 Bram W. van Os1,2,3
Bram W. van Os1,2,3 Pascal J. H. Kusters1,2,3Myrthe den Toom1,2,3Linda Beckers1,2,3
Pascal J. H. Kusters1,2,3Myrthe den Toom1,2,3Linda Beckers1,2,3 Claudia M. van Tiel1,2,3
Claudia M. van Tiel1,2,3 Winnie G. Vos1,2,3
Winnie G. Vos1,2,3 Elize de Jong1
Elize de Jong1 Arnd Kieser4
Arnd Kieser4 Cindy van Roomen1,2,3
Cindy van Roomen1,2,3 Christoph J. Binder5
Christoph J. Binder5 Myrthe E. Reiche1,2,3
Myrthe E. Reiche1,2,3 Menno P. de Winther1,2,3
Menno P. de Winther1,2,3 Laura A. Bosmans1,2,3*†Esther Lutgens1,6,7,8,†
Laura A. Bosmans1,2,3*†Esther Lutgens1,6,7,8,†
- 1Department of Medical Biochemistry, Amsterdam UMC Location University of Amsterdam, Amsterdam, Netherlands
- 2Amsterdam Cardiovascular Sciences, Atherosclerosis & Ischemic Syndromes, Amsterdam, Netherlands
- 3Amsterdam Immunity and Infection, Amsterdam UMC, Amsterdam, Netherlands
- 4Research Unit Signaling and Translation, Helmholtz Zentrum München-German Research Center for Environmental Health, Neuherberg, Germany
- 5Department of Laboratory Medicine, Medical University of Vienna, Vienna, Austria
- 6Institute for Cardiovascular Prevention (IPEK), Ludwig-Maximilians-Universität, Munich, Germany
- 7German Center for Cardiovascular Research (DZHK), Partner site Munich Heart Alliance, Ludwig-Maximilians-Universität München, Germany
- 8Department of Cardiovascular Medicine and Immunology, Mayo Clinic, Rochester, MN, United States
Background: Atherosclerosis is the underlying cause of many cardiovascular diseases, such as myocardial infarction or stroke. B cells, and their production of pro- and anti-atherogenic antibodies, play an important role in atherosclerosis. In B cells, TRAF2 and NCK-interacting Kinase (TNIK), a germinal center kinase, was shown to bind to TNF-receptor associated factor 6 (TRAF6), and to be involved in JNK and NF-κB signaling in human B cells, a pathway associated with antibody production.
Objective: We here investigate the role of TNIK-deficient B cells in atherosclerosis.
Results: ApoE−/−TNIKfl/fl (TNIKBWT) and ApoE−/−TNIKfl/flCD19-cre (TNIKBKO) mice received a high cholesterol diet for 10 weeks. Atherosclerotic plaque area did not differ between TNIKBKO and TNIKBWT mice, nor was there any difference in plaque necrotic core, macrophage, T cell, α-SMA and collagen content. B1 and B2 cell numbers did not change in TNIKBKO mice, and marginal zone, follicular or germinal center B cells were unaffected. Total IgM and IgG levels, as well as oxidation specific epitope (OSE) IgM and IgG levels, did not change in absence of B cell TNIK. In contrast, plasma IgA levels were decreased in TNIKBKO mice, whereas the number of IgA+ B cells in intestinal Peyer's patches increased. No effects could be detected on T cell or myeloid cell numbers or subsets.
Conclusion: We here conclude that in hyperlipidemic ApoE−/− mice, B cell specific TNIK deficiency does not affect atherosclerosis.
1. Introduction
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality world-wide, with one in four deaths being linked to CVD (1, 2). Atherosclerosis (AS) is the most frequent underlying pathology of CVD, causing clinical features, such as myocardial infarction, stroke or heart failure. In recent years AS has been recognized as a chronic inflammatory lipid-driven disease of mid-sized and large arteries, characterized by the formation of lumen encroaching plaques (3, 4).
B cells are an important player in the adaptive immune system and various B cell subsets have different roles in the initiation and progression of atherosclerosis. Two distinct B cell subsets are identified, the innate like B1 cell and the conventional B2 subset (5). B1 B cells are recognized for their anti-atherogenic properties through the production of protective immunoglobulin (Ig)M antibodies, reactive against oxidation specific epitopes (OSE) (6). Most B2 cell subsets, but not marginal zone B cells, and plasma cells are considered pro-atherogenic, as their deficiency reduces atherosclerosis (7). Consequently, B cell depletion in atherosclerotic mice via an anti-CD20 antibody, reduced cardiovascular burden (8, 9).
The TRAF2 and NCK-interacting Kinase (TNIK) is a ubiquitously expressed member of the germinal center kinase family (10). In B cells, TNIK was identified as an interaction partner of the latent membrane protein (LMP1) signalosome after EBV infection, as well as of LMP1's cellular counterpart, the immune checkpoint CD40. Specifically, TNIK was reported to directly bind TRAF6, thereby bridging its interaction with the C-terminus of LMP1/CD40. In this complex, TNIK's C-terminal kinase part is essential for JNK activation, whereas its N-terminal part induces IKKβ/NF-κB activation, both essential for B cell activation (11).
As a regulatory component of the Wnt signaling pathways β-catenin and T-cell factor-4 (TCF-4) complex, TNIK displays divergent functions in different cell types (12). Studies reveal that TNIK is essential for the activation of Wnt target genes, allowing for proliferation of tumor cells (13), which induces tumor progression of myelogenous leukemia, colorectal cancer and lung non-small cell carcinoma (14–16). Similar results were observed in CD8+ TNIK deficient T cells, as these cells lost the capability to expand and have impeded memory formation, caused by TNIK-CD27-mediated activation of the Wnt signaling pathway (17).
In our previous research, we have shown that whole body deficiency of CD40, as well as cell-specific deficiency of CD40 in dendritic cells, adipocytes and macrophages, reduces atherosclerosis and especially plaque inflammation (18–21). We have also shown a pivotal role for the macrophage CD40-TRAF6-NF-kB pathway in driving atherosclerosis (22). Moreover, mixed chimeric Ldlr−/− mice whose B cells are deficient in CD40 show a reduction in atherosclerosis (23). As TNIK seems to be involved in the CD40-TRAF6 signaling complex, and as B cell CD40 activation seems to accelerate atherosclerosis, we here aimed to elucidate the role of TNIK signaling in B cells in atherosclerosis.
2. Materials and methods
2.1. Mice
ApoE−/−TNIKfl/fl (TNIKBWT) and ApoE−/−TNIKfl/fl CD19-cre (TNIKBKO) mice were generated and bred at the animal facility of the Amsterdam University Medical Centers, University of Amsterdam, the Netherlands. Eight-week-old female TNIKBWT and TNIKBKO littermates received a high cholesterol diet, containing 0.15% cholesterol (Arie Blok Animal Nutrition, Woerden, NL) for 10 weeks. The mice had access to food and water ad libitum and were housed according to institutional guidelines.
Mice were humanely killed by a combination of carbon dioxide and exsanguination as blood samples were collected via intracardiac puncture in ethylenediaminetetraacetic acid (EDTA)-coated syringes. Mice were perfused with phosphate buffered saline (PBS) to reduce any blood contamination and organs were harvested for analysis. All animal experiments were conducted at the Amsterdam University Medical Center, location Academic Medical Center, University of Amsterdam, NL and approved by the committee for animal welfare and the Dutch Centrale Commissie Proefdieren (AVD1180020171666).
2.2. TNIK knockout efficiency and NF-κB signaling
B cells were isolated by magnetic bead isolation with mouse Pan B Cell Isolation beads (Miltenyi Biotec, #130-095-813) using the manufacturers provided protocol. Isolated B cells were exposed to 10 µg/ml αCD40 (BioXCell, #BE0016-2) for 4 h. Proteins were measured by western blot by staining with αTNIK (Abcam, ab95887), αNF-κB p65 (cell Signaling, #3033) and αPhospho-NF-κB p65 (Cell signaling, #6956). αAlpha-tubulin (Sanbio, CLT9002) or αActin (Sigma-Aldrich A3853) were used as a loading control. TNIK and NF-κB p65 were visualized by staining with HRP conjugated Goat anti-Rabbit (Invitrogen, #32260) and Goat anti-Mouse (Invitrogen, #32330). Phospho-NF-κB p65 and alpha-tubulin were staining with IRDye 800CW conjugated Goat anti-Mouse (Odyssey, #926-32210) and Goat anti-Rabbit IRDye 680RD (Odyssey, #926-68071). Data was analyzed using FiJi.
