 Xinmin Zhang
Xinmin Zhang He Cai
He Cai He Xu4
He Xu4 Su Dong
Su Dong Haichun Ma
Haichun Ma- 1Department of Anesthesiology, The First Hospital of Jilin University, Changchun, China
- 2The Public Laboratory Platform of the First Hospital of Jilin University, Changchun, China
- 3The Cardiovascular Center, The First Hospital of Jilin University, Changchun, China
- 4Department of Integrative Medicine, Lequn Branch, The First Hospital of Jilin University, Changchun, China
Cardiovascular diseases (CVDs) have been established as a major cause of mortality globally. However, the exact pathogenesis remains obscure. N6-methyladenosine (m6A) methylation is the most common epigenetic modification on mRNAs regulated by methyltransferase complexes (writers), demethylase transferases (erasers) and binding proteins (readers). It is now understood that m6A is a major player in physiological and pathological cardiac processes. m6A methylation are potentially involved in many mechanisms, for instance, regulation of calcium homeostasis, endothelial function, different forms of cell death, autophagy, endoplasmic reticulum stress, macrophage response and inflammation. In this review, we will summarize the molecular functions of m6A enzymes. We mainly focus on m6A-associated mechanisms and functions in CVDs, especially in heart failure and ischemia heart disease. We will also discuss the potential application and clinical transformation of m6A modification.
1. Introduction
Cardiovascular disease (CVD) is a leading major cause of death, responsible for 31.5% of mortalities globally (1). Current evidence suggests that China and India have the highest burdens of CVD worldwide (2).
The past decade has witnessed significant progress achieved in research on CVD, which has led to the development of new therapeutic approaches, such as medications (e.g., angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), and statins) and interventional methods [e.g., coronary artery bypass surgery (CABG) and percutaneous coronary intervention (PCI)]. However, current preventive and therapeutic options for CVD remain limited.
With significant inroads achieved in science and technology over the past couple of years, investigations on diseases have progressed to the genetic and epigenetic levels (3, 4). Most of the traditional treatments for patients with CVD are protein-targeting drugs. It has been established that mRNA is subject to tight regulation at the transcriptional and post-transcriptional levels before translation into proteins. Therefore, post-transcriptional targeting has huge prospects for drug development. Above 150 post-transcriptional modifications have hitherto been documented in RNAs in living organisms. A frequent RNA epigenetic modification at the post-transcriptional stage is observed at the N6 position of adenosine, which undergoes N6-m6A RNA methylation. Although Desrosiers et al. (5, 6) first reported m6A in the 1970s, it is only recently that the mechanisms underlying the specificity of m6A modification and biogenesis in cells have been uncovered. m6A RNA methylation usually occurs at the RRm6ACH consensus motif, which is enriched in internal long exons and 3′ untranslated regions (3′UTRs) near stop codons. Furthermore, m6A occurs in precursor mRNAs (pre-RNAs) and long noncoding RNAs (lncRNAs).
In recent years, N6-methyladenosine methylation has been associated with important processes in mammals, such as embryonic development (7), sex determination (8), circadian rhythm (9), neurogenesis (10), stress responses (11) and cancers (12). The functions of m6A methylation in cardiovascular diseases have been recognized (13, 14), however, further studies are warranted.
Herein, we discuss the m6A proteins respectively. Secondly, we provide a comprehensive overview of the roles of m6A methylation in CVDs, laying emphasis on cardiac hypertrophy, heart failure and ischemic heart disease (IHD). Also encompassing atherosclerosis, aortic dissecting aneurysm (ADA), aortic valve calcification, hypoxic pulmonary hypertension (HPH), dilated cardiomyopathy (DCM) and cardiotoxicity. Finally, we discuss future research directions of the application of m6A for CVD treatment.
2. m6A RNA methylation
The m6A modification process involves methyltransferase complexes (writers), demethylase transferases (erasers), and binding proteins (readers) (15) (Figure 1). m6A modification can be catalyzed by “writers” and removed by “erasers”. Thus this methylation-dependent process can be reversed and controlled. “Readers” can specifically identify and link RNA methylation modification sites to perform specific biological functions (16), such as RNA splicing, translation and stability (Figure 1).
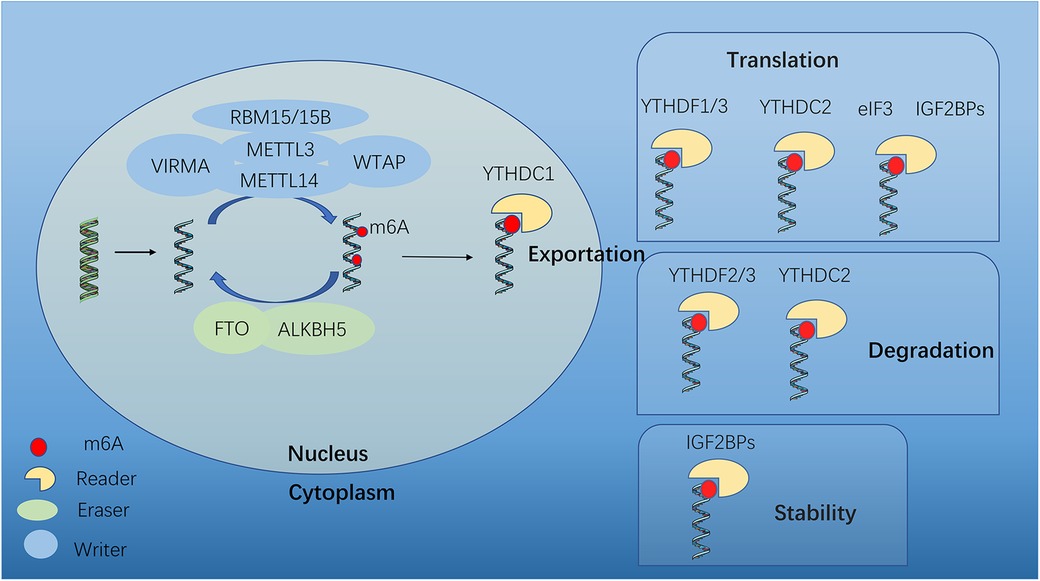
Figure 1. Dynamic m6A modification and mediated functions. m6A mRNA methylation is regulated by methyltransferases (“writers”), demethylases (“erasers”) and m6A-binding proteins (“readers”). METTL3, methyltransferase-like 3; METTL14, methyltransferase-like 14; WTAP, Wilms tumor 1- associated protein; VIRMA, KIAA1429; METTL16, methyltransferase-like 16; RBM15, RNA binding motif protein 15; FTO, fat mass and obesity-associated; ALKBH5, AlkB homologue 5; YTHDF1/2/3, YTH N6-methyladenosine RNA binding protein 1/2/3; YTHDC1, YTH domain containing 1; YTHDC2, YTH domain containing 2; eIF3, Eukaryotic translation initiation factor 3 subunit A; IGF2BP2, insulin-like growth factor 2 mRNA binding protein 2.
2.1. Methyltransferases/writers
The m6A methyltransferase complex is comprised of Methyltransferase-like 3 and 14 (METTL3 and METTL14), which form a stable heterodimer to serve as the catalytic core. Although both METTL3 and METTL14 contain a methyltransferase domain, crystal structure analysis shows that only METTL3 functions as catalytic core, while METTL14 serves as an allosteric adapter to stabilize interaction with RNA binding and improve methylation efficiency. Several auxiliary cofactors facilitate m6A deposition as adaptor proteins, by interacting with the core to guide methylase specificity, localization, binding, and activity. These cofactors include Wilms' tumor 1-associating protein (WTAP), VRIMA (KIAA1429), RBM15 (RNA-binding motif protein 15) and ZC3H13. WTAP helps recruit and anchor the methylases to target RNAs, interacts with both METTL3 and METTL14, and guides the specificity and levels of methylation. Methyltransferase-like 16 (METTL16), as a newly identified m6A methyltransferase, is the homolog of METTL3 (17). Besides, VIRMA, RBM15 and ZC3H13 are required for m6A methylation.
2.2. Demethylases/erasers
The m6A modification can be removed by “erasers”, including fat mass and obesity-associated (FTO) and AlkB Homolog 5 (ALKBH5). FTO is predominantly expressed in the nucleus, suggesting that any demethylation will occur before mRNA export, and even suggesting that FTO prevents m6A addition rather than actively erasing the mark. It has been observed that FTO demethylation in the cytoplasm during cancerous states, DNA damage responses and heat shock, indicating that this may be a specialized pathway. The second known demethylase ALKBH5 was discovered in 2013 (18), which is more likely to serve as a specific eraser of m6A, because the expression of ALKBH5 consistently correlates with reduced methylation both in human tissues and mice. Similar to FTO, ALKBH5 exhibits nuclear localization, indicating that cytoplasmic demethylation is largely nonexistent.
