 Erica Gianazza1
Erica Gianazza1 Chiara Macchi
Chiara Macchi Massimiliano Ruscica
Massimiliano Ruscica
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 12 June 2023
Sec. Precision Cardiology
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1191303
This article is part of the Research Topic Clinical Implementation of Genetic Risk Scores in Cardiovascular Medicine View all 7 articles
Proprotein convertase subtilisin/kexin type 9 (PCSK9), one of the key regulators of the low-density lipoprotein receptor (LDLR), can play a direct role in atheroma development. Although advances in the understandings of genetic PCSK9 polymorphisms have enabled to reveal the role of PCSK9 in the complex pathophysiology of cardiovascular diseases (CVDs), increasing lines of evidence support non-cholesterol-related processes mediated by PCSK9. Owing to major improvements in mass spectrometry-based technologies, multimarker proteomic and lipidomic panels hold the promise to identify novel lipids and proteins potentially related to PCSK9. Within this context, this narrative review aims to provide an overview of the most significant proteomics and lipidomics studies related to PCSK9 effects beyond cholesterol lowering. These approaches have enabled to unveil non-common targets of PCSK9, potentially leading to the development of novel statistical models for CVD risk prediction. Finally, in the era of precision medicine, we have reported the impact of PCSK9 on extracellular vesicles (EVs) composition, an effect that could contribute to an increased prothrombotic status in CVD patients. The possibility to modulate EVs release and cargo could help counteract the development and progression of the atherosclerotic process.
Since its discovery in 2003, as the third gene critical for low-density lipoprotein cholesterol (LDL-C) regulation (1), increasing lines of evidence supported non-cholesterol-related processes mediated by PCSK9 (proprotein convertase subtilisin/kexin type 9). Although mainly derived from the liver, PCSK9 is expressed in many tissues (e.g., intestine, pancreas and central nervous system) and cells types (e.g., macrophages) (2–4).
PCSK9 is the ninth member of a family of endoprotease enzymes. Seven of the nine are core members that are structurally and biochemically similar to each other and to the bacterial (subtilisin) and yeast (kexin) proteins from which they were derived. Proprotein convertases (PC) are synthesized as zymogens (inactive precursors) that are chaperoned through the cell by their prodomains. These contribute to protein folding and activation, favouring the egress of the core proprotein convertases from the endoplasmic reticulum (5). Differently from the first seven members of the PC (PC1, PC2, furin, PC4, PC5, paired basic amino acid cleaving enzyme 4 (PACE4) and PC7) (6) PCSK9 cleaves itself at its internal VFAQ152↓ sequence, and then no longer functions as a protease is allowed. PCSK9 acts as a binding protein to specific cell surface receptors and in particular fosters the endosomal and lysosomal degradation of LDL receptor (LDLR) (7). The three-dimensional structure of PCSK9 revealed the existence of three distinct structural domains, the prodomain (aa 31–152), the catalytic subunit (aa 152–421), and the C-terminal Cys/His-rich domain (aa 453–692), each playing critical roles in the regulation of PCSK9 and its intracellular trafficking (8, 9). Degradation of the LDLR by PCSK9 depends on the internalization of the PCSK9-LDLR complex into clathrin-coated acidic endosomes. Through its catalytic subunit, secreted/plasma PCSK9 binds the epidermal growth factor precursor homology domain A (EGF-A) of the LDLR fostering its degradation in endosomes/lysosomes, rather than being recycled (10). In particular, Gly293, Asp299, Arg329, and Glu332 in EGF-A of the LDLR are pillar to PCSK9 binding at the cell surface (11).
The expression of D374Y gain-of-function variant of human PCSK9 in mice has led to accelerated atherosclerosis, a process beyond the reduced activity of LDLR (12). Conversely, the lack of PCSK9 was linked to the reduction of neointima formation in atherosclerotic plaques in full knockout mice (13). Similar con-clusions were raised by Sun et al. showing that hepatic reduction of PCSK9 affects atherogenesis by modulating the levels and properties of apolipoprotein B-containing lipoproteins, an effect independent of PCSK9-driven modulation of LDLR (14).
Mainly produced by the liver (15), circulating PCSK9 may enter the intimal space (16), where it may stimulate macrophages to produce proinflammatory cytokines and vascular smooth muscle cells (VSMCs) to migrate. In particular, PCSK9 seemed to promote M1 macrophage polarization at least in vitro (17). Migration of VSMCs from the media into the intima can contribute to the accumulation of VSMCs in the growing atherosclerotic plaque. These cells can proliferate over the years and elaborate extracellular matrix macromolecules that comprise much of the bulk of an established atherosclerotic plaque. Atherosclerotic macrophages and vascular smooth muscle cells also secrete PCSK9 within the plaque, contributing to increased inflammation and fatty deposition (18, 19). Chronic inflammation is a hallmark of atherosclerotic cardiovascular disease contributing to the initiation, progression and rupture of an atherosclerotic plaque (20). Within this context, the overexpression of human PCSK9 in mice altered plaque morphology and increased the infiltration of inflammatory bone marrow-derived Ly6Chigh monocytes into the lesion, leading to their differentiation into macrophages (21). Pro-inflammatory stimuli such as tumour-necrosis factor-α and lipopolysaccharides (LPS) enhance the expression of PCSK9 in vascular endothelial cells and VSMCs (22, 23). In line with this evidence, the expression of PCSK9 was increased in mouse macrophages after stimulation with LPS, an effect mediated by the involvement of NLRP3 (NOD-Like Receptor Protein 3) inflammasome (24). Conversely, PCSK9 has been considered a trigger for the expression of pro-inflammatory cytokines, e.g., it raises the inflammatory milieu in macrophages (25). The overexpression of PCSK9 in Ldlr−/− mice induced pro-inflammatory macrophage activation, namely, immune responses, proliferation, and migration, all via LDLR-independent mechanisms (26). Accordingly, a lower number of macrophages and vascular inflammatory regulators was found in atherosclerotic lesions of mice injected with short hairpin RNA targeting PCSK9 (27). However, it is worth mentioning that haemodynamic shear stress, a major determinant of atherogenesis, affects the expression of PCSK9. A greater expression was found in aortic arch branch points and aorta-iliac bifurcation regions where the shear stress is low, than in the thoracic aorta and iliac arteries where shear stress is higher (23) (Figure 1). Considering that the role of platelets in atherothrombosis cannot be overlooked, Pcsk9−/− mice were protected from occlusion of the carotid artery induced by the topical application of FeCl3. Roughly 70% of Pcsk9−/− mice formed unstable nonocclusive thrombi whereas 57% of Pcsk9+/+ littermates developed a total occlusion with stable thrombi (28). In line with these findings, PCSK9 enhanced platelet activation in a CD36 receptor-manner (29).
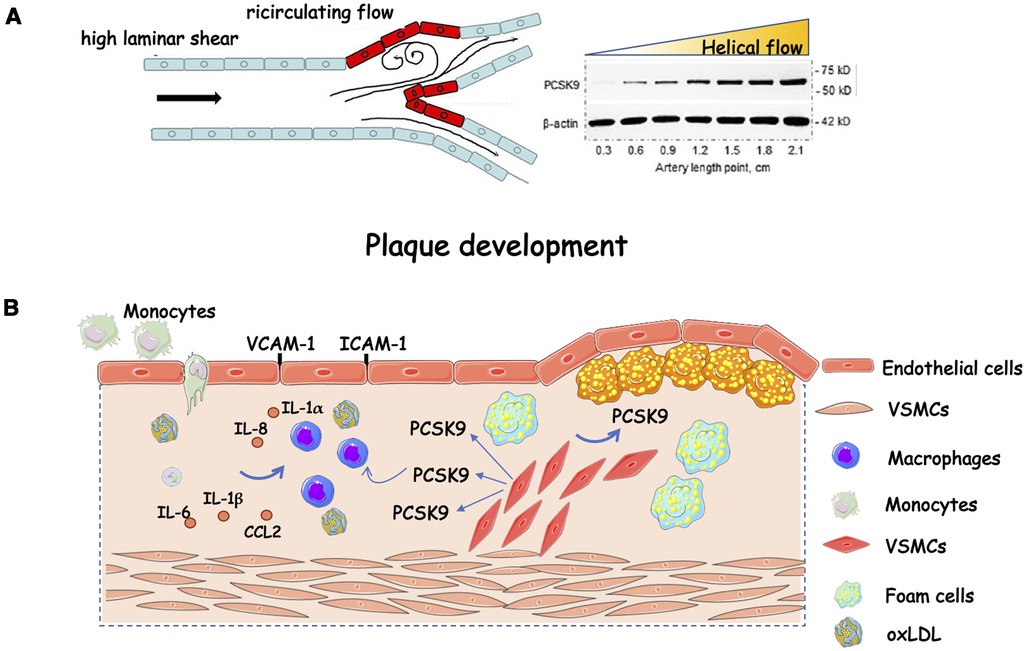
Figure 1. Role of PCSK9 in plaque progression. (A) Helical flow inhibits whereas low flow induces protein PCSK9 expression; (B) Schematic representation of the effect of PCSK9 on plaque progression fuelling. Compelling evidence implicates PCSK9 in plaque development. It is expressed in endothelial cells, vascular smooth muscle cells, and macrophages. In these latter, there is a stepwise increase in PCSK9 gene expression while transitioning from monocytes, to differentiating monocytes, to fully differentiated macrophages. Exposure to human recombinant PCSK9 up-regulates the mRNA expression of proinflammatory cytokines and chemokines (e.g., IL-1β, IL-6, TNF-α, CXCL2, and monocyte chemoattractant protein-1). CCL2, C-C motif chemokine ligand 2; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; oxLDL, oxidized low-density lipoprotein; VCAM-1, vascular cell adhesion protein 1; VSMCs, vascular smooth muscle cells. The figure has been partially modified by permission of Oxford press (30).
Carriers of loss-of-function (LOF) variants Y142X and C679X leading to ∼40% reductions in LDL-C and those carrying R46L leading to 15% reduction in LDL-C showed, respectively, astonishing drops of 88% and 47% in the incidence of coronary heart disease during a 15-year follow-up period (31). E144K and C378W mutations are two new LOF mutations that impair the self-cleavage and endoplasmic reticulum exit of PCSK9, respectively, thus preventing PCSK9 secretion and PCSK9-induced degradation of the LDLR (32).
A genome-wide association study and a following Mendelian Randomization analysis, conducted on a cohort of 3,290 individuals of the LIFE-Heart study receiving coronary angiography for suspected coronary artery disease (CAD), with a mean age of 61.7 years, supported the hypothesis of a causal effect of PCSK9 on coronary artery disease status and severity, carotid plaques, and intima-media thickness (33). A different conclusion was reached in Caucasian carriers of the T allele at R46l (34). In this PROSPER (Prospective Study of Pravastatin in the Elderly at Risk) study, 5,783 elderly participants were enrolled with an age between 70 and 82 years, of whom 43% had a history of vascular disease at baseline and randomized to pravastatin or placebo with follow-up. In these individuals, the minor allele at the R46l locus in PCSK9 did not significantly reduce coronary heart disease or vascular disease risk. The ATHEROREMO-IVUS (The European Collaborative Project on Inflammation and Vascular Wall Remodeling in Atherosclerosis—Intravascular Ultrasound) study, performed on 581 patients (mean age 61.5 years) who underwent coronary angiography for acute coronary syndrome (ACS) or stable angina, showed that PCSK9 levels were linearly associated with higher necrotic core fraction in coronary atheromas (35). In line with these findings, a positive association was found between PCSK9 levels and a 10-year progression of carotid atherosclerosis beyond LDL-C (36). Conversely, neutral conclusions were reached when the relationship between serum PCSK9 concentrations and measures of vascular health, and subclinical atherosclerosis were assessed in a cohort consisting of 1,527 middle-aged men (mean age 49.4 years), who were in good health and free of vascular disease, enrolled in the Firefighters and Their Endothelium (FATE) study (37).
