 mRNA expression and ACE protein expression. This increased ACE2 expression is expected to increase intracellular Ang (1-7) formation (153). This data supports the hypothesis that the intracellular RAS, especially ACE2 and Ang (1-7), and estrogen cooperate in a manner that protects against the development of 2K1C renal hypertension, most likely due to increased Ang (1-7) production and AT2 receptor activation.
The MasR is another component of the alternative vasoprotective RAS pathway that demonstrates sex-dependent properties. Previous studies have solidified the hypothesis that NO release is mediated by Ang (1-7) activation of MasR (64, 154, 155). Sobrino et al. used HUVEC to demonstrate that estradiol increased the intracellular expression of enzymes responsible for Ang (1-7) and NO production (156). Their data showed that estradiol treatment increased ACE and cathepsin A expression which are ultimately responsible to produce Ang (1-7). These authors also reported that eNOS and cytosolic guanylate cyclase expression was increased, indicating that NO synthesis was promoted by estradiol treatment. When MasR was blocked, they found that NO levels were decreased, supporting their hypothesis that estradiol mediates increased NO production via the activation of MasR (156). Mompéon et al. also used HUVEC to show that estradiol increased Ang (1-7) production via ER-α activation and increased ACE2 mRNA expression (153). One limitation of these studies, however, is the tissue-specific characteristics of intracellular RAS. It would be beneficial to utilize human or animal kidney cells to fully determine the relationship between estrogen treatment and intracellular RAS responses in the kidney.
In addition to in vitro cell culture studies, animal studies have also demonstrated estrogen effects on MasR function. When subjected to Ang II infusion, female rats demonstrated reduced renal blood flow responses, but only in the context of dual MasR and AT1 blockade (157, 158). With AT1 blockade, there is an increased concentration of circulating Ang II, possibly allowing for increased Ang (1-7) formation via the ACE2 pathway. Saberi et al. compared the effects of estrogen supplementation in response to Ang (1-7) infusion and MasR blockade. They found that estradiol-treated ovariectomized rats had decreased renal blood flow in response to Ang (1-7) after MasR blockade when compared to their untreated counterparts (159). These studies suggest that one of estradiol's antihypertensive mechanisms operates via MasR activation. When MasR is blocked, there are fewer opportunities for estrogen to exert protective effects leading to decreased renal blood flow and worsening hypertension.
Finally, an additional protective axis of the RAAS consisting of Ang III/AT2 receptor activation is also modified by estrogen. Female mice have been shown to utilize the AT2 receptor pathway to attenuate the effects of Ang II via AT1 receptors; however, this effect diminishes with increased age (160, 161). Another study demonstrated that exogenous estrogen replacement reinstituted this protective pathway and attenuated Ang II-induced hypertension (162). Together, these studies support the hypothesis that estrogen affects the RAS primarily through activation of the vasoprotective signaling pathways, rather than the attenuation of the classical RAAS signaling pathway. This evidence could result in novel therapeutics for estrogen-deficient individuals who are suffering from resistant hypertension.
Sex differences in Ang II-induced hypertension and the roles of testosterone and estrogen
There is no question that testosterone contributes to sex differences in cardiovascular and kidney diseases and hypertension, but its contribution to sex differences is not as well-studied as that of estrogen. Historically, there are animal studies showing mild adverse effects of testosterone on hypertensive outcomes in young spontaneously hypertensive rats (138, 139, 163, 164). Dalmasso et al. have suggested that in aging SHRs, testosterone supplementation causes a reduction of blood pressure, indicating that age, in concordance with testosterone status, affects hypertensive outcomes rather than testosterone alone (164). A more recent animal study determined that testosterone played a permissive role in the development of hypertension since Ang II-induced hypertension was worsened when castrated males were supplemented with exogenous testosterone (165). They also noted that castrated males demonstrated a reduced AT1/AT2 receptor ratio, which favors the vasoprotective axis of the RAS. This ratio was restored when testosterone was re-administered (165). A mendelian randomization model concluded that high testosterone states could lead to increased rates of hypertension (166). Studies utilizing human subjects present only mildly convincing data. In women specifically, one study showed some evidence that high testosterone states were correlated with increased carotid-femoral pulse wave velocities, which is an indicator of arterial stiffness (167). One review article summarizing the effects of testosterone therapy on various laboratory markers of transgender men concluded that there was only weak evidence supporting the correlation between increased blood pressure and testosterone administration (168). Interestingly, some studies have correlated testosterone-deficient states to the development of hypertension, which would appear to be contrary to the trends observed in previous studies. One such study investigated the effects of free testosterone and biologically available testosterone on blood pressure. It found that free testosterone is essentially inversely correlated with systolic and diastolic blood pressure in men (169). Given the evidence, it is likely that increased testosterone levels in conjunction with decreased estrogen levels, like those found in PCOS, work synergistically to facilitate the development of hypertension. Further research is necessary to characterize the mechanisms by which testosterone regulates blood pressure and its role in the development of hypertension.
Whether there are sex differences in Ang II-dependent or Ang II-induced hypertension remains to be further studied. Some inconsistencies have been reported in the roles of sex differences in Ang II-induced hypertension in animal models (160–162, 170, 171). These inconsistencies range from complete reversal, attenuated responses, or no effect at all in female rats or mice, based on the doses of Ang II infusion (low pressor or high pressor), animal models (rat or mouse, global AT1a or AT2 receptor knockout), or routes of administration (subcutaneous or intraperitoneal infusion) (160–162, 170, 171). It is difficult to directly compare these studies and draw a clear conclusion on whether sex differences contribute to the development of Ang II-induced hypertension. Indeed, no significant sex differences in basal blood pressure levels in age-matched adult male and female Sprague-Dawley rats, wild-type, or AT2 receptor knockout mice in which Ang II induced similar increases in blood pressure, natriuretic, or diuretic responses (172–175).
Recently, we have determined whether there are sex differences in the blood pressure, renal excretory, and fibrotic responses to Ang II between male and female wild-type mice, and between male and female proximal tubule-specific AT1a receptor knockout mice (PT-Agtr1a−/−) (170, 171). Although we found sex differences in some minor phenotypic responses, deletion of AT1a receptors selectively in the proximal tubules decreased basal arterial blood pressure similarly in both male and female wild-type and PT-Agtr1a−/− mice. Both male and female wild-type and PT-Agtr1a−/− mice responded to Ang II infusion and developed hypertension to the similar magnitudes (Figure 4) (170, 171). The maximal pressor responses remained to be ∼20 mmHg lower in male and female PT-Agtr1a−/− mice than male and female wild-type mice. Furthermore, concurrent blockade of AT1 receptors with losartan decreased the pressor response to Ang II to similar extents in male and female wild-type and PT-Agtr1a−/− mice (170, 171). Thus, no significant sexual dimorphism or sex differences in blood pressure phenotypes were discovered in wild-type and PT-Agtr1a−/− mice in response to Ang II or AT1 receptor blockage. However, we did uncover sex differences in Ang II-induced hypertension in a mutant mouse model with deletion of the Na+/H+ exchanger 3 (NHE3) selectively in the proximal tubules of the kidney (PT-Nhe3−/−) (36). In male wild-type and PT-Nhe3−/− mice infused with a high pressor dose of Ang II, systolic, diastolic, and mean arterial blood pressure increased in a time-dependent manner reaching a peak response within a week of Ang II infusion (Figure 5). In female PT-Nhe3−/− mice, however, systolic, diastolic, and mean arterial blood pressure responses to Ang II began to decrease 4 days after Ang II infusion, suggesting that estrogen (and/or other female hormones) may contribute to these sex differences in Ang II-induced hypertension in this mutant mouse model (Figure 5).
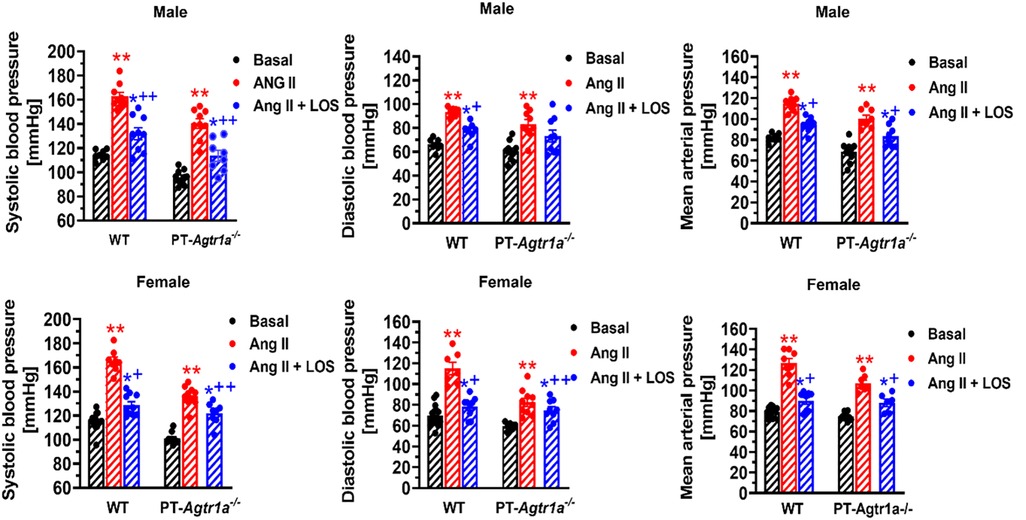
Figure 4. Comparisons of basal systolic, diastolic, and mean arterial blood pressure and their responses to Ang II infusion with or without AT1 (AT1a) receptor blocker losartan between male and female wild-type (WT) and PT-Agtr1a−/− mice. Proximal tubule-specific deletion of AT1a receptors significantly decreased basal blood pressure similarly in male and female PT-Agtr1a−/− mice under basal conditions, and significantly attenuated the hypertensive responses to Ang II similarly in both male and female PT-Agtr1a−/− mice. No significant sex differences were found in basal blood pressure and its responses to Ang II with or without losartan treatment between male and female WT or between male and female PT-Agtr1a−/− mice. *P < 0.05 or **P < 0.01 vs. control WT or PT-Agtr1a−/− mice; +P < 0.05 or ++P < 0.01 vs. Ang II-infused male or female wild-type or PT-Agtr1a−/− mice. Reproduced from reference (171) with permission.
