 Anna Kovilakath
Anna Kovilakath George Wohlford
George Wohlford L. Ashley Cowart
L. Ashley Cowart- 1Department of Human and Molecular Genetics, Virginia Commonwealth University, Richmond, VA, United States
- 2Pauley Heart Center, Virginia Commonwealth University, Richmond, VA, United States
- 3Department of Biochemistry and Molecular Biology and the Massey Cancer Center, Virginia Commonwealth University, Richmond, VA, United States
- 4Richmond Veteran's Affairs Medical Center, Richmond, VA, United States
Lack of significant advancements in early detection and treatment of heart failure have precipitated the need for discovery of novel biomarkers and therapeutic targets. Over the past decade, circulating sphingolipids have elicited promising results as biomarkers that premonish adverse cardiac events. Additionally, compelling evidence directly ties sphingolipids to these events in patients with incident heart failure. This review aims to summarize the current literature on circulating sphingolipids in both human cohorts and animal models of heart failure. The goal is to provide direction and focus for future mechanistic studies in heart failure, as well as pave the way for the development of new sphingolipid biomarkers.
Introduction
Heart failure (HF) is a debilitating medical condition where the heart is unable to meet the metabolic demands of the body. It is a widespread clinical syndrome affecting at least 64 million people globally, and this number is expected to rise (1, 2). HF poses a significant economic and clinical burden and dramatically impacts patients' quality of life (3). Different classifications of HF exist, with the most widely accepted based on the percentage of blood volume ejected by the left ventricle (LV) during systole, i.e., heart failure with reduced ejection fraction (HFrEF, LVEF ≤40%), heart failure with preserved ejection fraction (HFpEF, LVEF ≥50%), and as of 2016, heart failure with borderline/mildly reduced ejection fraction (HFbEF/HFmrEF, LVEF 41%–49%) (4–6). Though, there is some controversy related to the cut-off points in this HF classification. Currently, no human studies link HFbEF/HFmrEF with sphingolipids and will not be discussed further in this review.
Advances in medicine over the past decade have improved the quality of life and outcomes for patients with HFrEF. These include pharmacotherapies such as Aldosterone/Mineralocorticoid Receptor Antagonists (ARA/MRA, e.g., spironolactone), Angiotensin Receptor-Neprilysin Inhibitor (ARNI, e.g., Sacubitril-valsartan), ventricular assist devices (VAD), cardiac resynchronization therapy (CRT), and implantable cardioverter-defibrillator (ICDs) (4, 7–10). However, HFpEF has now become the most prevalent form of HF, and clinical trials of HFpEF pharmacotherapies, such as Sodium-GLucose co-Transporter 2 (SGLT2) inhibitors (e.g., empagliflozin), ARNI, and ARA/MRA, and novel device therapies like InterAtrial Shunt Devices (IASD), have demonstrated only modest reductions in the risk of hospitalization (4, 9, 11–16). This is likely due to the highly heterogenous nature of the HFpEF population with respect to pathogenesis, pathophysiology, and non-cardiac comorbidities. Despite these advancements, the median 5-year mortality rates upon clinical diagnosis of HFrEF and HFpEF are 75.3% and 75.7%, respectively (17).
Sphingolipids are a class of lipids, conserved across all eukaryotic organisms, characterized by an amino alcohol headgroup and fatty acid carbon chain. Though sphingolipids serve essential structural roles in membranes, many sphingolipid metabolites are considered bioactive lipids and play a central roles in inflammation, autophagy, apoptosis, immune cell trafficking, cell survival, metabolism, mitochondrial function, and many other critical cell processes (18, 19). Disrupted sphingolipid metabolism has been implicated in HF pathophysiology, and thus, enzymes involved in sphingolipid metabolism (or the protein signaling effectors) have become potential therapeutic targets. Consequently, over the past 15 years, there has been a surge of interest in the study of sphingolipid metabolism in HF (19). Sphingolipids are synthesized de novo in the endoplasmic reticulum via condensation of an amino acid, typically serine, and a fatty acid, catalyzed by serine palmitoyltransferase (SPT) (20). This pathway is depicted in Figure 1. This reaction produces a sphingoid base, the molecular scaffold on which all sphingolipids are constructed. SPT exists as a heterooligomeric enzyme consisting of a dimer of tetramers, with SPTLC1 being essential, and SPTLC2 or 3 required for catalytic activity. Two small subunits, SPTssa and SPTssb, differentially regulate Acyl-CoA chain length utilization by SPT1 and 2/3 (21). A fourth subunit, ORMDL, acts as three isoforms (ORMDLs 1–3) to sense membrane sphingolipids to attenuate sphingolipid synthesis in the presence of high membrane ceramide (Cer) (22). Differences between these three isoforms, ORMDL 1, 2, and 3, are very poorly understood at present. Nonetheless, the composition of the SPT complex determines Acyl-CoA selectivity and thus determines the catalytic product. For example, when the subunits SPTLC1/SPTLC2/SPTssa are present in SPT, then palmitoyl-CoA is condensed with an amino acid to synthesize all downstream sphingolipid species with an 18-carbon sphingoid backbone, i.e., d18 (23). This generates the canonical sphingoid bases that have been studied for over 150 years. Alternatively, SPTLC3 can substitute for SPTLC2, and SPTssb can substitute for SPTssa, leading to production of sphingolipid species with alternative carbon sphingoid backbones (e.g., d16, and d20) (24–26). The complexity of SPT and the variety of sphingoid bases has only recently become appreciated (largely driven by the discovery of SPTLC3 and the small subunits). Recent research has shown that the SPT enzyme and the sphingoid bases it produces are crucial components of cardiovascular health, with several studies supporting their relevance (27–30). For example, we previously demonstrated in mice hearts where SPTLC3 is expressed, the SPT complex can generate a d16-derived sphingolipid from myristoyl-CoA, which was shown to stimulate cardiomyocyte apoptosis (25).
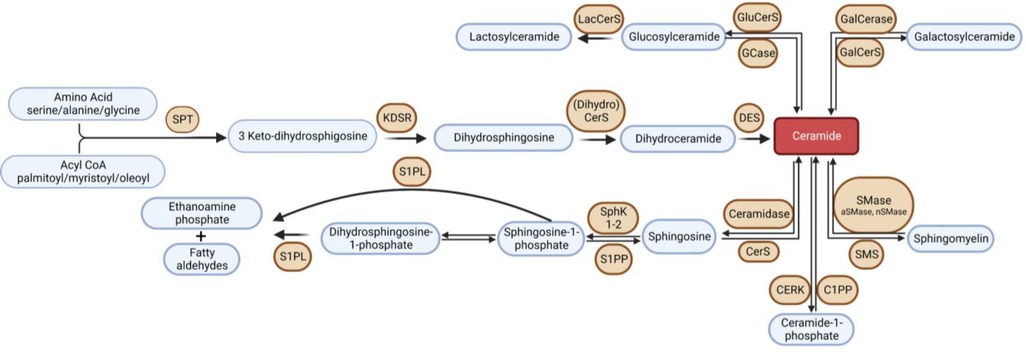
Figure 1. Overview of sphingolipid metabolism. SPT, serine palmitoyltransferase; KDSR, 3-keto-dihydrosphingosine reductase; DES, dihydroceramide desaturase; GCase, glucocerebrosidase; GluCerS; glucosylceramide synthase; LacCerS, lactosylceramide synthase; GalCerS, galactosylceramide synthase; SMase, sphingomyelinase; SMS, sphingomyelin synthase; CerS, ceramide synthase; C1PP, ceramide-1-phosphate phosphatase; CERK,; S1PP, sphingosine-1-phosphate phosphatase; SphK, sphingosine kinase; S1PL, sphingosine-1-phosphate lyase.
The first detectable sphingolipid to be synthesized is the sphingoid base, dihydrosphingosine (DHS), which is then N-acetylated by one of 6 isoforms of (dihydro) ceramide synthase (CerS), with different chain length fatty acids to synthesize dihydroceramides (DHC) (31). CerS1 typically adds 18-carbon acyl chain length (C18:0, and C18:1); CerS2 adds C22:0, C24:0, C24:1, C26:0 and C26:1; CerS3 adds C22:0 to C36:0; CerS4 adds C18:0 to C22:0; CerS5 adds C14:0 to C18:0; and CerS6 adds C14:0 to C18:0 (Figure 2). The acyl chain length ranges from medium MCFA (C12–14), long LCFA (C16–20), very long VLCFA (C22–26), and ultra-long chain fatty acids ULCFA (>C26) (32–38). DHC desaturase then converts DHC to Cer, the backbone of all complex sphingolipids (39). Acid or alkaline ceramidase enzymes (AC or ACER 1–3) can then hydrolyze Cer to yield sphingosine (Sph), which can be phosphorylated by Sph kinase (SphK) 1 or 2 to generate sphingosine-1-phosphate (S1P) or dihydrosphingosine-1-phosphate (DHS1P) (Figure 1). S1P is a potent signaling molecule that can bind to one of its five G protein-coupled receptors (S1PR 1–5) to elicit downstream effector cell activity or be exported extracellularly by Spinster 2 (Spns2). Sph can also be “salvaged” and re-incorporated into Cer through the action of CerS enzymes. This mechanism allows cells to “remodel” Cer pools. If Cer is not hydrolyzed, it can be further metabolized into sphingomyelin (SM) or glycosphingolipids (GSL) through the addition of various headgroups. SM can then be hydrolyzed by acid or neutral sphingomyelinase (aSMase, nSMase) to yield Cer. GSLs containing Cer and glucose are the predominant precursors of globosides and gangliosides, while those with Cer and galactose synthesize downstream sulfatides. To exit the sphingolipid de novo pathway, S1P lyase (S1PL) catabolizes S1P or DHS1P in lysosomes and plasma membranes to yield a fatty aldehyde and phosphoethanolamine (40). The heart requires a constant supply of energy to maintain its contractile function, which is primarily provided through fatty acid oxidation (41). Studies have shown that disruptions in sphingolipid metabolism contribute to impaired fatty acid oxidation, with the Cer-S1P rheostat playing a particularly important role (42). The dogma being Cer accumulation is harmful, while S1P accumulation is beneficial (42–45). Therefore, interconversion between these sphingolipid species is a highly regulated process, and any small deviation in anabolism, catabolism or substrate availability can lead to abnormal accumulation of one or more sphingolipid species, resulting in dysregulated fatty acid supply in the heart (46). As such, alterations in sphingolipid content and profiles have become a key area of investigation in HF research.