PCSK9 was also associated with arterial stiffness (38), an index linked to the presence of carotid plaques (39). This evidence was supported in two cohorts of Italian ancestry in which short-term therapy with monoclonal antibodies improved endothelial function (40) and arterial stiffness (41). Pertaining to platelet activation, the PCSK9-REACT study (Association of PCSK9 Serum levels and Platelet Reactivity in Patients with Acute Coronary Syndrome Treated with Prasugrel or Ticagrelor), performed on consecutive ACS patients undergoing percutaneous coronary intervention with a mean age of 57 years, found a positive association between circulating PCSK9 and platelet reactivity in patients with ACS undergoing percutaneous coronary intervention (42).
PCSK9 is also recognized as an atherothrombotic risk factor because is involved in inflammatory cytokines production, endothelial dysfunction, atherosclerotic plaque formation and rupture, as well as in the pathogenesis of atherothrombosis (43). PCSK9 promotes platelet activation and aggregation, leukocyte recruitment and clot formation (44) and a positive association between circulating PCSK9 levels and platelet markers exists (28, 42, 45). Besides that, the presence of PCSK9 on platelet was found in thrombi aspirated from the coronary arteries of patients with ST-segment-elevation myocardial infarction during primary percutaneous coronary intervention (29).
In a large cohort of 707 consecutive patients with symptomatic CAD, the association of platelet-derived PCSK9 and platelet aggregation was evaluated demonstrating that PCSK9 inhibition significantly reduced platelet-dependent thrombus formation (46). They observed a significant reduction of the thrombus area in whole blood samples treated with anti PCSK9 antibodies. In addition, they observed that circulating platelets expressed and released PCSK9 in high amount promoting platelet aggregation and thrombus formation, monocyte migration and differentiation into macrophages/foam cells, thus further contributing to the atherosclerotic process. In patients with atrial fibrillation, PCSK9 induced platelet activation by Nox2 via binding with CD36 receptor (47).
However, the clinical relevance of measuring PCSK9 circulating levels as a predictor or qualifier for cardiovascular (CV) risk estimation remains unanswered. Several studies did not associate PCSK9 levels with the number of events (48), as was in the case of acute coronary syndrome patients in which PCSK9 was associated with inflammation and hypercholesterolaemia without predicting the mortality at one year (49); conversely, different conclusions were reached by others (50, 51).
The advances in mass spectrometry (MS) technologies could allow gaining additional information on various PCSK9 targets involved in the development and progression of cardiac and cardiovascular complications. Mass spectrometry coupled with liquid chromatography (LC-MS) is a powerful and sensitive approach that can be used to identify and quantify proteins in complex biological samples, providing a global proteome view of the sample (untargeted proteomics) or a targeted detection of a specific protein of interest.
Proteomics methods have evolved from gel-based techniques to gel-free MS-based approaches, obtaining convenient and in-depth proteome coverage (52). PCSK9 is a low abundant plasma protein and, for this reason, a pre-treatment step such as immunoenrichment or solid-phase extraction (SPE) before the MS analysis is often required to allow an accurate detection. Quantitative MS analysis can be performed both using a label-free approach, mainly applied for a discovery analysis (53), and also using protein/peptide labelling with stable isotopes (54). Label-based quantification involves the artificial labelling of peptides or proteins before liquid chromatography-tandem mass spectrometry (LC-MS/MS), which generates a mass difference between the endogenous peptide and its labelled counterpart, and their intensity ratio allows a measurement of the protein abundance (55).
Targeted proteomics, instead, limits the analysis to a specific number of proteins for screening or validation of candidate protein biomarkers that could be useful for diagnosis, prognosis, and characterization of pathophysiological mechanisms. Multiple reaction monitoring (MRM) is an example of targeted MS-based strategy, which guarantees high sensitivity and selectivity, giving reproducible results at very low levels of quantitation (56). Furthermore, due to its great high-throughput features, it is possible to use this targeted MS technique in several proteomics areas to understand better the abundances and activities of individual proteins, as well as to validate their potential role as biomarkers.
In this context, Croyal et al. measured plasma PCSK9, as an example of a low abundant protein, by LC-MS/MS and compared the results to those obtained by using standard enzyme-linked immunosorbent assay (ELISA) (57). The authors showed that the calculated PCSK9 plasma concentrations were not statistically different between the two methodologies. They also found that a SPE clean up allowed to avoid a sample concentration by immunoaffinity before sample proteolysis, and the subsequent analysis of a PCSK9 proteotypic peptide, by using MRM, successfully provided a targeted quantification of plasma PCSK9. A proteomics approach enables to discriminate among different forms of PCSK9. The PCSK9 PAC-qMS assay allows to measure seven PCSK9 peptides based on prodomain, the catalytic domain before and after furin cleavage, the hinge-, the cysteine- and histidine-rich domain, and the C-terminal domain encompassing S688 in both its unmodified and phosphorylated form. By using this method, it has been possible to demonstrate that analysis of prodomain and the phosphorylated state of S688 may represent novel cardiovascular risk biomarkers (58).
MS is successfully applied also in lipidomics for the study of cellular lipids on a large scale. Lipids are highly complex molecular species with important roles in cellular functions and several metabolic pathways (59). Therefore, the identification and accurate quantification of lipids provide a powerful tool to understand the mechanism of lipid-related diseases and disorders (59). A typical MS-based workflow for lipidomics of biological samples includes sample preparation, MS-based acquisition and data processing. The major challenge in lipidomics is lipid extraction from samples and several strategies were developed in the past decades due to the different nature of lipid species (59, 60). However, there is no extraction method that can completely extract all lipid species, and several preparative modifications have been made to improve and increase the efficiency of extracting specific types of lipids (61). Certainly, the aim is to optimize a simple method for extracting all major classes of lipids by using a minimal amount of sample and for obtaining a high recovery rate and reproducibility. The typically used methods for lipidomic analysis are nuclear magnetic resonance spectroscopy (NMR), gas chromatography (GC) after derivatization of polar groups, and LC-MS. The advances in MS with the development of highly sensitive MS detectors allowed higher resolution, sensitivity, and throughput (59). Furthermore, along with the analysis of lipids with LC-MS, it is possible to perform either a direct analysis without previous chromatographic separation with MS imaging (MSI) or a direct infusion of the total lipid extracts using shotgun lipidomics. Likewise, to proteomics analysis, MS-based lipidomics can be performed with untargeted or targeted objectives. For untargeted lipidomics, non-selective extraction protocols are required that can extract all detectable lipid classes in a sample without contaminating non-lipid molecules such as proteins and carbohydrates (60). Instead, the main focus of targeted lipidomic analysis is the analysis of specific types of lipids and thus the selection of the extraction method is crucial to obtain the correct recovery.
In addition, solid phase extraction is a purification strategy that can help in the isolation of selected lipids or the enrichment of small lipid classes using specific cartridges packed with different sorbents able to selectively hold the desirable fractions and to let undesirable compounds pass through (59).
Besides regulating LDL-C homeostasis, PCSK9 non-cholesterol-related processes have emerged since its discovery. Mainly secreted from the liver, PCSK9 is expressed in many tissues (e.g., intestine, pancreas and central nervous system) (Figure 2). Thus, to unveil new molecular targets of PCSK9 is mandatory to foresee possible drawbacks related to the long-term PCSK9 pharmacological inhibition.
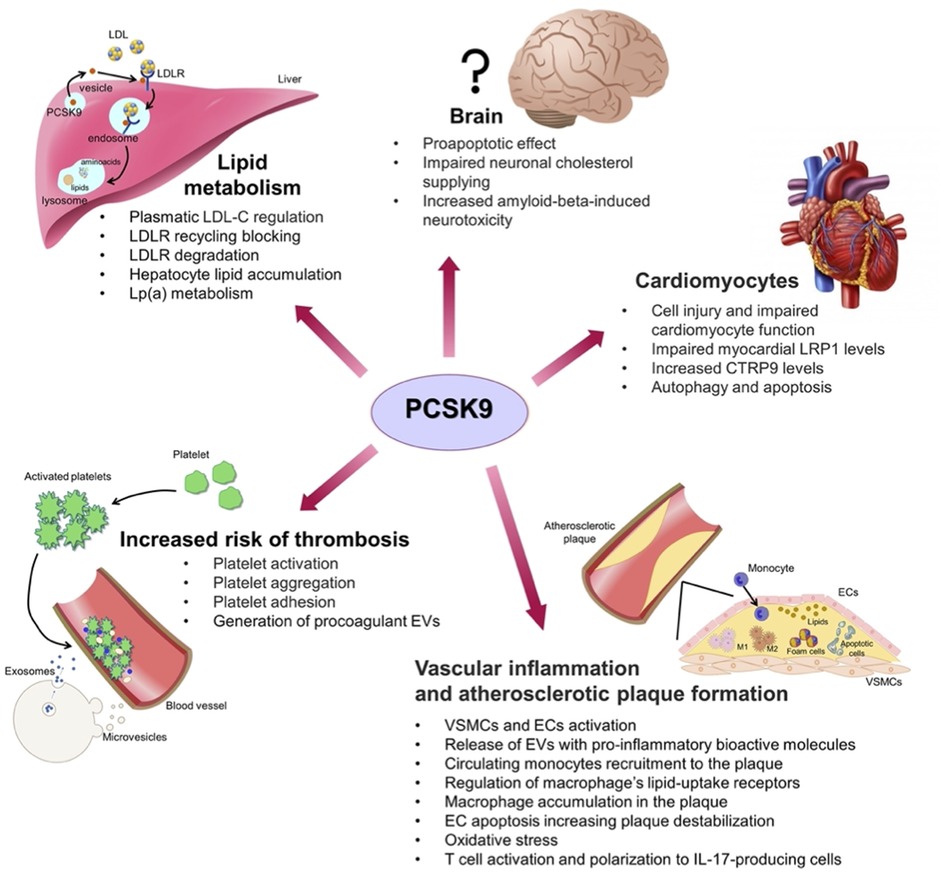
Figure 2. Summary of the non-classic effects played by PCSK9 on various cell types in different organs: liver and lipid metabolism; brain; cardiomyocytes; vascular system with inflammation, atherosclerotic plaque formation and increased risk of thrombosis; CTRP9, C1q/tumour necrosis factor-related protein 9; ECs, endothelial cells; EVs, extracellular vesicles; LDL, low-density lipoprotein; LDL-C, LDL cholesterol; LDLR, low-density lipoprotein receptor; LRP1, low density lipoprotein receptor-related protein 1; M1, macrophages type 1; M2, macrophages type 2; VSMCs, vascular smooth muscle cells.
