
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 01 September 2023
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1165302
This article is part of the Research Topic The Role of Stem Cells, RNA Binding Proteins and Non-Coding RNAs in Diabetes and Vascular Diseases View all 8 articles
 Koray N. Potel1
Koray N. Potel1 Victoria A. Cornelius1
Victoria A. Cornelius1 Andrew Yacoub1
Andrew Yacoub1 Ali Chokr2
Ali Chokr2 Clare L. Donaghy1Sophia Kelaini1
Clare L. Donaghy1Sophia Kelaini1 Magdalini Eleftheriadou1
Magdalini Eleftheriadou1 Andriana Margariti1*
Andriana Margariti1*
Vascular complications are the main cause of diabetes mellitus-associated morbidity and mortality. Oxidative stress and metabolic dysfunction underly injury to the vascular endothelium and myocardium, resulting in diabetic angiopathy and cardiomyopathy. Mitochondrial dysfunction has been shown to play an important role in cardiomyopathic disruptions of key cellular functions, including energy metabolism and oxidative balance. Both non-coding RNAs and RNA-binding proteins are implicated in diabetic cardiomyopathy, however, their impact on mitochondrial dysfunction in the context of this disease is largely unknown. Elucidating the effects of non-coding RNAs and RNA-binding proteins on mitochondrial pathways in diabetic cardiomyopathy would allow further insights into the pathophysiological mechanisms underlying diabetic vascular complications and could facilitate the development of new therapeutic strategies. Stem cell-based models can facilitate the study of non-coding RNAs and RNA-binding proteins and their unique characteristics make them a promising tool to improve our understanding of mitochondrial dysfunction and vascular complications in diabetes.
Diabetes mellitus (DM) and its complications are a major global health burden, affecting millions of patients worldwide (1). Amongst the various macro- and microvascular complications of DM, diabetic cardiomyopathy (DCM) is a critical cause of DM-associated morbidity and mortality. DCM is characterized by cardiac dysfunction and heart failure in the absence of hypertension, coronary artery disease or other cardiac pathologies (2, 3). This dysfunction is driven by the molecular changes seen in DM, including hyperglycemia, increased fatty acid metabolism, inflammation and fibrosis, which collectively contribute to the pathophysiology of DCM (4). In recent years, a role for mitochondrial dysfunction in DCM has emerged also.
Whilst mitochondria are regarded primarily for their central role in bioenergetic and biosynthetic pathways, within the last few decades a greater recognition for the role of mitochondria in a range of cellular processes such as regulation of ion homeostasis, cell growth, redox status, signal transduction, immunity and cell survival has been gained (5–7). Accordingly, an appreciation for the role of mitochondrial dysfunction as a major contributing factor to multiple disease states, including neurodegenerative disorders, diabetes and cardiovascular diseases, has emerged (8, 9). Modulated by genetics, lifestyle and environment, mitochondrial dysfunction is therefore now accepted to not only underlie mitochondrial diseases through the mutation of either genes encoding mitochondrial proteins or non-mitochondrial genes involved in mitochondrial biology, but also a repertoire of complex disease states. Due to the heart's high demand for adenosine triphosphate (ATP), mitochondria are found more abundantly within cardiomyocytes. A study revealed between 20% and 40% of the volume within adult cardiomyocytes to consist of mitochondria (10). It is therefore not surprising, that mitochondrial dysfunction, and the associated disruptions in energy metabolism, oxidative balance, calcium handling and apoptosis, have emerged as key drivers of cardiac dysfunction and the pathophysiological changes in DCM (Figure 1) (3, 11, 12).
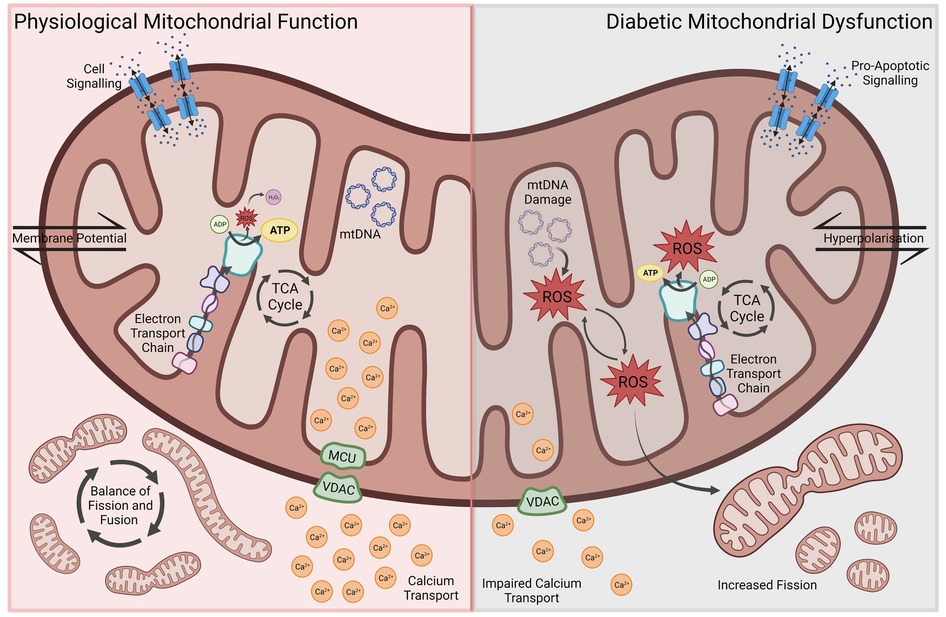
Figure 1. Aspects of mitochondrial dysfunction seen in diabetic cardiomyopathy, including reduced energy metabolic efficiency, ROS-associated oxidative stress and mitochondrial DNA damage, impaired Ca2+ transport, increased mitochondrial fission, membrane hyperpolarization and pro-apoptotic signaling. ROS, reactive oxygen species; MCU, mitochondrial calcium uniporter; VDAC, voltage-dependent anion channel.
In the healthy heart, about 70% of energy is derived from the oxidation of fatty acids. The source of the remaining 30% comprise glucose, amino acids and other nutrients. While the oxidation of fatty acids is not as efficient as the metabolism of glucose or other substrates, requiring more oxygen to generate the same amount of ATP, there exists a physiological balance between energy need, oxygen consumption and ATP synthesis. In the diabetic heart, however, a shift towards increased fatty acid metabolism results in an increased oxygen demand and reduced cardiac efficiency (11, 13). This metabolic shift can be already observed in the hearts of obese human subjects, manifesting as increased oxygen consumption alongside enhanced uptake and oxidation of fatty acids (14). Montaigne et al. showed, however, that contractile impairment and mitochondrial dysfunction can develop in DCM irrespective of weight. Furthermore, they found mitochondrial dysfunction to be directly and independently associated with hemoglobin A1c (HbA1c) levels—highlighting the importance of hyperglycemia and decreased glucose uptake in the development of these metabolic changes (15). In various animal models of DCM, a reduced mitochondrial oxidative capacity and efficiency can be observed. Additionally, cardiomyocytes seem to be unable to sufficiently adapt their substrate utilization in response to shifting levels of glucose and fatty acids, resulting in increased fatty acid metabolism and oxygen demand irrespective of substrate availability (16–19). The peroxisome proliferator-activated receptor ɑ (PPARɑ) plays a central role in shifting mitochondrial metabolism towards increased fatty acid oxidation (12, 20). PPARɑ is a member of the ligand-activated nuclear receptor superfamily and major gene expression regulator of enzymes involved in fatty acid β-oxidation. Increased lipid uptake induces PPARɑ which in turn upregulates enzyme expression (21). In mice with cardiac-specific overexpression of PPARɑ, a DCM-like phenotype can be observed, including characteristic metabolic shifts and cardiac dysfunction (22). Further elucidating the role of PPARɑ in the development of DCM, Buchanan et al. found increased fatty acid metabolism and the development of cardiac dysfunction to precede PPARɑ-overexpression in obese and diabetic mice, which indicates that there are co-existing mechanisms driving early metabolic and phenotypic changes (23).
Alongside the reduced mitochondrial oxidative capacity and efficiency, increased oxidative stress has been identified as a key driver of DCM pathogenesis. During mitochondrial ATP production through the respiratory chain, reactive oxygen species (ROS) are produced, such as superoxide anion (O2−). Whilst normal amounts of ROS can be converted to more stable forms including O2− into hydrogen peroxide, overproduction of ROS creates oxidative stress resulting in damage to mitochondrial DNA via mutations, altered membrane permeability, calcium homeostasis and mitochondrial defense systems. Moreover, damage to mitochondrial DNA has been shown to increase ROS and oxidative stress creating a harmful cycle of distress (24, 25). Due to the abundance of mitochondria in cardiac tissue, mitochondrial dysfunction and ROS production are thought to contribute significantly to cardiac pathologies (26–29). In diabetic mice, increased myocardial oxygen consumption and cardiac dysfunction coincide with augmented generation of ROS as well as lipid and protein peroxidation products (30). Similarly, mitochondrial dysfunction and increased oxidative stress are seen in rats with DCM (31). This increase in oxidative stress promotes cardiac cell death (32). Insulin treatment, on the other hand, can prevent free radical damage and reverse mitochondrial dysfunction in diabetic hearts (33).
Besides their role in energy metabolism and oxidative balance, mitochondria are also involved in the intracellular handling and storage of calcium. Ca2+ is a central component of cardiomyocyte contraction and signaling, which is why disruptions of Ca2+ homeostasis secondary to mitochondrial dysfunction contribute to the pathophysiological features of DCM. In a reciprocal regulatory relationship, cytosolic Ca2+ mediates both ATP production and utilization, by influencing mitochondrial oxidation, enhancing aerobic respiration and modulating ATPase activity (34–36). Cytosolic and mitochondrial Ca2+ levels fluctuate throughout the cardiac contraction cycle. Mitochondria take up Ca2+ during systole and release it into the cytoplasm throughout diastole (37). Allowing the flow of Ca2+ from the cytoplasm into the mitochondrial matrix is the mitochondrial Ca2+ uniporter (MCU), which is downregulated in diabetes. The MCU plays a vital role in intracellular Ca2+ homeostasis and signaling. Its downregulation in diabetes leads to impaired mitochondrial Ca2+ uptake and was found to be associated with a decreased capacity to upregulate ATP synthesis in response to increased workload, resulting in contractile dysfunction (38). Upregulation of MCU in diabetic animal models restored normal energy metabolism and mitochondrial Ca2+ handling, while decreasing oxidative stress and cardiomyocyte apoptosis (39, 40). Other key ion channels found to be downregulated in diabetes include sarco-endoplasmic reticulum calcium ATPase (SERCA) and Ryanodine Receptor 2 (RYR2), which are responsible for sarcoplasmic Ca2+ uptake and release, respectively. Their downregulation directly impacts cytosolic and mitochondrial Ca2+ levels, disrupting intracellular Ca2+ shifts and energy metabolism. This impairs mitochondrial energy metabolism, increases oxidative stress, promotes cardiomyocyte death and results in cardiac dysfunction (3, 41–44). These disruptions in Ca2+ homeostasis develop in response to prolonged hyperglycemia, and studies showed that impaired mitochondrial Ca2+ handling and its pathophysiological consequences can be reversed by insulin therapy (45, 46).
Mitochondrial morphology and DM-induced cardiomyocyte dysfunction have a reciprocal influence on each other, with DCM altering mitochondrial morphology, and altered morphology in turn promoting the progression of DCM (11). Mitochondrial morphology is controlled by a complex balance of fusion- and fission-promoting proteins. This balance is disrupted in DM, with downregulation of mitofusins 1 and 2 (MFN1 and MFN2) resulting in decreased mitochondrial fusion. MFN1 expression was found to be inversely proportional to HbA1c levels and enhancement of MFN2 in a diabetic mouse model prevented DCM progression (15, 47). ROS are able to upregulate fission proteins such as DRP1, further contributing to mitochondrial fragmentation (48).
Besides morphological changes, intracellular processes responsible for mitochondrial regeneration and quality-control were found to be impaired in DM. Mitophagy plays an important role in the healthy heart by removing dysfunctional mitochondria. Impaired mitophagy and mitochondrial biogenesis lead to persistent mitochondrial dysfunction, thereby contributing to the development and progression of DCM (49–51).
In recent years, studies have tried to elucidate the posttranscriptional changes underlying DM and its complications. Due to the association of mitochondrial function and regulation of gene expression, non-coding RNAs (ncRNAs) and RNA-binding proteins (RBPs) have emerged as key regulators of mitochondrial health and have also been implicated in the pathophysiology of DCM. With about 74% of transcribed DNA being non-coding, ncRNAs play a central role in modulating cellular responses by acting as posttranscriptional inhibitors of their target genes. While some have been shown to be critical to cellular health, others seem to contribute to various diseases, including DCM (52). Regulatory ncRNAs have a vital role in cellular homeostatic and adaptive mechanisms and are categorized according to their length: short ncRNAs are less than 200 nucleotides long and include microRNAs (miRNAs), RNAs comprising more than 200 nucleotides are referred to as long ncRNAs (lncRNAs). They are important regulators of DNA replication and transcription, as well as mRNA translation. Similarly, RBPs use RNA-binding domains to regulate gene expression through various posttranscriptional mechanisms, including alternative splicing, as well as RNA degradation, and have been shown to play a role in cardiovascular health and disease (53, 54). RBPs modulate RNA fate in a cell dependent manner to result in a tissue-specific protein repertoire and cellular function. As such, proper functioning of these intricate posttranscriptional manipulations is essential to cell health, with defects in both RBPs or RBP-regulated RNA networks showing to be central in the onset and progression of pathological disorders. Due to the involvement of RBPs in deciphering between health and disease states, RBPs have emerged as key research targets to enhance our understanding pathophysiological processes.