2.3. Histology
Isolated aortic arches were fixed in 1% paraformaldehyde and embedded in paraffin. To analyze plaque size in the aortic arch, the arch was cut in 4 µm sections, where every 5th section was stained with hematoxylin and eosin (H&E; Merck, Kenilworth, USA and Klinipath/VWR International, Radnor, USA).
Plaque collagen content was visualized by picro Sirius Red staining (Sigma-Aldrich, Saint Louis, Missouri, USA). Immunohistochemical staining for alpha smooth muscle actin (α-SMA; 1:3,000; Sigma-Aldrich), macrophages (Mac-3; 1:100, BD Pharmingen) and CD3+ T cells (CD3, 1:100, AbD Serotec) were applied and counterstaining was performed with hematoxylin.
Images were taken using a Leica CRT6 with LAS X software and analyzed using Adobe Photoshop (2021), where positively stained area (for Sirius Red, Mac-3 and α-SMA) were quantified using color threshold measurements while cells positive for CD3 were counted by a researcher blinded for the experimental conditions. Necrotic regions were defined as plaque regions negative for cells or stromal tissue.
2.4. Plasma lipid levels
Blood was isolated and spun down (2,100 rpm, 10 min at 4°C), and plasma was collected. Total cholesterol and triglycerides were measured by standard enzymatic methods according to manufacturer's instructions (CHOD and GPO BIOLABO).
2.5. Immunoglobulins
Blood was obtained via intracardial puncture and spun down (2,100 rpm, 10 min at 4°C). Total IgM, IgA and IgG concentrations of plasma were detected using Enzyme-Linked Immuno Sorbent Assay (ELISA), IgG, IgA and IgM (total) mouse uncoated ELISA kits (ThermoFisher). IgM and IgG antibodies specific to CuOx-LDL and MDA-LDL and reactivity of the AB1-2 antibody (against the T15/E06 anti-idiotype) were determined as described previously (24).
2.6. In vitro generation of IgG and IgM isotype switched B cells
Naive B cells were isolated from Peyer's patches and spleen using magnetic CD43+ bead isolation (Miltenyi Biotec, #130-049-801) following the manufacturer's provided protocol. To generate IgG producing plasma cells, naive B cells were exposed to 50 µg/ml LPS (InvivoGen, tlrl-pb5lps) and 10 ng/ml IL-4 (Peprotech, #214-14) for 3 days. To generate IgA producing plasma cells, naive B cells were exposed to 20 μg/ml αIgD (ThermoFisher scientific, 16-5924), 5 ng/ml IL-4 (Peprotech, #214-14), 1 ng/ml IL-5 (R&D systems, 405-ML), 10 ng/ml TGF-β (R&D systems, 7666-MB) and 20 μg/ml αCD40 (BioXcell, BE0016-2). Cells were stained in FACS buffer (0.5% bovine serum albumin, 5 mM EDTA in PBS; pH 7.4) with the following antibodies: αCD3 (1:100, APC, Biolegend, #100312), αCD11b (1:100, APC, BD Biosciences, #553312), αCD45 (1:100, APC-Cy7, Biolegend, #103115), αIgA (1:100, PE, ThermoFisher scientific, #12-4201), αIgG (1:200, FITC, Biolegend, #406001), αIgM (1:1,000, PE-Cy7, ThermoFisher scientific, #25-5790).
2.7. Flow cytometry
Single cell suspensions of lymph nodes, Peyer's patches and spleens were prepared by crushing the tissue through a 70 µm cell strainer (Corning). Erythrocytes in blood and spleen were removed by incubation with hypotonic lysis buffer (HLB) (160 mM ammonium chloride, 10 mM sodium bicarbonate, 1.3 mM EDTA; pH 7.4). Cells were washed with PBS and resuspended and stained in FACS buffer. Gating strategy can be found in Supplementary Methods.
To identify pre-B cells and discriminate between follicular and marginal zone cells, the following antibodies were used: αB220 (1:200, APC-eFluor780, ThermoFisher Scientific, #47-0452), αCD16/CD32 (1:1,000, Biolegend, #101330), αCD19 (1:200, PE, ThermoFisher Scientific, #12-0193), αCD21 (1:100, BV421, BD Biosciences, #562756), αCD23 (1:100, BV510, BD biosciences, #563200), αCD38 (1:100, FITC, BD Biosciences, #558813), αCD93 (1:100, APC, ThermoFisher scientific, #17-5892), αIgM (1:1600, PE-Cy7, ThermoFisher scientific, #25-5790), Prior to analysis 7-AAD (final concentration of 1 μg/ml, ThermoFisher scientific, A1310) was added to exclude dead cells.
To identify germinal center and plasma cells, the following antibodies were used: αB220 (1:200, APC-eFluor780, ThermoFisher Scientific, #47-0452), αCD16/CD32 (1:1,000, Biolegend, #101330), αCD19 (1:200, PE, ThermoFisher Scientific, #12-0193), αCD23 (1:100, BV510, BD biosciences, #563200), αCD95 (1:100, AF647, BD biosciences, #563647), αCD138 (1:100, BV421, Biolegend, #562610), αGL7 (1:100, AF488, ThermoFisher scientific, #53-5902), αIgM (1:1,600, PE-Cy7, ThermoFisher scientific, #25-5790), Prior to analysis 7-AAD (final concentration of 1μg/ml, ThermoFisher scientific, A1310) was added to exclude dead cells.
Identification of T cell naive and memory populations, the following antibodies were used: αCD3 (1:200, APC-Cy7, Biolegend, #100222), αCD4 (1:1,000, BV650, Biolegend, #100469), αCD8 (1:400, BV605, Biolegend, #100744), αCD16/CD32 (1:1,000, Biolegend, #101330), αCD44 (1:800, FITC, Biolegend, #103006), αCD62l (1:1,000, PE-Cy7, Biolegend, 104418). Prior to analysis 7-AAD (final concentration of 1μg/ml, ThermoFisher scientific, A1310) was added to exclude dead cells.
Follicular T cells and dendritic cells, were identified using the following antibodies: αCD3 (1:300, FITC, Biolegend, #100306), αCD4 (1:1,000, APC, Biolegend, #100516), αCD11c (1:100, BV421, ThermoFisher scientific, #48-0114), αCD16/CD32 (1:1,000, Biolegend, #101330), αCD23 (1:100, BV510, BD biosciences, #563200), αCXCR5 (1:100, PE-Cy7, Biolegend, 145516), αPD-1 (1:100, PE, Biolegend, 109104). Prior to analysis 7-AAD (final concentration of 1μg/ml, ThermoFisher scientific, A1310) was added to exclude dead cells.
Populations from the myeloid lineage and their activation status was identified using the following antibodies: αCD11b (1:200, BV711, Biolegend, #101241), αCD11c (1:100, PE-Cy7, ThermoFisher scientific, #25-0114), αCD16/CD32 (1:1,000, Biolegend, #101330), αCD40 (1:100, PE, Biolegend, #124609), αCD45 (1:100, APC-Cy7, Biolegend, #103115), αCD86 (1:100, BV650, Biolegend, #105035), αLy6C (1:800, AF647, Biolegend, #128010), αLy6G (1:200, FITC, ThermoFisher scientific, #11-5931), αMHC class II (1:200, BV510, Biolegend, #107635), αSiglec- F (1:100, BV421, BD Biosciences, #562681).
Bone marrow was isolated by gently scraping away the bone at the joint and flushing ice-cold PBS through. Samples were incubated 5 min on ice in HLB followed by a PBS washing step. Samples were resuspended in FACS buffer. To identify stem cell populations, samples were stained in FACS buffer with the following antibodies: αCD16/32 (1:50, BV711, Biolegend, #101337), αCD27 (1:100, BUV395, BD Biosciences, #740247), αCD34 (1:50, eFluor450, ThermoFisher scientific, #48-0341), αCD48 (1:100, APC-Cy7, Biolegend, #103432), αCD127 (1:50, PE-Cy7, ThermoFisher scientific #25-1271), αCD135 (1:100, APC, Biolegend, #135310), αCD150 (1:100, PerCP-eFluor710, ThermoFisher scientific, #46-1502), αc-kit (1:100, PE, Biolegend, #105808), αlineage cocktail (1:5, FITC, ThermoFisher scientific, #22-7770) and αSca-1 (1:100, V500, BD Biosciences, #561228). Prior to analysis DAPI (final concentration of 15 ng/ml, ThermoFisher scientific, D21490) was added to exclude dead cells.
