 Weifeng Lu1,†
Weifeng Lu1,† Lixin Wang
Lixin Wang- 1Department of Vascular Surgery, Zhongshan Hospital (Xiamen), Fudan University, Xiamen, China
- 2Division of Vascular Surgery, Department of Surgery, University of Arizona, Tucson, AZ, United States
- 3Department of Vascular Surgery, Zhongshan Hospital, Fudan University, Shanghai, China
Resistin has been shown to play a key role in inducing vascular smooth muscle cells (VSMCs) malfunction in the atherosclerosis progression. Ginsenoside Rb1 is the main component of ginseng, which has been used for thousands of years and has been reported to have a powerful vascular protective effect. The aim of this study was to explore the protective effect of Rb1 on VSMCs dysfunction induced by resistin. In the presence or absence of Rb1, human coronary artery smooth muscle cells (HCASMC) were treated at different time points with or without 40 ng/ml resistin and acetylated low-density lipoprotein (acetylated LDL). Cell migration and proliferation were analyzed using wound healing test and CellTiter Aqueous Cell Proliferation Assay (MTS) test, respectively. Intracellular reactive oxygen species (ROS) (H2DCFDA as a dye probe) and superoxide dismutase (SOD) activities were measured by a microplate reader and the differences between groups were compared. Rb1 significantly reduced resistin-induced HCASMC proliferation. Resistin increased HCASMC migration time-dependently. At 20 µM, Rb1 could significantly reduce HCASMC migration. Resistin and Act-LDL increased ROS production to a similar level in HCASMCs, while Rb1 pretreated group reversed the effects of resistin and acetyl-LDL. Besides, the mitochondrial SOD activity was significantly reduced by resistin but was restored when pretreated with Rb1. We confirmed the protection of Rb1 on HCASMC and suggested that the mechanisms involved might be related to the reduction of ROS generation and increased activity of SOD. Our study clarified the potential clinical applications of Rb1 in the control of resistin-related vascular injury and in the treatment of cardiovascular disease.
Introduction
Ginseng is a traditional herbal adaptation, which has a history of more than 5,000 years. It is widely used in Asia and also in western medicine. At present, it is simply made into a drink to improve physical performance and treat many chronic diseases, such as cancer and cardiovascular, inflammatory, neuronal, and metabolic diseases (1). In the United States of America, ginseng ranks second and fifth in use among adult best-selling natural products in 2002 and 2007, respectively (2). Ginsenoside, a sterol glycoside, is found only in ginseng, which are divided into two groups: Rb1 group and Rg1 group. Ginsenoside Rb1 (Rb1) is the main active components from ginseng, the pharmacological effects of which attract much attention in the endocrine, cardiovascular, and immune systems in recent years (3–9).
Cardiovascular diseases (CVD) are now one of the leading causes of disability and death in the world, especially in the elderly population. Recently, accumulating evidence has proven that Rb1 demonstrated its cardioprotective effect both in vitro and in vivo. We first reported that Rb1 could block homocysteine-induced endothelial dysfunction and superoxide anion production effectively in porcine coronary arteries by in vitro studies (5). It was reported that Rb1 could also protect human endothelial cells from oxidized low-density lipoprotein (oxLDL) (6). In vivo studies showed that pretreatment with Rb1 reduced cardiomyocyte apoptosis induced by myocardial ischemia/reperfusion injury in diabetic rats and alleviated cardiac dysfunction (7). In a rat model, Rb1 was proved to inhibit the carotid intima hyperplasia induced by balloon dilation (8). Rb1 administration promoted atherosclerotic plaque stability along with increased macrophage autophagy and M2 phenotype in the atherosclerosis model in ApoE−/− mice (4, 9). However, few studies examined the protective effect of Rb1 on vascular smooth muscle cells (VSMCs), especially on human VSMCs.
The key feature of vascular proliferation diseases is the abnormal proliferation and migration of VSMC, including atherosclerosis, restenosis after vascular injury and vascular wall hypertrophy caused by hypertension.(10–12) Resistin is an adipokine mainly expressed in human monocyte/macrophage lineage cells, which is closely related to cardiovascular diseases and adverse clinical outcomes (13). Resistin has been proved to act in a variety of cells including VSMCs, indicating that resistin has a certain role in atherosclerosis (14). We and others demonstrated that resistin at a pathological concentration promotes vascular smooth muscle cell (VSMC) proliferation and migration, as well as reactive oxygen species (ROS) production (15–17). We also demonstrated that inflammation was induced in VSMCs and VSMCs dysfunction by resistin through protein kinase C epsilon (PKCε)-mediated NADPH oxidase (Nox) activation (17). However, the role of Rb1 in VSMCS dysfunction and related mechanisms by resitin has not been investigated and needs further confirmation. Considering the protective role of Rb1 in cardiovascular disease, we hypothesized that Rb1 may protect the progression of atherosclerosis by reversing resistin induced VSMCs dysfunction and oxidative stress. To test the hypothesis, we tested the proliferation and migration of VSMCs induced by resistin with or without Rb1. We also validated the related mechanism was associated with enhancing antioxidation property of Rb1 by reducing ROS production and increasing superoxide dismutase (SOD) activity in VSMCs.
Methods
In vitro treatment and cell culture
Experiments were performed using human coronary artery smooth muscle cells (HCASMC) or VSMCs from Genlantis (18) at passage 5–7. Before each experiment, the medium was replaced to Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 2% FBS for at least 8 h (hs) to starve the cells. In our study, we selected a pathological resistin level (40 ng/ml) which based on the published data of resistin in human subjects and our previous study, indicating that this concentration leads to VSMCs dysfunction and intimal hyperplasia (17, 19). Cells were managed with or without resistin at 40 ng/ml in the presence or absence of ginsenoside Rb1 (G0777-5MG, Sigma-Aldrich) for various time points at 20 µM according to previous research (n = 5) (4). Acetylated low-density lipoprotein (acetyl-LDL) was also used to treat cells in proliferation and ROS experiments because it is known to accumulate in macrophages to form foam cells and play a critical role in the mechanism of atherosclerosis by stimulating the proliferation of VSMCs (20).
