 Tauben Averbuch
Tauben Averbuch James A. White1,2
James A. White1,2 Nowell M. Fine
Nowell M. Fine
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 02 June 2023
Sec. Heart Failure and Transplantation
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1152568
This article is part of the Research Topic Infiltrative Cardiomyopathy and Heart Failure View all 5 articles
Anderson-Fabry disease (AFD) is an X-linked lysosomal storage disorder caused by deficient activity of the enzyme alpha-galactosidase. While AFD is recognized as a progressive multi-system disorder, infiltrative cardiomyopathy causing a number of cardiovascular manifestations is recognized as an important complication of this disease. AFD affects both men and women, although the clinical presentation typically varies by sex, with men presenting at a younger age with more neurologic and renal phenotype and women developing a later onset variant with more cardiovascular manifestations. AFD is an important cause of increased myocardial wall thickness, and advances in imaging, in particular cardiac magnetic resonance imaging and T1 mapping techniques, have improved the ability to identify this disease non-invasively. Diagnosis is confirmed by the presence of low alpha-galactosidase activity and identification of a mutation in the GLA gene. Enzyme replacement therapy remains the mainstay of disease modifying therapy, with two formulations currently approved. In addition, newer treatments such as oral chaperone therapy are now available for select patients, with a number of other investigational therapies in development. The availability of these therapies has significantly improved outcomes for AFD patients. Improved survival and the availability of multiple agents has presented new clinical dilemmas regarding disease monitoring and surveillance using clinical, imaging and laboratory biomarkers, in addition to improved approaches to managing cardiovascular risk factors and AFD complications. This review will provide an update on clinical recognition and diagnostic approaches including differentiation from other causes of increased ventricular wall thickness, in addition to modern strategies for management and follow-up.
Anderson-Fabry disease (AFD) is an X-linked glycogen storage disorder, characterized by the accumulation of globotriaosylceramide (Gb3) due to a deficiency of α-galactosidase. First described independently 1898 by Johannes Fabry and William Anderson in two patients with angiokeratoma (1), the GLA gene responsible for the production of α-galactosidase was identified in 1974 (1). Over time, AFD has been identified as a multi-system disease, with characteristic cardiac, renal, neurologic, dermatologic, and gastrointestinal manifestations. Additionally, the variable presentation of AFD has increasingly been recognized, with both classical and non-classical (or late onset) phenotypes (2). Patients with classical AFD are more commonly males in early adulthood who present with painful peripheral neuropathy, cutaneous lesions, and gastrointestinal upset in childhood or the early teenage years, with gradual development of heart failure (HF) and chronic kidney disease (3). Late-onset AFD is considerably more variable, presents in middle-aged or older patients, may be limited to cardiac or renal involvement (4) and has been described as a more common presentation for affected female patients. In contrast to classical AFD, α-galactosidase levels are higher in late-onset AFD, accounting for the delayed presentation (5).
Patients with AFD face significant morbidity and mortality, with reduced life expectancy. Early diagnosis of AFD is essential in order to start therapy, however, the diagnosis of AFD is frequently delayed by several years after the development of symptoms (6). Treatment has historically been limited to enzyme replacement therapy with recombinant α-galactosidase, however, novel therapies are also now available with new agents in development (7). In this comprehensive review, we describe the genetic basis of AFD and its pathophysiology, as well as the presentation and natural history of AFD with a focus on sex-based on phenotypic differences and a particular emphasis on the natural history of cardiac involvement. We review the diagnostic process in AFD, as well as non-invasive techniques to differentiate AFD from mimics, such as hypertrophic cardiomyopathy. Lastly, we review the evidence for enzyme replacement therapy in AFD as well as novel therapeutic agents.
Anderson-Fabry disease (AFD) is caused by mutations of the GLA gene, located at Xq22 on the X chromosome (8). Mutations result in a deficiency or absence of the enzyme α-galactosidase, resulting in the accumulation of globotriaosylceramide (Gb3), which is trafficked to the lysosomes in numerous tissues, including the heart, kidneys, skin, and vascular endothelium (Figure 1) (8). In female patients, random inactivation of the X-chromosome can lead to residual α-galactosidase activity, with variable phenotypic presentation. Deposition of Gb3 into the vascular endothelium is a significant contributor to the end-organ manifestations of AFD (9). Gb3 binds to lipoprotein receptors on the vascular endothelium, where it is then deposited within the vascular intima and media. Binding of Gb3 leads to increased leukocyte migration and adhesion, leading to the production of pro-inflammatory as well as vascular smooth muscle hypertrophy (10). This process ultimately leads to reactive oxygen species production and a decrease in nitric oxide, resulting in endothelial injury and dysfunction (11). The inflammatory cytokine production also contributes to a pro-thrombotic state (12). Vascular smooth muscle proliferation and microthrombi of the intramural coronary arterioles leads to reduced flow reserve and can ultimately cause infarction and fibrosis, whereas involvement of the cerebral vasculature contributes to the high risk of stroke in AFD (13, 14).
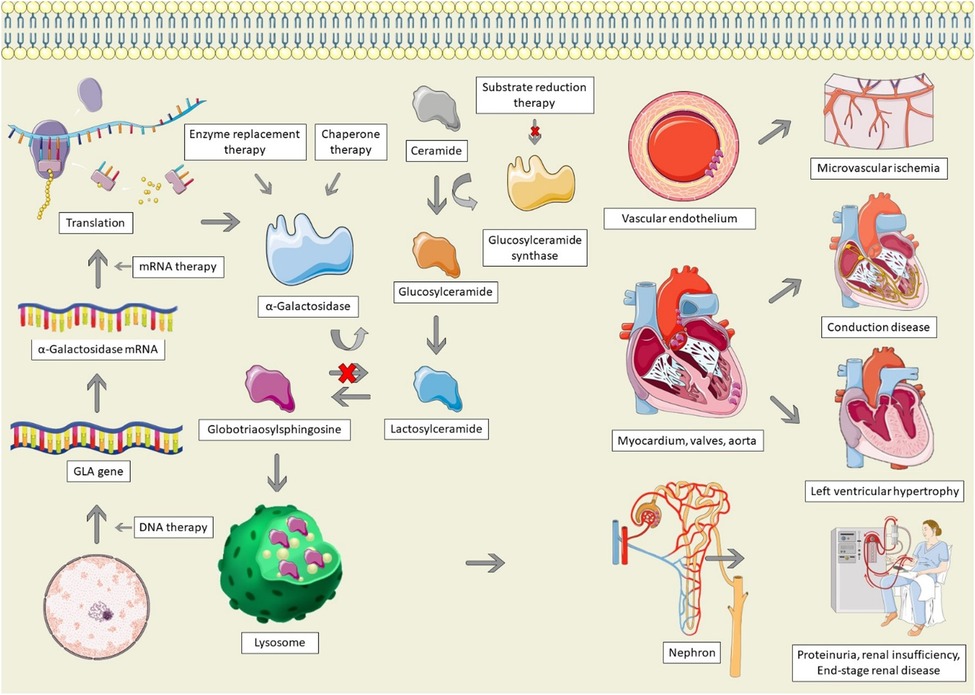
Figure 1. Pathophysiology and therapeutic options for Anderson-Fabry disease. AFD is characterized by GLA mutations, resulting in absent, insufficient, or dysfunctional α-galactosidase. Alpha-galactosidase catalyzes the conversion of globotriaosylsphingosine (Gb3) to Lactosylceramide; in its absence, Gb3 accumulates in lysosomes, particularly within the vascular endothelium, cardiac structures, and the nephron, leading to end-organ dysfunction. Therapeutic options for AFD involve increasing the production of α-galactosidase, replacing it, stabilizing misfolded galactosidase, or decreasing the production of glucosylceramide. The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license, as well as from freepik.com (https://www.freepik.com/free-vector/animal-cell-anatomy_26763764.htm).
Infiltration of Gb3 into the heart can lead to left ventricular hypertrophy (LVH), valvular disease, systolic and diastolic dysfunction, and conduction system disease (15). LVH is not solely caused by deposition of Gb3 (16). The deposition of Gb3 leads to a cascade of inflammation and oxidative stress, leading to extracellular matrix remodelling and hypertrophy, culminating in myocyte necrosis and fibrosis (17). In a study in which plasma from AFD patients was administered to rat vascular smooth muscle cells and neonatal mouse cardiomyocytes, there was a significant increase in cellular proliferation, which was correlated with the left ventricular mass index of the Fabry patient, suggesting that a circulating factor drives LVH in AFD (18). Similarly, another study found that patients with AFD have higher levels of sphingosine-1-phosphate (S1P) (19). Administration of S1P to mice led to cardiac remodelling resembling AFD, as well as increased vascular smooth muscle proliferation in a dose-dependent manner, suggestive that S1P may be one of the circulating factors responsible for LVH in AFD (19). Additionally, small vessel hypertrophy and Gb3 deposition can lead to microvascular ischemia, causing severe diastolic dysfunction, contributing to LVH (20). Lastly, hypertension is common among AFD patients, which may result from activation of the renin-angiotensin-aldosterone system due to direct renal injury, further contributing to LVH (21).
To date, more than 1,100 mutations of the GLA gene associated with development of AFD have been identified (22). Mutations can be classified as pathological, likely pathological, or variants of unknown significance (22). The type of mutation can be missense (23), nonsense (24), or frame shift (25); although the majority of documented mutations are missense (23). While several mutations have been identified, there does not appear to be one dominant mutation; the mutations observed appear to be limited to individuals and families (26). Mutations typically affect active binding sites of the GLA protein, or impaired trafficking to the lysosome, leading to early degradation within the endoplasmic reticulum (26). In addition to causing loss of function of the GLA protein, missense variants may also interfere with the wild-type product through dominant negative effects (24). The specific mutation involved has implications for the natural history of AFD in each patient, as well as the relevant organ systems involved.
Classical AFD is early-onset, with characteristic multi-system involvement, including cardiac, renal, and dermatologic manifestations. However, later onset AFD may predominantly affect a single system, as in the case of cardiac variants. It can be challenging to predict the clinical manifestations of a given genotype. However, in a population of Italian AFD patients, a decision tree model was derived to predict classical or variant AFD, based on the sequential and structural properties of the mutation (27). In a separate study of Chinese AFD patients, among males, patients with frameshift and nonsense mutations had a classical phenotype, while missense mutations could lead to classical or variant phenotypes (28). Among female patients, the majority of patients with frameshift mutations had a classical phenotype (28). Prediction tools for specific organ involvement for a given mutation have yet to be developed. Cardiac specific variants of AFD are being increasingly recognized (29, 30). In order to address the challenge for correlating genotype with phenotype, the AFD Genotype-Phenotype Working Group developed a 5-stage iterative system for previously unassigned GLA variants, which involved clinical assessment of affected patients, published literature, expert consensus, and a 2-point scoring system, with final validation by Kaplan-Maier curve analysis for severe event-free survival (31). The 2-point GLA scoring system is based on the proportion of males with the GLA variant who have either angiokeratomas or cornea verticillata, and has been shown to discriminate between classic and late-onset AFD (31). Using this iterative process, 32 of 33 previously unclassified GLA variants were determined to be pathogenic, which led to classification of previously unclassified GLA variants as pathogenic in 17.5% and 16.6% of male and female patients, respectively (31).
The overall prevalence of AFD causing clinical disease is estimated as 1 in 40,000 to 1 in 170,000 births, affecting all ethnic groups, with specific ethnic predispositions (32). The prevalence of mutations known or thought to be related to the development of AFD is considerable higher, although the proportion of individuals with GLA variants who develop clinical disease is unknown. In a study of the 200,643 individuals in the UK Biobank, the prevalence of likely pathogenic late-onset variant mutations was 1 in 5,732, and 1 in 200,643 for mutations causing classic AFD (33). Similarly, studies of the prevalence of GLA variants in newborn screening programs have found a high prevalence of disease-causing mutations, ranging from 1 in 1,250 male newborns in Taiwan to 1 in 21,973 in the USA (34, 35). Overall, later-onset variants were more common in studies involving newborn screening; the ratio of late-onset to classic variants ranged from 7:1 in Italy to 16:1 in Taiwan (2). While mutations associated with late-onset variant AFD are more common than mutations causing classical AFD; classical AFD is more common in cohorts of patients with established disease, suggesting that late-onset variant AFD is significantly underrecognized (36). The prevalence of AFD within specifically selected clinical populations with manifestations of AFD is considerably higher. Among patients with LVH, two recent cohort studies found a prevalence of AFD of 0.9% and 0.3% (37, 38). The prevalence of AFD in otherwise unexplained LVH is higher, ranging from 2% in Canada and Hong Kong (39) to 4% in the Czech Republic (40).
AFD is classically a multisystem disease with prominent cardiac involvement, including LVH, systolic and diastolic dysfunction, valvular disease, and small vessel ischemia. Although male and female patients may present with similar symptoms and systems involved, the proportion affected and age at development of each manifestation differs (Table 1). In the largest cohort study of AFD, the mean (SD) age of symptom onset was 9 (13.2) for male and 13 (19.1) for female patients, while the median age at diagnosis was 23 and 32, respectively (41). Cardiac symptoms at presentation were similar between male and female patients (13% vs. 10%), but the median age of onset was far younger for male than female patients (12 vs. 32 years) (41). Cardiovascular events, defined as arrhythmia, myocardial infarction, angina, HF, or requirement for a cardiac procedure were more common in male than female patients (19% vs. 14%) and occurred at a lower median age (41 vs. 47) (41). While cardiac symptoms were not as common at diagnosis, the prevalence of cardiac involvement overall is considerably higher. In the Fabry Outcome Survey, 61.4% of male and 46.3% of female patients with AFD had cardiac symptoms (42), including dyspnea (23.1%), angina (22.4%), palpitations (26.5%), and syncope (3.2%) (42).
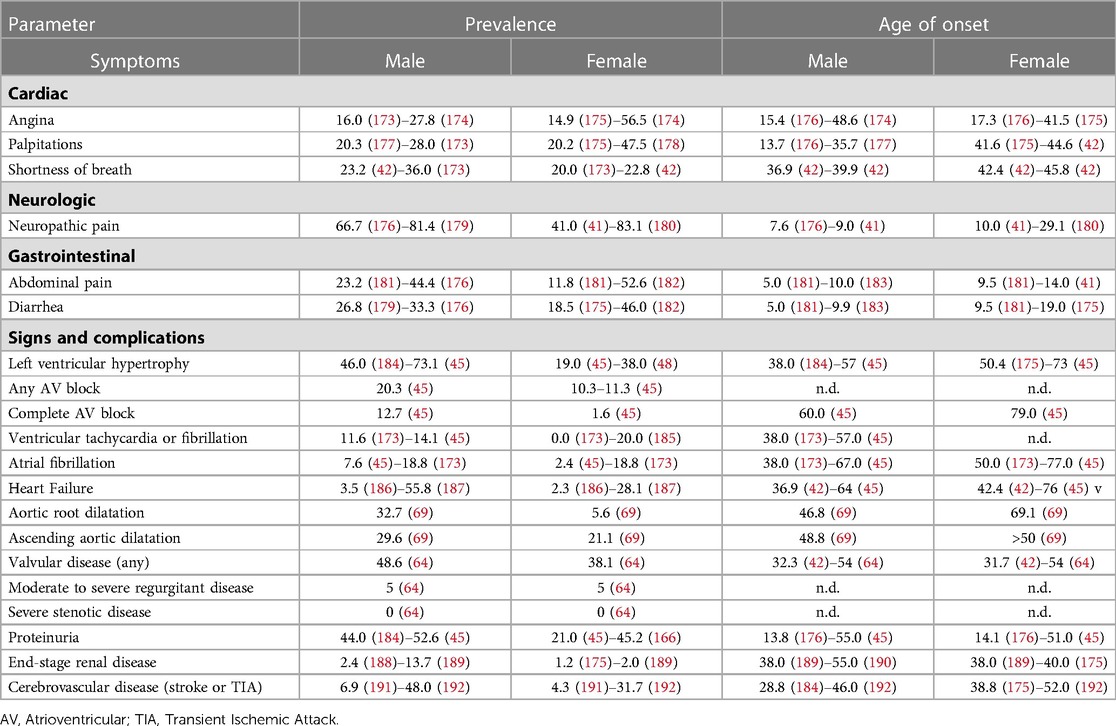
Table 1. Natural history of symptoms, signs, and complications of Anderson-Fabry disease.