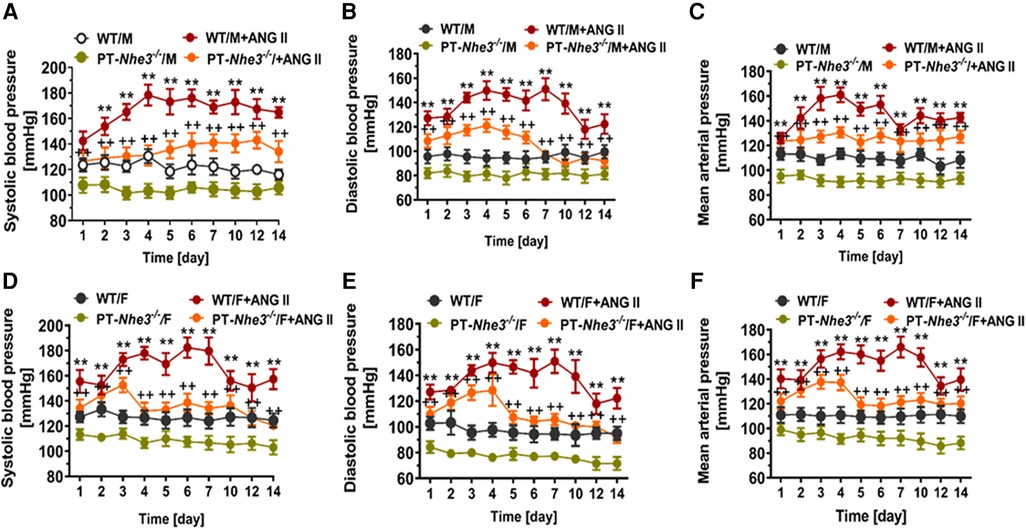
Figure 5. Sex differences in basal systolic, diastolic, and mean arterial blood pressure and their responses to a high pressor dose of Ang II infusion, 1.5 mg/kg per day, intraperitoneal via osmotic minipump in conscious, adult male and female wild-type (WT) and PT-Nhe3−/− (proximal tubule-specific NHE3 knockout) mice, as measured using the direct implanted telemetry technique. Please note the time-dependent increases in systolic, diastolic, and mean arterial blood pressure responses to Ang II infusion in male WT mice and significantly attenuated hypertensive responses to Ang II in male PT-Nhe3−/− mice. However, systolic, diastolic, and mean arterial blood pressure responses to Ang II began to decrease 4 days after Ang II infusion in female PT-Nhe3−/− mice, revealing significant sex differences in these mutant mice. (A–C) Male mice; whereas (D–F) female mice. **P < 0.01 vs. WT time-control group; ++P < 0.01 vs. PT-Nhe3−/− time-control group, respectively. Reproduced from reference (36) with permission.
Sex differences in antihypertensive treatments or managements
In 2017, the American College of Cardiology published new guidelines for the treatment of hypertension. They stratified blood pressure into five categories with different treatment strategies or approaches. Non-pharmacological interventions are an integral part of controlling hypertension of all categories. Lifestyle changes that promote blood pressure reduction include weight loss, DASH diet, sodium intake reduction, dietary potassium supplementation, increased physical activity, and reduced alcohol consumption (1–4). These lifestyle changes are recommended to every patient, regardless of blood pressure status. Patients are initiated on BP-lowering medications once they are diagnosed with Stage 1 and have ASCVD or a 10-year CVD risk ≥10% (1–4). Primary agents for the treatment of hypertension include thiazide diuretics, ACE inhibitors, angiotensin II receptor blockers (ARBs), and calcium channel blockers (CCBs). Secondary agents include loop diuretics, potassium-sparing diuretics, aldosterone antagonists, beta-blockers, direct renin inhibitors, alpha-blockers, and direct vasodilators (1–4).
The INTERHEART study established that elevated blood pressures presented an increased risk for adverse cardiac events for female subjects when compared to male subjects (176). Regarding control, there has been an ongoing debate about the risks and benefits of intensive vs. less intensive therapy. The 2021 SPRINT trial concluded that patients with increased cardiovascular risk were less likely to experience a major adverse cardiac event when their target systolic blood pressure was <120 mmHg when compared to the less intensive <140 mmHg target that was previously established by clinical guidelines (177). When the data is analyzed by sex, the hazard ratio is not statistically significant in the female subgroup. It is important to note that this outcome could be attributed to small female sample size within the trial and lower baseline cardiovascular risk (177). Although the data on blood pressure control is not unanimous, it is generally accepted in clinical practice that a more intensive approach to BP control yields better long-term outcomes (178). Indeed, a study examining worldwide rates of hypertensive control found that blood pressure control rates were significantly worse in women (34.0%) when compared to men (37.7%) (179).
However, current guidelines still do not have sex-specific recommendations when it comes to hypertension management, with an exception for women who are pregnant, breastfeeding, or of childbearing age. One meta-analysis comparing the treatment benefits of ACE inhibitors, CCBs, ARBs, and diuretics/beta-blockers concluded that these blood pressure-lowering regimens all have similar protection against major cardiovascular events between men and women (180). Another study determined that women who have been prescribed losartan were more likely to be hospitalized for angina than their male counterparts receiving the same treatment (181). The ACCOMPLISH trial compared multidrug therapy consisting of ACE inhibitors + CCBs to ACE inhibitors + HCTZ. Their data demonstrated that the ACEI + CCB combination was more effective in reducing adverse cardiovascular events and death, but this same significance was not demonstrated in the female subject subgroup. These findings were likely limited by the fact that only 39.5% of study subjects were women (182). Generally, data demonstrating the relationship between specific antihypertensive regimens and cardiovascular outcomes is lacking when it comes to comparing female and male subjects.
Sex differences have been identified in drug bioavailability, an important factor when it comes to dosing considerations. Women generally have higher gastric pH, slower gastric emptying, and longer gastrointestinal transit time (183). All these features would promote absorption, causing increased drug absorption in women compared to men. After a drug is absorbed, it is distributed around the body into different compartments which can alter bioavailability. Sex differences in body composition such as higher body fat percentage and decreased plasma volume in females could affect drug availability and create higher levels of lipid-soluble drugs in men and hydrophilic drugs in women. Increased bioavailability usually results in increased risk of adverse outcomes, when not accounted for in dosing regimens.
Adverse outcomes to hypertension treatment are an important consideration when trying to optimize cardiovascular outcomes in patients. Rabi et al. reviewed controlled trials of ACE inhibitors and ARBs and found that only 43% of studies reported sex-specific outcomes (184). A comparative study by Rydberg et al. concluded that women had an increased prevalence of adverse drug reactions to ACEIs, thiazides, diuretics, and potassium-sparing agents. When it comes to ACE inhibitor adverse drug reactions (ADRs), female patients were 1.31 times more likely to report adverse reactions (185). The most reported symptoms in both sexes were cough and angioedema (185). Male subjects were more likely to report adverse drug reactions while taking aldosterone antagonists, with the most common reported reaction being hyperkalemia (186). No statistical difference was found between males and females for ARBS, sulfonamides, and selective beta-blockers in the prevalence of adverse drug reactions (187). Overall, female patients are more likely to experience adverse drug reactions while undergoing treatment for hypertension (187–192).
Concluding remarks
In summary, hypertension remains a critical area of research due to its prevalence and strong association with adverse cardiovascular events. Historically, female subjects have been excluded from in vivo animal experiments and clinical trials in humans, leaving half of the population unaccounted for in health, hypertension, cardiovascular, and kidney research. However, recent efforts have increased our understanding of sex differences in the physiological and pathological development of hypertension.
The data summarized in this review highlights the protective effect of estrogen on hypertension. After menopause, women are more likely to develop hypertension due to decreased estrogen levels. Estrogen exerts inhibitory effects on the classical RAAS while promoting non-classical RAS pathways, resulting in an overall vasodilatory and antihypertensive response. However, the mechanisms through which testosterone influences blood pressure remain unclear, and further research is necessary to elucidate its interaction with the RAAS.