2.3. Readers
Reader proteins can recognize and bind to the mark, thus paly different downstream effects. They employ effectors to specify transcript splicing, processing, stability, translation, and localization (19). YT521-B homology (YTH) domain containing proteins bind to methyl moiety on the RNA molecule directly and mediate methylated transcript regulation, including YTH domain family proteins (YTHDF1, YTHDF2 and YTHDF3), YTH domain containing 1 and 2 (YTHDC1 and YTHDC2). Other m6A regulators bind indirectly, by weaking binding to m6A or via m6A structural switches, such as eukaryotic translation initiation factor 3H (Eif3), insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) and heterogeneous nuclear ribonucleoproteins (HNRNPs) (20, 21).
3. Cardiac hypertrophy and heart failure (HF)
3.1. Pathological hypertrophy and heart failure
Heart failure represents the terminal stage of various cardiovascular diseases, featuring poor cardiac performance and left ventricular dilatation. Besides, pathological cardiac hypertrophy often results in HF. Clinically, HF is classified into two major subtypes: HF with preserved ejection fraction (HfpEF, EF ≥ 50%) and HF with reduced ejection fraction (HfrEF, EF ≤ 40%). The mechanisms underlying HFpEF are largely obscure. Recently, m6A has been closely associated with heart failure in many studies. In this respect, it has been reported that FTO was upregulated in HFpEF patients and mice (22), while FTO was downregulated in HfrEF (13). This differential expression might be caused by pathophysiological differences. Mathiyalagan et al. (13) demonstrated that FTO was downregulated in failing mammalian hearts and hypoxic primary cardiomyocyte (CM) cells. Overexpression of FTO can reportedly increase the m6A level in failing hearts and improve cardiac contractile function, as indicated by higher ejection fraction, fractional shortening, and improved wall motion. Similarly, Berulava et al.'s study showed that m6A methylation levels were altered in the myocardium of HF and hypertrophy. In cardiac-specific FTO knockout mice, heart failure progression was accelerated, ejection fraction was decreased, and cardiac dilation was increased. METTL3 and FTO knockout may impair the myocardial response to stress load (23). Another study based on a transverse aortic constriction (TAC) model consistently showed that the m6A expression gradually increased with time. FTO is the main contributor to increased m6A levels. FTO plays a predominant role in increased m6A levels. FTO expression was significantly reduced at 8 weeks after TAC, and minimal changes were observed at 1, 3, and 7 days after TAC. FTO overexpression could attenuate cardiac hypertrophy and remodeling and improve cardiac dysfunction and stamina compared with TAC mice (24). Furthermore, FTO plays an important role in cardiomyocyte metabolic homeostasis; the loss of function of FTO has been reported to reduce the glycolytic capacity of cardiomyocytes (24). Mechanistically, FTO targets SERCA2a (a contractile protein) and demethylates SERCA2a resulting in increased SERCA2a expression and cardiac function improvement in mice failing hearts (13) (Table 1). Erkens et al. (52) found that SERCA2a was downregulated in Nrf2 KO mice, associated with LV dysfunction and cardiac hypertrophy, while FTO demethylates SERCA2a, suggesting that FTO is a key factor in Nrf2-associated cardiac hypertrophy. As for cardiac energy metabolism, PGAM2 is involved to a certain extent in the glycolytic changes in vitro and in vivo. Thus, FTO regulates glycolysis in an m6A-dependent way while regulating glucose uptake, possibly by modulating the AKT–GLUT4 axis (24). METTL3, as a “writer” protein, yields an opposite effect compared with “erasers” on cardiac hypertrophy in an m6A-dependent manner. Dorn et al. (14) demonstrated that METTL3 expression was increased in hypertrophic cardiomyocytes. Significant m6A modification was observed in genes associated with protein kinase mRNAs and intracellular signaling pathways, including members of the MAPK signaling cascade. METTL3 knockout myocardium showed morphological and functional changes of heart failure. Inhibition of METTL3 was sufficient to block hypertrophy in vitro, while enhancing METTL3 expression could induce cardiomyocyte hypertrophy without additional stimuli in vitro and in vivo (14). Kmietzyk et al. (35) found that METTL3 overexpression reduced pathological cardiac hypertrophy, myocardial fibrosis and collagen transcription. FTO knockout attenuated cardiomyocyte hypertrophy in phenylephrine-stimulated cardiomyocyte hypertrophy, while METTL3 knockout increased cell size. These studies overlap in their assertion that METTL3 plays an important role in supporting cardiac homeostasis and hypertrophic stress responses in mice. A subsequent study by Lu et al. (36) validated the effects of METTL3 overexpression on myocardial hypertrophy. A post-translational process termed ubiquitination has been reported to mediate protein stability, intracellular trafficking, and enzyme activity. Lu et al. revealed that USP12 promoted Ang II-induced cardiomyocyte hypertrophy; METTL3 expression was induced by Ang II but was downregulated in USP12 knockdown neonatal rat cardiomyocytes (NRCMs). Upregulation of METTL3 reversed the decrease in myocardial hypertrophy induced by AngII in USP12-silenced NRCMs. Similarly, METTL3 was interacted with Parp10 mRNA, and participate in the prohypertrophic effect of CHAPIR (53). Xu et al. (54) found that the m6A reader YTHDF2 was increased in both human and mice HF samples. Furthermore, YTHDF2 suppressed cardiac hypertrophy via m6A-mediated degradation of Myh7 mRNA. These studies indicate the potential roles of m6A in pathological cardiac hypertrophy and HF (Figure 2).
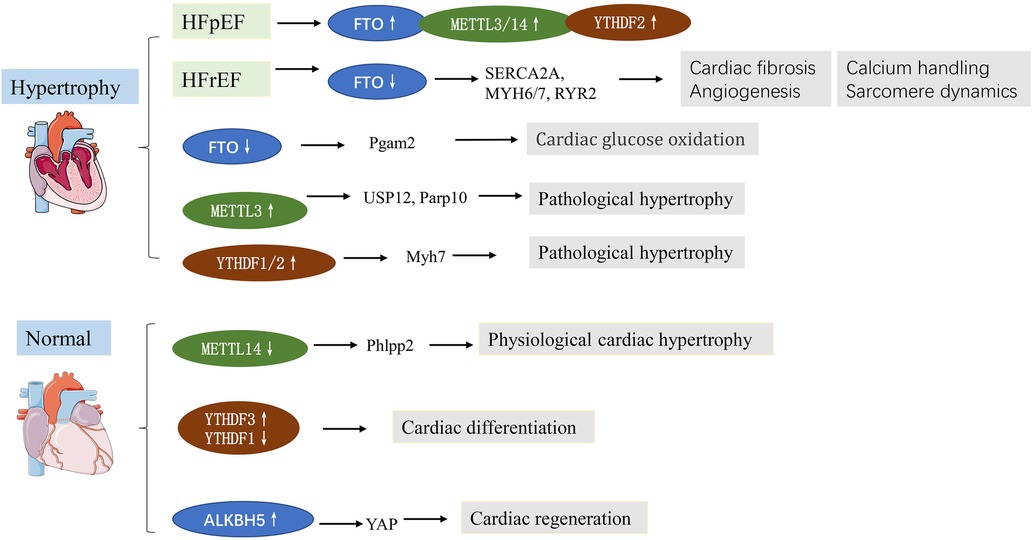
Figure 2. m6A methylation in cardiac hypertrophy and heart failure (HF). See main text for more details.
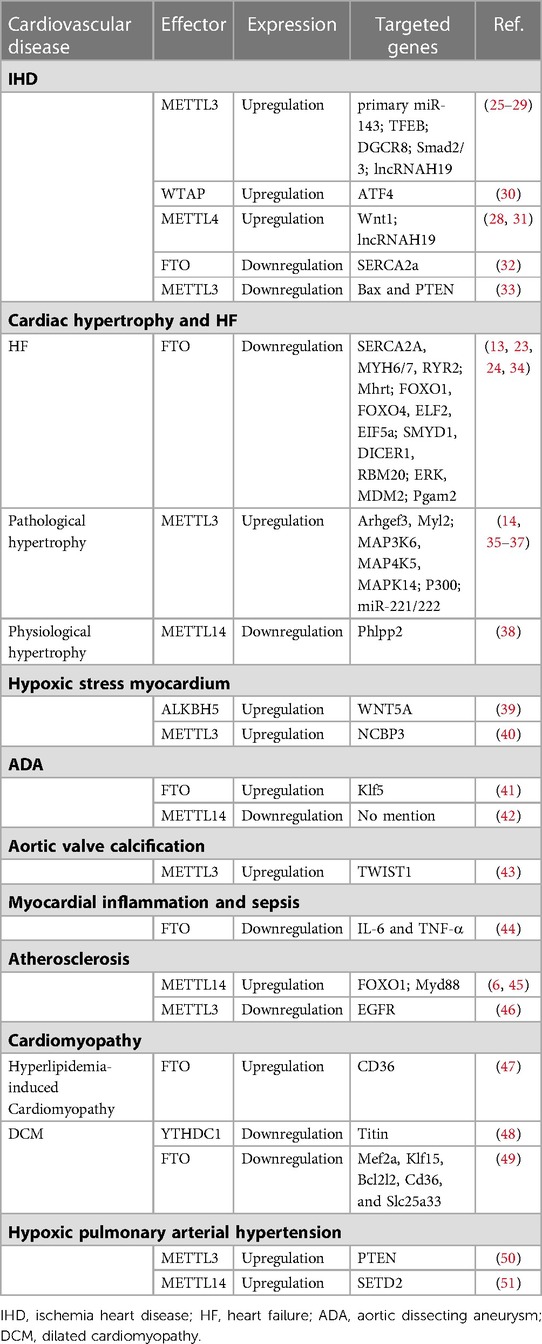
Table 1. Roles of m6A enzymes in the cardiovascular system.