While their influence on mitochondrial health or dysfunction in DCM needs to be further uncovered, ncRNAs and RBPs could prove critical to understanding the exact molecular mechanisms driving DM. Here we will present the current knowledge of the roles of ncRNAs and RBPs in mitochondrial dysfunction and DCM. We explore how ncRNA/RBP-driven disruption of mitochondrial dysfunction could contribute to the development and progression of DCM, proposing key regulatory pathways and highlighting gaps in our current understanding of these mechanisms. Finally, we present established and emerging models of mitochondrial dysfunction and DM-associated vascular complications, which could provide further insights into the functions of ncRNAs and RBPs, thereby paving the way for new therapies and improved outcomes for patients suffering from DCM.
ncRNAs play a vital role in mitochondrial regulation. As 99% of mitochondrial proteins are nuclear-encoded, mitochondrial ncRNAs—called mitoRNAs—can derive from the nucleus or the mitochondria (55, 56). Adequate mitochondrial function and metabolic adaptation requires constant mito-nuclear crosstalk, which is facilitated by ncRNAs (57).
Looking at miRNAs, there are three distinct types which can influence mitochondrial function: cytoplasmic miRNAs, nuclear-encoded mitochondrial miRNAs (mitomiRs) and mitochondria-encoded mitomiRs. Cytoplasmic miRNAs have been found to play a crucial role in regulating mito-nuclear crosstalk by binding to mitochondria-associated transcripts in the cytoplasm and have been reviewed elsewhere (58). Briefly, they influence key aspects of mitochondrial function, including oxidative phosphorylation and fatty acid metabolism, as well as mitochondrial dynamics and autophagy. Cytoplasmic miR-378, for example, plays an important role in mitochondrial fatty acid metabolism, by regulating the effects of PGC-1β — a transcriptional coactivator and key regulator of mitochondrial metabolism and biogenesis. In a mouse model, miR-378 knockout increased oxidative capacity and resulted in resistance to high-fat diet-induced obesity (59). Similarly, miR-23a was shown to downregulate mitofusins and promote mitochondrial dysfunction by targeting the related transcriptional coactivator PGC-1α (60). Cytoplasmic members of the miR-30 family, on the other hand, can prevent mitochondrial fission and apoptosis by inhibiting p53 signaling and its downstream pro-apoptotic targets (61). Other miRNAs can regulate mitochondrial Ca2+ handling, such as miR-25, which is known to silence the expression of MCU (62). Besides acting on mitochondria-related transcripts in the cytoplasm, nuclear miRNAs and Argonaute proteins, which play a key role in miRNA-mediated gene silencing, can easily translocate into mitochondria and influence their function through downregulation of target genes (63). One example is cardiac miR-181c, which is transcribed in the nucleus and later translocates together with its silencing complex into mitochondria, where their inhibition of mitochondrial COX1 results in mitochondrial dysfunction (64). Other mitomiRs have been associated with similar regulatory mechanisms influencing mitochondrial transcripts, including miR-1 and miR-2392 (65, 66). MiR-378, which regulates mito-nuclear crosstalk and energy metabolism in the cytoplasm, is also found within mitochondria, where it was shown to inhibit the translation of ATP synthase (67, 68). While the roles of mitomiRs in mitochondrial dysfunction remain to be further uncovered, nuclear- and mitochondrial-encoded miRNAs, both within the cytoplasm and mitochondria, undoubtedly play a central role regulating mitochondrial metabolism and dynamics and might be important drivers of regulatory dysregulations see in mitochondrial dysfunction.
LncRNAs can promote or prevent mitochondrial dysfunction through multiple mechanisms and have been implicated in various cardiovascular diseases. Interestingly, Yang et al. found expression profiles of lncRNAs, but not mRNAs or miRNAs, to characterize various pathologies of the failing heart. Much like miRNAs, lncRNAs can be encoded by nuclear or mitochondrial DNA and act on mitochondria-related transcripts both in the cytoplasm and within mitochondria (58). Some lncRNAs are known to dysregulate mitochondrial energy metabolism, such as the lncRNA AK055347, which controls the expression of ATP synthase in the heart (69). Others regulate the complex balances between mitochondrial fission and fusion as well as mitophagy and biogenesis. LncRNA HOTAIR regulates mitoribosomal proteins and its inhibition has been shown to result in mitochondrial dysfunction (70, 71). While lncRNA H19 reduces the expression of fusion protein MFN2 through miR675-mediated gene silencing, lncRNA CARL sponges miR-539, thereby suppressing mitochondrial fission and apoptosis (72, 73). Other lncRNAs have been shown to regulate gene expression by inhibiting miRNA-associated gene silencing, including CEROX1, which increases expression and activity of complexes I and IV by preventing their miR-488-3p-driven downregulation (74). Similarly, lncRNA KCNQ1OT1sponges miR-378 and prevents its inhibition of ATP synthase translation (67). Other lncRNAs interact with gene promoter regions, such as TUG1, which has been shown to induce PGC-1α and regulate mitochondrial bioenergetics (75). LncRNA UIHTC has also been shown to improve mitochondrial function in cardiomyocyte by enhancing PGC-1α expression (76). Though the exact effects and underlying mechanisms of many lncRNAs remain poorly understood, they have been found to play important regulatory roles in mitochondrial health and dysfunction, potentially contributing to the pathogenesis of cardiovascular and other diseases.
Other ncRNAs have been shown to play crucial roles in mitochondrial function. Ro et al., for example, identified mitochondrial small RNAs (mitosRNAs) which are derived from sense transcripts of mitochondrial genes and seem to regulate mitochondrial gene expression (77). Studies also identified hundreds of mitochondria encoded circRNAs (mecciRNAs) (78). Similar to lncRNAs, circRNAs are able to sponge miRNAs, thereby increasing the translation of their target genes (79). CircRNA 0000495, for example, is able to sponge and inhibit the silencing action of miR-488-3p, which is known to target electron transport chain complexes (74, 80). While circRNAs seem to play an important physiological function and are involved in various mitochondrial functions, their impact on mitochondrial dysfunction in diseases such as DCM remains unclear (81). Besides ncRNAs, other posttranscriptional regulators of gene expression are known to be involved in mitochondrial dysfunction, including RBPs (Figure 2).
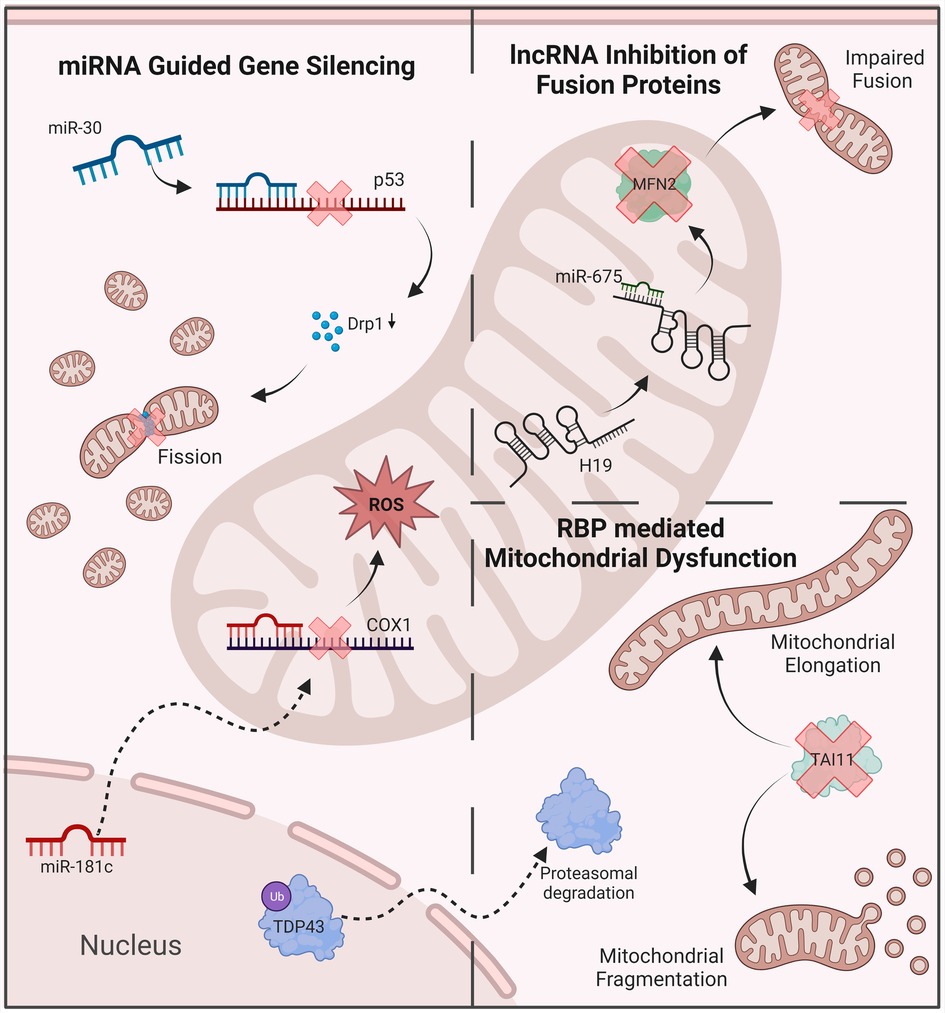
Figure 2. Examples of ncRNAs anr RBPs’ involvement in mitochondrial dysfunction. MiRNA-mediated gene silencing contributes to the disruption of oxidative balance and mitochondrial dynamics. LncRNas inhibit key regulatory proteins, including mitofusin 2. RBPs disrupt mitochondrial dynamics, leading to abnormal morphology and function. ROS, reactive oxygen species; MFN2, mitofusin 2; DRP1, dynamin-related protein 1.
Despite the involvement of RBPs in various disease states, the effect of mitochondrial dysfunction on RBP involvement in cardiac phenotypes has not been fully investigated. Nevertheless, a connection between RBPs and oxidative stress has been uncovered in the recent years, albeit with a focus on neurological diseases, primarily Amyotrophic lateral sclerosis (ALS). Although studied under different physiological conditions, RBPs identified to have oxidative stress induced posttranscriptional modifications similarly have known fundamental roles in cardiac cells also. TAR DNA binding protein 43 (TDP-43), for example, is a versatile RNA/DNA binding protein involved in RNA-related metabolism. Moreover, TDP-43 is known to interact with several mitochondrial proteins and is thought to have a role in the stabilization of mitochondrial transcripts (82, 83). Dysfunctional TDP-43 has therefore been linked to various mechanisms of mitochondrial dysfunction, previously reviewed elsewhere (84). In brief, overexpression of TDP-43 results in abnormal mitochondrial morphology, disrupted mitochondrial dynamics and altered mitochondrial-ER contacts (85–87). Furthermore, a recent study identified the mitochondrial DNA release as a result of TDP-43 expression to drive an inflammatory response mediated via the cGAS-STING pathway (88). Knockdown, on the other hand, reduced the number of mitochondria, mitochondrial membrane potential as well as the expression of mitochondrial transcripts (89). Inhibition of TDP-43 mediated dysfunction may serve as a beneficial therapeutic approach in certain disease states (90).
Oxidative stress has been shown to cause the acetylation and cysteine oxidation of TDP-43, both of which result in increased TDP-43 aggregation as well as its phosphorylation (91–95). The phosphorylation of TDP-43 is heavily associated with deleterious functional changes including mis-localization, decreased turnover, changes in solubility, and altered splicing activity, to name a few (96). Mutations in TDP-43, including its aggregation and depletion, is heavily associated with ALS. Studies to investigate the effect of loss of function of TDP-43, a consequence of phosphorylation, have also uncovered cardiovascular complications, of which further investigation is needed. For example, a recent study investigating the loss of TDP-43 function in mice found these mice to have short lifespans, accredited to cardiac failure due to TDP-43 knockdown mice having enlarged hearts (97). Similarly, in zebrafish, silencing of TDP-43 resulted in early death attributed to severely reduced blood circulation and resulted in the mis-patterning of blood vessels, alongside muscle degeneration (98). Since these studies, phosphorylated TDP-43 has shown to aggregate within cardiac muscle as well as blood vessels (99, 100). Likewise, the RBP TAI1, has been found to undergo cysteine oxidation in response to oxidative stress (101). TAI1 is commonly known for regulating alternative splicing of select pre-mRNAs and promoting the assembly of stress granules, however, roles for TAI1 in mitochondria fission have also been identified. Moreover, downregulation of TAI1 results in mitochondrial elongation or overexpression and increased abundance of proteins involved in mitochondrial fission (102, 103). Similarly, the RBP HuD, known to regulate MFN2, is downregulated in diabetes contributing to mitochondrial dysfunction (104). It is widely accepted that interruption of mitochondrial fission and fusion contributes to cardiomyopathies, further investigation of the impact mitochondrial dysfunction has on RBPs regulating fission/fusion could therefore provide further insight. Likewise, as the prevalence of mitochondrial dysfunction in cardiomyopathies is already established, as in the case of other diseases, the evaluation of oxidative stress on RBPs in cardiomyopathies will undoubtedly identify novel pathogenic mechanisms and potential therapeutic targets.
Multiple miRNAs have been found to be differentially expressed in in vitro and animal models of DCM (Table 1). Diao et al. investigated miRNA expression in streptozotocin-induced diabetic mice and identified ten up- and six downregulated miRNAs which were linked to genes associated with cardiac hypertrophy and fibrosis, including TGFB3 and COL1A1 (132). Similarly, 43 miRNAs were found to be differentially expressed in a rat model of DCM. This model showed characteristic features of DCM, including changes in sarcomeric and mitochondrial structure, as well as increased oxidative stress and contractile dysfunction. Antioxidant treatment reversed these pathological features and restored the levels of previously downregulated miRNAs, including miR-1 and miR-133a (105). MiR-1 and miR-133a are highly expressed in healthy cardiomyocytes and have known cardioprotective properties. Their downregulation in diabetic myocardium results in increased cardiac autophagy and hypertrophy, which are commonly seen in heart failure (111). Overexpression of miR-133a, on the other hand, protects from cardiac fibrosis by downregulating TGF-β1 and preventing the phosphorylation of ERK1/2 and SMAD2 (112). In the same DCM rat model, miR-21, which is known to promote high-glucose induced cardiac fibrosis, was found to be upregulated (105, 120). Similarly, miR-155 is known to enhance inflammatory signaling and contribute to adverse inflammatory responses in cardiovascular diseases (133). Therapeutic downregulation of miR-155 in ovariectomized diabetic mice led to a reduction in cell apoptosis and restoration of cardiac function (134). Other miRNAs are known to prevent pyroptosis but are downregulated in DCM. miR-9 levels, for example, are reduced in hyperglycemic cardiomyocytes. miR-9 targets ELAV-like protein 1 (ELAVL1) — a mRNA-stabilizing protein which enhances TNF-ɑ-induced cardiac cell death. In an in vitro model, upregulation of miR-9 prevented cardiomyocyte pyroptosis in response to hyperglycemia (106).