Regarding clinical management, there has been some progress in including female subjects in clinical trials. However, research on the clinical outcomes of female and male subjects on specific antihypertensive regimens remains limited. Female patients have been shown to be more prone to adverse drug reactions while undergoing treatment, likely due to sex differences in pharmacokinetics and pharmacodynamics. As such, hypertension treatment that accounts for biological sex might provide better patient outcomes and fewer adverse drug reactions.
Looking towards the future, sex differences in hypertension, cardiovascular and kidney pathogenesis might provide new opportunities to develop novel therapies that not only suppress the classical AGT/renin/ACE/Ang II/AT1 receptor responses, but also restore the vasoprotective axis of the ACE2/Ang (1-7)/MasR/AT2 receptor responses. For example, therapies that promote Ang (1-7) binding with MasR or activate AT2 receptors might be beneficial for postmenopausal women with poorly controlled hypertension, cardiovascular and kidney diseases. Several clinical trials are currently underway to investigate these as viable treatment targets for hypertension.
In conclusion, while some progresses have been made in studying and understanding sex differences in hypertension, cardiovascular and kidney diseases, further research is necessary to develop more effective and personalized treatments that account for biological sex. Inclusion of female subjects in clinical studies is especially critical to help promote clinical decisions that take into account sex-specific factors in the future.
Author contributions
SN, AL, XL, and JZ: contributions to the conception or design of the work; draft manuscript, and interpretation of data, revision of the manuscript, and approval of manuscript submission, and agreement to be accountable for all aspects of the work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by grants from National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK123144-01; 2R01DK067299-10A1; and 2R01DK102429-03A1) to JZ.
Acknowledgments
Most of authors' recent studies were carried out in the laboratory of JZ at the University of Mississippi Medical Center in Jackson, Mississippi, and Tulane University School of Medicine, in New Orleans, Louisiana, respectively. SN was a DeBakey Scholar and medical student at Tulane University School of Medicine and supported by Warren R. Bourgeois, III, M.D., and Usha Ramadhyani, M.D., Student Research Endowed Fund.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor HW declared a past co-authorship with the author JZ.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yusuf S, Joseph P, Rangarajan S, Islam S, Mente A, Hystad P, et al. Modifiable risk factors, cardiovascular disease, and mortality in 155 722 individuals from 21 high-income, middle-income, and low-income countries (PURE): a prospective cohort study. Lancet. (2020) 395(10226):795–808. doi: 10.1016/S0140-6736(19)32008-2
2. Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Hypertension. (2018) 71(6):e13–e115. doi: 10.1161/HYP.0000000000000065
3. Fuchs FD, Whelton PK. High blood pressure and cardiovascular disease. Hypertension. (2020) 75(2):285–92. doi: 10.1161/HYPERTENSIONAHA.119.14240
4. Kjeldsen SE. Hypertension and cardiovascular risk: general aspects. Pharmacol Res. (2018) 129:95–9. doi: 10.1016/j.phrs.2017.11.003
5. Geller SE, Koch AR, Roesch P, Filut A, Hallgren E, Carnes M. The more things change, the more they stay the same: a study to evaluate compliance with inclusion and assessment of women and minorities in randomized controlled trials. Acad Med J Assoc Am Med Coll. (2018) 93(4):630–5. doi: 10.1097/ACM.0000000000002027
6. Clayton JA. Studying both sexes: a guiding principle for biomedicine. FASEB J. (2016) 30(2):519–24. doi: 10.1096/fj.15-279554
7. Klein SL, Schiebinger L, Stefanick ML, Cahill L, Danska J, de Vries GJ, et al. Opinion: sex inclusion in basic research drives discovery. Proc Natl Acad Sci U S A. (2015) 112(17):5257–8. doi: 10.1073/pnas.1502843112
8. Mauvais-Jarvis F, Bairey Merz N, Barnes PJ, Brinton RD, Carrero JJ, DeMeo DL, et al. Sex and gender: modifiers of health, disease, and medicine. Lancet Lond Engl. (2020) 396(10250):565–82. doi: 10.1016/S0140-6736(20)31561-0
9. Li XC, Zhang J, Zhuo JL. The vasoprotective axes of the renin-angiotensin system: physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacol Res. (2017) 125(Pt A):21–38. doi: 10.1016/j.phrs.2017.06.005
10. Carey RM, McGrath HE, Pentz ES, Gomez RA, Barrett PQ. Biomechanical coupling in renin-releasing cells. J Clin Invest. (1997) 100(6):1566–74. doi: 10.1172/JCI119680
11. Bock HA, Hermle M, Brunner FP, Thiel G. Pressure dependent modulation of renin release in isolated perfused glomeruli. Kidney Int. (1992) 41(2):275–80. doi: 10.1038/ki.1992.39
12. Bell PD, Lapointe JY, Sabirov R, Hayashi S, Peti-Peterdi J, Manabe K, et al. Macula densa cell signaling involves ATP release through a maxi anion channel. Proc Natl Acad Sci U S A. (2003) 100(7):4322–7. doi: 10.1073/pnas.0736323100
13. Lorenz JN, Weihprecht H, He XR, Skøtt O, Briggs JP, Schnermann J. Effects of adenosine and angiotensin on macula densa-stimulated renin secretion. Am J Physiol. (1993) 265(2 Pt 2):F187–94.8368332
14. Lorenz JN, Weihprecht H, Schnermann J, Skøtt O, Briggs JP. Renin release from isolated juxtaglomerular apparatus depends on macula densa chloride transport. Am J Physiol. (1991) 260(4 Pt 2):F486–93.2012204
15. Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical renin-angiotensin system in kidney physiology. Compr Physiol. (2014) 4(3):1201–28. doi: 10.1002/cphy.c130040
16. Urata H, Boehm KD, Philip A, Kinoshita A, Gabrovsek J, Bumpus FM, et al. Cellular localization and regional distribution of an angiotensin II-forming chymase in the heart. J Clin Invest. (1993) 91(4):1269–81. doi: 10.1172/JCI116325
17. Li M, Liu K, Michalicek J, Angus JA, Hunt JE, Dell'Italia LJ, et al. Involvement of chymase-mediated angiotensin II generation in blood pressure regulation. J Clin Invest. (2004) 114(1):112–20. doi: 10.1172/JCI200420805
18. Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase-dependent generation of angiotensin II from angiotensin-(1-12) in human atrial tissue. PLoS One. (2011) 6(12):e28501. doi: 10.1371/journal.pone.0028501
19. Ahmad S, Wei CC, Tallaj J, Dell'Italia LJ, Moniwa N, Varagic J, et al. Chymase mediates angiotensin-(1-12) metabolism in normal human hearts. J Am Soc Hypertens. (2013) 7(2):128–36. doi: 10.1016/j.jash.2012.12.003
20. Dell'Italia LJ, Collawn JF, Ferrario CM. Multifunctional role of chymase in acute and chronic tissue injury and remodeling. Circ Res. (2018) 122(2):319–36. doi: 10.1161/CIRCRESAHA.117.310978
21. Li J, Jubair S, Janicki JS. Estrogen inhibits mast cell chymase release to prevent pressure overload-induced adverse cardiac remodeling. Hypertension. (2015) 65(2):328–34. doi: 10.1161/HYPERTENSIONAHA.114.04238
22. Domenig O, Manzel A, Grobe N, Königshausen E, Kaltenecker CC, Kovarik JJ, et al. Neprilysin is a mediator of alternative renin-angiotensin-system activation in the murine and human kidney. Sci Rep. (2016) 6:33678. doi: 10.1038/srep33678
23. Kaltenecker CC, Domenig O, Kopecky C, Antlanger M, Poglitsch M, Berlakovich G, et al. Critical role of neprilysin in kidney angiotensin metabolism. Circ Res. (2020) 127(5):593–606. doi: 10.1161/CIRCRESAHA.119.316151
24. Pendergrass KD, Pirro NT, Westwood BM, Ferrario CM, Brosnihan KB, Chappell MC. Sex differences in circulating and renal angiotensins of hypertensive mRen(2). Lewis but not normotensive lewis rats. Am J Physiol Heart Circ Physiol. (2008) 295(1):H10–20. doi: 10.1152/ajpheart.01277.2007
25. Ahmad S, Sun X, Lin M, Varagic J, Zapata-Sudo G, Ferrario CM, et al. Blunting of estrogen modulation of cardiac cellular chymase/RAS activity and function in SHR. J Cell Physiol. (2018) 233(4):3330–42. doi: 10.1002/jcp.26179
26. da Silva JS, Gabriel-Costa D, Wang H, Ahmad S, Sun X, Varagic J, et al. Blunting of cardioprotective actions of estrogen in female rodent heart linked to altered expression of cardiac tissue chymase and ACE2. J Renin Angiotensin Aldosterone Syst. (2017) 18(3):1470320317722270. doi: 10.1177/1470320317722270
27. Elton TS, Stephan CC, Taylor GR, Kimball MG, Martin MM, Durand JN, et al. Isolation of two distinct type I angiotensin II receptor genes. Biochem Biophys Res Commun. (1992) 184(2):1067–73. doi: 10.1016/0006-291X(92)90700-U
28. Iwai N, Inagami T. Isolation of preferentially expressed genes in the kidneys of hypertensive rats. Hypertension. (1991) 17(2):161–9.1991648
29. Murphy TJ, Alexander RW, Griendling KK, Runge MS, Bernstein KE. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature. (1991) 351(6323):233–6. doi: 10.1038/351233a0
30. Lin H, Geurts F, Hassler L, Batlle D, Mirabito Colafella KM, Denton KM, et al. Kidney angiotensin in cardiovascular disease: formation and drug targeting. Pharmacol Rev. (2022) 74(3):462–505. doi: 10.1124/pharmrev.120.000236
31. Kawai T, Forrester SJ, O’Brien S, Baggett A, Rizzo V, Eguchi S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol Res. (2017) 125(Pt A):4–13. doi: 10.1016/j.phrs.2017.05.008
32. Zhuo JL, Allen AM, Alcorn D, MacGregor D, Aldred GP, Mendelsohn FAO. The distribution of angiotensin II receptors. In: Laragh JH, Brenner BM, editors. Hypertension: Pathology, diagnosis, & management. 2nd ed. New York: Raven Press (1995). p. 1739–62.