To identify mature cells that returned to the bone marrow, samples were stained in FACS buffer with the following antibodies: αCD3 (1:200, APC, Biolegend, #100312), αCD4 (1:400, BV650, Biolegend, #100469), αCD8 (1:400, BV605, Biolegend, #100744), αCD16/CD32 (1:1,000, Biolegend, #101330), αCD19 (1:100, PerCP-Cy5.5, ThermoFisher scientific, #45-0193), αCD44 (1:300, FITC, Biolegend, #103006), αCD45 (1:100, APC-Cy7, Biolegend, #103115), αCD62l (1:800, PE-Cy7, Biolegend, 104418), αCD138 (1:200, BV421, Biolegend, #562610), αc-kit (1:100, PE, Biolegend, #105808). Prior to analysis DAPI (final concentration of 15 ng/ml, ThermoFisher scientific, D21490) was added to exclude dead cells.
Cells were measured on a LSRFortessa Cell Analyzer (BD Biosciences) or a Symphony A1 Cell analyzer (BD Biosciences) and analyzed using FCS Express software, version 7 (De Novo Software).
2.8. Statistics
Data are presented as mean ± standard deviation, unless stated otherwise. Statistical analyses were performed using GraphPad Prism 9.3.1 software. Data was analyzed with an unpaired t-test or unpaired non-parametric Mann-Whitney U test depending on data distribution. Two-way ANOVA was used where appropriate, and outliers were identified using Grubbs test (α = 0.05). *p ≤ 0.05, **p ≤ 0.01.
3. Results
3.1. B cell specific TNIK deficiency does not affect atherosclerosis
To determine the role of B cell TNIK in atherosclerosis, we generated ApoE−/−TNIKfl/fl (TNIKBWT) and ApoE−/−TNIKfl/fl CD19-cre (TNIKBKO) mice, which were fed a high cholesterol diet for 10 weeks. The mice had a knock-down efficiency of B-cell TNIK of ∼90% (Supplementary Figure S1).
Atherosclerotic plaque area in the aortic arch and its main branch points did not differ between TNIKBWT and TNIKBKO mice (Figure 1A). Similar results were found in the aortic root (Supplementary Figure S2). Apart from a trend towards a decrease in macrophage content in TNIKBKO mice (Figure 1B), other plaque composition features, including necrotic core content (Figure 1C), CD3+ T cell content (Figure 1D), α-SMA+ smooth muscle cell content (Figure 1E) and collagen content (Figure 1F), were not affected by B cell specific TNIK deficiency, as were plasma cholesterol and triglyceride levels (Figures 1G–H). These data indicate that B cell specific TNIK deletion does not affect atherosclerotic lesion development or composition.
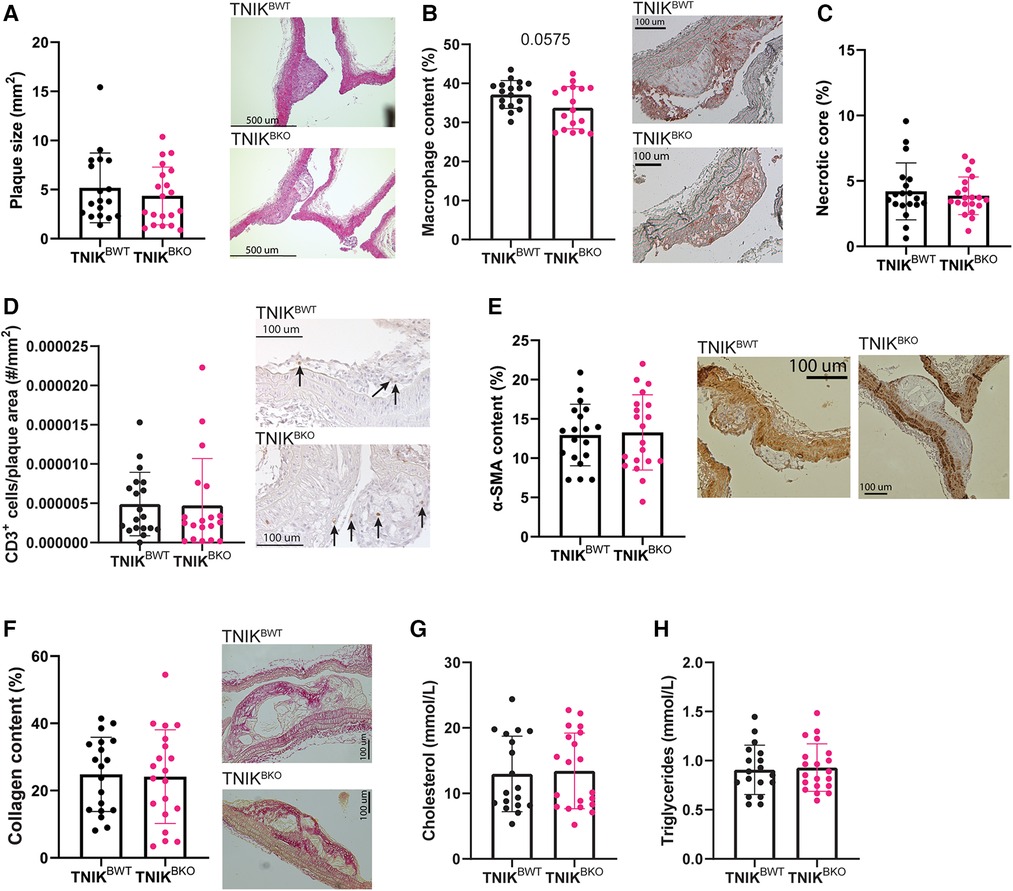
Figure 1. B cell TNIK deficiency does not alter plaque size, nor composition. Aortic arches of ApoE−/−TNIKflf (TNIKBWT) and ApoE−/−TNIKflflCD19-cre (TNIKBKO) mice were isolated and analyzed for (A) atherosclerotic plaque size (n = 19/20) and (B–F) plaque composition. Representative images are shown. (B) Necrotic core content as percentage of plaque area (n = 19/20), (C) Mac3+ macrophage content (n = 17/17), (D) CD3+ T cells in plaques (n = 19/19), (E) α-SMA+ smooth muscle cells (n = 19/20) and (F) collagen content (n = 19/20) did not differ between TNIKBWT and TNIKBKO mice. (G) Plasma cholesterol (n = 18/20) and (H) triglyceride (n = 19/20) concentrations were unaltered in TNIKBWT and TNIKBKO mice.
3.2. TNIK deletion does not alter B cell subsets and function
Next, we examined the effects of B cell TNIK deficiency on composition of B cell subsets in spleen (Figure 2) and blood (Supplementary Figures S3A–I). The total number of splenic B cells and of B1 and B2 B cells in spleens was similar in TNIKBKO and TNIKBWT mice (Figures 2A–E), as was the fraction of marginal zone (MZ), follicular (FO), germinal center (GC) B cells, and plasma cells (Figures 2F–I). Splenic transitional stage T-1, T-2 and T-3 B cells were also unaltered (Figures 2J–L). Moreover, the fraction of follicular helper T cells (Tfh), associated with germinal center cell transition, was similar in TNIKBWT and TNIKBKO mice (Figure 2M). Our analyses in blood phenocopied those from spleen (Supplementary Figures 3A–I).

Figure 2. Splenic B cell differentiation and subsets unaltered due to B cell specific TNIK deficiency in atherogenic mice. The differentiation and subsets of B cells are measured by flow cytometry in the spleen of TNIKBWT and TNIKBKO mice. (A) The total number of splenic B cells (CD19+B220+) are similar between TNIKBWT and TNIKBKO mice (n = 9/11). The (B) relative fraction of splenic B1 cells (CD19lowB220+) (n = 17/15), (C) total splenic B1 cell (n = 8/8), (D) relative number of B2 cells (CD19+B220+) (n = 19/17), (E) and total splenic B2 cells (n = 9/11)are unaffected in TNIKBKO compared to TNIKBWT mice. Further subdivision of B2 cells into (F) marginal zone (CD19+B220+CD21+CD23−) (n = 19/18), (G) follicular (CD19+B220+CD21lowCD23+) (n = 19/18) and (H) germinal center B cells (CD19+B220+CD95+GL7+) (n = 19/20), as well as (I) plasma cells (CD19−B220−CD238+) (n = 18/19) showed no differences between genotypes. Transitional stage (CD19+B220+CD93+) (J) T-1 (IgM+CD23−) (n = 19/18), (K) T-2 (IgM+CD23+) (n = 18/17), and (L) T-3 (IgMlowCD23+) (n = 19/17) B cells were similar between TNIKBWT and TNIKBKO mice. (M) Follicular helper T cells (CD3+CD4+CXCR5+PD-1+) are also unchanged between TNIKBWT and TNIKBKO mice (n = 19/19).
