 Hashum Sum
Hashum Sum Alison C. Brewer
Alison C. Brewer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 25 May 2023
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1183181
This article is part of the Research Topic The Role of Stem Cells, RNA Binding Proteins and Non-Coding RNAs in Diabetes and Vascular Diseases View all 8 articles
Significant progress in the diagnosis and treatment of cardiovascular disease (CVD) has been made in the past decade, yet it remains a leading cause of morbidity and mortality globally, claiming an estimated 17.9 million deaths per year. Although encompassing any condition that affects the circulatory system, including thrombotic blockage, stenosis, aneurysms, blood clots and arteriosclerosis (general hardening of the arteries), the most prevalent underlying hallmark of CVD is atherosclerosis; the plaque-associated arterial thickening. Further, distinct CVD conditions have overlapping dysregulated molecular and cellular characteristics which underlie their development and progression, suggesting some common aetiology. The identification of heritable genetic mutations associated with the development of atherosclerotic vascular disease (AVD), in particular resulting from Genome Wide Association Studies (GWAS) studies has significantly improved the ability to identify individuals at risk. However, it is increasingly recognised that environmentally-acquired, epigenetic changes are key factors associated with atherosclerosis development. Increasing evidence suggests that these epigenetic changes, most notably DNA methylation and the misexpression of non-coding, microRNAs (miRNAs) are potentially both predictive and causal in AVD development. This, together with their reversible nature, makes them both useful biomarkers for disease and attractive therapeutic targets potentially to reverse AVD progression. We consider here the association of aberrant DNA methylation and dysregulated miRNA expression with the aetiology and progression of atherosclerosis, and the potential development of novel cell-based strategies to target these epigenetic changes therapeutically.
Epigenetics is the study of mechanisms that regulate mitotically stable patterns of gene expression and hence cellular phenotype without altering the genomic sequence of the cell. The field has converged upon the locus-specific regulation of chromatin structure and accessibility by DNA methylation, the covalent and non-covalent modification of histones together with non-coding RNA-based mechanisms (1). Epigenetic changes are typically reversible, enzymatically-regulated processes which are modulated by the intracellular milieu. Consequently, they are critical drivers of cellular differentiation, development and homeostasis (2) but can lead to disease states when influenced by pathological environmental factors (3). Understanding the mechanistic causes and pathological consequences of aberrant epigenetic regulation might therefore offer the possibility of novel curative therapeutic treatments for human diseases. This is particularly true in the case of atherosclerosis, where it remains the case that current (predominantly lipid-lowering) treatments can only lessen, but not reverse, disease progression (4).
DNA methylation in mammals usually occurs at the 5th position of cytosine (5mC) in the context of CpG dinucleotides and is controlled by enzymatic systems which add or remove methyl groups, termed writers and erasers. CpG dinucleotides are distributed unevenly across the genome, and regions rich in CpG dinucleotides are known as CpG islands. Characteristically, CpG islands have a GC content >50% and are at least 200 bp in size. They contain around 7% of total CpG dinucleotides present in the human genome and are commonly found within promoter regions such that 60% of human gene promoter sites contain CpG islands (5). The DNA-methyl transferase (DNMT) family of enzymes add methyl groups to (predominantly cytosine) residues, while methyl groups can be removed passively via DNA replication or actively via the sequential oxidation of the methyl group, catalysed by the family of three Ten Eleven Translocase enzymes (TET1/2/3), producing 5-hydroxymethyl-cytosine (5hmC), 5-formyl-cytosine (5fC) and 5-carboxyl-cytosine (5caC). 5hmC is the most stable (and therefore most abundant) of these oxidation derivatives, while both 5fC and 5caC can be actively removed by base excision repair (BER) mechanisms to restore unmodified cytosines (6). The covalent modification of DNA affects both the structure of chromatin and the affinity for DNA-binding trans-acting factors and hence is a critical regulator of transcriptional gene expression (7). DNA methylation is usually associated with transcriptional repression and serves as a major cellular regulatory mechanism. It has numerous well-characterised roles across the biological spectrum, from embryonic development to cellular ageing and death and the manifestation of numerous diseases (8). By contrast, the precise functions of the individual TET-generated oxidised derivatives remain to be fully elucidated. Nevertheless, with the generation of TET mutants that uncouple these sequential oxidative steps, distinct regulatory functions for 5hmC, 5fC and 5caC are now becoming uncovered (9, 10).
Studies have shown that DNA methylation signatures are highly dynamic and can be influenced by different types of environmental stimuli that the individual had been exposed to, such as those known to be risk factors for the development of AVD (11, 12). In this regard it is noteworthy that the activities of both the DNMTs and TETs are known to be subject to modulation by risk factors associated with atherosclerosis, including oxidative stress and hyperglycaemia associated with diabetes (13, 14). Functional phenotypic changes may occur because of methylation-driven alterations in gene expression leading to improper regulation of homeostatic biochemical pathways. Many studies have investigated both changes in global levels of methylation, and changes in methylation at specific CpGs which link to vascular diseases including atherosclerosis. At the global level, genome-wide hypomethylation appears to associate with atherosclerosis, while hypermethylation of some specific genes are observed as the disease progresses (15). Chronic inflammation and the development of fatty plaques are hallmarks of atherosclerosis. Perhaps accordingly, aberrant DNA (hyper)methylation has commonly been observed, associated with downregulation of key proteins that are essential in regulating the inflammatory response or those that are involved in the modulation of lipid pathways (Table 1). It should be noted that most of these studies to date have been retrospective and the methylation changes observed may therefore be a consequence rather than a cause of the pathologies. Crucially however, recent epigenome-wide association studies (EWASs) designed to investigate the association between incident CVD events associated with stable or unstable atherosclerotic plaques (such as stable coronary heart disease and myocardial infarction) have begun to identify characteristic methylation signatures which may be predictive of future risk and therefore causal with respect to cardiovascular outcomes (44). Reversing specific aberrant methylation marks, potentially in combination with other treatments, therefore represents a promising future clinical therapeutic strategy.
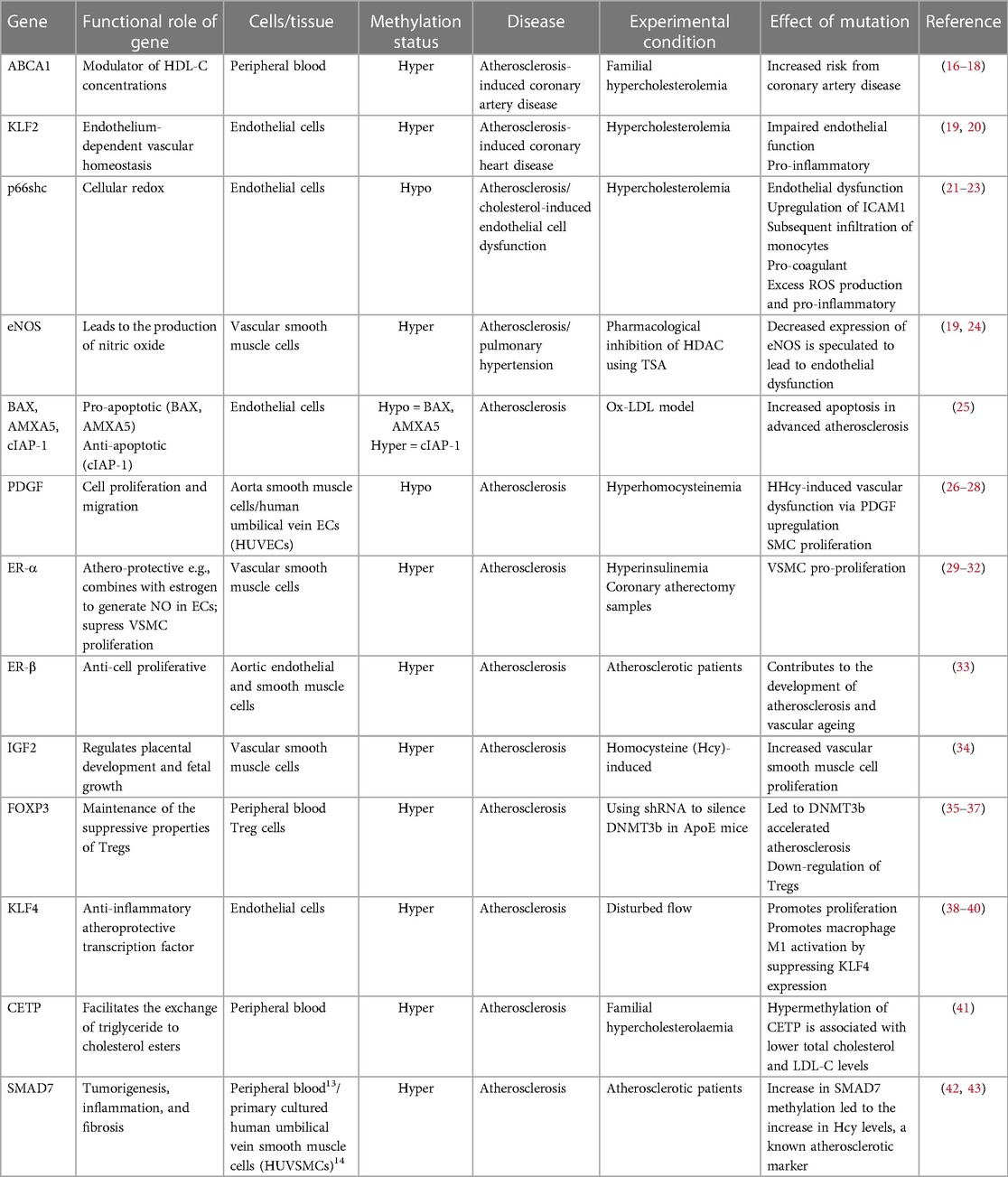
Table 1. Vascular disease-related genes that are regulated by DNA methylation.