A deep molecular phenotyping of samples from homozygous carriers can help to discriminate between silent and functional variants. Combining whole-genome sequencing with proteomics and metabolomics have allowed to discover a homozygous PCSK9 genetic variant (rs746442570) which was associated to low PCSK9 protein levels and LDL-C levels (62).
Denis et al. performed a quantitative MS analysis with a metabolic stable isotope labelling by amino acids in cell culture (SILAC) to study proteomic changes occurring in different subcellular compartments of the human hepatocyte cell line (HuH7) after a stable overexpression of a gain-of-function PCSK9 membrane-bound chimaera (PCSK9-V5-ACE2) (63). After SILAC labelling, subcellular fractionation was performed before the protein separation by Sodium Dodecyl Sulphate—PolyAcrylamide Gel Electrophoresis (SDS-PAGE), tryptic digestion and LC-MS/MS analysis. As mentioned before, gain- and loss-of function variants in human PCSK9 modulate the levels of circulating LDL (31). More than 5,700 peptides were quantified in the lysosomal/endosomal, Golgi and endoplasmic reticulum compartments, among which 327 showed significant changes in the expression levels following stable overexpression of PCSK9-V5-ACE2 in comparison to the control cells transfected with a mock vector. These proteins were involved in multiple functions including cytoskeletal organization, vesicle transport, membrane receptor recycling, lipid and cholesterol homeostasis, cell signalling and protein folding. In particular, it was confirmed a significant downregulation of the levels of EH-domain binding protein 1 (EHBP1), a protein involved in receptor recycling from internal vesicles back to the cell surface. This suggests a reduced recycling of the LDLR and its relocation to the late endosome and lysosome for degradation (63). Moreover, several proteins of the Rab family of GTPases, that control vesicle transport, vesicle movement along cytoskeleton, and membrane networks, showed both higher and lower expression by PCSK9-V5-ACE2 overexpression in HuH7 cells (63). Among the proteins that were upregulated upon PCSK9-V5-ACE2 overexpression, A-kinase anchor protein-12 (AKAP-12) is a potent scaffold protein that interacts with many signalling factors and facilitates the internalization and the recycling of the β2-adrenergic receptor through calcium-dependent interactions (63). Its overexpression leads to an upregulation of the expression of sterol regulatory element-binding protein-2 (SREBP-2) and liver 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCoA), resulting in an enhancement of cholesterol efflux from hepatic cells (64). Overall, this study found that an endosomal-related protein was affected by PCSK9 providing insight into novel functions and pathways affected by PCSK9 and unrelated to LDLR processing.
In the context of CVDs, PCSK9 is overexpressed in the ischaemic mouse hearts, mostly in the region bordering the infarct area, and promotes the development of infarct size, cardiac contractile function, and autophagy (65). Cultured mouse cardiomyocytes were examined for the correlation between myocardial ischaemia and PCSK9 expression and autophagy, and PCSK9 levels increased when cardiomyocytes were in hypoxia condition, in a hypoxia duration-dependent manner. Moreover, proteins from hearts and primary cardiomyocytes were purified and analysed by WB demonstrating that deletion of hypoxia inducible factor-1 α (HIF-1α) significantly reduced the expression of PCSK9, microtubule-associated protein 1 light chain 3 (LC3), and beclin-1 during ischaemia. Therefore, a possible association between PCSK9 and cardiac diseases and their risk factors is now well-defined in the literature. Independently of LDL-C regulation, PCSK9 exerts various effects in the heart and its higher levels promote reactive oxygen species production, excessive autophagy and apoptosis in cardiomyocytes leading to a cardiac insufficiency following the acute myocardial infarction (66). In the attempt to reduce the CVD burden driven by LDL-C, the use of inhibitors of PCSK9 has provided a very powerful therapy for patients at highest risk (67). Despite the well-demonstrated benefits of statins, a large number of patients does not reach adequate levels of LDL-C recommended by guidelines, as well as blood triglyceride-rich lipoproteins, or lipoprotein(a), thus still remaining at significant risk of CVD. For this reason, alternative cholesterol lowering approaches to use in combination with statin, such as inhibitors of PCSK9, have been developed to obtain better efficacy and tolerability.
Among the risk factors accounting for CVD is now recognized that lipid and lipoprotein abnormalities play a key role in this process. So said, the implication of PCSK9 inhibition on lipid classes, besides LDL, has been only recently investigated. Hilvo et al. performed a lipidomic analysis in patients with established coronary heart disease after administration of a RG7652, a fully human PCSK9 monoclonal antibody, to characterize the lipidome alterations of plasma and major lipoprotein particles (67). Lipoprotein fractions were isolated by sequential density ultracentrifugation and analysed in MS to obtain a lipidomic profiling. A significant reduction of sphingolipids, cholesteryl esters and free cholesterol was found in plasma upon PCSK9 inhibition, as was for lipids in lipoprotein classes. A global decrease of total lipid species was reported in LDL, very low-density lipoprotein/intermediate low-density lipoprotein (VLDL-IDL) particles, whereas high-density lipoproteins (HDL)-associated phospholipids rose (e.g., linoleic acid and arachidonic acid containing phosphatidylcholines (PCs) and phosphatidylethanolamines). In particular, the most pronounced reduction in both LDL and VLDL-IDL was reported for cholesteryl esters and sphingolipids including dihydroceramides (Cer d18:0), ceramides (Cer d18:1), globotriasoylceramides (Gb3), glucosyl/galactosylceramides (Glc/GalCer), lactosylceramides (LacCer), and sphingomyelins. In addition, in LDL fraction also PCs showed a significant decrease, as well as phosphatidylethanolamines and phosphatidylinositol.
Ceramides containing very long saturated fatty acid (22:0, 24:0 and 26:0) showed a more extensive decrease in plasma levels following inhibition of PCSK9 compared to the other ceramide species. To be noted that Cer(d18:1/24:1)/Cer(d18:1/24:0) ratios are able to predict fatal cardiovascular events in patients with stable coronary artery disease and acute coronary syndrome (68). This study was the first characterization of the lipidome of plasma and isolated lipoprotein fractions following therapy with a PCSK9 inhibitor (67).
Similarly, a ceramide- and phospholipid-based risk stratification score, known as CERT2, can successfully predict residual CVD event risk in CAD patients demonstrating an excellent and potentially clinically relevant prognostic value for future personalized medicine (67). The lipidomic profile in patients with a very high risk of atherosclerotic CVD was also evaluated by ultra-performance LC-MS (UPLC-MS) (69). In patients receiving evolocumab, besides the evaluation of total cholesterol, LDL-C, and lipoprotein(a), also changes in cholesteryl esters, triacylglycerols, and sphingomyelins were investigated by MS. Compared to individuals given atorvastatin alone, the addition of evolocumab led to a drop in sphingolipids, phospholipids, triacylglyceride, and diacylglycerol. However, it should be pointed out that this study is a two-centre study with a small cohort of selected patients (n = 64) and only changes in the blood lipidomic profile at short-term have been studied, not focusing also on long-term follow-up data about the effects of treatment and the potential cardiovascular impact. In addition, the authors did not evaluate the effects of atorvastatin and evolocumab on the specific lipid molecules in each lipid class, which could provide a far more comprehensive overview of what was happening on the lipidomic profile. Furthermore, the PCSK9-related genes, which could affect the therapeutic efficacy of PCSK9 inhibitors, were not identified.
Familial hypercholesterolaemia is one of the most common inherited metabolic diseases in humans and can affect individuals from all ethnic groups. It associates with mutations in several genes that result in the inability of the liver to sufficiently remove LDL particles from the circulation. Autosomal dominant mutations in the LDLR, APOB (apolipoprotein B100) and PCSK9 genes account for most cases of familial hypercholesterolaemia (70). Plasma lipidome of familial hypercholesterolemic patients before and during therapy with evolocumab was studied by an ultra-high-performance LC-MS (UHPLC-MS) to obtain an untargeted lipidomic analysis and to evaluate the effect of PCSK9 inhibition on whole lipid metabolism (71). After lipid extraction using a chloroform/methanol solution added with butylhydroxytoluene, lipid samples were analysed by MS and, after treatment, significant low plasma levels were observed in several lipid classes belonging to sphingomyelin, ceramide, cholesteryl ester, phosphatidylcholine, triacylglycerol and phosphatidylinositol, as compared to baseline. Instead, four classes, including three ceramides and phosphatidylinositol, were increased following evolocumab. In accordance with Hilvo et al. (67), sphingomyelin, ceramide, and cholesteryl ester were the species most affected by the treatment (71). It is possible to assume that these lipidomic changes may be due to an indirect mechanism that involved the de novo synthesis of sphingomyelin and ceramide through the activation of sphingomyelin synthase by oxidized LDL, thus increasing cholesterol ester production. Therefore, the upregulation of LDLR and the lipidome modifications seem to determine an improvement of vascular state in patients upon the pharmacological inhibition of PCSK9. However, further targeted analysis, based on lipid fraction isolation, is needed to strengthen these conclusions and the identification of antibodies against oxidized LDL will be necessary to deepen the role of oxidized LDL in the modulation of ACS.
In the last few years, numerous studies have shown that PCSK9 LOFs variants are associated with higher fasting plasma glucose levels, possibly leading to an increased risk of type 2 diabetes (72, 73), a side effect not confirmed in clinical trials (74). Recently, Saitoski et al. investigated the role of PCSK9 in insulin-producing pancreatic cells to clarify its function and to evaluate potential other protein targets beyond LDLR (75). The authors studied PCSK9 regulation and function in the human pancreatic beta cell line EndoC-βH1, showing that PCSK9 is expressed and secreted by these cells and its expression is regulated by cholesterol, lipoproteins, and sterol regulatory element-binding protein transcription factors. Western blot analyses showed that cholesterol and lipoproteins, except for oxidized LDL that had no effects, reduced intracellular and secreted PCSK9 levels. Following PCSK9 knockdown by using a siRNA approach, several deregulations in the transcriptome, proteome, and secretome of these cells were detected, along with rises in basal and glucose-stimulated insulin secretion (75). Ingenuity pathway enrichment analysis showed impaired mitochondrial function, which is essential for insulin secretion by beta cells, and differential expression of several markers associated with glucose-stimulated insulin secretion. By using Tandem Mass Tag labelling and data-dependent acquisition MS on cell lysates, together with other data-independent acquisition cell proteomic and secretome experiments, higher levels of the enzyme proprotein convertase subtilisin/kexin type 1 (PCSK1) involved in proinsulin to insulin processing were noticed at 72 h after PCSK9 knockdown. MHC-I complex and Golgi/ER proteins were also overexpressed. The authors also performed loss- and gain-of-function experiments in the EndoC-βH1 cells using siPCSK9 or recombinant PCSK9 treatments to discover proteins regulated by PCSK9 in human beta cells. By Western blot, they observed low levels of LDLR and very low-density lipoprotein receptor (VLDLR) only through an extracellular signalling mechanism involving exogenous PCSK9, while CD36 via an intracellular signalling mechanism. The specific mechanism of CD36 regulation by PCSK9 was not clarified by the authors but maybe involved both proteasomal and lysosomal degradation, so future experiments by using proteasome or lysosome inhibitors will be necessary to better understand the role of PCSK9 on CD36 regulation in the pancreatic β cells.