3.2. Physiological cardiac growth and regeneration
Loss of cardiomyocytes following cardiac injuries plays a key role in the development of heart failure. In mammals, cardiomyocytes have long been considered as permanent cells with no ability to proliferate. However, recent studies show that, in fact, cardiomyocytes in mammals have some degree of regenerative capacity during development and very soon after birth (55). m6A is involved in cardiomyocyte proliferation and differentiation (56). A study showed that m6A modification and m6A peaks were lowest in 1-day-old mouse hearts than at 7 and 28 days after birth (57). Zhenbo Han et al. (58) investigated the importance of m6A alteration in heart regeneration during postnatal and adult injury. As expected, m6A demethylase ALKBH5 was downregulated, while the global m6A level was increased after birth. Cardiac function and regeneration ability decreased significantly in ALKBH5 knockout mice after neonatal apex resection. After induction of ALKBH5 expression, the myocardial infarction area was significantly reduced, cardiac function was restored, and CM proliferation was promoted after myocardial infarction in young and adult mice. ALKBH5 promoted YAP translation by increasing YTHDF1 levels. Interestingly, two similar proteins, YTHDF1 and YTHDF3, reportedly yield distinct effects on the same biological process (59). Recent evidence revealed that although YTHDF1 and YTHDF3 play diverse roles in embryonic stem cell-derived cardiac differentiation, they exhibit decreased levels. Loss of YTHDF1 could downregulate cardiomyocyte-specific genes and impair their differentiation. In contrast, YTHDF3 knockdown promoted differentiation by upregulating CM-specific genes (59).
Multiple studies have demonstrated that the endogenous regenerative potential of cardiomyocytes in the adult heart can be activated by interventions like exercise, and exercise can induce physiological cardiac hypertrophy in the heart. Although pathological cardiac hypertrophy and physiological cardiac hypertrophy appear similar, the underlying mechanisms that exist are fundamentally different. m6A is reportedly essential for exercise-induced physiological cardiac hypertrophy (38). METTL14 is downregulated during exercise-induced physiological cardiac hypertrophy, while METTL14 knockdown impairs cardiac dysfunction during ischemia-reperfusion remodeling. Mechanistically, METTL14 mediates cardiomyocyte development and apoptosis by suppressing Phlpp2 mRNA m6A modifications and activates Akt-S473. microRNAs are important in both cardiac pathologies and physiologies. microRNA-222 (miR-222) was upregulated in exercise-induced physiological cardiac hypertrophy, protecting heart against adverse remodeling (60). It has been reported METTL3 directly regulate miR-221/222 by promoting miR-221/222 maturation in Ang-II-induced cardiac hypertrophy, subsequently activating the Wnt/β-catenin pathway (37). Overall, these studies indicate m6A is essential in preserving cardiac homeostasis and associated with cardiac regeneration.
The above findings support the important roles of m6A modification in cardiac hypertrophy (physiological and pathological) and heart failure, highlighting that it is a promising therapeutic strategy for the diagnosis and therapy of HF. In addition, finding the targets of physiological cardiac hypertrophy may be of great significance for the treatment of pathological cardiac hypertrophy, and the m6A-dependant way may be an important target (Figure 2).
4. Ischemic heart disease
4.1. Myocardial infarction (MI)
IHD represents a significant threat to public health worldwide. Gong et al. (25) demonstrated that myocardial infarction was decreased and cardiac function improved in METTL3 knockout mice, consistent with findings reported by Song et al. (26), who demonstrated that METTL3 is a negative regulator of autophagy in cardiomyocytes; however, ALKBH5 has the opposite effect. There is an increasing consensus that METTL3 and METTL14 are increased in hypoxic and reoxygenated(H/R) cardiomyocytes and IR myocardium (26, 31, 40). The upregulation of METTL3 was found to suppress autophagic flux and potentiate apoptosis in H/R-treated cardiomyocytes. TFEB is a key downstream target gene of METTL3, and TFEB mRNA expression decreases after METTL3 overexpression. Overexpression of TFEB or ALKBH5 reversed the effect of METTL3 on H/R cardiomyocytes. In addition, METTL3 interacted with NCBP3, facilitating the translational process in the myocardium under hypoxia stress (40). Thus, METTL3-mediated m6A modification represents a pivotal hypoxic stress response.
m6A level is increased in fibrotic tissues after an MI and in TGF-β1-treated CFs. METTL3 is the most significantly altered protein. It has been reported that METTL3 overexpression activated CF deposition and promoted collagen synthesis and deposition, while its inhibition improved myocardial fibrosis and cardiac function (27).
Unlike retina angiogenesis (61), no METTL3 upregulation has been observed in hypoxic cardiac microvascular endothelial cells (CMECs) (39). In contrast, significant ALKBH5 upregulation was observed in hypoxic CMECs, which impaired their proliferation, migration, and tube formation, while m6A levels were decreased. ALKBH5 knockdown increased angiogenic phenotypes in hypoxic but not in normoxic CMECs. ALKBH5 regulated postischemic angiogenesis by post-transcriptional modulating and destabilizing WNT5A mRNA in an m6A-dependent manner (39). In addition, sympathetic hyperactivity after myocardial infarction is related to METTL3 (62), which supports the old concept of the brain-heart axis in neurocardiology (63).
4.2. Myocardial ischemia-reperfusion (MI/R)
Timely revascularization is the standard care treatment of IHD, but the recovery of blood flow after myocardial ischemia can cause further tissue damage [myocardial ischemia-reperfusion (MI/R) injury] (64). An increasing body of evidence from recently published studies suggests that m6A modification is present in many important pathological processes, such as cell death (apoptosis, pyroptosis), autophagy, and endoplasmic reticulum stress, which have played significant roles on MI/R and were validated by bioinformatics analysis (65).
Oxidative stress and apoptosis are important pathological processes in ischemia-reperfusion injury (IRI) (66). The pathophysiological processes can be fine-tuning via epigenetic post-transcriptional modifications, such as m6A methylation, by regulating post-transcriptional RNA levels. METTL14, as a “writer” protein, is upregulated during ischemia-reperfusion and oxidative stress-induced cardiomyocyte injury. METTL14 deficiency aggravates myocardial injury and dysfunction. It has been reported that METTL14 overexpression significantly alleviated infarct size and apoptosis and improved cardiac function during I/R injury. Further study found that METTL14 activated the Wnt/β-catenin signaling pathways through methylating Wnt1 mRNA. Wnt1 knockout eliminated the METTL14-mediated protective effect against myocardial injury and apoptosis (31). Shen et al. (34) demonstrated that FTO inhibits H/R cardiomyocyte apoptosis by regulating Mhrt mRNA. Hypoxic preconditioning/ischemic preconditioning(HPC/IPC) is reportedly protective against myocardial ischemia/reperfusion (MIRI) (67). H19, an imprinted lncRNA, participates in MIRI and cardiomyocyte hypertrophy (68, 69). Y. Su et al. (28) demonstrated that METTL3 and METTL14 interact with lncRNA H19 to reduce H9c2 cell apoptosis, highlighting their importance in HPC treatment.
Current evidence suggests that during myocardial IRI, endoplasmic reticulum (ER) stress is important in I/R-induced damage mediated by m6A. Wang et al. (30) demonstrated that WTAP targeted activating transcription factor 4 (ATF4), one of the stress-responsive transcription factors, by activating ATF4 mRNA stability. Mechanistically, WTAP knockdown downregulated ATF4 mRNA stability and protected cardiomyocytes against apoptosis and ER stress. In contrast, overexpression of WTAP induced apoptosis and ER stress.
Pyroptosis is involved in many pathological processes, including MI/R injury, METTL3 aggravated cardiomyocyte pyroptosis through promoting DGCR8 binding to pri-miR-143-3p, thus inhibiting PRKCE transcripton (29).
Xuan Su found that the global level of m6A methylation and METTL3 protein were down-regulated both in young and elderly hearts after I/R injury, while FTO was only decreased in aging myocardium (33). In line with this, in vitro studies revealed that FTO was decreased in hypoxic cardiomyocytes (13, 70). Overexpression FTO reversed MI-induced high levels of m6A, reduced the myocardial infarction area and the degree of fibrosis, and enhanced angiogenesis (13). FTO targets SERCA2a and plays an important role in calcium homeostasis, enhancing the energy metabolism of H/R cardiomyocytes and cardiac contraction (13, 32). FTO enhanced the stability of YAP1 mRNA in cardiomyocytes following H/R injury by disrupting the m6A modification of YAP1 mRNA (70) (Figure 3). myocardial fibrosis was relieved after FTO overexpression. It has been confirmed the interaction between FTO demethylates and a series of mRNAs, such as Mef2a, Klf15, Bcl2l2, Cd36, Slc25a33 (49).