Cell proliferation assay
The cells treated with resistin (40 ng/ml) or acetyl-LDL (20 µg/ml) in the presence or absence of Rb1 (20 µM) for 1 h were seeded in a 96-well plate (5,000 cells/well) and incubated for an extra 24 h. Cell proliferation was evaluated by the usage of CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS assay, Promega). Briefly, 20 ml MTS was added to the cells in each well containing the samples in 100 µl of culture medium and cultured for 4 h at 37°C in a humidified 5% CO2 atmosphere. The cell plate covered with tinfoil was then subjected to a plate reader to fetch the absorbance at 490 nm. The formula of cell viability was calculated as follows: Cell Viability = (ODsample−ODbackground)/(ODcontrol−ODbackground) × 100%.
Cell migration assay
VSMCs migrations were evaluated by wound healing assay and transwell assay. VSMCs were grown and converged in a 6-well plate. A transverse scratch wound on each monolayer of VSMC was made by using a sterilized 200 µl-tip. The scratched VSMCs were then stimulated with or without resistin (40 ng/ml) in the presence or absence of Rb1 (20 µM) for an additional 6, 18, and 24 h, then the transverse scratch wounds were reexamined for cell migration. Pictures were captured with a phase-contrast microscope, and cell migration was quantified using ImageJ software, which was calculated as the percent of the wound closure area relative to that at the start point (t = 0).
Transwell assay was performed in Transwell chambers (HTS Transwell-96 Well Plate, 5 µm pore size; Corning Inc., USA). Briefly, 1 × 105/ml cells were seeded on the Transwell Inserts in 75 µl of serum-free medium with or without Rb1 (20 µM). The bottom plate contained 235 µl of complete growth medium with resistin at 40 ng/ml. Cells migrated at 37°C for 24 h. After 24 h of culture, the non-migrated cells on the membrane top surface were removed with a cotton swab. After that, the migrated cells on the bottom side of the membrane were stained with 0.1% crystal violet and then destained with PBS.
Intracellular ROS evaluation
Intracellular ROS were evaluated using the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (ab113851, Abcam) according to the supplier's protocol. Briefly, cells were seeded in a 96-well plate (2.5 × 104/well) overnight, then incubated with the probe (25 µM final concentration) for 45 min at 37°C in a humidified, 5% CO2 atmosphere. Then the cells were washed in PBS and treated with or without resistin or acetyl-LDL in supplemented buffer containing 10% fetal bovine serum (FBS) for 2 h. Rb1 was added at the same time (Co-treatment group) or 24 h before the addition of resistin or acetyl-LDL (Pre-treatment group). The pretreatment group was set because we found the exposure time of 2 h was not enough for Rb1 to take effect. The fluorescence of the cell homogenate was measured on a fluorescence plate reader at Ex/Em = 485/535 nm at the end point. The data are expressed as the ratio of fluorescence/fluorescence of the unstimulated control. Tert-Butyl Hydrogen Peroxide (TBHP) was used as a positive control.
Measurement of antioxidant enzyme activity
In order to elucidate the effects of Rb1 after resistin exposure, SOD activities in the homogenate were measured by an enzymatic assay using a commercial kit (706,002, Cayman Chemical) according to the manufacturer's instructions. In brief, cells seeded in a 6-well plate were pretreated with Rb1 for 24 h and then exposed to resistin for 2 h. The cells in each group were harvested using a rubber policeman and sonicated in cold 20 mM Hepes (PH 7.2, containing 1 mM EGTA, 210 mM mannitol, and 70 mM sucrose). Then the cell lysates were used for assaying total SOD activity (cytosolic and mitochondrial). To separate these two enzymes, the cell lysates were centrifugated at 10,000×g for 15 min at 4°C. The resulting 10,000×g supernatant contained cytosolic SOD and the pellet contained mitochondrial SOD. For SOD assay, 200 µl of radical detector and 10 µl of SOD standard with different concentrations or 10 µl of sample were added to each designated well. The reactions were initiated by adding 20 µl of xanthine oxidase to all wells except the sample background. The 96-well plate was incubated on a shaker for 30 min at room temperature. The absorbance was read at 450 nm by a plate reader. The SOD activity of the samples was calculated from the linear regression of the standard curve. One unit of SOD is defined as the amount of enzyme needed to exhibit 50% dismutation of the superoxide radical.
Statistical analysis
The numeric data are presented as mean ± SD. Differences between groups were analyzed by one-way ANOVA or an independent sample t-test. If the p-value is less than 0.05, the results are considered to be statistically different.
Results
Rb1 ameliorated resistin-stimulated HCASMC proliferation
We first examined the effect of Rb1 on resistin-stimulated HCASMC proliferation by MTS assay. Data on the percentage of viable cells after treatment revealed that both resistin and acetyl-LDL stimulated HCASMC proliferation significantly. Rb1 alone had no effect on the proliferation of HCASMC, however, it significantly reduced resistin-induced HCASMC proliferation (Figure 1). Rb1 also ameliorated acetyl-LDL induced proliferation of HCASMC, although no statistical significance had been found in our data.