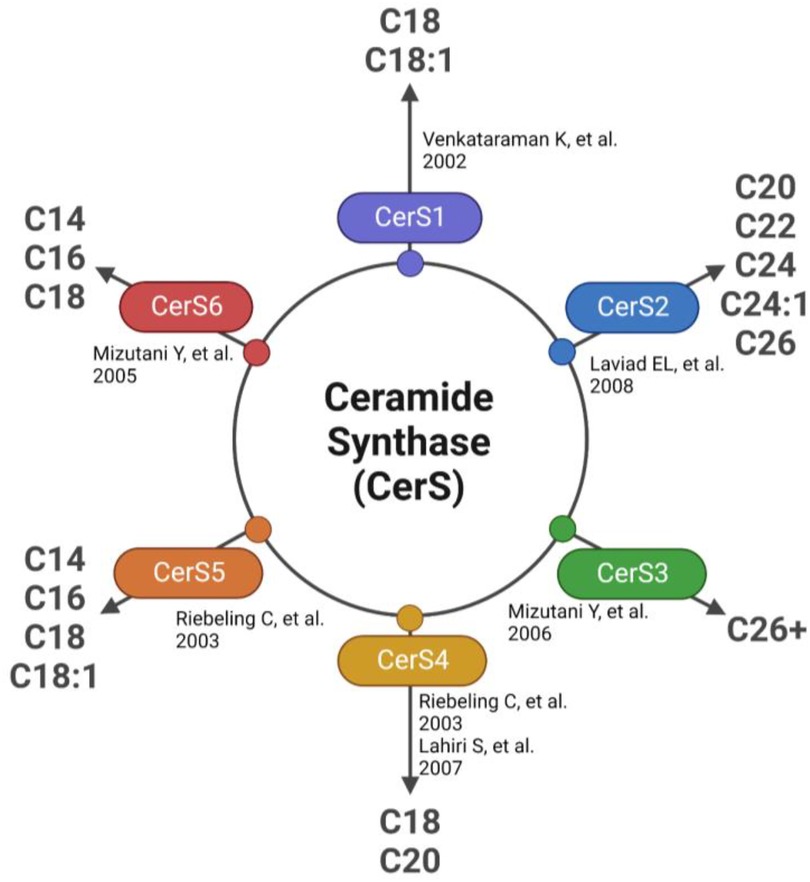
Figure 2. Ceramide synthase (CerS) substrate selectivity.
In this review, we examine the relationship between sphingolipids and HF in both human study cohorts and animal models. We analyze the current literature to identify potential biomarkers and druggable targets for the detection and treatment of HF.
Sphingolipids in heart failure
Many studies, such as the Framingham Heart Study (FHS), have been ongoing for decades and have analyzed traditional risk factors like obesity, smoking status, and LDL-cholesterol from these patient cohorts. However, only in recent years have investigators started to address circulating sphingolipids in subjects from these studies through post hoc analyses. This involves mass spectrometry to identify and quantify sphingolipids in blood serum, blood plasma, or myocardial tissue. This method yields measurements for numerous sphingolipids, which can then be correlated to disease and other clinical parameters to gain a better understanding of their relationship. Current literature on sphingolipid associations with HF can broadly be divided into two types: sphingolipid associations in patients with incident HF (summarized in Table 1), and secondary endpoints in patients with prevalent HF (summarized in Table 2).
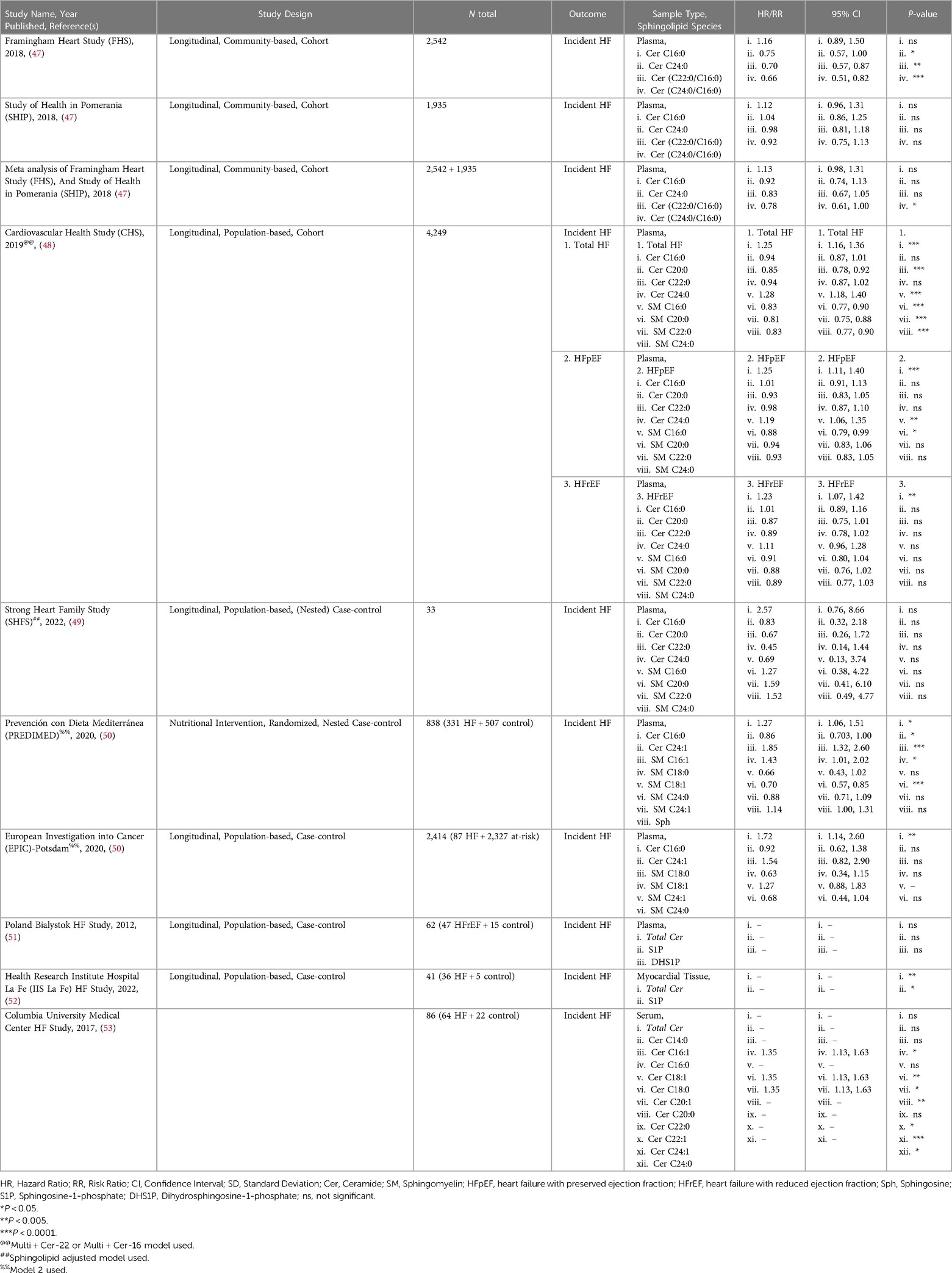
Table 1. Studies on sphingolipids associated with incident heart failure.
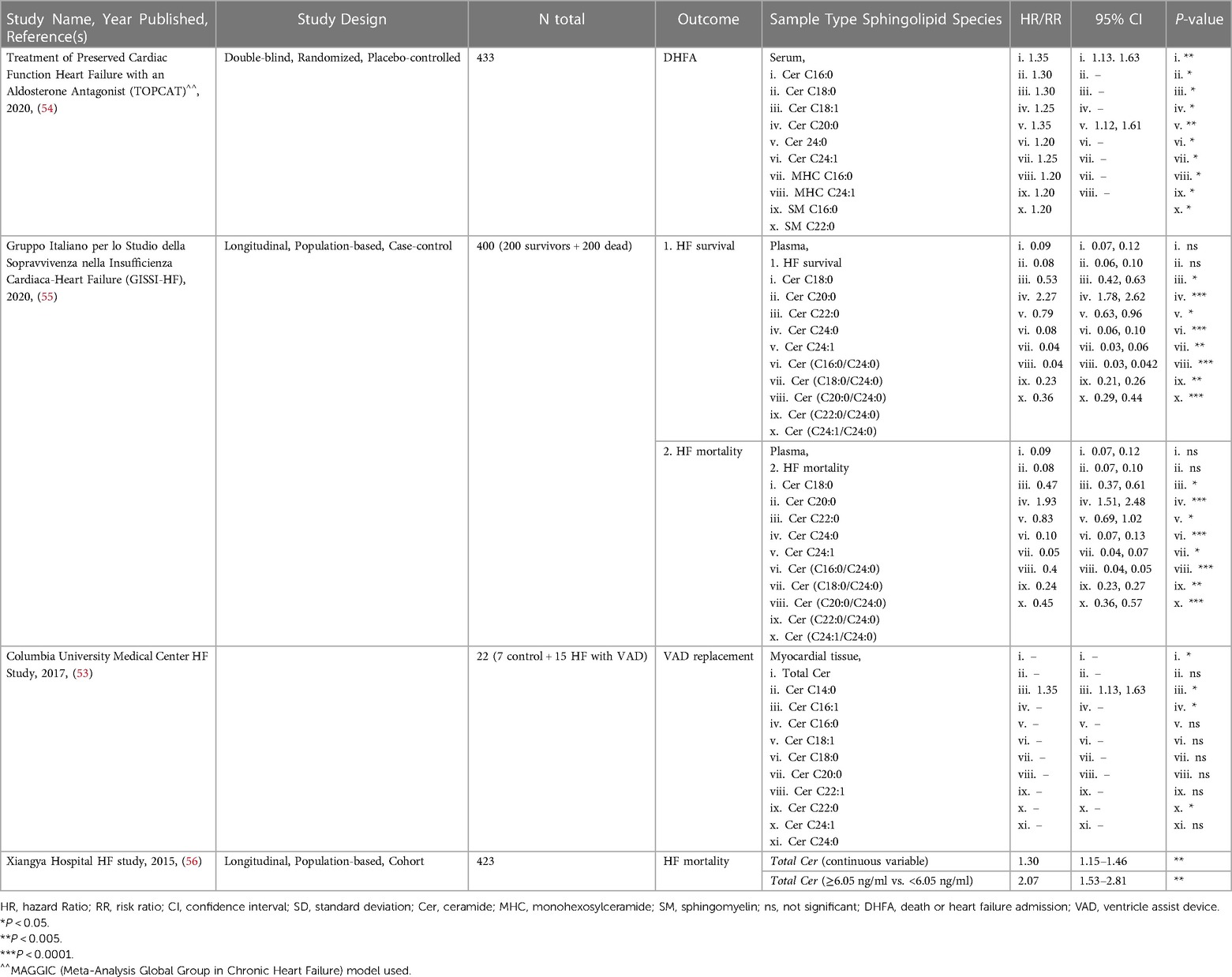
Table 2. Studies on sphingolipids associated with secondary endpoints in patients with prevalent HF.