In considering the aetiology, development and potential treatment of AVD, it is crucial to identify and fully understand the specific roles of different cell types. The development of atherosclerosis involves the dysregulation of vascular cells (endothelial, vascular smooth muscle and adventitial fibroblasts) (45), together with haematopoietic cells of both the innate and adaptive immune system (46). Most associations between aberrant methylation and (either retrospective or incident) cardiovascular events have been observed in total circulating blood cells as this can be obtained in the most non-invasive way. However, it is possible to isolate and purify both sub populations of leukocytes and the small numbers of circulating endothelial cells (ECs) (47) and minimally invasive cell biopsy techniques are also being developed to isolate small numbers of cardiovascular cells (48, 49). This, together with the use and development of microfluidic techniques to enable “omic” analyses, including DNA methylation maps of single-cells selected from a tissue (50), will enable a better understanding of the tissue-specific roles of methylation changes in the development of AVD.
Age is a potent and independent risk factor for atherosclerosis (51), in part due to the age-related accumulation of somatic mutations. Crucially, the clonal expansion of haematopoietic stem cells harbouring mutations (which confer a selective proliferative and/or survival to the cell) can result not only in haematopoietic malignancy but also an increased risk for CVD including atherosclerosis (52). Thus, atherosclerotic lesions typically display clonal growth and are postulated to be able to arise from neoplastic processes consequent upon critical somatic mutations within expanded haematopoietic cell populations (53). Strikingly, approximately 50% of all mutations which result in the clonal expansion of haematopoietic cells are within genetic loci of specific members of the DNA methylation-regulatory genes; Dnmt3a and Tet2 (54). Further, whole-exome sequencing of samples obtained from coronary heart disease patients revealed that clonal hematopoietic mutations in these genes conferred a significantly greater (approximately 2-fold) risk of atherosclerosis-associated incident heart disease compared to patients without such mutations (55). These data suggest that loss of function of these genes provides (potentially tumour-promoting) selective advantage and clearly underline the functional significance of DNA methylation in the development of AVD. The causal effect of both DNMT3A and TET2 loss-of-function within myeloid cells upon clonal expansion and the development of atherosclerosis has also been demonstrated unequivocally in atherosclerosis-prone mice (56–58). Thus, in bone marrow transplantation studies, DNMT3A- and TET2-deficient haematopoietic stem/progenitor cells (HSPCs) were both shown to expand preferentially (and most notably into the macrophage population) leading to a marked increase in atherosclerotic plaque size (56, 58). Strikingly, despite exerting opposing catalytic effects with respect to DNA methylation, the lack of either DNMT3A or TET2 in these mice resulted in similar downstream transcriptomic and pathogenic cellular effects. This perhaps highlights the crucial importance of correctly balanced methylation and demethylation and should be taken into consideration in designing therapeutic strategies.
It is now well-recognised from studies in mice that vascular smooth muscle cells (VSMCs) within the atherosclerotic plaque arise from the clonal expansion of a few select cells within the medial arterial wall and although not conclusively proven, much evidence suggests that this is also the case in the human pathology (53). This raises the question as to whether similar mechanisms might underlie the clonal expansion of VSMCs in atherosclerosis to those which drive the expansion of (age-related) somatic cell mutations in haematopoietic cells. VSMCs in atherosclerosis exhibit considerable plasticity and can adopt phenotypes resembling foam cells, macrophages, mesenchymal stem cells and osteochondrogenic cells (59). Evidence suggests that DNA methylation is a mediator of phenotypic switching in VSMCs (60) and that the catalytic (5-hmC-generating) activity of TET2, in particular, plays an active role in this (61). Whether clonally-expanded VSMCs in atherosclerotic plaques commonly harbour somatic mutations in Tet2 or Dnmt3a is not currently known, but is a clear possibility.
The involvement of aberrant DNA methylation in ECs, associated with atherosclerotic development has also been demonstrated. DNMT-dependent changes in EC DNA methylation, associated with disturbed flow and endothelial inflammation, have been shown in vitro and in animal models in vivo (38, 62) and a role for EC-expressed TET2 in atherosclerosis progression has been demonstrated in mice (57). Although technically challenging to demonstrate, recent studies involving single-cell sequencing have also identified clonal expansion of ECs, associated with cardiovascular pathologies (63–66). Further, segmental endothelial dysfunction (ED) (particularly associated with disturbed flow at the branch point of arteries) is a significant contributor to arterial remodelling resulting in atherosclerosis (67) although it remains to be determined whether this is due to a clonally-expanded EC population. The possibility that circulating endothelial progenitor cells (EPCs, derived from the haematopoietic stem cell population) might be recruited, incorporated and expanded into atherosclerotic lesions has been widely investigated (68). The origin and functional significance of these cells remains highly controversial (69). Nevertheless, in a comparative mutational profiling study, common somatic mutations in tet2 were identified in some subjects between a mature population of these circulating endothelial (CD105/146+ve/CD45−ve) cells and CD34+ve HSPCs, strongly suggesting the existence of a common precursor cell (70). This raises the possibility that critical somatic mutations, observed within expanded haematopoietic cell populations, may also impact upon EC function in the development of the atherosclerotic plaque. Again, it would clearly be of interest to determine whether mutations in DNA methylation modifying enzymes (TETs or DNMTs) associate with any clonal expansion of ECs in the context of atherosclerosis.
Given the clear association of DNA methylation changes and AVD, therapeutic strategies involving natural and synthetic pharmacological inhibitors of epigenetic modifications have, and continue to be, intensely investigated (71, 72). Further, the use of methyl-containing nutrients, notably folic acid and methionine, particularly in the prevention of CVD (including atherosclerosis), is of clear clinical benefit (73). However, more targeted strategies are required to reverse specific methylation changes shown to be causal in the disease.
Genetically-programmed cell-based strategies to both generate disease models and treat atherosclerosis are increasingly being investigated and developed. Gene editing, in particular using the clustered regularly interspaced palindromic repeat (CRISPR)-associated protein (CRISPR/Cas9) system is now widely implemented towards the goal of correcting specific mutations to treat a wide range of diseases (74). Many experimental studies have demonstrated the proof of principle that the correction of a single gene defect can alleviate the severity of atherosclerosis in animal models [reviewed in (75)]. With regard to DNA methylation, mutations in Tet2 and Dnmt3a, associated with haematopoietic cell clonal expansion and CVD might therefore be considered therapeutic targets for correctional gene editing. An increasingly popular strategy for delivering edited genes for targeting cardiovascular dysfunction in vivo, in is by using some form of stem cell, such as mesenchymal stem cells (for example, haematopoietic stem cells), embryonic stem cells or induced pluripotent stem cells (iPSCs) as a vector, all of which have associated advantages and drawbacks [reviewed in (76)]. iPSCs derived from the patient offer advantages in terms of availability and reduced immuno-reactivity but have previously proved difficult to engineer for CRISPR/Cas9 action. However, a recent study has shown that this could be overcome following lentiviral transduction in an iPSC-derived macrophage model with almost 100% efficiency (77). There is also an increasing use of the large variety of mesenchymal stem cells that can be derived from the umbilical cord (UC-MSCs). These also have advantages of low immunogenicity, easy collection and isolation together with high paracrine potential (78).
A clear problem with any cell-based gene delivery system for addressing cardiovascular abnormalities, is that the site of dysfunction often involves multiple cell types and restoration of normal function may require site-specific changes in tissue architecture. In the case of the corrections of Tet2 and Dnmt3a mutations within haematopoietic cells this may not present as an issue. However, as suggested above, mutations within these genes will also possibly be found to associate with EC and/or VSMC pathology. In this context, it is promising that ECs derived from EPCs in vitro can occupy the perivascular space when introduced into immuno-suppressed mice and anastomose with the host vasculature (79). Further, bone marrow-derived stem cells can also traverse the vessel walls of atherosclerotic plaques and form the smooth muscle cells that are enriched there in this condition (80).
In addition to genetic editing, the CRISPR/Cas9 system has been developed to edit epigenetic (DNA and histone) marks. By fusing “dead” Cas9 to the catalytic domains of epigenetic writers and erasers, epigenetic changes can be introduced at specific sites, guided by CRISPR guide-RNAs (gRNAs) (81). Both site-specific DNA methylation and demethylation, by fusing to DNMT and TET catalytic activity respectively, has been achieved in mammalian cells (82, 83). Moreover, the CRISPR/Cas9 technology has been adapted to target multiple sites simultaneously, with the use of a polycistronic-tRNA-gRNA (84). Going forward, as (tissue-specific) methylation signatures that are shown to be predictive and causal in atherosclerosis become identified, these would represent potential targets of epigenetic editing in appropriate stem cell-based therapies.