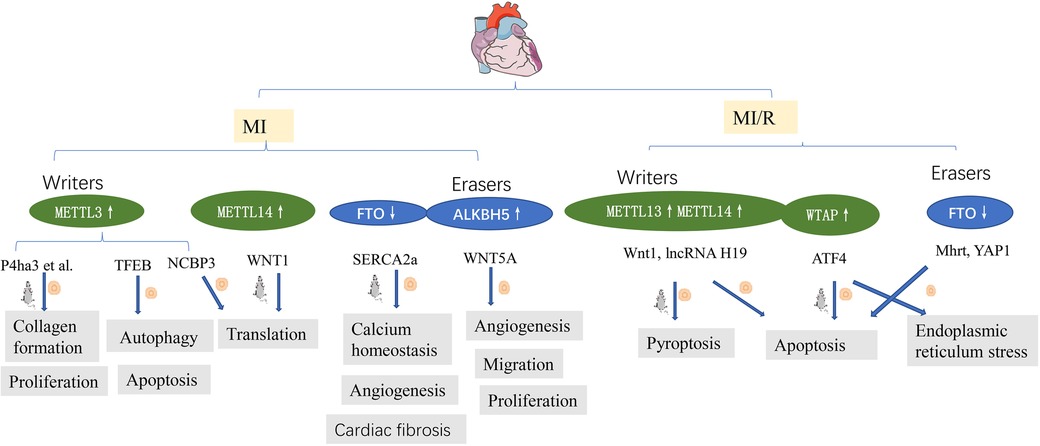
Figure 3. m6A methylation in ischemia heart disease. See main text for more details.
Overall, m6A expression in myocardial ischemia-reperfusion injury is complex. Similar proteins often have different functions in the heart and exhibit significant heterogeneity in distribution with age and body part. Even the distribution of m6A proteins varied in different parts of the heart (71). Further research is warranted to uncover the different functions of m6A modifications in MI/R.
5. Aortic dissecting aneurysm (ADA)
It is widely acknowledged that ADA features an intimal flap separating the true and false lumens (72). Growing evidence suggests that the m6A methylation is significantly altered in ADA tissue (41, 42), although significant inconsistencies have been reported in the literature.
In Ma et al.'s study (41), FTO expression was significantly upregulated in human aortic dissection (AD) tissues compared to the aortic aneurysm (AA) group. Another study reported significantly decreased FTO expression in AD tissue samples compared with normal samples, while METTL14 was significantly upregulated (42).
Current evidence suggests that forced expression of FTO potentiates vascular smooth muscle cell proliferation and migration and upregulation of its target gene, Klf5 (41). m6A levels play a determining role in the abdominal aortic aneurysm (AAA) and ADA (73), regulating the metabolism and stability of mRNA, suggesting that m6A represents a potential target to prevent aortic diseases.
6. Aortic valve calcification (AVC)
Aortic valve calcification is one of the most common cardiac valvulopathies. Current evidence suggests that human aortic valve interstitial cells (hVICs) are predominantly found in the aortic valve. It has been shown that METTL3 is upregulated, and twist-related protein 1 (TWIST1) is downregulated in AVC. Luciferase reporter assays and MeRIP-qRT-PCR confirmed the interaction between METTL3 and TWIST1. Further study showed that METTL3 inhibited TWIST1 and promoted osteogenic differentiation of human aortic valve interstitial cells via an m6A-YTHDF2-dependent pathway (43).
7. Inflammation and sepsis
Sepsis is an organ dysfunction-related systemic inflammatory response to infection associated with high morbidity and mortality rates globally (74). Cardiovascular dysfunction attributed to sepsis was first documented in 1951 by Waisbren (75). A higher mortality rate has been observed in sepsis patients presenting with cardiovascular dysfunction than those without (76). Although many pathways and mediators have been associated with myocardial depression in sepsis, the exact cause remains obscure (77).
A study on sepsis-induced myocardial dysfunction revealed that in septic heart tissues, the global m6A levels were significantly decreased; The changes of the m6A modification levels were significantly in mRNAs and lncRNAs. Pathway analyses revealed significant enrichment in immune and inflammatory response pathways (78). In contrast, Dubey et al.'s research showed increased m6A modification with downregulated FTO in LPS-induced myocardium in vitro and in vivo (44). Although the two studies yielded contrasting findings, m6A modification remains crucial in inflammatory signaling pathways of the sepsis myocardial injury model. The genes of inflammatory cytokines (IL-6, TNF-α, IL-1β) were upregulated, and left ventricular function was reduced in the sepsis hearts (44). Feng et al. found that by mediating the alternative splicing of MyD88, METTL3 could inhibit the inflammatory response triggered by lipopolysaccharides (79). In another study by Jian et al. (6), METTL14 expression but not METTL3 expression was significantly increased in endothelial cell inflammation induced by TNF-α, suggesting that METTL3 and METTL14 play different regulatory roles in m6A modification although they work synergistically. Macrophages represent a vital immune system component and are crucial in the inflammatory process. Rui Yu et al. revealed upregulation of YTHDF2 during the LPS-induced inflammatory response of macrophages (80). YTHDF2 knockdown promotes the release of proinflammatory cytokines and exacerbates inflammation in LPS-stimulated RAW 264.7 cells by activating MAPK and NF-κB signaling pathways.
Overall, m6A modification represents a potential target to attenuate cardiac inflammation and dysfunction during endotoxemia or sepsis.
8. Atherosclerosis
Atherosclerosis is the dominant cause of cardiovascular diseases, characterized by lipid accumulation in the walls of arteries (81). Overwhelming literature substantiates atherosclerosis as a chronic inflammatory disease associated with lipid accumulation (82, 83). There is a rich literature available substantiating that epigenetic processes, including DNA methylation, histone modification and m6A RNA methylation, play an important role in atherosclerosis (6, 46, 47, 84).
Growth factor receptors (EGFR) can accelerate the formation of atherosclerotic lesions (85). It has been reported that METTL3 is decreased in atherosclerosis regions. Further studies demonstrated that m6A modification of the EGFR mRNA 3′UTR contributes to atherogenesis (46). A mechanistic analysis revealed that m6A could interact with the EGFR mRNA; m6A modification of the EGFR 3′UTR accelerated its mRNA degradation leading to endothelial dysfunction. Moreover, METTL3 overexpression significantly reduced EGFR activation and endothelial dysfunction during oscillatory stress (OS). Thrombospondin-1 (TSP-1), a shear-sensitive protein, is vital in regulating vascular remodeling. Interestingly, TSP-1/EGFR inhibition using shRNA and AG1478 prevented atherosclerosis development. These results suggest that METTL3 and m6A modifications could alleviate endothelial activation and atherogenesis by accelerating the degradation of oscillatory flow-induced EGFR mRNA expression (46). Jian et al. (6) found that downregulated METTL14 expression could suppress endothelial inflammation and atherosclerotic progression. Furthermore, in vivo experiments were carried out in METTL14 knockout mice. After 12 weeks of western diet (WD) feeding, a significant decrease in lesion size was observed in METTL14 knockout mice. METTL14 interacted with FOXO1 enhancing its translation by increasing m6A modification, thus upregulating the expression of adhesion molecules, regulating endothelial monocyte adhesion, and participating in atherosclerosis progression. Similarly, it has been reported that the knockdown of METTL14 mitigates the macrophage inflammatory response by promoting M2 polarization via the NF-κB pathway (45). Since METTL3 and METTL14 are “writers”, they should theoretically have similar functions. However, current evidence suggests synergistic m6A proteins play diverse roles in different models and pathways.
As mentioned above, lipid accumulation is an important factor in atherosclerosis progression. It is now understood that a high-fat diet (HFD) leads to cardiac lipid deposition in obesity cardiomyopathy. In contrast, intermittent fasting (IF) (ad libitum feeding alternated with fasting periods) has been reported to yield cardioprotective effects (86). A study showed that IF could ameliorate the effects of HFD-induced cardiac dysfunction and serum lipid metabolic disorder (87). IF downregulated the mRNA levels of genes associated with fatty acid uptake and synthesis, upregulated fatty acid catabolism genes, and decreased the m6A methylation levels (decreased METTL3 expression and increased FTO expression) in HFD-induced obesity cardiomyopathy (87). Consistently, the LuHui Derivative (LHD), a novel synthetic anthraquinone compound, has been reported to reduce lipid deposition in cardiomyocytes (47). Interestingly, the cluster of differentiation 36 (CD36) as a downstream target of LHD participates in treating cardiac inflammation triggered by palmitic acid. LDH can bind to FTO and elevate intracellular m6A levels, alleviating hyperlipidemia-induced inflammation in cardiomyocytes. Moreover, FTO overexpression significantly upregulated CD36 expression and inhibited LHD's anti-inflammatory effects. Conversely, silencing FTO inhibited cardiac inflammation induced by palmitic acid by decreasing the stability of CD36 mRNA (47) (Figure 4).