Sphingolipids associated with incident heart failure are discussed in this section. Meta-analysis of PREDIMED and EPIC-Potsdam cohorts showed that Cer C16:0 was significantly increased but also ranked as one of the topmost lipid cluster networks associated with HF (50). A 2012 study by Knapp et al., found no significant association of plasma S1P, DHS1P or total Cer in HFrEF and HFpEF patients (51). However, the plasma of both HFpEF and HFrEF patients was characterized by significantly lower levels of free Sph and DHS, but stable S1P levels. This observation suggests that diminished action of S1P does not contribute to cardiac dysfunction in patients with chronic HF (51). A small yet more recent study by Pérez-Carrillo et al., showed that myocardial tissue from HFrEF and HFpEF patients had double S1P and triple total Cer levels compared to controls (52). After mRNA-sequencing, 12 sphingolipid-specific genes were differentially expressed. SPTssa, SPTssb, SPTLC1, and SPTLC3 genes were downregulated suggesting de novo sphingolipid synthesis is reduced. However, CerS1, which can act both de novo and in the salvage pathway, was upregulated, potentially explaining the increased Cer. Additionally, S1P phosphatase (S1PP), the enzyme converting S1P to Sph, and S1PR3 were downregulated in HF patients. Though this should result in decreased S1P in myocardial tissue and not the observed increase in levels (52). While this data is interesting and suggests S1P levels are not indicative of HF status, they do not provide the full picture of sphingolipid de novo synthesis dynamics and leave us with more questions instead of answers. A 2019 study, using the Cardiovascular Health Study (CHS) cohort, identified higher levels of SM C16:0, and Cer C16:0 associated with higher risk and SM C20:0, C22:0, C24:0, and Cer C22:0 with lower risk of developing HF, independently. Interestingly, these species had similar associations regardless of whether the patient had HFpEF or HFrEF (48).
In contrast to studies that assessed the correlation of individual sphingolipid species level with HF, some more recent studies have analyzed ratios between specific sphingolipids. Analysis of sphingolipid ratios may not only be more effective in predicting incident HF, but also adverse cardiac events. This sphingolipid ratio score is more reliable as it removes complications that arise from altered sphingolipid concentrations postprandially and/or with hyperlipidemia. Newly developed high throughput assays to quantify ratios of VLCFA/LCFA Cer or vice versa in the plasma were applied to the FHS and SHIP (Study of Health in Pomerania) studies. These assays were able to show a higher ratio in plasma Cer(C24:0/C16:0) was inversely associated with incident HF (47). Outcomes from these studies paved the way for the launch of the MI-Heart Ceramide Risk Score (CERAM) blood test used by Mayo Clinic to predict unfavorable cardiovascular events in patients (57). The test measures concentrations of plasma Cer C16:0, C18:0, C24:1 and the plasma ratios Cer(C16:0/24:0), Cer(18:0/24:0), and Cer(24:1/24:0). The risks conferred by Cer ratios are independent of sex, age, gender, LDL and other traditional factors, and continually outperform cholesterol testing.
Sphingolipids associated with secondary endpoints in patients with prevalent HF such as major adverse cardiac event(s), Ventricle Assist Device (VAD) placement or replacement, death, or heart failure admission (DHFA), and HF-related mortality are described in this section. A 2015 study by Yu et al., concluded that plasma from patients with HF who died had total Cer >6.05 ng/ml, and HF patients that survived (at least up to the 4 years during the study) had circulating total Cer <6.05 ng/ml (56). There has been no follow-up study to verify whether this or another total Cer threshold can predict survival in HF patients. A 2017 study from Christian Schulze's lab revealed that hearts from advanced HF patients showed significantly increased total Cer driven by increased Cer C16:0, C16:1 and C24:1 and increased SPTLC2 protein expression (irreconcilably no change in CerS1, CerS2 or CerS5 expression was noted) undergoing placement of VAD (53). Interestingly, there was no significant change in circulating total Cer, but significant increases in circulating Cer C16:0, C18:0, C20:1, C20:0, C22:1, C24:1, and C24:0. Though, after VAD implantation, these changes showed partial reversibility in myocardial tissue but not circulating Cer (53). A study by Javaheri et al., linked increased serum sphingolipids Cer C16:0 and C18:0 with death or HF admission (DHFA) in a TOPCAT study of 433 HFpEF patients (54). In a study with Italian HF patients, associated increased plasma ratios of Cer(C16:0/C24:0), Cer(C18:0/C24:0), Cer(C18:0/C24:0), Cer(C22:0/C24:0), and Cer(C24:1/C24:0), along with higher Cer C16:0, C18:0, C20:0, C22:0, and Cer C24:1 individually with increased cardiovascular mortality in ambulatory patients with chronic HF. However, these Cer to HF associations became non-significant after adjustment for established cardiovascular risk factors, medication use, and plasma N-terminal pro b-type natriuretic peptide (NT-proBNP) concentrations (55). Though not a study of secondary outcomes in patients with prevalent HF, A subset of the major findings in the 2018 study using the community-based cohorts, FHS and SHIP, also showed that Cer C16:0 is inversely correlated while Cer C24:0, Cer(C22:0/C16:0), and Cer(C24:0/C16:0) were positively correlated with predictive information about CVD and all-cause mortality in the general population 6 years before the actual onset of disease (47).
Similar approaches have been used not in incident HF populations but in populations presenting with pre-HF etiologies such as type two diabetes (T2D), obesity or incident coronary heart disease (CHD), including but not limited to myocardial infarction (MI), atrial fibrillation, coronary insufficiency, adverse cardiac remodeling, and angina pectoris. The SHS (Strong Heart Study) and SHFS (Strong Heart Family Study) fare population-based longitudinal studies addressed cardiovascular disease (CVD) in several American Indian communities in Arizona, North and South Dakota, and Oklahoma. It is interesting to note that increased Cer C16:0, though not significant, correlated with increased CVD risk after diabetes onset, while the inverse was true of increased SM C22:0, C24:1, C24:0, C26:1, and SM C26:0 (49). A study by Fretts et al., using the SHS and SHFS plasma samples, showed that higher Cer C18:0, C20:0, and Cer C22:0 were associated with higher risk of diabetes (58). Mikhalkova et al., sought to determine whether there were changes in circulating Cer and circulating SM species in women with obesity and HFpEF before and after bariatric surgery. After the surgery-induced weight loss the patients showed improved symptoms, reverse cardiac and remodeling and improved relaxation. Though the weight loss was associated with reduced plasma SM C23:1, cardiac function improvement was not associated with sphingolipidomic changes, which may have been due to the relatively small sample size (N = 12) (59). Another study, determined that Cer C18:0, but not Cer C16:0, C24:0, or C24:1 were associated with incident of major adverse cardiovascular events (MACE), and showed stronger correlation for recurrent and fatal events than for first events (60). In contrast to the MACE study, plasma concentrations of 74 ischemic heart disease patients showed no association of Cer C16:0 Cer with LVEF. However, total SM and S1P were significantly lower in patients with HFrEF compared to patients with HFpEF (61). The LUdwigshafen RIsk and Cardiovascular Health (LURIC) study established well defined phenotypes for CVD, metabolic disorders, and their progression to cardiovascular complications including HF (62). Independent of traditional risk factors, SM C23:0, and SM C24:0 were the most protective sphingolipid species, intermediate protection conferred by Cer C23:0, Cer C24:0, SM C16:0, and SM C24:1, while Cer C16:0, and Cer C24:1 had the strongest positive association with CVD and mortality (63). Another study put diabetic patients on diets rich with either LCFA or MCFA. Patients on the MCFA diet showed improved systolic function with concomitant decrease of circulating sphingolipids, while subjects on the LCFA diet showed reduced stroke volume, cardiac output, and no change in systolic function which was associated with increased SM C15:0, and SM C22:1 (64). Newly developed high throughput assays to quantify ratios of VLCFA/LCFA Cer in the plasma were applied to the Framingham Heart Study and the SHIP (Study of Health in Pomerania). These assays were able to show a higher ratio in plasma Cer(C24:0/C16:0) was inversely associated with incident HF (47). A more recent study of the FHS cohort associated higher plasma Cer(C16:0/C24:0) with detrimental cardiac structural and functional changes that can lead to HF (65). A 2020 study analyzing the CHS cohort, determined that Cer C20:0, Cer C22:0, and Cer C24:0 were associated with reduced atrial fibrillation risk (66).
Taken together, these studies suggest the dogma implicating total Cer accumulation in HF and HF-related outcomes, holds true. Though these results are from vastly different studies, they consistently show inverse association of the LCFA Cer C16:0, synthesized by CerS5 or CerS6, and the unsaturated VLCFA Cer C24:1, synthesized by CerS2, to HF, regardless of HF classification and population ethnicity. While the saturated VLCFA Cer C24:0 and in some studies the SM C24:0 is positively correlated with HF. Though, ratios of sphingolipid LCFA/VLCFA seem to be better prognostic markers of adverse cardiac events, HF and prediction of HF-mortality up to 5 years.
Insights from Mendelian randomization analysis studies
Developing new pharmacotherapies is a costly and time-consuming process, and drug companies are incentivized to speed up the path to market approval due to the time-limits on drug patents. However, testing agents that may augment a specific disease biomarker without fully understanding their potential mechanistic roles has led to failed clinical trials. To increase the likelihood of identifying causal biomarker-disease associations, researchers have started to employ Mendelian randomization (MR). This approach involves a genome-wide association study (GWAS) analysis to identify candidate genetic variants associated with variations in the biomarker, which are then compared to those associated with differences in the population. When a biomarker is identified and a causal effect is confirmed by MR, the probability of clinical trial success for interventions targeting that biomarker increases significantly (67).
Although MR analyses have not been reported in the literature for the association between circulating sphingolipids and HF, a GWAS analysis has been conducted for circulating sphingolipids associated with a reduction in HF events. MR analyses for sphingolipids linked to incident coronary heart disease (CHD) have also been reported, which is relevant since CHD and HF can coexist in the same patient, and patients with CHD are at increased risk for development of HF (4, 68, 69). Inclusion of these investigations in this focused review is justified given the robustness of the MR methodology, the lack of direct evidence reported in the literature for HF and sphingolipids tested with the MR methodology and the proximity of CHD to HF across the landscape of cardiovascular disease.
VLCFA Cer measured in the plasma, has been shown to be prognostic for incident HF events (relative risk of 0.75 for every 3 unit increase in plasma Cer C24:0 for HF) despite traditionally being implicated in the etiology of T2DM (30). Analyses of patient samples obtained from the FHS identified 19 genetic variants associated with lower levels of Cer C22:0 and 9 variants associated with lower levels of Cer C24:0. These 28 variants were located on chromosome 20, near the SPTLC3 encoding gene. The identified lead variant (rs4814175) was associated with 3% lower Cer C22:0 and 10% lower Cer C24:0 concentrations, though, MR analysis for this variant was not conducted (30).