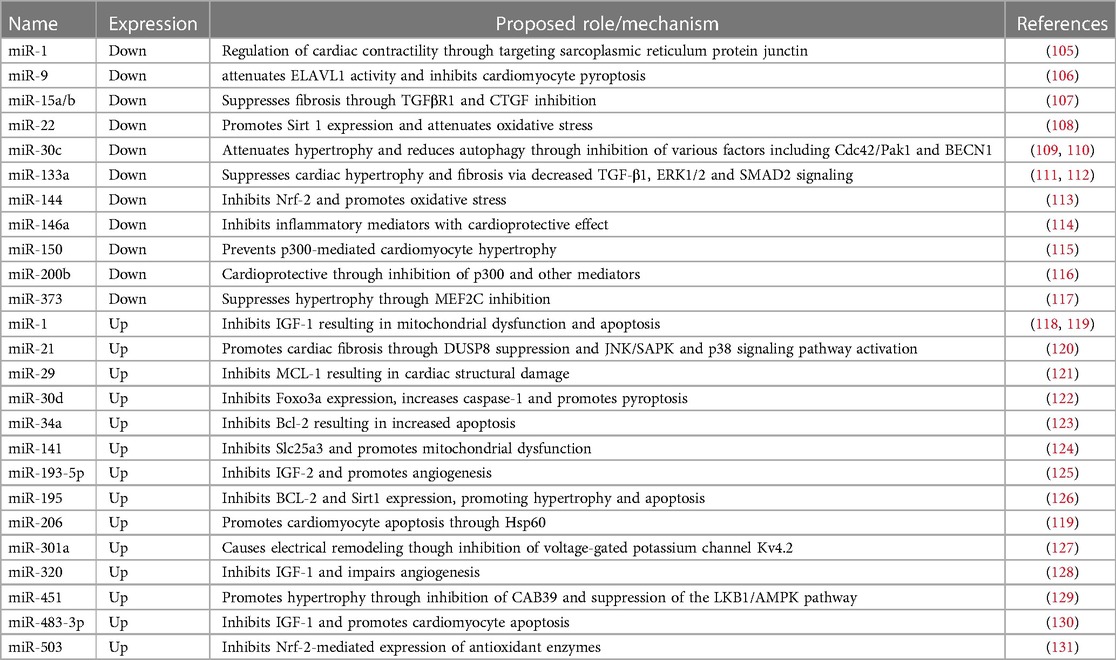
Table 1. Dysregulated miRNAs identified in models of diabetic cardiomyopathy.
Other miRNAs modulate cellular metabolism and promote or prevent the shifts in molecular mechanisms underlying DCM pathogenesis. As described, PPARα signaling facilitates the increase in fatty acid metabolism and contributes to lipotoxicity in diabetic cardiomyocytes. PGC-1β induces PPARα and is targeted by miR-30c. Overexpression of miR-30c resulted in reduced transcriptional activity of PPARα and reversed the metabolic shift towards increased uptake and use of fatty acids (135). Conversely, miR-503 might contribute to DCM development through inhibiting Nrf-2 mediated expression of antioxidant enzymes. In a diabetic rat model, cardiomyocytes exhibited low levels of Nrf2 and increased expression of miR-503 (131). MiR-451 is also upregulated in response to high fatty acid levels in the hearts of Type 2 diabetic mice. Knockout of miR-451 in this model reduced lipotoxicity and improved features of DCM, including cardiac hypertrophy (129). Similarly, miR-195 is increased in mouse models of Type 1 and Type 2 DM and its knockdown was shown to improve cardiac function while reducing oxidative stress and hypertrophy (126). Decreased expression of miR-30c in DCM, on the other hand, results in increased hypertrophy (109). Other miRNAs, such as miR-143, are regulating insulin action and may contribute to insulin resistance seen in Type 2 DM (136).
Similar to miRNAs, lncRNAs have been shown to posttranscriptionally regulate key cellular mechanisms, including energy metabolism as well as inflammation, and have been found to be implicated in the pathophysiological changes seen in DCM (Table 2, Figure 3). In a diabetic mouse model, DCM development was accompanied by significant changes in lncRNA expression. Differentially expressed lncRNAs were found to be associated with myofilament development and motion, as well as inflammation, immunity and apoptosis (152). The lncRNA H19 is a precursor of miR-675, which targets VDAC1—a voltage-gated ion channel and critical component of mitochondria-mediated apoptosis. H19 regulates cardiomyocyte apoptosis through VDAC1 inhibition. In DCM, however, it is found to be downregulated, leading to increased apoptosis, which eventually results in cardiac dysfunction. H19 overexpression was found to reduce oxidative stress, inflammation and apoptosis, as well as improve left ventricular function (138, 139). Similarly, DCM-associated upregulation of lncRNA MALAT1 results in increased cardiomyocyte apoptosis. Downregulation of MALAT1 in a DCM rat model improved cardiac function by attenuating apoptosis (146). Other lncRNA are modulating cardiac fibroblast-to-myofibroblast differentiation and are involved in the fibrotic response seen in DCM. The expression of lncRNA CRNDE negatively correlates with profibrotic genes, for example, and its overexpression has been shown to reduce cardiac fibrosis and improve contractile function in DCM mice (143).

Table 2. Dysregulated lncRNAs identified in models of diabetic cardiomyopathy.
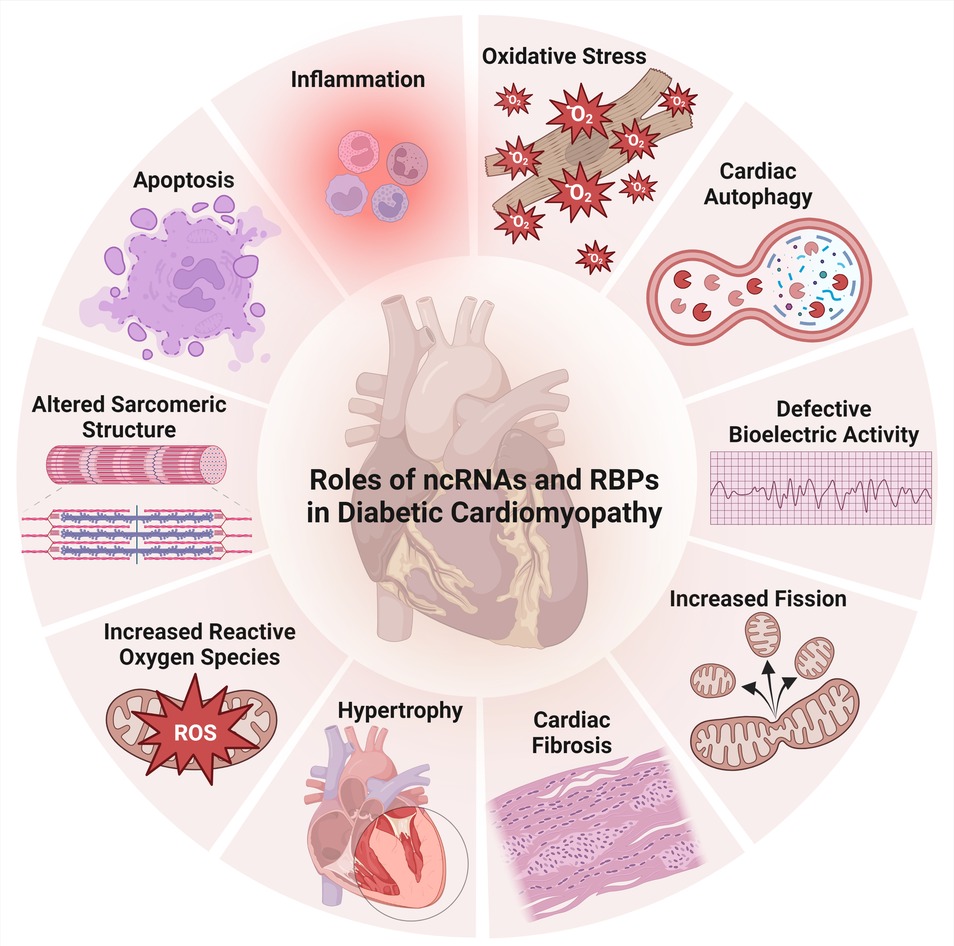
Figure 3. Aspects of ncRNAs and RBPs’ involvement in diabetic cardiomyopathy.
Adding another layer of posttranscriptional regulation of gene expression, some lncRNAs are inhibiting gene silencing by sponging miRNAs. HOTAIR lncRNAs acts by sponging miR-34a, thereby inhibiting inflammation and apoptosis. Its expression is downregulated in DCM and overexpression in a diabetic animal model has been shown to reduce cardiac inflammation, oxidative injury and cardiomyocyte apoptosis (140). Other lncRNAs are upregulated in DCM, leading to increased miRNA-sponging and reduced silencing of target genes. MIAT lncRNA was found to sponge miR-22-3p in a DM rat model, and its upregulation in DCM promoted excessive cardiomyocyte apoptosis (148). Similarly, overexpression of lncRNA KCNQ1OT1 in DCM inhibits the miR-214-3p-regulated silencing of caspase-1, resulting in increased inflammatory signaling and pyroptosis (145).
Other ncRNAs, such as circRNAs are known to be involved in the posttranscriptional signaling shifts seen in DM and its complications, including DCM (Table 3) (153, 154). Several circRNAs which are upregulated in DCM are known to promote myocardial fibrosis by regulating miRNAs and their downstream targets. Sponging of miRNAs by these circRNAs results in upregulation of associated miRNA target genes, including TGF-β1 and collagen (157, 160, 161). Similarly, circRNA CACR, which is upregulated in high-glucose-treated cardiomyocytes and in the serum of DM patients, promotes pyroptosis by sponging miR-214-3p, leading to an increase in caspase-1 activity (156). Conversely, circRNA DICAR can alleviate pyroptosis and other hallmarks of DCM. However, DICAR was found to be reduced in both diabetic mouse hearts and the blood of DM patients (155).

Table 3. Dysregulated circRNAs identified in models of diabetic cardiomyopathy.
Altogether, these results show that while many ncRNAs have been implicated in DCM, their complex mechanistic interactions and modes of regulation remain to be further studied and better understood.
Numerous proteomic studies have demonstrated the depth of RBP involvement in cardiac health and disease through isolating hundreds of cardiomyocyte-specific RBPs (162, 163). Moreover, assessment of RBP splicing patterns within adult hearts has been heavily linked to cardiomyocyte function. Specifically, the expression of crucial splicing factors SF1, ZRSR2, SRSF4, and SRSF5 is downregulated in dysfunctional cardiomyocytes. Within the heart, binding of RBP CIRP to mature RNAs has been shown to enhance the translation of KCND2 and KCND3, inhibition of which results in reduced voltage-gated potassium channel function and defective bioelectric activity (164). Likewise, RBP MBLN1 was shown to regulate the expression of voltage-gated sodium channel SCN5A (165). Moreover, RBP PCBP2 was found to promote GPR56 mRNA degradation in cardiomyocytes, inhibiting angiotensin II-induced hypertrophy (166).
Although the function for the majority of cardiac specific RBPs remains unknown, several studies have revealed dysfunctional RBPs to contribute to cardiomyopathies, in particular diabetic cardiomyopathy (Table 4). Whilst this review focuses on critical RBPs which in the recent years have been heavily implicated in diabetic cardiomyopathy, it is noteworthy to mention a role for RBPs within the blood vascular system and cardiovascular disease onset and progression has also been established and previously reviewed elsewhere (54). Human genetic studies have revealed polymorphisms and mutations in RBPs to be linked with diabetes, as such the role of RBPs in diabetic cardiomyopathy has been a recent topic of interest. Assessment of diabetic hearts revealed the RBP CUG-BP, also referred to as CELF1, a highly conserved RBP that regulates alternative splicing, polyadenylation, mRNA stability, and translation, for instance, to be upregulated (169). Moreover, in mice, overexpression of CUG-BP caused dilated cardiomyopathy and heart failure and interestingly a reactivation of embryonic splicing patterns (169–172). Accordingly, numerous studies have also isolated fetal specific RBPs in adult heart failure tissues, indicating that during heart failure fetal specific RBPs are reactivated and suggesting a role for fetal RBPs in cardiac disease development, including in DM (173). Thus, suggesting a role for fetal RBPs in cardiac disease development, including in diabetes.

Table 4. Dysregulated RBPs identified in models of diabetic cardiomyopathy.
Moreover, within diabetic endothelial cells, overexpression of CUG-BP enhances the expression of the alternatively spliced isoform QKI-7, orchestrating endothelial cell dysfunction (174). QKI-7 belongs to the Signal Transduction and Activation of RNA (STAR) RBP Quaking family, alternative splicing of which generates three major isoforms known as QKI-5, QKI-6 and QKI-7. Both major isoforms QKI-5 and QKI-6 were shown to have pivotal roles in regulating and maintaining cardiovascular health, deficiency of which triggers embryonic lethality (175). In addition, studies have demonstrated both QKI-5 and QKI-6 to be anti-apoptotic in cardiomyocytes, favoring cell survival via the elevation of pro-apoptotic factors during cardiac injury (176). Furthermore, QKI-5 was recently shown to be integral to the regulation of cardiac myofibrillogenesis and its deficiency promotes apoptosis and atrophy in cardiomyocytes (177, 178).