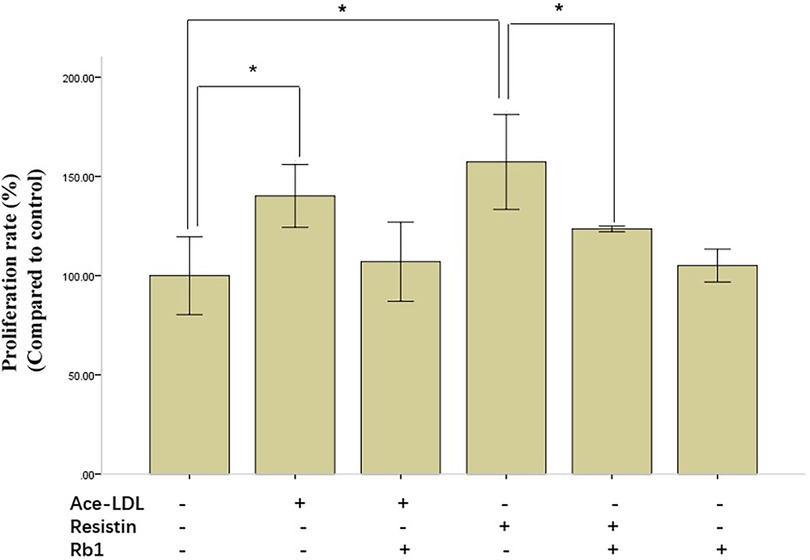
Figure 1. Resistin-induced human coronary artery smooth muscle cells (HCASMC) proliferation was ameliorated by Rb1. HCASMC proliferation, evaluated by MTS assay, was significantly increased by resistin and acetyl-LDL, which could be restored by Rb1. Note that Rb1 alone had no effect on proliferation of HCASMC. Cell Viability = (ODs−ODb)/(ODc−ODb) × 100%. ODs, the OD value of the samples; ODb, the OD value of the background; ODc, the OD value of the negative control. Values were means and SD (n = 5). *P < 0.05.
Rb1 suppressed resistin-induced HCASMC migration
To elucidate the effects of Rb1 on HCASMC migration, a wound healing assay was used. The wound healing assay revealed resistin at 40 ng/ml treatment time dependently increased VSMCs migration, which was attenuated significantly by Rb1 at 20 µM at 18 h and 24 h (Figure 2). A transwell migration chamber assay was also used to further identify the chemotaxis effect of resistin with or without Rb1. As shown in Figure 2, resistin increased the migratory ability of HCASMCs, whereas Rb1 inhibited the resistin-induced HCASMC migration.
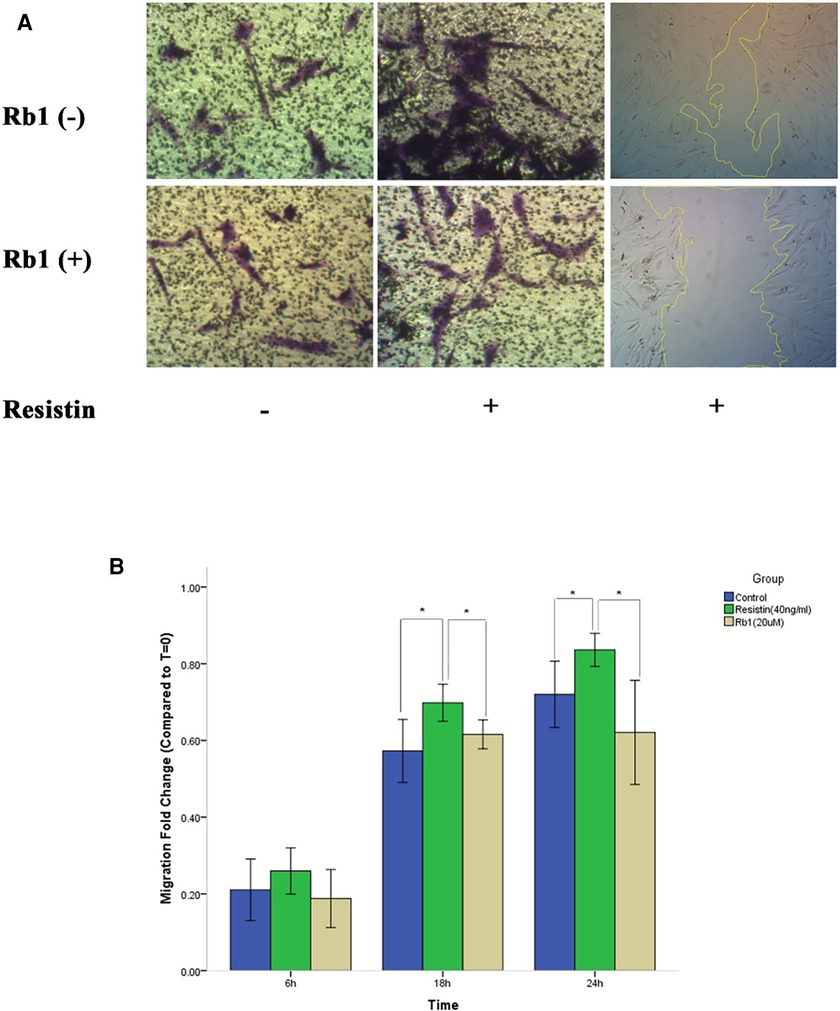
Figure 2. Rb1 inhibited resistin-induced HCASMC migration. (A) Transwell migration chamber assay and wound healing assay showed resistin-induced HCASMC migration was inhibited by Rb1. (B) Cell migration was calculated as the fold change of wound closure relative to controls (T = 0). Resistin (40 ng/ml) treatment time dependently increased HCASMC migration, which could be significantly attenuated by Rb1 (20 µM) at 18 h and 24 h (B). Values were means and SD (n = 5). * P < 0.05.