The findings from another GWAS utilizing data from two different large epidemiologic datasets (N = 1,094 and N = 4,034), were congruent with the FHS findings. Variants near SPTLC3 were confirmed to be contributing factors in the variations of circulating Cer linked to CVD and T2DM (70). However, after MR analysis was performed, the lead SNP (rs680379) tested was different than what was previously identified. The new variant was used to assess the risk for incident T2DM and the resulting change in risk for incident CVD for individuals that carried the SNP. The investigators also linked circulating Cer C16:0 and DHC C22:2 with an increased risk of incident CVD in these separate patient populations. However, subsequent GWAS and MR analysis for the assessment of the association with CVD or HF events was not completed for these Cer. Reported in 2014, investigators from Sweden pooled samples and clinical data from three longitudinal registry trials (N = 3,668). SM C28:1, one of the biomarkers identified, showed an inverse relationship with incidence of CHD, but was not found to have a causal relationship by MR analysis (68).
However, definitive evidence for a causal role of circulating sphingolipids in HF, when assessed by MR analysis, remains elusive in the published literature. Possibly due to the contribution of the gut microbiome to circulating lipids and metabolites (71). Therefore, to identify causal factors by MR analysis, deeper genotyping that includes additional procedures for characterizing the microbiome may be required.
Sphingolipids in animal models of heart failure
Animal models play a crucial role in studying HF and developing novel treatment strategies. Small animal models generally utilize pharmacological, surgical, and genetic modifications either alone or in combination while large animal models rely on surgical or pharmacological methods (Figure 3) (72, 73). A comprehensive list of animal models of HF with the respective sphingolipid alterations in blood serum, blood plasma, and/or myocardial tissue can be found in Table 3.
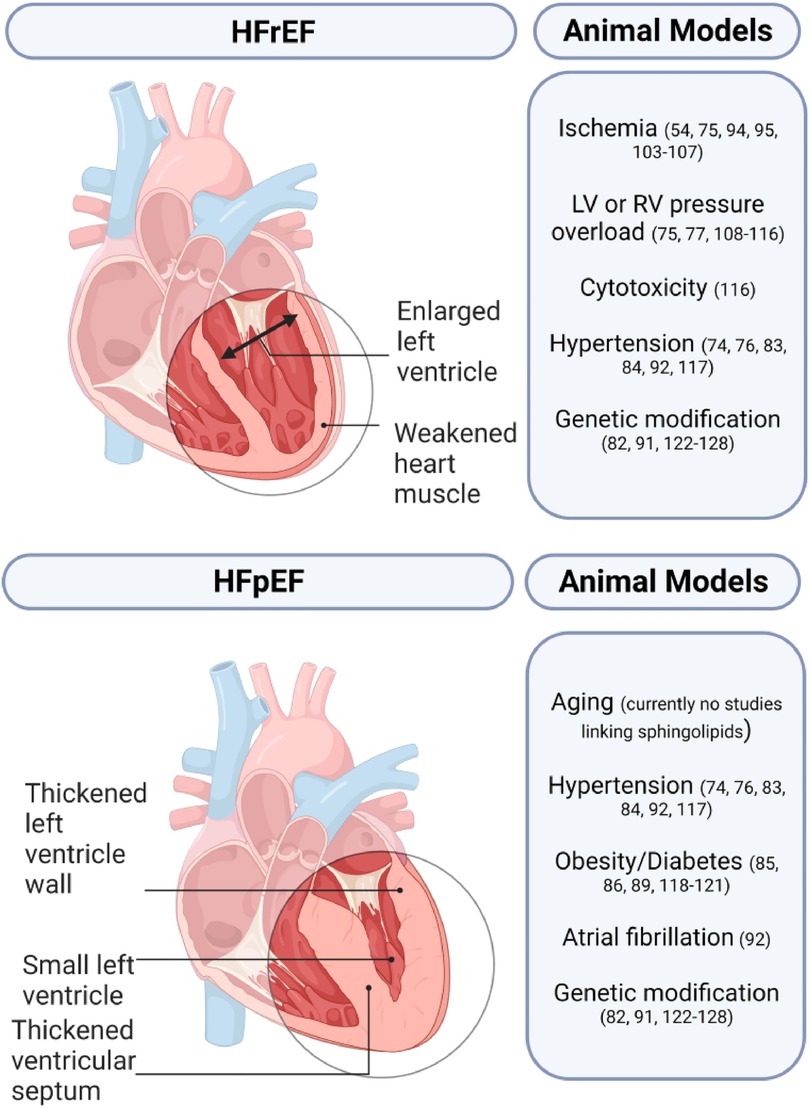
Figure 3. Sphingolipids in animal models of heart failure.
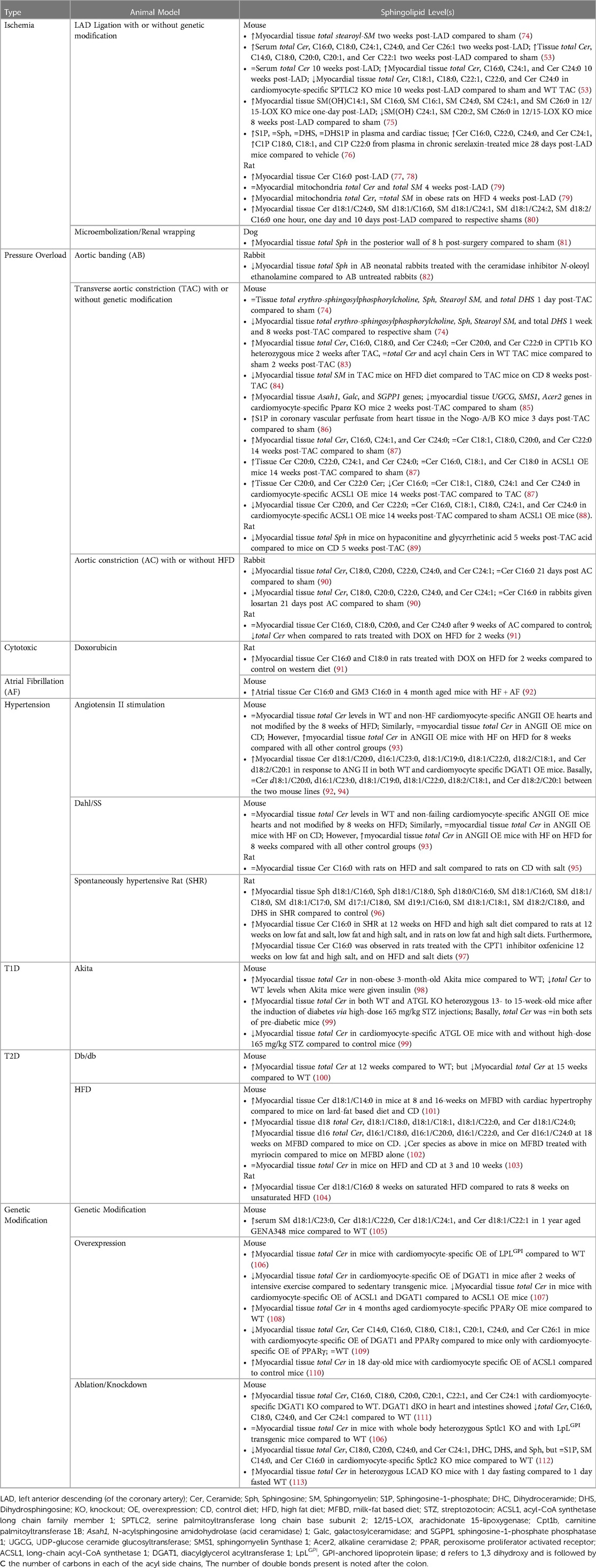
Table 3. Sphingolipids associated with animal models of heart failure.
Heart failure with preserved ejection fraction
HFpEF is a complex condition that only a few animal models have been able to replicate successfully. Animal models of HFpEF are typically pressure overload models as 55%–86% of patients with HFpEF have hypertension. The Dahl salt-sensitive (Dahl/SS) rat is the most popular HFpEF model and is characterized by hypersensitivity to sodium intake. Other popular models include animals with transverse aortic constriction (TAC), and gene knockout (KO), or overexpression (OE) that develop left ventricular hypertrophy (LVH).
A 2019 study observed an increase of the following sphingolipids prior to HF in Dahl/SS rats—1.27× N-palmitoyl-Sph C16:0, 1.67× glycosyl-N-stearoyl-Sph C18:0, 1.34× DHS, 1.72× N-palmitoyl-DHS C16:0, 1.24× palmitoyl SM C16:0, 1.28× stearoyl SM C18:0, 1.21× SM (d18:1/C17:0, d17:1/C18:0, d19:1/C16:0), 1.44× SM (d18:1/C18:1, d18:2/C18:0), and 1.32× Sph (96). Another study analyzed the myocardial sphingolipids at multiple timepoints in post-TAC mice compared to sham mice (74). Erythro-sphingosylphosphorylcholine was 1.04 at 1 day, 0.68 at 1 week and 0.69 at 8 weeks post-TAC. Sph and stearoyl SM levels decreased by half 8 weeks post-TAC. While DHS sharply increased at 1 day, then stayed relatively the same up to 8 weeks post-TAC (74). Another study determined that mice with HF could not adapt to excessive fatty acid supply vs. mice with LVH, both cohorts having angiotensin II (ANG II) OE (93). The hearts from HF mice on high fat diet (HFD), accumulated 50% more total Cer, aggravating contractile dysfunction, whereas the LVH mice on HFD showed a similar phenotype as the WT and no accumulation of total Cer, suggesting that impaired fatty acid oxidation in this model is associated with Cer lipotoxicity (93). CPT1b controls uptake of LCFAs in mitochondrial β-oxidation. Hearts from heterozygous CPT1b knockout (CPT1b+/−) mice subjected to TAC-induced pressure overload, showed substantially elevated total Cer levels driven by the Cer C16:0, C18:0, and C24:0 species, while sham WT and CPT1b+/− mice showed no differences with respect to Cer content (83). While we cannot conclude that pressure overload is the sole driver of Cer accumulation, we can assume accumulated Cer in this model further aggravates the pressure overload induced hypertrophy caused by CPT1b deficiency.
Obesity and diabetes markedly increase the risk of HF, and alteration of sphingolipid metabolism contributes either directly or indirectly to metabolic stress in diabetes leading to diabetic cardiomyopathy and eventually HF (114–116). Diacylglycerol acyltransferase 1 (DGAT1) converts diacylglycerol (DAG) to triglyceride. Failing human hearts have severely reduced DGAT1 levels with concomitant accumulation of Cer and DAGs (117). Ablation of Dgat in mice led to no known cardiac dysfunction, and these mice express normal levels of circulating Cer. However, half of the cardiomyocyte-specific DGAT1 KO mice died within 9 months and their hearts showed an 85% and 95% increase in total Cer and DAGs, respectively, compared to littermate controls (111). Further studies are needed to delineate the role accumulated Cer, and accumulated DAGs play in causing HF followed by mortality in the cardiomyocyte-specific deletion of DGAT1. Another study found that decreased Cer d18:1/C20:0, increased Cer d16:1/C23:0, d18:1/C19:0, d18:1/C22:0, and d18:2/C20:1, were observed between control and DGAT1 OE animals, whereas 3% were responsive to ANG II administration. ANG II treatment in the OE mice resulted in a marked increase in heart size, systolic dysfunction, and cardiac fibrosis, with major reduction of the above-mentioned Cer, compared with control littermates (94). This could suggest Cer reduction renders cardiomyocytes more vulnerable to other pathological stresses.