The life expectancy of patients with AFD is significantly reduced. Within registry studies of AFD, the mean age at death ranged from 51.8 to 54.3 years for male patients and 62.0–64.4 years for female patients, with 36%–53% of deaths as a result of cardiac disease (43). In the Fabry Outcome Survey, LVH was present in 69% of deceased patients; conduction disease in 45%, and valvular disease in 38% (45). The life expectancy for male patients in the Fabry Registry was estimated as 58.2 years, while for female patients was 75.4 years (44). In the Fabry Registry, deceased patients were diagnosed later in life, at a mean age of 39.8 vs. 24.4 years for male and 55.4 vs. 32.7 years for female patients, and had only received enzyme replacement therapy for a median of 12 vs. 4 months for male and female patients, respectively (44). The short life-expectancy in this registry likely reflects delays in diagnosis and therapy; the life-expectancy of contemporary patients receiving early administration of enzyme replacement therapy has yet to be determined but is likely much higher.
Characterization of outcomes in late-onset AFD is less well-established. In a study of 203 patients with late-onset AFD due to the p.F113l GLA mutation, the mean (SD) age at diagnosis was 49 (15) for male patients and 44 (19) for female patients (45). LVH was found in 73.1% of male vs. 19.0% of female patients, and increased with age; all male patients older than 60 and all female patients older than 80 had LVH (45). HF was more common in male than female patients (32.9% vs. 14.8%), while mean survival free from HF was higher for female than male patients (64 vs. 76 years) (45). The late-onset phenotype of AFD appears to present with a greater burden of cardiovascular manifestations and longer life expectancy.
LVH is the most common cardiac manifestation of AFD and is an important determinant of clinical outcomes. In a contemporary study of 560 patients treated with enzyme replacement therapy, 55% of patients had LVH at baseline (46). LVH has been found to correlate with α-galactosidase activity levels; consequently, male patients more frequently have LVH than female patients (47). LVH develops early in the course of classic AFD; in a longitudinal study of 76 patients, LVH was present in 11.1% of patients between the age of 20–29, 40.6% of patients between 30 and 39, and 76.5% of patients between 50 and 59 (48). Male patients experienced a greater increase in left ventricular mass index over time compared to female patients (4.07 ([1.03] vs. 2.13 [0.81]g/m2.7 per year) (48). While LVH is common in both classical and non-classical AFD, left ventricular mass index is higher in classic AFD (36).
While concentric LVH is the most common pattern of hypertrophy, a variety of patterns have been described, including concentric, apical, septal, and eccentric (49). In a study of 166 patients with AFD the pattern was concentric remodeling in 21.6%, concentric LVH is 30.9%, eccentric LVH in 5.2%, and asymmetrical septal hypertrophy in 6.2% (48). Left ventricular outflow tract obstruction is infrequently described in AFD due to asymmetric septal hypertrophy or systolic anterior motion of the mitral valve; AFD may be identified in the context of evaluation of hypertrophic cardiomyopathy (50). In studies of patients initially diagnosed with hypertrophic cardiomyopathy, the prevalence of AFD ranged from 0.5 to 1.0% (29, 51). A recent case series has also identified left ventricular mid-cavitary obstruction due to papillary muscle hypertrophy as an infrequent but significant manifestation of advanced AFD (52).
Deposition of glycosphingolipids into the conducting fibers of the heart can lead to conduction system disease and dysrhythmias. In a human stem-cell model of AFD, AFD cardiomyocytes had enhanced sodium and calcium channel function, with higher spontaneous action potential frequency and shorter action potential duration (53). Similarly, electrophysiologic studies of conduction tissue from AFD patients found shorter conduction and a prolonged refractory period; additionally, the burden of conduction tissue infiltration correlated with the burden of conduction disease and arrhythmia (54). Patients with AFD are at particular risk of atrial fibrillation due to both left atrial dilatation and dysfunction (55). While left atrial dilatation in AFD can develop as a consequence of left ventricular diastolic dysfunction and LVH, the development of left atrial dilatation is independent of LVH or left ventricular diastolic dysfunction, and often predates LVH (56).
In a cohort study of patients with late-onset AFD due to the p.F113l GLA mutation, atrial fibrillation occurred in 4.4% of patients who underwent Holter monitoring, while non-sustained ventricular tachycardia (VT) occurred in 14.1% of male and 5.6% of female patients which increased in frequency with age, such that 33.3% of both male and female patients older than 70 years had non-sustained VT (45). The mean (SD) age of onset for atrial fibrillation was 67 (11) for male patients and 77 (3) years for female patients (45). Any degree of atrioventricular (AV) block was present in 20.3% and 10.3% of male and female patients (45). Male patients more commonly required a permanent pacemaker (12.7% vs. 2.4%) or implantable cardioverter-defibrillator (1.3% vs. 0.8%) than female patients (45). Complete AV block developed at a mean (SD) age of 60 (7) for male patients and 79 (6) years for female patients. Patients with AFD also face an increased risk of sudden cardiac death (SCD): in a systematic review and meta-analysis, SCD was the cause of cardiac death in 62% of cases (57). Risk factors for SCD included male sex, increasing age, LVH, non-sustained VT, and late-gadolinium enhancement on cardiac magnetic resonance imaging (CMR) (57).
AFD is associated with abnormalities in both diastolic and systolic function, due to a combination of direct infiltration of the myocardium by glycosphingolipids, fibrosis, and microvascular ischemia, and may result in overt, symptomatic heart failure. The prevalence of systolic dysfunction in AFD varies depending on the method of assessment; overt systolic dysfunction with reduced left ventricular ejection fraction (LVEF) is uncommon; in a cohort study from 2010, the prevalence of LVEF < 55% was 6.8% (47); the prevalence has not been reassessed in more recent cohort studies. Global longitudinal systolic strain (GLS), a more sensitive marker of systolic function, is frequently abnormal in AFD. In a recent study of 160 patients with AFD, patients with AFD had worse mean [SD] GLS than controls, particularly among those with LVH (−16.5%[4.2] vs. −21.2% [2.7]), with no differences between sexes (58). Patients with AFD without LVH had worse longitudinal strain at the mid to apical, anterior, and inferolateral left ventricular walls relative to controls (58). GLS may allow for the identification of early disease progression in AFD prior to the onset of overt LVH. Similarly, strain imaging can identify early right ventricular involvement; in one study right ventricular systolic function by standard echocardiographic assessment was reduced in 8%, while 3- or 6-segment right ventricular strain was reduced in 40%, with worse right ventricular strain in patients with LVH than those without (59). Comparatively, diastolic dysfunction is significantly more common than systolic dysfunction in AFD. In one study, diastolic dysfunction was found in 43.8% overall, and higher in patients with late gadolinium enhancement on CMR than those without (moderate: 28.1% vs. 2.1%, severe: 14.0% vs. 0%) (60). Diastolic dysfunction may develop with or without LVH, and is an important contributor to the development of left atrial enlargement and atrial fibrillation (15). While overt, symptomatic HF is less common than abnormal strain, an elevation in amino-terminal pro-B-type natriuretic peptide (NT-proBNP) has been found in 57% of patients with AFD, and correlates with diastolic dysfunction (61, 62).
AFD is associated with small rather than medium or large-vessel ischemia (20). Small vessel ischemia is common in AFD. In one cohort study of patients with HCM and AFD, myocardial infarction with non-obstructive coronary arteries (MINOCA) occurred in 7.5% of AFD patients compared to 0.5% of HCM patients over a mean follow-up of 4.5 years (63). Furthermore, MINOCA was more common among patients with LVH, with MINOCA occurring in 17.7% of patients with LVH compared with 1.7% of patients without LVH (63). AFD was independently associated with MINOCA (OR 6.12) suggesting that MINOCA may represent a red flag for AFD (63).
AFD is associated with the development of valvular stenosis and regurgitation, although valvular heart disease is generally uncommon. Regurgitant lesions are thought to develop due to glycosphingolipid deposition within the valve leaflets and sub-valvular apparatus, as well as geometric distortion of the atria, valvular annulus, or aortic root dilatation (64). In a contemporary study of predominantly classic-phenotype AFD (80%), the prevalence of moderate to severe valvular disease was 10%, with mitral and tricuspid regurgitation being the most common (64). Thickening of valve leaflets and the sub-valvular apparatus are more common than severe valvular disease, affecting the mitral and aortic valve in 57% and 47% of patients, respectively (65, 66). Valvular disease is more commonly regurgitant than stenotic; in one cohort, mitral, and tricuspid regurgitation were common (15.3%, 51.4%, and 34.2% respectively), but no patients had moderate or greater aortic stenosis (65). Owing to the relative rarity of severe valvular lesions, there are few case reports of transcatheter or surgical management of severe aortic stenosis (67, 68) in AFD patients.
Aortic root dilatation, where present, is typically at the level of the sinus of Valsalva or ascending aorta. There are no contemporary estimates of the prevalence of aortic involvement; in an older cohort study of 106 AFD patients, male patients more commonly had dilatation at the level of the sinus of Valsalva (32.7% vs. 5.6%) than female patients, with a similar proportion at the ascending aorta (29.6% vs. 21.1%) (69). The prevalence of aortic dilatation increased with age, and developed at a younger age in male patients, similar to other manifestations of AFD (69). Aortic dilatation is progressive in AFD; in one study of patients followed by CMR, the mean [SD] annual expansion rate at the sinotubular junction [0.41 (0.16)mm/year] and proximal ascending aorta [0.41 (0.26) mm/year] was significantly higher than the expected progression in normal patients [0.14 (0.11)mm/year], whereas the annual growth in female patients was no greater than expected for normal patients (70). The optimal size threshold to intervene on aortic aneurysms has not been determined in AFD; acute aortic syndromes in AFD are limited to case reports (71).
Confirmatory diagnosis of AFD is dependent on α-galactosidase activity and GLA mutation analysis. In male patients, the first step is to measure α-galactosidase activity, which can be diagnostic and can be measured in leukocytes, plasma, or dried blood spot (72). Characteristically, in male patients with classic AFD, α-galactosidase activity levels are very low to undetectable (73, 74), and usually less than 3% of normal (3). By contrast, α-galactosidase activity levels are usually higher in late-onset variants of AFD, but typically less than 30% of normal (3). In one series of patients, male patients with late-onset variants of AFD had a mean α-galactosidase activity of 20% of normal, compared to 3% for male patients with the classic phenotype (5). Among female patients, due to selective X-inactivation, α-galactosidase activity has a more limited role in the diagnosis of AFD. In one study of female AFD patients using traditional reference values for α-galactosidase activity, the positive predictive value for low plasma α-galactosidase activity was 94%, with a negative predictive value of 74% (73).
GLA gene analysis is essential for both male and female patients; however, in male patients it is confirmatory after detecting low measure α-galactosidase activity, whereas in female patients, GLA analysis is typically the first step in establishing the diagnosis of AFD (73). For patients with genetic variants of unknown significance (GVUS), a detailed clinical and biochemical review for disease manifestations, a review of family history, and multidisciplinary input, particularly with a geneticist, is essential (74). Following GLA analysis in female patients, if the mutation identified does not have established pathogenicity, then measurement of lyso-Gb3 can demonstrate the clinical significance of the genetic variant of unknown significance (75–77). In one study, elevated lyso-Gb3 offered a sensitivity and specificity of 98% and 100%, respectively, for male patients, and 97% and 100% for female patients (73). By comparison; the sensitivity and specificity of α-galactosidase activity in female patients was 49% and 91%, respectively (73). More recently, the ratio between α-galactosidase activity and lyso-Gb3 has shown promise in differentiating between AFD and controls among female patients, with 100% sensitivity for differentiating AFD, compared to a sensitivity of 8.6% and 74.4% for α-galactosidase activity and lyso-Gb3 alone (78). GLA gene analysis offers disease confirmation, can be prognostic with respect to the disease manifestations associated with the specific mutation, and is important in pedigree analysis. It is also worth noting that proteinuria is an important renal manifestation of AFD, although relatively nonspecific. Detection of proteinuria can increase suspicion for AFD in the correct clinical context and is also used in follow-up to assess disease therapy.
Endomyocardial biopsy is an important modality in the diagnosis of AFD, albeit with a limited role. Endomyocardial biopsy in AFD demonstrates myocytes containing fine-granulated vacuoles positive at Sudan-black staining, indicative of lipid accumulation (116). Electron microscopy demonstrates the characteristic finding of concentric lamellar bodies composed of Gb3. Consensus guidelines on when to perform endomyocardial biopsy, or biopsy of alternative sites specifically in the context of AFD are lacking. Per the 2007 European Society of Cardiology guidelines on endomyocardial biopsy (EMB), EMB can be considered in heart failure with unexplained HCM (Class IIb) (114). Similarly, the 2021 ESC HF guidelines suggest EMB can be considered in the diagnosis of forms of cardiomyopathy where a specific treatment exists (i.e., storage diseases), where the diagnosis cannot be reached by non-invasive means (115). In light of the advances in biochemical and genetic diagnosis of AFD, EMB is usually not necessary but can be performed in cases of diagnostic uncertainty, as in the case of female patients with a late-onset phenotype with a GLA VUS.
One of the earliest electrocardiographic (ECG) manifestations of AFD is shortening of the PQ interval, due to shortening of the duration of the P wave (79). Although PQ shortening is well described in AFD, it is uncommon; two separate cohort studies found a prevalence of 7.5% and 14% (80, 81). By contrast, shortening of the PendQ (PQ—duration of P wave) is more common in AFD, with one study finding a prevalence of 47.5% (80). Both QT and QRS prolongation have been described in AFD, with one study finding that QT prolongation was limited to male patients (80). The ECG findings correlate with the extent of cardiac involvement. In one study, patients with AFD but without LVH underwent CMR, and those with a low T1 signal (suggestive of glycosphingolipid deposition) had a greater maximum Q-wave amplitude, Sokolow-Lyon index, and Cornell index than AFD patients with a normal T1 signal. In another study, an ECG-based nomogram was developed to predict cardiac involvement, defined as a low T1 on CMR, using four variables: Sokolow-Lyon Index, ratio between the P wave and PR segment duration, QRS duration, and QT (82). Over time, patients develop ECG features of LVH, which is present in 50.5% of patients in one study (58). The ECG is valuable in the diagnosis of AFD and differentiation from mimics (Table 2): in one study, an algorithm was developed using ECG features to differentiate between AFD, hypertensive heart disease, hypertrophic cardiomyopathy, aortic stenosis, and amyloidosis (83). The combination of QTc < 440 ms with a PendQ < 40 ms was highly sensitive and specific for AFD (83). Additionally, an index comprised of PendQ, QTc, and the Sokolow-Lyon Index was able to differentiate between AFD and amyloidosis (83).