Thus, both intracellular and extracellular PCSK9 regulate β-cell fatty acid homeostasis. Finally, intracellular PCSK9 was found to modulate the cell surface expression of programmed death-ligand 1 (PDL1) and human leukocyte antigen (HLA)-ABC, which are proteins involved in cell-lymphocytes interaction. Therefore, this study confirms that PCSK9 is a regulator of multiple cell surface receptors in pancreatic beta cells (75). The authors did not discuss the potential effect of PCSK9 monoclonal antibodies or siRNA-based therapies against PCSK9 on the expression of LDLR, VLDLR, CD36, PDL1, and HLA-ABC in pancreatic β cells, as was in the case of cotreatment with statins that could modify the concentrations of these proteins.
However, a recent study by Peyot et al. reported contradictory results regarding the glucose metabolism regulation following the inactivation of PCSK9, demonstrating that in beta cells in mice the reduction of PCSK9 mRNA and protein expression did not affect glucose homeostasis and β-cell function (76). The authors created the first β-cell-specific KO of PCSK9 (βKO) and showed that both whole body Pcsk9−/− KO and βKO mice presented an increased cholesterol uptake, but normal glucose tolerance, insulin release in response to glucose load and insulin sensitivity as compared to their respective control wild type mice. Moreover, ex vivo glucose-stimulated insulin secretion was similar in wild type and KO islets both in presence or absence of fatty acids. According to these results, another study showed that liver-specific KO of PCSK9 in mice did not alter the expression of LDLR in β-cells and maintained insulin secretion and glucose homeostasis (72). Conversely, mice specifically lacking Pcsk9 in pancreas and those with whole loss of all local PCSK9 in islets showed a moderate impairment of insulin response and glucose tolerance (73). This suggests the importance of local PCSK9 expression in pancreatic islets to maintain a β-cell function. Similarly, impairment of glucose metabolism is also variable in human subjects carrying PCSK9 LOF variants, an effect depending on the genetic background of the studied population, in which the risk to develop diabetes is also associated to the individual degree of functional loss (77).
Therefore, several PCSK9 LOF variants have been reported in literature with different effects on glucose homeostasis and many controversies exist (78). Some variants result in a significant increase in glucose levels and higher risk of type 2 diabetes, other variants do not cause alteration of glucose metabolism (74).
Hypercholesterolemia can cause neuroinflammation in midlife and is associated with a higher risk of age-related neurological disorders including Alzheimer’s disease (79). Cholesterol is important for maintaining normal brain functions and most of it is synthesized de novo and separated from peripheral cholesterol through the blood-brain-barrier (80).
The role of PCSK9 in neurodegenerative disorders is still unclear and conflicting data on this subject have been recently published, as elsewhere reviewed (81) An in vitro study on PCSK9 modulation of human astrocytes and neurons showed an altered cholesterol metabolism and neuronal cholesterol supplying, which resulted to be significantly reduced following the PCSK9 overexpression, thus impairing the normal neuronal functions (82). Moreover, PCSK9-overexpressing cells displayed increased amyloid-beta-induced neurotoxicity compared to control cells. Although Mendelian randomization analyses and meta-analyses on early studies have hypothesized a PCSK9-related neurocognitive impairment (83–85), analyses of cardiovascular outcomes trials (CVOT) did not show a risk of neurocognitive adverse effects following the use of PCSK9 inhibitors (86, 87), actually, prolonged exposure was associated with a decreased risk (88).
The evidence that also serum cholesterol levels represent a risk factor for dementia and cognitive impairment is still unclear. Many side-chain hydroxylated metabolites of cholesterol, such as 24S-hydroxycholesterol (24S-OHC) and 27-hydroxycholesterol (27-OHC), can pass the brain-blood-barrier and are implicated in neuroinflammation (89). In particular, it was assumed that 27-OHC in the brain reduces central nervous system cholesterol synthesis and 24S-OHC production (90).
The effects of evolocumab and alirocumab on lipidome profile were evaluated in hypercholesterolemic patients responding insufficiently to maximally tolerated statin and/or ezetimibe therapy. The brain-specific oxysterols 24S-OHC and 27-OHC were assessed by isotope dilution GC-MS selected ion-monitoring (GC–MS-SIM) by using the corresponding deuterium-labelled oxysterols as internal standards, before, after 1-month and after 3-month of treatment (91). 24S-OHC ratios to cholesterol and to 27-OHC in serum or plasma are thought to be potential biomarkers for brain cholesterol metabolism (92). After 1-month of treatment, evolocumab and alirocumab significantly reduced serum total cholesterol, LDL-C and 27-OHC concentrations, whereas serum ratios of 24S-OHC to cholesterol and 24S-OHC to 27-OHC increased, reaching even higher levels within 3 months of treatment (91). The authors suggested that this specific brain cholesterol turnover was probably due to lower levels of 27-OHC in the brain, resulting in increased synthesis of brain cholesterol, higher production of 24S-OHC and its secretion across the blood-brain barrier. Therefore, this study demonstrated how brain excretion of 24S-OHC is improved upon anti-PCSK9 antibody treatment. Anyway, in this study, the cohort of hypercholesterolemic patients was small with large variability in sex, pre-treatment and PCSK9 inhibiting agent, and the percentages of increment of 24S-OHC and 24S-OHC to 27-OHC after 1-month treatment were statistically significant even if small. Of course, wider and longer-term demographic studies are needed. The measurement of 7α-hydroxy-3-oxo-4-cholestenoic acid, which is formed from the conversion of 27-OHC in the brain, could also be useful to obtain more information on brain cholesterol metabolism because this conversion may affect the inhibition of brain cholesterol synthesis by 27-OHC. Moreover, the authors would have to measure the arteriovenous difference of oxysterols 24S-OHC and 27-OHC between the internal jugular vein and an artery also in patients during PCSK9 treatment and not only in healthy normocholesterolaemic subjects to have a more complete view of the flux of these oxysterols.
The major critical function of PCSK9 deduced from human and mouse studies, as well as cellular and structural analyses, is its role in increasing the levels of circulating LDL-C, via its ability to enhance the sorting and escort of the LDLR to lysosomes (93). However, in this complex scenario, many unsolved questions remain, e.g., it was demonstrated that PCSK9 lacking its C-terminal Cys/His-rich domain was still able to regulate LDLR internalization, but PCSK9-LDLR complex did not follow the endosome/lysosome pathway for degradation suggesting the presence of additional interactors (94, 95). Therefore, it is important to investigate the presence of additional membrane-bound or secreted cellular proteins involved in the LDLR internalization and degradation mediated by PCSK9.
By affinity purification and shotgun LC-MS/MS analysis, Xu et al. identified 22 potential PCSK9 binding proteins (96), including endoplasmic reticulum-localised proteins, proteins associated with the ubiquitination pathway (e.g., cellular inhibitor of apoptosis protein 1 and TNF receptor-associated factor 2), mitochondrial carriers, and molecular chaperones. In particular, they focused the attention on cellular inhibitor of apoptosis protein 1 (c-IAP1) showing that this protein binds and processes PCSK9 (96). Indeed, in carriers of the S127R, a gain-of-function variation, PCSK9 was not able to bind to c-IAP1, impairing its autocatalytic activity. However, the specific mechanism by which the S127R mutant proprotein stimulates LDLR degradation causing hypercholesterolaemia still remains unresolved. Therefore, further clarification of this aspect will be necessary.
Using a siRNA-c-IAP1 construct to knock down endogenous c-IAP1 in a human T-Rex-293 stable cell line overexpressing FLAG-tagged wild-type PCSK9, the same authors showed a significant increase of pro-PCSK9 in comparison to the functionally mature PCSK9 that is the predominant form in non-silencing RNA control; this suggests that c-IAP1 impairs PCSK9 processing and autocatalytic cleavage. In the same study, there were found lower levels of secreted mature PCSK9 protein in the medium from c-IAP1 siRNA treated vs. control samples and a higher concentration of pro-PCSK9 protein aggregates in c-IAP1 siRNA cells. In c-IAP1 null mouse embryonic fibroblasts, there was a reduction in the secreted mature form of PCSK9 together with a significant increase in the levels of LDLR. Finally, the study reported a role of c-IAP1 in the PCSK9 ubiquitination mediated by the only lysine-27, which led to the lysosomal localization of the PCSK9/LDLR complex and its possible degradation (96). Therefore, this study provided new insights into the roles of c-IAP1 in regulating the activities of PCSK9, giving a clue to develop more efficient treatments against cardiovascular and infectious diseases.
Ly et al. performed a co-immunoprecipitation of PCSK9 combined with an UHPLC-MS/MS analyses in HepG2 secretome to identify PCSK9-binding proteins able to control the PCSK9-LDLR complex formation and LDLR degradation (97). The authors identified a total of 42 secreted proteins and, among the proteins linked to cholesterol metabolism, many potential PCSK9 interactors were selected, such as glypican-3 (GPC3), matrilin-3, phospholipid transfer protein, fibrinogen-like 1, tissue factor pathway inhibitor, and plasminogen activator inhibitor-1. By silencing each of these proteins in HepG2 cells, they demonstrated also that only GPC3 and phospholipid transfer protein were able to significantly increase LDL uptake and led to higher total LDLR protein levels in these cells compared to control cells (97). Focusing on GPC3, data from this study showed that PCSK9 Cys/His-rich domain is not involved in the interaction with extracellular GPC3, which instead happens with the PCSK9 prodomain and/or catalytic subunit. Moreover, to investigate whether intracellular GPC3 is also implicated in the interaction with the PCSK9-LDLR complex, the authors analysed both HepG2 and Huh7 cells and concluded that GPC3 interacts with pro-PCSK9 and immature LDLR intracellularly most likely in the endoplasmic reticulum. In conclusion, the findings led the authors to demonstrate a competitive interaction between GPC3 and PCSK9 which can reduce the PCSK9 extracellular activity on LDLR degradation (97). However, further studies are needed to verify which is the exact region or residues involved in the GPC3-PCSK9 interaction, because the authors in this study did not deepen this structural aspect.
Another aspect to be worth considering is that the understanding of the protein-protein interaction that governs the PCSK9-LDLR interaction could be fundamental to developing new therapeutic strategies in the near future.
A combination of affinity chromatography, high-resolution LC-MS/MS proteomics, gene expression, and ELISA assays (98) allowed to identify three proteins, alpha-1-antitrypsin, alpha-1-microglobulin/bikunin precursor, and apolipoprotein H that interact with PCSK9. Alpha-1-antitrypsin and apolipoprotein H showed a higher expression both at the mRNA and protein levels in human hepatic C3A cells treated with MITO+ medium, which is a medium able to significantly affect PCSK9 function lowering PCSK9/LDL receptor complexes levels as compared to cells exposed to standard medium. Conversely, alpha-1-microglobulin/bikunin precursor did not statistically change its expression in response to the treatment. In particular, the authors noticed that under MITO+ condition the secretion of apolipoprotein H was lower, while its internalization was increased. In addition, they suggested an interaction with PCSK9 at the plasma membrane and within the endosome for alpha-1-antitrypsin and alpha-1-microglobulin/bikunin precursor, while apolipoprotein H may interact with PCSK9 mainly in the endosome (98). Previous evidence described the formation of complexes between oxidized alpha-1-antitrypsin and LDL in atherosclerotic lesions, although their mechanisms underlying the formation and clinical significance are still unclear (99). Similarly, apolipoprotein H is an important player in atherosclerotic plaque and contributes to atherosclerosis progression by binding oxidized LDL (100, 101).