HFD are the most popular metabolic-disease induced-HF model, generally considered beneficial in the setting of non-ischemic HF (97). Different types of HFD can be generated for rodents that mimic human diets with respect to the content of carbohydrate, protein, and saturated and unsaturated fatty acids, with relative ease and economical cost. Most HF patients in the human trials discussed above present with a form of diabetes and or obesity, making this model even more illuminating. A few studies from our lab showed that mice fed on a 60% milk-fat based diet (MFBD) as opposed to a 60% lard (oleate)-fat based diet and control diet (CD), had much higher levels of CerS2 derived Cer C:20 to C:26 in earlier stages of diabetes and diabetic cardiomyopathy. Further along the diet, the mice now presented with LV hypertrophy and CerS5 derived Cer C14:0 was significantly higher (101, 102). Thus, these diets give us the ability to identify specific sphingolipid species over the course of the events leading up to HF and in incident HF of animals. A new model of HFpEF uses metabolic and hypertensive stress elicited by HFD coupled with NOS inhibition and showed differentially methylated RNAs were enriched in sphingolipid metabolism (118, 119).
Akita mice (Ins2Akita+/−) have a mutation in the Insulin2 gene and are a good non-obese type 1 diabetes mellitus (T1DM) model. At adulthood, the hearts of these mice had accumulated Cer C18:0 and exhibited preserved systolic function, reduced diastolic function, and increased inflammation. This lipotoxic cardiomyopathy phenotype was reversed with insulin replacement therapy (98). GENA348 mice, develop LVH and HF around 6 months of age (120). In 12-month GENA348 mice with systolic, diastolic dysfunction and LVH the following fold-change increases in serum samples were also observed—2.65× of SM C23:0, and a 2.26×, 1.80× and 1.45× of Cer C22:0, C24:1, and C22:1, respectively, compared to control mice (105).
A mouse model of atrial fibrillation (AF)-induced HF (cardiac-specific dnPI3K-Mst1 KO) showed significant increases of Cer C16:0 and the most abundant mammalian ganglioside, GM3 C16:0, in atria of AF + HF mice compared to control (92). These changes were also associated with increased atrial mass, though, treatment with the small molecule, a hydroximic acid derivative named BGP-15, reduced atrial mass and was positively correlated with reduced GM3 C16:0. Furthermore, BGP-15 treatment concomitantly increased insulin-like growth factor 1 binding to caveolins, a cardioprotective signaling pathway, thought to be inhibited by GM3 (92). This study suggests GM3 C16:0 may contribute to atrial pathology in the context of HF.
Heart failure with reduced ejection fraction
Most HF animal models are used in research to study HFrEF due to the clear clinical diagnostic criteria, and parameters. The two main animal models for HFrEF are ischemia/infarction and pacing models (72). Other less common models include surgically induced mitral regurgitation, arteriovenous fistula creation, and administration of doxorubicin (72, 121).
In the ischemia/infarction, microsphere beads are injected intracoronarily down selected arteries under fluoroscopy or there is temporary or permanent occlusion of the left anterior descending (LAD) coronary artery. A study on this model found that stearoyl-SM was reduced by less than 50% in heart tissue 5 days post permanent-LAD ligation in mice compared to mice with sham surgery (74). Rats with HF induced via permanent LAD ligation showed higher Cer C16:0 in the heart tissue compared to sham-operated mice (77). 8 weeks post MI, these rats went on a 45% kcal saturated and unsaturated combination HFD but showed no further exacerbation of LVH and no further increase in tissue total Cer. Thus, suggesting Cer myocardial content is not dependent on availability of fatty acids in the context of the failing heart. Another study also showed a similar increase in total Cer content after MI, interestingly, once the rats were placed on a 60% kcal high-saturated fat diet, unlike the previous study, the myocardial total Cer content increased, though there was no further progression of HF (78, 122). Isolated hearts from male rats subject to ischemia led to 14.1% increase in myocardial total Cer, 48.4% increase with ischemic reperfusion (IR), and partial reversal with ischemic preconditioning. The most significant being that of Cer C16:0, C18:0, C18:1, C18:2, C20:4, C22:5, and Cer C22:6 (123). These studies have shown that increased release of MCFA, LCFA, and VLCFA of Cer into circulation and found within cardiac tissue are often associated with negative effects on cardiac function. Although it is essential to develop pharmacological inhibitors of specific CerS, there have been multiple roadblocks in this process.
Conclusions
Great strides have been made in understanding the sphingolipidome in patients with HF. The Mayo Clinic's CERAM panel's ratios of the VLCFA to MCFA Cer are becoming increasingly popular for assessing the risk of HF development and progression. Evidence suggests these novel Cer biomarkers will continue be more successful than conventional risk factors in predicting adverse cardiac events and the onset of HF, especially when applied on a larger scale. However, there is still no clear evidence to show mechanistically how Cer C24:0 is linked to beneficial pathways or Cer C16:0 to adverse pathways in HF. More studies are needed to determine how Cer (and other sphingolipids) with different acyl chain lengths regulate various signaling pathways. Despite these developments, prognosis for HFrEF and HFpEF patients remain poor. Thus, when considering future studies in humans and animal models of HF some points should be considered.
The first point to consider: different results can be obtained from different materials analyzed, and while tissue sphingolipid levels are useful in patients receiving non-pharmacological intervention such as LVAD, it is of more clinical benefit to discover potent biomarkers in circulation, i.e., serum or plasma. This is due to ease of accessibility, and ability to collect samples more frequently in follow-up appointments. Nonetheless, tissue sphingolipid levels are important In fact, tissue from HF patients prior to LVAD showed induction of SPTLC3 at the protein level, a follow-up showed reduced SPTLC3 levels post-VAD (53).
Which leads to the second point for consideration, it is becoming more apparent that the de novo sphingolipid synthesis pathway can make very diverse sphingolipids in turn triggering unique signaling cascades. While d18 backbone sphingolipids are the most abundant and well-studied, there is mounting evidence to suggest substrate availability is the major determinant for the type of sphingolipids synthesized. For example, variable amino acid substitutes for serine, such as alanine or glycine, result in loss of the hydroxyl group at the alpha carbon to produce deoxysphingolipids. Deoxysphingolipids have been shown to play important roles in pathogenesis of both T1 and T2D. SPTLC3 can use isoleucine, a branched chain amino acid, to create methyl-branched long chain based sphingolipids, the implications of these in HF have yet to be elucidated (124). Though, it was recently revealed that transgenic mice with pressure-overload induced HFrEF showed impaired catabolism of myocardial branched-chain amino acids, upon which restoration of these amino acids reversed the dysfunction in this HFrEF animal model (125). Another way substrate availability can determine which sphingolipids to synthesize is availability of myristoyl- or stearoyl-CoA over palmitoyl-CoA. SPTLC3 can readily use myristoyl-CoA in place of palmitoyl CoA (25). Thus, more recent evidence points towards less abundant sphingolipid species with d16, d19 and d20 backbones associated with events leading up to and in HF (27, 30). A few animal studies highlighted in this review observed significant differences of SPTLC3-derived sphingolipid species. This could provide a new avenue of research in developing specific inhibitors of CerS and SMS.
The third point to consider: Which sphingolipidomic analyses technique to use. The omics field has seen a rapid increase in both the number of studies and the size of datasets. In parallel, liquid chromatography coupled with mass spectrometry (LC-MS) approaches have advanced yielding thousands of distinct MS peaks representing individual sphingolipid species and their metabolites (126, 127). Targeted LC-MS measurements are more sensitive, accurate, and quantitative than untargeted ones. However, most studies on human cohorts only focus on d18 Cer or SM species, and it would be of more beneficial consequence to include targeted LC-MS runs for d16, d19 and d20 backboned sphingolipids with acyl chains ranging from C14 to C26, as well as other species such as Sph, S1P, DHS1P, monohexosylceramides (MHC), and the glucosylceramides. While using targeted LC-MS may overlook associations with many known and unknown species in HF, untargeted mass spectrometric techniques can detect species that were not considered in targeted runs and associate them with HF (128). Though untargeted techniques are still being fine-tuned and require more starting sample per run. It is likely that multiple sphingolipids unique to each HF group (HFrEF, HFpEF, and HFbEF/HFmrEF) are involved in different pathogenesis, and these sphingolipid signatures could provide more concise cut-off values for classifying patients.