Total plasma IgM and IgG levels did not differ between TNIKBWT and TNIKBKO mice (Figures 3A–B). Levels of IgM and IgG antibodies directed against oxidation-specific epitopes (OSE), including anti-copper-oxide containing low density lipoprotein (CuOx-LDL), anti-malondialdehyde-modified LDL (MDA-LDL), anti-phosphocholine (PC-BSA) and E06/T15 idiotype (AB1-2) (Figure 3C) were also similar in both genotypes.
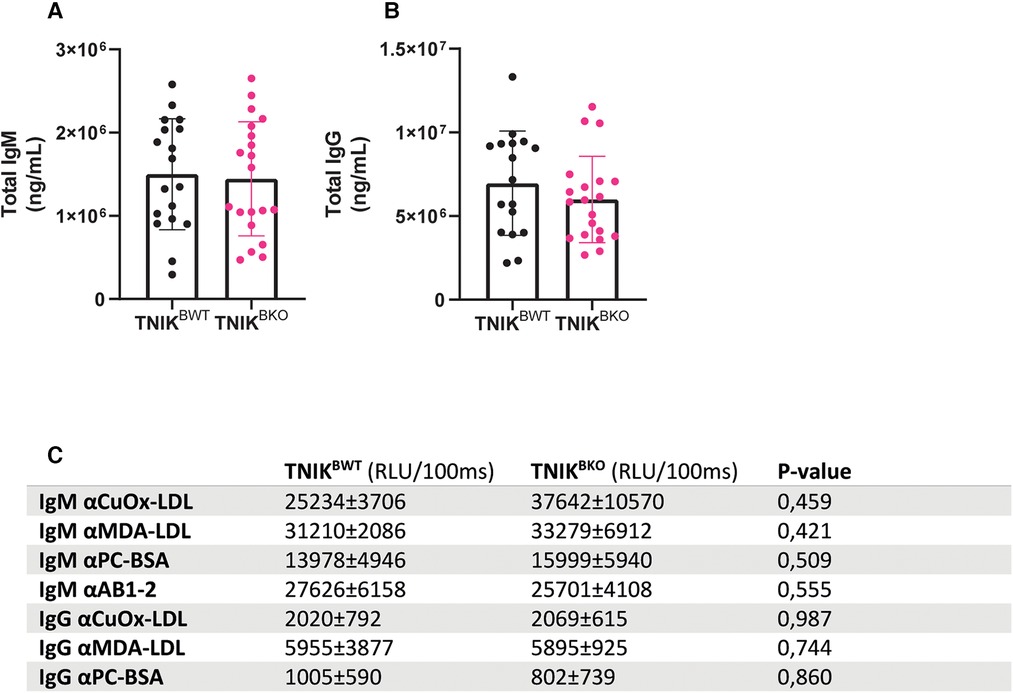
Figure 3. Plasma levels of IgM and IgG and anti-oxLDL immunoglobulin levels are unaffected due to B cell specific TNIK deficiency. Blood from female TNIKBWT and TNIKBKO mice, fed a high cholesterol diet for 10 weeks, was analyzed for plasma levels of total IgM and IgG and their lipid oxidation-specific epitopes (OSE). (A) Total plasma IgM (n = 18/20) and (B) IgG levels (n = 17/20) were unaffected in TNIKBKO compared to TNIKBWT. Concentrations of OSE specific (C) IgM and IgG were similar in TNIKBKO compared to TNIKBWT (n = 8/8). Data expressed as relative light units per 100 ms ± SD.
TNIK was identified as an essential part of the Wnt signaling pathway (12) and it was shown to also activate NF-κB via TRAF2, an important mediator in B cell signaling (25). Furthermore, it has been suggested that TNIK could signal via TRAF6 (11). Yet, when we activate CD40, no differences were observed in protein levels of NF-κB and phosphorylated NF-κB, nor in the ratio between phosphorylated NF-κB of total NF-κB in TNIKBKO mice (Supplementary Figures S4A–C).
3.3. Intestinal IgA is increased in B cell deficient TNIK atherosclerotic mice
Interestingly, TNIKBKO mice displayed a reduction in total plasma IgA levels (Figure 4A). IgA is an antibody-subtype involved in mucosal homeostasis, and is produced mainly in the gut. The intestinal Peyer's patches, small lymphoid aggregates, are known to be the main producers of gut IgA (26). However, in contrast to the observed reduction of plasma IgA, Peyer's patches in TNIKBKO mice contained an increased number of IgA+ B cells (Figure 4B). The numbers of IgM+ and IgG+ B cells did not differ (Figures 4C–D). This is in contrast to the spleen, where the number of IgA producing B cells was unchanged, suggesting a gut specific effect in the induction of IgA production in the intestine, but not spleen. In Peyer's patches, B cells isotype switch to IgA under the influence of transforming growth factor β (TGF-β) (27). And indeed, treatment of naive B cells isolated from TNIKBKOmice showed an increase isotype switching compared to B cells from TNIKBWT mice. (Figure 4E). These data suggest that TNIK deficient B cells in the gut exhibit enhanced TGF-β mediated IgA isotype switching. However, this TNIK-deficiency induced increase in mucosal IgA did not affect atherosclerosis in our model.
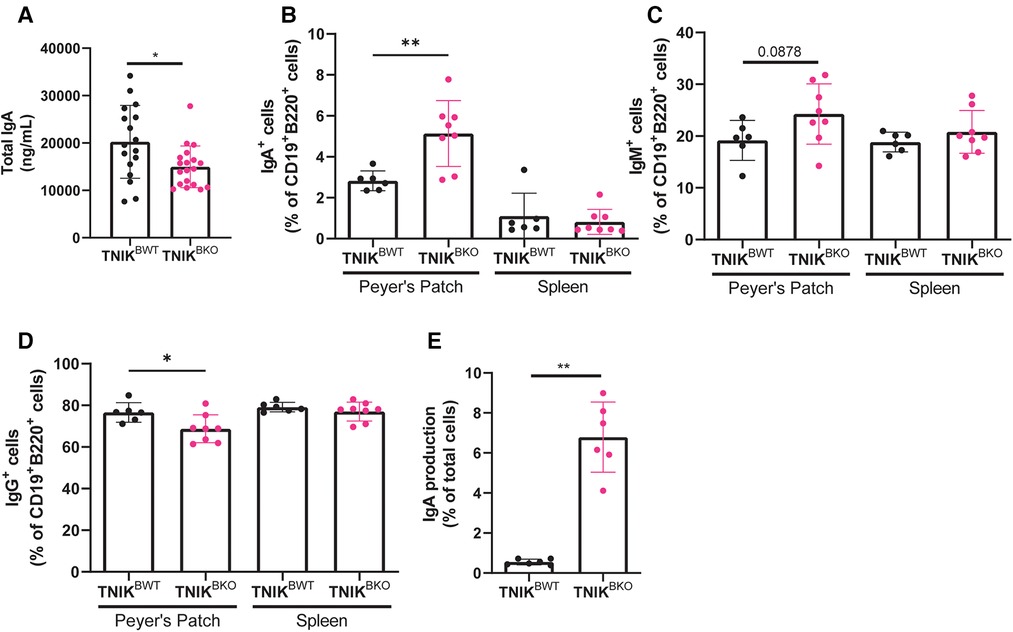
Figure 4. TNIK B cell deficiency increases IgA production. (A) Blood plasma was isolated and analyzed for IgA levels, which were decreased in TNIKBKO compared to TNIKBWT mice (n = 17/19). (B–D) Cells from intestinal Peyer's Patches and spleen were analyzed via flow cytometry for IgA+, IgM+ and IgG+ expressing cells (CD45+) (n = 6/8). (E) Splenic resting B cells (CD43−) were isolated and stimulated with αCD40, IL-4, IL-5, TGF-β and αIgD to differentiate into plasma cells, which were analyzed via flow cytometry for IgA expression (n = 6 per genotype).