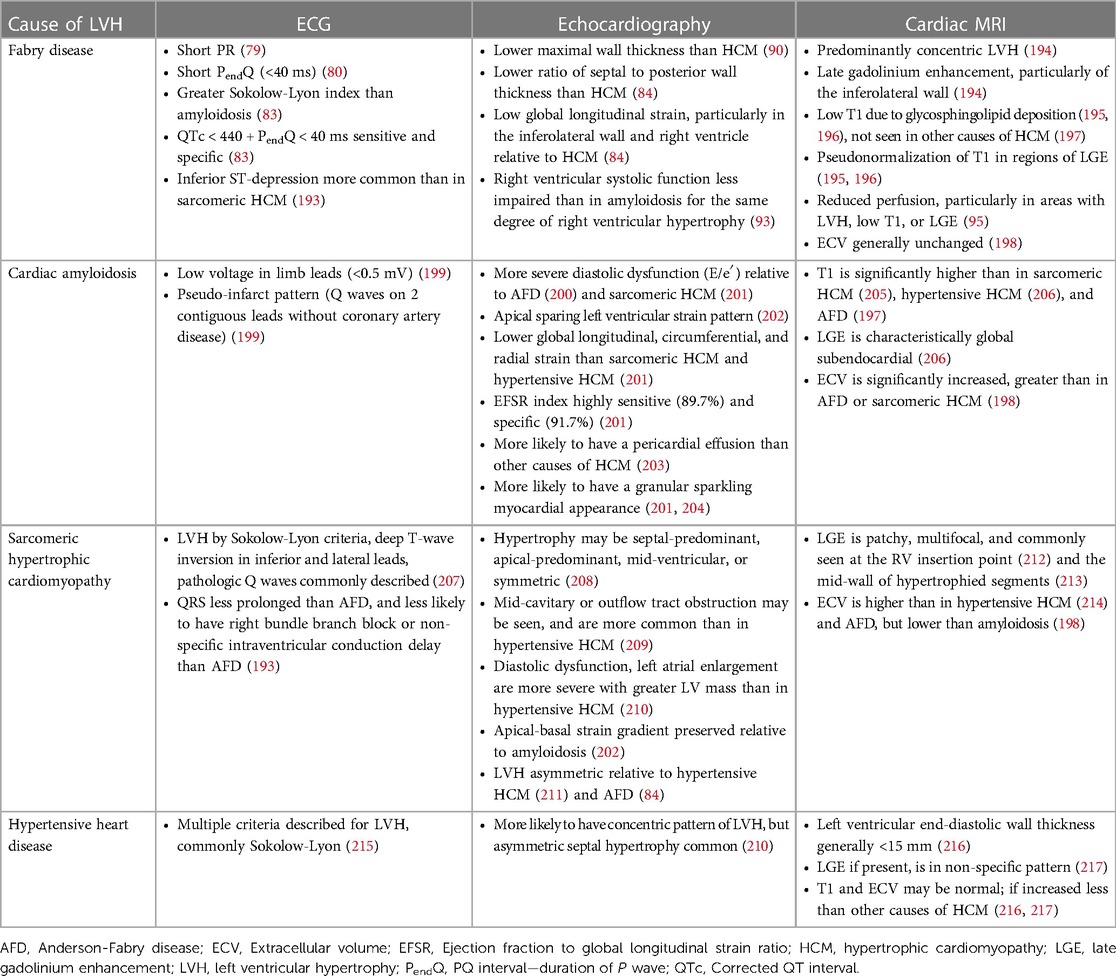
Table 2. Characteristic features and differentiation between causes of left ventricular hypertrophy.
The hallmark echocardiographic abnormality of AFD is LVH, with or without diastolic dysfunction (Figure 2) (84). The binary sign, previously thought to be specific to AFD, is the finding of a hyperechogenic endocardial layer adjacent to a hypoechogenic subendocardial layer, due to relative sparing of the outer myocardium from glycosphingolipid deposition (85). Subsequent analyses have found this to be insensitive and non-specific for AFD (86, 87).
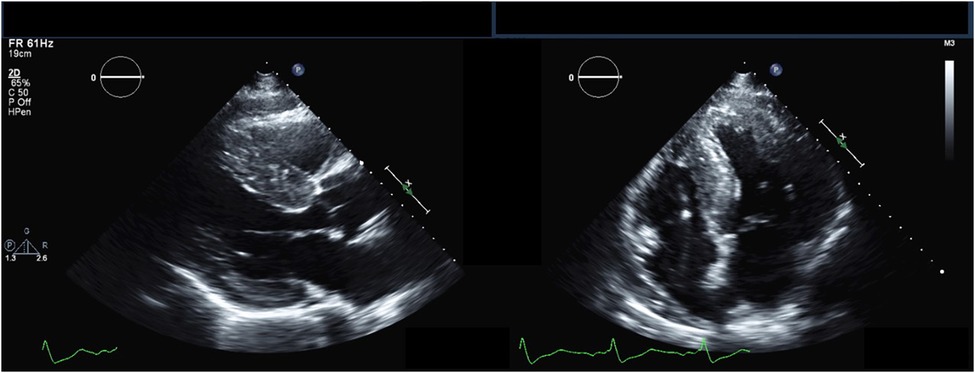
Figure 2. Echocardiography findings in Anderson-Fabry disease. Parasternal long-axis (left) and apical 4-chamber (right) transthoracic echocardiography views demonstrating increased left ventricular wall thickness.
In an effort to identify AFD prior to the develop of overt LVH, a variety of techniques have been utilized to identify early myocardial involvement on echocardiography. Tissue doppler imaging allows for characterization of diastolic function and has been found to be abnormal prior to the development of LVH. In one study, patients with AFD but without LVH had a lower early diastolic mitral valve inflow (Ea) and a shorter isovolumic relaxation time than controls (88). Patients with LVH had a significantly longer isovolumic relaxation time, with a reduced systolic myocardial velocity, which was negatively correlated with septal and posterior wall thickness (88). Another older study had shown abnormalities in mitral annulus e′ even in the absence of LVH (89); this was not found in a larger, contemporary study (58). Strain imaging using speckle-tracking echocardiography, a newer technique, can allow for earlier identification of abnormalities in systolic and diastolic function in both ventricular and atrial function. In the same study of 160 patients with AFD with or without LVH, GLS was abnormal in both LVH+ and LVH- patients, with regional differences noted, and a correlation between GLS and LV mass index (58). LVH+ had the lowest longitudinal strain among all segments; however, compared to controls, LVH- patients had lower peak longitudinal strain in the apical, mid, anterior, and inferolateral segments (58). Speckle-tracking echocardiography has also been used to assess atrial function in AFD; patients with AFD have a reduction in all three phasic functions of the left atrium: reservoir, conduit, and contractile function (55).
Echocardiography has a role in differentiating between AFD and other causes of LVH (Table 2). In one study comparing the echocardiograms of patients with AFD and hypertrophic cardiomyopathy (HCM), patients with AFD had a significantly lower maximal wall thickness, and greater indexed sinus of Valsalva diameter (90). In another study, compared to patients with HCM, patients with AFD had a significantly lower interventricular septum to posterior wall thickness ratio, and had worse regional longitudinal strain in the inferolateral left ventricular wall and right ventricular free wall (84). Both right ventricular free wall strain and GLS are significantly more impaired in AFD than HCM (91). By contrast, a study of left atrial function found no differences in left atrial reservoir, conduit, and contractile function between AFD and HCM patients, although HCM patients had significantly larger left atria (92). Echocardiography has also been used to compare AFD and cardiac amyloidosis; compared to patients with cardiac amyloidosis, patients with AFD have relatively preserved right ventricular systolic function at the same degree of right ventricular hypertrophy, with higher TAPSE, right ventricular S′, and right ventricular fractional shortening (93).
Cardiac MRI is a comprehensive diagnostic modality that allows for the assessment of a variety of cardiac sequelae of AFD, including LVH, particularly with papillary muscle involvement; subtle abnormalities in systolic function, sphingolipid deposition, and fibrosis. Patients with AFD commonly have enlarged atria and reduced left atrial strain prior to the onset of LVH, representing an atrial myopathy that correlates with disease severity (94). Perfusion is often abnormal, particularly in areas with LVH, low T1, or late gadolinium enhancement (LGE) (95). The pattern of LGE in AFD is often non-specific, but most commonly involves the basal posterior wall and corresponds with areas of collagen deposition and fibrosis (Figure 3) (96). The presence of LGE is associated with increased risk of cardiac events, with the highest risk among patients with LGE > 15% of left ventricular mass (97). LGE is correlated with elevated troponin in AFD, which is persistently elevated in up to 21% of patients (111, 112).
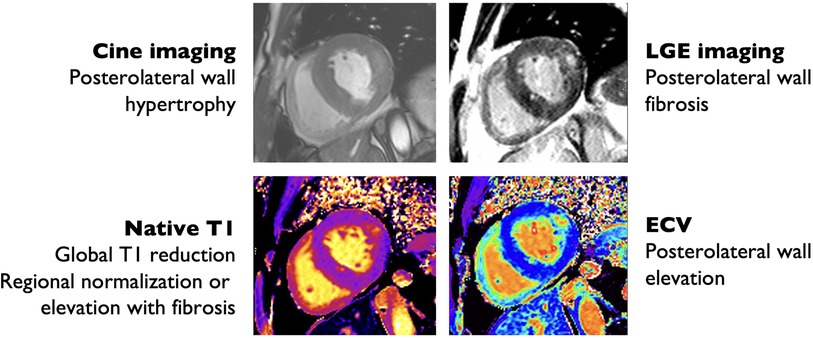
Figure 3. Cardiac magnetic resonance imaging findings in Anderson-Fabry disease. Hypertrophic with prominent hypertrophy of the posterolateral and lateral wall (top left) with posterolateral wall replacement fibrosis by late gadolinium enhancement (LGE) imaging (top right), also evident by increased extra-cellular volume (ECV) quantification (bottom right). There is reduced interventricular septum native T1 time consistent with glycosphingolipid deposition (bottom left).
Cardiac MRI is the gold-standard for the assessment of left ventricular mass index (LVMI) and maximal wall thickness (MWT), and offers greater inter-rater reliability than echocardiography (98). In one study comparing CMR and echocardiography in patients with AFD, LVMI and MWT were higher on echocardiography, with discrepant measurements between echocardiography and cardiac MRI in 26% (98). As a result, 26% of patients had conflicting recommendations regarding whether to start ERT (98). In addition to providing comprehensive assessment of cardiac involvement in AFD, CMR can also be used to differentiate between causes of cardiomyopathy, and assess the clinical stage of AFD.
T1 is particularly useful for differentiating between AFD and other causes of cardiomyopathy (Figure 3). T1 is low in the setting of fat infiltration (arrhythmogenic RV dysplasia), glycosphingolipid infiltration (AFD), and iron deposition (hemochromatosis), and high in the setting of fibrosis or amyloid protein deposition (99). T1-mapping has previously been used to differentiate between AFD and cardiac amyloidosis and HCM (100, 101). AFD can be differentiated from iron deposition using T2*-weighted imaging (99). An important limitation to the use of T1-mapping is pseudo-normalization of T1 due to the combination of glycosphingolipid deposition and fibrosis (102). Pseudo-normalization can be suspected when T1 values are normal in areas with late gadolinium enhancement (LGE) (102).
A novel application of cardiac MRI is in disease staging. Cardiac MRI has identified four phenotypes of cardiac involvement in AFD: a pre-deposition phase, where sphingolipid deposition is undetectable; a detectable accumulation phase; a hypertrophic phase; and a fibrosis phase (103). On cardiac MRI, the detectable deposition phase can be identified by a low T1 value. However, patients with AFD in the pre-detectable deposition phase have observable abnormalities on cardiac MRI, including reduced GLS and myocardial blood flow (103). In one cohort study of pre-hypertrophic AFD, 59% of patients with AFD but without LVH had a low T1 value compared to 0% of healthy controls, which was associated with the presence of LGE and ECG abnormalities (104). Although MWT and LVMI were normal in AFD patients, subtle abnormalities were present, including a greater papillary muscle mass and longer anterior mitral valve leaflet (104). Over the natural history of AFD, T1 decreases due to glycosphingolipid deposition, and then begins to stabilize in female and increase in male patients with LVH (105). The increase in T1 later in the course of illness can be explained in part by fibrosis, as well as dilution due to the normal T1 of hypertrophied myocardium, as only a minority of LVH in AFD is caused by glycosphingolipid deposition (16, 111). Cardiac MRI offers promise in identifying patients with very early cardiac involvement, who may derive the greatest benefit from early pharmacologic therapy.
A variety of modalities have been used to assess myocardial perfusion in AFD, including CT perfusion (80), SPECT (106), and CMR (95). Perfusion abnormalities are generally attributed to LVH and small vessel ischemia (20, 113). On perfusion CMR, myocardial blood flow is correlated with wall thickness, T2, LGE, and extracellular volume (95). The use of positron-emission tomography (PET) in AFD is increasingly being described (107, 108). Increased uptake in the basal inferolateral wall in AFD on PET has been well-documented, similar to the distribution of LGE on CMR (107, 109). In one study of hybrid PET-CMR, patients with focal 18F-FDG uptake had higher T1 values, suggestive of pseudo-normalization (108). PET has also been used to evaluate other causes of cardiomyopathy such as amyloidosis and sarcoidosis (110); however, features on PET al.lowing for the differentiation of cardiomyopathies have yet to be identified. PET has also been used to characterize the perfusion abnormalities in AFD; in one cohort, coronary microvascular dysfunction was evident in all AFD patients, with the greatest reduction in myocardial blood flow in patients with LVH (14). Perfusion abnormalities were global, with a marked reduction in myocardial blood flow at the apical segments (14). PET, and PET-CMR may allow for earlier recognition of cardiac involvement in AFD as evidenced by reduced perfusion prior to the development of overt LVH, and identify early fibrosis (108).
The mainstay of therapy for AFD is enzyme replacement therapy. There are two formulations available: agalsidase-alfa, derived from human fibroblasts, and agalsidase-beta, derived from hamster ovarian cells. In the United States, only agalsidase-beta is licensed, whereas both formulations have been licensed for use in Canada and throughout Europe (117). Both formulations are structurally similar and thought to have a similar efficacy. Although a systematic review of observational cohort studies suggested a significantly lower incidence of cerebrovascular events in patients treated with agalsidase-alfa than those treated with agalsidase-beta (118), a Cochrane review of randomized trial data found no difference in renal deterioration, cardiac events, death, acroparesthesia, or reduction in Gb3 levels in patients treated with agalsidase-alfa compared to agalsidase-beta (119). Administration of ERT is generally well tolerated; pertinent adverse effects include infusion reactions and development of anti-drug antibodies (ADAs).
Given that ERT is comprised of a foreign recombinant protein, a humoral response leading to the development of ADAs is common, generally within 3–6 months of treatment initiation (120, 121). Once ADAs develop, they are generally thought to be permanent (122, 123). Up to 40% of male patients treated with ERT develop ADAs; patients with minimal or absent production of α-galactosidase are at particularly high risk as there is no endogenous α-galactosidase for the immune system to develop tolerance to (120). In one observational study, patients treated with agalsidase-beta had a greater risk of developing ADAs than those treated with agalsidase-alfa (52% vs. 28% of male patients), which may relate to the greater dose of agalsidase-beta administered (1 mg/kg every 2 weeks for agalsidase beta compared to 0.2 mg/kg every 2 weeks for agalsidase alfa) (124). The risk of antibody development also appears to be higher in those with a more severe, classic disease phenotype (125), and patients with a higher Gb3 level at treatment initiation (126). Antibodies to either formulation are thought to be cross-reactive, such that switching when antibodies develop may be of little benefit (121). However, the clinical significance of the development of ADAs is unclear. In one study, patients with ADAs had higher Gb3 levels than those treated with ERT but without antibodies, however there was no difference in the progression of renal impairment (125). In a separate retrospective study comparing patients with or without ADAs found that antibody-positive patients had a deterioration in renal function, increased MSSI and disease severity index, whereas disease activity remained stable in the antibody-negative group (123). The optimal strategy for monitoring for the development of ADAs is unclear, as well as how to respond to the development of ADAs. ERT can be dosed to achieve saturation of ADAs; in one observational study, compared to patients who achieved saturation of ADAs, those that did not had a greater decline in eGFR, an increase in interventricular septal thickness, and a lesser reduction in lyso-Gb3 (127).