Approximately 99% of the human genome does not encode proteins (85). Although once considered to be “junk DNA”, much of it is now understood to be highly transcribed into different sub-categories of long and short non-coding RNAs (ncRNAs) with regulatory and structural functions (86). MicroRNAs (miRNAs) are a class of short (18–26 nucleotides) ncRNAs which act as post-transcriptional regulators of gene expression (87). More than 1,500 miRNAs, comprising about 1%–5% of the human genome have so far been annotated (88, 89) which are estimated to regulate more than 60% of the mRNA transcriptome (90). They act by binding to a homologous sequence (typically within the 3′ untranslated region) of their target gene(s) and destabilising the mRNA and/or repressing translation [see (91, 92) for detailed reviews of their synthesis and mechanisms of silencing]. They therefore affect protein levels and hence cellular phenotype without modifying the primary DNA sequence and are thus considered as epigenetic modulators. A single miRNA may suppress the expression of multiple functionally-related genes in critical cellular signalling pathways (93). Consequently, miRNAs have emerged as important regulators of physiological processes in development and cellular homeostasis (94). Further, their dysregulation is increasingly shown to be associated with pathological states, most notably cancers, in many cases causally (95). Accordingly, many miRNAs are now known to play critical roles in normal cardiovascular physiology and to be key players in the development of CVD, including atherosclerosis (96, 97). A table of key miRNAs associated with functions relevant to atherosclerosis development and/or known to be misexpressed in atherosclerosis is shown (Table 2).
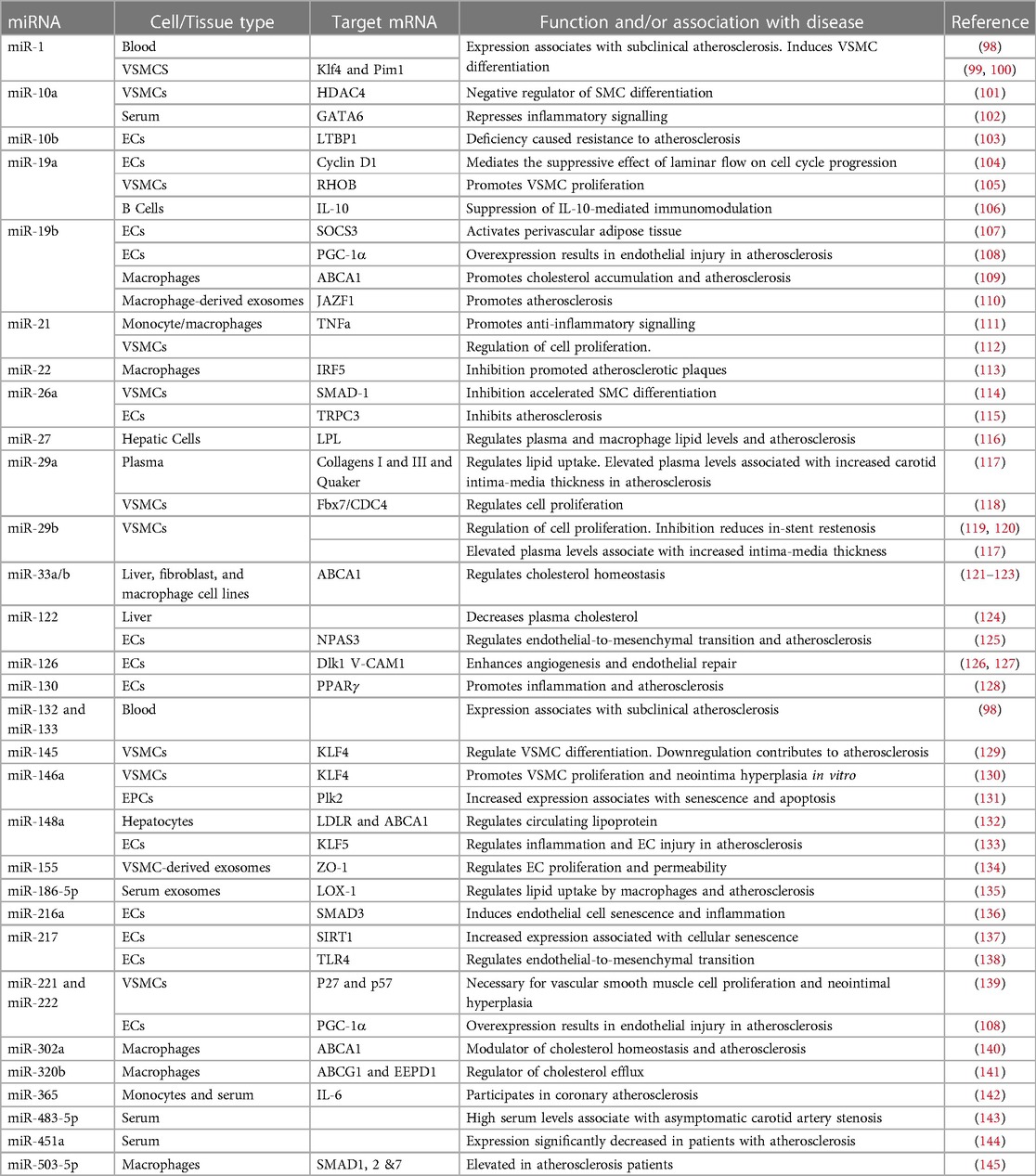
Table 2. Selected miRNAs dysregulated in atherosclerosis and/or involved in pathways relevant to atherosclerosis development.
Perhaps unexpectedly, miRNAs have been found to be relatively stable and can readily be detected in body fluids including blood, urine and saliva. This is now believed to be predominantly due to their encapsulation within microvesicles and particularly exosomes (146). These are small (30–150 nm in size) membrane vesicles which originate from intracellular compartments and are secreted by most cells (147). They carry bioactive (tissue-specific) cargo from their host cell, including proteins, lipids and nucleic acids (notably miRNAs) and are involved in intercellular communication and thus a wide range of physiological processes (148). The biological components of exosomes can therefore influence both physiological and pathophysiological processes. Further, it has been shown that the molecular constituents of the exosomes (including the miRNAs they carry) are themselves modulated by disease states (and responses to treatments) (149). Accordingly, there is now considerable investigation into the clinical use of levels of specific miRNAs as biomarkers for disease onset and progression. In the case of atherosclerosis, an increasing number of circulating miRNAs have been identified whose levels have been shown to associate with a thickening of the arterial wall in the very early stages of the of the disease and it is hoped that these will prove diagnostically useful in future [reviewed in (150)].
The numerous causal associations between dysregulated miRNAs and the onset and progression of AVD (tabled above) has driven the use of miRNA-based therapeutic approaches to either supress or restore their misexpression (151). Thus, miRNA mimics and anti-miRNAs (antagomirs) have been used to counteract the functional pathological consequences of reduced or upregulated miRNA expression respectively. However, there are considerable challenges associated with their use. Unmodified, naked nucleic acid oligonucleotides are subject to degradation by serum nucleases and are immunoreactive. Considerable progress has been achieved in the development of chemical modifications to stabilise therapeutic oligonucleotides including the use of 2′-O-methyl (2′-OMe), phosphorothioate, 2′-O-methyoxyethyl (2′-MOE), 2′-fluoro (2′-F) and N,N–diethyl–4-(4–nitronaphthalen–1–ylazo)- phenylamine (ZEN) modifications and locked nucleic acid (LNA) technologies. These are all aimed at increasing the stability and reducing the immune recognition of oligonucleotides while maintaining their binding affinity and specificity and functional efficacy [reviewed in (151)].
Other major challenges in delivering small RNA therapeutics include the specificity with which these molecules can identify and associate with their target cell(s) and the efficiency with which they can cross cellular membranes (152). In this regard there is increasing interest in the use of exosomes as vehicles for their delivery, which have inherent cell-type specificity and are naturally taken up by the target cell by endocytosis (153). Typically, such vesicles are isolated and purified from cultured cells in vitro before injecting them either into the circulation or directly into the target site (153). As required, the cells generating the exosomes in culture can be engineered to overexpress the desired small RNAs, for example by transfection or transduction, to become encapsulated into the secreted exosomes.