Another RBP known to regulate many genes involved in cardiac function via alternative splicing is RBFOX2. In multiple animal models, a downregulation of RBFOX2 has been shown to lower heart rate, cause myofibrillar disarray and result in heart failure (179, 180). Moreover, in individuals with cardiac disease, mutations in RBFOX2 have been identified (181). Unsurprisingly, a role for RBFOX2 has also been identified in diabetic cardiomyopathies. Although studies have shown RBFOX2 levels to be high in diabetic hearts, its alternative splicing activity is low (169, 170). Like the alternative splicing of the QKI family, analysis of RBFOX2 in the diabetic heart revealed diabetic hearts to express a dominant negative alternatively spliced isoform of RBFOX2 responsible for blocking RBFOX2-mediated alternative splicing (170). Further analysis of the mis-spliced transcripts in diabetic hearts revealed 73% to have RBFOX2-binding sites, including genes associated with the expression of cytoskeleton and intracellular calcium handling (170). Accordingly, RBFOX2 dysregulation contributes to diabetic complications in the heart. A reduction of LIN28, an RBP predominantly known for its roles in promoting pluripotency, in cardiomyocytes has shown to decrease contractile function and cell death (167). LIN28 was found to be significantly reduced in diabetic hearts and exacerbate cardiac symptoms observed through a decrease in left ventricular ejection fraction, increased apoptotic index and mitochondrial dysfunction (167, 182). Overexpression of LIN28, however, prevented cardiomyopathy in diabetic mice, demonstrating its protective role in cardiac function (167, 168). Comparably, knockdown of the RBP ELAVL1, a known major contributor to diabetic complications, which has also been found to be upregulated in diabetic hearts and correspond with cardiomyocyte death, resulted in smaller infarct size and fibrosis area following a myocardial infarction (106, 183). Extensive evidence therefore demonstrates a primary role for RBPs and their dysregulation in the development and progression of diabetic cardiomyopathies; revelations of such roles may aid the development of novel therapeutic interventions.
Mitochondrial dysfunction has emerged as a key driver of cardiovascular diseases, including DCM. While mitochondria are mainly known for their metabolic roles and properties, they are integral to other important cellular processes, including oxidative balance, Ca2+ handling and apoptosis. Hence, mitochondrial dysfunction goes far beyond alterations of energy metabolism and manifests in disruptions of both intra- and extramitochondrial cellular mechanisms. Mitochondrial dysfunction has been thoroughly studied and implicated in DCM pathogenesis, its hallmarks including a shift towards increased fatty acid uptake and oxidation leading to decreased cardiac efficiency, increased oxidative stress causing mitochondrial damage, disruption of Ca2+ homeostasis resulting in contractile dysfunction as well as imbalance in fission and fusion giving rise to mitochondrial fragmentation (3, 11, 12).
In recent years, ncRNAs and RBPs were studied in many diseases and have been implicated in both mitochondrial dysfunction and DCM. Some seem to serve physiological or protective functions and their downregulation is found to be associated with various disease states. Conversely, others contribute to the disruption of mitochondrial function and pathophysiological changes seen in DCM (58, 81, 82, 132, 152, 163). This review aimed to collect and newly synthesize current insights into ncRNA and RBP involvement in mitochondrial dysfunction in the context of DCM.
Many ncRNAs and RBPs which are known to play a key role in mitochondrial health and dysfunction, have also been implicated in DCM (Table 5). For the majority of these, their impact on mitochondrial health or dysfunction has been assessed in diseases other than DCM—most studies using cancer-related in vitro models—and their specific function in DCM remains to be uncovered. Only for few ncRNAs, including miR-1 and miR-141, a specific role and associated mechanistic pathway impacting on mitochondrial dysregulation in the context of DCM has been determined (118, 124).
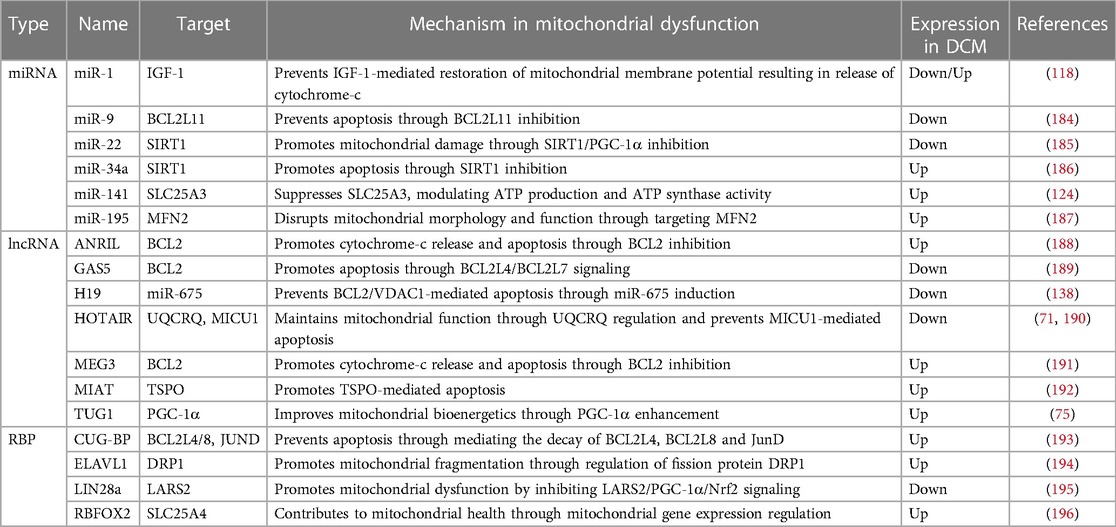
Table 5. ncRNAs and RBPs involved in mitochondrial health/dysfunction and found to be differentially expressed in DCM.
Overall, there have been few studies looking at unifying pathways that would link ncRNA/RBP-associated dysregulation of mitochondrial function to DCM development and progression. While this fascinating link remains to be uncovered, the required research is complicated by the complex mechanisms and interactions underlying ncRNA/RBP-driven posttranscriptional regulation of gene expression. The associated processes are multifaceted and comprise different layers of regulation, impacting every stage of gene expression, including transcription, splicing and translation.
MiRNAs, such as miR-675, can regulate mitochondrial function through inhibition of multiple key regulators, including the fusion protein MFN2 and the anion channel VDAC1 (138, 197). MiR-675's potential role in DCM highlights the complex and sometimes contradicting mechanisms that need to be understood in order to fully appreciate the underlying regulatory pathways. Studies observed downregulation of miR-675 in the context of hyperglycemia which led to increased cardiomyocyte apoptosis. While excessive cardiomyocyte apoptosis can be seen in cardiomyopathies, miR-675 downregulation also results in increased expression of MFN2, which is contrary to what many studies observed in DM. This not only highlights the complexity of one protein, such as MFN2, being targeted by multiple posttranscriptional regulators, but might also exemplify the difficulty of deciphering the exact role of each player in the coexisting health-protecting and disease-promoting processes. While some expression changes might actively promote DCM development and progression, others might be a protective response aimed at maintaining cellular homeostasis and function. Additionally, effects of molecular expression changes might differ in the acute and chronic setting, as seen in other cardiovascular diseases (198). Some protective responses in the context of acute hyperglycemia might become pathological if maintained for too long. With multiple targets, both within and outside of mitochondria, miRNAs can be involved in mitochondrial and extra-mitochondrial processes, such as miR-214, which targets MFN2, but also the tumor suppressor protein PTEN and various other genes (199, 200). Furthermore, lncRNAs and other ncRNAs can influence and modulate miRNA action. MiR-214, for example is known to be inhibited by lncR KCNQ1OT1, which promotes cardiomyocyte pyroptosis and is upregulated in DM (145). However, the role of KCNQ1OT1 and many other miRNA-regulating ncRNAs in mitochondrial dysfunction has never been explicitly investigated. Considering that gene expression changes are not dependent on a single mediator, but are influenced by the complex, interconnected relationships of various posttranscriptional regulators, it is not surprising that the exact role of individual ncRNAs or RBPs in mitochondrial dysfunction and DCM is difficult to ascertain. Nevertheless, the emerging evidence linking ncRNAs and RBPs with mitochondrial dysfunction and DCM independently, justifies further studies into unifying pathways and mechanisms that could underly mitochondrial dysfunction in DCM.
Looking beyond these complex regulatory mechanisms, certain genes seem to emerge as particularly prominent targets. These include regulators of mitochondrial metabolism, ion channels, and mitochondrial fusion/fission proteins. PGC-1α/β, for example, seem to be targeted by various ncRNAs associated with both mitochondrial dysfunction and DCM (60, 135, 201). Given their central role in mediating the uptake and use of fatty acid, their crucial role in DCM-associated metabolic shifts appear plausible. Additionally, they have been shown to impact on proinflammatory and prooxidant signaling, further highlighting their potential importance for the development of mitochondrial dysfunction in the context of DCM (198, 202). Similarly, mitofusins appear to be targeted by various ncRNAs and RBPs implicated in DCM, linking DM-associated pathophysiological changes to disrupted mitochondrial dynamics.
Since ncRNAs and RBPs are secreted into the systemic circulation and their blood concentrations have been shown to differ in patients compared to heathy controls, they have been proposed as circulating biomarkers for various diseases. For instance, a large number of cardiac and non-cardiac miRNAs, including miRs 1 and 133a, have shown biomarker potential due to their differential serum concentrations in cardiovascular disease (203). Various ncRNAs have also been proposed as biomarkers for Type 2 DM and its complications, such as diabetic retinopathy (204–206). In the context of DCM, lncRNA serum levels were found to be directly associated with diastolic dysfunction and cardiac remodelling in patients with Type 2 DM (207).
Furthermore, some circulating ncRNAs have been shown to correlate with commonly used clinical markers, such as serum glucose and HbA1c, in patients with Type 1 and 2 DM (208, 209). However, there is an ongoing need for prospective studies which track the expression levels of ncRNAs or RBPs and correlate those with DCM severity and progression in order to adequately assess the potential of these mediators as diagnostic or prognostic biomarkers.
Therapies trying to build upon these insights, might focus on posttranscriptional regulators, including ncRNAs and RBPs, or their targets, such as PGC-1α/β or MFN1/2. Deciding on the most promising or effective therapeutic target is made difficult by the fact that each regulator has multiple targets, and each target has multiple regulators. Potential RNA-based therapies can either use ncRNAs as therapeutic agents to re-establish physiological expression levels of key genes, or directly target ncRNAs known to be involved in pathogenic processes. While multiple such agents have been approved or are currently undergoing clinical trials, there remain challenges in therapeutically using or targeting ncRNAs and other transcriptional regulators, which include agent immunogenicity, target specificity, and effective delivery (210). To improve our understanding and allow therapeutic application of new insights, further studies need to uncover the exact mechanisms and identify key drivers underlying mitochondrial dysfunction in the context of DCM.
While in vivo animal models, such as obese (ob/ob) or diabetic (db/db) mice, are a great source of insight into the pathophysiological manifestations of DCM and the effect of interventions, such as the administration of insulin, the further investigation and thorough understanding of the underlying molecular mechanisms require adequate and reliable in vitro models. Such models should display key features of DCM, including insulin resistance, increased fatty acid metabolism and oxidative stress, as well as disrupted Ca2+ handling and contractile dysfunction (211). In recent years, stem cell models have emerged as a promising tool for studying DM and its vascular complications.
Other in vitro models have been frequently used in the past, each, however, with its own challenges. While H9C2 rat cardiac cells or HL-1 immortalized mouse cardiomyocytes are easy to culture and study, their applicability in the context of DCM is limited due to significant differences in their expression of key cardiac genes and phenotypic features. Primary adult cardiomyocytes, on the other hand, are difficult to culture and while neonatal cells are easier to maintain, their phenotype remains immature (212, 213).
Induced pluripotent stem cells (iPSCs)-derived cardiomyocytes have been used to study various cardiovascular diseases, including myocardial hypertrophy, arrythmias and cardiomyopathies, with some of their features, such as being of human origin as well as the possibility of generating patient-specific models, making them a particularly useful model system (214, 215). The use of models comprising a single cell type, such as cardiomyocytes, offers a chance to study cell specific mechanisms, but cannot capture the complex interactions of different cells types seen in vivo. Therefore, organoids and other three-dimensional models using multiple cell types, are an interesting and rapidly developing resource for future studies into the mechanisms underlying vascular complications in DM (216). While iPSC-derived cardiomyocytes have been used as models of DCM and offer fascinating insights into disease- and patient-specific molecular changes, further validation and characterization of differentiation protocols are needed to ensure adequate reproducibility and applicability (214, 217).
iPSCs have also been widely used to model mitochondrial diseases, since their metabolic profile allows for manipulation of mitochondrial function while preserving cell viability (218). Assessing key mitochondrial properties, such as oxidative capacity, Ca2+ handling and fission/fusion, in these cells provide further insights into the mitochondrial changes resulting from and possibly contributing to disease development and progression. Li et al., for instance, investigated mitochondrial dysfunction in hypertrophic cardiomyopathy using iPSC-derived cardiomyocytes. Measuring mitochondrial morphology, mitochondrial membrane potentials and Ca2+ flux, they were able to correlate mitochondrial functional disruptions with pathophysiological changes, including electrophysiological abnormalities (219).