33. Li XC, Zhou X, Zhuo JL. Evidence for a physiological mitochondrial angiotensin II system in the kidney proximal tubules. Hypertension. (2020) 76(1):121–32. doi: 10.1161/HYPERTENSIONAHA.119.13942
34. Kemp BA, Howell NL, Keller SR, Gildea JJ, Padia SH, Carey RM. AT2 receptor activation prevents sodium retention and reduces blood pressure in angiotensin II-dependent hypertension. Circ Res. (2016) 119(4):532–43. doi: 10.1161/CIRCRESAHA.116.308384
35. Nwia SM, Li XC, Leite APO, Hassan R, Zhuo JL. The Na+/H+ exchanger 3 in the intestines and the proximal tubule of the kidney: localization, physiological function, and key roles in angiotensin II-induced hypertension. Front Physiol. (2022) 13:861659. doi: 10.3389/fphys.2022.861659
36. Li XC, Zhu D, Chen X, Zheng X, Zhao C, Zhang J, et al. Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) in the kidney attenuates ang II (angiotensin II)-induced hypertension in mice. Hypertension. (2019) 74(3):526–35. doi: 10.1161/HYPERTENSIONAHA.119.13094
37. Kemp BA, Bell JF, Rottkamp DM, Howell NL, Shao W, Navar LG, et al. Intrarenal angiotensin III is the predominant agonist for proximal tubule angiotensin type 2 receptors. Hypertension. (2012) 60(2):387–95. doi: 10.1161/HYPERTENSIONAHA.112.191403
38. Padia SH, Howell NL, Siragy HM, Carey RM. Renal angiotensin type 2 receptors mediate natriuresis via angiotensin III in the angiotensin II type 1 receptor-blocked rat. Hypertension. (2006) 47(3):537–44. doi: 10.1161/01.HYP.0000196950.48596.21
39. Padia SH, Kemp BA, Howell NL, Fournie-Zaluski MC, Roques BP, Carey RM. Conversion of renal angiotensin II to angiotensin III is critical for AT2 receptor-mediated natriuresis in rats. Hypertension. (2008) 51(2):460–5. doi: 10.1161/HYPERTENSIONAHA.107.103242
40. Siragy HM, Inagami T, Ichiki T, Carey RM. Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proc Natl Acad Sci U S A. (1999) 96(11):6506–10. doi: 10.1073/pnas.96.11.6506
41. Hakam AC, Hussain T. Angiotensin II AT2 receptors inhibit proximal tubular Na+-K+-ATPase activity via a NO/cGMP-dependent pathway. Am J Physiol Renal Physiol. (2006) 290(6):F1430–6. doi: 10.1152/ajprenal.00218.2005
42. Hakam AC, Hussain T. Angiotensin II type 2 receptor agonist directly inhibits proximal tubule sodium pump activity in obese but not in lean zucker rats. Hypertension. (2006) 47(6):1117–24. doi: 10.1161/01.HYP.0000220112.91724.fc
43. Zhuo JL, Li XC. Angiotensin III/AT2 receptor/NHE3 signaling pathway in the proximal tubules of the kidney: a novel natriuretic and antihypertensive mechanism in hypertension. J Am Heart Assoc. (2019) 8(9):e012644. doi: 10.1161/JAHA.119.012644
44. Kemp BA, Howell NL, Keller SR, Gildea JJ, Shao W, Navar LG, et al. Defective renal angiotensin III and AT2 receptor signaling in prehypertensive spontaneously hypertensive rats. J Am Heart Assoc. (2019) 8(9):e012016.31039659
45. Balla T, Baukal AJ, Eng S, Catt KJ. Angiotensin II receptor subtypes and biological responses in the adrenal cortex and medulla. Mol Pharmacol. (1991) 40(3):401–6.1654513
46. Yatabe J, Yoneda M, Yatabe MS, Watanabe T, Felder RA, Jose PA, et al. Angiotensin III stimulates aldosterone secretion from adrenal gland partially via angiotensin II type 2 receptor but not angiotensin II type 1 receptor. Endocrinology. (2011) 152(4):1582–8. doi: 10.1210/en.2010-1070
47. Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, et al. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci. (1999) 96(5):2514–9. doi: 10.1073/pnas.96.5.2514
48. Soundararajan R, Pearce D, Ziera T. The role of the ENaC-regulatory complex in aldosterone-mediated sodium transport. Mol Cell Endocrinol. (2012) 350(2):242–7. doi: 10.1016/j.mce.2011.11.003
49. Mazzocchi G, Gottardo G, Macchi V, Malendowicz LK, Nussdorfer GG. The AT2 receptor-mediated stimulation of adrenal catecholamine release may potentiate the AT1 receptor-mediated aldosterone secretagogue action of angiotensin-II in rats. Endocr Res. (1998) 24(1):17–28. doi: 10.3109/07435809809031866
50. Cavadas C, Grand D, Mosimann F, Cotrim MD, Fontes Ribeiro CA, Brunner HR, et al. Angiotensin II mediates catecholamine and neuropeptide Y secretion in human adrenal chromaffin cells through the AT1 receptor. Regul Pept. (2003) 111(1–3):61–5. doi: 10.1016/S0167-0115(02)00253-7
51. Gammelgaard I, Wamberg S, Bie P. Systemic effects of angiotensin III in conscious dogs during acute double blockade of the renin-angiotensin-aldosterone-system. Acta Physiol Oxf Engl. (2006) 188(2):129–38. doi: 10.1111/j.1748-1716.2006.01595.x
52. Oki K, Kopf PG, Campbell WB, Luis Lam M, Yamazaki T, Gomez-Sanchez CE, et al. Angiotensin II and III metabolism and effects on steroid production in the HAC15 human adrenocortical cell line. Endocrinology. (2013) 154(1):214–21. doi: 10.1210/en.2012-1557
53. Wamberg C, Plovsing RR, Sandgaard NCF, Bie P. Effects of different angiotensins during acute, double blockade of the renin system in conscious dogs. Am J Physiol Regul Integr Comp Physiol. (2003) 285(5):R971–80. doi: 10.1152/ajpregu.00262.2003
54. Plovsing RR, Wamberg C, Sandgaard NCF, Simonsen JA, Holstein-Rathlou NH, Hoilund-Carlsen PF, et al. Effects of truncated angiotensins in humans after double blockade of the renin system. Am J Physiol Regul Integr Comp Physiol. (2003) 285(5):R981–91. doi: 10.1152/ajpregu.00263.2003
55. Freel EM, Connell JMC. Mechanisms of hypertension: the expanding role of aldosterone. J Am Soc Nephrol JASN. (2004) 15(8):1993–2001. doi: 10.1097/01.ASN.0000132473.50966.14
56. Nagata D, Takahashi M, Sawai K, Tagami T, Usui T, Shimatsu A, et al. Molecular mechanism of the inhibitory effect of aldosterone on endothelial NO synthase activity. Hypertension. (2006) 48(1):165–71. doi: 10.1161/01.HYP.0000226054.53527.bb
57. Serfozo P, Wysocki J, Gulua G, Schulze A, Ye M, Liu P, et al. Ang II (angiotensin II) conversion to angiotensin-(1-7) in the circulation is POP (prolyloligopeptidase)-dependent and ACE2 (angiotensin-converting enzyme 2)-independent. Hypertension. (2020) 75(1):173–82. doi: 10.1161/HYPERTENSIONAHA.119.14071
58. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. (2000) 87(5):e1–9. doi: 10.1161/01.RES.87.5.e1
59. Santos RA, Simoes e Silva ACS, Maric C, Silva DMR, Machado RP, de Buhr I, et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. (2003) 100(14):8258–63. doi: 10.1073/pnas.1432869100
60. Sasaki S, Higashi Y, Nakagawa K, Matsuura H, Kajiyama G, Oshima T. Effects of angiotensin-(1-7) on forearm circulation in normotensive subjects and patients with essential hypertension. Hypertension. (2001) 38(1):90–4. doi: 10.1161/01.HYP.38.1.90
61. Schinzari F, Tesauro M, Veneziani A, Mores N, Di Daniele N, Cardillo C. Favorable vascular actions of angiotensin-(1–7) in human obesity. Hypertension. (2018) 71(1):185–91. doi: 10.1161/HYPERTENSIONAHA.117.10280
62. Ueda S, Masumori-Maemoto S, Wada A, Ishii M, Brosnihan KB, Umemura S. Angiotensin(1-7) potentiates bradykinin-induced vasodilatation in man. J Hypertens. (2001) 19(11):2001–9. doi: 10.1097/00004872-200111000-00010
63. Cm F HN, Sb F, Dl R EW, Rh D. Angiotensin-(1-7) and nitric oxide interaction in renovascular hypertension. Hypertension. (1995) 25(4 Pt 2):796–802. Available at: https://pubmed.ncbi.nlm.nih.gov/7536715/7536715
64. Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension. (2007) 49(1):185–92. doi: 10.1161/01.HYP.0000251865.35728.2f