In contrast, alpha-1-microglobulin/bikunin precursor has not yet been associated with atherosclerosis, but no evidence suggests otherwise since α1-microglobulin has been suggested to be involved in the protection against atherosclerosis, inhibiting the oxidation of LDL and counteracting heme toxicity and reactive oxygen species (102).
In humans, PCSK9 circulates in a free and lipoprotein-bound form (103), possibly bound to LDL (104), lipoprotein(a) [Lp(a)] (105) and HDL (106). Burnap et al. evaluated the associations between PCSK9 and apolipoproteins in fasted plasma from two prospective, community-based cohort studies by nuclear magnetic resonance spectroscopy and targeted MS analysis (107). A significant positive correlation was observed between plasma PCSK9 levels and the particle number of small dense HDL, as well as a strong association between PCSK9 and apolipoprotein C3 (apoC3), an inhibitor of lipoprotein lipase that can hydrolyse plasma triglycerides. In addition, the authors investigated the PCSK9 kinetics in healthy volunteers during postprandial lipemia and characterized the HDL composition by label-free MS. They confirmed a decrement of PCSK9 levels during the postprandial lipemia peak within 5 h compared to the fasted condition, while a negative correlation was found between the postprandial PCSK9 and triglyceride responses. The postprandial reduction of HDL-bound PCSK9 was also confirmed by proteomics, and a decrease of apoC3 from HDL was observed at the same time, as well as an increase of ApoA1 and inflammatory-related proteins, respectively, at 8 and 4 h postprandially. Using crosslinking MS to study plasma and HDL interactome, PCSK9 was identified to interact with HDL through apoA1 binding, and the further quantitative analysis of HDL proteome and lipidome in a cohort of patients with CAD vs. control subjects revealed a strong distinct cluster of PCSK9, phospholipid transfer protein, clusterin, and apolipoprotein E that was statistically altered by sex and positively associated with sphingomyelin content (107). Finally, the authors determined whether HDL was able to alter PCSK9 function, demonstrating that HDL promoted PCSK9-driven LDLR degradation through the modulation of PCSK9 cellular uptake and multimerization. Therefore, this study demonstrated that the interaction between PCSK9 and HDL impairs PCSK9 functionality. In future studies, it will be interesting to unravel the mechanisms between PCSK9 function and lipoprotein binding to evaluate the possibility to use small molecule inhibitors instead of monoclonal antibodies to inhibit PCSK9 activity with higher therapeutic efficacy and lower costs.
In search of new mechanisms linking PCSK9 with cardiovascular risk, the role of extracellular vesicles (EVs) should be considered. EVs are membrane-bound particles released by cells in biological fluids, both in physiological and pathological contexts and seem to participate in each stage of the atherosclerotic process. They carry a cargo composed of microRNAs, proteins, lipids, and organelles that vary according to environmental stimuli and risk factors (108). The content of ceramides and phosphatidylcholines in plasma EVs assessed in preoperative state was independently associated with an increased 3-year risk of major adverse cardiac events in patients undergoing carotid endarterectomy (109). PCSK9 influences the release of EVs, in particular those derived from macrophages (CD14+), endothelium (CD105+), and neutrophils (CD66+) (110). PCSK9 seems to mediate the generation of procoagulant EVs, a process that could contribute to an increased prothrombotic status in patients with CVDs (111).
So said, it becomes of interest to explore the relationship between PCSK9 and the release of EVs from cells relevant to the atherosclerotic process to hypothesize the potential use of these EVs as a pharmacological vehicle. As an example, in patients with obesity, PCSK9 has a key role on the release of EVs-derived from different atherosclerotic components (i.e., platelets, monocytes/macrophages, endothelium, and neutrophils) and EVs-derived miRNA linked to inflammation and atherosclerosis (112). In particular, the PCSK9 effect on platelet-derived EVs was affected by platelet count in a significant and negative way.
EVs are vehicles of bioactive molecules involved in several atherosclerotic processes, among which lesion development, plaque progression, hemostasis and thrombin regulation (113). In this regard, a recent study (111) confirmed the PCSK9-mediated generation of procoagulant EVs from human mononuclear cells (PBMCs) and THP-1 cells via TLR4/NF-κB activation, thus increasing the prothrombotic status in patients with CVDs. The pharmacological inhibition of PCSK9 with evolocumab showed a reduction of PCSK9-induced EV generation and extracellular-bound tissue factor. Therefore, PCSK9 stimulates the generation of prothrombotic EVs by cells of monocytic lineage, and these EVs are characterized by a TF-dependent procoagulant activity. These data demonstrate the important role of PCK9 in the atherothrombotic process.
In addition, PCSK9 seems to confer procalcific and inflammatory properties to EVs released by VSMCs, which are cells relevant for the atheroma formation (114, 115). Since PCSK9 is expressed and secreted as functionally active by VSMCs, an association between PCSK9 and the release of EVs was recently described (110). PCSK9 is released in significant amounts from of VSMCs, more than from endothelial cells (116), and it is abundantly expressed in VSMCs of human atherosclerotic lesions and abdominal aortic aneurysm tissues (117) suggesting its potential role in plaque instability and increased cardiovascular events. The impact of PCSK9 on the composition of EVs released from human VSMCs and how these EVs regulated the intercellular communication among atherosclerotic components have been the leverage to study a model of human VSMCs overexpressing PCSK9 (VSMCPCSK9) (114). Using untargeted proteomic analysis, VSMCPCSK9-EVs showed a different and specific cargo of proteins compared to EVs derived from their counterpart released by VSMCs wildtype, mainly represented by extracellular matrix structural constituents that are involved in different aspects of cardiovascular pathophysiology. The isolated EVs were then subjected to functional experiments by using both in vitro and in vivo models, demonstrating that when VSMCs expressed higher levels of PCSK9, the released EVs carried pro-inflammatory bioactive molecules. The impact of EVs on pro-inflammatory markers in endothelial cells, monocytes and monocyte-derived macrophages was evaluated and a significantly raised expression of adhesion molecules was observed in endothelial cells exposed to VSMCPCSK9-EVs, as well as an increase of pro-inflammatory cytokines and proteins in monocytes and macrophages. All these proteins are clear promoters of vascular inflammation and atherosclerosis initiation or progression. Using a label-free mass spectrometry-based approach, also the secretome of monocytes exposed to EVs was analysed to confirm these data, reporting proteins involved in immune response and immune effector processes. Moreover, VSMCPCSK9-EVs enhanced the migratory capacity of monocytes and glycolytic activity while decreasing oxidative phosphorylation. Macrophages showed a reduced migratory capacity, as a mirror of their accumulation in atherosclerotic plaques, and higher oxLDL-derived cholesterol uptake. To better understand the role of macrophages as mediators of the pro-inflammatory state, when VSMCPCSK9-EVs were microinjected into an in vivo model of zebrafish embryos, there was an increased recruitment of macrophages toward the local injection site (114). This study highlighted the pro-inflammatory role of PCSK9 through the content of EVs released from VSMCs, pointing out the cholesterol-independent function played by PCSK9 in atheroma formation. In addition, the mitochondrial dysfunction following the treatment of monocytes with VSMCPCSK9-EVs confirmed the key role of mitochondria in the pathogenesis of inflammatory diseases such as atherosclerosis, thus representing a potential target for therapeutic agents.
Although the clinical benefits of PCSK9 inhibition are clear, large gaps remain in the understanding of PCSK9 biology (e.g., molecular mechanisms that regulate circulating PCSK9 levels). Within this context, advancement in high-throughput approaches (e.g., proteomics and lipidomics) can allow to unravel unsuspected pathways providing knowledge on possible new therapeutic targets (Table 1). In the era of personalized medicine, identification of specific biomarkers (e.g., the phosphorylated form of PCSK9) may reveal important insights in the pathophysiology of CVDs. Considering that improvements in risk stratification is fundamental to reduce the burden of CVDs, the assessment of the proteins’ expression in a specific state and the evaluation of how these proteins change over time can identify patients at greatest risk of CV events. Although these approaches hold the promise to be assessed in a high-throughput routine, several limitations (e.g., cost-effectiveness, rigorous prospective studies, validation in rigorous clinical studies) need to be addressed before a wider clinical use (118).
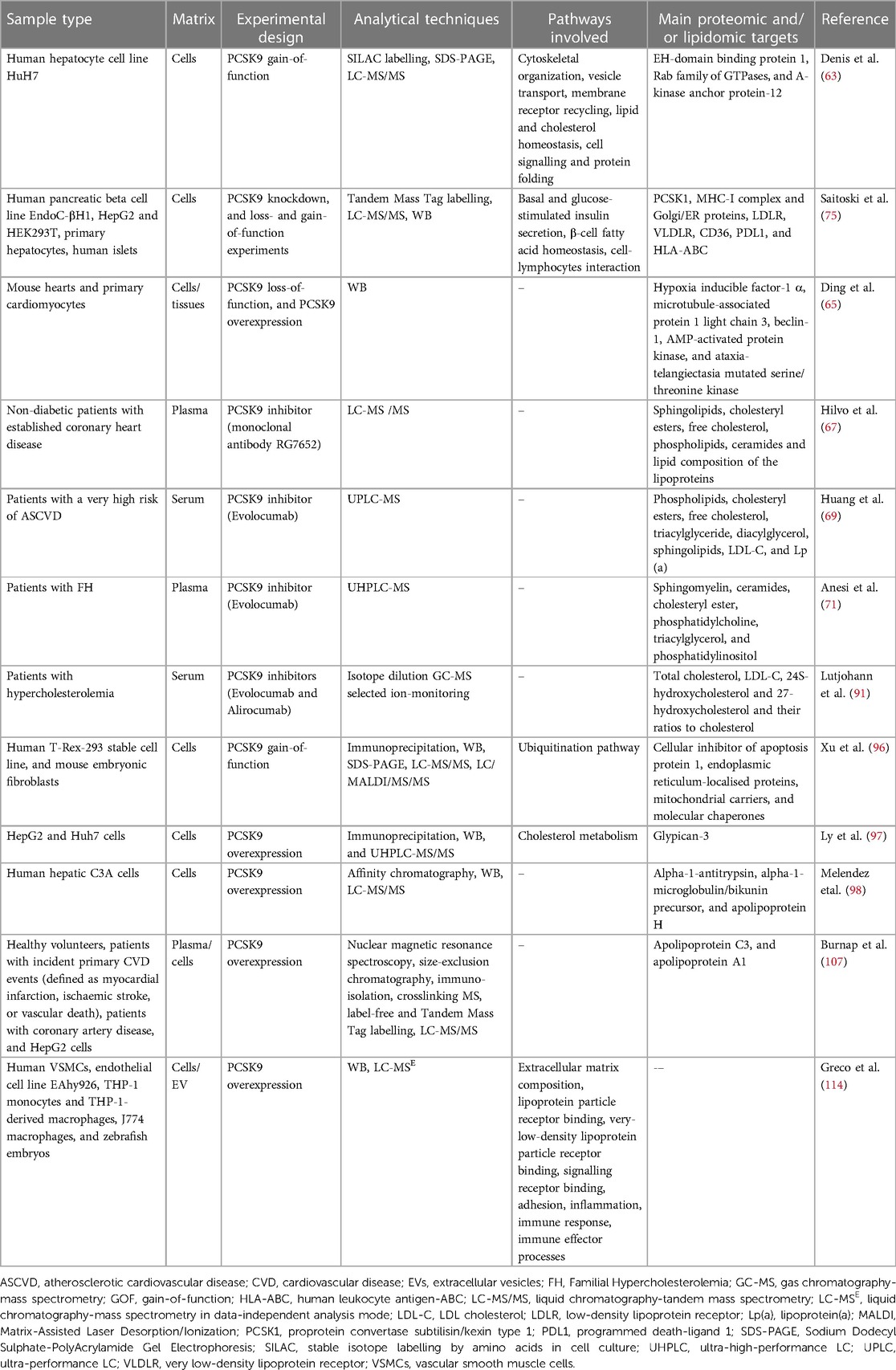
Table 1. Summary of the main proteomic and lipidomic studies on the molecular changes associated with pleiotropic effects of PCSK9.