Similar iPSC-models could provide valuable insights when investigating the role of ncRNAs and RBPs in regulating mitochondrial health and dysfunction in DCM. Their unique features, including the patient-specific genetic profile, make iPSC-based models suitable to study aspects of DCM which have been challenging to determine using other in vitro models, such as the importance of ncRNA/RBP expression differences in type 1 and type 2 DM-associated DCM and their potential contribution to variabilities in underlying pathological mechanisms. Building on the current understanding of key regulators and their associated targets, future in vitro studies should further characterize the complex changes to posttranscriptional mechanisms contributing to mitochondrial dysfunction and driving DCM pathogenesis. Continuously expanding analytic capabilities, such as the transcriptomic and proteomic analysis of organoids used in the context of neurodevelopment and precision medicine, could be applied to DCM to further characterize pathological changes in ncRNA- and RBP-mediated posttranscriptional regulation (220, 221). While available evidence clearly highlights the crucial role of mitochondrial dysfunction in DCM and indicates the importance of ncRNA/RBP-mediated posttranscriptional regulation of gene expression in both mitochondrial dysfunction and DCM, future studies will need to bring together these insights and further define the underlying mechanisms and pathways. Identifying key posttranscriptional regulators and their targets would not only improve our understanding of the complex drivers of DCM pathogenesis but would also allow the study of new therapies which could prove effective in treating DCM and other DM-associated cardiovascular complications by modulating these complex regulatory mechanisms.
In summary, mitochondrial dysfunction has been shown to be a key driver of DCM, by disrupting cellular mechanisms in cardiomyocytes, including energy metabolism, Ca2+ homeostasis and autophagy. ncRNAs and RBPs are implicated in mitochondrial health and disease and have been shown to be significantly dysregulated in DCM. Synthesizing the evidence from studies looking at ncRNAs and RBPs in DCM and other diseases, we provide an overview of key regulators and targets, but also highlight the lack of evidence regarding the impact of ncRNAs and RBPs on mitochondrial dysfunction in the context of DCM. Future studies will need to investigate the role of individual ncRNAs and RBPs and their impact on gene expression of important cardiac proteins, such as mitochondrial enzymes and ion channels. iPSC-derived cardiomyocytes emerge as a promising in vitro model allowing the study of disease- and patient-specific cells of human origin. Gaining further insights into the exact posttranscriptional mechanisms driving mitochondrial dysfunction in DCM could identify promising drug targets, thereby contributing to the vital search for new therapies to improve clinical outcomes for patients suffering from DCM and other DM-associated complications.
Conceptualization: KP and AM; writing: KP, VC, AY, AC, and ME; figures and tables: KP and VC; review and editing: VC, CD, SK, ME, and AM. All authors contributed to the article and approved the submitted version.
This work was supported by grants from MRC (MR/X00533X/1), British Heart Foundation (PG/18/29/33731), and Northern Ireland Department for the Economy (USI-159).
Figures were created using BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ingelfinger JR, Jarcho JA. Increase in the incidence of diabetes and its implications. N Engl J Med. (2017) 376(15):1473–4. doi: 10.1056/NEJMe1616575
2. Aneja A, Tang WH, Bansilal S, Garcia MJ, Farkouh ME. Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am J Med. (2008) 121(9):748–57. doi: 10.1016/j.amjmed.2008.03.046
3. Dillmann WH. Diabetic cardiomyopathy. Circ Res. (2019) 124(8):1160–2. doi: 10.1161/CIRCRESAHA.118.314665
4. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. (2014) 57(4):660–71. doi: 10.1007/s00125-014-3171-6
5. Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell Dev Biol. (2020) 8:467. doi: 10.3389/fcell.2020.00467
6. Collier JJ, Olahova M, McWilliams TG, Taylor RW. Mitochondrial signalling and homeostasis: from cell biology to neurological disease. Trends Neurosci. (2023) 46(2):137–52. doi: 10.1016/j.tins.2022.12.001
7. Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res. (2012) 111(9):1198–207. doi: 10.1161/CIRCRESAHA.112.268946
8. Norat P, Soldozy S, Sokolowski JD, Gorick CM, Kumar JS, Chae Y, et al. Mitochondrial dysfunction in neurological disorders: exploring mitochondrial transplantation. NPJ Regen Med. (2020) 5(1):22. doi: 10.1038/s41536-020-00107-x
9. Zhunina OA, Yabbarov NG, Grechko AV, Starodubova AV, Ivanova E, Nikiforov NG, et al. The role of mitochondrial dysfunction in vascular disease, tumorigenesis, and diabetes. Front Mol Biosci. (2021) 8:671908. doi: 10.3389/fmolb.2021.671908
10. Schaper J, Meiser E, Stämmler G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res. (1985) 56(3):377–91. doi: 10.1161/01.RES.56.3.377
11. Bai J, Liu C, Zhu P, Li Y. Novel insights into molecular mechanism of mitochondria in diabetic cardiomyopathy. Front Physiol. (2020) 11:609157. doi: 10.3389/fphys.2020.609157
12. Bugger H, Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res. (2010) 88(2):229–40. doi: 10.1093/cvr/cvq239
13. How OJ, Aasum E, Severson DL, Chan WYA, Essop MF, Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes. (2006) 55(2):466–73. doi: 10.2337/diabetes.55.02.06.db05-1164
14. Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation. (2004) 109(18):2191–6. doi: 10.1161/01.CIR.0000127959.28627.F8
15. Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. (2014) 130(7):554–64. doi: 10.1161/CIRCULATIONAHA.113.008476
16. Pham T, Loiselle D, Power A, Hickey AJ. Mitochondrial inefficiencies and anoxic ATP hydrolysis capacities in diabetic rat heart. Am J Physiol Cell Physiol. (2014) 307(6):C499–507. doi: 10.1152/ajpcell.00006.2014
17. Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. (2005) 112(17):2686–95. doi: 10.1161/CIRCULATIONAHA.105.554360
18. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. (2009) 54(20):1891–8. doi: 10.1016/j.jacc.2009.07.031
19. Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. (2004) 53(9):2366–74. doi: 10.2337/diabetes.53.9.2366
20. Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, et al. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem. (1998) 273(10):5678–84. doi: 10.1074/jbc.273.10.5678
21. Barger PM, Kelly DP. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med. (2000) 10(6):238–45. doi: 10.1016/S1050-1738(00)00077-3
22. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, et al. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J Clin Invest. (2002) 109(1):121–30. doi: 10.1172/JCI0214080
23. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. (2005) 146(12):5341–9. doi: 10.1210/en.2005-0938
24. Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. (1997) 94(2):514–9. doi: 10.1073/pnas.94.2.514
25. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. (2019) 51(12):1–13. doi: 10.1038/s12276-019-0355-7
26. Maulik SK, Kumar S. Oxidative stress and cardiac hypertrophy: a review. Toxicol Mech Methods. (2012) 22(5):359–66. doi: 10.3109/15376516.2012.666650
27. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301(6):H2181–90. doi: 10.1152/ajpheart.00554.2011
28. Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit. (2009) 15(10):Ra209–219.19789524
29. Kaludercic N, Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med. (2020) 7:12. doi: 10.3389/fcvm.2020.00012
30. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. (2007) 56(10):2457–66. doi: 10.2337/db07-0481
31. Raza H, John A, Howarth FC. Alterations in glutathione redox metabolism, oxidative stress, and mitochondrial function in the left ventricle of elderly zucker diabetic fatty rat heart. Int J Mol Sci. (2012) 13(12):16241–54. doi: 10.3390/ijms131216241
32. Sun X, Chen RC, Yang ZH, Sun GB, Wang M, Ma XJ, et al. Taxifolin prevents diabetic cardiomyopathy in vivo and in vitro by inhibition of oxidative stress and cell apoptosis. Food Chem Toxicol. (2014) 63:221–32. doi: 10.1016/j.fct.2013.11.013
33. Lashin OM, Szweda PA, Szweda LI, Romani AM. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic Biol Med. (2006) 40(5):886–96. doi: 10.1016/j.freeradbiomed.2005.10.040
34. Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol. (2002) 34(10):1259–71. doi: 10.1006/jmcc.2002.2082
35. Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F0/F1-ATPase. Am J Physiol Cell Physiol. (2000) 278(2):C423–35. doi: 10.1152/ajpcell.2000.278.2.C423
36. Hansford RG, Zorov D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol Cell Biochem. (1998) 184(1–2):359–69. doi: 10.1023/A:1006893903113
37. Isenberg G, Han S, Schiefer A, Wendt-Gallitelli MF. Changes in mitochondrial calcium concentration during the cardiac contraction cycle. Cardiovasc Res. (1993) 27(10):1800–9. doi: 10.1093/cvr/27.10.1800
38. Flarsheim CE, Grupp iL, Matlib MA. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am J Physiol. (1996) 271(1 Pt 2):H192–202. doi: 10.1152/ajpheart.1996.271.1.H192
39. Diaz-Juarez J, Suarez J, Cividini F, Scott BT, Diemer T, Dai A, et al. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am J Physiol Cell Physiol. (2016) 311(6):C1005–13. doi: 10.1152/ajpcell.00236.2016
40. Suarez J, Cividini F, Scott BT, Lehmann K, Diaz-Juarez J, Diemer T, et al. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J Biol Chem. (2018) 293(21):8182–95. doi: 10.1074/jbc.RA118.002066
41. Belke DD, Dillmann WH. Altered cardiac calcium handling in diabetes. Curr Hypertens Rep. (2004) 6(6):424–9. doi: 10.1007/s11906-004-0035-3
42. Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes. (2004) 53(12):3201–8. doi: 10.2337/diabetes.53.12.3201
43. Salin Raj P, Swapna SUS, Raghu KG. High glucose induced calcium overload via impairment of SERCA/PLN pathway and mitochondrial dysfunction leads to oxidative stress in H9c2 cells and amelioration with ferulic acid. Fundam Clin Pharmacol. (2019) 33(4):412–25. doi: 10.1111/fcp.12452
44. Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, et al. Mechanisms of [Ca2+] i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. (2006) 55(3):608–15. doi: 10.2337/diabetes.55.03.06.db05-1284
45. Fauconnier J, Lanner JT, Zhang SJ, Tavi P, Bruton JD, Katz A, et al. Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca2+ transients and reveal mitochondrial Ca2+ handling defects in cardiomyocytes of ob/ob mice. Diabetes. (2005) 54(8):2375–81. doi: 10.2337/diabetes.54.8.2375
46. Ganguly PK, Pierce GN, Dhalla KS, Dhalla NS. Defective sarcoplasmic reticular calcium transport in diabetic cardiomyopathy. Am J Physiol. (1983) 244(6):E528–35. doi: 10.1152/ajpendo.1983.244.6.E528
47. Hu L, Ding M, Tang D, Gao E, Li C, Wang K, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics. (2019) 9(13):3687–706. doi: 10.7150/thno.33684
48. Watanabe T, Saotome M, Nobuhara M, Sakamoto A, Urushida T, Katoh H, et al. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp Cell Res. (2014) 323(2):314–25. doi: 10.1016/j.yexcr.2014.02.027
49. Tao LC, Wang TT, Zheng L, Hua F, Li JJ. The role of mitochondrial biogenesis dysfunction in diabetic cardiomyopathy. Biomol Ther. (2022) 30(5):399–408. doi: 10.4062/biomolther.2021.192
50. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. (2019) 124(9):1360–71. doi: 10.1161/CIRCRESAHA.118.314607
51. Zheng H, Zhu H, Liu X, Huang X, Huang A, Huang Y. Mitophagy in diabetic cardiomyopathy: roles and mechanisms. Front Cell Dev Biol. (2021) 9:750382. doi: 10.3389/fcell.2021.750382
52. Xia L, Song M. Role of non-coding RNA in diabetic cardiomyopathy. Adv Exp Med Biol. (2020) 1229:181–95. doi: 10.1007/978-981-15-1671-9_10
53. de Bruin RG, Rabelink TJ, van Zonneveld AJ, van der Veer EP. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur Heart J. (2017) 38(18):1380–8. doi: 10.1093/eurheartj/ehw567
54. Cornelius VA, Naderi-Meshkin H, Kelaini S, Margariti A. RNA-binding proteins: emerging therapeutics for vascular dysfunction. Cells. (2022) 11(16):2494. doi: 10.3390/cells11162494