Author contributions
LC conceived of the content and scope of the review. GW and AK gathered relevant literature and wrote the manuscript. AK prepared figures and tables. LC and AK edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors would like to acknowledge support from National Heart, Lung, and Blood Institute (T32HL149645 to GW, F31HL156529 to AK, and R01HL151243 to LC). LC also wishes to acknowledge support from Veterans' Affairs, I01BX0002000.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GM, Coats AJ. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res. (2022) 118(17):3272–87. doi: 10.1093/cvr/cvac013
2. Conrad N, Judge A, Tran J, Mohseni H, Hedgecott D, Crespillo AP, et al. Temporal trends and patterns in heart failure incidence: a population-based study of 4 million individuals. Lancet. (2018) 391(10120):572–80. doi: 10.1016/S0140-6736(17)32520-5
3. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics—2021 update: a report from the American heart association. Circulation. (2021) 143(8):e254–743. doi: 10.1161/CIR.0000000000000950
4. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. J Am Coll Cardiol. (2022) 79(17):e263–421. doi: 10.1016/j.jacc.2021.12.012
5. Ponikowski P, Voors A, Anker S, Bueno H, Cleland J, Coats A, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European society of cardiology (ESC) developed with the special contribution of the heart failure association (HFA) of the ESC. Eur Heart J. (2016) 37(27):2129–200. doi: 10.1093/eurheartj/ehw128
6. Savarese G, Stolfo D, Sinagra G, Lund LH. Heart failure with mid-range or mildly reduced ejection fraction. Nat Rev Cardiol. (2022) 19(2):100–16. doi: 10.1038/s41569-021-00605-5
7. Reed BN, Sueta CA. A practical guide for the treatment of symptomatic heart failure with reduced ejection fraction (HFrEF). Curr Cardiol Rev. (2015) 11(1):23–32. doi: 10.2174/1574884708666131117125508
8. Balmforth C, Simpson J, Shen L, Jhund PS, Lefkowitz M, Rizkala AR, et al. Outcomes and effect of treatment according to etiology in HFrEF: an analysis of PARADIGM-HF. JACC Heart Fail. (2019) 7(6):457–65. doi: 10.1016/j.jchf.2019.02.015
9. Tang R, Chang Y, Song J. Advances in novel devices for the treatment of heart failure. Heart Fail Rev. (2023) 28(2):331–45. doi: 10.1007/s10741-022-10293-z
10. Wang Y, Zhou R, Lu C, Chen Q, Xu T, Li D. Effects of the angiotensin-receptor neprilysin inhibitor on cardiac reverse remodeling: meta-analysis. J Am Heart Assoc. (2019) 8(13):e012272. doi: 10.1161/JAHA.119.012272
11. Shah SJ, Borlaug BA, Chung ES, Cutlip DE, Debonnaire P, Fail PS, et al. Atrial shunt device for heart failure with preserved and mildly reduced ejection fraction (REDUCE LAP-HF II): a randomised, multicentre, blinded, sham-controlled trial. Lancet. (2022) 399(10330):1130–40. doi: 10.1016/S0140-6736(22)00016-2
12. Giannopoulos G, Kousta M, Anagnostopoulos I, Karageorgiou S, Myrovali E, Deftereos G, et al. Advances in heart failure with preserved ejection fraction management-the role of sacubitril-valsartan, pirfenidone, spironolactone and empagliflozin: is success a series of small victories? Curr Pharm Des. (2023) 29(7):502–8. doi: 10.2174/1381612829666230202141437
13. Gronda E, Vanoli E, Iacoviello M. The PARAGON-HF trial: the sacubitril/valsartan in heart failure with preserved ejection fraction. Eur Heart J Suppl. (2020) 22(Suppl L):L77–81. doi: 10.1093/eurheartj/suaa140
14. Solomon SD, McMurray JJ, Anand IS, Ge J, Lam CS, Maggioni AP, et al. Angiotensin–neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. (2019) 381(17):1609–20. doi: 10.1056/NEJMoa1908655
15. Nassif ME, Windsor SL, Borlaug BA, Kitzman DW, Shah SJ, Tang F, et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat Med. (2021) 27(11):1954–60. doi: 10.1038/s41591-021-01536-x
16. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. (2014) 370:1383–92. doi: 10.1056/NEJMoa1313731
17. Shah KS, Xu H, Matsouaka RA, Bhatt DL, Heidenreich PA, Hernandez AF, et al. Heart failure with preserved, borderline, and reduced ejection fraction: 5-year outcomes. J Am Coll Cardiol. (2017) 70(20):2476–86. doi: 10.1016/j.jacc.2017.08.074
18. Chiurchiù V, Leuti A, Maccarrone M. Bioactive lipids and chronic inflammation: managing the fire within. Front Immunol. (2018) 9:38. doi: 10.3389/fimmu.2018.00038
19. Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. (2014) 510(7503):58–67. doi: 10.1038/nature13475
20. Pagano RE, Sleight RG. Defining lipid transport pathways in animal cells. Science. (1985) 229(4718):1051–7. doi: 10.1126/science.4035344
21. Zhao L, Spassieva S, Gable K, Gupta SD, Shi L-Y, Wang J, et al. Elevation of 20-carbon long chain bases due to a mutation in serine palmitoyltransferase small subunit b results in neurodegeneration. Proc Natl Acad Sci USA. (2015) 112(42):12962–7. doi: 10.1073/pnas.1516733112
22. Hjelmqvist L, Tuson M, Marfany G, Herrero E, Balcells S, Gonzàlez-Duarte R. ORMDL proteins are a conserved new family of endoplasmic reticulum membrane proteins. Genome Biol. (2002) 3:1–16. doi: 10.1186/gb-2002-3-6-research0027
23. Nagiec MM, Baltisberger JA, Wells GB, Lester RL, Dickson RC. The LCB2 gene of saccharomyces and the related LCB1 gene encode subunits of serine palmitoyltransferase, the initial enzyme in sphingolipid synthesis. Proc Natl Acad Sci USA. (1994) 91(17):7899–902. doi: 10.1073/pnas.91.17.7899
24. Hornemann T, Richard S, Rütti MF, Wei Y, von Eckardstein A. Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. J Biol Chem. (2006) 281(49):37275–81. doi: 10.1074/jbc.M608066200
25. Russo SB, Tidhar R, Futerman AH, Cowart LA. Myristate-derived d16: 0 sphingolipids constitute a cardiac sphingolipid pool with distinct synthetic routes and functional properties. J Biol Chem. (2013) 288(19):13397–409. doi: 10.1074/jbc.M112.428185
26. Lone MA, Bourquin F, Hornemann T. Serine palmitoyltransferase subunit 3 and metabolic diseases. Zurich, Switzerland: Springer (2022). 47–56.
27. Hicks AA, Pramstaller PP, Johansson Å, Vitart V, Rudan I, Ugocsai P, et al. Genetic determinants of circulating sphingolipid concentrations in European populations. PLoS Genet. (2009) 5(10):e1000672. doi: 10.1371/journal.pgen.1000672
28. Othman A, Saely CH, Muendlein A, Vonbank A, Drexel H, von Eckardstein A, et al. Plasma C20-sphingolipids predict cardiovascular events independently from conventional cardiovascular risk factors in patients undergoing coronary angiography. Atherosclerosis. (2015) 240(1):216–21. doi: 10.1016/j.atherosclerosis.2015.03.011
29. Tabassum R, Rämö JT, Ripatti P, Koskela JT, Kurki M, Karjalainen J, et al. Genetic architecture of human plasma lipidome and its link to cardiovascular disease. Nat Commun. (2019) 10(1):4329. doi: 10.1038/s41467-019-11954-8
30. Cresci S, Zhang R, Yang Q, Duncan MS, Xanthakis V, Jiang X, et al. Genetic architecture of circulating very-long-chain (C24: 0 and C22: 0) ceramide concentrations. J Lipid Atheroscler. (2020) 9(1):172–83. doi: 10.12997/jla.2020.9.1.172
31. Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do lasses (longevity assurance genes) become CerS (ceramide synthases)? insights into the regulation of ceramide synthesis. J Biol Chem. (2006) 281(35):25001–5. doi: 10.1074/jbc.R600010200
32. Shimeno H, Soeda S, Sakamoto M, Kouchi T, Kowakame T, Kihara T. Partial purification and characterization of sphingosine N-acyltransferase (ceramide synthase) from bovine liver mitochondrion-rich fraction. Lipids. (1998) 33:601–5. doi: 10.1007/s11745-998-0246-2
33. Laviad EL, Albee L, Pankova-Kholmyansky I, Epstein S, Park H, Merrill AH, et al. Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem. (2008) 283(9):5677–84. doi: 10.1074/jbc.M707386200
34. Mizutani Y, Kihara A, Igarashi Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem J. (2005) 390(1):263–71. doi: 10.1042/BJ20050291
35. Mizutani Y, Kihara A, Igarashi Y. LASS3 (longevity assurance homologue 3) is a mainly testis-specific (dihydro) ceramide synthase with relatively broad substrate specificity. Biochem J. (2006) 398(3):531–8. doi: 10.1042/BJ20060379
36. Riebeling C, Allegood JC, Wang E, Merrill AH, Futerman AH. Two mammalian longevity assurance gene (LAG1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-CoA donors. J Biol Chem. (2003) 278(44):43452–9. doi: 10.1074/jbc.M307104200
37. Venkataraman K, Riebeling C, Bodennec J, Riezman H, Allegood JC, Sullards MC, et al. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulatesN-stearoyl-sphinganine (C18-(dihydro) ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J Biol Chem. (2002) 277(38):35642–9. doi: 10.1074/jbc.M205211200
38. Lahiri S, Lee H, Mesicek J, Fuks Z, Haimovitz-Friedman A, Kolesnick RN, et al. Kinetic characterization of mammalian ceramide synthases: determination of km values towards sphinganine. FEBS Lett. (2007) 581(27):5289–94. doi: 10.1016/j.febslet.2007.10.018
39. Merrill M Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. (2011) 111(10):6387–422. doi: 10.1021/cr2002917
40. Aguilar A, Saba JD. Truth and consequences of sphingosine-1-phosphate lyase. Adv Biol Regul. (2012) 52(1):17. doi: 10.1016/j.advenzreg.2011.09.015
41. Bing R, Siegel A, Ungar I, Gilbert M. Metabolism of the human heart. II. Metabolism of fats, proteins and ketones. Am J Med. (1954) 16:504. doi: 10.1016/0002-9343(54)90365-4
42. Schulze PC, Drosatos K, Goldberg IJ. Lipid use and misuse by the heart. Circ Res. (2016) 118(11):1736–51. doi: 10.1161/CIRCRESAHA.116.306842
43. Park T-S, Rosebury W, Kindt EK, Kowala MC, Panek RL. Serine palmitoyltransferase inhibitor myriocin induces the regression of atherosclerotic plaques in hyperlipidemic ApoE-deficient mice. Pharmacol Res. (2008) 58(1):45–51. doi: 10.1016/j.phrs.2008.06.005
44. Zhang DX, Fryer RM, Hsu AK, Zou A-P, Gross GJ, Campbell WB, et al. Production and metabolism of ceramide in normal and ischemic-reperfused myocardium of rats. Basic Res Cardiol. (2001) 96:267–74. doi: 10.1007/s003950170057