3.4. B cell specific TNIK deletion does not affect other immune cell types or subsets
TNIKBKO mice displayed similar amounts of CD4+ T cells, CD8+ T cells, naive, effector and central memory CD4+ and CD8+ T cells in blood, spleen and lymph nodes (Figures 5A–D; Supplementary Figure 5). Furthermore, the myeloid compartment, containing neutrophils, dendritic cells, Ly6Clow and Ly6Chigh monocytes, and eosinophils, were not affected in spleen and blood by TNIK deficiency in B cells (Figures 5E–K; Supplementary Figure S6). In addition, stem cells and mature immune cells present in the bone marrow were unaffected by B-cell deficiency of TNIK (Supplementary Figure S7).
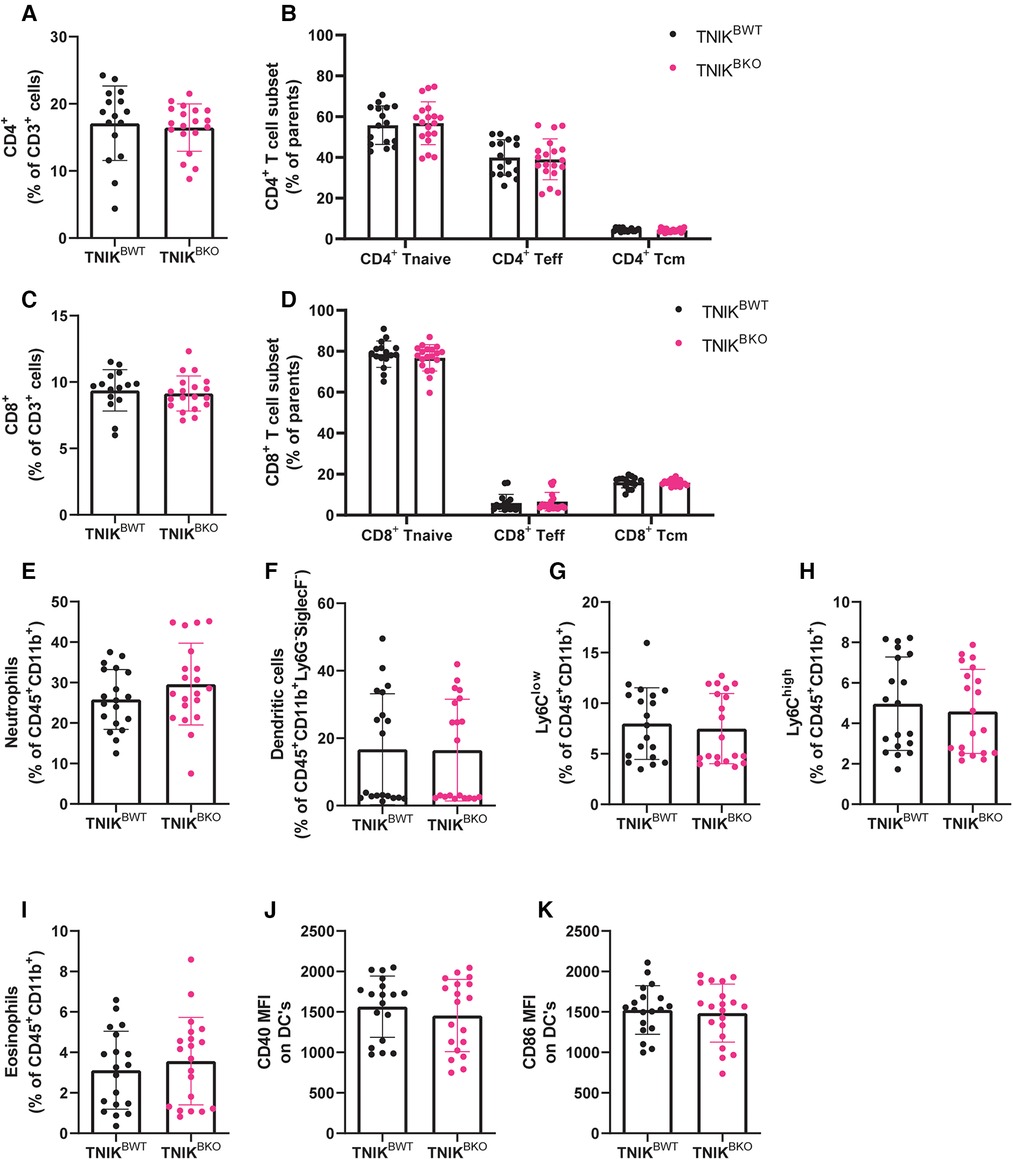
Figure 5. B cell TNIK deficiency does not affect T cell ratios or myeloid populations in the spleen. The CD4+ and CD8+ T cells from the spleens of TNIKBWT and TNIKBKO mice were analyzed via flow cytometry. (A) Total CD4+ T cells, alongside (B) their subsets, naive (CD44−CD62L+), effector (CD44+/−CD62L−) and central memory T cells (CD44+ CD62L+), are unaffected by B cell TNIK deficiency (n = 16/20). (C) Total CD8+ T cells and (D) its subsets, naive (CD44−CD62L+), effector (CD44+/−CD62L−) and central memory (CD44+ CD62L+), are unaltered in the TNIKBKO compared to TNIKBWT mice (n = 15/19). Splenic myeloid cells (CD45+ CD11b+) were also analyzed by flow cytometry. (E) Neutrophils (ly6G+SiglecF−), (F) dendritic cells (MHCII+CD11c+), (G) non-classical Ly6Clow monocytes, (H) classical Ly6Chigh monocytes and (I) eosinophils (Ly6GlowSiglecF+) were unaffected by genotype. Further activation status of dendritic cells by measuring (J) CD40 and (K) CD86 expression revealed no differences between TNIKBWT and TNIKBKO mice [(E,G–K) n = 19/20, and (F) n = 10/10].
4. Discussion
In the current paper, we investigated the role of B cell specific TRAF2- and NCK-interacting kinase (TNIK) in atherosclerosis. We conclude that TNIK signaling in B cells does not affect atherogenesis, nor did it affect immunological pathways relevant for atherosclerosis.
There is limited research on the role of TNIK in B cells (11). TNIK has been shown to be a crucial partner in the signaling pathway of latent membrane protein 1 (LMP1), an Epstein-Barr virus (EBV) oncogene with many similarities to CD40 (28). Here TNIK was shown to bind TRAF6 and TRAF2 (11). Thereby activating the nuclear factor kappa B (NF-κB) signaling pathway inducing proliferation and activation of B cells (29). Furthermore, the authors suggest that TNIK activation via LMP1 might not induce Wnt signaling. However, in our TNIKBKO mice, no reduction in NF-κB phosphorylation was measured after activating CD40 signaling in B cells, despite a 90% reduction in B-cell specific TNIK expression. One limitation of the study was the use of HEK293 cells as a model for CD40 and LMP1 signaling (11). This suggests that while TNIK may be associated with TRAF6 when co-transfected with LMP1 in HEK293 cells, this pathway may not be functional in primary B cells. Furthermore, these outcomes highlight functional differences between LMP1 and CD40. While it was shown that LMP1 and CD40 have many similarities in their signaling pathways, LMP1's intracellular signaling domain differs from CD40 (30). Even though both mainly signal via TRAFs, they bind these with different affinities. Furthermore, LMP1 but not CD40 engages tumor necrosis factor receptor-associated death domain (TRADD) for its signaling (31). While CD40 signaling promotes germinal center formation, LMP1 inhibits this, instead promoting extrafollicular B cell activation (30). Another key difference between CD40 and LMP1 is the dependence on (TNF)-receptor 1–associated death domain protein (TRADD) for its TRAF6 signaling. While CD40 can directly bind TRAF6, LMP1 cannot (28, 32), and is dependent on TRADD (33, 34). Further, LMP1 and CD40 downstream signaling differs with regard to the functions of the kinases TAK1, IKK2 and TPL2 (35). This is contrasted by CD40, which does not bind TRADD. This suggest that results found in EBV transformed B cells expressing LMP1 are not directly translatable to CD40 signaling in primary B cells.