Effect of Rb1 on intracellular ROS production of HCASMC
It has been well-documented that high expressed ROS could lead to destructive impact on both differentiated EC and VSMC (21). Since ROS generations are key instigators in the pathophysiology of resistin-associated VSMC dysfunction and intimal hyperplasia (14), we determined whether Rb1 was able to inhibit resistin-induced ROS production in HCASMC by DCF-DA fluorescence assay. We found that both resistin and acetyl-LDL could increase ROS production in HCASMC to a similar level, and while pretreated with Rb1, ROS production induced by resistin and acetyl-LDL were reduced significantly (Figure 3). In addition, we proved that both Rb1 and resistin have dose-dependent effects on ROS production in HCASMC, suggesting Rb1 at a concentration of 20 µM could significantly reduce resistin (40 ng/ml) induced cytosolic ROS in VSMCs (Figure 4). It was interesting to find that Rb1 pretreatment was more effective than co-treatment in restoring ROS levels induced by resistin in HCASMC (Figure 3).
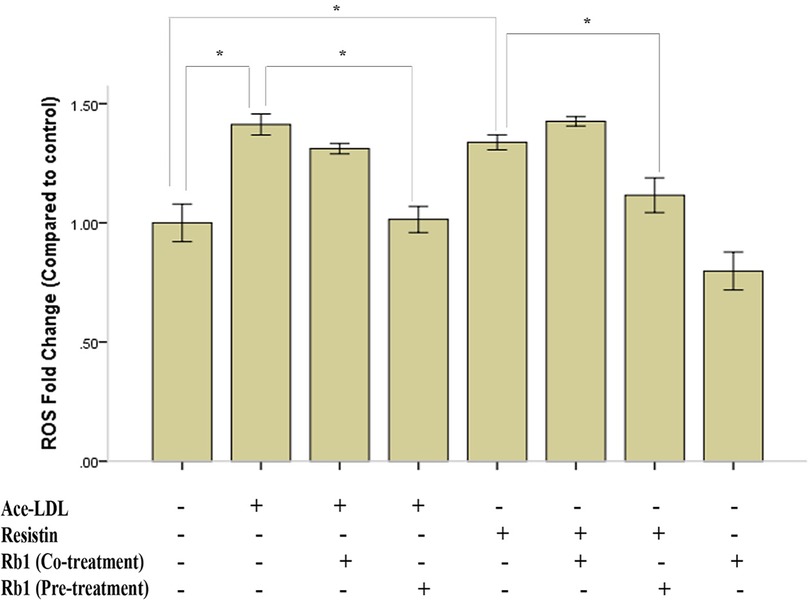
Figure 3. Rb1 pretreatment reduced resistin-induced ROS production in HCASMC. HCASMC ROS production was detected by DCF-DA fluorescence assay. After treated with resistin (40 ng/ml) or acetyl-LDL (20 µg/ml) for 2 h, the intracellular ROS were significantly increased to a similar level. Rb1 pretreatment (24 h) significantly reduced HCASMC ROS generation induced by resistin and acetyl-LDL, while Rb1 co-treatment (2 h) had no such effect. Values were means and SD (n = 5). *P < 0.05.
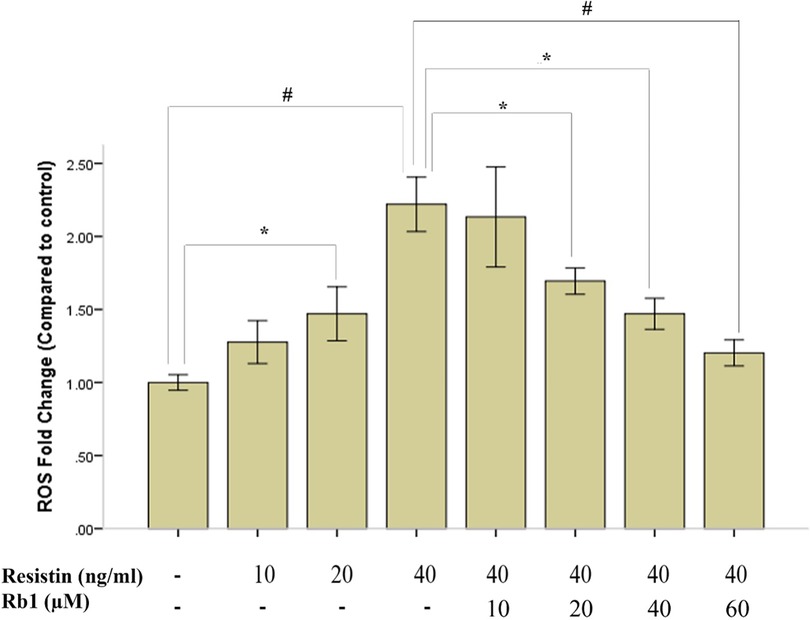
Figure 4. Rb1 pretreatment dose dependently inhibited resistin-induced HCASMC ROS generation. Resistin dose dependently increased ROS generation in HCASMC, which could be significantly inhibited by Rb1 (>20 µM) in a dose-dependent manner. Values were means and SD (n = 5). *P < 0.05, #P < 0.01.