Existing guidelines emphasize the early introduction of ERT, prior to the development of irreversible end-stage organ damage. In both the Canadian guidelines and European Fabry Working Group, patients with renal (elevated creatinine, proteinuria), cardiac (LV mass, septal thickness, arrhythmia), neurologic (stroke, hearing loss, pain), or gastrointestinal involvement qualify for initiation of ERT (128, 129). The European guidelines are less conservative, also advising initiating ERT in all male patients with classical AFD older than 16 years prior to the development of symptoms or organ involvement (129). Both guidelines also address which patients should not initiate ERT, and in whom therapy can be discontinued. Patients with end-stage renal disease who are not transplant candidates, patients with advanced HF, patients with significant myocardial fibrosis, and those with comorbid conditions with a life-expectancy less than 1 year who are not expected to derive a clinical or quality of life benefit from treatment (128, 129).
Enzyme replacement therapy with agalsidase-alfa or -beta has demonstrated efficacy in the reduction of Gb3 and improvement in cardiac, renal, and neurologic outcomes. Treatment with ERT has been shown to reduce plasma and urinary Gb3 and lyso-Gb3, a deacetylated form of Gb3 (7, 129–132). Treatment with ERT offers benefit for both male and female patients, as well as those with classical and late-onset AFD phenotypes. In a meta-analysis of both randomized and observational trials that separated results by sex, ERT led to stabilization in LVMI in male patients with AFD and LVH at baseline; among male patients without LVH at baseline, the increase in LVMI over time was lower ERT-treated patients (133). Among female patients, ERT led to a reduction in LVMI among patients with LVH at baseline, and stabilization of the LVMI among female patients without LVH at baseline (133). A more recent systematic review and pooled analysis that included a balance of male and female patients found a reduction in LVMI with ERT (134). Early initiation of treatment is paramount; patients with existing LGE on CMR are unlikely to experience regression of LVH with ERT (135, 136). In a study comparing patients with AFD who started ERT within 24 months of diagnosis and those who started after 24 months, earlier initiation of ERT was associated with reduced risk of cardiovascular events, defined as HF, arrhythmia, cardiac surgery, LVH, conduction abnormality, and myocardial infarction (137).
Treatment with ERT is limited by its short half-life, immunogenicity, and limited tissue penetration. Novel therapies for AFD aim to address these shortcomings. Pegunigalsidase-alfa is a pegylated, covalently cross-linked form of agalsidase-alfa whose role in the treatment of AFD is being evaluated in ongoing trials (NCT03180840, NCT04552691). The pegylated, cross-linked structure is thought to increase plasma-half life, and reduce immunogenicity (138). In one study, pre-existing ADAs to agalsidase-alfa or -beta demonstrated reduced affinity for pegunigalsidase-alfa, with a 30% reduction in inhibitory capacity (139). In a small study of 18 patients, patients treated with pegunigalsidase-alfa exhibited stability of renal function over the year of treatment (140). The effect of pegunigalsidase-alfa on cardiac outcomes has yet to be determined. Another novel agent is moss-derived agalsidase-alfa (Moss aGal). Moss-derived agalsidase-alfa differs from traditional ERT in its glycosylation profile and cellular uptake, relying on mannose receptors rather than mannose-6-phosphate receptor-mediated endocytosis (141). Mannose receptors are expressed on vascular endothelial, smooth muscle, dendritic and renal mesangial cells, making Moss-aGal attractive for treatment of AFD, and may help overcome limitations in the tissue distribution of ERT (141). Treatment with Moss-aGal has been shown to reduce urinary Gb3 and is well-tolerated, however the effect on hard clinical outcomes has not been established (141). In a mouse model of AFD, treatment with Moss-aGal led to similar distribution in the heart and spleen compared to agalsidase-alfa treated mice, with better distribution in the kidney, and similar efficacy for reduction in Gb3 (142). The risk of ADA development with Moss-aGal has not previously been explored owing to the preclinical nature of Moss-aGal trials, however, ADAs to agalsidase-alfa or -beta have similar affinity to Moss-aGal (143).
Chaperone therapy is alternative to ERT for the treatment of AFD that aims to correct misfolded α-galactosidase and improve its stability, allowing it to be delivered to the lysosomes, where it can degrade Gb3 (144). Chaperone therapy is only effective in amenable mutations where unstable but functional α-galactosidase is produced; mutations resulting in the absence of production of α-galactosidase would not be amenable to chaperone therapy. Missense mutations in particular may respond to treatment with chaperone therapy, whereas large deletions, frameshift mutations, splicing mutations, and insertions are not responsive (144). In-vitro assays of responsiveness should be performed prior to initiation of treatment, defined as an increase in α-galactosidase activity ≥1.2-fold, or an absolute α-galactosidase activity of ≥3% of normal (145).
Migalastat is the only chaperone agent that is currently licensed for the treatment of AFD. The ATTRACT trial, which randomized patients with AFD to continue ERT or switch to migalastat found that migalastat, but not ERT led to a reduction in LVMI; among patients with existing LVH, the effect size was greatest (146). The composite of renal (worsening of eGFR, proteinuria), cardiac (myocardial infarction, arrhythmia, HF, unstable angina) and cerebrovascular events (stroke, transient ischemic attack) was numerically but not significantly lower among patients treated with migalastat (29% vs. 44%, p = 0.36) (146). Similarly, the FACETS trial found that patients treated with migalastat experienced a significant reduction in LVMI at 24 months, particularly among patients with LVH at baseline (147). In the more recent MAIORA study, an uncontrolled before-after study of treatment-naïve AFD patients, migalastat was associated with stabilization of LVMI at 18 months, a trend towards increased septal T1 on cardiac MRI, and improved exercise tolerance (148). As is often seen with ERT, patients with the earliest stage of disease derived the greatest benefit from migalastat, emphasizing the role of early treatment for AFD (148). Chaperone therapy has the advantages of oral administration and less immunogenicity than ERT but is only effective in a subset of patients. Observational studies have also demonstrated a reduction in LVMI with migalastat, including in patients switched from ERT to migalastat (149–151).
Novel agents have been developed and are now available for the treatment of AFD that do not rely on enzyme replacement. Substrate reduction therapy (SRT) is one such method that involves reducing the production of Gb3 through the inhibition of glucosylceramide synthase, an enzyme involved in the production of glycosphingolipids. SRT has established efficacy in Gaucher disease, another glycogen storage disease (152). Three agents have been studied in AFD: eliglustat, venglustat and lucerastat. In a mouse model of AFD, treatment with eliglustat reduced levels of Gb3, and was more effective than ERT in reducing levels of Gb3 in the kidney, but less effective at reducing Gb3 in the heart or liver (153). The combination of SRT and ERT was most effective at reducing Gb3 in all tissues (153). Similarly, venglustat has been shown to reduce levels of Gb3 and lyso-Gb3 in a mouse model of AFD, and unlike ERT, lowered Gb3 and lyso-Gb3 in the brain (154). In a small study of 10 patients receiving ERT with lucerastat and 4 patients receiving ERT alone, the combination led to a greater decrease in plasma Gb3 and urinary Gb3, but no change in LVEF or LVMI at 12 weeks (155). While larger studies with longer follow-up are needed, SRT is promising as an addition to ERT given its tolerability, oral route of administration, lack of anti-drug antibodies, and ability to cross the blood brain barrier (153). Trials are underway to determine the efficacy of SRT monotherapy in AFD (NCT03425539, NCT05280548).
Another advance in the treatment of AFD is mRNA therapy encoding recombinant α-galactosidase. In a mouse model of AFD, mRNA therapy led to a significant reduction in Gb3 in cardiac and renal tissue, with similar efficacy to ERT over two months of treatment (156). Administration of mRNA to non-human primates has been shown to increase the activity of α-galactosidase in the heart, liver, and spleen, but not kidney (157). The effect of mRNA therapy in AFD patients has yet to be established. Another investigational treatment for AFD is gene therapy which can take several forms: direct administration of adenovirus/adeno-associated virus containing GLA DNA to the target organ; infusion and engraftment of autologous hematopoietic stem/progenitor cells with lentivirus-mediated transduction of GLA DNA into host DNA; and gene editing with CRISPR/Cas systems (158). Studies involving the administration of lentivirus-transduced hematopoietic stem/progenitor cells to a limited number of patients with AFD have demonstrated sustained increases in α-galactosidase activity; large-scale studies with hard clinical endpoints are needed (159, 160).
Owing to the myriad of organ systems involved in AFD, multidisciplinary care is essential to manage the complications (Figure 4). Multidisciplinary care models have been developed for the care of AFD patients, typically involving a geneticist, nephrologist, cardiologist, neurologist, nurse specialists, and psychologists (161, 162). Hypertension is common in AFD; and in one cohort, was uncontrolled in 57% of male and 47% of female patients (163). Given the prevalence of proteinuria and renal dysfunction in AFD, ACE-inhibitors or angiotensin receptor blockers should be considered for the treatment of hypertension (164, 165). In one study, proteinuria was present in 91% of male and 72% of female patients with AFD and eGFR < 60 ml/min/1.73 m2 (166). Beta-blockers or non-dihydropyridine calcium channel blockers should be used with caution in the presence of conduction system disease (165). HF in AFD should be managed according to standard guidelines; beta-blockers and ivabradine should be used with caution given the prevalence of conduction disease in AFD (167). Similarly, the management of atrial fibrillation or flutter are similar in AFD and the general population, with the caveat that amiodarone should generally be avoided given its ability to alter lysosomal pH, thereby reducing the activity of endogenous and exogenous α-galactosidase (168). Additionally, anticoagulation is recommended in all patients with atrial fibrillation or flutter where the bleeding risk is not prohibitive (167). Furthermore, maintenance of sinus rhythm over a rate control strategy is recommended (169). Bradyarrhythmias requiring anti-bradycardia pacing are common; patients with prolonged PR or QRS should be monitored closely (170). Proposed indications for the insertion of an implantable cardioverter-defibrillator mirror that of hypertrophic cardiomyopathy; for patients with unexplained syncope, sustained ventricular fibrillatory, cardiac arrest, non-sustained ventricular tachycardia, and extensive cardiac fibrosis (169, 171). Where possible, patients with AFD should be referred to centers of excellence that allow for comprehensive care in multidisciplinary clinics.
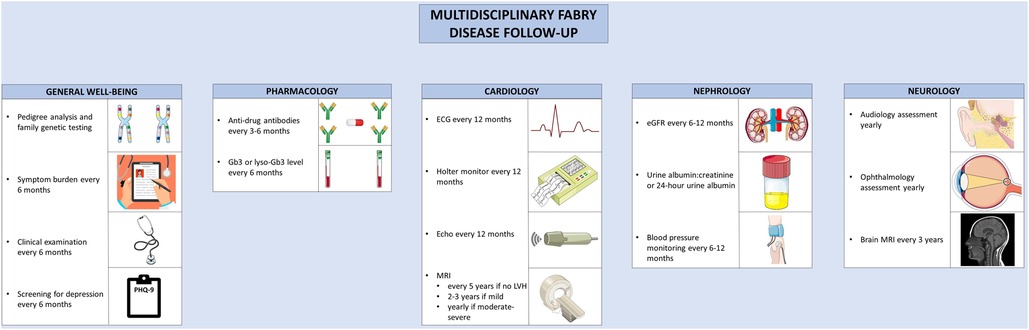
Figure 4. Multidisciplinary follow-up for Anderson-Fabry disease. Recommended follow-up for Anderson-Fabry disease involves co-ordinated care between disciplines. A variety of clinical, biochemical, and imaging-based techniques are used to screen for complications. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license, as well as from freepik.com (https://www.freepik.com/free-vector/doctor-writing-clipboard_2095625.htm.
Guidelines have been developed for the routine clinical follow-up of patients with AFD (169). Routine follow-up for patients with AFD involves monitoring for treatment efficacy and disease progression. Patients should be monitored for symptoms of AFD, ideally using a validated assessment tool; the Brief Pain Inventory, EQ-5D, or MSSI can be used to assess pain, quality of life, and overall disease activity, respectively (172). Clinical examination and assessment of symptom burden should be performed every 6 months (172). To assess treatment efficacy, glycolipid burden should be monitored with lyso-Gb3 or Gb3 levels, performed at baseline, every 6 months, and after treatment switches (169). ADAs should be tested for at baseline, every 3–6 months during the first 18 months of therapy, and then every 6 months thereafter (132, 169). Echocardiography should be performed at diagnosis, at the time of development of new or worsening symptoms, and annually thereafter. In the presence of exertional intolerance, exercise echocardiography can be performed to evaluate for provoked left ventricular outflow tract obstruction or mitral regurgitation, and cardiopulmonary exercise testing or treadmill ECG can be performed to assess for chronotropic incompetence (169). Where possible, CMR should be performed in all adult patients at the time of the initial evaluation (169). If the initial CMR is normal, it can be repeated every 2–5 years (169, 172). In the presence of LVH, the CMR should be repeated every 2–3 years if mild, or annually in the presence of moderate to severe LVH (169, 172). Yearly ECG and 24 h Holter monitoring should be performed to assess the burden of arrhythmia (171). For patients with unexplained palpitations or left atrial enlargement, more frequent 48-hour monitoring should be considered. In the case of unexplained syncope, longer-term monitoring with an implantable loop recorder should be considered (169). The follow-up and surveillance of AFD and its diverse manifestations can be better coordinated in specialized disease-management clinic, where available.
AFD is an X-linked, heritable disease caused by mutation of GLA, leading to reduced production of α-galactosidase, and the accumulation of glycosphingolipids. There is no one characteristic mutation causing AFD; a variety have been described with varying phenotypes. Patients can present with classic disease, characterized by early onset of peripheral neuropathy, LVH, and proteinuric chronic kidney disease; or may present with a late-onset, cardiac-predominant phenotype. Patients with AFD face a significant burden of cardiovascular involvement, including HF, microvascular ischemia, brady- and tachyarrhythmias, valvular dysfunction, and aortic dilatation. The diagnosis of AFD relies on multidisciplinary input, and involves assessment of α-galactosidase activity, measurement of glycosphingolipids, and GLA mutation testing. Non-invasive modalities such as echocardiography and CMR can be used to assess the burden of disease and monitor response to therapy. The backbone of contemporary therapy is ERT; chaperone therapy represents an alternative for patients with amenable mutations. Novel forms of enzyme replacement therapy have been developed to address concerns of immunogenicity, tissue penetration, and duration; however, larger clinical trials are needed. A variety of experimental therapies have been developed for AFD, including substrate reduction therapy, mRNA therapy, and gene therapy, with ongoing randomized clinical trials to establish their role in the care of AFD patients. Long-term, multi-disciplinary care is needed to address the multi-organ complications of AFD and monitor disease activity.