Communication between different cell types, mediated by exosomes, has increasingly been demonstrated to be important in both the prevention and the progression of atherosclerosis, dependent upon the physiological state of the donor cell [reviewed in (154)]. Exosomes generated from a variety of differentiated cell types have been used to deliver (usually detrimental) miRNAs in experimental studies of atherosclerosis, evidencing proof of principle. These are typically cells that are associated with the formation of atherosclerotic plaques and include macrophages, ECs and VSMCs among others (154). The functions targeted by the miRNAs in these studies usually represent different components of the atherosclerotic process such as inflammation, lesion formation or cholesterol biosynthesis. For example, purified exosomes, secreted by human aortic smooth muscle cells, which had been engineered to overexpress miR-155, were shown to transfer their miR cargo to endothelial cells within the aorta and promote plaque formation in atherosclerotic-prone (ApoE-/-) mice after injection into the tail vein (135). Endothelial-derived exosome cargos have been shown to modulate both VSMC plasticity and monocyte/macrophage phenotype. Accordingly, miR-92a secreted from ECs into exosomes was shown to regulate the contractile-to-synthetic phenotypic switch of VSMCs, contributing to arterial stiffness (155) and to enhance the proinflammatory responses and low-density lipoprotein uptake of cocultured macrophages (156). The ability of exosomes derived from macrophages to affect the VSMC phenotype in vivo has also been demonstrated. Thus exosomes isolated from nicotine-treated macrophages induced VSMC proliferation and migration via the miR-21-3p-dependent targeting of PTEN in vitro, while in vivo they promoted the generation of atherosclerotic lesions in mice (157). Going forward, and based on these studies, the use of genetically engineered exosomes to deliver miRNAs known to be beneficial in the treatment of atherosclerosis is therefore very promising. It should be noted that cell-free methodologies can also be used to load vesicles with miRNAs after their isolation. Thus, miRNAs can be introduced into exosomes that have been isolated directly from biological fluids, by a number of chemical and physical methods including transfection and electroporation. miRNAs were shown to be introduced successfully into exosomes isolated from a number of different sources, and furthermore these exosomes retained their abilities to deliver the miRNAs to the target cell (158).
Stem cell transplantation also represents a promising strategy to repair and regenerate diseased tissue in CVD, including atherosclerosis and has been shown to be effective in animal models (159). However, this approach has limitations due to the compromised survival and differentiation, together with potential immunoreactivity of (allogenic) stem cells (160). The use of stem cell-derived exosomes represents an acellular therapeutic strategy (which avoids the issues often seen with stem cell transplantation) and atherosclerosis is now being widely investigated using stem cell-derived exosomes carrying miRNAs (161). Further, much evidence now suggests that many of the beneficial effects of stem cell administration can, in fact, be attributed to the secretion of paracrine factors by the transplanted cells, including exosomal miRNAs (162). Thus, exosomes derived from progenitor cells, such as mesenchymal stem cells (MSCs), endothelial progenitor cells (EPCs), bone marrow derived macrophages (BMDM) and platelets have been shown to express endogenous miRNAs that ameliorate and inhibit atherosclerosis progression [reviewed in (154)]. For instance, MSC-derived exosomes containing miR-21a-5p were shown to attenuate atherosclerosis in the ApoE-/- mouse model, via promoting M2 macrophage polarization and infiltration (163). In another recent study, miR-100-5p in human umbilical cord mesenchymal stem cell-derived exosomes was shown to reduce plaque formation in atherosclerosis-prone mice via alleviation of eosinophil inflammatory responses (164). The treatment of diabetic-associated atherosclerotic mice with EPC-derived exosomes was also demonstrated to be beneficial in reducing the disease, although in this study the therapeutic activity was not assigned to a specific miRNA (165).
The development of vascular disorders including atherosclerosis has increasingly often been demonstrated to be driven by environmentally-acquired, aberrant changes in DNA methylation. In some cases these may be due to (clonal expansion of) somatic mutations within the epigenetic modifying enzymes, Tet2 and Dnmt3a. Genetic editing, to reverse mutations known to be causal in the development of atherosclerosis represents a promising stem cell-based therapeutic strategy in the elusive struggle to reverse the progression of atherosclerosis. Tet2 and Dnmt3a may therefore be potential therapeutic targets for gene editing in this context. Alternatively, epigenetic editing, involving the CRISPR/Cas9-dependent guiding of epigenetic modifiers to target the aberrant methylation marks themselves represents another potential therapeutic strategy.
The misexpression of specific miRNA(s), when shown to be causal in atherosclerosis development, also represents a promising therapeutic target to treat the disease. Genetically-programmed cell-based therapies involving the generation of exosomes loaded with specific small RNA cargo are therefore also being developed. Further, the clinical use of (naturally-occurring) exosomes expressing therapeutically-beneficial miRNAs, isolated from stem cell populations offers great promise.
Both the above approaches have significant inherent challenges to overcome and will rely on a fuller understanding of the methylation/miRNA changes involved, their causal relationship with the disease, and the involvement of specific cell-types in atherosclerosis pathology. This will be necessary both to inform the epigenetic changes to target and the choice of stem cell to engineer.
HS and AB: both contributed to the text and generation of the tables in this paper. Both authors contributed to the article and approved the submitted version.
This work was supported by the British Heart Foundation, Student grant FS/4yPhD/F/21/34154.
We thank Dr. John Pizzey for critical reading of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. (2007) 128(4):635–8. doi: 10.1016/j.cell.2007.02.006
2. Atlasi Y, Stunnenberg HG. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet. (2017) 18(11):643–58. doi: 10.1038/nrg.2017.57
3. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. (2019) 571(7766):489–99. doi: 10.1038/s41586-019-1411-0
4. Brassington K, Selemidis S, Bozinovski S, Vlahos R. Chronic obstructive pulmonary disease and atherosclerosis: common mechanisms and novel therapeutics. Clin Sci (Lond). (2022) 136(6):405–23. doi: 10.1042/CS20210835
5. Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. (2007) 128(4):669–81. doi: 10.1016/j.cell.2007.01.033
6. Parry A, Rulands S, Reik W. Active turnover of DNA methylation during cell fate decisions. Nat Rev Genet. (2021) 22(1):59–66. doi: 10.1038/s41576-020-00287-8
7. Shimbo T, Wade PA. Proteins that read DNA methylation. Adv Exp Med Biol. (2016) 945:303–20. doi: 10.1007/978-3-319-43624-1_13
8. Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. (2018) 392(10149):777–86. doi: 10.1016/S0140-6736(18)31268-6
9. Caldwell BA, Liu MY, Prasasya RD, Wang T, DeNizio JE, Leu NA, et al. Functionally distinct roles for TET-oxidized 5-methylcytosine bases in somatic reprogramming to pluripotency. Mol Cell. (2021) 81(4):859–69 e8. doi: 10.1016/j.molcel.2020.11.045
10. Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. (2017) 18(9):517–34. doi: 10.1038/nrg.2017.33
11. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. (2005) 102(30):10604–9. doi: 10.1073/pnas.0500398102
12. Zaina S, Lindholm MW, Lund G. Nutrition and aberrant DNA methylation patterns in atherosclerosis: more than just hyperhomocysteinemia? J Nutr. (2005) 135(1):5–8. doi: 10.1093/jn/135.1.5