Interestingly, when we analyzed atherosclerosis in B cell TNIK deficient atherosclerotic mice, we did not observe any effects on plaque size or composition, with the exception of a trend towards a decrease in plaque macrophage content. We have no solid explanation for this, but the increase of intestinal IgA may have altered the microbiome and it can be speculated that this may have shifted the immune system towards a slightly more immunomodulatory state, thereby reducing leukocyte recruitment. Furthermore, no B cell subsets differences were detected, nor any indirect effects on other cell types. This is unexpected, as TNIK plays a role key role in Wnt signaling, a pathway with a role in several immune related diseases such as Rheumatoid Arthritis and Ankylosing Spondylitis (36). Moreover, this pathways has a known role in atherogenesis (37). In acute lymphocytic choriomeningitis virus (LCMV), deletion of TNIK on CD8+ T cells via the UBC-Cre tamoxifen system impaired the formation of memory cells due to defective signaling in the Wnt pathway (17). Furthermore, TNIK deficient effector CD8+ T cells displayed increased proliferation, apoptosis and reprogramming towards a higher glycolytic metabolic profile (17). In colorectal cancer, over 90% of tumors exhibit mutations in the Wnt/β-catenin signaling pathway. TNIK was identified as a promising target to inhibit Wnt-signaling due to its binding to TCF-4/β-catenin complex (15). In colorectal cancer, it was found that TNIK is necessary for tumor-initiating function of stem cells in the intestines, and in acute myeloid leukemia blasts TNIK reduces survival rate due to the activation of Wnt signaling (38, 39). As B cells are regulated by the Wnt signaling pathway to induce proliferation (40), we anticipated similar effects within our B cell specific mouse model. However, we could not identify any differences in B cell differentiation and subset distribution. Furthermore, no secondary changes were observed in T cell or myeloid population in our atherosclerotic model, indicating that B cell TNIK deficiency does not indirectly affect other immune cells.
Interestingly, we found a strong increase in IgA producing B cells in the small intestine's Peyer's patches. Something which is in contrast to the observed decrease in blood IgA while no difference in splenic IgA producing B cells was observed. While significant redundancy exists, in vivo isotype switching to IgA is mostly controlled by transforming growth factor-β (TGF-β) (41, 42). One key difference between the splenic environment and that on the Payer's patches is the local production of TGF-β by stimulation of immune cells by the microbiome (43). The potential role of TNIK on TGF-β signaling was highlighted by in vitro experiments where naïve splenic B cells were isolated and found to more efficiently isotype switch to IgA production under the influence of TGF-β. It has been shown that the kinase family where TNIK belongs to can interact with, and inhibit the binding of Smad proteins to the TGF-β receptor, thereby inhibiting TGF-β signaling (44).
There are some limitations to our current experimental setup. First, we only used female mice, who develop atherosclerosis faster than their male counterparts, and of course, have different estrogen levels than male mice. Second, we used mice that were fed a high cholesterol diet, and did not do a normal chow diet group, which would have developed atherosclerosis more slowly. We have only investigated relatively advanced atheroma's and did not elucidate the role of B cell TNIK deficiency on early plaque formation, yet we do feel that the effects of B cell TNIK on atherogenesis are minor.
Although our data do not show any effect of B cell TNIK deficiency on atherosclerosis, B cell TNIK may play an important role in IgA mediated diseases. The majority of IgA is produced by B cells in Peyer's patches (45). The fact that absence of TNIK in B cells increases IgA production in Peyer's patches suggests that inhibition of TNIK could increase microbiota diversity and metabolism of gut microbiota (46). Increases in intestinal IgA production are correlated with IgA-mediated diseases, including IgA nephropathies (47) and inflammatory bowel diseases (48). Activating B cell TNIK could be a potential target to lower IgA levels and ameliorate IgA-associated diseases.
In conclusion, TNIK signaling in B cells does not play a role in the pathogenesis of atherosclerosis. However, our data suggest that B-cell TNIK signaling may be relevant for inhibiting TGF-β mediated IgA switching of B cells.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Centrale Commissie Proefdieren, Nederland. Animal Welfare Committee Amsterdam UMC, location AMC.
Author contributions
BvO, PK, LB and EL designed the experiments. BvO, PK, MdT, LB, CvT, WV, EdJ, AK, CvR, CB, MR, LB executed the experiments and/or analyzed the data. MdW provided input for the manuscript. BvO, LB and EL wrote and revised the manuscript. EL received funding for this project. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the European Research Council (ERC consolidator grant to EL).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1171764/full#supplementary-material.
Supplementary Figure 1
B cell specific TNIK deficiency. TNIK deficiency of ApoE−/−TNIKfl/fl (TNIKBWT) and ApoE−/−TNIKfl/flCD19-cre (TNIKBKO) mice was validated in isolated splenic B cells (n = 2 or 3/genotype of 4 individual experiments).
Supplementary Figure 2
B cell TNIK deficiency does not affect aortic root plaque size. Aortic root of TNIKBWT and TNIKBKO mice were isolated and examined for the atherosclerotic plaque size. Area under the curve of the plaque volume throughout the atherosclerotic plaque formation did not differ between genotypes (n = 19/20).
Supplementary Figure 3
Blood B cell differentiation and subsets unaffected in TNIKBKO compared to TNIKBWT mice. The differentiation and subsets of B cells are measured by flow cytometry in blood from TNIKBWT and TNIKBKO mice. (A) The total number of leukocytes (CD45+) per µl blood was unaffected between TNIKBWT and TNIKBKO mice (n = 9/10). (B) Transitional stage T-1 (CD19+B220+IgM+CD23−) (n = 19/19) and (C) T-2 (CD19+B220+IgM+CD23−) (n = 19/20) are unaffected in TNIKBKO compared to TNIKBWT mice. The (D) number of B1 cell per μl blood (CD19lowB220+) (n = 9/8), (E) fraction of B1 cells (n = 17/19), (F) number of B2 cells (CD19+B220+) per μl blood, (n = 9/10), (G) fraction of B2 cell (n = 19/20) are similar between the genotypes, as are the (H) marginal zone (CD19+B220+CD21+CD23−) (n = 19/20) and (I) follicular B cells (CD19+B220+CD21lowCD23+) (n = 19/20).
Supplementary Figure 4
B cell specific TNIK deficiency does not affect canonical NF-κB pathway by activation of CD40. (A) Expression of the p65 subunit from NF-κB expression the (B) phosphorylated P65 expression was measured in TNIKBWT and TNIKBKO isolated splenic B cells (n = 5/6). (C) Ratio of phosphorylated NF-κB over NF-κB measured in TNIKBWT and TNIKBKO isolated splenic B cells.
Supplementary Figure 5
T cell subsets from lymph nodes and blood are not altered due to B cell TNIK deficiency. (A) Fraction of CD4+ T cells in lymph nodes (gated from total cells) (n = 19/20) and (B) CD4+ naive (CD44−CD62L+), effector (CD44+/−CD62L−) and central memory T cells (CD44+CD62L+) (n = 19/20), are unaffected by B cell TNIK deficiency (n = 16/20). (C) Fraction of CD8+ T cells in lymph nodes (gated from total cells) (n = 19/20) and (D) CD8+ naive (CD44−CD62L+), effector (CD44+/−CD62L−) and central memory T cells (CD44+CD62L+) (n = 19/20), are unaffected by B cell TNIK deficiency (n = 16/20). (E) Fraction of CD4+ T cells in blood (gated from total cells) (n = 19/20) and (F) CD4+ naive (CD44−CD62L+), effector (CD44+/−CD62L−) and central memory T cells (CD44+CD62L+) (n = 19/20), are unaffected by B cell TNIK deficiency (n = 16/20). (G) Fraction of CD8+ T cells in blood (gated from total cells) (n = 19/20) and (H) CD8+ naive (CD44−CD62L+), effector (CD44+/−CD62L−) and central memory T cells (CD44+CD62L+) (n = 19/20), are unaffected by B cell TNIK deficiency (n = 16/20).
Supplementary Figure 6
Myeloid immune cells in the blood largely unaffected by TNIK B cell deficiency. Blood myeloid (CD45+CD11b+) cells were analyzed by flow cytometry from TNIKBWT and TNIKBKO mice. (A) Neutrophils (ly6G+SiglecF−) are unaffected in TNIKBKO compared to TNIKBWT mice (n = 9/10). (B) Eosinophils (Ly6GlowSiglecF+) (n = 9/10), (C) non-classical Ly6Clow monocytes (n = 8/10), (D) classical Ly6Chigh monocytes (n = 9/10), (E) dendritic cells (MHCII+CD11c+), (n = 9/10) are unaffected by genotype. Further activation status of dendritic cells by measuring (G) CD40 and (H) CD86 expression revealed no differences between TNIKBWT and TNIKBKO mice (n = 9/10).