55. Jusic A, Devaux Y, EU-CardioRNA COST Action (CA17129). Mitochondrial noncoding RNA-regulatory network in cardiovascular disease. Basic Res Cardiol. (2020) 115(3):23. doi: 10.1007/s00395-020-0783-5
56. Barrey E, Saint-Auret G, Bonnamy B, Damas D, Boyer O, Gidrol X. Pre-microRNA and mature microRNA in human mitochondria. PLoS One. (2011) 6(5):e20220. doi: 10.1371/journal.pone.0020220
57. Dong Y, Yoshitomi T, Hu JF, Cui J. Long noncoding RNAs coordinate functions between mitochondria and the nucleus. Epigenetics Chromatin. (2017) 10(1):41. doi: 10.1186/s13072-017-0149-x
58. Gusic M, Prokisch H. ncRNAs: new players in mitochondrial health and disease? Front Genet. (2020) 11:95. doi: 10.3389/fgene.2020.00095
59. Carrer M, Liu N, Grueter CE, Williams AH, Frisard MI, Hulver MW, et al. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc Natl Acad Sci U S A. (2012) 109(38):15330–5. doi: 10.1073/pnas.1207605109
60. Russell AP, Wada S, Vergani L, Hock MB, Lamon S, Leger B, et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol Dis. (2013) 49:107–17. doi: 10.1016/j.nbd.2012.08.015
61. Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. (2010) 6(1):e1000795. doi: 10.1371/journal.pgen.1000795
62. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol. (2013) 23(1):58–63. doi: 10.1016/j.cub.2012.11.026
63. Bandiera S, Ruberg S, Girard M, Cagnard N, Hanein S, Chretien D, et al. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One. (2011) 6(6):e20746. doi: 10.1371/journal.pone.0020746
64. Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res. (2012) 110(12):1596–603. doi: 10.1161/CIRCRESAHA.112.267732
65. Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y, et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. (2014) 158(3):607–19. doi: 10.1016/j.cell.2014.05.047
66. Fan S, Tian T, Chen W, Lv X, Lei X, Zhang H, et al. Mitochondrial miRNA determines chemoresistance by reprogramming metabolism and regulating mitochondrial transcription. Cancer Res. (2019) 79(6):1069–84. doi: 10.1158/0008-5472.CAN-18-2505
67. Durr AJ, Hathaway QA, Kunovac A, Taylor AD, Pinti MV, Rizwan S, et al. Manipulation of the miR-378a/mt-ATP6 regulatory axis rescues ATP synthase in the diabetic heart and offers a novel role for lncRNA Kcnq1ot1. Am J Physiol Cell Physiol. (2022) 322(3):C482–95. doi: 10.1152/ajpcell.00446.2021
68. Jagannathan R, Thapa D, Nichols CE, Shepherd DL, Stricker JC, Croston TL, et al. Translational regulation of the mitochondrial genome following redistribution of mitochondrial microRNA in the diabetic heart. Circ Cardiovasc Genet. (2015) 8(6):785–802. doi: 10.1161/CIRCGENETICS.115.001067
69. Chen G, Guo H, Song Y, Chang H, Wang S, Zhang M, et al. Long non-coding RNA AK055347 is upregulated in patients with atrial fibrillation and regulates mitochondrial energy production in myocardiocytes. Mol Med Rep. (2016) 14(6):5311–7. doi: 10.3892/mmr.2016.5893
70. Delhaye L, De Bruycker E, Volders PJ, Fijalkowska D, De Sutter D, Degroeve S, et al. Orthogonal proteomics methods to unravel the HOTAIR interactome. Sci Rep. (2022) 12(1):1513. doi: 10.1038/s41598-022-05405-6
71. Zheng P, Xiong Q, Wu Y, Chen Y, Chen Z, Fleming J, et al. Quantitative proteomics analysis reveals novel insights into mechanisms of action of long noncoding RNA hox transcript antisense intergenic RNA (HOTAIR) in heLa cells. Mol Cell Proteomics. (2015) 14(6):1447–63. doi: 10.1074/mcp.M114.043984
72. Sultan HK, El-Ayat WM, AbouGhalia AH, Lasheen NN, Moustafa AS. Study of long non-coding RNA and mitochondrial dysfunction in diabetic rats. Tissue Cell. (2021) 71:101516. doi: 10.1016/j.tice.2021.101516
73. Wang K, Long B, Zhou LY, Liu F, Zhou QY, Liu CY, et al. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat Commun. (2014) 5:3596. doi: 10.1038/ncomms4596
74. Sirey TM, Roberts K, Haerty W, Bedoya-Reina O, Rogatti-Granados S, Tan JY, et al. The long non-coding RNA Cerox1 is a post transcriptional regulator of mitochondrial complex I catalytic activity. Elife. (2019) 8:e45051. doi: 10.7554/eLife.45051
75. Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest. (2016) 126(11):4205–18. doi: 10.1172/JCI87927
76. Zhang J, Yu L, Xu Y, Liu Y, Li Z, Xue X, et al. Long noncoding RNA upregulated in hypothermia treated cardiomyocytes protects against myocardial infarction through improving mitochondrial function. Int J Cardiol. (2018) 266:213–7. doi: 10.1016/j.ijcard.2017.12.097
77. Ro S, Ma HY, Park C, Ortogero N, Song R, Hennig GW, et al. The mitochondrial genome encodes abundant small noncoding RNAs. Cell Res. (2013) 23(6):759–74. doi: 10.1038/cr.2013.37
78. Liu X, Wang X, Li J, Hu S, Deng Y, Yin H, et al. Identification of mecciRNAs and their roles in the mitochondrial entry of proteins. Sci China Life Sci. (2020) 63(10):1429–49. doi: 10.1007/s11427-020-1631-9
79. Altesha MA, Ni T, Khan A, Liu K, Zheng X. Circular RNA in cardiovascular disease. J Cell Physiol. (2019) 234(5):5588–600. doi: 10.1002/jcp.27384
80. Hao Y, Zhang D, Guo Y, Fu Z, Yu D, Guan G. miR-488-3p sponged by circ-0000495 and mediated upregulation of TROP2 in head and neck squamous cell carcinoma development. J Cancer. (2020) 11(11):3375–86. doi: 10.7150/jca.40339
81. Liu X, Shan G. Mitochondria encoded non-coding RNAs in cell physiology. Front Cell Dev Biol. (2021) 9:713729. doi: 10.3389/fcell.2021.713729
82. Davis SA, Itaman S, Khalid-Janney CM, Sherard JA, Dowell JA, Cairns NJ, et al. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci Lett. (2018) 678:8–15. doi: 10.1016/j.neulet.2018.04.053
83. Izumikawa K, Nobe Y, Yoshikawa H, Ishikawa H, Miura Y, Nakayama H, et al. TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci Rep. (2017) 7(1):7709. doi: 10.1038/s41598-017-06953-y
84. Gao J, Wang L, Yan T, Perry G, Wang X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol Cell Neurosci. (2019) 100:103396. doi: 10.1016/j.mcn.2019.103396
85. Xu YF, Gendron TF, Zhang YJ, Lin WL, D'Alton S, Sheng H, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. (2010) 30(32):10851–9. doi: 10.1523/JNEUROSCI.1630-10.2010
86. Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, et al. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet. (2013) 22(23):4706–19. doi: 10.1093/hmg/ddt319
87. Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF, et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun. (2014) 5:3996. doi: 10.1038/ncomms4996
88. Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. (2020) 183(3):636–649.e18. doi: 10.1016/j.cell.2020.09.020
89. Briese M, Saal-Bauernschubert L, Lüningschrör P, Moradi M, Dombert B, Surrey V, et al. Loss of tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol Commun. (2020) 8(1):116. doi: 10.1186/s40478-020-00987-6
90. Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. (2016) 22(8):869–78. doi: 10.1038/nm.4130
91. Cohen TJ, Hwang AW, Unger T, Trojanowski JQ, Lee VM Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. Embo J. (2012) 31(5):1241–52. doi: 10.1038/emboj.2011.471
92. Chang CK, Chiang MH, Toh EK, Chang CF, Huang TH. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. (2013) 587(6):575–82. doi: 10.1016/j.febslet.2013.01.038
93. Cohen TJ, Hwang AW, Restrepo CR, Yuan CX, Trojanowski JQ, Lee VM. An acetylation switch controls TDP-43 function and aggregation propensity. Nat Commun. (2015) 6:5845. doi: 10.1038/ncomms6845
94. Iguchi Y, Katsuno M, Takagi S, Ishigaki S, Niwa J, Hasegawa M, et al. Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiol Dis. (2012) 45(3):862–70. doi: 10.1016/j.nbd.2011.12.002
95. Zuo X, Zhou J, Li Y, Wu K, Chen Z, Luo Z, et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat Struct Mol Biol. (2021) 28(2):132–42. doi: 10.1038/s41594-020-00537-7
96. Eck RJ, Kraemer BC, Liachko NF. Regulation of TDP-43 phosphorylation in aging and disease. Geroscience. (2021) 43(4):1605–14. doi: 10.1007/s11357-021-00383-5
97. Yang C, Wang H, Qiao T, Yang B, Aliaga L, Qiu L, et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. (2014) 111(12):E1121–9. doi: 10.1073/pnas.1322641111
98. Schmid B, Hruscha A, Hogl S, Banzhaf-Strathmann J, Strecker K, van der Zee J, et al.. Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc Natl Acad Sci U S A. (2013) 110(13):4986–91. doi: 10.1073/pnas.1218311110
99. Mori F, Tada M, Kon T, Miki Y, Tanji K, Kurotaki H, et al. Phosphorylated TDP-43 aggregates in skeletal and cardiac muscle are a marker of myogenic degeneration in amyotrophic lateral sclerosis and various conditions. Acta Neuropathol Commun. (2019) 7(1):165. doi: 10.1186/s40478-019-0824-1
100. Ferrer I, Andrés-Benito P, Carmona M, Assialioui A, Povedano M. TDP-43 Vasculopathy in the spinal cord in sporadic amyotrophic lateral sclerosis (sALS) and frontal cortex in sALS/FTLD-TDP. J Neuropathol Exp Neurol. (2021) 80(3):229–39. doi: 10.1093/jnen/nlaa162
101. Arimoto-Matsuzaki K, Saito H, Takekawa M. TIA1 Oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat Commun. (2016) 7:10252. doi: 10.1038/ncomms10252
102. Sánchez-Jiménez C, Izquierdo JM. T-cell intracellular antigen (TIA)-proteins deficiency in murine embryonic fibroblasts alters cell cycle progression and induces autophagy. PLoS One. (2013) 8(9):e75127. doi: 10.1371/journal.pone.0075127
103. Tak H, Eun JW, Kim J, Park SJ, Kim C, Ji E, et al. T-cell-restricted intracellular antigen 1 facilitates mitochondrial fragmentation by enhancing the expression of mitochondrial fission factor. Cell Death Differ. (2017) 24(1):49–58. doi: 10.1038/cdd.2016.90
104. Hong Y, Tak H, Kim C, Kang H, Ji E, Ahn S, et al. RNA binding protein HuD contributes to β-cell dysfunction by impairing mitochondria dynamics. Cell Death Differ. (2020) 27(5):1633–43. doi: 10.1038/s41418-019-0447-x
105. Yildirim SS, Akman D, Catalucci D, Turan B. Relationship between downregulation of miRNAs and increase of oxidative stress in the development of diabetic cardiac dysfunction: junctin as a target protein of miR-1. Cell Biochem Biophys. (2013) 67(3):1397–408. doi: 10.1007/s12013-013-9672-y
106. Jeyabal P, Thandavarayan RA, Joladarashi D, Suresh Babu S, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun. (2016) 471(4):423–9. doi: 10.1016/j.bbrc.2016.02.065
107. Rawal S, Munasinghe PE, Nagesh PT, Lew JKS, Jones GT, Williams MJA, et al. Down-regulation of miR-15a/b accelerates fibrotic remodelling in the type 2 diabetic human and mouse heart. Clin Sci. (2017) 131(9):847–63. doi: 10.1042/CS20160916
108. Tang Q, Len Q, Liu Z, Wang W Overexpression of miR-22 attenuates oxidative stress injury in diabetic cardiomyopathy via sirt 1. Cardiovasc Ther. (2018) 36(2). doi: 10.1111/1755-5922.12318 [Epub]
109. Raut SK, Kumar A, Singh GB, Nahar U, Sharma V, Mittal A, et al. miR-30c mediates upregulation of Cdc42 and Pak1 in diabetic cardiomyopathy. Cardiovasc Ther. (2015) 33(3):89–97. doi: 10.1111/1755-5922.12113
110. Chen C, Yang S, Li H, Yin Z, Fan J, Zhao Y, et al. Mir30c is involved in diabetic cardiomyopathy through regulation of cardiac autophagy via BECN1. Mol Ther Nucleic Acids. (2017) 7:127–39. doi: 10.1016/j.omtn.2017.03.005
111. Nandi SS, Duryee MJ, Shahshahan HR, Thiele GM, Anderson DR, Mishra PK. Induction of autophagy markers is associated with attenuation of miR-133a in diabetic heart failure patients undergoing mechanical unloading. Am J Transl Res. (2015) 7(4):683–96.26064437
112. Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW 2nd, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med. (2014) 18(3):415–21. doi: 10.1111/jcmm.12218
113. Yu M, Liu Y, Zhang B, Shi Y, Cui L, Zhao X. Inhibiting microRNA-144 abates oxidative stress and reduces apoptosis in hearts of streptozotocin-induced diabetic mice. Cardiovasc Pathol. (2015) 24(6):375–81. doi: 10.1016/j.carpath.2015.06.003
114. Feng B, Chen S, Gordon AD, Chakrabarti S miR-146a mediates inflammatory changes and fibrosis in the heart in diabetes. J Mol Cell Cardiol. (2017) 105:70–6. doi: 10.1016/j.yjmcc.2017.03.002
115. Duan Y, Zhou B, Su H, Liu Y, Du C. miR-150 regulates high glucose-induced cardiomyocyte hypertrophy by targeting the transcriptional co-activator p300. Exp Cell Res. (2013) 319(3):173–84. doi: 10.1016/j.yexcr.2012.11.015
116. Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S. miR-200b mediates endothelial-to-mesenchymal transition in diabetic cardiomyopathy. Diabetes. (2016) 65(3):768–79. doi: 10.2337/db15-1033
117. Shen E, Diao X, Wang X, Chen R, Hu B. MicroRNAs involved in the mitogen-activated protein kinase cascades pathway during glucose-induced cardiomyocyte hypertrophy. Am J Pathol. (2011) 179(2):639–50. doi: 10.1016/j.ajpath.2011.04.034
118. Yu XY, Song YH, Geng YJ, Lin QX, Shan ZX, Lin SG, et al. Glucose induces apoptosis of cardiomyocytes via microRNA-1 and IGF-1. Biochem Biophys Res Commun. (2008) 376(3):548–52. doi: 10.1016/j.bbrc.2008.09.025
119. Shan ZX, Lin QX, Deng CY, Zhu JN, Mai LP, Liu JL, et al. miR-1/miR-206 regulate Hsp60 expression contributing to glucose-mediated apoptosis in cardiomyocytes. FEBS Lett. (2010) 584(16):3592–600. doi: 10.1016/j.febslet.2010.07.027
120. Liu S, Li W, Xu M, Huang H, Wang J, Chen X. Micro-RNA 21Targets dual specific phosphatase 8 to promote collagen synthesis in high glucose-treated primary cardiac fibroblasts. Can J Cardiol. (2014) 30(12):1689–99. doi: 10.1016/j.cjca.2014.07.747
121. Arnold N, Koppula PR, Gul R, Luck C, Pulakat L. Regulation of cardiac expression of the diabetic marker microRNA miR-29. PLoS One. (2014) 9(7):e103284. doi: 10.1371/journal.pone.0103284
122. Li X, Du N, Zhang Q, Li J, Chen X, Liu X, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. (2014) 5(10):e1479. doi: 10.1038/cddis.2014.430
123. Zhao F, Li B, Wei YZ, Zhou B, Wang H, Chen M, et al. MicroRNA-34a regulates high glucose-induced apoptosis in H9c2 cardiomyocytes. J Huazhong Univ Sci Technolog Med Sci. (2013) 33(6):834–9. doi: 10.1007/s11596-013-1207-7
124. Baseler WA, Thapa D, Jagannathan R, Dabkowski ER, Croston TL, Hollander JM. miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am J Physiol Cell Physiol. (2012) 303(12):C1244–51. doi: 10.1152/ajpcell.00137.2012
125. Yi F, Shang Y, Li B, Dai S, Wu W, Cheng L, et al. MicroRNA-193-5p modulates angiogenesis through IGF2 in type 2 diabetic cardiomyopathy. Biochem Biophys Res Commun. (2017) 491(4):876–82. doi: 10.1016/j.bbrc.2017.07.108
126. Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G, et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia. (2015) 58(8):1949–58. doi: 10.1007/s00125-015-3622-8
127. Panguluri SK, Tur J, Chapalamadugu KC, Katnik C, Cuevas J, Tipparaju SM MicroRNA-301a mediated regulation of Kv4.2 in diabetes: identification of key modulators. PLoS One. (2013) 8(4):e60545. doi: 10.1371/journal.pone.0060545
128. Wang XH, Qian RZ, Zhang W, Chen SF, Jin HM, Hu RM. MicroRNA-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clin Exp Pharmacol Physiol. (2009) 36(2):181–8. doi: 10.1111/j.1440-1681.2008.05057.x
129. Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S, et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res. (2015) 116(2):279–88. doi: 10.1161/CIRCRESAHA.116.304707
130. Qiao Y, Zhao Y, Liu Y, Ma N, Wang C, Zou J, et al. miR-483-3p regulates hyperglycaemia-induced cardiomyocyte apoptosis in transgenic mice. Biochem Biophys Res Commun. (2016) 477(4):541–7. doi: 10.1016/j.bbrc.2016.06.051
131. Miao Y, Wan Q, Liu X, Wang Y, Luo Y, Liu D, et al. miR-503 is involved in the protective effect of phase II enzyme inducer (CPDT) in diabetic cardiomyopathy via Nrf2/ARE signaling pathway. Biomed Res Int. (2017) 2017:9167450. doi: 10.1155/2017/9167450
132. Diao X, Shen E, Wang X, Hu B. Differentially expressed microRNAs and their target genes in the hearts of streptozotocin-induced diabetic mice. Mol Med Rep. (2011) 4(4):633–40. doi: 10.3892/mmr.2011.489
133. Corsten MF, Papageorgiou A, Verhesen W, Carai P, Lindow M, Obad S, et al. MicroRNA profiling identifies microRNA-155 as an adverse mediator of cardiac injury and dysfunction during acute viral myocarditis. Circ Res. (2012) 111(4):415–25. doi: 10.1161/CIRCRESAHA.112.267443
134. Jia C, Chen H, Wei M, Chen X, Zhang Y, Cao L, et al. Gold nanoparticle-based miR155 antagonist macrophage delivery restores the cardiac function in ovariectomized diabetic mouse model. Int J Nanomedicine. (2017) 12:4963–79. doi: 10.2147/IJN.S138400
135. Yin Z, Zhao Y, He M, Li H, Fan J, Nie X, et al. MiR-30c/PGC-1beta protects against diabetic cardiomyopathy via PPARalpha. Cardiovasc Diabetol. (2019) 18(1):7. doi: 10.1186/s12933-019-0811-7
136. Blumensatt M, Greulich S, Herzfeld de Wiza D, Mueller H, Maxhera B, Rabelink MJ, et al. Activin A impairs insulin action in cardiomyocytes via up-regulation of miR-143. Cardiovasc Res. (2013) 100(2):201–10. doi: 10.1093/cvr/cvt173
137. Xu Y, Fang H, Xu Q, Xu C, Yang L, Huang C. LncRNA GAS5 inhibits NLRP3 inflammasome activation-mediated pyroptosis in diabetic cardiomyopathy by targeting miR-34b-3p/AHR. Cell Cycle. (2020) 19(22):3054–65. doi: 10.1080/15384101.2020.1831245
138. Li X, Wang H, Yao B, Xu W, Chen J, Zhou X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci Rep. (2016) 6:36340. doi: 10.1038/srep36340
139. Zhuo C, Jiang R, Lin X, Shao M LncRNA H19 inhibits autophagy by epigenetically silencing of DIRAS3 in diabetic cardiomyopathy. Oncotarget. (2017) 8(1):1429–37. doi: 10.18632/oncotarget.13637
140. Gao L, Wang X, Guo S, Xiao L, Liang C, Wang Z, et al. LncRNA HOTAIR functions as a competing endogenous RNA to upregulate SIRT1 by sponging miR-34a in diabetic cardiomyopathy. J Cell Physiol. (2019) 234(4):4944–58. doi: 10.1002/jcp.27296
141. Zhang Y, Zhang YY, Li TT, Wang J, Jiang Y, Zhao Y, et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J Mol Cell Cardiol. (2018) 115:64–72. doi: 10.1016/j.yjmcc.2018.01.001
142. Thomas AA, Feng B, Chakrabarti S. ANRIL regulates production of extracellular matrix proteins and vasoactive factors in diabetic complications. Am J Physiol Endocrinol Metab. (2018) 314(3):E191–200. doi: 10.1152/ajpendo.00268.2017
143. Zheng D, Zhang Y, Hu Y, Guan J, Xu L, Xiao W, et al. Long noncoding RNA crnde attenuates cardiac fibrosis via smad3-crnde negative feedback in diabetic cardiomyopathy. FEBS J. (2019) 286(9):1645–55. doi: 10.1111/febs.14780
144. Feng Y, Xu W, Zhang W, Wang W, Liu T, Zhou X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics. (2019) 9(15):4558–66. doi: 10.7150/thno.31052
145. Yang F, Qin Y, Wang Y, Li A, Lv J, Sun X, et al. LncRNA KCNQ1OT1 mediates pyroptosis in diabetic cardiomyopathy. Cell Physiol Biochem. (2018) 50(4):1230–44. doi: 10.1159/000494576
146. Zhang M, Gu H, Chen J, Zhou X. Involvement of long noncoding RNA MALAT1 in the pathogenesis of diabetic cardiomyopathy. Int J Cardiol. (2016) 202:753–5. doi: 10.1016/j.ijcard.2015.10.019
147. Chen Y, Zhang Z, Zhu D, Zhao W, Li F. Long non-coding RNA MEG3 serves as a ceRNA for microRNA-145 to induce apoptosis of AC16 cardiomyocytes under high glucose condition. Biosci Rep. (2019) 39(6):BSR20190444. doi: 10.1042/BSR20090096
148. Zhou X, Zhang W, Jin M, Chen J, Xu W, Kong X. lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22-3p in diabetic cardiomyopathy. Cell Death Dis. (2017) 8(7):e2929. doi: 10.1038/cddis.2017.321
149. Zou G, Zhong W, Wu F, Wang X, Liu L. Catalpol attenuates cardiomyocyte apoptosis in diabetic cardiomyopathy via Neat1/miR-140-5p/HDAC4 axis. Biochimie. (2019) 165:90–9. doi: 10.1016/j.biochi.2019.05.005
150. Yu M, Shan X, Liu Y, Zhu J, Cao Q, Yang F, et al. RNA-Seq analysis and functional characterization revealed lncRNA NONRATT007560.2 regulated cardiomyocytes oxidative stress and apoptosis induced by high glucose. J Cell Biochem. (2019) 120(10):18278–87. doi: 10.1002/jcb.29134
151. Zhao L, Li W, Zhao H. Inhibition of long non-coding RNA TUG1 protects against diabetic cardiomyopathy induced diastolic dysfunction by regulating miR-499-5p. Am J Transl Res. (2020) 12(3):718–30.32269707
152. Pant T, Dhanasekaran A, Zhao M, Thorp EB, Forbess JM, Bosnjak ZJ, et al. Identification and analysis of circulating long non-coding RNAs with high significance in diabetic cardiomyopathy. Sci Rep. (2021) 11(1):2571. doi: 10.1038/s41598-021-82345-7
153. Fan W, Pang H, Xie Z, Huang G, Zhou Z. Circular RNAs in diabetes mellitus and its complications. Front Endocrinol. (2022) 13:885650. doi: 10.3389/fendo.2022.885650
154. Wan H, Zhao S, Zeng Q, Tan Y, Zhang C, Liu L, et al. CircRNAs in diabetic cardiomyopathy. Clin Chim Acta. (2021) 517:127–32. doi: 10.1016/j.cca.2021.03.001
155. Yuan Q, Sun Y, Yang F, Yan D, Shen M, Jin Z, et al. CircRNA DICAR as a novel endogenous regulator for diabetic cardiomyopathy and diabetic pyroptosis of cardiomyocytes. Signal Transduct Target Ther. (2023) 8(1):99. doi: 10.1038/s41392-022-01306-2
156. Yang F, Li A, Qin Y, Che H, Wang Y, Lv J, et al. A novel circular RNA mediates pyroptosis of diabetic cardiomyopathy by functioning as a competing endogenous RNA. Mol Ther Nucleic Acids. (2019) 17:636–43. doi: 10.1016/j.omtn.2019.06.026
157. Wang W, Zhang S, Xu L, Feng Y, Wu X, Zhang M, et al. Involvement of circHIPK3 in the pathogenesis of diabetic cardiomyopathy in mice. Diabetologia. (2021) 64(3):681–92. doi: 10.1007/s00125-020-05353-8
158. Tang Y, Bao J, Hu J, Liu L, Xu DY. Circular RNA in cardiovascular disease: expression, mechanisms and clinical prospects. J Cell Mol Med. (2021) 25(4):1817–24. doi: 10.1111/jcmm.16203
159. Fu L, Zhang J, Lin Z, Li Y, Qin G. CircularRNA circ_0071269 knockdown protects against from diabetic cardiomyopathy injury by microRNA-145/gasdermin a axis. Bioengineered. (2022) 13(2):2398–411. doi: 10.1080/21655979.2021.2024688
160. Zhou B, Yu JW. A novel identified circular RNA, circRNA_010567, promotes myocardial fibrosis via suppressing miR-141 by targeting TGF-beta1. Biochem Biophys Res Commun. (2017) 487(4):769–75. doi: 10.1016/j.bbrc.2017.04.044
161. Tang CM, Zhang M, Huang L, Hu ZQ, Zhu JN, Xiao Z, et al. CircRNA_000203 enhances the expression of fibrosis-associated genes by derepressing targets of miR-26b-5p, Col1a2 and CTGF, in cardiac fibroblasts. Sci Rep. (2017) 7:40342. doi: 10.1038/srep40342
162. Riechert E, Kmietczyk V, Stein F, Schwarzl T, Sekaran T, Jürgensen L, et al. Identification of dynamic RNA-binding proteins uncovers a Cpeb4-controlled regulatory cascade during pathological cell growth of cardiomyocytes. Cell Rep. (2021) 35(6):109100. doi: 10.1016/j.celrep.2021.109100
163. Liao Y, Castello A, Fischer B, Leicht S, Föehr S, Frese CK, et al. The cardiomyocyte RNA-binding proteome: links to intermediary metabolism and heart disease. Cell Rep. (2016) 16(5):1456–69. doi: 10.1016/j.celrep.2016.06.084
164. Li J, Xie D, Huang J, Lv F, Shi D, Liu Y, et al. Cold-inducible RNA-binding protein regulates cardiac repolarization by targeting transient outward potassium channels. Circ Res. (2015) 116(10):1655–9. doi: 10.1161/CIRCRESAHA.116.306287
165. Freyermuth F, Rau F, Kokunai Y, Linke T, Sellier C, Nakamori M, et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat Commun. (2016) 7:11067. doi: 10.1038/ncomms11067
166. Zhang Y, Si Y, Ma N, Mei J. The RNA-binding protein PCBP2 inhibits ang II-induced hypertrophy of cardiomyocytes though promoting GPR56 mRNA degeneration. Biochem Biophys Res Commun. (2015) 464(3):679–84. doi: 10.1016/j.bbrc.2015.06.139
167. Sun S, Zhang M, Lin J, Hu J, Zhang R, Li C, et al. Lin28a protects against diabetic cardiomyopathy via the PKA/ROCK2 pathway. Biochem Biophys Res Commun. (2016) 469(1):29–36. doi: 10.1016/j.bbrc.2015.11.065
168. You P, Cheng Z, He X, Deng J, Diao J, Chen H, et al. Lin28a protects against diabetic cardiomyopathy through Mst1 inhibition. J Cell Physiol. (2020) 235(5):4455–65. doi: 10.1002/jcp.29321
169. Verma SK, Deshmukh V, Liu P, Nutter CA, Espejo R, Hung ML, et al. Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling. J Biol Chem. (2013) 288(49):35372–86. doi: 10.1074/jbc.M113.507426
170. Nutter CA, Jaworski EA, Verma SK, Deshmukh V, Wang Q, Botvinnik OB, et al. Dysregulation of RBFOX2 is an early event in cardiac pathogenesis of diabetes. Cell Rep. (2016) 15(10):2200–13. doi: 10.1016/j.celrep.2016.05.002
171. Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum Mol Genet. (2010) 19(6):1066–75. doi: 10.1093/hmg/ddp570