EG, CB, and MR conceived and wrote the review. CM wrote the part pertaining PCSK9 biology and preclinical evidence. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. (2003) 34(2):154–6. doi: 10.1038/ng1161
2. Macchi C, Ferri N, Sirtori CR, Corsini A, Banach M, Ruscica M. Proprotein convertase subtilisin/kexin type 9: a view beyond the canonical cholesterol-lowering impact. Am J Pathol. (2021) 191(8):1385–97. doi: 10.1016/j.ajpath.2021.04.016
3. Wu NQ, Shi HW, Li JJ. Proprotein convertase subtilisin/kexin type 9 and inflammation: an updated review. Front Cardiovasc Med. (2022) 9:763516. doi: 10.3389/fcvm.2022.763516
4. Seidah NG, Garcon D. Expanding biology of PCSK9: roles in atherosclerosis and beyond. Curr Atheroscler Rep. (2022) 24(10):821–30. doi: 10.1007/s11883-022-01057-z
5. Artenstein AW, Opal SM. Proprotein convertases in health and disease. N Engl J Med. (2011) 365(26):2507–18. doi: 10.1056/NEJMra1106700
6. Seidah NG, Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov. (2012) 11(5):367–83. doi: 10.1038/nrd3699
7. Park SW, Moon YA, Horton JD. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem. (2004) 279(48):50630–8. doi: 10.1074/jbc.M410077200
8. Cunningham D, Danley DE, Geoghegan KF, Griffor MC, Hawkins JL, Subashi TA, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. (2007) 14(5):413–9. doi: 10.1038/nsmb1235
9. Piper DE, Jackson S, Liu Q, Romanow WG, Shetterly S, Thibault ST, et al. The crystal structure of PCSK9: a regulator of plasma LDL-cholesterol. Structure. (2007) 15(5):545–52. doi: 10.1016/j.str.2007.04.004
10. Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. (2007) 282(25):18602–12. doi: 10.1074/jbc.M702027200
11. Gu HM, Adijiang A, Mah M, Zhang DW. Characterization of the role of EGF-A of low density lipoprotein receptor in PCSK9 binding. J Lipid Res. (2013) 54(12):3345–57. doi: 10.1194/jlr.M041129
12. Herbert B, Patel D, Waddington SN, Eden ER, McAleenan A, Sun XM, et al. Increased secretion of lipoproteins in transgenic mice expressing human D374Y PCSK9 under physiological genetic control. Arterioscler Thromb Vasc Biol. (2010) 30(7):1333–9. doi: 10.1161/ATVBAHA.110.204040
13. Ferri N, Marchiano S, Tibolla G, Baetta R, Dhyani A, Ruscica M, et al. PCSK9 knock-out mice are protected from neointimal formation in response to perivascular carotid collar placement. Atherosclerosis. (2016) 253:214–24. doi: 10.1016/j.atherosclerosis.2016.07.910
14. Sun H, Krauss RM, Chang JT, Teng BB. PCSK9 Deficiency reduces atherosclerosis, apolipoprotein B secretion, and endothelial dysfunction. J Lipid Res. (2018) 59(2):207–23. doi: 10.1194/jlr.M078360
15. Zaid A, Roubtsova A, Essalmani R, Marcinkiewicz J, Chamberland A, Hamelin J, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. (2008) 48(2):646–54. doi: 10.1002/hep.22354
16. Rosenson RS, Hegele RA, Fazio S, Cannon CP. The evolving future of PCSK9 inhibitors. J Am Coll Cardiol. (2018) 72(3):314–29. doi: 10.1016/j.jacc.2018.04.054
17. Wang F, Li M, Zhang A, Li H, Jiang C, Guo J. PCSK9 modulates macrophage polarization-mediated ventricular remodeling after myocardial infarction. J Immunol Res. (2022) 2022:7685796.35832650
18. Luquero A, Badimon L, Borrell-Pages M. PCSK9 functions in atherosclerosis are not limited to plasmatic LDL-cholesterol regulation. Front Cardiovasc Med. (2021) 8:639727. doi: 10.3389/fcvm.2021.639727
19. Punch E, Klein J, Diaba-Nuhoho P, Morawietz H, Garelnabi M. Effects of PCSK9 targeting: alleviating oxidation, inflammation, and atherosclerosis. J Am Heart Assoc. (2022) 11(3):e023328.35048716
20. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5(1):56. doi: 10.1038/s41572-019-0106-z
21. Giunzioni I, Tavori H, Covarrubias R, Major AS, Ding L, Zhang Y, et al. Local effects of human PCSK9 on the atherosclerotic lesion. J Pathol. (2016) 238(1):52–62. doi: 10.1002/path.4630
22. Ding Z, Liu S, Wang X, Deng X, Fan Y, Shahanawaz J, et al. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc Res. (2015) 107(4):556–67. doi: 10.1093/cvr/cvv178
23. Ding Z, Liu S, Wang X, Deng X, Fan Y, Sun C, et al. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid Redox Signal. (2015) 22(9):760–71. doi: 10.1089/ars.2014.6054
24. Ding Z, Wang X, Liu S, Zhou S, Kore RA, Mu S, et al. NLRP3 Inflammasome via IL-1beta regulates PCSK9 secretion. Theranostics. (2020) 10(16):7100–10. doi: 10.7150/thno.45939
25. Ricci C, Ruscica M, Camera M, Rossetti L, Macchi C, Colciago A, et al. PCSK9 Induces a pro-inflammatory response in macrophages. Sci Rep. (2018) 8(1):2267. doi: 10.1038/s41598-018-20425-x
26. Katsuki S, Jha PK, Lupieri A, Nakano T, Passos LSA, Rogers MA, et al. Proprotein convertase subtilisin/kexin 9 (PCSK9) promotes macrophage activation via LDL receptor-independent mechanisms. Circ Res. (2022) 131(11):873–89. doi: 10.1161/CIRCRESAHA.121.320056
27. Tang ZH, Peng J, Ren Z, Yang J, Li TT, Li TH, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-kappaB pathway. Atherosclerosis. (2017) 262:113–22. doi: 10.1016/j.atherosclerosis.2017.04.023
28. Camera M, Rossetti L, Barbieri SS, Zanotti I, Canciani B, Trabattoni D, et al. PCSK9 As a positive modulator of platelet activation. J Am Coll Cardiol. (2018) 71(8):952–4. doi: 10.1016/j.jacc.2017.11.069
29. Qi Z, Hu L, Zhang J, Yang W, Liu X, Jia D, et al. PCSK9 (proprotein convertase subtilisin/kexin 9) enhances platelet activation, thrombosis, and myocardial infarct expansion by binding to platelet CD36. Circulation. (2021) 143(1):45-–61. doi: 10.1161/CIRCULATIONAHA.120.046290
30. Liu S, Deng X, Zhang P, Wang X, Fan Y, Zhou S, et al. Blood flow patterns regulate PCSK9 secretion via MyD88-mediated pro-inflammatory cytokines. Cardiovasc Res. (2020) 116(10):1721–32. doi: 10.1093/cvr/cvz262
31. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. (2006) 354(12):1264–72. doi: 10.1056/NEJMoa054013
32. Meng FH, Liu S, Xiao J, Zhou YX, Dong LW, Li YF, et al. New loss-of-function mutations in PCSK9 (proprotein convertase subtilisin/kexin type 9) reduce plasma LDL (low-density lipoprotein) cholesterol. Arterioscler Thromb Vasc Biol. (2023). doi: 10.1161/ATVBAHA.122.318839. [Epub ahead of print].