TA contributed to the design of the review, drafted and edited the manuscript. JW critically reviewed and edited the manuscript. NF conceived of the study, contributed to its design, critically reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Figures 1 and 2 were partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unsupported license, as well as from freepik.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bartolotta C, Filogamo M, Colomba P, Zizzo C, Albeggiani G, Scalia S, et al. FP907History of Anderson—Fabry disease. Nephrol Dial Transplant. (2015) 30(suppl_3):iii379. doi: 10.1093/ndt/gfv186.08
2. Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, et al. High incidence of later-onset Fabry disease revealed by newborn screening*. Am J Hum Genet. (2006) 79(1):31–40. doi: 10.1086/504601
3. Michaud M, Mauhin W, Belmatoug N, Garnotel R, Bedreddine N, Catros F, et al. When and how to diagnose Fabry disease in clinical pratice. Am J Med Sci. (2020) 360(6):641–9. doi: 10.1016/j.amjms.2020.07.011
4. Germain DP, Brand E, Cecchi F, Kempf J, Laney DA, Linhart A, et al. The phenotypic characteristics of the p.N215S Fabry disease genotype in male and female patients: a multi-center Fabry Registry study. Mol Genet Metab. (2017) 120(1–2):S51–2. doi: 10.1002/mgg3.389
5. Duro G, Zizzo C, Cammarata G, Burlina A, Burlina A, Polo G, et al. Mutations in the GLA gene and LysoGb3: is it really Anderson-Fabry disease? IJMS. (2018) 19(12):3726. doi: 10.3390/ijms19123726
6. Andrikos E, Iatrou C, Boletis JN, Diamandopoulos A, Katsinas C, Kalaitzidis K, et al. Evolution of Fabry disease in male patients: the greek experience. CN. (2010) 73(01):58–63. doi: 10.5414/CNP73058
7. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. Safety and efficacy of recombinant human α-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. (2001) 345(1):9–16. doi: 10.1056/NEJM200107053450102
8. Clarke JTR. Narrative review: Fabry disease. Ann Intern Med. (2007) 146(6):425. doi: 10.7326/0003-4819-146-6-200703200-00007
9. Olivera-González S, Josa-Laorden C, Torralba-Cabeza MA. The pathophysiology of Fabry disease. Rev Clín Española (English Edition). (2018) 218(1):22–8. doi: 10.1016/j.rceng.2017.09.001
10. Mauhin W, Lidove O, Masat E, Mingozzi F, Mariampillai K, Ziza JM, et al. Innate and adaptive immune response in fabry disease. In: Zschocke J, Baumgartner M, Morava E, Patterson M, Rahman S, Peters V, editors. JIMD Reports, Vol. 22. Berlin, Heidelberg: Springer Berlin Heidelberg (2015). p. 1–10. (JIMD Reports; vol. 22). doi: 10.1007/8904_2014_371
11. Shu L, Vivekanandan-Giri A, Pennathur S, Smid BE, Aerts JMFG, Hollak CEM, et al. Establishing 3-nitrotyrosine as a biomarker for the vasculopathy of Fabry disease. Kidney Int. (2014) 86(1):58–66. doi: 10.1038/ki.2013.520
12. DeGraba T, Azhar S, Dignat-George F, Brown E, Boutière B, Altarescu G, et al. Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol. (2000) 47(2):229–33. doi: 10.1002/1531-8249(200002)47:2%3C229::AID-ANA13%3E3.0.CO;2-T
13. Dubuc V, Moore DF, Gioia LC, Saposnik G, Selchen D, Lanthier S. Prevalence of Fabry disease in young patients with cryptogenic ischemic stroke. J Stroke Cerebrovasc Dis. (2013) 22(8):1288–92. doi: 10.1016/j.jstrokecerebrovasdis.2012.10.005
14. Tomberli B, Cecchi F, Sciagrà R, Berti V, Lisi F, Torricelli F, et al. Coronary microvascular dysfunction is an early feature of cardiac involvement in patients with Anderson–Fabry disease. Eur JHeart Fail. (2013) 15(12):1363–73. doi: 10.1093/eurjhf/hft104
15. Akhtar MM, Elliott PM. Anderson-Fabry disease in heart failure. Biophys Rev. (2018) 10(4):1107–19. doi: 10.1007/s12551-018-0432-5
16. Morrissey RP, Philip KJ, Schwarz ER. Cardiac abnormalities in Anderson-Fabry disease and Fabry’s cardiomyopathy. Cardiovasc J Afr. (2011) 22(1):38–44.21298206
17. Putko BN, Wen K, Thompson RB, Mullen J, Shanks M, Yogasundaram H, et al. Anderson-Fabry cardiomyopathy: prevalence, pathophysiology, diagnosis and treatment. Heart Fail Rev. (2015) 20(2):179–91. doi: 10.1007/s10741-014-9452-9
18. Barbey F, Brakch N, Linhart A, Rosenblatt-Velin N, Jeanrenaud X, Qanadli S, et al. Cardiac and vascular hypertrophy in Fabry disease: evidence for a new mechanism independent of blood pressure and glycosphingolipid deposition. ATVB. (2006) 26(4):839–44. doi: 10.1161/01.ATV.0000209649.60409.38
19. Brakch N, Dormond O, Bekri S, Golshayan D, Correvon M, Mazzolai L, et al. Evidence for a role of sphingosine-1 phosphate in cardiovascular remodelling in Fabry disease. Eur Heart J. (2010) 31(1):67–76. doi: 10.1093/eurheartj/ehp387
20. Chimenti C, Morgante E, Tanzilli G, Mangieri E, Critelli G, Gaudio C, et al. Angina in Fabry disease reflects coronary small vessel disease. Circ Heart Fail. (2008) 1(3):161–9. doi: 10.1161/CIRCHEARTFAILURE.108.769729
21. Rossi F, Svarstad E, Elsaid H, Binaggia A, Roggero L, Auricchio S, et al. Elevated ambulatory blood pressure measurements are associated with a progressive form of Fabry disease. High Blood Press Cardiovasc Prev. (2021) 28(3):309–19. doi: 10.1007/s40292-021-00450-0
22. Dai X, Zong X, Pan X, Lu W, Jiang GR, Lin F. Identification and functional characterization of the first deep intronic GLA mutation (IVS4+1326C>T) causing renal variant of Fabry disease. Orphanet J Rare Dis. (2022) 17(1):237. doi: 10.1186/s13023-022-02377-8
23. Citro V, Cammisa M, Liguori L, Cimmaruta C, Lukas J, Cubellis M, et al. The large phenotypic spectrum of Fabry disease requires graduated diagnosis and personalized therapy: a meta-analysis can help to differentiate missense mutations. IJMS. (2016) 17(12):2010. doi: 10.3390/ijms17122010
24. Lombardi S, Ferrarese M, Marchi S, Pinton P, Pinotti M, Bernardi F, et al. Translational readthrough of GLA nonsense mutations suggests dominant-negative effects exerted by the interaction of wild-type and missense variants. RNA Biol. (2020) 17(2):254–63. doi: 10.1080/15476286.2019.1676115
25. Kim DH, Kim SY, Im M, Lee Y, Seo YJ, Lee JH. A novel frameshift mutation of galactosidase-alpha in Fabry disease restricted to dermatologic manifestations. Ann Dermatol. (2013) 25(1):95. doi: 10.5021/ad.2013.25.1.95
26. Onay H, Bolat H, Kılıç Yıldırım G, Kose E, Kalkan Uçar S, Aşıkovalı S, et al. Analysis of the alpha galactosidase gene: mutation profile and description of two novel mutations with extensive literature review in Turkish population. J Pediatr Endocrinol Metab. (2020) 33(10):1245–50. doi: 10.1515/jpem-2020-0056
27. Saito S, Ohno K, Sese J, Sugawara K, Sakuraba H. Prediction of the clinical phenotype of Fabry disease based on protein sequential and structural information. J Hum Genet. (2010) 55(3):175–8. doi: 10.1038/jhg.2010.5
28. Pan X, Ouyang Y, Wang Z, Ren H, Shen P, Wang W, et al. Genotype: a crucial but not unique factor affecting the clinical phenotypes in Fabry disease. PLoS One. (2016) 11(8):e0161330. doi: 10.1371/journal.pone.0161330
29. Savostyanov K, Pushkov A, Zhanin I, Mazanova N, Trufanov S, Pakhomov A, et al. The prevalence of Fabry disease among 1009 unrelated patients with hypertrophic cardiomyopathy: a Russian nationwide screening program using NGS technology. Orphanet J Rare Dis. (2022) 17(1):199. doi: 10.1186/s13023-022-02319-4
30. Fan Y, Chan TN, Chow JTY, Kam KKH, Chi WK, Chan JYS, et al. High prevalence of late-onset Fabry cardiomyopathy in a cohort of 499 non-selective patients with left ventricular hypertrophy: the Asian Fabry cardiomyopathy high-risk screening study (ASIAN-FAME). JCM. (2021) 10(10):2160. doi: 10.3390/jcm10102160
31. Germain DP, Oliveira JP, Bichet DG, Yoo HW, Hopkin RJ, Lemay R, et al. Use of a rare disease registry for establishing phenotypic classification of previously unassigned GLA variants: a consensus classification system by a multispecialty Fabry disease genotype–phenotype workgroup. J Med Genet. (2020) 57(8):542–51. doi: 10.1136/jmedgenet-2019-106467
32. Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. (2008) 372(9647):1427–35. doi: 10.1016/S0140-6736(08)61589-5
33. Gilchrist M, Casanova F, Tyrrell JS, Cannon S, Wood AR, Fife N, et al. Prevalence of Fabry disease-causing variants in the UK biobank. J Med Genet. (2022) 60(4):jmedgenet-2022-108523. doi: 10.1136/jmg-2022-108523
34. Burton BK, Charrow J, Hoganson GE, Waggoner D, Tinkle B, Braddock SR, et al. Newborn screening for lysosomal storage disorders in Illinois: the initial 15-month experience. J Pediatr. (2017) 190:130–5. doi: 10.1016/j.jpeds.2017.06.048
35. Hwu WL, Chien YH, Lee NC, Chiang SC, Dobrovolny R, Huang AC, et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum Mutat. (2009) 30(10):1397–405. doi: 10.1002/humu.21074
36. Arends M, Wanner C, Hughes D, Mehta A, Oder D, Watkinson OT, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. JASN. (2017) 28(5):1631–41. doi: 10.1681/ASN.2016090964
37. Kim WS, Kim HS, Shin J, Park JC, Yoo HW, Takenaka T, et al. Prevalence of Fabry disease in Korean men with left ventricular hypertrophy. J Korean Med Sci. (2019) 34(7):e63. doi: 10.3346/jkms.2019.34.e63
38. Terryn W, Deschoenmakere G, De Keyser J, Meersseman W, Van Biesen W, Wuyts B, et al. Prevalence of Fabry disease in a predominantly hypertensive population with left ventricular hypertrophy. Int J Cardiol. (2013) 167(6):2555–60. doi: 10.1016/j.ijcard.2012.06.069
39. Palecek T, Honzikova J, Poupetova H, Vlaskova H, Kuchynka P, Golan L, et al. Prevalence of Fabry disease in male patients with unexplained left ventricular hypertrophy in primary cardiology practice: prospective Fabry cardiomyopathy screening study (FACSS). J Inherit Metab Dis. (2014) 37(3):455–60. doi: 10.1007/s10545-013-9659-2
40. Sadasivan C, Chow JTY, Sheng B, Chan DKH, Fan Y, Choi PCL, et al. Screening for Fabry Disease in patients with unexplained left ventricular hypertrophy. PLoS ONE. (2020) 15(9):e0239675. doi: 10.1371/journal.pone.0239675
41. Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inher Metab Dis. (2007) 30(2):184–92. doi: 10.1007/s10545-007-0521-2
42. Linhart A, Kampmann C, Zamorano JL, Sunder-Plassmann G, Beck M, Mehta A, et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. (2007) 28(10):1228–35. doi: 10.1093/eurheartj/ehm153
43. Mehta A, Clarke JTR, Giugliani R, Elliott P, Linhart A, Beck M, et al. Natural course of Fabry disease: changing pattern of causes of death in FOS—Fabry outcome survey. J Med Genet. (2009) 46(8):548–52. doi: 10.1136/jmg.2008.065904
44. Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet Med. (2009) 11(11):790–6. doi: 10.1097/GIM.0b013e3181bb05bb
45. Azevedo O, Gago MF, Miltenberger-Miltenyi G, Robles AR, Costa MA, Pereira O, et al. Natural history of the late-onset phenotype of Fabry disease due to the p.F113l mutation. Mol Genet Metab Rep. (2020) 22:100565. doi: 10.1016/j.ymgmr.2020.100565
46. Feriozzi S, Linhart A, Ramaswami U, Kalampoki V, Gurevich A, Hughes D. Effects of baseline left ventricular hypertrophy and decreased renal function on cardiovascular and renal outcomes in patients with Fabry disease treated with agalsidase alfa: a Fabry outcome survey study. Clin Ther. (2020) 42(12):2321–30.e0. doi: 10.1016/j.clinthera.2020.10.007
47. Wu JC, Ho CY, Skali H, Abichandani R, Wilcox WR, Banikazemi M, et al. Cardiovascular manifestations of Fabry disease: relationships between left ventricular hypertrophy, disease severity, and alpha-galactosidase A activity. Eur Heart J. (2010) 31(9):1088–97. doi: 10.1093/eurheartj/ehp588
48. Kampmann C, Linhart A, Baehner F, Palecek T, Wiethoff CM, Miebach E, et al. Onset and progression of the Anderson–Fabry disease related cardiomyopathy. Int J Cardiol. (2008) 130(3):367–73. doi: 10.1016/j.ijcard.2008.03.007
49. Yeung DF, Sirrs S, Tsang MYC, Gin K, Luong C, Jue J, et al. Echocardiographic assessment of patients with Fabry disease. J Am Soc Echocardiogr. (2018) 31(6):639–49.e2. doi: 10.1016/j.echo.2018.01.016
50. Raju B, Roberts CS, Sathyamoorthy M, Schiffman R, Swift C, McCullough PA. Ventricular septal myectomy for the treatment of left ventricular outflow tract obstruction due to Fabry disease. Am J Cardiol. (2020) 132:160–4. doi: 10.1016/j.amjcard.2020.07.020
51. Monserrat L, Gimeno-Blanes JR, Marín F, Hermida-Prieto M, García-Honrubia A, Pérez I, et al. Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. (2007) 50(25):2399–403. doi: 10.1016/j.jacc.2007.06.062
52. Graziani F, Lillo R, Panaioli E, Spagnoletti G, Pieroni M, Ferrazzi P, et al. Evidence of evolution towards left midventricular obstruction in severe Anderson-Fabry cardiomyopathy. ESC Heart Fail. (2021) 8(1):725–8. doi: 10.1002/ehf2.13101
53. Birket MJ, Raibaud S, Lettieri M, Adamson AD, Letang V, Cervello P, et al. A human stem cell model of Fabry disease implicates LIMP-2 accumulation in cardiomyocyte pathology. Stem Cell Rep. (2019) 13(2):380–93. doi: 10.1016/j.stemcr.2019.07.004