Rb1 attenuated resistin-induced suppression of mitochondrial SOD activity in HCASMC
In order to clarify the pathogenesis of the antioxidation of Rb1 in HCASMC exposed to resistin, the catalytic activity of SOD was analyzed by the measurement of dismutation of superoxide radical in HCASMC that had been treated with Rb1 for 24 h followed by 40 ng/ml of resistin for additional 2 h. Resistin reduced the total SOD activity in HCASMC which was shown in Figure 5; however, pretreatment with Rb1 (20 µM) significantly attenuated the resistin-induced suppression of the total SOD activity. Rb1 alone was capable to exert its antioxidation by increasing the SOD activity in HCASMC. To further target the specific part of HCASMC on which resistin and Rb1 took effect, we analyzed the cytosolic and mitochondrial SOD activity within HCASMC. Our data revealed that the mitochondrial SOD activity was significantly reduced by resistin but was significantly increased when pretreated by Rb1 (P < 0.01). This suggested resistin might increase ROS production of HCASMC partially through reducing the mitochondrial SOD activity, which could be significantly reversed by Rb1.
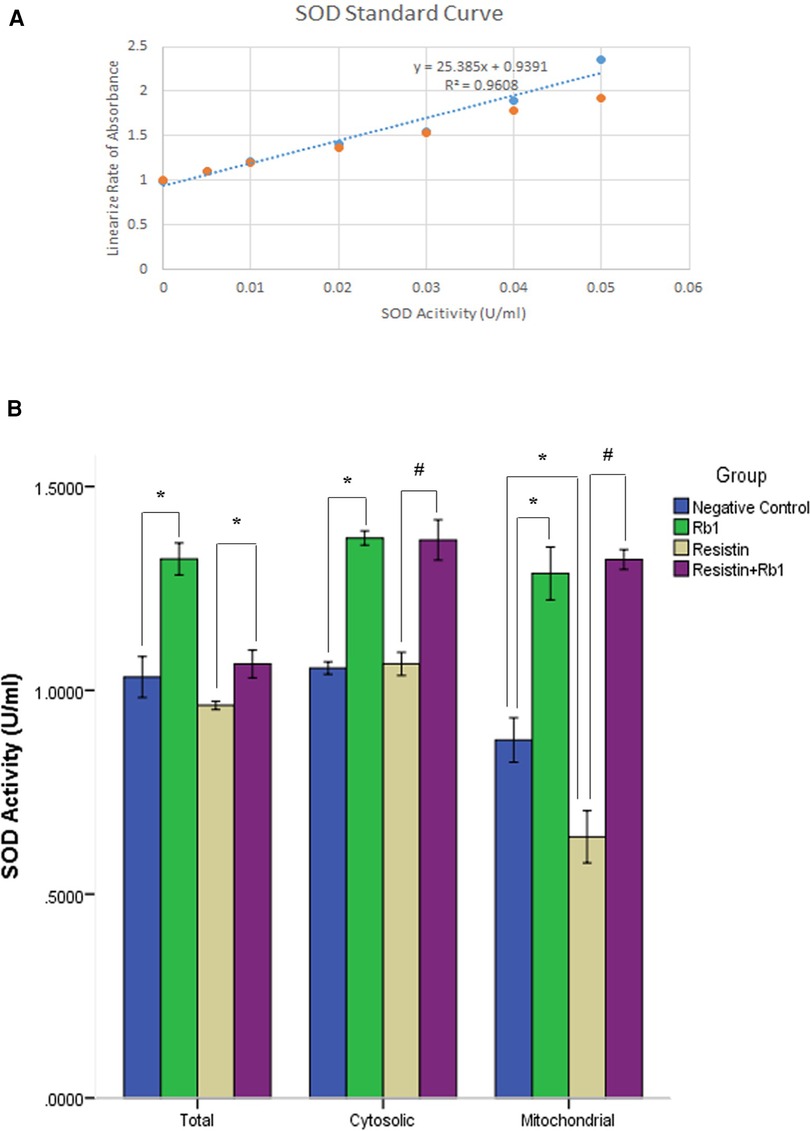
Figure 5. Rb1 attenuated resistin-induced suppression of HCASMC SOD activity. (A) The SOD activity of the samples was calculated from the linear regression of the standard curve. One unit of SOD is defined as the amount of enzyme needed to exhibit 50% dismutation of superoxide radical. (B) The mitochondrial SOD activity was significantly reduced by resistin. In contrast, Rb1 significantly increased both cytosolic and mitochondrial SOD activity in HCASMC, which attenuated resistin-induced reduction of mitochondrial SOD activity. Values were means and SD (n = 5). *P < 0.05, #P < 0.01.
Discussion
Dysfunction of VSMCs and macrophages are the main causes of atherosclerotic plaque progression. Recent studies suggest that the crosstalk between VSMC and monocytes/ macrophages represents a great role for the atherosclerosis and leads to up-regulation of resistin in monocytes (18, 22). Resistin is a type of cytokine produced by monocytes/macrophages in human atheroma, and a grow number of clinical evidence indicates that resistin may independently predict major adverse cardiovascular events (18, 23). Additionally, resistin has been demonstrated to play a critical role in inflammation and intracellular ROS production of VSMCS that are closely relevant in the pathogenesis of cardiovascular disease (17, 18). However, the relevant mechanism and new therapeutic strategies remain uncertain. Our study represents a new potential treatment option by demonstrating that resistin and acetyl-LDL induced HCASMCs migration and proliferation could be significantly attenuated by Rb1, which was related to the reduction of ROS and the increase of SOD activity in HCASMCs. Our data suggested ginsenoside Rb1 has the potential of protecting against resistin-induced pathophysiological changes of atherosclerosis.