33. Pott J, Schlegel V, Teren A, Horn K, Kirsten H, Bluecher C, et al. Genetic regulation of PCSK9 (proprotein convertase subtilisin/kexin type 9) plasma levels and its impact on atherosclerotic vascular disease phenotypes. Circ Genom Precis Med. (2018) 11(5):e001992. doi: 10.1161/CIRCGEN.117.001992
34. Polisecki E, Peter I, Robertson M, McMahon AD, Ford I, Packard C, et al. Genetic variation at the PCSK9 locus moderately lowers low-density lipoprotein cholesterol levels, but does not significantly lower vascular disease risk in an elderly population. Atherosclerosis. (2008) 200(1):95–101. doi: 10.1016/j.atherosclerosis.2007.12.005
35. Cheng JM, Oemrawsingh RM, Garcia-Garcia HM, Boersma E, van Geuns RJ, Serruys PW, et al. PCSK9 In relation to coronary plaque inflammation: results of the ATHEROREMO-IVUS study. Atherosclerosis. (2016) 248:117–22. doi: 10.1016/j.atherosclerosis.2016.03.010
36. Xie W, Liu J, Wang W, Wang M, Qi Y, Zhao F, et al. Association between plasma PCSK9 levels and 10-year progression of carotid atherosclerosis beyond LDL-C: a cohort study. Int J Cardiol. (2016) 215:293–8. doi: 10.1016/j.ijcard.2016.04.103
37. Zhu YM, Anderson TJ, Sikdar K, Fung M, McQueen MJ, Lonn EM, et al. Association of proprotein convertase subtilisin/kexin type 9 (PCSK9) with cardiovascular risk in primary prevention. Arterioscler Thromb Vasc Biol. (2015) 35(10):2254–9. doi: 10.1161/ATVBAHA.115.306172
38. Ruscica M, Ferri N, Fogacci F, Rosticci M, Botta M, Marchiano S, et al. Circulating levels of proprotein convertase subtilisin/kexin type 9 and arterial stiffness in a large population sample: data from the brisighella heart study. J Am Heart Assoc. (2017) 6(5). doi: 10.1161/JAHA.117.005764
39. Chirinos JA, Segers P, Hughes T, Townsend R. Large-artery stiffness in health and disease: JACC state-of-the-art review. J Am Coll Cardiol. (2019) 74(9):1237–63. doi: 10.1016/j.jacc.2019.07.012
40. Maulucci G, Cipriani F, Russo D, Casavecchia G, Di Staso C, Di Martino L, et al. Improved endothelial function after short-term therapy with evolocumab. J Clin Lipidol. (2018) 12(3):669–73. doi: 10.1016/j.jacl.2018.02.004
41. Cicero AFG, Toth PP, Fogacci F, Virdis A, Borghi C. Improvement in arterial stiffness after short-term treatment with PCSK9 inhibitors. Nutr Metab Cardiovasc Dis. (2019) 29(5):527–9. doi: 10.1016/j.numecd.2019.01.010
42. Navarese EP, Kolodziejczak M, Winter MP, Alimohammadi A, Lang IM, Buffon A, et al. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: the PCSK9-REACT study. Int J Cardiol. (2017) 227:644–9. doi: 10.1016/j.ijcard.2016.10.084
43. Sotler T, Sebestjen M. PCSK9 as an atherothrombotic risk factor. Int J Mol Sci. (2023) 24(3). doi: 10.3390/ijms24031966
44. Barale C, Melchionda E, Morotti A, Russo I. PCSK9 biology and its role in atherothrombosis. Int J Mol Sci. (2021) 22(11). doi: 10.3390/ijms22115880
45. Barale C, Bonomo K, Frascaroli C, Morotti A, Guerrasio A, Cavalot F, et al. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: a 12-month follow-up. Nutr Metab Cardiovasc Dis. (2020) 30(2):282–91. doi: 10.1016/j.numecd.2019.09.012
46. Petersen-Uribe A, Kremser M, Rohlfing AK, Castor T, Kolb K, Dicenta V, et al. Platelet-derived PCSK9 is associated with LDL metabolism and modulates atherothrombotic mechanisms in coronary artery disease. Int J Mol Sci. (2021) 22(20). doi: 10.3390/ijms222011179
47. Cammisotto V, Pastori D, Nocella C, Bartimoccia S, Castellani V, Marchese C, et al. PCSK9 Regulates Nox2-mediated platelet activation via CD36 receptor in patients with atrial fibrillation. Antioxidants (Basel. (2020) 9(4). doi: 10.3390/antiox9040296
48. Ridker PM, Rifai N, Bradwin G, Rose L. Plasma proprotein convertase subtilisin/kexin type 9 levels and the risk of first cardiovascular events. Eur Heart J. (2016) 37(6):554–60. doi: 10.1093/eurheartj/ehv568
49. Gencer B, Montecucco F, Nanchen D, Carbone F, Klingenberg R, Vuilleumier N, et al. Prognostic value of PCSK9 levels in patients with acute coronary syndromes. Eur Heart J. (2016) 37(6):546–53. doi: 10.1093/eurheartj/ehv637
50. Qiu C, Zhou Q, Li X, Zhang Z, Zeng P, Cao Z, et al. High circulating proprotein convertase subtilisin/kexin type 9 concentration associates with cardiovascular risk: a meta-analysis of cohort studies. Medicine (Baltimore). (2017) 96(48):e8848. doi: 10.1097/MD.0000000000008848
51. Macchi C, Ferri N, Favero C, Cantone L, Vigna L, Pesatori AC, et al. Long-term exposure to air pollution raises circulating levels of proprotein convertase subtilisin/kexin type 9 in obese individuals. Eur J Prev Cardiol. (2019) 26(6):578–88. doi: 10.1177/2047487318815320
52. Meyer JG. Qualitative and quantitative shotgun proteomics data analysis from data-dependent acquisition mass spectrometry. Methods Mol Biol. (2021) 2259:297–308. doi: 10.1007/978-1-0716-1178-4_19
53. Guan S, Taylor PP, Han Z, Moran MF, Ma B. Data dependent-independent acquisition (DDIA) proteomics. J Proteome Res. (2020) 19(8):3230–7. doi: 10.1021/acs.jproteome.0c00186
54. Chahrour O, Cobice D, Malone J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J Pharm Biomed Anal. (2015) 113:2–20. doi: 10.1016/j.jpba.2015.04.013
55. Rozanova S, Barkovits K, Nikolov M, Schmidt C, Urlaub H, Marcus K. Quantitative mass spectrometry-based proteomics: an overview. Methods Mol Biol. (2021) 2228:85–116. doi: 10.1007/978-1-0716-1024-4_8
56. Banfi C, Baetta R, Gianazza E, Tremoli E. Technological advances and proteomic applications in drug discovery and target deconvolution: identification of the pleiotropic effects of statins. Drug Discov Today. (2017) 22(6):848–69. doi: 10.1016/j.drudis.2017.03.001
57. Croyal M, Fall F, Krempf M, Thedrez A, Ouguerram K, Ferchaud-Roucher V, et al. Plasma PCSK9 measurement by liquid chromatography-tandem mass spectrometry and comparison with conventional ELISA. J Chromatogr B Analyt Technol Biomed Life Sci. (2017) 1044–1045:24–9. doi: 10.1016/j.jchromb.2016.12.040
58. Gauthier MS, Awan Z, Bouchard A, Champagne J, Tessier S, Faubert D, et al. Posttranslational modification of proprotein convertase subtilisin/kexin type 9 is differentially regulated in response to distinct cardiometabolic treatments as revealed by targeted proteomics. J Clin Lipidol. (2018) 12(4):1027–38. doi: 10.1016/j.jacl.2018.03.092
59. Agatonovic-Kustrin S, Morton DW, Smirnov V, Petukhov A, Gegechkori V, Kuzina V, et al. Analytical strategies in lipidomics for discovery of functional biomarkers from human Saliva. Dis Markers. (2019) 2019:6741518. doi: 10.1155/2019/6741518
60. Saini RK, Prasad P, Shang X, Keum YS. Advances in lipid extraction methods-a review. Int J Mol Sci. (2021) 22(24). doi: 10.3390/ijms222413643
61. Zhao Z, Xu Y. An extremely simple method for extraction of lysophospholipids and phospholipids from blood samples. J Lipid Res. (2010) 51(3):652–9. doi: 10.1194/jlr.D001503
62. Belkadi A, Thareja G, Abbaszadeh F, Badii R, Fauman E, Albagha OME, et al. Identification of PCSK9-like human gene knockouts using metabolomics, proteomics, and whole-genome sequencing in a consanguineous population. Cell Genom. (2023) 3(1):100218. doi: 10.1016/j.xgen.2022.100218
63. Denis N, Palmer-Smith H, Elisma F, Busuttil A, Wright TG, Bou Khalil M, et al. Quantitative proteomic analysis of PCSK9 gain of function in human hepatic HuH7 cells. J Proteome Res. (2011) 10(4):2011–26. doi: 10.1021/pr2000072
64. Choi MC, Lee YU, Kim SH, Lee JH, Park JH, Streb JW, et al. Overexpression of A-kinase anchoring protein 12A activates sterol regulatory element binding protein-2 and enhances cholesterol efflux in hepatic cells. Int J Biochem Cell Biol. (2008) 40(11):2534–43. doi: 10.1016/j.biocel.2008.04.020
65. Ding Z, Wang X, Liu S, Shahanawaz J, Theus S, Fan Y, et al. PCSK9 Expression in the ischaemic heart and its relationship to infarct size, cardiac function, and development of autophagy. Cardiovasc Res. (2018) 114(13):1738–51. doi: 10.1093/cvr/cvy128
66. Guo Y, Yan B, Tai S, Zhou S, Zheng XL. PCSK9: associated with cardiac diseases and their risk factors? Arch Biochem Biophys. (2021) 704:108717. doi: 10.1016/j.abb.2020.108717
67. Hilvo M, Simolin H, Metso J, Ruuth M, Oorni K, Jauhiainen M, et al. PCSK9 Inhibition alters the lipidome of plasma and lipoprotein fractions. Atherosclerosis. (2018) 269:159–65. doi: 10.1016/j.atherosclerosis.2018.01.004
68. Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J. (2016) 37(25):1967–76. doi: 10.1093/eurheartj/ehw148
69. Huang K, Wen XQ, Ren N, Yang L, Gao B. Lipidomic profile in patients with a very high risk of atherosclerotic cardiovascular disease on PCSK9 inhibitor therapy. Rev Cardiovasc Med. (2021) 22(2):461–7. doi: 10.31083/j.rcm2202052
70. Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers. (2017) 3:17093. doi: 10.3390/biomedicines9121941
71. Anesi A, Di Minno A, Calcaterra I, Cavalca V, Tripaldella M, Porro B, et al. An untargeted lipidomic analysis reveals depletion of several phospholipid classes in patients with familial hypercholesterolemia on treatment with evolocumab. Biomedicines. (2021) 9(12).