54. Frustaci A, Morgante E, Russo MA, Scopelliti F, Grande C, Verardo R, et al. Pathology and function of conduction tissue in Fabry disease cardiomyopathy. Circ Arrhythm Electrophysiol. (2015) 8(4):799–805. doi: 10.1161/CIRCEP.114.002569
55. Pichette M, Serri K, Pagé M, Di LZ, Bichet DG, Poulin F. Impaired left atrial function in Fabry disease: a longitudinal speckle-tracking echocardiography study. J Am Soc Echocardiogr. (2017) 30(2):170–9.e2. doi: 10.1016/j.echo.2016.10.014
56. Boyd AC, Lo Q, Devine K, Tchan MC, Sillence DO, Sadick N, et al. Left atrial enlargement and reduced atrial compliance occurs early in Fabry cardiomyopathy. J Am Soc Echocardiogr. (2013) 26(12):1415–23. doi: 10.1016/j.echo.2013.08.024
57. Baig S, Edward NC, Kotecha D, Liu B, Nordin S, Kozor R, et al. Ventricular arrhythmia and sudden cardiac death in Fabry disease: a systematic review of risk factors in clinical practice. Europace. (2018) 20(FI2):f153–61. doi: 10.1093/europace/eux261
58. Lu DY, Huang WM, Wang WT, Hung SC, Sung SH, Chen CH, et al. Reduced global longitudinal strain as a marker for early detection of Fabry cardiomyopathy. Eur Heart J Cardiovasc Imaging. (2022) 23(4):487–95. doi: 10.1093/ehjci/jeab214
59. Lillo R, Graziani F, Panaioli E, Mencarelli E, Pieroni M, Camporeale A, et al. Right ventricular strain in Anderson-Fabry disease. Int J Cardiol. (2021) 330:84–90. doi: 10.1016/j.ijcard.2021.02.038
60. Liu D, Oder D, Salinger T, Hu K, Müntze J, Weidemann F, et al. Association and diagnostic utility of diastolic dysfunction and myocardial fibrosis in patients with Fabry disease. Open Heart. (2018) 5(2):e000803. doi: 10.1136/openhrt-2018-000803
61. Torralba-Cabeza MÁ, Olivera S, Hughes DA, Pastores GM, Mateo RN, Pérez-Calvo JI. Cystatin C and NT-proBNP as prognostic biomarkers in Fabry disease. Mol Genet Metab. (2011) 104(3):301–7. doi: 10.1016/j.ymgme.2011.06.021
62. Coats CJ, Parisi V, Ramos M, Janagarajan K, O’Mahony C, Dawnay A, et al. Role of serum N-terminal pro-brain natriuretic peptide measurement in diagnosis of cardiac involvement in patients with Anderson-Fabry disease. Am J Cardiol. (2013) 111(1):111–7. doi: 10.1016/j.amjcard.2012.08.055
63. Graziani F, Lillo R, Biagini E, Limongelli G, Autore C, Pieroni M, et al. Myocardial infarction with non-obstructive coronary arteries in hypertrophic cardiomyopathy vs Fabry disease. Int J Cardiol. (2022) 369:29–32. doi: 10.1016/j.ijcard.2022.07.046
64. Yogasundaram H, Nikhanj A, Chatur S, Qi A, Hagen L, Bailey L, et al. Burden of valvular heart disease in patients with Fabry disease. J Am Soc Echocardiogr. (2022) 35(2):236–8. doi: 10.1016/j.echo.2021.09.013
65. Weidemann F, Strotmann JM, Niemann M, Herrmann S, Wilke M, Beer M, et al. Heart valve involvement in Fabry cardiomyopathy. Ultrasound Med Biol. (2009) 35(5):730–5. doi: 10.1016/j.ultrasmedbio.2008.10.010
66. Linhart A, Paleček T, Bultas J, Ferguson JJ, Hrudová J, Karetová D, et al. New insights in cardiac structural changes in patients with Fabry’s disease. Am Heart J. (2000) 139(6):1101–8. doi: 10.1067/mhj.2000.105105
67. Saccheri MC, Cianciulli TF, Blanco FL, Blanco RI. Rapidly progressive aortic stenosis treated with transcatheter aortic valve implantation in a patient with Fabry disease: a case report. Eur Heart J Case Rep. (2021) 5(6):ytab124. doi: 10.1093/ehjcr/ytab124
68. Vlachou A, Gogakos AS, Cheva A, Drossos G. Surgical treatment of severe aortic valve stenosis with concomitant Fabry disease. Cardiovasc Med. (2019) 22. doi: 10.4414/cvm.2019.02066
69. Barbey F, Qanadli SD, Juli C, Brakch N, Palacek T, Rizzo E, et al. Aortic remodelling in Fabry disease. Eur Heart J. (2010) 31(3):347–53. doi: 10.1093/eurheartj/ehp426
70. Monney P, Qanadli SD, Hajdu S, Tran C, Schwitter J, Dormond O, et al. Ascending aortic remodelling in Fabry disease after long-term enzyme replacement therapy. Swiss Med Wkly. (2017) 147(4142):w14517. doi: 10.4414/smw.2017.14517
71. Dobrowolski MK, Marczak M, Dąbrowski R. Aortic dissection four months after SARS-CoV-2 infection in a patient with Fabry disease whose targeted treatment was stopped 2 months earlier. Kardiol Pol. (2022) 80(6):713–4. doi: 10.33963/KP.a2022.0133
72. Kok K, Zwiers KC, Boot RG, Overkleeft HS, Aerts JMFG, Artola M. Fabry disease: molecular basis, pathophysiology, diagnostics and potential therapeutic directions. Biomolecules. (2021) 11(2):271. doi: 10.3390/biom11020271
73. Stiles AR, Zhang H, Dai J, McCaw P, Beasley J, Rehder C, et al. A comprehensive testing algorithm for the diagnosis of Fabry disease in males and females. Mol Genet Metab. (2020) 130(3):209–14. doi: 10.1016/j.ymgme.2020.04.006
74. Fuller M, Lovejoy M, Brooks DA, Harkin ML, Hopwood JJ, Meikle PJ. Immunoquantification of α-galactosidase: evaluation for the diagnosis of Fabry disease. Clin Chem. (2004) 50(11):1979–85. doi: 10.1373/clinchem.2004.037937
75. Pasqualim G, Simon L, Sperb-Ludwig F, Burin MG, Michelin-Tirelli K, Giugliani R, et al. Fabry disease: a new approach for the screening of females in high-risk groups. Clin Biochem. (2014) 47(7–8):657–62. doi: 10.1016/j.clinbiochem.2014.02.014
76. Germain DP, Levade T, Hachulla E, Knebelmann B, Lacombe D, Seguin VL, et al. Challenging the traditional approach for interpreting genetic variants: lessons from Fabry disease. Clin Genet. (2022) 101(4):390–402. doi: 10.1111/cge.14102
77. Ouyang Y, Chen B, Pan X, Wang Z, Ren H, Xu Y, et al. Clinical significance of plasma globotriaosylsphingosine levels in Chinese patients with Fabry disease. Exp Ther Med. (2018) 15(4):3733–42. doi: 10.3892/etm.2018.5889
78. Rombach SM, Dekker N, Bouwman MG, Linthorst GE, Zwinderman AH, Wijburg FA, et al. Plasma globotriaosylsphingosine: diagnostic value and relation to clinical manifestations of Fabry disease. Biochim Biophys Acta. (2010) 1802(9):741–8. doi: 10.1016/j.bbadis.2010.05.003
79. Niemann M, Rolfs A, Störk S, Bijnens B, Breunig F, Beer M, et al. Gene mutations versus clinically relevant phenotypes: lyso-Gb3 defines Fabry disease. Circ Cardiovasc Genet. (2014) 7(1):8–16. doi: 10.1161/CIRCGENETICS.113.000249
80. Baydakova GV, Ilyushkina AA, Moiseev S, Bychkov IO, Nikitina NV, Buruleva ТА, et al. α-Galactosidase A/lysoGb3 ratio as a potential marker for Fabry disease in females. Clin Chim Acta. (2020) 501:27–32. doi: 10.1016/j.cca.2019.10.031
81. Tschöpe C, Dominguez F, Canaan-Kühl S, Blaschke D, Kühl U, Pieske B, et al. Endomyocardial biopsy in Anderson–Fabry disease: the key in uncertain cases. Int J Cardiol. (2015) 190:284–6. doi: 10.1016/j.ijcard.2015.04.130
82. Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American heart association, the American college of cardiology, and the European society of cardiology endorsed by the heart failure society of America and the heart failure association of the European society of cardiology. Eur Heart J. (2007) 28(24):3076–93. doi: 10.1093/eurheartj/ehm456
83. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2021) 42(36):3599–726. doi: 10.1093/eurheartj/ehab368
84. Namdar M. Electrocardiographic changes and arrhythmia in Fabry disease. Front Cardiovasc Med. (2016) 3. doi: 10.3389/fcvm.2016.00007
85. Morimoto S, Nojiri A, Fukuro E, Anan I, Kawai M, Sakurai K, et al. Characteristics of the electrocardiogram in Japanese Fabry patients under long-term enzyme replacement therapy. Front Cardiovasc Med. (2021) 7:614129. doi: 10.3389/fcvm.2020.614129
86. Namdar M, Kampmann C, Steffel J, Walder D, Holzmeister J, Lüscher TF, et al. PQ Interval in patients with Fabry disease. Am J Cardiol. (2010) 105(5):753–6. doi: 10.1016/j.amjcard.2009.10.056
87. Figliozzi S, Camporeale A, Boveri S, Pieruzzi F, Pieroni M, Lusardi P, et al. ECG-based score estimates the probability to detect Fabry disease cardiac involvement. Int J Cardiol. (2021) 339:110–7. doi: 10.1016/j.ijcard.2021.07.022
88. Namdar M, Steffel J, Jetzer S, Schmied C, Hürlimann D, Camici GG, et al. Value of electrocardiogram in the differentiation of hypertensive heart disease, hypertrophic cardiomyopathy, aortic stenosis, amyloidosis, and Fabry disease. Am J Cardiol. (2012) 109(4):587–93. doi: 10.1016/j.amjcard.2011.09.052
89. Militaru S, Jurcuț R, Adam R, Roşca M, Ginghina C, Popescu BA. Echocardiographic features of Fabry cardiomyopathy—comparison with hypertrophy-matched sarcomeric hypertrophic cardiomyopathy. Echocardiography. (2019) 36(11):2041–9. doi: 10.1111/echo.14508
90. Pieroni M, Chimenti C, De Cobelli F, Morgante E, Del Maschio A, Gaudio C, et al. Fabry’s disease cardiomyopathy: echocardiographic detection of endomyocardial glycosphingolipid compartmentalization. J Am Coll Cardiol. (2006) 47(8):1663–71. doi: 10.1016/j.jacc.2005.11.070
91. Koskenvuo JW, Engblom E, Kantola IM, Hartiala JJ, Saraste A, Kiviniemi TO, et al. Echocardiography in Fabry disease: diagnostic value of endocardial border binary appearance. Clin Physiol Funct Imaging. (2009) 29(3):177–80. doi: 10.1111/j.1475-097X.2008.00851.x
92. Mundigler G, Gaggl M, Heinze G, Graf S, Zehetgruber M, Lajic N, et al. The endocardial binary appearance (“binary sign”) is an unreliable marker for echocardiographic detection of Fabry disease in patients with left ventricular hypertrophy. Eur J Echocardiogr. (2011) 12(10):744–9. doi: 10.1093/ejechocard/jer112
93. Toro R, Perez-Isla L, Doxastaquis G, Barba MA, Rivera Gallego A, Pintos G, et al. Clinical usefulness of tissue Doppler imaging in predicting preclinical Fabry cardiomyopathy. Int J Cardiol. (2009) 132(1):38–44. doi: 10.1016/j.ijcard.2008.04.075
94. Pieroni M, Chimenti C, Ricci R, Sale P, Russo MA, Frustaci A. Early detection of Fabry cardiomyopathy by tissue Doppler imaging. Circulation. (2003) 107(15):1978–84. doi: 10.1161/01.CIR.0000061952.27445.A0
95. Junqua N, Legallois D, Segard S, Lairez O, Réant P, Goizet C, et al. The value of electrocardiography and echocardiography in distinguishing Fabry disease from sarcomeric hypertrophic cardiomyopathy. Arch Cardiovasc Dis. (2020) 113(8–9):542–50. doi: 10.1016/j.acvd.2020.04.008
96. Meucci MC, Lillo R, Lombardo A, Lanza GA, Bootsma M, Butcher SC, et al. Comparative analysis of right ventricular strain in Fabry cardiomyopathy and sarcomeric hypertrophic cardiomyopathy. Eur Heart J Cardiovasc Imaging. (2022) 24(4):jeac151. doi: 10.1093/ehjci/jeac151
97. Saccheri MC, Cianciulli TF, Challapa Licidio W, Lax JA, Beck MA, Morita LA, et al. Comparison of left atrial size and function in hypertrophic cardiomyopathy and in Fabry disease with left ventricular hypertrophy. Echocardiography. (2018) 35(5):643–50. doi: 10.1111/echo.13829
98. Graziani F, Laurito M, Pieroni M, Pennestrì F, Lanza GA, Coluccia V, et al. Right ventricular hypertrophy, systolic function, and disease severity in Anderson-Fabry disease: an echocardiographic study. J Am Soc Echocardiogr. (2017) 30(3):282–91. doi: 10.1016/j.echo.2016.11.014
99. Bernardini A, Camporeale A, Pieroni M, Pieruzzi F, Figliozzi S, Lusardi P, et al. Atrial dysfunction assessed by cardiac magnetic resonance as an early marker of Fabry cardiomyopathy. JACC Cardiovasc Imaging. (2020) 13(10):2262–4. doi: 10.1016/j.jcmg.2020.05.011
100. Knott KD, Augusto JB, Nordin S, Kozor R, Camaioni C, Xue H, et al. Quantitative myocardial perfusion in Fabry disease. Circ Cardiovasc Imaging. (2019) 12(7):e008872. doi: 10.1161/CIRCIMAGING.119.008872
101. Moon J, Sheppard M, Reed E, Lee P, Elliott P, Pennell D. The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. J Cardiovasc Magn Reson. (2006) 8(3):479–82. doi: 10.1080/10976640600605002
102. Hanneman K, Karur GR, Wasim S, Wald RM, Iwanochko RM, Morel CF. Left ventricular hypertrophy and late gadolinium enhancement at cardiac MRI are associated with adverse cardiac events in Fabry disease. Radiology. (2020) 294(1):42–9. doi: 10.1148/radiol.2019191385
103. Feustel A, Hahn A, Schneider C, Sieweke N, Franzen W, Gündüz D, et al. Continuous cardiac troponin I release in Fabry disease. PLoS ONE. (2014) 9(3):e91757. doi: 10.1371/journal.pone.0091757
104. Seydelmann N, Liu D, Krämer J, Drechsler C, Hu K, Nordbeck P, et al. High-sensitivity troponin: a clinical blood biomarker for staging cardiomyopathy in Fabry disease. JAHA. (2016) 5(6):e002839. doi: 10.1161/JAHA.115.002839
105. O’Brien C, Britton I, Karur GR, Iwanochko RM, Morel CF, Nguyen ET, et al. Left ventricular mass and wall thickness measurements using echocardiography and cardiac MRI in patients with Fabry disease: clinical significance of discrepant findings. Radiol Cardiothorac Imaging. (2020) 2(3):e190149. doi: 10.1148/ryct.2020190149
106. Aquaro GD, De Gori C, Faggioni L, Parisella ML, Aringhieri G, Cioni D, et al. Cardiac magnetic resonance in Fabry disease: morphological, functional, and tissue features. Diagnostics. (2022) 12(11):2652. doi: 10.3390/diagnostics12112652
107. Karur GR, Robison S, Iwanochko RM, Morel CF, Crean AM, Thavendiranathan P, et al. Use of myocardial T1 mapping at 3.0 T to differentiate Anderson-Fabry disease from hypertrophic cardiomyopathy. Radiology. (2018) 288(2):398–406. doi: 10.1148/radiol.2018172613