45. Waeber C, Walther T. Sphingosine-1-phosphate as a potential target for the treatment of myocardial infarction. Circ J. (2014) 78(4):795–802. doi: 10.1253/circj.CJ-14-0178
46. Pitson SM. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci. (2011) 36(2):97–107. doi: 10.1016/j.tibs.2010.08.001
47. Peterson LR, Xanthakis V, Duncan MS, Gross S, Friedrich N, Völzke H, et al. Ceramide remodeling and risk of cardiovascular events and mortality. J Am Heart Assoc. (2018) 7(10):e007931. doi: 10.1161/JAHA.117.007931
48. Lemaitre RN, Jensen PN, Hoofnagle A, McKnight B, Fretts AM, King IB, et al. Plasma ceramides and sphingomyelins in relation to heart failure risk: the cardiovascular health study. Circ Heart Fail. (2019) 12(7):e005708. doi: 10.1161/CIRCHEARTFAILURE.118.005708
49. Jensen PN, Fretts AM, Hoofnagle AN, McKnight B, Howard BV, Umans J, et al. Circulating ceramides and sphingomyelins and the risk of incident cardiovascular disease among people with diabetes: the strong heart study. (2022) 21(1):167. doi: 10.1186/s12933-022-01596-4
50. Wittenbecher C, Eichelmann F, Toledo E, Guasch-Ferré M, Ruiz-Canela M, Li J, et al. Lipid profiles and heart failure risk: results from two prospective studies. Circ Res. (2021) 128(3):309–20. doi: 10.1161/CIRCRESAHA.120.317883
51. Knapp M, Baranowski M, Lisowska A, Musiał W. Decreased free sphingoid base concentration in the plasma of patients with chronic systolic heart failure. Adv Med Sci. (2012) 57(1):100–5. doi: 10.2478/v10039-011-0057-4
52. Pérez-Carrillo L, Giménez-Escamilla I, Martínez-Dolz L, Sánchez-Lázaro IJ, Portolés M, Roselló-Lletí E, et al. Implication of sphingolipid metabolism gene dysregulation and cardiac sphingosine-1-phosphate accumulation in heart failure. Biomedicines. (2022) 10(1):135. doi: 10.3390/biomedicines10010135
53. Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI insight. (2017) 2(14):e96203. doi: 10.1172/jci.insight.96203
54. Javaheri A, Allegood JC, Cowart LA, Chirinos JA. Circulating ceramide 16: 0 in heart failure with preserved ejection fraction. J Am Coll Cardiol. (2020) 75(17):2273–5. doi: 10.1016/j.jacc.2020.02.062
55. Targher G, Lunardi G, Mantovani A, Meessen J, Bonapace S, Temporelli PL, et al. Relation between plasma ceramides and cardiovascular death in chronic heart failure: a subset analysis of the GISSI-HF trial. ESC Heart Fail. (2020) 7(6):3288–97. doi: 10.1002/ehf2.12885
56. Yu J, Pan W, Shi R, Yang T, Li Y, Yu G, et al. Ceramide is upregulated and associated with mortality in patients with chronic heart failure. Can J Cardiol. (2015) 31(3):357–63. doi: 10.1016/j.cjca.2014.12.007
58. Fretts AM, Jensen PN, Hoofnagle A, McKnight B, Howard BV, Umans J, et al. Plasma ceramide species are associated with diabetes risk in participants of the strong heart study. J Nutr. (2020) 150(5):1214–22. doi: 10.1093/jn/nxz259
59. Mikhalkova D, Holman SR, Jiang H, Saghir M, Novak E, Coggan AR, et al. Bariatric surgery–induced cardiac and lipidomic changes in obesity-related heart failure with preserved ejection fraction. Obesity. (2018) 26(2):284–90. doi: 10.1002/oby.22038
60. Havulinna AS, Sysi-Aho M, Hilvo M, Kauhanen D, Hurme R, Ekroos K, et al. Circulating ceramides predict cardiovascular outcomes in the population-based FINRISK 2002 cohort. Arterioscler Thromb Vasc Biol. (2016) 36(12):2424–30. doi: 10.1161/ATVBAHA.116.307497
61. Polzin A, Piayda K, Keul P, Dannenberg L, Mohring A, Gräler M, et al. Plasma sphingosine-1-phosphate concentrations are associated with systolic heart failure in patients with ischemic heart disease. J Mol Cell Cardiol. (2017) 110:35–7. doi: 10.1016/j.yjmcc.2017.07.004
62. Winkelmann BR, März W, Boehm BO, Zotz R, Hager J, Hellstern P, et al. Rationale and design of the LURIC study-a resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics. (2001) 2(1):S1–73. doi: 10.1517/14622416.2.1.S1
63. Sigruener A, Kleber ME, Heimerl S, Liebisch G, Schmitz G, Maerz W. Glycerophospholipid and sphingolipid species and mortality: the ludwigshafen risk and cardiovascular health (LURIC) study. PLoS One. (2014) 9(1):e85724. doi: 10.1371/journal.pone.0085724
64. Airhart S, Cade WT, Jiang H, Coggan AR, Racette SB, Korenblat K, et al. A diet rich in medium-chain fatty acids improves systolic function and alters the lipidomic profile in patients with type 2 diabetes: a pilot study. J Clin Endocrinol Metab. (2016) 101(2):504–12. doi: 10.1210/jc.2015-3292
65. Nwabuo CC, Duncan M, Xanthakis V, Peterson LR, Mitchell GF, McManus D, et al. Association of circulating ceramides with cardiac structure and function in the community: the framingham heart study. J Am Heart Assoc. (2019) 8(19):e013050. doi: 10.1161/JAHA.119.013050
66. Jensen PN, Fretts AM, Hoofnagle AN, Sitlani CM, McKnight B, King IB, et al. Plasma ceramides and sphingomyelins in relation to atrial fibrillation risk: the cardiovascular health study. J Am Heart Assoc. (2020) 9(4):e012853. doi: 10.1161/JAHA.119.012853
67. Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. (2015) 47(8):856–60. doi: 10.1038/ng.3314
68. Ganna A, Salihovic S, Sundström J, Broeckling CD, Hedman ÅK, Magnusson PK, et al. Large-scale metabolomic profiling identifies novel biomarkers for incident coronary heart disease. PLoS Genet. (2014) 10(12):e1004801. doi: 10.1371/journal.pgen.1004801
69. Wittenbecher C, Cuadrat R, Johnston L, Eichelmann F, Jäger S, Kuxhaus O, et al. Dihydroceramide-and ceramide-profiling provides insights into human cardiometabolic disease etiology. Nat Commun. (2022) 13(1):1–13. doi: 10.1038/s41467-022-28496-1
70. Luo C, Liu H, Wang X, Xia L, Huang H, Peng X, et al. The associations between individual plasma SFAs, serine palmitoyl-transferase long-chain base subunit 3 gene rs680379 polymorphism, and type 2 diabetes among Chinese adults. Am J Clin Nutr. (2021) 114(2):704–12. doi: 10.1093/ajcn/nqab102
71. Liu X, Tong X, Zou Y, Lin X, Zhao H, Tian L, et al. Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome. Nat Genet. (2022) 54(1):52–61. doi: 10.1038/s41588-021-00968-y
72. Charles CJ, Rademaker MT, Scott NJ, Richards AM. Large animal models of heart failure: reduced vs. preserved ejection fraction. Animals (Basel). (2020) 10(10):1906. doi: 10.3390/ani10101906
73. Conceicao G, Heinonen I, Lourenco A, Duncker D, Falcao-Pires I. Animal models of heart failure with preserved ejection fraction. Neth Heart J. (2016) 24(4):275–86. doi: 10.1007/s12471-016-0815-9
74. Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail. (2014) 7(4):634–42. doi: 10.1161/CIRCHEARTFAILURE.114.001151
75. Halade GV, Kain V, Tourki B, Jadapalli JK. Lipoxygenase drives lipidomic and metabolic reprogramming in ischemic heart failure. Metab Clin Exp. (2019) 96:22–32. doi: 10.1016/j.metabol.2019.04.011
76. Devarakonda T, Valle Raleigh J, Mauro AG, Lambert JM, Cowart LA, Salloum FN. Chronic treatment with serelaxin mitigates adverse remodeling in a murine model of ischemic heart failure and modulates bioactive sphingolipid signaling. Sci Rep. (2022) 12(1):8897. doi: 10.1038/s41598-022-12930-x
77. Morgan EE, Rennison JH, Young ME, McElfresh TA, Kung TA, Tserng K-Y, et al. Effects of chronic activation of peroxisome proliferator-activated receptor-α or high-fat feeding in a rat infarct model of heart failure. Am J Physiol Heart Circ Physiol. (2006) 290(5):H1899–904. doi: 10.1152/ajpheart.01014.2005
78. Rennison JH, McElfresh TA, Okere IC, Vazquez EJ, Patel HV, Foster AB, et al. High-fat diet postinfarction enhances mitochondrial function and does not exacerbate left ventricular dysfunction. Am J Physiol Heart Circ Physiol. (2007) 292(3):H1498–506. doi: 10.1152/ajpheart.01021.2006
79. Marín-Royo G, Ortega-Hernández A, Martínez-Martínez E, Jurado-López R, Luaces M, Islas F, et al. The impact of cardiac lipotoxicity on cardiac function and mirnas signature in obese and non-obese rats with myocardial infarction. Sci Rep. (2019) 9(1):1–11. doi: 10.1038/s41598-018-36914-y
80. Nam M, Jung Y, Hwang G-S. A metabolomics-driven approach reveals metabolic responses and mechanisms in the rat heart following myocardial infarction. Int J Cardiol. (2017) 227:239–46. doi: 10.1016/j.ijcard.2016.11.127
81. Thielmann M, Dörge H, Martin C, Belosjorow S, Schwanke U, van De Sand A, et al. Myocardial dysfunction with coronary microembolization: signal transduction through a sequence of nitric oxide, tumor necrosis factor-α, and sphingosine. Circ Res. (2002) 90(7):807–13. doi: 10.1161/01.RES.0000014451.75415.36
82. Stamm C, Friehs I, Cowan DB, Moran AM, Cao-Danh H, Duebener LF, et al. Inhibition of tumor necrosis factor-α improves postischemic recovery of hypertrophied hearts. Circulation. (2001) 104(Suppl_1):I-350–5. doi: 10.1161/circ.104.suppl_1.I-350
83. He L, Kim T, Long Q, Liu J, Wang P, Zhou Y, et al. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload–induced cardiac hypertrophy caused by lipotoxicity. Circulation. (2012) 126(14):1705–16. doi: 10.1161/CIRCULATIONAHA.111.075978
84. Muthuramu I, Amin R, Singh N, Postnov A, Gheysens O, Van Veldhoven P, et al. Dietary saturated fatty acids aggravate pressure overload-induced cardiomyopathy in mice in the absence of cardiac steatosis. Atherosclerosis. (2016) 252(e119) doi: 10.1016/j.atherosclerosis.2016.07.632
85. Wang X, Zhu X-X, Jiao S-Y, Qi D, Yu B-Q, Xie G-M, et al. Cardiomyocyte peroxisome proliferator-activated receptor α is essential for energy metabolism and extracellular matrix homeostasis during pressure overload-induced cardiac remodeling. Acta Pharmacol Sin. (2022) 43(5):1231–42. doi: 10.1038/s41401-021-00743-z
86. Zhang Y, Huang Y, Cantalupo A, Azevedo PS, Siragusa M, Bielawski J, et al. Endothelial Nogo-B regulates sphingolipid biosynthesis to promote pathological cardiac hypertrophy during chronic pressure overload. JCI insight. (2016) 1(5). doi: 10.1172/jci.insight.85484