72. Da Dalt L, Ruscica M, Bonacina F, Balzarotti G, Dhyani A, Di Cairano E, et al. PCSK9 Deficiency reduces insulin secretion and promotes glucose intolerance: the role of the low-density lipoprotein receptor. Eur Heart J. (2019) 40(4):357–68. doi: 10.1093/eurheartj/ehy357
73. Marku A, Da Dalt L, Galli A, Dule N, Corsetto P, Rizzo AM, et al. Pancreatic PCSK9 controls the organization of the beta-cell secretory pathway via LDLR-cholesterol axis. Metab Clin Exp. (2022) 136:155291. doi: 10.1016/j.metabol.2022.155291
74. Carugo S, Sirtori CR, Corsini A, Tokgozoglu L, Ruscica M. PCSK9 Inhibition and risk of diabetes: should we worry? Curr Atheroscler Rep. (2022) 24(12):995–1004. doi: 10.1007/s11883-022-01074-y
75. Saitoski K, Ryaboshapkina M, Hamza GM, Jarnuczak AF, Berthault C, Carlotti F, et al. Proprotein convertase PCSK9 affects expression of key surface proteins in human pancreatic beta cells via intracellular and extracellular regulatory circuits. J Biol Chem. (2022) 298(7):102096. doi: 10.1016/j.jbc.2022.102096
76. Peyot ML, Roubtsova A, Lussier R, Chamberland A, Essalmani R, Murthy Madiraju SR, et al. Substantial PCSK9 inactivation in beta-cells does not modify glucose homeostasis or insulin secretion in mice. Biochim Biophys Acta Mol Cell Biol Lipids. (2021) 1866(8):158968. doi: 10.1016/j.bbalip.2021.158968
77. Path G, Perakakis N, Mantzoros CS, Seufert J. PCSK9 Inhibition and cholesterol homeostasis in insulin producing beta-cells. Lipids Health Dis. (2022) 21(1):138. doi: 10.1186/s12944-022-01751-6
78. Tcheoubi SER, Akpovi CD, Coppee F, Decleves AE, Laurent S, Agbangla C, et al. Molecular and cellular biology of PCSK9: impact on glucose homeostasis. J Drug Target. (2022) 30(9):948–60. doi: 10.1080/1061186X.2022.2092622
79. Liu JP, Tang Y, Zhou S, Toh BH, McLean C, Li H. Cholesterol involvement in the pathogenesis of neurodegenerative diseases. Mol Cell Neurosci. (2010) 43(1):33–42. doi: 10.1016/j.mcn.2009.07.013
80. Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. (2001) 12(2):105–12. doi: 10.1097/00041433-200104000-00003
81. Adorni MP, Ruscica M, Ferri N, Bernini F, Zimetti F. Proprotein convertase subtilisin/kexin type 9, brain cholesterol homeostasis and potential implication for Alzheimer’s disease. Front Aging Neurosci. (2019) 11:120. doi: 10.3389/fnagi.2019.00120
82. Papotti B, Adorni MP, Marchi C, Zimetti F, Ronda N, Panighel G, et al. PCSK9 Affects astrocyte cholesterol metabolism and reduces neuron cholesterol supplying in vitro: potential implications in Alzheimer’s disease. Int J Mol Sci. (2022) 23(20). doi: 10.3390/ijms232012192
83. Lipinski MJ, Benedetto U, Escarcega RO, Biondi-Zoccai G, Lhermusier T, Baker NC, et al. The impact of proprotein convertase subtilisin-kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta-analysis. Eur Heart J. (2016) 37(6):536–45. doi: 10.1093/eurheartj/ehv563
84. Benn M, Nordestgaard BG, Frikke-Schmidt R, Tybjaerg-Hansen A. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer’s disease and Parkinson’s disease: mendelian randomisation study. BMJ. (2017) 357:j1648. doi: 10.1136/bmj.j1648
85. Khan AR, Bavishi C, Riaz H, Farid TA, Khan S, Atlas M, et al. Increased risk of adverse neurocognitive outcomes with proprotein convertase subtilisin-kexin type 9 inhibitors. Circ Cardiovasc Qual Outcomes. (2017) 10(1). doi: 10.1161/CIRCOUTCOMES.116.003153
86. Gencer B, Mach F, Guo J, Im K, Ruzza A, Wang H, et al. Cognition after lowering LDL-cholesterol with evolocumab. J Am Coll Cardiol. (2020) 75(18):2283–93. doi: 10.1016/j.jacc.2020.03.039
87. Janik MJ, Urbach DV, van Nieuwenhuizen E, Zhao J, Yellin O, Baccara-Dinet MT, et al. Alirocumab treatment and neurocognitive function according to the CANTAB scale in patients at increased cardiovascular risk: a prospective, randomized, placebo-controlled study. Atherosclerosis. (2021) 331:20–7. doi: 10.1016/j.atherosclerosis.2021.06.913
88. Raccah B H, Yanovsky A, Treves N, Rotshild V, Renoux C, Danenberg H, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and the risk for neurocognitive adverse events: a systematic review, meta-analysis and meta-regression. Int J Cardiol. (2021) 335:7–14 doi: 10.1016/j.ijcard.2021.04.025
89. Testa G, Staurenghi E, Zerbinati C, Gargiulo S, Iuliano L, Giaccone G, et al. Changes in brain oxysterols at different stages of Alzheimer’s disease: their involvement in neuroinflammation. Redox Biol. (2016) 10:24–33. doi: 10.1016/j.redox.2016.09.001
90. Bjorkhem I, Leoni V, Svenningsson P. On the fluxes of side-chain oxidized oxysterols across blood-brain and blood-CSF barriers and origin of these steroids in CSF (review). J Steroid Biochem Mol Biol. (2019) 188:86–9. doi: 10.1016/j.jsbmb.2018.12.009
91. Lutjohann D, Stellaard F, Bolukbasi B, Kerksiek A, Parhofer KG, Laufs U. Anti-PCSK 9 antibodies increase the ratios of the brain-specific oxysterol 24S-hydroxycholesterol to cholesterol and to 27-hydroxycholesterol in the serum. Br J Clin Pharmacol. (2021) 87(11):4252–61. doi: 10.1111/bcp.14841
92. Wang HL, Wang YY, Liu XG, Kuo SH, Liu N, Song QY, et al. Cholesterol, 24-Hydroxycholesterol, and 27-hydroxycholesterol as surrogate biomarkers in cerebrospinal fluid in mild cognitive impairment and Alzheimer’s disease: a meta-analysis. J Alzheimers Dis. 2016;51(1):45–55. doi: 10.3233/JAD-150734
93. Seidah NG, Prat A. The multifaceted biology of PCSK9. Endocr Rev. (2022) 43(3):558–82. doi: 10.1210/endrev/bnab035
94. Ni YG, Condra JH, Orsatti L, Shen X, Di Marco S, Pandit S, et al. A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J Biol Chem. (2010) 285(17):12882–91. doi: 10.1074/jbc.M110.113035
95. Holla OL, Cameron J, Tveten K, Strom TB, Berge KE, Laerdahl JK, et al. Role of the C-terminal domain of PCSK9 in degradation of the LDL receptors. J Lipid Res. (2011) 52(10):1787–94. doi: 10.1194/jlr.M018093
96. Xu W, Liu L. Hornby D. c-IAP1 binds and processes PCSK9 protein: linking the c-IAP1 in a TNF-alpha pathway to PCSK9-mediated LDLR degradation pathway. Molecules. (2012) 17(10):12086–101. doi: 10.3390/molecules171012086
97. Ly K, Essalmani R, Desjardins R, Seidah NG, Day R. An unbiased mass spectrometry approach identifies glypican-3 as an interactor of proprotein convertase subtilisin/kexin type 9 (PCSK9) and low density lipoprotein receptor (LDLR) in hepatocellular carcinoma cells. J Biol Chem. (2016) 291(47):24676–87. doi: 10.1074/jbc.M116.746883
98. Melendez QM, Wooten CJ, Krishnaji ST, Knagge K, Kirchner D, Lopez D. Identification of novel proteins interacting with proprotein convertase subtilisin/kexin 9. Int J Biomed Investig. (2020) 3(1). doi: 10.31531/2581-4745.1000123
99. Mashiba S, Wada Y, Takeya M, Sugiyama A, Hamakubo T, Nakamura A, et al. In vivo complex formation of oxidized alpha(1)-antitrypsin and LDL. Arterioscler Thromb Vasc Biol. (2001) 21(11):1801–8. doi: 10.1161/hq1101.098232
100. George J, Harats D, Gilburd B, Afek A, Levy Y, Schneiderman J, et al. Immunolocalization of beta2-glycoprotein I (apolipoprotein H) to human atherosclerotic plaques: potential implications for lesion progression. Circulation. (1999) 99(17):2227–30. doi: 10.1161/01.cir.99.17.2227
101. Harats D, George J. Beta2-glycoprotein I and atherosclerosis. Curr Opin Lipidol. (2001) 12(5):543–6. doi: 10.1097/00041433-200110000-00010
102. Kristiansson A, Gram M, Flygare J, Hansson SR, Akerstrom B, Storry JR. The role of alpha(1)-microglobulin (A1M) in erythropoiesis and erythrocyte homeostasis-therapeutic opportunities in hemolytic conditions. Int J Mol Sci. (2020) 21(19). doi: 10.3390/ijms21197234
103. Ferri N, Corsini A, Macchi C, Magni P, Ruscica M. Proprotein convertase subtilisin kexin type 9 and high-density lipoprotein metabolism: experimental animal models and clinical evidence. Transl Res. (2016) 173:19–29. doi: 10.1016/j.trsl.2015.10.004
104. Tavori H, Giunzioni I, Linton MF, Fazio S. Loss of plasma proprotein convertase subtilisin/kexin 9 (PCSK9) after lipoprotein apheresis. Circ Res. (2013) 113(12):1290–5. doi: 10.1161/CIRCRESAHA.113.302655
105. Tavori H, Christian D, Minnier J, Plubell D, Shapiro MD, Yeang C, et al. PCSK9 association with lipoprotein(a). Circ Res. (2016) 119(1):29–35. doi: 10.1161/CIRCRESAHA.116.308811
106. Burnap SA, Joshi A, Tsimikas S, Fernandez-Hernando C, Kiechl S, Berry SE, et al. High-density lipoproteins are the main carriers of PCSK9 in the circulation. J Am Coll Cardiol. (2020) 75(12):1495–7. doi: 10.1016/j.jacc.2020.01.033
107. Burnap SA, Sattler K, Pechlaner R, Duregotti E, Lu R, Theofilatos K, et al. PCSK9 activity is potentiated through HDL binding. Circ Res. (2021) 129(11):1039–53. doi: 10.1161/CIRCRESAHA.121.319272
108. Giro O, Jimenez A, Pane A, Badimon L, Ortega E, Chiva-Blanch G. Extracellular vesicles in atherothrombosis and cardiovascular disease: friends and foes. Atherosclerosis. (2021) 330:61–75. doi: 10.1016/j.atherosclerosis.2021.07.002
109. Timmerman N, Waissi F, Dekker M, de Borst GJ, van Bennekom J, de Winter RJ, et al. Ceramides and phospholipids in plasma extracellular vesicles are associated with high risk of major cardiovascular events after carotid endarterectomy. Sci Rep. (2022) 12(1):5521. doi: 10.1038/s41598-022-09225-6
110. Macchi C, Greco MF, Favero C, Dioni L, Cantone L, Hoxha M, et al. Associations among PCSK9 levels, atherosclerosis-derived extracellular vesicles, and their miRNA content in adults with obesity. Front Cardiovasc Med. (2021) 8:785250. doi: 10.3389/fcvm.2021.785250
111. Scalise V, Lombardi S, Sanguinetti C, Nieri D, Pedrinelli R, Celi A, et al. A novel prothrombotic role of proprotein convertase subtilisin kexin 9: the generation of procoagulant extracellular vesicles by human mononuclear cells. Mol Biol Rep. (2022) 49(5):4129–34. doi: 10.1007/s11033-022-07433-x
112. Ruscica M, Ferri N, Santos RD, Sirtori CR, Corsini A. Lipid lowering drugs: present status and future developments. Curr Atheroscler Rep. (2021) 23(5):17. doi: 10.1007/s11883-021-00918-3
113. Badimon L, Padro T, Arderiu G, Vilahur G, Borrell-Pages M, Suades R. Extracellular vesicles in atherothrombosis: from biomarkers and precision medicine to therapeutic targets. Immunol Rev. (2022) 312(1):6–19. doi: 10.1111/imr.13127
114. Greco MF, Rizzuto AS, Zara M, Cafora M, Favero C, Solazzo G, et al. PCSK9 confers inflammatory properties to extracellular vesicles released by vascular smooth muscle cells. Int J Mol Sci. (2022) 23(21). doi: 10.3390/ijms232113065
115. Lupo MG, Bressan A, Donato M, Canzano P, Camera M, Poggio P, et al. PCSK9 Promotes arterial medial calcification. Atherosclerosis. (2022) 346:86–97. doi: 10.1016/j.atherosclerosis.2022.01.015
116. Ferri N, Tibolla G, Pirillo A, Cipollone F, Mezzetti A, Pacia S, et al. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis. (2012) 220(2):381–6. doi: 10.1016/j.atherosclerosis.2011.11.026
117. Guo Y, Tang Z, Yan B, Yin H, Tai S, Peng J, et al. PCSK9 (proprotein convertase subtilisin/kexin type 9) triggers vascular smooth muscle cell senescence and apoptosis: implication of its direct role in degenerative vascular disease. Arterioscler Thromb Vasc Biol. (2022) 42(1):67–86. doi: 10.1161/ATVBAHA.121.316902
Keywords: proprotein convertase subtilisin/kexin type 9, atherosclerotic cardiovascular disease, proteomics, lipidomics, extracellular vesicles
Citation: Gianazza E, Macchi C, Banfi C and Ruscica M (2023) Proteomics and Lipidomics to unveil the contribution of PCSK9 beyond cholesterol lowering: a narrative review. Front. Cardiovasc. Med. 10:1191303. doi: 10.3389/fcvm.2023.1191303
Received: 21 March 2023; Accepted: 29 May 2023;
Published: 12 June 2023.
Edited by:
Maya S. Safarova, University of Kansas Medical Center, United StatesReviewed by:
Angelo Baldassare Cefalù, University of Palermo, Italy© 2023 Gianazza, Macchi, Banfi and Ruscica. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Banfi Y3Jpc3RpbmEuYmFuZmlAY2FyZGlvbG9naWNvbW9uemluby5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.