108. Ponsiglione A, Gambardella M, Green R, Cantoni V, Nappi C, Ascione R, et al. Cardiovascular magnetic resonance native T1 mapping in Anderson-Fabry disease: a systematic review and meta-analysis. J Cardiovasc Magn Reson. (2022) 24(1):31. doi: 10.1186/s12968-022-00859-z
109. Augusto JB, Johner N, Shah D, Nordin S, Knott KD, Rosmini S, et al. The myocardial phenotype of Fabry disease pre-hypertrophy and pre-detectable storage. Eur Heart J Cardiovasc Imaging. (2021) 22(7):790–9. doi: 10.1093/ehjci/jeaa101
110. Nordin S, Kozor R, Baig S, Abdel-Gadir A, Medina-Menacho K, Rosmini S, et al. Cardiac phenotype of prehypertrophic Fabry disease. Circ Cardiovasc Imaging. (2018) 11(6):e007168. doi: 10.1161/CIRCIMAGING.117.007168
111. Nordin S, Kozor R, Medina-Menacho K, Abdel-Gadir A, Baig S, Sado DM, et al. Proposed stages of myocardial phenotype development in Fabry disease. JACC Cardiovasc Imaging. (2019) 12(8):1673–83. doi: 10.1016/j.jcmg.2018.03.020
112. Djaileb L, Jankowski A, Riou L, Salvat M, Barone-Rochette G. Unusual SPECT myocardial perfusion imaging results in the setting of ventricular hypertrophy and non-obstructive coronary artery disease should suggest Anderson-Fabry disease. Eur Heart J Cardiovasc Imaging. (2019) 20(9):1073. doi: 10.1093/ehjci/jez062
113. Yuki H, Utsunomiya D, Izumiya Y, Oda S, Kidoh M, Takashio S, et al. Improvement in microvascular ischemia after enzyme replacement therapy in Anderson-Fabry disease — computed tomography myocardial perfusion imaging —. Circ J. (2017) 81(2):243–4. doi: 10.1253/circj.CJ-16-0873
114. Valtola K, Nino-Quintero J, Hedman M, Lottonen-Raikaslehto L, Laitinen T, Maria M, et al. Cardiomyopathy associated with the Ala143Thr variant of the α-galactosidase A gene. Heart. (2020) 106(8):609–15. doi: 10.1136/heartjnl-2019-315933
115. Manabe O, Oyama-Manabe N, Tamaki N. Positron emission tomography/MRI for cardiac diseases assessment. BJR. (2020) 93(1113):20190836. doi: 10.1259/bjr.20190836
116. Schaefer RM, Tylki-Szymańska A, Hilz MJ. Enzyme replacement therapy for Fabry disease: a systematic review of available evidence. Drugs. (2009) 69(16):2179–205. doi: 10.2165/11318300-000000000-00000
117. El Dib R, Gomaa H, Ortiz A, Politei J, Kapoor A, Barreto F. Enzyme replacement therapy for Anderson-Fabry disease: a complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS ONE. (2017) 12(3):e0173358. doi: 10.1371/journal.pone.0173358
118. El Dib R, Gomaa H, Carvalho RP, Camargo SE, Bazan R, Barretti P, et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane cystic fibrosis and genetic disorders group, editor. Cochrane Database Syst Rev. (2016) 2017(6):CD006663. doi: 10.1002/14651858.CD006663.pub4
119. Lenders M, Brand E. Effects of enzyme replacement therapy and antidrug antibodies in patients with Fabry disease. JASN. (2018) 29(9):2265–78. doi: 10.1681/ASN.2018030329
120. Linthorst GE, Hollak CEM, Donker-Koopman WE, Strijland A, Aerts JMFG. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. (2004) 66(4):1589–95. doi: 10.1111/j.1523-1755.2004.00924.x
121. Rombach SM, Aerts JMFG, Poorthuis BJHM, Groener JEM, Donker-Koopman W, Hendriks E, et al. Long-term effect of antibodies against infused alpha-galactosidase A in Fabry disease on plasma and urinary (lyso)Gb3 reduction and treatment outcome. PLoS One. (2012) 7(10):e47805. doi: 10.1371/journal.pone.0047805
122. Lenders M, Stypmann J, Duning T, Schmitz B, Brand SM, Brand E. Serum-Mediated inhibition of enzyme replacement therapy in Fabry disease. JASN. (2016) 27(1):256–64. doi: 10.1681/ASN.2014121226
123. Arends M, Biegstraaten M, Wanner C, Sirrs S, Mehta A, Elliott PM, et al. Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: an international cohort study. J Med Genet. (2018) 55(5):351–8. doi: 10.1136/jmedgenet-2017-104863
124. Mauhin W, Lidove O, Amelin D, Lamari F, Caillaud C, Mingozzi F, et al. Deep characterization of the anti-drug antibodies developed in Fabry disease patients, a prospective analysis from the French multicenter cohort FFABRY. Orphanet J Rare Dis. (2018) 13(1):127. doi: 10.1186/s13023-018-0877-4
125. van der Veen SJ, Vlietstra WJ, van Dussen L, van Kuilenburg ABP, Dijkgraaf MGW, Lenders M, et al. Predicting the development of anti-drug antibodies against recombinant alpha-galactosidase A in male patients with classical Fabry disease. IJMS. (2020) 21(16):5784. doi: 10.3390/ijms21165784
126. Lenders M, Neußer LP, Rudnicki M, Nordbeck P, Canaan-Kühl S, Nowak A, et al. Dose-Dependent effect of enzyme replacement therapy on neutralizing antidrug antibody titers and clinical outcome in patients with Fabry disease. JASN. (2018) 29(12):2879–89. doi: 10.1681/ASN.2018070740
127. Sirrs S, Bichet DG, Iwanochko RM, Khan A, Moore D, Oudit GY, et al. Canadian Fabry Disease Treatment Guidelines 2017. (2017). Available from: https://www.fabrycanada.com/content/uploads/Final-Can-FD-Treatment-Guidelines-2017Oct18.pdf [cited November 28, 2022].
128. Biegstraaten M, Arngrímsson R, Barbey F, Boks L, Cecchi F, Deegan PB, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry working group consensus document. Orphanet J Rare Dis. (2015) 10(1):36. doi: 10.1186/s13023-015-0253-6
129. Clarke JTR, West ML, Bultas J, Schiffmann R. The pharmacology of multiple regimens of agalsidase alfa enzyme replacement therapy for Fabry disease. Genet Med. (2007) 9(8):504–9. doi: 10.1097/GIM.0b013e318133fb1b
130. van Breemen MJ, Rombach SM, Dekker N, Poorthuis BJ, Linthorst GE, Zwinderman AH, et al. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim Biophys Acta. (2011) 1812(1):70–6. doi: 10.1016/j.bbadis.2010.09.007
131. Sakuraba H, Togawa T, Tsukimura T, Kato H. Plasma lyso-Gb3: a biomarker for monitoring fabry patients during enzyme replacement therapy. Clin Exp Nephrol. (2018) 22(4):843–9. doi: 10.1007/s10157-017-1525-3
132. Nowak A, Dormond O, Monzambani V, Huynh-Do U, Barbey F. Agalsidase-β should be proposed as first line therapy in classic male Fabry patients with undetectable α-galactosidase A activity. Mol Genet Metab. (2022) 137(1–2):173–8. doi: 10.1016/j.ymgme.2022.08.003
133. Lee CL, Lin SP, Niu DM, Lin HY. Fabry disease and the effectiveness of enzyme replacement therapy (ERT) in left ventricular hypertrophy (LVH) improvement: a review and meta-analysis. Int J Med Sci. (2022) 19(1):126–31. doi: 10.7150/ijms.66448
134. Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Störk S, et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. (2009) 119(4):524–9. doi: 10.1161/CIRCULATIONAHA.108.794529
135. Beer M, Weidemann F, Breunig F, Knoll A, Koeppe S, Machann W, et al. Impact of enzyme replacement therapy on cardiac morphology and function and late enhancement in Fabry’s cardiomyopathy. Am J Cardiol. (2006) 97(10):1515–8. doi: 10.1016/j.amjcard.2005.11.087
136. Hughes D, Linhart A, Gurevich A, Kalampoki V, Jazukeviciene D, Feriozzi S, et al. Prompt agalsidase alfa therapy initiation is associated with improved renal and cardiovascular outcomes in a Fabry outcome survey analysis. Drug Des Devel Ther. (2021) 15:3561–72. doi: 10.2147/DDDT.S313789
137. Ruderfer I, Shulman A, Kizhner T, Azulay Y, Nataf Y, Tekoah Y, et al. Development and analytical characterization of pegunigalsidase alfa, a chemically cross-linked plant recombinant human α-galactosidase-A for treatment of Fabry disease. Bioconjugate Chem. (2018) 29(5):1630–9. doi: 10.1021/acs.bioconjchem.8b00133
138. Lenders M, Pollmann S, Terlinden M, Brand E. Pre-existing anti-drug antibodies in Fabry disease show less affinity for pegunigalsidase alfa. Mol Ther Methods Clin Dev. (2022) 26:323–30. doi: 10.1016/j.omtm.2022.07.009
139. Schiffmann R, Goker-Alpan O, Holida M, Giraldo P, Barisoni L, Colvin RB, et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: a 1-year phase 1/2 clinical trial. J Inherit Metab Dis. (2019) 42(3):jimd.12080. doi: 10.1002/jimd.12080
140. Hennermann JB, Arash-Kaps L, Fekete G, Schaaf A, Busch A, Frischmuth T. Pharmacokinetics, pharmacodynamics, and safety of moss-aGalactosidase A in patients with Fabry disease. J Inherit Metab Dis. (2019) 42(3):527–33. doi: 10.1002/jimd.12052
141. Shen JS, Busch A, Day TS, Meng XL, Yu CI, Dabrowska-Schlepp P, et al. Mannose receptor-mediated delivery of moss-made α-galactosidase A efficiently corrects enzyme deficiency in Fabry mice. J Inherit Metab Dis. (2016) 39(2):293–303. doi: 10.1007/s10545-015-9886-9
142. Lenders M, Scharnetzki D, Heidari A, Di Iorio D, Wegner SV, Brand E. Generation and characterization of a polyclonal human reference antibody to measure anti-drug antibody titers in patients with Fabry disease. IJMS. (2021) 22(5):2680. doi: 10.3390/ijms22052680
143. Weidemann F, Jovanovic A, Herrmann K, Vardarli I. Chaperone therapy in Fabry disease. IJMS. (2022) 23(3):1887. doi: 10.3390/ijms23031887
144. Benjamin ER, Della Valle MC, Wu X, Katz E, Pruthi F, Bond S, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. (2017) 19(4):430–8. doi: 10.1038/gim.2016.122
145. Hughes DA, Nicholls K, Shankar SP, Sunder-Plassmann G, Koeller D, Nedd K, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. (2017) 54(4):288–96. doi: 10.1136/jmedgenet-2016-104178
146. Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med. (2016) 375(6):545–55. doi: 10.1056/NEJMoa1510198
147. Camporeale A, Bandera F, Pieroni M, Pieruzzi F, Spada M, Bersano A, et al. Effect of migalastat on cArdiac involvement in FabRry disease: mAIORA study. J Med Genet. (2023):jmg-2022-108768. doi: 10.1136/jmg-2022-108768
148. Riccio E, Zanfardino M, Ferreri L, Santoro C, Cocozza S, Capuano I, et al. Switch from enzyme replacement therapy to oral chaperone migalastat for treating fabry disease: real-life data. Eur J Hum Genet. (2020) 28(12):1662–8. doi: 10.1038/s41431-020-0677-x
149. Müntze J, Gensler D, Maniuc O, Liu D, Cairns T, Oder D, et al. Oral chaperone therapy migalastat for treating Fabry disease: enzymatic response and serum biomarker changes after 1 year. Clin Pharmacol Ther. (2019) 105(5):1224–33. doi: 10.1002/cpt.1321
150. Lenders M, Nordbeck P, Kurschat C, Eveslage M, Karabul N, Kaufeld J, et al. Treatment of Fabry disease management with migalastat—outcome from a prospective 24 months observational multicenter study (FAMOUS). Eur Heart J Cardiovasc Pharmacother. (2022) 8(3):272–81. doi: 10.1093/ehjcvp/pvab025
151. Charrow J, Fraga C, Gu X, Ida H, Longo N, Lukina E, et al. Once- versus twice-daily dosing of eliglustat in adults with Gaucher disease type 1: the phase 3, randomized, double-blind EDGE trial. Mol Genet Metab. (2018) 123(3):347–56. doi: 10.1016/j.ymgme.2017.12.001
152. Marshall J, Ashe KM, Bangari D, McEachern K, Chuang WL, Pacheco J, et al. Substrate reduction augments the efficacy of enzyme therapy in a mouse model of Fabry disease. PLoS ONE. (2010) 5(11):e15033. doi: 10.1371/journal.pone.0015033
153. Ashe KM, Budman E, Bangari DS, Siegel CS, Nietupski JB, Wang B, et al. Efficacy of enzyme and substrate reduction therapy with a novel antagonist of glucosylceramide synthase for Fabry disease. Mol Med. (2015) 21(1):389–99. doi: 10.2119/molmed.2015.00088
154. Guérard N, Oder D, Nordbeck P, Zwingelstein C, Morand O, Welford RWD, et al. Lucerastat, an iminosugar for substrate reduction therapy: tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry disease on enzyme replacement. Clin Pharmacol Ther. (2018) 103(4):703–11. doi: 10.1002/cpt.790
155. DeRosa F, Smith L, Shen Y, Huang Y, Pan J, Xie H, et al. Improved efficacy in a Fabry disease model using a systemic mRNA liver depot system as compared to enzyme replacement therapy. Mol Ther. (2019) 27(4):878–89. doi: 10.1016/j.ymthe.2019.03.001
156. Zhu X, Yin L, Theisen M, Zhuo J, Siddiqui S, Levy B, et al. Systemic mRNA therapy for the treatment of Fabry disease: preclinical studies in wild-type mice, Fabry mouse model, and wild-type non-human primates. Am J Hum Genet. (2019) 104(4):625–37. doi: 10.1016/j.ajhg.2019.02.003
157. Domm JM, Wootton SK, Medin JA, West ML. Gene therapy for Fabry disease: progress, challenges, and outlooks on gene-editing. Mol Genet Metab. (2021) 134(1–2):117–31. doi: 10.1016/j.ymgme.2021.07.006
158. Medin JA, Khan A, Huang J, Barber D, Anthony Rupar C, Auray-Blais C, et al. FACTs Fabry gene therapy clinical trial: two-year data. Mol Genet Metab. (2019) 126(2):S99. doi: 10.1016/j.ymgme.2018.12.248
159. Khan A, Barber DL, Huang J, Rupar CA, Rip JW, Auray-Blais C, et al. Lentivirus-mediated gene therapy for Fabry disease. Nat Commun. (2021) 12(1):1178. doi: 10.1038/s41467-021-21371-5
160. Hughes DA, Evans S, Milligan A, Richfield L, Mehta A. Chapter 35: a multidisciplinary approach to the care of patients with Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry disease: Prespectives from 5 years of FOS. Oxford: Oxford Pharmagenesis (2006).