65. Shefer G, Marcus Y, Knoll E, Dolkart O, Foichtwanger S, Nevo N, et al. Angiotensin 1–7 is a negative modulator of aldosterone secretion in vitro and in vivo. Hypertension. (2016) 68(2):378–84. doi: 10.1161/HYPERTENSIONAHA.116.07088
66. Achard V, Tassistro V, Boullu-Ciocca S, Grino M. Expression and nutritional regulation of the (pro)renin receptor in rat visceral adipose tissue. J Endocrinol Invest. (2011) 34(11):840–6. doi: 10.3275/7627
67. Achard V, Boullu-Ciocca S, Desbriere R, Nguyen G, Grino M. Renin receptor expression in human adipose tissue. Am J Physiol Regul Integr Comp Physiol. (2007) 292(1):R274–82. doi: 10.1152/ajpregu.00439.2005
68. Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. (2002) 109(11):1417–27. doi: 10.1172/JCI0214276
69. Feldman DL, Jin L, Xuan H, Contrepas A, Zhou Y, Webb RL, et al. Effects of aliskiren on blood pressure, albuminuria, and (pro)renin receptor expression in diabetic TG(mRen-2)27 rats. Hypertension. (2008) 52(1):130–6. doi: 10.1161/HYPERTENSIONAHA.107.108845
70. Contrepas A, Walker J, Koulakoff A, Franek KJ, Qadri F, Giaume C, et al. A role of the (pro)renin receptor in neuronal cell differentiation. Am J Physiol Regul Integr Comp Physiol. (2009) 297(2):R250–7. doi: 10.1152/ajpregu.90832.2008
71. Burcklé CA, Jan Danser AH, Müller DN, Garrelds IM, Gasc JM, Popova E, et al. Elevated blood pressure and heart rate in human renin receptor transgenic rats. Hypertension. (2006) 47(3):552–6. doi: 10.1161/01.HYP.0000199912.47657.04
72. Nguyen G. The (pro)renin receptor: pathophysiological roles in cardiovascular and renal pathology. Curr Opin Nephrol Hypertens. (2007) 16(2):129–33. doi: 10.1097/MNH.0b013e328040bfab
73. Ramkumar N, Stuart D, Calquin M, Quadri S, Wang S, Van Hoek AN, et al. Nephron-specific deletion of the prorenin receptor causes a urine concentration defect. Am J Physiol Ren Physiol. (2015) 309(1):F48–56. doi: 10.1152/ajprenal.00126.2015
74. Wang F, Lu X, Peng K, Fang H, Zhou L, Su J, et al. Antidiuretic action of collecting duct (pro)Renin receptor downstream of vasopressin and PGE2 receptor EP4. J Am Soc Nephrol JASN. (2016) 27(10):3022–34. doi: 10.1681/ASN.2015050592
75. Ramkumar N, Stuart D, Mironova E, Abraham N, Gao Y, Wang S, et al. Collecting duct principal, but not intercalated, cell prorenin receptor regulates renal sodium and water excretion. Am J Physiol Ren Physiol. (2018) 315(3):F607–17. doi: 10.1152/ajprenal.00122.2018
76. Prieto MC, Reverte V, Mamenko M, Kuczeriszka M, Veiras LC, Rosales CB, et al. Collecting duct prorenin receptor knockout reduces renal function, increases sodium excretion, and mitigates renal responses in ANG II-induced hypertensive mice. Am J Physiol Ren Physiol. (2017) 313(6):F1243–53. doi: 10.1152/ajprenal.00152.2017
77. Li C, Wang W, Rivard CJ, Lanaspa MA, Summer S, Schrier RW. Molecular mechanisms of angiotensin II stimulation on aquaporin-2 expression and trafficking. Am J Physiol Ren Physiol. (2011) 300(5):F1255–61. doi: 10.1152/ajprenal.00469.2010
78. Gonzalez AA, Lara LS, Luffman C, Seth DM, Prieto MC. Soluble form of the (pro)renin receptor is augmented in the collecting duct and urine of chronic angiotensin II-dependent hypertensive rats. Hypertension. (2011) 57(4):859–64. doi: 10.1161/HYPERTENSIONAHA.110.167957
79. Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF, et al. Collecting duct (pro)renin receptor targets ENaC to mediate angiotensin II-induced hypertension. Am J Physiol Ren Physiol. (2017) 312(2):F245–53. doi: 10.1152/ajprenal.00178.2016
80. Ramkumar N, Stuart D, Mironova E, Bugay V, Wang S, Abraham N, et al. Renal tubular epithelial cell prorenin receptor regulates blood pressure and sodium transport. Am J Physiol Ren Physiol. (2016) 311(1):F186–94. doi: 10.1152/ajprenal.00088.2016
81. Arthur G, Osborn JL, Yiannikouris FB. (Pro)renin receptor in the kidney: function and significance. Am J Physiol Regul Integr Comp Physiol. (2021) 320(4):R377–83. doi: 10.1152/ajpregu.00259.2020
82. Li XC, Shao Y, Zhuo JL. AT1a receptor knockout in mice impairs urine concentration by reducing basal vasopressin levels and its receptor signaling proteins in the inner medulla. Kidney Int. (2009) 76(2):169–77. doi: 10.1038/ki.2009.134
83. Li H, Weatherford ET, Davis DR, Keen HL, Grobe JL, Daugherty A, et al. Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am J Physiol Regul Integr Comp Physiol. (2011) 301(4):R1067–77. doi: 10.1152/ajpregu.00124.2011
84. Li XC, Hopfer U, Zhuo JL. Novel signaling mechanisms of intracellular angiotensin II-induced NHE3 expression and activation in mouse proximal tubule cells. Am J Physiol Renal Physiol. (2012) 303(12):F1617–28. doi: 10.1152/ajprenal.00219.2012
85. Gurley SB, Riquier ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, et al. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. (2011) 13(4):469–75. doi: 10.1016/j.cmet.2011.03.001
86. Mendelsohn FA. Angiotensin II: evidence for its role as an intrarenal hormone. Kidney Int Suppl. (1982) 12:S78–81.6182339
87. Navar LG, Kobori H, Prieto-Carrasquero M. Intrarenal angiotensin II and hypertension. Curr Hypertens Rep. (2003) 5(2):135–43. doi: 10.1007/s11906-003-0070-5
88. Carey RM. The intrarenal renin-angiotensin system in hypertension. Adv Chronic Kidney Dis. (2015) 22(3):204–10. doi: 10.1053/j.ackd.2014.11.004
89. Zhuo JL, Ferrao FM, Zheng Y, Li XC. New frontiers in the intrarenal renin-angiotensin system: a critical review of classical and new paradigms. Front Endocrinol. (2013) 4:166. doi: 10.3389/fendo.2013.00166
90. Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. (2006) 86(3):747–803. doi: 10.1152/physrev.00036.2005
91. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. (2007) 59(3):251–87. doi: 10.1124/pr.59.3.3
92. Ellis B, Li XC, Miguel-Qin E, Gu V, Zhuo JL. Evidence for a functional intracellular angiotensin system in the proximal tubule of the kidney. Am J Physiol Regul Integr Comp Physiol. (2012) 302(5):R494–509. doi: 10.1152/ajpregu.00487.2011
93. Ingelfinger JR, Zuo WM, Fon EA, Ellison KE, Dzau VJ. In situ hybridization evidence for angiotensinogen messenger RNA in the rat proximal tubule. An hypothesis for the intrarenal renin angiotensin system. J Clin Invest. (1990) 85(2):417–23. doi: 10.1172/JCI114454
94. Harrison-Bernard LM, Zhuo J, Kobori H, Ohishi M, Navar LG. Intrarenal AT1 receptor and ACE binding in ANG II-induced hypertensive rats. Am J Physiol Renal Physiol. (2002) 282(1):F19–25. doi: 10.1152/ajprenal.0335.2000
95. Harrison-Bernard LM, El-Dahr SS, O’Leary DF, Navar LG. Regulation of angiotensin II type 1 receptor mRNA and protein in angiotensin II-induced hypertension. Hypertension. (1999) 33(1 Pt 2):340–6.9931127
96. Eggena P, Zhu JH, Clegg K, Barrett JD. Nuclear angiotensin receptors induce transcription of renin and angiotensinogen mRNA. Hypertension. (1993) 22(4):496–501.8406654
97. Booz GW, Conrad KM, Hess AL, Singer HA, Baker KM. Angiotensin-II-binding sites on hepatocyte nuclei. Endocrinology. (1992) 130(6):3641–9. doi: 10.1210/endo.130.6.1597161
98. Eggena P, Zhu JH, Sereevinyayut S, Giordani M, Clegg K, Andersen PC, et al. Hepatic angiotensin II nuclear receptors and transcription of growth-related factors. J Hypertens. (1996) 14(8):961–8. doi: 10.1097/00004872-199608000-00005