Recently, the positive effects of ginsenosides on CVD were confirmed including their ability of controlling ROS, production of nitric oxide (NO), and the activation of various receptors in endothelial cells (24). We first reported ginsenoside Rb1 could cut off superoxide anion production, endothelial dysfunction and eNOS down-regulation of porcine coronary arteries by homocysteine (5). Other studies also suggested that Rb1 protected human aortic and umbilical vein endothelial cells of human through the production and release of nitric oxide in the endothelium via PI3K/Akt activation and PKC inhibition (25, 26). Zhang et al. proved that Rb1 restricted the inflammatory response and promoted M2 macrophage polarization, which further helps to stabilize atherosclerosis progression in ApoE−/− mice (4). These reports, however, are limited mostly to data of endothelial cells, with few studies examining the protective effect of Rb1 on VSMCs. Li et al. found Rb1 could inhibit TNF-α-evoked inflammatory responses and FBS-induced proliferation in vitro experiment of rats (28). In a rat model, Zhang demonstrated that Rb1 could inhibited neointimal hyperplasia after balloon dilation by inhibition of VSMC proliferation (8). However, the aforementioned studies are confined to the level of rat VSMCs. In the present study, the protective effect of Rb1 on HCASMCs dysfunction induced by resistin and acetyl-LDL was demonstrated by wound healing assay and MTS assay. As far as we know, this is the first study to evaluate the role of Rb1 in human VSMC dysfunction induced by resistin.
It is worth noting that the protective effect of Rb1 was related to the ROS reduction within VSMCs as shown in our study. ROS are key signaling molecules that regulate vascular function and structure under physiological conditions. However, excessive ROS expression may be associated with the progression of several adverse cardiovascular diseases, such as VSMC proliferation and vascular remodeling caused by inflammatory reaction (21). At the intracellular level, the activation of mitogen activated protein kinases (MAPKs) in ROS-dependent signaling pathways in VSMCs involves p38, extracellular signal-regulated kinases (ERK1/2), c-Jun N-terminal kinases and the serine/threonine kinase Akt/protein kinase, which play a critical role in cell proliferation and migration and pathological remodeling (21, 28). We previously have proved that resistin-related intimal hyperplasia and VSMC dysfunction were associated with ROS generation and PKCε-dependent Nox activation (17). In the present study, we further demonstrated resistin-induced ROS production, which was partially through suppression of SOD activity, could be reversed by Rb1 pretreatment. Interestingly, we found that Rb1 pretreatment was more effective in reversing this disorder than co-treatment. In the co-treatment group of ROS experiment, the exposure time of 2 h was not long enough for Rb1 to take effect on VSMCs but was sufficient for resistin to generate a large amount of ROS, which resulted in the dysfunction of VSMCs afterwards. This suggested Rb1 requires a relatively long period of time (more than 2 h) to exert its antioxidative effect on VSMCs.
Besides NADPH oxidase, it is generally believed that ROS in living cells including vascular cells mainly comes from mitochondria (29). In the present study, we observed that resistin significantly triggered ROS generation by targeting the mitochondrion SOD activity and causing an imbalance of oxidative stress within VSMCs, which could be reversed by Rb1. The mitochondrion SOD is comprised of three isoforms: cytosolic Cu/Zn-SOD (SOD1), mitochondrial Mn-SOD (SOD2) and extracellular EC-SOD (SOD3) (21). The regulation of Rb1 on mitochondrial ROS involves the regulation of oxidase and antioxidant enzymes to inhibit the excessive production and accumulation of ROS. As previously mentioned, after treatment with Rb1, through activation of the PI3K/Akt/Nrf2 signaling pathway, serum or tissue level of SOD were increased in spinal cord injury and intestinal ischemia/reperfusion model rats (30–32). In vitro study, Rb1 could markedly increase intracellular SOD activity in ritonavir-treated human endothelial cells and inhibit the production of intracellular ROS (34). Thus, in consistent with this, our results showed Rb1 effectively exerted antioxidation by regulating SOD activity to relieve resistin-induced mitochondrial damage in VSMCs.
There are some limitations in our study. Although we confirmed the protection of Rb1 on VSMCs was related to the increase of SOD activity, the dynamic equilibrium of ROS production and elimination is far more complicated and needs to be further elucidated. For example, the elimination of superoxide anions is the consequence of coaction of several antioxidant enzymes. SOD catalyzes the dismutation of superoxide anions into H2O2 and oxygen, and H2O2 can be metabolized rapidly to water and oxygen by enzyme-linked reactions (34, 35). Whether Rb1 has a positive effect on other antioxidant enzymes, such as catalase, remains to be investigated. Secondly, the exact molecular mechanism for the protective effect of Rb1 has not been fully revealed in the present study. Numerous studies have identified that Rb1 exhibits antioxidant effects by activating the PI3K/Akt pathway with subsequent Nrf2 nuclear translocation and induction of antioxidant enzymes (30–32, 36–38). Since we have shown the protective effect of Rb1 was related to the antioxidation of SOD, which is one of the downstream productions of Nrf2 gene, we speculated that the part of the mechanism underlying the beneficial effects of Rb1 on resistin-induced VSMC dysfunction might be related to ROS dependent PI3K/Akt/Nrf2 signaling pathway. However, further studies will be required to examine this hypothesis.
Conclusion
This study provides new evidence supporting the hypothesis that ginsenoside Rb1can inhibit resistin-induced proliferation and migration in HCASMCs. Furthermore, we showed that the mechanisms involved in these effects exerted by Rb1 might be related to the reduction of ROS generation and increased activity of SOD. However, the molecular mechanism for the protective effect of Rb1 is far more complicated and needs to be further elucidated. The findings presented here also highlight the potential clinical applications of Rb1 in controlling resistin-associated vascular injury and the possible therapeutic use in cardiovascular disease.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
WZ and LXW designed and supervised the study; WFL, NH, and PM did the experiments; WFL, NH, and WZ analyzed the data; WFL and YL wrote the paper; LXW, and WZ scientifically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Xiamen Medical and Health Guidance Project (No. 3502Z20209050) and Fujian Provincial Natural Science Foundation Project (No. 2022J011422).