172. Nutter CA, Jaworski E, Verma SK, Perez-Carrasco Y, Kuyumcu-Martinez MN. Developmentally regulated alternative splicing is perturbed in type 1 diabetic skeletal muscle. Muscle Nerve. (2017) 56(4):744–9. doi: 10.1002/mus.25599
173. D'Antonio M, Nguyen JP, Arthur TD, Matsui H, Donovan MKR, DȧAntonio-Chronowska A, et al. In heart failure reactivation of RNA-binding proteins is associated with the expression of 1,523 fetal-specific isoforms. PLoS Comput Biol. (2022) 18(2):e1009918. doi: 10.1371/journal.pcbi.1009918
174. Yang C, Eleftheriadou M, Kelaini S, Morrison T, González MV, Caines R, et al. Targeting QKI-7 in vivo restores endothelial cell function in diabetes. Nat Commun. (2020) 11(1):3812. doi: 10.1038/s41467-020-17468-y
175. Noveroske JK, Lai L, Gaussin V, Northrop JL, Nakamura H, Hirschi KK, et al. Quaking is essential for blood vessel development. Genesis. (2002) 32(3):218–30. doi: 10.1002/gene.10060
176. Guo W, Shi X, Liu A, Yang G, Yu F, Zheng Q, et al. RNA binding protein QKI inhibits the ischemia/reperfusion-induced apoptosis in neonatal cardiomyocytes. Cell Physiol Biochem. (2011) 28(4):593–602. doi: 10.1159/000335755
177. Chen X, Liu Y, Xu C, Ba L, Liu Z, Li X, et al. QKI is a critical pre-mRNA alternative splicing regulator of cardiac myofibrillogenesis and contractile function. Nat Commun. (2021) 12(1):89. doi: 10.1038/s41467-020-20327-5
178. Gupta Shashi K, Garg A, Bär C, Chatterjee S, Foinquinos A, Milting H, et al. Quaking inhibits doxorubicin-mediated cardiotoxicity through regulation of cardiac circular RNA expression. Circ Res. (2018) 122(2):246–54. doi: 10.1161/CIRCRESAHA.117.311335
179. Wei C, Qiu J, Zhou Y, Xue Y, Hu J, Ouyang K, et al. Repression of the central splicing regulator RBFox2 is functionally linked to pressure overload-induced heart failure. Cell Rep. (2015) 10(9):1521–33. doi: 10.1016/j.celrep.2015.02.013
180. Gallagher TL, Arribere JA, Geurts PA, Exner CR, McDonald KL, Dill KK, et al. Rbfox-regulated alternative splicing is critical for zebrafish cardiac and skeletal muscle functions. Dev Biol. (2011) 359(2):251–61. doi: 10.1016/j.ydbio.2011.08.025
181. Verma SK, Deshmukh V, Nutter CA, Jaworski E, Jin W, Wadhwa L, et al. Rbfox2 function in RNA metabolism is impaired in hypoplastic left heart syndrome patient hearts. Sci Rep. (2016) 6(1):30896. doi: 10.1038/srep30896
182. Zhang M, Sun D, Li S, Pan X, Zhang X, Zhu D, et al. Lin28a protects against cardiac ischaemia/reperfusion injury in diabetic mice through the insulin-PI3K-mTOR pathway. J Cell Mol Med. (2015) 19(6):1174–82. doi: 10.1111/jcmm.12369
183. Krishnamurthy P, Lambers E, Verma S, Thorne T, Qin G, Losordo DW, et al. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. Faseb J. (2010) 24(7):2484–94. doi: 10.1096/fj.09-149815
184. Li Y, Peng T, Li L, Wang X, Duan R, Gao H, et al. MicroRNA-9 regulates neural apoptosis in methylmalonic acidemia via targeting BCL2L11. Int J Dev Neurosci. (2014) 36:19–24. doi: 10.1016/j.ijdevneu.2014.04.005
185. Du JK, Cong BH, Yu Q, Wang H, Wang L, Wang CN, et al. Upregulation of microRNA-22 contributes to myocardial ischemia-reperfusion injury by interfering with the mitochondrial function. Free Radic Biol Med. (2016) 96:406–17. doi: 10.1016/j.freeradbiomed.2016.05.006
186. Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. (2008) 105(36):13421–6. doi: 10.1073/pnas.0801613105
187. Purohit PK, Edwards R, Tokatlidis K, Saini N. MiR-195 regulates mitochondrial function by targeting mitofusin-2 in breast cancer cells. RNA Biol. (2019) 16(7):918–29. doi: 10.1080/15476286.2019.1600999
188. Zhu H, Li X, Song Y, Zhang P, Xiao Y, Xing Y. Long non-coding RNA ANRIL is up-regulated in bladder cancer and regulates bladder cancer cell proliferation and apoptosis through the intrinsic pathway. Biochem Biophys Res Commun. (2015) 467(2):223–8. doi: 10.1016/j.bbrc.2015.10.002
189. Gao J, Liu M, Zou Y, Mao M, Shen T, Zhang C, et al. Long non-coding RNA growth arrest-specific transcript 5 is involved in ovarian cancer cell apoptosis through the mitochondria-mediated apoptosis pathway. Oncol Rep. (2015) 34(6):3212–21. doi: 10.3892/or.2015.4318
190. Kong L, Zhou X, Wu Y, Wang Y, Chen L, Li P, et al. Targeting HOTAIR induces mitochondria related apoptosis and inhibits tumor growth in head and neck squamous cell carcinoma in vitro and in vivo. Curr Mol Med. (2015) 15(10):952–60. doi: 10.2174/1566524016666151123112716
191. Wang M, Huang T, Luo G, Huang C, Xiao XY, Wang L, et al. Long non-coding RNA MEG3 induces renal cell carcinoma cells apoptosis by activating the mitochondrial pathway. J Huazhong Univ Sci Technolog Med Sci. (2015) 35(4):541–5. doi: 10.1007/s11596-015-1467-5
192. Bai X, Yang C, Jiao L, Diao H, Meng Z, Wang L, et al. LncRNA MIAT impairs cardiac contractile function by acting on mitochondrial translocator protein TSPO in a mouse model of myocardial infarction. Signal Transduct Target Ther. (2021) 6(1):172. doi: 10.1038/s41392-021-00538-y
193. Talwar S, Balasubramanian S, Sundaramurthy S, House R, Wilusz CJ, Kuppuswamy D, et al. Overexpression of RNA-binding protein CELF1 prevents apoptosis and destabilizes pro-apoptotic mRNAs in oral cancer cells. RNA Biol. (2013) 10(2):277–86. doi: 10.4161/rna.23315
194. Bae JE, Park SJ, Hong Y, Jo DS, Lee H, Park NY, et al. Loss of RNA binding protein, human antigen R enhances mitochondrial elongation by regulating Drp1 expression in SH-SY5Y cells. Biochem Biophys Res Commun. (2019) 516(3):713–8. doi: 10.1016/j.bbrc.2019.06.091
195. Chen J, Liu W. Lin28a induced mitochondrial dysfunction in human granulosa cells via suppressing LARS2 expression. Cell Signal. (2023) 103:110536. doi: 10.1016/j.cellsig.2022.110536
196. Cao J, Verma SK, Jaworski E, Mohan S, Nagasawa CK, Rayavara K, et al. RBFOX2 is critical for maintaining alternative polyadenylation patterns and mitochondrial health in rat myoblasts. Cell Rep. (2021) 37(5):109910. doi: 10.1016/j.celrep.2021.109910
197. Xu X, Sun S. H19 promotes vascular smooth muscle cell proliferation by releasing miR-675-5p to target mitofusin-2. Atheroscler Suppl. (2018) 32:103. doi: 10.1016/j.atherosclerosissup.2018.04.315
198. Aggarwal R, Potel KN, McFalls EO, Butterick TA, Kelly RF. Novel therapeutic approaches enhance PGC1-alpha to reduce oxidant stress-inflammatory signaling and improve functional recovery in hibernating myocardium. Antioxidants. (2022) 11(11):2155. doi: 10.3390/antiox11112155
199. Bucha S, Mukhopadhyay D, Bhattacharyya NP. Regulation of mitochondrial morphology and cell cycle by microRNA-214 targeting Mitofusin2. Biochem Biophys Res Commun. (2015) 465(4):797–802. doi: 10.1016/j.bbrc.2015.08.090
200. Maity S, Das F, Ghosh-Choudhury N, Kasinath BS, Ghosh Choudhury G. High glucose increases miR-214 to power a feedback loop involving PTEN and the akt/mTORC1 signaling axis. FEBS Lett. (2019) 593(16):2261–72. doi: 10.1002/1873-3468.13505
201. Wang K, Lin ZQ, Long B, Li JH, Zhou J, Li PF. Cardiac hypertrophy is positively regulated by MicroRNA miR-23a. J Biol Chem. (2012) 287(1):589–99. doi: 10.1074/jbc.M111.266940
202. Eisele PS, Salatino S, Sobek J, Hottiger MO, Handschin C. The peroxisome proliferator-activated receptor gamma coactivator 1alpha/beta (PGC-1) coactivators repress the transcriptional activity of NF-kappaB in skeletal muscle cells. J Biol Chem. (2013) 288(4):2246–60. doi: 10.1074/jbc.M112.375253
203. Viereck J, Thum T. Circulating noncoding RNAs as biomarkers of cardiovascular disease and injury. Circ Res. (2017) 120(2):381–99. doi: 10.1161/CIRCRESAHA.116.308434
204. Chi T, Lin J, Wang M, Zhao Y, Liao Z, Wei P. Non-coding RNA as biomarkers for type 2 diabetes development and clinical management. Front Endocrinol. (2021) 12:630032. doi: 10.3389/fendo.2021.630032
205. Liu B, Cong C, Ma Y, Ma X, Zhang H, Wang J. Potential value of lncRNAs as a biomarker for proliferative diabetic retinopathy. Eye. (2022) 36(3):575–84. doi: 10.1038/s41433-021-01507-z
206. Biswas S, Coyle A, Chen S, Gostimir M, Gonder J, Chakrabarti S. Expressions of serum lncRNAs in diabetic retinopathy—a potential diagnostic tool. Front Endocrinol. (2022) 13:851967. doi: 10.3389/fendo.2022.851967
207. de Gonzalo-Calvo D, Kenneweg F, Bang C, Toro R, van der Meer RW, Rijzewijk LJ, et al. Circulating long-non coding RNAs as biomarkers of left ventricular diastolic function and remodelling in patients with well-controlled type 2 diabetes. Sci Rep. (2016) 6:37354. doi: 10.1038/srep37354
208. Satake E, Pezzolesi MG, Md Dom ZI, Smiles AM, Niewczas MA, Krolewski AS. Circulating miRNA profiles associated with hyperglycemia in patients with type 1 diabetes. Diabetes. (2018) 67(5):1013–23. doi: 10.2337/db17-1207
209. Higuchi C, Nakatsuka A, Eguchi J, Teshigawara S, Kanzaki M, Katayama A, et al. Identification of circulating miR-101, miR-375 and miR-802 as biomarkers for type 2 diabetes. Metab Clin Exp. (2015) 64(4):489–97. doi: 10.1016/j.metabol.2014.12.003
210. Winkle M, El-Daly SM, Fabbri M, Calin GA. Noncoding RNA therapeutics—challenges and potential solutions. Nat Rev Drug Discov. (2021) 20(8):629–51. doi: 10.1038/s41573-021-00219-z
211. Granéli C, Hicks R, Brolén G, Synnergren J, Sartipy P. Diabetic cardiomyopathy modelling using induced pluripotent stem cell derived cardiomyocytes: recent advances and emerging models. Stem Cell Rev Rep. (2019) 15(1):13–22. doi: 10.1007/s12015-018-9858-1
212. Purnama U, Castro-Guarda M, Sahoo OS, Carr CA. Modelling diabetic cardiomyopathy: using human stem cell-derived cardiomyocytes to complement animal models. Metabolites. (2022) 12(9):832. doi: 10.3390/metabo12090832
213. Wong LY, Glatz JFC, Wang S, Geraets IME, Vanherle S, Wijngaard AVD, et al.. Comparison of human and rodent cell models to study myocardial lipid-induced insulin resistance. Prostaglandins Leukot Essent Fatty Acids. (2021) 167:102267. doi: 10.1016/j.plefa.2021.102267
214. Davis RP, van den Berg CW, Casini S, Braam SR, Mummery CL. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol Med. (2011) 17(9):475–84. doi: 10.1016/j.molmed.2011.05.001
215. Parrotta EI, Lucchino V, Scaramuzzino L, Scalise S, Cuda G. Modeling cardiac disease mechanisms using induced pluripotent stem cell-derived cardiomyocytes: progress, promises and challenges. Int J Mol Sci. (2020) 21(12):4354. doi: 10.3390/ijms21124354
216. Wimmer RA, Leopoldi A, Aichinger M, Wick N, Hantusch B, Novatchkova M, et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature. (2019) 565(7740):505–10. doi: 10.1038/s41586-018-0858-8
217. Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, Weber M, et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. (2014) 9(3):810–21. doi: 10.1016/j.celrep.2014.09.055
218. McKnight CL, Low YC, Elliott DA, Thorburn DR, Frazier AE. Modelling mitochondrial disease in human pluripotent stem cells: what have we learned? Int J Mol Sci. (2021) 22(14):7730. doi: 10.3390/ijms22147730
219. Li S, Pan H, Tan C, Sun Y, Song Y, Zhang X, et al. Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient iPSC-derived cardiomyocytes with MT-RNR2 mutation. Stem Cell Rep. (2018) 10(3):808–21. doi: 10.1016/j.stemcr.2018.01.013
220. Sidhaye J, Trepte P, Sepke N, Novatchkova M, Schutzbier M, Durnberger G, et al. Integrated transcriptome and proteome analysis reveals posttranscriptional regulation of ribosomal genes in human brain organoids. Elife. (2023) 12:e85135. doi: 10.7554/eLife.85135
Keywords: diabetes mellitus, diabetic cardiomyopathy, mitochondrial dysfunction, noncoding RNAs, RNA-binding proteins, stem cell modelling
Citation: Potel KN, Cornelius VA, Yacoub A, Chokr A, Donaghy CL, Kelaini S, Eleftheriadou M and Margariti A (2023) Effects of non-coding RNAs and RNA-binding proteins on mitochondrial dysfunction in diabetic cardiomyopathy. Front. Cardiovasc. Med. 10:1165302. doi: 10.3389/fcvm.2023.1165302
Received: 13 February 2023; Accepted: 15 August 2023;
Published: 1 September 2023.
Edited by:
Paola Campagnolo, University of Surrey, United KingdomReviewed by:
Yasuhide Kuwabara, Cincinnati Children’s Hospital Medical Center, United States© 2023 Potel, Cornelius, Yacoub, Chokr, Donaghy, Kelaini, Eleftheriadou and Margariti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andriana Margariti YS5tYXJnYXJpdGlAcXViLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.