161. Paim-Marques L, de Oliveira RJ, Appenzeller S. Multidisciplinary management of Fabry disease: current perspectives. JMDH. (2022) 15:485–95. doi: 10.2147/JMDH.S290580
162. Kleinert J, Dehout F, Schwarting A, Delorenzo A, Ricci R, Kampmann C, et al. Prevalence of uncontrolled hypertension in patients with Fabry disease. Am J Hypertens. (2006) 19(8):782–7. doi: 10.1016/j.amjhyper.2006.01.011
163. Jain G, Warnock DG. Blood pressure, proteinuria and nephropathy in Fabry disease. Nephron Clin Pract. (2010) 118(1):c43–8. doi: 10.1159/000320903
164. Yim J, Yau O, Yeung DF, Tsang TSM. Fabry cardiomyopathy: current practice and future directions. Cells. (2021) 10(6):1532. doi: 10.3390/cells10061532
165. Ortiz A, Oliveira JP, Waldek S, Warnock DG, Cianciaruso B, Wanner C, et al. Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant. (2008) 23(5):1600–7. doi: 10.1093/ndt/gfm848
166. Linhart A, Germain DP, Olivotto I, Akhtar MM, Anastasakis A, Hughes D, et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur J Heart Fail. (2020) 22(7):1076–96. doi: 10.1002/ejhf.1960
167. Fine NM, Wang Y, Khan A. Acute decompensated heart failure after initiation of amiodarone in a patient with Anderson-Fabry disease. Can J Cardiol. (2019) 35(1):104.e5–e7. doi: 10.1016/j.cjca.2018.10.004
168. Germain DP, Altarescu G, Barriales-Villa R, Mignani R, Pawlaczyk K, Pieruzzi F, et al. An expert consensus on practical clinical recommendations and guidance for patients with classic Fabry disease. Mol Genet Metab. (2022) 137(1–2):49–61. doi: 10.1016/j.ymgme.2022.07.010
169. O’Mahony C, Coats C, Cardona M, Garcia A, Calcagnino M, Murphy E, et al. Incidence and predictors of anti-bradycardia pacing in patients with Anderson-Fabry disease. Europace. (2011) 13(12):1781–8. doi: 10.1093/europace/eur267
170. Sené T, Lidove O, Sebbah J, Darondel JM, Picard H, Aaron L, et al. Cardiac device implantation in Fabry disease: a retrospective monocentric study. Medicine (Baltimore). (2016) 95(40):e4996. doi: 10.1097/MD.0000000000004996
171. Azevedo O, Cordeiro F, Gago MF, Miltenberger-Miltenyi G, Ferreira C, Sousa N, et al. Fabry disease and the heart: a comprehensive review. IJMS. (2021) 22(9):4434. doi: 10.3390/ijms22094434
172. Shah JS, Hughes DA, Sachdev B, Tome M, Ward D, Lee P, et al. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol. (2005) 96(6):842–6. doi: 10.1016/j.amjcard.2005.05.033
173. Liu HC, Perrin A, Hsu TR, Yang CF, Lin HY, Yu WC, et al. Age at first cardiac symptoms in fabry disease: association with a Chinese hotspot fabry mutation (IVS4+919G>A), classical fabry mutations, and sex in a Taiwanese population from the fabry outcome survey (FOS). In: Zschocke J, Baumgartner M, Morava E, Patterson M, Rahman S, Peters V, editors. JIMD Reports, Vol 22. Berlin, Heidelberg: Springer Berlin Heidelberg (2015). p. 107–13. (JIMD Reports; vol. 22). doi: 10.1007/8904_2015_418
174. Deegan PB. Natural history of Fabry disease in females in the Fabry outcome survey. J Med Genet. (2005) 43(4):347–52. doi: 10.1136/jmg.2005.036327
175. Ramaswami U, Whybra C, Parini R, Pintos-Morell G, Mehta A, Sunder-Plassmann G, et al. Clinical manifestations of Fabry disease in children: data from the Fabry outcome survey. Acta Paediatr. (2006) 95(1):86–92. doi: 10.1080/08035250500275022
176. Beck M. Demographics of FOS—the fabry outcome survey. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry disease: Perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis. (2006). Available from: http://www.ncbi.nlm.nih.gov/books/NBK11581/ (cited January 11, 2023).
177. Bouwman MG, Rombach SM, Schenk E, Sweeb A, Wijburg FA, Hollak CEM, et al. Prevalence of symptoms in female Fabry disease patients: a case-control survey. J Inherit Metab Dis. (2012) 35(5):891–8. doi: 10.1007/s10545-011-9447-9
178. Hoffmann B, Schwarz M, Mehta A, Keshav S. Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. (2007) 5(12):1447–53. doi: 10.1016/j.cgh.2007.08.012
179. Martins AM, Cabrera G, Molt F, Suárez-Obando F, Valdés RA, Varas C, et al. The clinical profiles of female patients with Fabry disease in Latin America: a Fabry registry analysis of natural history data from 169 patients based on enzyme replacement therapy status. JIMD Rep. (2019) 49(1):107–17. doi: 10.1002/jmd2.12071
180. Hopkin RJ, Bissler J, Banikazemi M, Clarke L, Eng CM, Germain DP, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry registry. Pediatr Res. (2008) 64(5):550–5. doi: 10.1203/PDR.0b013e318183f132
181. Gupta S, Ries M, Kotsopoulos S, Schiffmann R. The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. Medicine (Baltimore). (2005) 84(5):261–8. doi: 10.1097/01.md.0000178976.62537.6b
182. Hopkin RJ, Feldt-Rasmussen U, Germain DP, Jovanovic A, Martins AM, Nicholls K, et al. Improvement of gastrointestinal symptoms in a significant proportion of male patients with classic Fabry disease treated with agalsidase beta: a Fabry registry analysis stratified by phenotype. Mol Genet Metab Rep. (2020) 25:100670. doi: 10.1016/j.ymgmr.2020.100670
183. Mehta A, Ricci R, Widmer U, Dehout F, de Lorenzo A G, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry outcome survey. Eur J Clin Invest. (2004) 34(3):236–42. doi: 10.1111/j.1365-2362.2004.01309.x
184. Pinderski LJ, Strotmann J. Congestive heart failure in Fabry cardiomyopathy: natural history experience in an international cohort of 1,448 patients. J Heart Lung Transplant. (2006) 25(2):S70. doi: 10.1016/j.healun.2005.11.080
185. Patel MR, Cecchi F, Cizmarik M, Kantola I, Linhart A, Nicholls K, et al. Cardiovascular events in patients with Fabry disease. J Am Coll Cardiol. (2011) 57(9):1093–9. doi: 10.1016/j.jacc.2010.11.018
186. Rob D, Marek J, Dostalova G, Linhart A. Heart failure in Fabry disease revisited: application of current heart failure guidelines and recommendations. ESC Heart Fail. (2022) 9(6):4043–52. doi: 10.1002/ehf2.14091
187. Lavalle L, Thomas AS, Beaton B, Ebrahim H, Reed M, Ramaswami U, et al. Phenotype and biochemical heterogeneity in late onset Fabry disease defined by N215S mutation. PLoS ONE. (2018) 13(4):e0193550. doi: 10.1371/journal.pone.0193550
188. Ortiz A, Cianciaruso B, Cizmarik M, Germain DP, Mignani R, Oliveira JP, et al. End-stage renal disease in patients with Fabry disease: natural history data from the Fabry registry. Nephrol Dial Transplant. (2010) 25(3):769–75. doi: 10.1093/ndt/gfp554
189. Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, et al. Natural history of Fabry renal disease: influence of α-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). (2002) 81(2):122–38. doi: 10.1097/00005792-200203000-00003
190. Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry registry. Stroke. (2009) 40(3):788–94. doi: 10.1161/STROKEAHA.108.526293
191. Vedder AC, Linthorst GE, van Breemen MJ, Groener JEM, Bemelman FJ, Strijland A, et al. The Dutch Fabry cohort: diversity of clinical manifestations and Gb3 levels. J Inherit Metab Dis. (2007) 30(1):68–78. doi: 10.1007/s10545-006-0484-8
192. Vitale G, Ditaranto R, Graziani F, Tanini I, Camporeale A, Lillo R, et al. Standard ECG for differential diagnosis between Anderson-Fabry disease and hypertrophic cardiomyopathy. Heart. (2022) 108(1):54–60. doi: 10.1136/heartjnl-2020-318271
193. Małek LA, Chojnowska L, Spiewak M, Kłopotowski M, Miśko J, Petryka J, et al. Cardiac magnetic resonance imaging in patients with Fabry’s disease. Kardiol Pol. (2010) 68(8):929–34.
194. Thompson RB, Chow K, Khan A, Chan A, Shanks M, Paterson I, et al. T 1 mapping with cardiovascular MRI is highly sensitive for Fabry disease independent of hypertrophy and sex. Circ Cardiovasc Imaging. (2013) 6(5):637–45. doi: 10.1161/CIRCIMAGING.113.000482
195. Roller FC, Fuest S, Meyer M, Harth S, Gündüz D, Bauer P, et al. Assessment of cardiac involvement in Fabry disease (FD) with native T1 mapping. Fortschr Röntgenstr. (2019) 191(10):932–9. doi: 10.1055/a-0836-2723
196. Sado DM, White SK, Piechnik SK, Banypersad SM, Treibel T, Captur G, et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging. (2013) 6(3):392–8. doi: 10.1161/CIRCIMAGING.112.000070
197. Sado DM, Flett AS, Banypersad SM, White SK, Maestrini V, Quarta G, et al. Cardiovascular magnetic resonance measurement of myocardial extracellular volume in health and disease. Heart. (2012) 98(19):1436–41. doi: 10.1136/heartjnl-2012-302346
198. Cheng Z, Zhu K, Tian Z, Zhao D, Cui Q, Fang Q. The findings of electrocardiography in patients with cardiac amyloidosis: the electrocardiography of cardiac amyloidosis. Ann Noninvasive Electrocardiol. (2013) 18(2):157–62. doi: 10.1111/anec.12018
199. Marek J, Palecek T, Magne J, Lavergne D, Boulogne C, Fadel BM, et al. Comparison of echocardiographic parameters in Fabry cardiomyopathy and light-chain cardiac amyloidosis. Echocardiography. (2018) 35(11):1755–63. doi: 10.1111/echo.14144
200. Pagourelias ED, Mirea O, Duchenne J, Van Cleemput J, Delforge M, Bogaert J, et al. Echo parameters for differential diagnosis in cardiac amyloidosis: a head-to-head comparison of deformation and nondeformation parameters. Circ Cardiovasc Imaging. (2017) 10(3):e005588. doi: 10.1161/CIRCIMAGING.116.005588
201. Baccouche H, Maunz M, Beck T, Gaa E, Banzhaf M, Knayer U, et al. Differentiating cardiac amyloidosis and hypertrophic cardiomyopathy by use of three-dimensional speckle tracking echocardiography: 3D speckle tracking in cardiac amyloidosis and hypertrophic cardiomyopathy. Echocardiography. (2012) 29(6):668–77. doi: 10.1111/j.1540-8175.2012.01680.x
202. Hoigné P, Attenhofer Jost CH, Duru F, Oechslin EN, Seifert B, Widmer U, et al. Simple criteria for differentiation of Fabry disease from amyloid heart disease and other causes of left ventricular hypertrophy. Int J Cardiol. (2006) 111(3):413–22. doi: 10.1016/j.ijcard.2005.08.023
203. Eriksson P, Backman C, Eriksson A, Eriksson S, Karp K, Olofsson BO. Differentiation of cardiac amyloidosis and hypertrophic cardiomyopathy. A comparison of familial amyloidosis with polyneuropathy and hypertrophic cardiomyopathy by electrocardiography and echocardiography. Acta Med Scand. (1987) 221(1):39–46. doi: 10.1111/j.0954-6820.1987.tb01243.x
204. Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging. (2014) 7(2):157–65. doi: 10.1016/j.jcmg.2013.10.008
205. Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. (2005) 111(2):186–93. doi: 10.1161/01.CIR.0000152819.97857.9D
206. Finocchiaro G, Sheikh N, Biagini E, Papadakis M, Maurizi N, Sinagra G, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. (2020) 17(1):142–51. doi: 10.1016/j.hrthm.2019.07.019
207. Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol. (1995) 26(7):1699–708. doi: 10.1016/0735-1097(95)00390-8
208. Doi YL, Deanfield JE, McKenna WJ, Dargie HJ, Oakley CM, Goodwin JF. Echocardiographic differentiation of hypertensive heart disease and hypertrophic cardiomyopathy. Heart. (1980) 44(4):395–400. doi: 10.1136/hrt.44.4.395
209. de la Rosa A, Shah M, Shiota T, Siegel R, Rader F. Comparing echocardiographic characteristics in genotype positive–phenotype positive hypertrophic cardiomyopathy and hypertensive left ventricular hypertrophy. Eur Heart J Cardiovasc Imaging. (2022) 23(3):340–8. doi: 10.1093/ehjci/jeab217
210. Mirza SJ, Radaideh GA. Pattern of left ventricular hypertrophy seen on transthoracic echo in patients with hypertensive cardiomyopathy when compared with idiopathic hypertrophic cardiomyopathy. J Pak Med Assoc. (2013) 63(1):16–9.23865125
211. Teraoka K, Hirano M, Ookubo H, Sasaki K, Katsuyama H, Amino M, et al. Delayed contrast enhancement of MRI in hypertrophic cardiomyopathy. Magn Reson Imaging. (2004) 22(2):155–61. doi: 10.1016/j.mri.2003.08.009
212. Choudhury L, Mahrholdt H, Wagner A, Choi KM, Elliott MD, Klocke FJ, et al. Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. (2002) 40(12):2156–64. doi: 10.1016/S0735-1097(02)02602-5
213. Hinojar R, Varma N, Child N, Goodman B, Jabbour A, Yu CY, et al. T1 mapping in discrimination of hypertrophic phenotypes: hypertensive heart disease and hypertrophic cardiomyopathy: findings from the international T1 multicenter cardiovascular magnetic resonance study. Circ Cardiovasc Imaging. (2015) 8(12):e003285. doi: 10.1161/CIRCIMAGING.115.003285
214. Antikainen RL, Grodzicki T, Palmer AJ, Beevers DG, Webster J, Bulpitt CJ, et al. Left ventricular hypertrophy determined by sokolow–Lyon criteria: a different predictor in women than in men? J Hum Hypertens. (2006) 20(6):451–9. doi: 10.1038/sj.jhh.1002006
215. Rodrigues JCL, Rohan S, Ghosh Dastidar A, Harries I, Lawton CB, Ratcliffe LE, et al. Hypertensive heart disease versus hypertrophic cardiomyopathy: multi-parametric cardiovascular magnetic resonance discriminators when end-diastolic wall thickness ≥15 mm. Eur Radiol. (2017) 27(3):1125–35. doi: 10.1007/s00330-016-4468-2
216. Rudolph A, Abdel-Aty H, Bohl S, Boyé P, Zagrosek A, Dietz R, et al. Noninvasive detection of fibrosis applying contrast-enhanced cardiac magnetic resonance in different forms of left ventricular hypertrophy. J Am Coll Cardiol. (2009) 53(3):284–91. doi: 10.1016/j.jacc.2008.08.064
Keywords: anderson-Fabry disease, heart failure, cardiac imaging, management, enzyme replacement therapy
Citation: Averbuch T, White JA and Fine NM (2023) Anderson-Fabry disease cardiomyopathy: an update on epidemiology, diagnostic approach, management and monitoring strategies. Front. Cardiovasc. Med. 10:1152568. doi: 10.3389/fcvm.2023.1152568
Received: 27 January 2023; Accepted: 9 May 2023;
Published: 2 June 2023.
Edited by:
Liza Thomas, University of New South Wales, AustraliaReviewed by:
Gionata Spagnoletti, Bambino Gesù Children’s Hospital (IRCCS), Italy© 2023 Averbuch, White and Fine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nowell M. Fine bm5maW5lQHVjYWxnYXJ5LmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.