13. Afanas’ev I. New nucleophilic mechanisms of ros-dependent epigenetic modifications: comparison of aging and cancer. Aging Dis. (2014) 5(1):52–62. doi: 10.14336/AD.2014.050052
14. Green HLH, Brewer AC. Dysregulation of 2-oxoglutarate-dependent dioxygenases by hyperglycaemia: does this link diabetes and vascular disease? Clin Epigenetics. (2020) 12(1):59. doi: 10.1186/s13148-020-00848-y
15. Aavik E, Babu M, Yla-Herttuala S. DNA methylation processes in atheosclerotic plaque. Atherosclerosis. (2019) 281:168–79. doi: 10.1016/j.atherosclerosis.2018.12.006
16. Guay S-P, Brisson D, Munger J, Lamarche B, Gaudet D, Bouchard L. ABCA1 gene promoter DNA methylation is associated with HDL particle profile and coronary artery disease in familial hypercholesterolemia. Epigenetics. (2012) 7:464–72. doi: 10.4161/epi.19633
17. Infante T, Franzese M, Ruocco A, Schiano C, Affinito O, Pane K, et al. ABCA1, TCF7, NFATC1, PRKCZ, and PDGFA DNA methylation as potential epigenetic-sensitive targets in acute coronary syndrome via network analysis. Epigenetics. (2022) 17:547–63. doi: 10.1080/15592294.2021.1939481
18. Lv Y-C, Tang Y-Y, Zhang P, Wan W, Yao F, He P-P, et al. Histone methyltransferase enhancer of zeste homolog 2-mediated ABCA1 promoter DNA methylation contributes to the progression of atherosclerosis. PLoS One. (2016) 11:e0157265. doi: 10.1371/journal.pone.0157265
19. Kumar A, Kumar S, Vikram A, Hoffman TA, Naqvi A, Lewarchik CM, et al. Histone and DNA methylation-mediated epigenetic downregulation of endothelial kruppel-like factor 2 by low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. (2013) 33:1936–42. doi: 10.1161/ATVBAHA.113.301765
20. Lee D-Y, Chiu J-J. Atherosclerosis and flow: roles of epigenetic modulation in vascular endothelium. J Biomed Sci. (2019) 26:56. doi: 10.1186/s12929-019-0551-8
21. Kim YR, Kim CS, Naqvi A, Kumar A, Kumar S, Hoffman TA, et al. Epigenetic upregulation of p66shc mediates low-density lipoprotein cholesterol-induced endothelial cell dysfunction. Am J Physiol Heart Circ Physiol. (2012) 303:H189–96. doi: 10.1152/ajpheart.01218.2011
22. Kim CS, Kim YR, Naqvi A, Kumar S, Hoffman TA, Jung SB, et al. Homocysteine promotes human endothelial cell dysfunction via site-specific epigenetic regulation of p66shc. Cardiovasc Res. (2011) 92:466–75. doi: 10.1093/cvr/cvr250
23. Shahzad K, Gadi I, Nazir S, Al-Dabet MDM, Kohli S, Bock F, et al. Activated protein C reverses epigenetically sustained p66Shc expression in plaque-associated macrophages in diabetes. Communications Biology. (2018) 1:104. doi: 10.1038/s42003-018-0108-5
24. Matouk CC, Marsden PA. Epigenetic regulation of vascular endothelial gene expression. Circ Res. (2008) 102:873–87. doi: 10.1161/circresaha.107.171025
25. Mitra S, Khaidakov M, Lu J, Ayyadevara S, Szwedo J, Wang XW, et al. Prior exposure to oxidized low-density lipoprotein limits apoptosis in subsequent generations of endothelial cells by altering promoter methylation. Am J Physiol Heart Circ Physiol. (2011) 301:H506–13. doi: 10.1152/ajpheart.00252.2011
26. Zhang D, Chen Y, Xie X, Liu J, Wang Q, Kong W, et al. Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet-derived growth factor in endothelial cells. J Mol Cell Cardiol. (2012) 53:487–96. doi: 10.1016/j.yjmcc.2012.07.010
27. Hiltunen MO, Turunen MP, Häkkinen TP, Rutanen J, Hedman M, Mäkinen K, et al. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med. (2002) 7:5–11. doi: 10.1191/1358863(02vm418oa
28. Han X-B, Zhang H-P, Cao C-J, Wang Y-H, Tian J, Yang X-L, et al. Aberrant DNA methylation of the PDGF gene in homocysteine-mediated VSMC proliferation and its underlying mechanism. Mol Med Rep. (2014) 10:947–54. doi: 10.3892/mmr.2014.2249
29. Min J, Weitian Z, Peng C, Yan P, Bo Z, Yan W, et al. Correlation between insulin-induced estrogen receptor methylation and atherosclerosis. Cardiovasc Diabetol. (2016) 15:156. doi: 10.1186/s12933-016-0471-9
30. Post WS, Goldschmidt-Clermont PJ, Wilhide CC, Heldman AW, Sussman MS, Ouyang P, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res. (1999) 43:985–91. doi: 10.1016/s0008-6363(99)00153-4
31. Ying AK, Hassanain HH, Roos CM, Smiraglia DJ, Issa JJ, Michler RE, et al. Methylation of the estrogen receptor-alpha gene promoter is selectively increased in proliferating human aortic smooth muscle cells. Cardiovasc Res. (2000) 46:172–9. doi: 10.1016/s0008-6363(00)00004-3
32. Huang YS, Zhi YF, Wang SR. Hypermethylation of estrogen receptor-alpha gene in atheromatosis patients and its correlation with homocysteine. Pathophysiology. (2009) 16:259–65. doi: 10.1016/j.pathophys.2009.02.010
33. Kim J, Kim JY, Song KS, Lee YH, Seo JS, Jelinek J, et al. Epigenetic changes in estrogen receptor beta gene in atherosclerotic cardiovascular tissues and in-vitro vascular senescence. Biochim Biophys Acta. (2007) 1772:72–80. doi: 10.1016/j.bbadis.2006.10.004
34. Li L, Xie J, Zhang M, Wang S. Homocysteine harasses the imprinting expression of IGF2 and H19 by demethylation of differentially methylated region between IGF2/H19 genes. Acta Biochim Biophys Sin. (2009) 41:464–71. doi: 10.1093/abbs/gmp033
35. Zhu L, Jia L, Liu N, Wu R, Guan G, Hui R, et al. DNA methyltransferase 3b accelerates the process of atherosclerosis. Oxid Med Cell Longev. (2022) 2022:5249367. doi: 10.1155/2022/5249367
36. Jia L, Zhu L, Wang JZ, Wang XJ, Chen JZ, Song L, et al. Methylation of FOXP3 in regulatory T cells is related to the severity of coronary artery disease. Atherosclerosis. (2013) 228:346–52. doi: 10.1016/j.atherosclerosis.2013.01.027
37. Lü CX, Xu RD, Cao M, Wang G, Yan FQ, Shang SS, et al. FOXP3 demethylation as a means of identifying quantitative defects in regulatory T cells in acute coronary syndrome. Atherosclerosis. (2013) 229:263–70. doi: 10.1016/j.atherosclerosis.2013.05.007
38. Jiang YZ, Jiménez JM, Ou K, McCormick ME, Zhang LD, Davies PF. Hemodynamic disturbed flow induces differential DNA methylation of endothelial kruppel-like factor 4 promoter in vitro and in vivo. Circ Res. (2014) 115:32–43. doi: 10.1161/circresaha.115.303883
39. Tang RZ, Zhu JJ, Yang FF, Zhang YP, Xie SA, Liu YF, et al. DNA methyltransferase 1 and krüppel-like factor 4 axis regulates macrophage inflammation and atherosclerosis. J Mol Cell Cardiol. (2019) 128:11–24. doi: 10.1016/j.yjmcc.2019.01.009
40. Wang Y, Xu Y, Yan S, Cao K, Zeng X, Zhou Y, et al. Adenosine kinase is critical for neointima formation after vascular injury by inducing aberrant DNA hypermethylation. Cardiovasc Res. (2021) 117:561–75. doi: 10.1093/cvr/cvaa040
41. Guay SP, Brisson D, Lamarche B, Marceau P, Vohl MC, Gaudet D, et al. DNA methylation variations at CETP and LPL gene promoter loci: new molecular biomarkers associated with blood lipid profile variability. Atherosclerosis. (2013) 228:413–20. doi: 10.1016/j.atherosclerosis.2013.03.033
42. Wei L, Zhao S, Wang G, Zhang S, Luo W, Qin Z, et al. SMAD7 methylation as a novel marker in atherosclerosis. Biochem Biophys Res Commun. (2018) 496:700–5. doi: 10.1016/j.bbrc.2018.01.121
43. Wei LH, Chao NX, Gao S, Yu YT, Shi L, Ma XB, et al. Homocysteine induces vascular inflammatory response via SMAD7 hypermethylation in human umbilical vein smooth muscle cells. Microvasc Res. (2018) 120:8–12. doi: 10.1016/j.mvr.2018.05.003
44. Xia Y, Brewer A, Bell JT. DNA methylation signatures of incident coronary heart disease: findings from epigenome-wide association studies. Clin Epigenetics. (2021) 13(1):186. doi: 10.1186/s13148-021-01175-6
45. Dasagrandhi D, Muthuswamy A, Swaminathan JK. Atherosclerosis: nexus of vascular dynamics and cellular cross talks. Mol Cell Biochem. (2022) 477(2):571–84. doi: 10.1007/s11010-021-04307-x
46. Abdolmaleki F, Gheibi Hayat SM, Bianconi V, Johnston TP, Sahebkar A. Atherosclerosis and immunity: a perspective. Trends Cardiovasc Med. (2019) 29(6):363–71. doi: 10.1016/j.tcm.2018.09.017
47. Benincasa G, Mansueto G, Napoli C. Fluid-based assays and precision medicine of cardiovascular diseases: the “hope” for pandora’s box? J Clin Pathol. (2019) 72(12):785–99. doi: 10.1136/jclinpath-2019-206178