Acknowledgments
An earlier version of this manuscript has been presented as POSTER ABSTRACT PRESENTATIONS in ABSTRACT 874: Role of Ginsenoside Rb1 in Resistin-induced Vascular smooth muscle cell Dysfunction (Circulation Research August 2, 2019, Volume 125, Suppl_1, p A874).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1164547/full#supplementary-material.
References
1. Yu SE, Mwesige B, Yi YS, Yoo BC. Ginsenosides: the need to move forward from bench to clinical trials. J Ginseng Res. (2019) 43:361–67. doi: 10.1016/j.jgr.2018.09.001
2. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Rep. (2008) 12:1–23.
3. Zhang XJ, He C, Tian K, Li P, Su H, Wan JB. Ginsenoside Rb1 attenuates angiotensin II-induced abdominal aortic aneurysm through inactivation of the JNK and p38 signaling pathways. Vascul Pharmacol. (2015) 73:86–95. doi: 10.1016/j.vph.2015.04.003
4. Zhang X, Liu MH, Qiao L, Zhang XY, Liu XL, Dong M, et al. Ginsenoside Rb1 enhances atherosclerotic plaque stability by skewing macrophages to the M2 phenotype. J Cell Mol Med. (2018) 22:409–16. doi: 10.1111/jcmm.13329
5. Zhou W, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. Ginsenoside Rb1 blocks homocysteine-induced endothelial dysfunction in porcine coronary arteries. J Vasc Surg. (2005) 41:861–68. doi: 10.1016/j.jvs.2005.01.054
6. He F, Guo R, Wu SL, Sun M, Li M. Protective effects of ginsenoside Rb1 on human umbilical vein endothelial cells in vitro. J Cardiovasc Pharmacol. (2007) 50:314–20. doi: 10.1097/FJC.0b013e3180cab12e
7. Wu Y, Xia ZY, Dou J, Zhang L, Xu JJ, Zhao B, et al. Protective effect of ginsenoside Rb1 against myocardial ischemia/reperfusion injury in streptozotocin-induced diabetic rats. Mol Biol Rep. (2011) 38:4327–35. doi: 10.1007/s11033-010-0558-4
8. Zhang S, Deng J, Gao Y, Yang DL, Gong QH, Huang XN. Ginsenoside Rb₁ inhibits the carotid neointimal hyperplasia induced by balloon injury in rats via suppressing the phenotype modulation of vascular smooth muscle cells. Eur J Pharmacol. (2012) 685:126–32. doi: 10.1016/j.ejphar.2012.04.032
9. Qiao L, Zhang X, Liu M, Liu X, Dong M, Cheng J, et al. Ginsenoside Rb1 enhances atherosclerotic plaque stability by improving autophagy and lipid metabolism in macrophage foam cells. Front Pharmacol. (2017) 8:727. doi: 10.3389/fphar.2017.00727
10. Braun-Dullaeus RC, Mann MJ, Dzau VJ. Cell cycle progression: new therapeutic target for vascular proliferative disease. Circulation. (1998) 98:82–9. doi: 10.1161/01.cir.98.1.82
11. Sriram V, Patterson C. Cell cycle in vasculoproliferative disease: potential interventions and routes of delivery. Circulation. (2001) 103:2414–19. doi: 10.1161/01.cir.103.19.2414
12. Rivard A, Andres V. Vascular smooth muscle cell proliferation in the pathogenesis of atherosclerotic cardiovascular diseases. Histol Histopathol. (2000) 15:557–71. doi: 10.14670/HH-15.557
13. Owens CD, Kim JM, Hevelone ND, Hamdan A, Raffetto JD, Creager MA, et al. Novel adipokines, high molecular weight adiponectin and resistin, are associated with outcomes following lower extremity revascularization with autogenous vein. J Vasc Surg. (2010) 51:1152–59. doi: 10.1016/j.jvs.2009.12.051
14. Burnett MS, Lee CW, Kinnaird TD, Stabile E, Durrani S, Dullum MK, et al. The potential role of resistin in atherogenesis. Atherosclerosis. (2005) 182:241–48. doi: 10.1016/j.atherosclerosis.2005.02.014
15. Ding Q, Chai H, Mahmood N, Tsao J, Mochly-Rosen D, Zhou W. Matrix metalloproteinases modulated by protein kinase Cε mediate resistin-induced migration of human coronary artery smooth muscle cells. J Vasc Surg. (2011) 53:1044–51. doi: 10.1016/j.jvs.2010.10.117
16. Calabro P, Samudio I, Willerson JT, Yeh ET. Resistin promotes smooth muscle cell proliferation through activation of extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase pathways. Circulation. (2004) 110:3335–40. doi: 10.1161/01.CIR.0000147825.97879.E7
17. Raghuraman G, Zuniga MC, Yuan H, Zhou W. PKCε mediates resistin-induced NADPH oxidase activation and inflammation leading to smooth muscle cell dysfunction and intimal hyperplasia. Atherosclerosis. (2016) 253:29–37. doi: 10.1016/j.atherosclerosis.2016.08.015
18. Gan AM, Pirvulescu MM, Stan D, Simion V, Calin M, Manduteanu I, et al. Monocytes and smooth muscle cells cross-talk activates STAT3 and induces resistin and reactive oxygen species and production. J Cell Biochem. (2013) 114:2273–83. doi: 10.1002/jcb.24571
19. Clinton SK, Underwood R, Hayes L, Sherman ML, Kufe DW, Libby P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol. (1992) 140:301–16.1739124