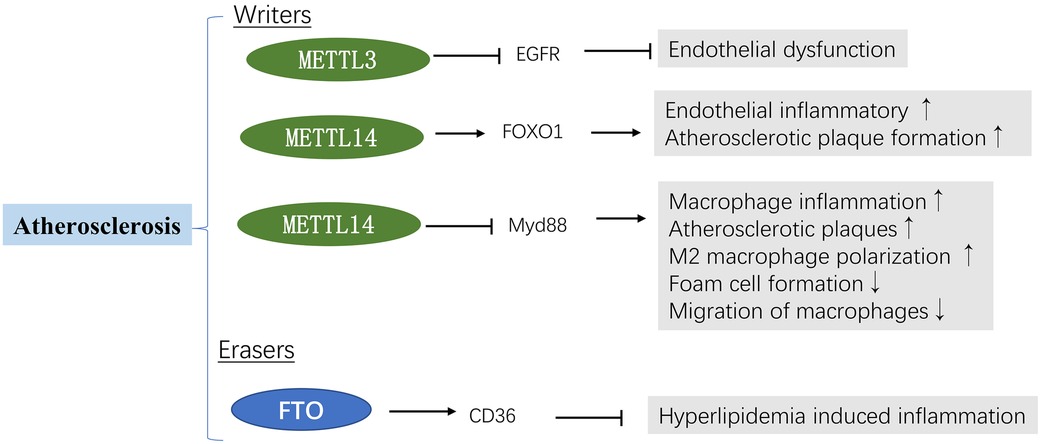
Figure 4. m6A methylation in atherosclerosis. See main text for more details.
9. Hypoxic pulmonary hypertension
HPH is a cardiopulmonary disease featuring increased pulmonary artery pressure and remodeled small pulmonary arteries conducive to right heart failure (88, 89). Epigenetic processes, such as DNA methylation, are pharmacologically reversible, making them an attractive target as therapeutic strategies for pulmonary arterial hypertension (PAH) (90). It is widely thought that m6A modifications mediate the development of HPH.
Qin et al. (50) and Hu et al. (91) used pulmonary artery smooth muscle cells (PASMCs) and hypoxic rat models to study m6A modifications in hypoxic pulmonary hypertension. The results showed that METTL3 and YTHDF2 were highly expressed in hypoxia-induced PASMCs and hypoxic pulmonary arteries (50), similar to YTHDF1 (91). Phosphatase and tensin homologue (PTEN) is reportedly the target gene of METTL3, and its increased degradation has been strongly associated with high YTHDF2 expression. Downregulation of METTL3 prevented PASMC proliferation and migration induced by hypoxia; however, downregulation of PTEN yielded the opposite effects by triggering the PI3K/Akt signaling pathway. The importance of the METTL3/YTHDF2/PTEN axis in HPH has been established (50). Similarly, Hu et al. confirmed that YTHDF1 knockdown could ameliorate proliferation phenotype switch and pulmonary hypertension development by targeting MAGED1 in vivo and in vitro. YTHDF1 could recognize and promote the translation of MAGED1. In addition, MAGED1 silencing mitigated pulmonary artery smooth muscle cell proliferation induced by hypoxia (91). Most importantly, Zhou et al. (51) found that SETD2 and METTL14 are promising targets in PAH. SETD2 deficiency could alleviate pulmonary arterial pressure and pathologic remodeling and improve right ventricular function and cardiac hypertrophy in hypoxia-induced PAH. Furthermore, silencing SETD2 in SMCs markedly decreased METTL14 and global m6A levels in PAH (Figure 5).
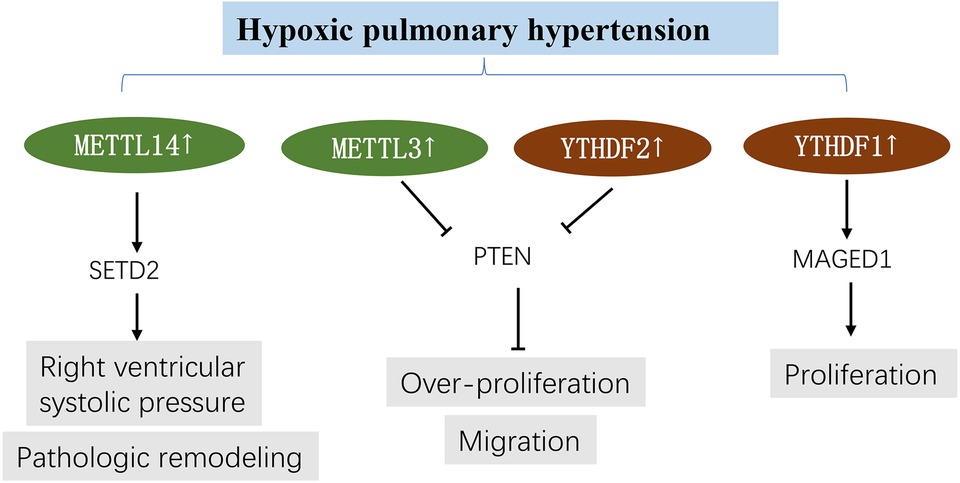
Figure 5. m6A methylation in hypoxic pulmonary hypertension. See main text for more details.
With a refined understanding of the epigenetic processes involved in PAH, m6A RNA methylation has huge prospects as a target to prevent and treat this patient population.
10. Dilated cardiomyopathy (DCM)
DCM is a condition whereby the left ventricle is dilated and associated with systolic dysfunction (92). In DCM, the total m6A levels were higher than normal hearts, while FTO protein were downregulated. FTO overexpression improved cardiac function in DCM mice (49). Genetic mutation of sarcomeric genes is an important cause of DCM (92). Although its mechanisms remain elusive, Gao et al. (48) established that YTHDC1 knockdown led to DCM. Current evidence suggests that Titin (TTN) mutations are responsible for 20%–25% of sarcomeric gene mutations, and an increased ratio of N2BA: N2B (two major Titin mRNA isoforms) is conducive to DCM. Dysregulated Titin pre-mRNA splicing results in an uneven N2BA: N2B ratio. Further research indicated that YTHDC1 deficiency causes aberrant splicing of Titin, increasing the ratio of N2BA: N2B isoform, ultimately leading to DCM. Overall, YTHDC1-dependent Titin splicing has huge prospects for treating DCM (48).
11. Cardiotoxicity
Cardiotoxicity is a well-known adverse effect of anticancer drugs (93, 94). Acute cardiotoxicity often presents with electrocardiogram (ECG) changes and arrhythmias, which lead to palpitations, presyncope and syncope, and even cardiac arrest. Chronic cardiotoxicity includes ventricular dysfunction, dilated cardiomyopathy, and heart failure (94). Tumor therapy drugs, such as doxorubicin (DOX) and cyclophosphamide (CYP), have raised significant concern, given their cardiotoxicity (95, 96). The past decade has witnessed a burgeoning interest in the molecular pathways of cardiotoxicity, including m6A modification.
Zhuang et al. found that ferroptosis is crucial in DOX-induced cardiotoxicity and may be associated with m6A RNA modification (95). The long noncoding RNA KCNQ1OT1, a miR-7-5p sponge, is modified by m6A through the action of METTL14. Besides, it has been established that miR-7-5p targets METTL14. Such a feed-forward mechanism emphasizes METTL14's crucial contribution to ferroptosis and cardiotoxicity attributed to DOX. Our findings suggest that a novel treatment strategy to manage DOX-induced cardiac injury may involve selectively reducing ferroptosis in cardiomyocytes, which is mediated by a METTL14/KCNQ1OT1/miR-7-5p positive feedback loop. Interestingly, cyclophosphamide has been associated with cardiac electrical and contractile alterations. In addition, cyclophosphamide can reportedly induce RNA m6A modification by upregulating METTL3 expression and suppressing JPH2 expression (96). These findings indicate that m6A is a potential target for preventing and treating drug-induced cardiotoxicity.
12. Conclusion and future perspectives
Epigenetics is a hot scientific research topic in recent years. In this review, we provided a comprehensive overview of the critical roles and mechanisms of m6A methylation in CVDs (Table 1). In general, m6A exists commonly with significant and complex roles. For example, the role of the same protein is distinct in different cells and exhibits heterogeneous levels in the same tissue (24, 61). In addition, different conclusions are gained from the same research topic, in Ma et al.'s study, FTO expression was elevated in human AD tissues (41), but downregulated in AD tissue in another study (42). Even different levels of METTL3 and METTL14 proteins have been documented in anatomical regions of mouse adult hearts, and these levels were altered with aging (71).
With the rapid development of detection methods and artificial intelligence (AI) are applied in discovery of m6A targeting drugs, m6A modulators will be the potential and promising therapeutic targets in the future.