48. Bienkowski M, Peksa R, Popeda M, Kolaczkowska M, Frankiewicz A, Zaczek AJ, et al. Liquid biopsy for minimally invasive heart transplant monitoring: a pilot study. J Clin Pathol. (2020) 73(8):507–10. doi: 10.1136/jclinpath-2019-205926
49. Olivan-Viguera A, Perez-Zabalza M, Garcia-Mendivil L, Mountris KA, Oros-Rodrigo S, Ramos-Marques E, et al. Minimally invasive system to reliably characterize ventricular electrophysiology from living donors. Sci Rep. (2020) 10(1):19941. doi: 10.1038/s41598-020-77076-0
50. Zong W, Kang H, Xiong Z, Ma Y, Jin T, Gong Z, et al. Scmethbank: a database for single-cell whole genome DNA methylation maps. Nucleic Acids Res. (2022) 50(D1):D380–6. doi: 10.1093/nar/gkab833
51. Jones J, Bolleddu SI, Vanthenapalli S, Rodgers LE, Shah K, et al. Cardiovascular risks associated with gender and aging. J Cardiovasc Dev Dis. (2019) 6(2):19. doi: 10.3390/jcdd6020019
52. Marnell CS, Bick A, Natarajan P. Clonal hematopoiesis of indeterminate potential (CHIP): linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J Mol Cell Cardiol. (2021) 161:98–105. doi: 10.1016/j.yjmcc.2021.07.004
53. Misra A, Rehan R, Lin A, Patel S, Fisher EA. Emerging concepts of vascular cell clonal expansion in atherosclerosis. Arterioscler Thromb Vasc Biol. (2022) 42(3):e74–e84. doi: 10.1161/ATVBAHA.121.316093
54. Cobo I, Tanaka T, Glass CK, Yeang C. Clonal hematopoiesis driven by DNMT3A and TET2 mutations: role in monocyte and macrophage biology and atherosclerotic cardiovascular disease. Curr Opin Hematol. (2022) 29(1):1–7. doi: 10.1097/MOH.0000000000000688
55. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377(2):111–21. doi: 10.1056/NEJMoa1701719
56. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. (2017) 355(6327):842–7. doi: 10.1126/science.aag1381
57. Peng J, Yang Q, Li AF, Li RQ, Wang Z, Liu LS, et al. Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE-/- mice. Oncotarget. (2016) 7(47):76423–36. doi: 10.18632/oncotarget.13121
58. Rauch PJ, Silver AJ, Gopakumar J, McConkey M, Sinha E, Fefer M, et al. Loss-of-function mutations in DNMT3A and TET2 lead to accelerated atherosclerosis and convergent macrophage phenotypes in mice. Blood. (2018) 132:745. doi: 10.1182/blood-2018-99-118288
59. Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16(12):727–44. doi: 10.1038/s41569-019-0227-9
60. Zhuang J, Luan P, Li H, Wang K, Zhang P, Xu Y, et al. The yin-yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler Thromb Vasc Biol. (2017) 37(1):84–97. doi: 10.1161/ATVBAHA.116.307923
61. Lin R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. (2013) 128(18):2047–57. doi: 10.1161/CIRCULATIONAHA.113.002887
62. Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW, et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest. (2014) 124(7):3187–99. doi: 10.1172/JCI74792
63. Li Z, Solomonidis EG, Meloni M, Taylor RS, Duffin R, Dobie R, et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur Heart J. (2019) 40(30):2507–20. doi: 10.1093/eurheartj/ehz305
64. Manavski Y, Lucas T, Glaser SF, Dorsheimer L, Gunther S, Braun T, et al. Clonal expansion of endothelial cells contributes to ischemia-induced neovascularization. Circ Res. (2018) 122(5):670–7. doi: 10.1161/CIRCRESAHA.117.312310
65. McDonald AI, Shirali AS, Aragon R, Ma F, Hernandez G, Vaughn DA, et al. Endothelial regeneration of large vessels is a biphasic process driven by local cells with distinct proliferative capacities. Cell Stem Cell. (2018) 23(2):210–25 e6. doi: 10.1016/j.stem.2018.07.011
66. Wakabayashi T, Naito H, Suehiro JI, Lin Y, Kawaji H, Iba T, et al. CD157 marks tissue-resident endothelial stem cells with homeostatic and regenerative properties. Cell Stem Cell. (2018) 22(3):384–97 e6. doi: 10.1016/j.stem.2018.01.010
67. Ganz P, Hsue PY. Endothelial dysfunction in coronary heart disease is more than a systemic process. Eur Heart J. (2013) 34(27):2025–7. doi: 10.1093/eurheartj/eht199
68. Zampetaki A, Kirton JP, Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res. (2008) 78(3):413–21. doi: 10.1093/cvr/cvn081
69. Fujisawa T, Tura-Ceide O, Hunter A, Mitchell A, Vesey A, Medine C, et al. Endothelial progenitor cells do not originate from the bone marrow. Circulation. (2019) 140(18):1524–6. doi: 10.1161/CIRCULATIONAHA.119.042351
70. Farina M, Bernardi S, Polverelli N, D'Adda M, Malagola M, Bosio K, et al. Comparative mutational profiling of hematopoietic progenitor cells and circulating endothelial cells (CECs) in patients with primary myelofibrosis. Cells. (2021) 10(10):2764. doi: 10.3390/cells10102764
71. Chistiakov DA, Orekhov AN, Bobryshev YV. Treatment of cardiovascular pathology with epigenetically active agents: focus on natural and synthetic inhibitors of DNA methylation and histone deacetylation. Int J Cardiol. (2017) 227:66–82. doi: 10.1016/j.ijcard.2016.11.204
72. Zhang Y, Mei J, Li J, Zhang Y, Zhou Q, Xu F. DNA methylation in atherosclerosis: a new perspective. Evid Based Complement Alternat Med. (2021) 2021:6623657. doi: 10.1155/2021/6623657
73. Liu G, Bin P, Wang T, Ren W, Zhong J, Liang J, et al. DNA methylation and the potential role of methyl-containing nutrients in cardiovascular diseases. Oxid Med Cell Longev. (2017) 2017:1670815. doi: 10.1155/2017/1670815
74. Sharma G, Sharma AR, Bhattacharya M, Lee SS, Chakraborty C. CRISPR-Cas9: a preclinical and clinical perspective for the treatment of human diseases. Mol Ther. (2021) 29(2):571–86. doi: 10.1016/j.ymthe.2020.09.028
75. Siew WS, Tang YQ, Kong CK, Goh BH, Zacchigna S, Dua K, et al. Harnessing the potential of CRISPR/Cas in atherosclerosis: disease modeling and therapeutic applications. Int J Mol Sci. (2021) 22(16). doi: 10.3390/ijms22168422
76. Hou L, Kim JJ, Woo YJ, Huang NF. Stem cell-based therapies to promote angiogenesis in ischemic cardiovascular disease. Am J Physiol Heart Circ Physiol. (2016) 310(4):H455–65. doi: 10.1152/ajpheart.00726.2015
77. Navarro-Guerrero E, Tay C, Whalley JP, Cowley SA, Davies B, Knight JC, et al. Genome-wide CRISPR/Cas9-knockout in human induced pluripotent stem cell (iPSC)-derived macrophages. Sci Rep. (2021) 11(1):4245. doi: 10.1038/s41598-021-82137-z
78. Wang M, Yuan Q, Xie L. Mesenchymal stem cell-based immunomodulation: properties and clinical application. Stem Cells Int. (2018) 2018:3057624. doi: 10.1155/2018/3057624
79. Chen X, Aledia AS, Popson SA, Him L, Hughes CC, George SC. Rapid anastomosis of endothelial progenitor cell-derived vessels with host vasculature is promoted by a high density of cotransplanted fibroblasts. Tissue Eng Part A. (2010) 16(2):585–94. doi: 10.1089/ten.tea.2009.0491
80. Caplice NM, Bunch TJ, Stalboerger PG, Wang S, Simper D, Miller DV, et al. Smooth muscle cells in human coronary atherosclerosis can originate from cells administered at marrow transplantation. Proc Natl Acad Sci U S A. (2003) 100(8):4754–9. doi: 10.1073/pnas.0730743100
81. Syding LA, Nickl P, Kasparek P, Sedlacek R. CRISPR/Cas9 epigenome editing potential for rare imprinting diseases: a review. Cells. (2020) 9(4). doi: 10.3390/cells9040993
82. Nguyen TV, Lister R. Genomic targeting of TET activity for targeted demethylation using CRISPR/Cas9. Methods Mol Biol. (2021) 2272:181–94. doi: 10.1007/978-1-0716-1294-1_10
83. Park H, Shin J, Kim Y, Saito T, Saido TC, Kim J. CRISPR/dCas9-DNMT3A-mediated targeted DNA methylation of APP rescues brain pathology in a mouse model of Alzheimer’s disease. Transl Neurodegener. (2022) 11(1):41. doi: 10.1186/s40035-022-00314-0
84. Dong F, Xie K, Chen Y, Yang Y, Mao Y. Polycistronic tRNA and CRISPR guide-RNA enables highly efficient multiplexed genome engineering in human cells. Biochem Biophys Res Commun. (2017) 482(4):889–95. doi: 10.1016/j.bbrc.2016.11.129
85. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. (2001) 291(5507):1304–51. doi: 10.1126/science.1058040
86. Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. (2006) 15(Spec No 1):R17–R29. doi: 10.1093/hmg/ddl046
87. Boyd SD. Everything you wanted to know about small RNA but were afraid to ask. Lab Invest. (2008) 88(6):569–78. doi: 10.1038/labinvest.2008.32
88. Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. (2005) 37(7):766–70. doi: 10.1038/ng1590
89. Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. (2005) 120(1):21–4. doi: 10.1016/j.cell.2004.12.031
90. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. (2005) 120(1):15–20. doi: 10.1016/j.cell.2004.12.035
91. Ipsaro JJ, Joshua-Tor L. From guide to target: molecular insights into eukaryotic RNA-interference machinery. Nat Struct Mol Biol. (2015) 22(1):20–8. doi: 10.1038/nsmb.2931
92. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. (2015) 16(7):421–33. doi: 10.1038/nrg3965
93. Chavali S, Bruhn S, Tiemann K, Saetrom P, Barrenas F, Saito T, et al. MicroRNAs act complementarily to regulate disease-related mRNA modules in human diseases. RNA. (2013) 19(11):1552–62. doi: 10.1261/rna.038414.113
94. Krutzfeldt J, Poy MN, Stoffel M. Strategies to determine the biological function of microRNAs. Nat Genet. (2006) 38(Suppl):S14–9. doi: 10.1038/ng1799
95. Reddy KB. MicroRNA (miRNA) in cancer. Cancer Cell Int. (2015) 15:38. doi: 10.1186/s12935-015-0185-1
96. Laffont B, Rayner KJ. MicroRNAs in the pathobiology and therapy of atherosclerosis. Can J Cardiol. (2017) 33(3):313–24. doi: 10.1016/j.cjca.2017.01.001
97. Lu Y, Thavarajah T, Gu W, Cai J, Xu Q. Impact of miRNA in atherosclerosis. Arterioscler Thromb Vasc Biol. (2018) 38(9):e159–70. doi: 10.1161/ATVBAHA.118.310227
98. Satrauskiene A, Navickas R, Laucevicius A, Krilavicius T, Uzupyte R, Zdanyte M, et al. Mir-1, miR-122, miR-132, and miR-133 are related to subclinical aortic atherosclerosis associated with metabolic syndrome. Int J Environ Res Public Health. (2021) 18(4). doi: 10.3390/ijerph18041483
99. Chen J, Yin H, Jiang Y, Radhakrishnan SK, Huang ZP, Li J, et al. Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler Thromb Vasc Biol. (2011) 31(2):368–75. doi: 10.1161/ATVBAHA.110.218149
100. Xie C, Huang H, Sun X, Guo Y, Hamblin M, Ritchie RP, et al. MicroRNA-1 regulates smooth muscle cell differentiation by repressing kruppel-like factor 4. Stem Cells Dev. (2011) 20(2):205–10. doi: 10.1089/scd.2010.0283
101. Huang H, Xie C, Sun X, Ritchie RP, Zhang J, Chen YE. miR-10a contributes to retinoid acid-induced smooth muscle cell differentiation. J Biol Chem. (2010) 285(13):9383–9. doi: 10.1074/jbc.M109.095612
102. Kuo JT, Tsai HE, Lin CT, Lee CI, Lee PL, Ruan YR, et al. Low levels of MicroRNA-10a in cardiovascular endothelium and blood Serum are related to human atherosclerotic disease. Cardiol Res Pract. (2021) 2021:1452917. doi: 10.1155/2021/1452917
103. Nazarenko MS, Koroleva IA, Zarubin AA, Sleptcov AA. miRNA regulome in different atherosclerosis phenotypes. Mol Biol. (2022) 56(2):227–43. doi: 10.1134/S0026893322020108
104. Qin X, Wang X, Wang Y, Tang Z, Cui Q, Xi J, et al. MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc Natl Acad Sci U S A. (2010) 107(7):3240–4. doi: 10.1073/pnas.0914882107
105. Sun G, Song H, Wu S. Mir-19a promotes vascular smooth muscle cell proliferation, migration and invasion through regulation of Ras homolog family member B. Int J Mol Med. (2019) 44(6):1991–2002. doi: 10.3892/ijmm.2019.4357
106. Ren ZQ, Liu N, Zhao K. Micro RNA-19a suppresses IL-10 in peripheral B cells from patients with atherosclerosis. Cytokine. (2016) 86:86–91. doi: 10.1016/j.cyto.2016.07.019
107. Li C, Li S, Zhang F, Wu M, Liang H, Song J, et al. Endothelial microparticles-mediated transfer of microRNA-19b promotes atherosclerosis via activating perivascular adipose tissue inflammation in apoE(-/-) mice. Biochem Biophys Res Commun. (2018) 495(2):1922–9. doi: 10.1016/j.bbrc.2017.11.195
108. Xue Y, Wei Z, Ding H, Wang Q, Zhou Z, Zheng S, et al. MicroRNA-19b/221/222 induces endothelial cell dysfunction via suppression of PGC-1alpha in the progression of atherosclerosis. Atherosclerosis. (2015) 241(2):671–81. doi: 10.1016/j.atherosclerosis.2015.06.031
109. Lv YC, Tang YY, Peng J, Zhao GJ, Yang J, Yao F, et al. MicroRNA-19b promotes macrophage cholesterol accumulation and aortic atherosclerosis by targeting ATP-binding cassette transporter A1. Atherosclerosis. (2014) 236(1):215–26. doi: 10.1016/j.atherosclerosis.2014.07.005
110. Wang Q, Dong Y, Wang H. microRNA-19b-3p-containing extracellular vesicles derived from macrophages promote the development of atherosclerosis by targeting JAZF1. J Cell Mol Med. (2022) 26(1):48–59. doi: 10.1111/jcmm.16938
111. Huang X, Yue Z, Wu J, Chen J, Wang S, Wu J, et al. MicroRNA-21 knockout exacerbates angiotensin II-induced thoracic aortic aneurysm and dissection in mice with abnormal transforming growth factor-beta-SMAD3 signaling. Arterioscler Thromb Vasc Biol. (2018) 38(5):1086–101. doi: 10.1161/ATVBAHA.117.310694
112. Sun P, Tang LN, Li GZ, Xu ZL, Xu QH, Wang M, et al. Effects of MiR-21 on the proliferation and migration of vascular smooth muscle cells in rats with atherosclerosis via the Akt/ERK signaling pathway. Eur Rev Med Pharmacol Sci. (2019) 23(5):2216–22. doi: 10.26355/eurrev_201903_17269
113. Wu Z, Geng J, Bai Y, Qi Y, Chang C, Jiao Y, et al. MicroRNA-22 inhibition promotes the development of atherosclerosis via targeting interferon regulator factor 5. Exp Cell Res. (2021) 409(2):112922. doi: 10.1016/j.yexcr.2021.112922
114. Leeper NJ, Raiesdana A, Kojima Y, Chun HJ, Azuma J, Maegdefessel L, et al. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J Cell Physiol. (2011) 226(4):1035–43. doi: 10.1002/jcp.22422
115. Feng M, Xu D, Wang L. miR-26a inhibits atherosclerosis progression by targeting TRPC3. Cell Biosci. (2018) 8:4. doi: 10.1186/s13578-018-0203-9
116. Xie W, Li L, Zhang M, Cheng HP, Gong D, Lv YC, et al. MicroRNA-27 prevents atherosclerosis by suppressing lipoprotein lipase-induced lipid accumulation and inflammatory response in apolipoprotein E knockout mice. PLoS One. (2016) 11(6):e0157085. doi: 10.1371/journal.pone.0157085
117. Liu CZ, Zhong Q, Huang YQ. Elevated plasma miR-29a levels are associated with increased carotid intima-media thickness in atherosclerosis patients. Tohoku J Exp Med. (2017) 241(3):183–8. doi: 10.1620/tjem.241.183
118. Zheng B, Zheng CY, Zhang Y, Yin WN, Li YH, Liu C, et al. Regulatory crosstalk between KLF5, miR-29a and Fbw7/CDC4 cooperatively promotes atherosclerotic development. Biochim Biophys Acta Mol Basis Dis. (2018) 1864(2):374–86. doi: 10.1016/j.bbadis.2017.10.021
119. Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, et al. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. (2007) 100(11):1579–88. doi: 10.1161/CIRCRESAHA.106.141986
120. McDonald RA, Halliday CA, Miller AM, Diver LA, Dakin RS, Montgomery J, et al. Reducing in-stent restenosis: therapeutic manipulation of miRNA in vascular remodeling and inflammation. J Am Coll Cardiol. (2015) 65(21):2314–27. doi: 10.1016/j.jacc.2015.03.549
121. Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. (2010) 328(5985):1566–9. doi: 10.1126/science.1189123
122. Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. (2010) 328(5985):1570–3. doi: 10.1126/science.1189862
123. Nishiga M, Horie T, Kuwabara Y, Nagao K, Baba O, Nakao T, et al. MicroRNA-33 controls adaptive fibrotic response in the remodeling heart by preserving lipid raft cholesterol. Circ Res. (2017) 120(5):835–47. doi: 10.1161/CIRCRESAHA.116.309528
124. Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. (2006) 3(2):87–98. doi: 10.1016/j.cmet.2006.01.005
125. Wu X, Du X, Yang Y, Liu X, Liu X, Zhang N, et al. Inhibition of miR-122 reduced atherosclerotic lesion formation by regulating NPAS3-mediated endothelial to mesenchymal transition. Life Sci. (2021) 265:118816. doi: 10.1016/j.lfs.2020.118816
126. Schober A, Nazari-Jahantigh M, Wei Y, Bidzhekov K, Gremse F, Grommes J, et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med. (2014) 20(4):368–76. doi: 10.1038/nm.3487
127. Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. (2008) 15(2):261–71. doi: 10.1016/j.devcel.2008.07.002
128. Liu F, Liu Y, Du Y, Li Y, et al. MiRNA-130a promotes inflammation to accelerate atherosclerosis via the regulation of proliferator-activated receptor gamma (PPARgamma) expression. Anatol J Cardiol. (2021) 25(9):630–7. doi: 10.5152/AnatolJCardiol.2021.56721