Supplementary Figure 7
Bone marrow differentiation unaffected due to B cell TNIK deficiency. Flow cytometric identification of bone marrow hematopoietic stem cells from TNIKBWT and TNIKBKO mice. (A) Total number of hematopoietic stem cells (Lineage−Sca-1+c-Kit+) showed a trend towards a decrease in TNIKBKO mice (n = 19/20). (B) Characterization of total number of long term stem cell (LT-SC: Lineage−Sca-1+c-Kit+CD150+CD48−), short term stem cell (ST-SC: Lineage−Sca-1+c-Kit+CD150−CD48−) showed no differences between genotypes (n = 19/20). Furthermore, no difference in multipotent progenitor cells (MPP: Sca-1+c-Kit+) or MPP subpopulations (MPP1a: CD48−CD150−135+, MPP1b: CD48−CD150−135+, MPP2: CD48+CD150+, MPP3:CD48+CD150−CD135+, MPP4:CD48+CD150−CD135+) was observed (n = 19/20). (C) Common lymphoid progenitors (CLP: Lineage−Sca-1lowc-Kit+CD48+CD16/CD32−) – early common lymphoid progenitor (CD127+CD135+), CLP (CD127+CD135−CD27−) and late CLP (CD127+CD135−CD27+) – did not reveal differences between TNIKBWT and TNIKBKO mice (n = 19/20). (D) Myeloid progenitors (Sca-1−cKit+) – granulocyte-monocyte progenitor (GMP: (CD16/32+), common myeloid progenitor (CMP: CD16/32low) and megakaryocyte-erythrocyte progenitor (MEP: CD16/32−) – also do not differ between TNIKBWT and TNIKBKO mice (n = 19/20). (E) Number of mature immune (CD45+) cells, Plasma B cells (CD138+), CD4+ and CD8+ T cells (CD3+) in the bone marrow are not affected by TNIK B cell deficiency (n = 9/10).
Supplementary Figure 8
Bone marrow differentiation unaffected due to B cell TNIK deficiency. Flow cytometric identification of bone marrow hematopoietic stem cells from TNIKBWT and TNIKBKO mice. (A) Total number of hematopoietic stem cells (Lineage−Sca-1+c-Kit+) showed a trend towards a decrease in TNIKBKO mice (n = 19/20). (B) Characterization of total number of long term stem cell (LT-SC: Lineage−Sca-1+c-Kit+CD150+CD48−), short term stem cell (ST-SC: Lineage−Sca-1+c-Kit+CD150−CD48−) showed no differences between genotypes (n = 19/20). Furthermore, no difference in multipotent progenitor cells (MPP: Sca-1+c-Kit+) or MPP subpopulations (MPP1a: CD48−CD150−135+, MPP1b: CD48−CD150−135+, MPP2: CD48+CD150+, MPP3:CD48+CD150−CD135+, MPP4:CD48+CD150−CD135+) was observed (n = 19/20). (C) Common lymphoid progenitors (CLP: Lineage−Sca-1lowc-Kit+CD48+CD16/CD32−) – early common lymphoid progenitor (CD127+CD135+), CLP (CD127+CD135−CD27−) and late CLP (CD127+CD135−CD27+) – did not reveal differences between TNIKBWT and TNIKBKO mice (n = 19/20). (D) Myeloid progenitors (Sca-1−cKit+) – granulocyte-monocyte progenitor (GMP: (CD16/32+), common myeloid progenitor (CMP: CD16/32low) and megakaryocyte-erythrocyte progenitor (MEP: CD16/32−) – also do not differ between TNIKBWT and TNIKBKO mice (n = 19/20). (E) Number of mature immune (CD45+) cells, Plasma B cells (CD138+), CD4+ and CD8+ T cells (CD3+) in the bone marrow are not affected by TNIK B cell deficiency (n = 9/10).
Supplementary Method 1
Gating strategy for flow cytometry on spleen, LN and blood samples. First three gating steps seen in figure (A) are applied to all other gating strategies. (A) Gating strategy for germinal center plasma cells. (B) Gating strategy for B cell differentiation subsets. (C) Gating strategy for CD4+ and CD8+ T cell subsets. (D) Gating strategy for myeloid cell populations.
Supplementary Method 2
Gating strategy for flow cytometry on Peyer’s patches and spleen. (A) Gating strategy for IgA+ cells in Peyer’s Patches and spleen. (B) Gating strategy for IgM+ and IgG+ plasma cells cultured in vitro.
Supplementary Method 3
Gating strategy for stem cells. Gating strategy for stem cells from bone marrow samples.
References
1. Centers for Disease Control and Prevention. Heart Disease in the United States. (2020). Available at: https://www.cdc.gov/heartdisease/facts.htm
2. de Boer A, van Dis I, Wimmers RH, Vaartjes I, Bots M. Hart- en vaatziekten in Nederland 2020. (2020). Available at: https://www.hartstichting.nl/getmedia/74a18eeb-e602-41bf-8a2c-c3932108e89b/cijfers-hart-vaatziekten-nederland-2020.pdf
3. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
4. Tabas I, Lichtman AH. Monocyte-Macrophages and T cells in atherosclerosis. Immunity. (2017) 47:621–34. doi: 10.1016/j.immuni.2017.09.008
5. Ma SD, Mussbacher M, Galkina EV. Functional role of B cells in atherosclerosis. Cells. (2021) 10(2):1–29. doi: 10.3390/cells10020270
6. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. (2016) 16(8):485–97. doi: 10.1038/nri.2016.63
7. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:180–96. doi: 10.1038/s41569-018-0106-9
8. Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. (2010) 207(8):1579–87. doi: 10.1084/jem.20100155
9. Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol. (2010) 185(7):4410–9. doi: 10.4049/jimmunol.1000033
10. Fu CA, Shen M, Huang BCB, Lasaga J, Payan DG, Luo Y. TNIK, a novel member of the germinal center kinase family that activates the c-jun N-terminal kinase pathway and regulates the cytoskeleton. J Biol Chem. (1999) 274(43):30729–37. doi: 10.1074/jbc.274.43.30729
11. Shkoda A, Town JA, Griese J, Romio M, Sarioglu H, Knöfel T, et al. The germinal center kinase TNIK is required for canonical NF-κB and JNK signaling in B-cells by the EBV oncoprotein LMP1 and the CD40 receptor. PLoS Biol. (2012) 10:e1001376. doi: 10.1371/journal.pbio.1001376
12. Mahmoudi T, Li VSW, Ng SS, Taouatas N, Vries RGJ, Mohammed S, et al. The kinase TNIK is an essential activator of wnt target genes. EMBO J. (2009) 28(21):3329–40. doi: 10.1038/emboj.2009.285
13. Masuda M, Sawa M, Yamada T. Therapeutic targets in the wnt signaling pathway: feasibility of targeting TNIK in colorectal cancer. Pharmacol Ther. (2015) 156:1–9. doi: 10.1016/j.pharmthera.2015.10.009
14. Kim J, Oh J, Peterson HM, Carlson JCT, Pittet MJ, Weissleder R, et al. TNIK Inhibition has dual synergistic effects on tumor and associated immune cells. Adv Biol. (2022) 6:2200030. doi: 10.1002/adbi.202200030
15. Yamada T, Masuda M. Emergence of TNIK inhibitors in cancer therapeutics. Cancer Sci. (2017) 108(5):818–23. doi: 10.1111/cas.13203
16. Puri N, Khramtsov A, Ahmed S, Nallasura V, Hetzel JT, Jagadeeswaran R, Karczmar G, Salgia R. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. (2007) 67:3529–34. doi: 10.1158/0008-5472.CAN-06-4416
17. Jaeger-Ruckstuhl CA, Hinterbrandner M, Höpner S, Correnti CE, Lüthi U, Friedli O, et al. TNIK Signaling imprints CD8+T cell memory formation early after priming. Nat Commun. (2020) 11:1632. doi: 10.1038/s41467-020-15413-7
18. Lutgens E, Lievens D, Beckers L, Wijnands E, Soehnlein O, Zernecke A, et al. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. (2010) 207(2):391–404. doi: 10.1084/jem.20091293