To date, many compounds targeting m6A methyltransferase and demethylases have been identified, especially in cancers. For example, METTL3 inhibitor is emerging as a new target to treat acute leukemia (97) and so is YTHDF2 inhibitor (98). Paris et al. (99) found a new lead treatment with FTO inhibitor in glioblastoma. FB23-2, an inhibitor of FTO, impairs proliferation and enhances differentiation of acute myeloid leukemia (AML) cells (100).
The cardiovascular regulating roles of m6A modulators are relatively fewer than cancers. IOX1, an inhibitor of ALKBH5, was loaded onto ferritin nanocage and it was found to effectively improve cardiac function (101). FTO is overexpressed in human artery specimens of obese individuals, and its inhibitor rhein or FB23-2 exerts protective effect with increasing prostaglandin D2 production and myogenic tone (102). Thus, m6A modulators are of translational and therapeutic interests in either cancers or cardiovascular diseases. But only a few of identified regulators are druggable and suitable for treatment. Due to the poor target specificity, pharmacokinetics, therapeutic safety and efficacy (99, 100), none of the regulators has been approved for clinical treatment.
Since m6A targeted drugs are widely used in tumors. As mentioned before, tumor drugs will cause cardiotoxicity in the application process. If an m6A regulated drug can be developed, it will kill two birds with one stone by acting on tumors while reducing their cardiotoxicity. Moreover, among all the studies of HF and ischemia heart, FTO and ALKBH5 have been proved to be therapeutic and studied most frequently, we speculate that compounds of m6A erasers could hold translational therapeutic value to treat ischemic hearts and HF. More experimental and clinical evidence is needed to substantiate the role of m6A methylation. Researching and developing targeted drugs for different diseases and various stages of the same disease can be challenging and promising, emphasizing the need for further studies.
Author contributions
XZ drafted the manuscript and developed the figures; HC, HX and SD edited the manuscript and developed the figures; HM conceived, supervised the study and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81970228), JiLin Province Science and Technology Development Plan (20210101353JC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Townsend N, Wilson L, Bhatnagar P, Wickramasinghe K, Rayner M, Nichols M. Cardiovascular disease in Europe: epidemiological update 2016. Eur Heart J. (2016) 37:3232–45. doi: 10.1093/eurheartj/ehw334
2. Zhao D, Liu J, Wang M, Zhang X, Zhou M. Epidemiology of cardiovascular disease in China: current features and implications. Nat Rev Cardiol. (2019) 16:203–12. doi: 10.1038/s41569-018-0119-4
3. Yi X, Jiang X, Li X, Jiang DS. Histone lysine methylation and congenital heart disease: from bench to bedside (review). Int J Mol Med. (2017) 40:953–64. doi: 10.3892/ijmm.2017.3115
4. Guo X, Fang ZM, Wei X, Huo B, Yi X, Cheng C. HDAC6 Is associated with the formation of aortic dissection in human.. Mol Med. (2019) 25:10. doi: 10.1186/s10020-019-0080-7
5. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from novikoff hepatoma cells. Proc Natl Acad Sci U S A. (1974) 71:3971–5. doi: 10.1073/pnas.71.10.3971
6. Jian D, Wang Y, Jian L, Tang H, Rao L, Chen K, et al. METTL14 Aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics. (2020) 10:8939–56. doi: 10.7150/thno.45178
7. Mendel M, Chen KM, Homolka D, Gos P, Pandey RR, McCarthy AA, et al. Methylation of structured RNA by the m(6)A writer METTL16 is essential for mouse embryonic development. Mol Cell 2018; 71: 986–1000.e1011 doi: 10.1016/j.molcel.2018.08.004
8. Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, et al. M(6)A modulates neuronal functions and sex determination in Drosophila. Nature. (2016) 540:242–7. doi: 10.1038/nature20568
9. Barajas JM, Lin CH, Sun HL, Alencastro F, Zhu AC, Aljuhani M, et al. METTL3 Regulates liver homeostasis, hepatocyte ploidy, and circadian rhythm-controlled gene expression in mice. Am J Pathol. (2022) 192:56–71. doi: 10.1016/j.ajpath.2021.09.005
10. Yoon KJ, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, et al. Temporal control of mammalian cortical neurogenesis by m(6)A methylation. Cell. (2017) 171:877–89.e817. doi: 10.1016/j.cell.2017.09.003
11. Engel M, Eggert C, Kaplick PM, Eder M, Röh S, Tietze L, et al. The role of m(6)A/m-RNA methylation in stress response regulation. Neuron. (2018) 99:389–403.e389. doi: 10.1016/j.neuron.2018.07.009
12. Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. (2018) 561:556–60. doi: 10.1038/s41586-018-0538-8
13. Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N, et al. FTO-Dependent N(6)-Methyladenosine regulates cardiac function during remodeling and repair. Circulation. (2019) 139:518–32. doi: 10.1161/CIRCULATIONAHA.118.033794
14. Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, et al. The N(6)-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation. (2019) 139:533–45. doi: 10.1161/CIRCULATIONAHA.118.036146
15. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m⁶A RNA methylation. Nat Rev Genet. (2014) 15:293–306. doi: 10.1038/nrg3724
16. Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. (2019) 74:640–50. doi: 10.1016/j.molcel.2019.04.025
17. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. (2017) 169:824–35.e814. doi: 10.1016/j.cell.2017.05.003
18. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 Is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
19. Patil DP, Pickering BF, Jaffrey SR. Reading M(6)A in the transcriptome: m(6)A-binding proteins. Trends Cell Biol. (2018) 28:113–27. doi: 10.1016/j.tcb.2017.10.001
20. Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR, et al. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun. (2018) 9:420. doi: 10.1038/s41467-017-02770-z
21. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
22. Zhang B, Xu Y, Cui X, Jiang H, Luo W, Weng X, et al. Alteration of m6A RNA methylation in heart failure with preserved ejection fraction. Front Cardiovasc Med. (2021) 8:647806. doi: 10.3389/fcvm.2021.647806
23. Berulava T, Buchholz E, Elerdashvili V, Pena T, Islam MR, Lbik D, et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur J Heart Fail. (2020) 22:54–66. doi: 10.1002/ejhf.1672
24. Zhang B, Jiang H, Wu J, Cai Y, Dong Z, Zhao Y, et al. M6a demethylase FTO attenuates cardiac dysfunction by regulating glucose uptake and glycolysis in mice with pressure overload-induced heart failure. Signal Transduct Target Ther. (2021) 6:377. doi: 10.1038/s41392-021-00699-w
25. Gong R, Wang X, Li H, Liu S, Jiang Z, Zhao Y, et al. Loss of m(6)A methyltransferase METTL3 promotes heart regeneration and repair after myocardial injury. Pharmacol Res. (2021) 174:105845. doi: 10.1016/j.phrs.2021.105845
26. Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, et al. METTL3 And ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy. (2019) 15:1419–37. doi: 10.1080/15548627.2019.1586246
27. Li T, Zhuang Y, Yang W, Xie Y, Shang W, Su S, et al. Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J. (2021) 35:e21162. doi: 10.1096/fj.201903169R
28. Su Y, Xu R, Zhang R, Qu Y, Zuo W, Ji Z, et al. N6-methyladenosine methyltransferase plays a role in hypoxic preconditioning partially through the interaction with lncRNA H19. Acta Biochim Biophys Sin (Shanghai). (2020) 52:1306–15. doi: 10.1093/abbs/gmaa130
29. Wang X, Li Y, Li J, Li S, Wang F. Mechanism of METTL3-mediated m(6)A modification in cardiomyocyte pyroptosis and myocardial ischemia-reperfusion injury. Cardiovasc Drugs Ther. (2022). doi: 10.1007/s10557-021-07300-0