129. Lovren F, Pan Y, Quan A, Singh KK, Shukla PC, Gupta N, et al. MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation. (2012) 126(11 Suppl 1):S81–S90. doi: 10.1161/CIRCULATIONAHA.111.084186
130. Sun SG, Zheng B, Han M, Fang XM, Li HX, Miao SB, et al. miR-146a and kruppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO Rep. (2011) 12(1):56–62. doi: 10.1038/embor.2010.172
131. Deng S, Wang H, Jia C, Zhu S, Chu X, Ma Q, et al. MicroRNA-146a induces lineage-negative bone marrow cell apoptosis and senescence by targeting polo-like kinase 2 expression. Arterioscler Thromb Vasc Biol. (2017) 37(2):280–90. doi: 10.1161/ATVBAHA.116.308378
132. Goedeke L, Rotllan N, Canfran-Duque A, Aranda JF, Ramirez CM, Araldi E, et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat Med. (2015) 21(11):1280–9. doi: 10.1038/nm.3949
133. Wang F, Ge J, Huang S, Zhou C, Sun Z, Song Y, et al. KLF5/LINC00346/miR-148a-3p axis regulates inflammation and endothelial cell injury in atherosclerosis. Int J Mol Med. (2021) 48(2). doi: 10.3892/ijmm.2021.4985
134. Zheng B, Yin WN, Suzuki T, Zhang XH, Zhang Y, Song LL, et al. Exosome-mediated miR-155 transfer from smooth muscle cells to endothelial cells induces endothelial injury and promotes atherosclerosis. Mol Ther. (2017) 25(6):1279–94. doi: 10.1016/j.ymthe.2017.03.031
135. Ding J, Li H, Liu W, Wang X, Feng Y, Guan H, et al. miR-186-5p dysregulation in serum exosomes from patients with AMI aggravates atherosclerosis via targeting LOX-1. Int J Nanomedicine. (2022) 17:6301–16. doi: 10.2147/IJN.S383904
136. Yang S, Mi X, Chen Y, Feng C, Hou Z, Hui R, et al. MicroRNA-216a induces endothelial senescence and inflammation via Smad3/IkappaBalpha pathway. J Cell Mol Med. (2018) 22(5):2739–49. doi: 10.1111/jcmm.13567
137. Menghini R, Casagrande V, Cardellini M, Martelli E, Terrinoni A, Amati F, et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation. (2009) 120(15):1524–32. doi: 10.1161/CIRCULATIONAHA.109.864629
138. Zhang B, Zhang YF, Li R, Zhao L, Qin SG, Pan LF, et al. MiR-217 inhibits apoptosis of atherosclerotic endothelial cells via the TLR4/PI3K/Akt/NF-kappaB pathway. Eur Rev Med Pharmacol Sci. (2020) 24(24):12867–77. doi: 10.26355/eurrev_202012_24190
139. Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. (2009) 104(4):476–87. doi: 10.1161/CIRCRESAHA.108.185363
140. Meiler S, Baumer Y, Toulmin E, Seng K, Boisvert WA. MicroRNA 302a is a novel modulator of cholesterol homeostasis and atherosclerosis. Arterioscler Thromb Vasc Biol. (2015) 35(2):323–31. doi: 10.1161/ATVBAHA.114.304878
141. Lu X, Yang B, Yang H, Wang L, Li H, Chen S, et al. MicroRNA-320b modulates cholesterol efflux and atherosclerosis. J Atheroscler Thromb. (2022) 29(2):200–20. doi: 10.5551/jat.57125
142. Lin B, Feng DG, Wang F, Wang JX, Xu CG, Zhao H, et al. MiR-365 participates in coronary atherosclerosis through regulating IL-6. Eur Rev Med Pharmacol Sci. (2016) 20(24):5186–92. Accession number: 28051250.28051250
143. Li R, Jiang L, Wang X. Aberrant expression of miR-483-5p in patients with asymptomatic carotid artery stenosis and its predictive value for cerebrovascular event occurrence. Exp Ther Med. (2021) 22(4):1101. doi: 10.3892/etm.2021.10536
144. Wang B, Duan X, Xu Q, Li Y. Diagnostic and prognostic significance of miR-451a in patients with atherosclerosis. Vascular. (2023) 31(1):47–53. doi: 10.1177/17085381211058571
145. Wang Y, Xu Z, Wang X, Zheng J, Peng L, Zhou Y, et al. Extracellular-vesicle containing miRNA-503-5p released by macrophages contributes to atherosclerosis. Aging (Albany NY). (2021) 13(8):12239–57. doi: 10.18632/aging.103855
146. Gallo A, Tandon M, Alevizos I, Illei GG. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS One. (2012) 7(3):e30679. doi: 10.1371/journal.pone.0030679
147. Thery C. Cancer: diagnosis by extracellular vesicles. Nature. (2015) 523(7559):161–2. doi: 10.1038/nature14626
148. Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. (2010) 73(10):1907–20. doi: 10.1016/j.jprot.2010.06.006
149. Zamani P, Fereydouni N, Butler AE, Navashenaq JG, Sahebkar A. The therapeutic and diagnostic role of exosomes in cardiovascular diseases. Trends Cardiovasc Med. (2019) 29(6):313–23. doi: 10.1016/j.tcm.2018.10.010
150. Sharma AR, Sharma G, Bhattacharya M, Lee SS, Chakraborty C. Circulating miRNA in atherosclerosis: a clinical biomarker and early diagnostic tool. Curr Mol Med. (2022) 22(3):250–62. doi: 10.2174/1566524021666210315124438
151. Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov. (2014) 13(8):622–38. doi: 10.1038/nrd4359
152. Winkle M, El-Daly SM, Fabbri M, Calin GA. Noncoding RNA therapeutics - challenges and potential solutions. Nat Rev Drug Discov. (2021) 20(8):629–51. doi: 10.1038/s41573-021-00219-z
153. Lakhal S, Wood MJ. Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. Bioessays. (2011) 33(10):737–41. doi: 10.1002/bies.201100076
154. Heo J, Kang H. Exosome-based treatment for atherosclerosis. Int J Mol Sci. (2022) 23(2). doi: 10.3390/ijms23021002
155. Wang C, Wu H, Xing Y, Ye Y, He F, Yin Q, et al. Endothelial-derived extracellular microRNA-92a promotes arterial stiffness by regulating phenotype changes of vascular smooth muscle cells. Sci Rep. (2022) 12(1):344. doi: 10.1038/s41598-021-04341-1
156. Chang YJ, Li YS, Wu CC, Wang KC, Huang TC, Chen Z, et al. Extracellular MicroRNA-92a mediates endothelial cell-macrophage communication. Arterioscler Thromb Vasc Biol. (2019) 39(12):2492–504. doi: 10.1161/ATVBAHA.119.312707
157. Zhu J, Liu B, Wang Z, Wang D, Ni H, Zhang L, et al. Exosomes from nicotine-stimulated macrophages accelerate atherosclerosis through miR-21-3p/PTEN-mediated VSMC migration and proliferation. Theranostics. (2019) 9(23):6901–19. doi: 10.7150/thno.37357
158. de Abreu RC, Ramos CV, Becher C, Lino M, Jesus C, da Costa Martins PA, et al. Exogenous loading of miRNAs into small extracellular vesicles. J Extracell Vesicles. (2021) 10(10):e12111. doi: 10.1002/jev2.12111
159. Li F, Guo X, Chen SY. Function and therapeutic potential of mesenchymal stem cells in atherosclerosis. Front Cardiovasc Med. (2017) 4:32. doi: 10.3389/fcvm.2017.00032
160. Samadi P, Saki S, Manoochehri H, Sheykhhasan M. Therapeutic applications of mesenchymal stem cells: a comprehensive review. Curr Stem Cell Res Ther. (2021) 16(3):323–53. doi: 10.2174/22123946MTA5vOTEa1
161. Ling H, Guo Z, Tan L, Cao Q, Song C. Stem cell-derived exosomes: role in the pathogenesis and treatment of atherosclerosis. Int J Biochem Cell Biol. (2021) 130:105884. doi: 10.1016/j.biocel.2020.105884
162. Moghaddam AS, Afshari JT, Esmaeili SA, Saburi E, Joneidi Z, Momtazi-Borojeni AA. Cardioprotective microRNAs: lessons from stem cell-derived exosomal microRNAs to treat cardiovascular disease. Atherosclerosis. (2019) 285:1–9. doi: 10.1016/j.atherosclerosis.2019.03.016
163. Ma J, Chen L, Zhu X, Li Q, Hu L, Li H. Mesenchymal stem cell-derived exosomal miR-21a-5p promotes M2 macrophage polarization and reduces macrophage infiltration to attenuate atherosclerosis. Acta Biochim Biophys Sin. (2021) 53(9):1227–36. doi: 10.1093/abbs/gmab102
164. Gao H, Yu Z, Li Y, Wang X. miR-100-5p in human umbilical cord mesenchymal stem cell-derived exosomes mediates eosinophilic inflammation to alleviate atherosclerosis via the FZD5/Wnt/beta-catenin pathway. Acta Biochim Biophys Sin. (2021) 53(9):1166–76. doi: 10.1093/abbs/gmab093
Keywords: atherosclerosis, DNA methylation, microRNAs, epigenetics, stem cell therapy, CRISPR/Cas9, TET2, DNMT3a
Citation: Sum H and Brewer AC (2023) Epigenetic modifications as therapeutic targets in atherosclerosis: a focus on DNA methylation and non-coding RNAs. Front. Cardiovasc. Med. 10:1183181. doi: 10.3389/fcvm.2023.1183181
Received: 9 March 2023; Accepted: 2 May 2023;
Published: 25 May 2023.
Edited by:
Andriana Margariti, Queen's University Belfast, United KingdomReviewed by:
Shafeeq Ahmed Mohammed, University of Zurich, Switzerland© 2023 Sum and Brewer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alison C. Brewer YWxpc29uLmJyZXdlckBrY2wuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.