99. Tang SS, Rogg H, Schumacher R, Dzau VJ. Characterization of nuclear angiotensin-II-binding sites in rat liver and comparison with plasma membrane receptors. Endocrinology. (1992) 131(1):374–80. doi: 10.1210/endo.131.1.1612017
100. Li XC, Zhuo JL. Intracellular ANG II directly induces in vitro transcription of TGF-beta1, MCP-1, and NHE-3 mRNAs in isolated rat renal cortical nuclei via activation of nuclear AT1a receptors. Am J Physiol Cell Physiol. (2008) 294(4):C1034–45. doi: 10.1152/ajpcell.00432.2007
101. Gwathmey TM, Shaltout HA, Pendergrass KD, Pirro NT, Figueroa JP, Rose JC, et al. Nuclear angiotensin II type 2 (AT2) receptors are functionally linked to nitric oxide production. Am J Physiol Ren Physiol. (2009) 296(6):F1484–93. doi: 10.1152/ajprenal.90766.2008
102. Gwathmey TM, Pendergrass KD, Reid SD, Rose JC, Diz DI, Chappell MC. Angiotensin-(1-7)-ACE2 attenuates reactive oxygen species formation to angiotensin II within the cell nucleus. Hypertension. (2010) 55(1):166. doi: 10.1161/HYPERTENSIONAHA.109.141622
103. Gwathmey TM, Westwood BM, Pirro NT, Tang L, Rose JC, Diz DI, et al. Nuclear angiotensin-(1–7) receptor is functionally coupled to the formation of nitric oxide. Am J Physiol Ren Physiol. (2010) 299(5):F983–90. doi: 10.1152/ajprenal.00371.2010
104. Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A. (2011) 108(36):14849–54. doi: 10.1073/pnas.1101507108
105. Wilson BA, Nautiyal M, Gwathmey TM, Rose JC, Chappell MC. Evidence for a mitochondrial angiotensin-(1–7) system in the kidney. Am J Physiol Ren Physiol. (2016) 310(7):F637–45. doi: 10.1152/ajprenal.00479.2015
106. Re RN. Role of intracellular angiotensin II. Am J Physiol Heart Circ Physiol. (2018) 314(4):H766–71. doi: 10.1152/ajpheart.00632.2017
107. Li XC, Zhu D, Zheng X, Zhang J, Roman RJ, Zhuo JL. Intratubular and intracellular renin-angiotensin system in the kidney: a unifying perspective in blood pressure control. Clin Sci Lond Engl 1979. (2018) 132(13):1383–401. doi: 10.1042/CS20180121
108. World Health Organization. Guideline for the pharmacological treatment of hypertension in adults. Geneva: World Health Organization (2021). Available at: https://apps.who.int/iris/handle/10665/344424 (Cited October 3, 2022).
109. Unger T, Borghi C, Charchar F, Khan NA, Poulter NR, Prabhakaran D, et al. 2020 international society of hypertension global hypertension practice guidelines. Hypertension. (2020) 75(6):1334–57. doi: 10.1161/HYPERTENSIONAHA.120.15026
110. Laurent S. Antihypertensive drugs. Pharmacol Res. (2017) 124:116–25. doi: 10.1016/j.phrs.2017.07.026
111. Chan You S, Krumholz HM, Suchard MA, Schuemie MJ, Hripcsak G, Chen R, et al. Comprehensive comparative effectiveness and safety of first-line β-blocker monotherapy in hypertensive patients. Hypertension. (2021) 77(5):1528–38. doi: 10.1161/HYPERTENSIONAHA.120.16402
112. Ghatage T, Goyal SG, Dhar A, Bhat A. Novel therapeutics for the treatment of hypertension and its associated complications: peptide- and nonpeptide-based strategies. Hypertens Res. (2021) 44(7):740–55. doi: 10.1038/s41440-021-00643-z
113. Arnold A. Cardiovascular effects of angiotensin 1-7 in obesity hypertension. clinicaltrials.gov Report No.: NCT03604289 (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT03604289 (Cited October 6, 2022).
114. University of Wisconsin, Madison. Angiotensin 2 receptor (AT2R) expression/activation in endothelial cells in Preeclampsia. clinicaltrials.gov. Report No.: NCT03806283 (2021). Available at: https://clinicaltrials.gov/ct2/show/NCT03806283 (Cited October 6, 2022).
115. Ostchega Y, Fryar CD, Nwankwo T, Nguyen DT. Hypertension prevalence among adults aged 18 and over: United States, 2017–2018. NCHS Data Brief. (2020) (364):1–8. 32487290.32487290
116. Connelly PJ, Currie G, Delles C. Sex differences in the prevalence, outcomes and management of hypertension. Curr Hypertens Rep. (2022) 24(6):185–92. doi: 10.1007/s11906-022-01183-8
117. Panza JA, Quyyumi AA, Brush JE, Epstein SE. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N Engl J Med. (1990) 323(1):22–7. doi: 10.1056/NEJM199007053230105
118. Node K, Kitakaze M, Yoshikawa H, Kosaka H, Hori M. Reduced plasma concentrations of nitrogen oxide in individuals with essential hypertension. Hypertension. (1997) 30(3 Pt 1):405–8.9314424
119. Fry BC, Edwards A, Layton AT. Impact of nitric-oxide-mediated vasodilation and oxidative stress on renal medullary oxygenation: a modeling study. Am J Physiol Ren Physiol. (2016) 310(3):F237–47. doi: 10.1152/ajprenal.00334.2015
120. Sullivan JC, Pardieck JL, Hyndman KA, Pollock JS. Renal NOS activity, expression, and localization in male and female spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. (2010) 298(1):R61–9. doi: 10.1152/ajpregu.00526.2009
121. Lopez-Ruiz A, Sartori-Valinotti J, Yanes LL, Iliescu R, Reckelhoff JF. Sex differences in control of blood pressure: role of oxidative stress in hypertension in females. Am J Physiol Heart Circ Physiol. (2008) 295(2):H466–474. doi: 10.1152/ajpheart.01232.2007
122. Zhen Y, Xiao S, Ren Z, Shen H, Su H, Tang Y, et al. Increased endothelial progenitor cells and nitric oxide in young prehypertensive women. J Clin Hypertens. (2015) 17(4):298–305. doi: 10.1111/jch.12493
123. Sader MA, Celermajer DS. Endothelial function, vascular reactivity and gender differences in the cardiovascular system. Cardiovasc Res. (2002) 53(3):597–604. doi: 10.1016/S0008-6363(01)00473-4
124. Forte P, Kneale BJ, Milne E, Chowienczyk PJ, Johnston A, Benjamin N, et al. Evidence for a difference in nitric oxide biosynthesis between healthy women and men. Hypertension. (1998) 32(4):730–4.9774371
125. Chen Y, Sullivan JC, Edwards A, Layton AT. Sex-specific computational models of the spontaneously hypertensive rat kidneys: factors affecting nitric oxide bioavailability. Am J Physiol Ren Physiol. (2017) 313(2):F174–83. doi: 10.1152/ajprenal.00482.2016
126. Jaimes EA, Galceran JM, Raij L. Angiotensin II induces superoxide anion production by mesangial cells. Kidney Int. (1998) 54(3):775–84. doi: 10.1046/j.1523-1755.1998.00068.x
127. de Cavanagh EMV, Inserra F, Ferder M, Ferder L. From mitochondria to disease: role of the renin-angiotensin system. Am J Nephrol. (2007) 27(6):545–53. doi: 10.1159/000107757
128. Banday AA, Lokhandwala MF. Oxidative stress-induced renal angiotensin AT1 receptor upregulation causes increased stimulation of sodium transporters and hypertension. Am J Physiol Ren Physiol. (2008) 295(3):F698–706. doi: 10.1152/ajprenal.90308.2008
129. Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertension. (2001) 38(3):635–8. doi: 10.1161/hy09t1.094234
130. Visniauskas B, Kilanowski-Doroh I, Ogola BO, Mcnally AB, Horton AC, Imulinde Sugi A, et al. Estrogen-mediated mechanisms in hypertension and other cardiovascular diseases. J Hum Hypertens. (2022) doi: 10.1038/s41371-022-00771-0. [Epub ahead of print].36319856
131. Thomas WG, Thekkumkara TJ, Baker KM. Molecular mechanisms of angiotensin II (AT1A) receptor endocytosis. Clin Exp Pharmacol Physiol Suppl. (1996) 3:S74–80. doi: 10.1111/j.1440-1681.1996.tb02817.x
132. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. (2005) 307(5715):1625–30. doi: 10.1126/science.1106943
133. Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F. Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids. (2014) 0:13–29. doi: 10.1016/j.steroids.2014.06.012
134. Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors alpha and beta. J Biol Chem. (2005) 280(20):19704–10. doi: 10.1074/jbc.M501244200
135. Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science. (2002) 295(5554):505–8. doi: 10.1126/science.1065250
136. Esqueda MD, Craig T, Hinojosa-Laborde C. Effect of ovariectomy on renal estrogen receptor-α and estrogen receptor-β in young salt-sensitive and -resistant rats. Hypertension. (2007) 50(4):768–72. doi: 10.1161/HYPERTENSIONAHA.107.095265
137. Masubuchi Y, Kumai T, Uematsu A, Komoriyama K, Hirai M. Gonadectomy-induced reduction of blood pressure in adult spontaneously hypertensive rats. Acta Endocrinol (Copenh). (1982) 101(1):154–60.7124289
138. Ganten U, Schröder G, Witt M, Zimmermann F, Ganten D, Stock G. Sexual dimorphism of blood pressure in spontaneously hypertensive rats: effects of anti-androgen treatment. J Hypertens. (1989) 7(9):721–6. doi: 10.1097/00004872-198909000-00005
139. Reckelhoff JF, Zhang H, Srivastava K. Gender differences in development of hypertension in spontaneously hypertensive rats. Hypertension. (2000) 35(1):480–3. doi: 10.1161/01.HYP.35.1.480
140. Silva-Antonialli MM, Tostes RCA, Fernandes L, Fior-Chadi DR, Akamine EH, Carvalho MHC, et al. A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc Res. (2004) 62(3):587–93. doi: 10.1016/j.cardiores.2004.01.020
141. Hinojosa-Laborde C, Craig T, Zheng W, Ji H, Haywood JR, Sandberg K. Ovariectomy augments hypertension in aging female dahl salt-sensitive rats. Hypertension. (2004) 44(4):405–9. doi: 10.1161/01.HYP.0000142893.08655.96
142. Fortepiani LA, Zhang H, Racusen L, Roberts LJ, Reckelhoff JF. Characterization of an animal model of postmenopausal hypertension in spontaneously hypertensive rats. Hypertension. (2003) 41(3):640–5. doi: 10.1161/01.HYP.0000046924.94886.EF
143. Reckelhoff JF, Fortepiani LA. Novel mechanisms responsible for postmenopausal hypertension. Hypertension. (2004) 43(5):918–23. doi: 10.1161/01.HYP.0000142893.08655.96
144. Proudler AJ, Ahmed AI, Crook D, Fogelman I, Rymer JM, Stevenson JC. Hormone replacement therapy and serum angiotensin-converting-enzyme activity in postmenopausal women. Lancet Lond Engl. (1995) 346(8967):89–90. doi: 10.1016/S0140-6736(95)92114-1
145. Schunkert H, Danser AH, Hense HW, Derkx FH, Kürzinger S, Riegger GA. Effects of estrogen replacement therapy on the renin-angiotensin system in postmenopausal women. Circulation. (1997) 95(1):39–45. doi: 10.1161/01.CIR.95.1.39
146. Gallagher PE, Li P, Lenhart JR, Chappell MC, Brosnihan KB. Estrogen regulation of angiotensin-converting enzyme mRNA. Hypertension. (1999) 33(1):323–8. doi: 10.1161/01.HYP.33.1.323
147. Lee SH, Lee YH, Jung SW, Kim DJ, Park SH, Song SJ, et al. Sex-related differences in the intratubular renin-angiotensin system in two-kidney, one-clip hypertensive rats. Am J Physiol Ren Physiol. (2019) 317(3):F670–82. doi: 10.1152/ajprenal.00451.2018
148. Yanes LL, Romero DG, Iles JW, Iliescu R, Gomez-Sanchez C, Reckelhoff JF. Sexual dimorphism in the renin-angiotensin system in aging spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. (2006) 291(2):R383–90. doi: 10.1152/ajpregu.00510.2005
149. Nickenig G, Bäumer AT, Grohè C, Kahlert S, Strehlow K, Rosenkranz S, et al. Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation. (1998) 97(22):2197–201. doi: 10.1161/01.CIR.97.22.2197
150. Roesch DM, Tian Y, Zheng W, Shi M, Verbalis JG, Sandberg K. Estradiol attenuates angiotensin-induced aldosterone secretion in ovariectomized rats. Endocrinology. (2000) 141(12):4629–36. doi: 10.1210/endo.141.12.7822
151. Toering TJ, Gant CM, Visser FW, van der Graaf AM, Laverman GD, Danser AHJ, et al. Sex differences in renin-angiotensin-aldosterone system affect extracellular volume in healthy subjects. Am J Physiol Renal Physiol. (2018) 314(5):F873–8. doi: 10.1152/ajprenal.00109.2017
152. Visniauskas B, Arita DY, Rosales CB, Feroz MA, Luffman C, Accavitti MJ, et al. Sex differences in soluble prorenin receptor in patients with type 2 diabetes. Biol Sex Differ. (2021) 12(1):33. doi: 10.1186/s13293-021-00374-3
153. Mompeón A, Lázaro-Franco M, Bueno-Betí C, Pérez-Cremades D, Vidal-Gómez X, Monsalve E, et al. Estradiol, acting through ERα, induces endothelial non-classic renin-angiotensin system increasing angiotensin 1-7 production. Mol Cell Endocrinol. (2016) 422:1–8. doi: 10.1016/j.mce.2015.11.004
154. Dibo P, Marañón RO, Chandrashekar K, Mazzuferi F, Silva GB, Juncos LA, et al. Angiotensin-(1-7) inhibits sodium transport via Mas receptor by increasing nitric oxide production in thick ascending limb. Physiol Rep. (2019) 7(5):e14015. doi: 10.14814/phy2.14015
155. Yang G, Istas G, Höges S, Yakoub M, Hendgen-Cotta U, Rassaf T, et al. Angiotensin-(1-7)-induced Mas receptor activation attenuates atherosclerosis through a nitric oxide-dependent mechanism in apolipoproteinE-KO mice. Pflüg Arch Eur J Physiol. (2018) 470(4):661–7. doi: 10.1007/s00424-018-2108-1
156. Sobrino A, Novella S, Monsalve E, Oviedo PJ, Laguna-Fernandez A, Bueno C, et al. Estradiol regulates renin–angiotensin system towards nitric oxide production through Mas receptor: PP.24.462. J Hypertens. (2010) 28:e385. doi: 10.1097/01.hjh.0000379388.11921.88
157. Safari T, Nematbakhsh M, Hilliard LM, Evans RG, Denton KM. Sex differences in the renal vascular response to angiotensin II involves the Mas receptor. Acta Physiol Oxf Engl. (2012) 206(2):150–6. doi: 10.1111/j.1748-1716.2012.02468.x
158. Pezeshki Z, Nematbakhsh M. Sex differences in the renal vascular responses of AT1 and Mas receptors in two-kidney-one-clip hypertension. Int J Hypertens. (2021) 2021:8820646. doi: 10.1155/2021/8820646
159. Saberi S, Dehghani A, Nematbakhsh M. Role of Mas receptor in renal blood flow response to angiotensin-(1-7) in ovariectomized estradiol treated rats. Res Pharm Sci. (2016) 11(1):65–72. 27051434 4794939.27051434
160. Brown RD, Hilliard LM, Head GA, Jones ES, Widdop RE, Denton KM. Sex differences in the pressor and tubuloglomerular feedback response to angiotensin II. Hypertension. (2012) 59(1):129–35. doi: 10.1161/HYPERTENSIONAHA.111.178715
161. Mirabito KM, Hilliard LM, Head GA, Widdop RE, Denton KM. Pressor responsiveness to angiotensin II in female mice is enhanced with age: role of the angiotensin type 2 receptor. Biol Sex Differ. (2014) 5(1):13. doi: 10.1186/s13293-014-0013-7
162. Barsha G, Mirabito Colafella KM, Walton SL, Gaspari TA, Spizzo I, Pinar AA, et al. In aged females, the enhanced pressor response to angiotensin II is attenuated by estrogen replacement via an angiotensin type 2 receptor-mediated mechanism. Hypertension. (2021) 78(1):128–37. doi: 10.1161/HYPERTENSIONAHA.121.17164
163. Reckelhoff JF, Zhang H, Granger JP. Testosterone exacerbates hypertension and reduces pressure-natriuresis in male spontaneously hypertensive rats. Hypertension. (1998) 31(1 Pt 2):435–9.9453341
164. Dalmasso C, Patil CN, Yanes Cardozo LL, Romero DG, Maranon RO. Cardiovascular and metabolic consequences of testosterone supplements in young and old male spontaneously hypertensive rats: implications for testosterone supplements in men. J Am Heart Assoc Cardiovasc Cerebrovasc Dis. (2017) 6(10):e007074. doi: 10.1161/JAHA.117.007074
165. Mishra JS, More AS, Gopalakrishnan K, Kumar S. Testosterone plays a permissive role in angiotensin II-induced hypertension and cardiac hypertrophy in male rats. Biol Reprod. (2019) 100(1):139–48. doi: 10.1093/biolre/ioy179