19. Lacy M, Burger C, Shami A, Ahmadsei M, Winkels H, Nitz K, et al. Cell-specific and divergent roles of the CD40l-CD40 axis in atherosclerotic vascular disease. Nat Commun. (2021) 12:3754. doi: 10.1038/s41467-021-23909-z
20. Bosmans LA, van Tiel CM, Aarts SABM, Willemsen L, Baardman J, van Os BW, et al. Myeloid CD40 deficiency reduces atherosclerosis by impairing macrophages’ transition into a pro-inflammatory state. Cardiovasc Res. (2022):cvac084. doi: 10.1093/cvr/cvac084. [Epub ahead of print]35587037
21. Reiche ME, Poels K, Bosmans LA, Vos WG, van Tiel CM, Gijbels MJJ, et al. Adipocytes control haematopoiesis and inflammation through CD40 signaling. Haematologica. (2020). doi: 10.3324/HAEMATOL.2022.281482. [Epub ahead of print]
22. Seijkens TTP, van Tiel C, Kusters P, Atzler D, Soehnlein O, Zarzycka B, et al. Targeting CD40-induced TRAF6 signaling in macrophages reduces atherosclerosis. J Am Coll Cardiol. (2018) 71(5):527–42. doi: 10.1016/j.jacc.2017.11.055
23. Tay C, Liu YH, Kanellakis P, Kallies A, Li Y, Cao A, et al. Follicular B cells promote atherosclerosis via T cell-mediated differentiation into plasma cells and secreting pathogenic immunoglobulin G. Arterioscler Thromb Vasc Biol. (2018) 38(5):e71–84. doi: 10.1161/ATVBAHA.117.310678
24. Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. (2009) 119(5):1335–49. doi: 10.1172/JCI36800
25. Etemadi N, Chopin M, Anderton H, Tanzer MC, Rickard JA, Abeysekera W, et al. TRAF2 Regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of sphingosine kinase. eLife. (2015) 4:1–27. doi: 10.7554/eLife.10592
26. Pabst O, Slack E. Iga and the intestinal microbiota: the importance of being specific. Mucosal Immunol. (2020) 13(1):12–21. doi: 10.1038/s41385-019-0227-4
27. Borsutzky S, Cazac BB, Roes J, Guzmán CA. TGF-beta receptor signaling is critical for mucosal IgA responses. J Immunol. (2004) 173(5):3305–9. doi: 10.4049/jimmunol.173.5.3305
28. Lam N, Sugden B. CD40 And its viral mimic, LMP1: similar means to different ends. Cell Signal. (2003) 15(1):9–16. doi: 10.1016/S0898-6568(02)00083-9
29. Gerondakis S, Siebenlist U. Roles of the NF-κB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol. (2010) 2:a000182–a000182. doi: 10.1101/cshperspect.a000182
30. Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N, et al. Mimicry of CD40 signals by epstein-barr virus LMP1 in B lymphocyte responses. Science. (1999) 286(5438):300–3. doi: 10.1126/science.286.5438.300
31. Izumi KM, Kieff ED. The epstein-barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc Natl Acad Sci U S A. (1997) 94(23):12592–7. doi: 10.1073/pnas.94.23.12592
32. Rastelli J, Hömig-Hölzel C, Seagal J, Müller W, Hermann AC, Rajewsky K, et al. LMP1 Signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood. (2008) 111(3):1448–55. doi: 10.1182/blood-2007-10-117655
33. Schneider F, Neugebauer J, Griese J, Liefold N, Kutz H, Briseño C, et al. The viral oncoprotein LMP1 exploits TRADD for signaling by masking its apoptotic activity. PLoS Biol. (2008) 6(1):0086–98. doi: 10.1371/journal.pbio.0060008
34. Kieser A. Signal transduction by the epstein-barr virus oncogene latent membrane protein 1 (LMP1). Signal Transduct. (2007) 7(1):20–33. doi: 10.1002/sita.200600116
35. Voigt S, Sterz KR, Giehler F, Mohr AW, Wilson JB, Moosmann A, et al. A central role of IKK2 and TPL2 in JNK activation and viral B-cell transformation. Nat Commun. (2020) 11(1):1–16. doi: 10.1038/s41467-020-14502-x
36. Shi J, Chi S, Xue J, Yang J, Li F, Liu X. Emerging role and therapeutic implication of wnt signaling pathways in autoimmune diseases. J Immunol Res. (2016) 2016:1–18. doi: 10.1155/2016/9392132
37. Boucher P, Matz RL, Terrand J. Atherosclerosis: gone with the wnt? Atherosclerosis. (2020) 301:15–22. doi: 10.1016/j.atherosclerosis.2020.03.024
38. Masuda M, Uno Y, Ohbayashi N, Ohata H, Mimata A, Kukimoto-Niino M, et al. TNIK Inhibition abrogates colorectal cancer stemness. Nat Commun. (2016) 7:12586. doi: 10.1038/ncomms12586
39. Riether C, Schürch CM, Bührer ED, Hinterbrandner M, Huguenin AL, Hoepner S, et al. CD70/CD27 Signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med. (2017) 214(2):359–80. doi: 10.1084/jem.20152008
40. Yu Q, Quinn WJ, Salay T, Crowley JE, Cancro MP, Sen JM. Role of beta-catenin in B cell development and function. J Immunol. (2008) 181(6):3777–83. doi: 10.4049/jimmunol.181.6.3777
41. Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, Cyster JG. Iga production requires B cell interaction with subepithelial dendritic cells in peyer’s patches. Science. (2016) 352(6287):aaf4822. doi: 10.1126/science.aaf4822
42. Cazac BB, Rgen Roes J. TGF-Receptor Controls B cell responsiveness and induction of IgA in vivo given the complexity of the TGF-1/phenotype, how-ever, it has been difficult to establish the effects of TGF-on individual leukocyte subsets in vivo. The B cell-specific function o. Immunity. (2000) 13:443–51. doi: 10.1016/S1074-7613(00)00044-3
43. Bauché D, Marie JC. Transforming growth factor β: a master regulator of the gut microbiota and immune cell interactions. Clin Transl Immunol. (2017) 6:e136. doi: 10.1038/cti.2017.9
44. Kaneko S, Chena X, Lua P, Yao X, Wright TG, Rajurkar M, et al. Smad inhibition by the Ste20 kinase misshapen. Proc Natl Acad Sci U S A. (2011) 108(27):11127–32. doi: 10.1073/pnas.1104128108
45. Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: t cell–dependent and T cell–independent IgA synthesis. Annu Rev Immunol. (2010) 28:243–73. doi: 10.1146/annurev-immunol-030409-101314
46. Huus KE, Petersen C, Finlay BB. Diversity and dynamism of IgA−microbiota interactions. Nat Rev Immunol. (2021) 21(8):514–25. doi: 10.1038/s41577-021-00506-1
47. Doumas SA, Tsironis C, Bolaji A-A, Garantziotis P, Frangou E. Glomerulonephritis and inflammatory bowel disease: a tale of gut-kidney axis dysfunction. Autoimmun Rev. (2023) 22(6):103327. doi: 10.1016/j.autrev.2023.103327
Keywords: TNIK, B cells, signaling, atherosclerosis, IgA
Citation: van Os BW, Kusters PJH, den Toom M, Beckers L, van Tiel CM, Vos WG, de Jong E, Kieser A, van Roomen C, Binder CJ, Reiche ME, de Winther MP, Bosmans LA and Lutgens E (2023) Deficiency of germinal center kinase TRAF2 and NCK-interacting kinase (TNIK) in B cells does not affect atherosclerosis. Front. Cardiovasc. Med. 10:1171764. doi: 10.3389/fcvm.2023.1171764
Received: 22 February 2023; Accepted: 6 April 2023;
Published: 5 May 2023.
Edited by:
Ayman Al Haj Zen, Hamad bin Khalifa University, QatarReviewed by:
Stephen Malin, Karolinska Institutet (KI), SwedenEmiel Van Der Vorst, University Hospital RWTH Aachen, Germany
© 2023 van Os, Kusters, den Toom, Beckers, van Tiel, Vos, de Jong, Kieser, van Roomen, Binder, Reiche, de Winther, Bosmans and Lutgens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura A. Bosmans bC5hLmJvc21hbnNAYW1zdGVyZGFtdW1jLm5s
†These authors share last authorship