30. Wang J, Zhang J, Ma Y, Zeng Y, Lu C, Yang F, et al. WTAP Promotes myocardial ischemia/reperfusion injury by increasing endoplasmic reticulum stress via regulating m(6)A modification of ATF4 mRNA. Aging (Albany NY). (2021) 13:11135–49. doi: 10.18632/aging.202770
31. Pang P, Qu Z, Yu S, Pang X, Li X, Gao Y, et al. Mettl14 attenuates cardiac ischemia/reperfusion injury by regulating Wnt1/β-catenin signaling pathway. Front Cell Dev Biol. (2021) 9:762853. doi: 10.3389/fcell.2021.762853
32. Deng W, Jin Q, Li L. Protective mechanism of demethylase fat mass and obesity-associated protein in energy metabolism disorder of hypoxia-reoxygenation-induced cardiomyocytes. Exp Physiol. (2021) 106:2423–33. doi: 10.1113/EP089901
33. Su X, Shen Y, Jin Y, Kim IM, Weintraub NL, Tang Y. Aging-associated differences in epitranscriptomic m6A regulation in response to acute cardiac ischemia/reperfusion injury in female mice. Front Pharmacol. (2021) 12:654316. doi: 10.3389/fphar.2021.654316
34. Shen W, Li H, Su H, Chen K, Yan J. FTO Overexpression inhibits apoptosis of hypoxia/reoxygenation-treated myocardial cells by regulating m6A modification of mhrt. Mol Cell Biochem. (2021) 476:2171–9. doi: 10.1007/s11010-021-04069-6
35. Kmietczyk V, Riechert E, Kalinski L, Boileau E, Malovrh E, Malone B, et al.. m(6)A-mRNA methylation regulates cardiac gene expression and cellular growth. Life Sci Alliance. (2019) 2:e201800233. doi: 10.26508/lsa.201800233
36. Lu P, Xu Y, Sheng ZY, Peng XG, Zhang JJ, Wu QH, et al. De-ubiquitination of p300 by USP12 critically enhances METTL3 expression and ang II-induced cardiac hypertrophy. Exp Cell Res. (2021) 406:112761. doi: 10.1016/j.yexcr.2021.112761
37. Zhang R, Qu Y, Ji Z, Hao C, Su Y, Yao Y, et al. METTL3 Mediates ang-II-induced cardiac hypertrophy through accelerating pri-miR-221/222 maturation in an m6A-dependent manner. Cell Mol Biol Lett. (2022) 27:55. doi: 10.1186/s11658-022-00349-1
38. Wang L, Wang J, Yu P, Feng J, Xu GE, Zhao X, et al. METTL14 Is required for exercise-induced cardiac hypertrophy and protects against myocardial ischemia-reperfusion injury. Nat Commun. (2022) 13:6762. doi: 10.1038/s41467-022-34434-y
39. Zhao Y, Hu J, Sun X, Yang K, Yang L, Kong L, et al. Loss of m6A demethylase ALKBH5 promotes post-ischemic angiogenesis via post-transcriptional stabilization of WNT5A. Clin Transl Med. (2021) 11:e402. doi: 10.1002/ctm2.402
40. Ye F, Wang X, Tu S, Zeng L, Deng X, Luo W, et al. The effects of NCBP3 on METTL3-mediated m6A RNA methylation to enhance translation process in hypoxic cardiomyocytes. J Cell Mol Med. (2021) 25:8920–8. doi: 10.1111/jcmm.16852
41. Ma D, Liu X, Zhang JJ, Zhao JJ, Xiong YJ, Chang Q, et al. Vascular smooth muscle FTO promotes aortic dissecting aneurysms via m6A modification of Klf5. Front Cardiovasc Med. (2020) 7:592550. doi: 10.3389/fcvm.2020.592550
42. Zhou X, Chen Z, Zhou J, Liu Y, Fan R, Sun T. Transcriptome and N6-methyladenosine RNA methylome analyses in aortic dissection and normal human aorta. Front Cardiovasc Med. (2021) 8:627380. doi: 10.3389/fcvm.2021.627380
43. Zhou T, Han D, Liu J, Shi J, Zhu P, Wang Y, et al. Factors influencing osteogenic differentiation of human aortic valve interstitial cells. J Thorac Cardiovasc Surg. (2021) 161:e163–185. doi: 10.1016/j.jtcvs.2019.10.039
44. Dubey PK, Patil M, Singh S, Dubey S, Ahuja P, Verma SK, et al. Increased m6A-RNA methylation and FTO suppression is associated with myocardial inflammation and dysfunction during endotoxemia in mice. Mol Cell Biochem. (2022) 477:129–41. doi: 10.1007/s11010-021-04267-2
45. Zheng Y, Li Y, Ran X, Wang D, Zheng X, Zhang M, et al. Mettl14 mediates the inflammatory response of macrophages in atherosclerosis through the NF-κB/IL-6 signaling pathway. Cell Mol Life Sci. (2022) 79:311. doi: 10.1007/s00018-022-04331-0
46. Li B, Zhang T, Liu M, Cui Z, Zhang Y, Liu M, et al. RNA N(6)-methyladenosine modulates endothelial atherogenic responses to disturbed flow in mice. eLife. (2022) 11:e69906. doi: 10.7554/eLife.69906
47. Yu Y, Pan Y, Fan Z, Xu S, Gao Z, Ren Z, et al. Luhui derivative, A novel compound that inhibits the fat mass and obesity-associated (FTO), alleviates the inflammatory response and injury in hyperlipidemia-induced cardiomyopathy. Front Cell Dev Biol. (2021) 9:731365. doi: 10.3389/fcell.2021.731365
48. Gao S, Sun H, Chen K, Gu X, Chen H, Jiang L, et al. Depletion of m(6) A reader protein YTHDC1 induces dilated cardiomyopathy by abnormal splicing of titin. J Cell Mol Med. (2021) 25:10879–91. doi: 10.1111/jcmm.16955
49. Ju W, Liu K, Ouyang S, Liu Z, He F, Wu J. Changes in N6-methyladenosine modification modulate diabetic cardiomyopathy by reducing myocardial fibrosis and myocyte hypertrophy. Front Cell Dev Biol. (2021) 9:702579. doi: 10.3389/fcell.2021.702579
50. Qin Y, Qiao Y, Li L, Luo E, Wang D, Yao Y, et al. The m(6)A methyltransferase METTL3 promotes hypoxic pulmonary arterial hypertension. Life Sci. (2021) 274:119366. doi: 10.1016/j.lfs.2021.119366
51. Zhou XL, Huang FJ, Li Y, Huang H, Wu QC. SEDT2/METTL14-mediated M6a methylation awakening contributes to hypoxia-induced pulmonary arterial hypertension in mice. Aging (Albany NY). (2021) 13:7538–48. doi: 10.18632/aging.202616
52. Erkens R, Kramer CM, Lückstädt W, Panknin C, Krause L, Weidenbach M, et al. Left ventricular diastolic dysfunction in Nrf2 knock out mice is associated with cardiac hypertrophy, decreased expression of SERCA2a, and preserved endothelial function. Free Radic Biol Med. (2015) 89:906–17. doi: 10.1016/j.freeradbiomed.2015.10.409
53. Gao XQ, Zhang YH, Liu F, Ponnusamy M, Zhao XM, Zhou LY, et al. The piRNA CHAPIR regulates cardiac hypertrophy by controlling METTL3-dependent N(6)-methyladenosine methylation of Parp10 mRNA. Nat Cell Biol. (2020) 22:1319–31. doi: 10.1038/s41556-020-0576-y
54. Xu H, Wang Z, Chen M, Zhao W, Tao T, Ma L, et al. YTHDF2 Alleviates cardiac hypertrophy via regulating Myh7 mRNA decoy. Cell Biosci. (2021) 11:132. doi: 10.1186/s13578-021-00649-7
55. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart.. Science. (2011) 331:1078–80. doi: 10.1126/science.1200708
56. Han Z, Xu Z, Yu Y, Cao Y, Bao Z, Gao X, et al. ALKBH5-mediated M(6)A mRNA methylation governs human embryonic stem cell cardiac commitment. Mol Ther Nucleic Acids. (2021) 26:22–33. doi: 10.1016/j.omtn.2021.05.019
57. Yang Y, Shen S, Cai Y, Zeng K, Liu K, Li S, et al. Dynamic patterns of N6-methyladenosine profiles of messenger RNA correlated with the cardiomyocyte regenerability during the early heart development in mice. Oxid Med Cell Longev. (2021) 2021:5537804.34413927
58. Han Z, Wang X, Xu Z, Cao Y, Gong R, Yu Y, et al. ALKBH5 Regulates cardiomyocyte proliferation and heart regeneration by demethylating the mRNA of YTHDF1. Theranostics. (2021) 11:3000–16. doi: 10.7150/thno.47354
59. Wang S, Zhang J, Wu X, Lin X, Liu XM, Zhou J. Differential roles of YTHDF1 and YTHDF3 in embryonic stem cell-derived cardiomyocyte differentiation. RNA Biol. (2021) 18:1354–63. doi: 10.1080/15476286.2020.1850628
60. Liu X, Xiao J, Zhu H, Wei X, Platt C, Damilano F, et al. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. (2015) 21:584–95. doi: 10.1016/j.cmet.2015.02.014
61. Yao MD, Jiang Q, Ma Y, Liu C, Zhu CY, Sun YN, et al. Role of METTL3-dependent N(6)-methyladenosine mRNA modification in the promotion of angiogenesis. Mol Ther. (2020) 28:2191–202. doi: 10.1016/j.ymthe.2020.07.022
62. Qi L, Hu H, Wang Y, Hu H, Wang K, Li P, et al. New insights into the central sympathetic hyperactivity post-myocardial infarction: roles of METTL3-mediated m(6) A methylation. J Cell Mol Med. (2022) 26(4):1264–80. doi: 10.1111/jcmm.17183
63. Natelson BH. Neurocardiology: an interdisciplinary area for the 80s. Arch Neurol. (1985) 42:178–84. doi: 10.1001/archneur.1985.04060020096022