166. Mohammadi-Shemirani P, Chong M, Pigeyre M, Morton RW, Gerstein HC, Paré G. Effects of lifelong testosterone exposure on health and disease using mendelian randomization. eLife. (2020). 9:e58914. doi: 10.7554/eLife.58914
167. Li N, Ma R, Wang S, Zhao Y, Wang P, Yang Z, et al. The potential role of testosterone in hypertension and target organ damage in hypertensive postmenopausal women. Clin Interv Aging. (2019) 14:743–52. doi: 10.2147/CIA.S195498
168. Velho I, Fighera TM, Ziegelmann PK, Spritzer PM. Effects of testosterone therapy on BMI, blood pressure, and laboratory profile of transgender men: a systematic review. Andrology. (2017) 5(5):881–8. doi: 10.1111/andr.12382
169. Yang Q, Li Z, Li W, Lu L, Wu H, Zhuang Y, et al. Association of total testosterone, free testosterone, bioavailable testosterone, sex hormone–binding globulin, and hypertension. Medicine (Baltimore). (2019) 98(20):e15628. doi: 10.1097/MD.0000000000015628
170. Li XC, Leite APO, Zheng X, Zhao C, Chen X, Zhang L, et al. Proximal tubule-specific deletion of angiotensin II type 1a receptors in the kidney attenuates circulating and intratubular angiotensin II-induced hypertension in PT-Agtr1a−/− mice. Hypertension. (2021) 77(4):1285–98. doi: 10.1161/HYPERTENSIONAHA.120.16336
171. Leite APO, Li XC, Hassan R, Zheng X, Alexander B, Casarini DE, et al. Sex differences in angiotensin II-induced hypertension and kidney injury: role of AT1a receptors in the proximal tubule of the kidney. Clin Sci (Lond). (2021) 135(15):1825–43. doi: 10.1042/CS20201574
172. Veiras LC, McFarlin BE, Ralph DL, Buncha V, Prescott J, Shirvani BS, et al. Electrolyte and transporter responses to angiotensin II induced hypertension in female and male rats and mice. Acta Physiol. (2020) 229(1):e13448. doi: 10.1111/apha.13448
173. Venegas-Pont M, Sartori-Valinotti JC, Glover PH, Reckelhoff JF, Ryan MJ. Sexual dimorphism in the blood pressure response to angiotensin II in mice after angiotensin-converting enzyme blockade. Am J Hypertens. (2010) 23(1):92–6. doi: 10.1038/ajh.2009.203
174. Wolf E, Diaz EJ, Hollis AN, Hoang TA, Azad HA, Bendt KM, et al. Vascular type 1 angiotensin receptors control blood pressure by augmenting peripheral vascular resistance in female mice. Am J Physiol Renal Physiol. (2018) 315(4):F997–1005. doi: 10.1152/ajprenal.00639.2017
175. Xue B, Pamidimukkala J, Hay M. Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am J Physiol Heart Circ Physiol. (2005) 288(5):H2177–84. doi: 10.1152/ajpheart.00969.2004
176. Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet Lond Engl. (2004) 364(9438):937–52. doi: 10.1016/S0140-6736(04)17018-9
177. SPRINT Research Group, Lewis CE, Fine LJ, Beddhu S, Cheung AK, Cushman WC, et al. Final report of a trial of intensive versus standard blood-pressure control. N Engl J Med. (2021) 384(20):1921–30. doi: 10.1056/NEJMoa1901281
178. Thomopoulos C, Parati G, Zanchetti A. Effects of blood pressure lowering on outcome incidence in hypertension: 7. Effects of more vs. less intensive blood pressure lowering and different achieved blood pressure levels—updated overview and meta-analyses of randomized trials. J Hypertens. (2016) 34(4):613–22. doi: 10.1097/HJH.0000000000000881
179. Thoenes M, Neuberger HR, Volpe M, Khan BV, Kirch W, Böhm M. Antihypertensive drug therapy and blood pressure control in men and women: an international perspective. J Hum Hypertens. (2010) 24(5):336–44. doi: 10.1038/jhh.2009.76
180. Turnbull F, Woodward M, Neal B, Barzi F, Ninomiya T, Chalmers J, et al. Do men and women respond differently to blood pressure-lowering treatment? Results of prospectively designed overviews of randomized trials. Eur Heart J. (2008) 29(21):2669–80. doi: 10.1093/eurheartj/ehn427
181. Os I, Franco V, Kjeldsen SE, Manhem K, Devereux RB, Gerdts E, et al. Effects of losartan in women with hypertension and left ventricular hypertrophy: results from the losartan intervention for endpoint reduction in hypertension study. Hypertension. (2008) 51(4):1103–8. doi: 10.1161/HYPERTENSIONAHA.107.105296
182. Jamerson K, Weber MA, Bakris GL, Dahlöf B, Pitt B, Shi V, et al. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med. (2008) 359(23):2417–28. doi: 10.1056/NEJMoa0806182
183. Freire AC, Basit AW, Choudhary R, Piong CW, Merchant HA. Does sex matter? The influence of gender on gastrointestinal physiology and drug delivery. Int J Pharm. (2011) 415(1–2):15–28. doi: 10.1016/j.ijpharm.2011.04.069
184. Rabi D, Khan N, Vallee M, Hladunewich M, Tobe S, Pilote L. Reporting on sex-based analysis in clinical trials of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker efficacy. Can J Cardiol. (2008) 24(6):491–6. doi: 10.1016/S0828-282X(08)70624-X
185. Bots SH, Schreuder MM, Roeters van Lennep JE, Watson S, van Puijenbroek E, Onland-Moret NC, et al. Sex differences in reported adverse drug reactions to angiotensin-converting enzyme inhibitors. JAMA Netw Open. (2022) 5(4):e228224. doi: 10.1001/jamanetworkopen.2022.8224
186. Rydberg DM, Mejyr S, Loikas D, Schenck-Gustafsson K, von Euler M, Malmström RE. Sex differences in spontaneous reports on adverse drug events for common antihypertensive drugs. Eur J Clin Pharmacol. (2018) 74(9):1165–73. doi: 10.1007/s00228-018-2480-y
187. Santoro N, Allshouse A, Neal-Perry G, Pal L, Lobo RA, Naftolin F, et al. Longitudinal changes in menopausal symptoms comparing women randomized to low-dose oral conjugated estrogens or transdermal estradiol plus micronized progesterone versus placebo: the kronos early estrogen prevention study. Menopause N Y N. (2017) 24(3):238–46. doi: 10.1097/GME.0000000000000756
188. Steingold KA, Laufer L, Chetkowski RJ, DeFazio JD, Matt DW, Meldrum DR, et al. Treatment of hot flashes with transdermal estradiol administration. J Clin Endocrinol Metab. (1985) 61(4):627–32. doi: 10.1210/jcem-61-4-627
189. Binder EF, Williams DB, Schechtman KB, Jeffe DB, Kohrt WM. Effects of hormone replacement therapy on serum lipids in elderly women. a randomized, placebo-controlled trial. Ann Intern Med. (2001) 134(9 Pt 1):754–60. doi: 10.7326/0003-4819-134-9_Part_1-200105010-00012
190. Darling GM, Johns JA, McCloud PI, Davis SR. Estrogen and progestin compared with simvastatin for hypercholesterolemia in postmenopausal women. N Engl J Med. (1997) 337(9):595–601. doi: 10.1056/NEJM199708283370903
191. Walsh BW, Schiff I, Rosner B, Greenberg L, Ravnikar V, Sacks FM. Effects of postmenopausal estrogen replacement on the concentrations and metabolism of plasma lipoproteins. N Engl J Med. (1991) 325(17):1196–204. doi: 10.1056/NEJM199110243251702
Keywords: cardiovascular, hypertension, kidney, renin-Angiotensin system, sex differences
Citation: Nwia SM, Leite APO, Li XC and Zhuo JL (2023) Sex differences in the renin-angiotensin-aldosterone system and its roles in hypertension, cardiovascular, and kidney diseases. Front. Cardiovasc. Med. 10:1198090. doi: 10.3389/fcvm.2023.1198090
Received: 31 March 2023; Accepted: 6 June 2023;
Published: 19 June 2023.
Edited by:
Hong Wang, Temple University, United StatesReviewed by:
Sarfaraz Ahmad, Wake Forest University, United StatesKazutaka Ueda, The University of Tokyo, Japan
© 2023 Nwia, Leite, Li and Zhuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Long Zhuo anpodW9AdHVsYW5lLmVkdQ==