20. Shen CM, Mao SJ, Huang GS, Yang PC, Chu RM. Stimulation of smooth muscle cell proliferation by ox-LDL- and acetyl LDL-induced macrophage-derived foam cells. Life Sci. (2001) 70:443–52. doi: 10.1016/s0024-3205(01)01428-x
21. Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. J Am Coll Cardiol. (2017) 70:212–29. doi: 10.1016/j.jacc.2017.05.035
22. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
23. On YK, Park HK, Hyon MS, Jeon ES. Serum resistin as a biological marker for coronary artery disease and restenosis in type 2 diabetic patients. Circ J. (2007) 71:868–73. doi: 10.1253/circj.71.868
24. Mohanan P, Subramaniyam S, Mathiyalagan R, Yang DC. Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J Ginseng Res. (2018) 42:123–32. doi: 10.1016/j.jgr.2017.01.008
25. Yu J, Eto M, Akishita M, Kaneko A, Ouchi Y, Okabe T. Signaling pathway of nitric oxide production induced by ginsenoside Rb1 in human aortic endothelial cells: a possible involvement of androgen receptor. Biochem Biophys Res Commun. (2007) 353:764–9. doi: 10.1016/j.bbrc.2006.12.119
26. Lan TH, Xu ZW, Wang Z, Wu YL, Wu WK, Tan HM. Ginsenoside Rb1 prevents homocysteine-induced endothelial dysfunction via PI3K/Akt activation and PKC inhibition. Biochem Pharmacol. (2011) 82:148–55. doi: 10.1016/j.bcp.2011.04.001
27. Li QY, Chen L, Fu WH, Li ZD, Wang B, Shi XJ, et al. Ginsenoside Rb1 inhibits proliferation and inflammatory responses in rat aortic smooth muscle cells. J Agric Food Chem. (2011) 59:6312–8. doi: 10.1021/jf200424k
28. Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. (2000) 20:2175–83. doi: 10.1161/01.atv.20.10.2175
29. Dromparis P, Michelakis ED. Mitochondria in vascular health and disease. Annu Rev Physiol. (2013) 75:95–126. doi: 10.1146/annurev-physiol-030212-183804
30. Chen S, Li X, Wang Y, Mu P, Chen C, Huang P. Ginsenoside Rb1 attenuates intestinal ischemia/reperfusion-induced inflammation and oxidative stress via activation of the PI3K/Akt/Nrf2 signaling pathway. Mol Med Rep. (2019) 19:3633–41. doi: 10.3892/mmr.2019.10018
31. Liu X, Gu X, Yu M, Zi Y, Yu H, Wang Y, et al. Effects of ginsenoside Rb1 on oxidative stress injury in rat spinal cords by regulating the eNOS/Nrf2/HO-1 signaling pathway. Exp Ther Med. (2018) 16:1079–86. doi: 10.3892/etm.2018.6286
32. Sun Q, Meng QT, Jiang Y, Xia ZY. Ginsenoside Rb1 attenuates intestinal ischemia reperfusion induced renal injury by activating Nrf2/ARE pathway. Molecules. (2012) 17:7195–205. doi: 10.3390/molecules17067195
33. Lü JM, Jiang J, Jamaluddin MS, Liang Z, Yao Q, Chen C. Ginsenoside Rb1 blocks ritonavir-induced oxidative stress and eNOS downregulation through activation of estrogen receptor-Beta and upregulation of SOD in human endothelial cells. Int J Mol Sci. (2019) 20:294. doi: 10.3390/ijms20020294
34. Villalpando-Rodriguez GE, Gibson SB. Reactive oxygen species (ROS) regulates different types of cell death by acting as a rheostat. Oxid Med Cell Longev. (2021) 2021:9912436, 17 pages. doi: 10.1155/2021/9912436
35. Bretón-Romero R, Lamas S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. (2014) 2:529–34. doi: 10.1016/j.redox.2014.02.005
36. Hwang YP, Jeong HG. Ginsenoside Rb1 protects against 6-hydroxydopamine-induced oxidative stress by increasing heme oxygenase-1 expression through an estrogen receptor-related PI3K/Akt/Nrf2-dependent pathway in human dopaminergic cells. Toxicol Appl Pharmacol. (2010) 242:18–28. doi: 10.1016/j.taap.2009.09.009
37. Zhuang CL, Mao XY, Liu S, Chen WZ, Huang DD, Zhang CJ, et al. Ginsenoside Rb1 improves postoperative fatigue syndrome by reducing skeletal muscle oxidative stress through activation of the PI3K/Akt/Nrf2 pathway in aged rats. Eur J Pharmacol. (2014) 740:480–7. doi: 10.1016/j.ejphar.2014.06.040
Keywords: ginsenoside Rb1, resistin, vascular smooth muscle cells (VMSCs), reactive oxygen species(ROS), superoxide dismutase(SOD)
Citation: Lu W, Lin Y, Haider N, Moly P, Wang L and Zhou W (2023) Ginsenoside Rb1 protects human vascular smooth muscle cells against resistin-induced oxidative stress and dysfunction. Front. Cardiovasc. Med. 10:1164547. doi: 10.3389/fcvm.2023.1164547
Received: 13 February 2023; Accepted: 5 May 2023;
Published: 25 May 2023.
Edited by:
Johnson Rajasingh, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Madhan Subramanian, Oklahoma State University, United StatesSelene Perales Sanchez, University of Tennessee Health Science Center, United States
Satish Ramalingam, SRM Institute of Science and Technology, India
© 2023 Lu, Lin, Haider, Moly, Wang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lixin Wang d2FuZy5saXhpbkB6cy1ob3NwaXRhbC5zaC5jbg== Wei Zhou emhvdXdAc3VyZ2VyeS5hcml6b25hLmVkdQ==
†These authors have contributed equally to this work and share first authorship