64. Binder A, Ali A, Chawla R, Aziz HA, Abbate A, Jovin IS. Myocardial protection from ischemia-reperfusion injury post coronary revascularization. Expert Rev Cardiovasc Ther. (2015) 13:1045–57. doi: 10.1586/14779072.2015.1070669
65. Shi X, Cao Y, Zhang X, Gu C, Liang F, Xue J, et al. Comprehensive analysis of N6-methyladenosine RNA methylation regulators expression identify distinct molecular subtypes of myocardial infarction. Front Cell Dev Biol. (2021) 9:756483. doi: 10.3389/fcell.2021.756483
66. Li H, Xia Z, Chen Y, Qi D, Zheng H. Mechanism and therapies of oxidative stress-mediated cell death in ischemia reperfusion injury. Oxid Med Cell Longev. (2018) 2018:2910643. doi: 10.1155/2018/2910643
67. Sivaraman V, Hausenloy DJ, Wynne AM, Yellon DM. Preconditioning the diabetic human myocardium. J Cell Mol Med. (2010) 14:1740–6. doi: 10.1111/j.1582-4934.2009.00796.x
68. Liu L, An X, Li Z, Song Y, Li L, Zuo S, et al. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc Res. (2016) 111:56–65. doi: 10.1093/cvr/cvw078
69. Luo H, Wang J, Liu D, Zang S, Ma N, Zhao L, et al. The lncRNA H19/miR-675 axis regulates myocardial ischemic and reperfusion injury by targeting PPARα. Mol Immunol. (2019) 105:46–54. doi: 10.1016/j.molimm.2018.11.011
70. Ke WL, Huang ZW, Peng CL, Ke YP. m(6)A demethylase FTO regulates the apoptosis and inflammation of cardiomyocytes via YAP1 in ischemia-reperfusion injury. Bioengineered. (2022) 13:5443–52. doi: 10.1080/21655979.2022.2030572
71. Arcidiacono OA, Krejčí J, Bártová E. The distinct function and localization of METTL3/METTL14 and METTL16 enzymes in cardiomyocytes. Int J Mol Sci. (2020) 21:E8139. doi: 10.3390/ijms21218139
72. Bossone E, Eagle KA. Epidemiology and management of aortic disease: aortic aneurysms and acute aortic syndromes. Nat Rev Cardiol. (2021) 18:331–48. doi: 10.1038/s41569-020-00472-6
73. He Y, Xing J, Wang S, Xin S, Han Y, Zhang J. Increased m6A methylation level is associated with the progression of human abdominal aortic aneurysm. Ann Transl Med. (2019) 7:797. doi: 10.21037/atm.2019.12.65
74. Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. (2001) 345:1368–77. doi: 10.1056/NEJMoa010307
75. Waisbren BA. Bacteremia due to gram-negative bacilli other than the Salmonella; a clinical and therapeutic study. AMA Arch Intern Med. (1951) 88:467–88. doi: 10.1001/archinte.1951.03810100051005
76. Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, et al. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. (1990) 113:227–42. doi: 10.7326/0003-4819-113-3-227
77. Merx MW, Weber C. Sepsis and the heart. Circulation. (2007) 116:793–802. doi: 10.1161/CIRCULATIONAHA.106.678359
78. Han YC, Xie HZ, Lu B, Xiang RL, Zhang HP, Li JY, et al. Lipopolysaccharide alters the m6A epitranscriptomic tagging of RNAs in cardiac tissue. Front Mol Biosci. (2021) 8:670160. doi: 10.3389/fmolb.2021.670160
79. Feng Z, Li Q, Meng R, Yi B, Xu Q. METTL3 Regulates alternative splicing of MyD88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J Cell Mol Med. (2018) 22:2558–68. doi: 10.1111/jcmm.13491
80. Yu R, Li Q, Feng Z, Cai L, Xu Q. M6a reader YTHDF2 regulates LPS-induced inflammatory response. Int J Mol Sci. (2019) 20:1323. doi: 10.3390/ijms20061323
81. Frostegård J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. (2013) 11:117. doi: 10.1186/1741-7015-11-117
82. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
83. Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res. (2014) 114:1867–79. doi: 10.1161/CIRCRESAHA.114.302699
84. Xu S, Pelisek J, Jin ZG. Atherosclerosis is an epigenetic disease. Trends Endocrinol Metab: TEM. (2018) 29:739–42. doi: 10.1016/j.tem.2018.04.007
85. Wang L, Huang Z, Huang W, Chen X, Shan P, Zhong P, et al. Inhibition of epidermal growth factor receptor attenuates atherosclerosis via decreasing inflammation and oxidative stress. Sci Rep. (2017) 8:45917. doi: 10.1038/srep45917
86. Dong TA, Sandesara PB, Dhindsa DS, Mehta A, Arneson LC, Dollar AL, et al. Intermittent fasting: a heart healthy dietary pattern? Am J Med. (2020) 133:901–7. doi: 10.1016/j.amjmed.2020.03.030
87. Xu Z, Qin Y, Lv B, Tian Z, Zhang B. Intermittent fasting improves high-fat diet-induced obesity cardiomyopathy via alleviating lipid deposition and apoptosis and decreasing m6A methylation in the heart. Nutrients. (2022) 14(251):1–12. doi: 10.3390/nu14020251
88. Nishihara T, Yamamoto E, Tokitsu T, Sueta D, Fujisue K, Usuku H, et al. New definition of pulmonary hypertension in patients with heart failure with preserved ejection fraction. Am J Respir Crit Care Med. (2019) 200:386–8. doi: 10.1164/rccm.201901-0148LE
89. Thompson AAR, Lawrie A. Targeting vascular remodeling to treat pulmonary arterial hypertension. Trends Mol Med. (2017) 23:31–45. doi: 10.1016/j.molmed.2016.11.005
90. Napoli C, Benincasa G, Loscalzo J. Epigenetic inheritance underlying pulmonary arterial hypertension. Arterioscler, Thromb, Vasc Biol. (2019) 39:653–64. doi: 10.1161/ATVBAHA.118.312262
91. Hu L, Wang J, Huang H, Yu Y, Ding J, Yu Y, et al. YTHDF1 Regulates pulmonary hypertension through translational control of MAGED1. Am J Respir Crit Care Med. (2021) 203:1158–72. doi: 10.1164/rccm.202009-3419OC
92. Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet (London, England). (2010) 375:752–62. doi: 10.1016/S0140-6736(09)62023-7
93. Mercurio V, Pirozzi F, Lazzarini E, Marone G, Rizzo P, Agnetti G, et al. Models of heart failure based on the cardiotoxicity of anticancer drugs. J Card Fail. (2016) 22:449–58. doi: 10.1016/j.cardfail.2016.04.008
94. Herrmann J. Adverse cardiac effects of cancer therapies: cardiotoxicity and arrhythmia. Nat Rev Cardiol. (2020) 17:474–502. doi: 10.1038/s41569-020-0348-1
95. Zhuang S, Ma Y, Zeng Y, Lu C, Yang F, Jiang N, et al. METTL14 Promotes doxorubicin-induced cardiomyocyte ferroptosis by regulating the KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biol Toxicol. (2021). doi: 10.1007/s10565-021-09660-7
96. Zhu M, Liu Y, Song Y, Zhang S, Hang C, Wu F, et al. The role of METTL3-mediated N6-methyladenosine (m6A) of JPH2 mRNA in cyclophosphamide-induced cardiotoxicity. Front Cardiovasc Med. (2021) 8:763469. doi: 10.3389/fcvm.2021.763469
97. Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. (2021) 593:597–601. doi: 10.1038/s41586-021-03536-w
98. Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the RNA m(6)A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. (2019) 25:137–48.e136. doi: 10.1016/j.stem.2019.03.021
99. Huff S, Tiwari SK, Gonzalez GM, Wang Y, Rana TM. m(6)A-RNA demethylase FTO inhibitors impair self-renewal in glioblastoma stem cells. ACS Chem Biol. (2021) 16:324–33. doi: 10.1021/acschembio.0c00841
100. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. (2019) 35:677–91.e610. doi: 10.1016/j.ccell.2019.03.006
101. Cheng P, Han H, Chen F, Cheng L, Ma C, Huang H, et al. Amelioration of acute myocardial infarction injury through targeted ferritin nanocages loaded with an ALKBH5 inhibitor. Acta Biomater. (2021) 140:481–91. doi: 10.1016/j.actbio.2021.11.041
Keywords: cardiovascular diseases, m6A methylation, epigenetic, cardiac hypertrophy, heart failure, ischemic heart disease, m6A methylation
Citation: Zhang X, Cai H, Xu H, Dong S and Ma H (2023) Critical roles of m6A methylation in cardiovascular diseases. Front. Cardiovasc. Med. 10:1187514. doi: 10.3389/fcvm.2023.1187514
Received: 16 March 2023; Accepted: 28 April 2023;
Published: 19 May 2023.
Edited by:
Qing Robert Miao, New York University, United StatesReviewed by:
Wenquan Hu, Chongqing Medical University, ChinaSuowen Xu, University of Science and Technology of China, China
© 2023 Zhang, Cai, Xu, Dong and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haichun Ma bWFoY0BqbHUuZWR1LmNu