87. Goldenberg JR, Carley AN, Ji R, Zhang X, Fasano M, Schulze PC, et al. Preservation of acyl coenzyme a attenuates pathological and metabolic cardiac remodeling through selective lipid trafficking. Circulation. (2019) 139(24):2765–77. doi: 10.1161/CIRCULATIONAHA.119.039610
88. Liu X, Wu J, Zhu C, Liu J, Chen X, Zhuang T, et al. Endothelial S1pr1 regulates pressure overload-induced cardiac remodelling through AKT-eNOS pathway. J Cell Mol Med. (2020) 24(2):2013–26. doi: 10.1111/jcmm.14900
89. Wang L, Deng H, Wang T, Qiao Y, Zhu J, Xiong M. Investigation into the protective effects of hypaconitine and glycyrrhetinic acid against chronic heart failure of the rats. BMC Complement Med Ther. (2022) 22(1):160. doi: 10.1186/s12906-022-03632-y
90. Itoi T, Oka T, Terada N. Modulation of C 16: 0-ceramide in hypertrophied immature hearts by losartan. Pediatr Int. (2013) 55(3):272–6. doi: 10.1111/ped.12052
91. Butler T, Ashford D, Seymour A-M. Western diet increases cardiac ceramide content in healthy and hypertrophied hearts. Nutr Metab Cardiovasc Dis. (2017) 27(11):991–8. doi: 10.1016/j.numecd.2017.08.007
92. Sapra G, Tham YK, Cemerlang N, Matsumoto A, Kiriazis H, Bernardo BC, et al. The small-molecule BGP-15 protects against heart failure and atrial fibrillation in mice. Nat Commun. (2014) 5(1):5705. doi: 10.1038/ncomms6705
93. Pellieux C, Montessuit C, Papageorgiou I, Pedrazzini T, Lerch R. Differential effects of high-fat diet on myocardial lipid metabolism in failing and nonfailing hearts with angiotensin II-mediated cardiac remodeling in mice. Am J Physiol Heart Circ Physiol. (2012) 302(9):H1795–805. doi: 10.1152/ajpheart.01023.2011
94. Glenn DJ, Cardema MC, Ni W, Zhang Y, Yeghiazarians Y, Grapov D, et al. Cardiac steatosis potentiates angiotensin II effects in the heart. Am J Physiol Heart Circ Physiol. (2015) 308(4):H339–50. doi: 10.1152/ajpheart.00742.2014
95. Okere IC, Chess DJ, McElfresh TA, Johnson J, Rennison J, Ernsberger P, et al. High-fat diet prevents cardiac hypertrophy and improves contractile function in the hypertensive dahl salt-sensitive rat. Clin Exp Pharmacol Physiol. (2005) 32(10):825–31. doi: 10.1111/j.1440-1681.2005.04272.x
96. Li J, Kemp BA, Howell NL, Massey J, Mińczuk K, Huang Q, et al. Metabolic changes in spontaneously hypertensive rat hearts precede cardiac dysfunction and left ventricular hypertrophy. J Am Heart Assoc. (2019) 8(4):e010926. doi: 10.1161/JAHA.118.010926
97. Okere IC, Young ME, McElfresh TA, Chess DJ, Sharov VG, Sabbah HN, et al. Low carbohydrate/high-fat diet attenuates cardiac hypertrophy, remodeling, and altered gene expression in hypertension. Hypertension. (2006) 48(6):1116–23. doi: 10.1161/01.HYP.0000248430.26229.0f
98. Kassiri Z. Type 1 diabetic cardiomyopathy in the akita (Ins2 WT/C96Y) mouse model is 6 characterized by lipotoxicity and diastolic dysfunction with preserved 7 systolic function 8. (2009) 297(6):H2096-108. doi: 10.1152/ajpheart.00452.2009
99. Pulinilkunnil T, Kienesberger PC, Nagendran J, Waller TJ, Young ME, Kershaw EE, et al. Myocardial adipose triglyceride lipase overexpression protects diabetic mice from the development of lipotoxic cardiomyopathy. Diabetes. (2013) 62(5):1464–77. doi: 10.2337/db12-0927
100. DeMarco VG, Ford DA, Henriksen EJ, Aroor AR, Johnson MS, Habibi J, et al. Obesity-related alterations in cardiac lipid profile and nondipping blood pressure pattern during transition to diastolic dysfunction in male db/db mice. Endocrinology. (2013) 154(1):159–71. doi: 10.1210/en.2012-1835
101. Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, et al. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest. (2012) 122(11):3919–30. doi: 10.1172/JCI63888
102. Law BA, Liao X, Moore KS, Southard A, Roddy P, Ji R, et al. Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J. (2018) 32(3):1403–16. doi: 10.1096/fj.201700300R
103. Zhang L, Ussher JR, Oka T, Cadete VJ, Wagg C, Lopaschuk GD. Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res. (2011) 89(1):148–56. doi: 10.1093/cvr/cvq266
104. Okere IC, Chandler MP, McElfresh TA, Rennison JH, Sharov V, Sabbah HN, et al. Differential effects of saturated and unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose distribution, and serum leptin. American Journal of Physiology-Heart and Circulatory Physiology. (2006) 291(1):H38–44. doi: 10.1152/ajpheart.01295.2005
105. Al-Maimani RA. Investigation into the underlying mechanisms of diabetic cardiomyopathy using a mouse model of diabetes. United Kingdom: The University of Manchester (2016).
106. Park T-S, Hu Y, Noh H-L, Drosatos K, Okajima K, Buchanan J, et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy*s⃞. J Lipid Res. (2008) 49(10):2101–12. doi: 10.1194/jlr.M800147-JLR200
107. Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, et al. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. (2009) 284(52):36312–23. doi: 10.1074/jbc.M109.049817
108. Son N-H, Park T-S, Yamashita H, Yokoyama M, Huggins LA, Okajima K, et al. Cardiomyocyte expression of PPARγ leads to cardiac dysfunction in mice. J Clin Invest. (2007) 117(10):2791–801. doi: 10.1172/JCI30335
109. Yu S, Khan RS, Homma S, Schulze PC, Blaner WS, Goldberg IJ. Diacylglycerol acyl transferase 1 overexpression detoxifies cardiac lipids in PPARγ transgenic mice [S]. J Lipid Res. (2012) 53(8):1482–92. doi: 10.1194/jlr.M024208
110. Chiu H-C, Kovacs A, Ford DA, Hsu F-F, Garcia R, Herrero P, et al. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. (2001) 107(7):813–22. doi: 10.1172/JCI10947
111. Liu L, Trent CM, Fang X, Son N-H, Jiang H, Blaner WS, et al. Cardiomyocyte-specific loss of diacylglycerol acyltransferase 1 (DGAT1) reproduces the abnormalities in lipids found in severe heart failure. J Biol Chem. (2014) 289(43):29881–91. doi: 10.1074/jbc.M114.601864
112. Lee S-Y, Kim JR, Hu Y, Khan R, Kim S-J, Bharadwaj KG, et al. Cardiomyocyte specific deficiency of serine palmitoyltransferase subunit 2 reduces ceramide but leads to cardiac dysfunction. J Biol Chem. (2012) 287(22):18429–39. doi: 10.1074/jbc.M111.296947
113. Bakermans AJ, Geraedts TR, van Weeghel M, Denis S, João Ferraz M, Aerts JM, et al. Fasting-induced myocardial lipid accumulation in long-chain acyl-CoA dehydrogenase knockout mice is accompanied by impaired left ventricular function. Circulation: Cardiovascular Imaging. (2011) 4(5):558–65. doi: 10.1161/CIRCIMAGING.111.963751
114. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the framingham study. Am J Cardiol. (1974) 34(1):29–34. doi: 10.1016/0002-9149(74)90089-7
115. Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmad MR, et al. Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest. (1977) 60(4):885–99. doi: 10.1172/JCI108843
116. Park T-S, Goldberg IJ. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail Clin. (2012) 8(4):633–41. doi: 10.1016/j.hfc.2012.06.003
117. Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation. (2012) 125(23):2844–53. doi: 10.1161/CIRCULATIONAHA.111.060889
118. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. (2019) 568(7752):351–6. doi: 10.1038/s41586-019-1100-z
119. Liu K, Ju W, Ouyang S, Liu Z, He F, Hao J, et al. Exercise training ameliorates myocardial phenotypes in heart failure with preserved ejection fraction by changing N6-methyladenosine modification in mice model. Front Cell Dev Biol. (2022) 10:954769. doi: 10.3389/fcell.2022.954769
120. Mamas M, Gibbons S, Zi M, Prehar S, Oceandy D, Cartwright E, et al. GENA348, a novel human relevant mouse model of diabetes displays spontaneous cardiac hypertrophy. Manchester, UK: BMJ Publishing Group Ltd and British Cardiovascular Society (2009).
121. Spannbauer A, Traxler D, Zlabinger K, Gugerell A, Winkler J, Mester-Tonczar J, et al. Large animal models of heart failure with reduced ejection fraction (HFrEF). Front Cardiovasc Med. (2019) 6:117. doi: 10.3389/fcvm.2019.00117
122. Stanley WC, Dabkowski ER, Ribeiro Jr RF, O'Connell KA. Dietary fat and heart failure: moving from lipotoxicity to lipoprotection. Circ Res. (2012) 110(5):764–76. doi: 10.1161/CIRCRESAHA.111.253104
123. Beresewicz A, Dobrzyń A, Gorski J. Accumulation of specific ceramides in ischemic/reperfused rat heart; effect of ischemic preconditioning. J Physiol Pharmacol. (2002) 53(3):371–82.12369735
124. Lone MA, Hülsmeier AJ, Saied EM, Karsai G, Arenz C, von Eckardstein A, et al. Subunit composition of the mammalian serine-palmitoyltransferase defines the spectrum of straight and methyl-branched long-chain bases. Proc Natl Acad Sci USA. (2020) 117(27):15591–8. doi: 10.1073/pnas.2002391117
125. Li Z, Xia H, Sharp TE III, LaPenna KB, Elrod JW, Casin KM, et al. Mitochondrial H2S regulates BCAA catabolism in heart failure. Circ Res. (2022) 131(3):222–35. doi: 10.1161/CIRCRESAHA.121.319817
126. Sullards MC, Liu Y, Chen Y, Merrill AH Jr. Analysis of mammalian sphingolipids by liquid chromatography tandem mass spectrometry (LC-MS/MS) and tissue imaging mass spectrometry (TIMS). Biochim Biophys Acta, Mol Cell Biol Lipids. (2011) 1811(11):838–53. doi: 10.1016/j.bbalip.2011.06.027
127. Scherer M, Leuthäuser-Jaschinski K, Ecker J, Schmitz G, Liebisch G. A rapid and quantitative LC-MS/MS method to profile sphingolipids. J Lipid Res. (2010) 51(7):2001–11. doi: 10.1194/jlr.D005322
Keywords: sphingolipid, ceramide, heart failure, HFrEF—heart failure with reduced ejection fraction, HFpEF—heart failure with preserved ejection fraction, serine palmitoyltransferase, lipidomics, ceramide score
Citation: Kovilakath A, Wohlford G and Cowart LA (2023) Circulating sphingolipids in heart failure. Front. Cardiovasc. Med. 10:1154447. doi: 10.3389/fcvm.2023.1154447
Received: 30 January 2023; Accepted: 4 April 2023;
Published: 9 May 2023.
Edited by:
Marco Piccoli, IRCCS San Donato Polyclinic, ItalyReviewed by:
Paola Signorelli, University of Milan, ItalyGregory Aubert, Vifor Pharma, Switzerland
Linda Ruth Peterson, Washington University in St. Louis, United States
© 2023 Kovilakath, Wohlford and Cowart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: L. Ashley Cowart bGF1cmVuLmNvd2FydEB2Y3VoZWFsdGgub3Jn