 Steven J. Humphreys
Steven J. Humphreys Claire S. Whyte
Claire S. Whyte Nicola J. Mutch
Nicola J. Mutch- Aberdeen Cardiovascular and Diabetes Centre, Institute of Medical Sciences, School of Medicine, Medical Sciences and Nutrition, University of Aberdeen, Aberdeen, United Kingdom
The superfamily of serine protease inhibitors (SERPINs) are a class of inhibitors that utilise a dynamic conformational change to trap and inhibit their target enzymes. Their powerful nature lends itself well to regulation of complex physiological enzymatic cascades, such as the haemostatic, inflammatory and complement pathways. The SERPINs α2-antiplasmin, plasminogen-activator inhibitor-1, plasminogen-activator inhibitor-2, protease nexin-1, and C1-inhibitor play crucial inhibitory roles in regulation of the fibrinolytic system and inflammation. Elevated levels of these SERPINs are associated with increased risk of thrombotic complications, obesity, type 2 diabetes, and hypertension. Conversely, deficiencies of these SERPINs have been linked to hyperfibrinolysis with bleeding and angioedema. In recent years SERPINs have been implicated in the modulation of the immune response and various thromboinflammatory conditions, such as sepsis and COVID-19. Here, we highlight the current understanding of the physiological role of SERPINs in haemostasis and inflammatory disease progression, with emphasis on the fibrinolytic pathway, and how this becomes dysregulated during disease. Finally, we consider the role of these SERPINs as potential biomarkers of disease progression and therapeutic targets for thromboinflammatory diseases.
Introduction
Serine proteases are powerful proteolytic enzymes that function in crucial pathways such as coagulation, fibrinolysis, inflammation, and the complement system (1–3). The general mechanism of action of these proteases is cleavage of peptide bonds, thereby converting proteins from inactive zymogens to active enzymes. The prolific nature of these serine proteases requires exquisite regulation which is the task of the superfamily of serine protease inhibitors (SERPINs). In most SERPINs the amino acid sequence in the reactive center loop (RCL) mimics that of the enzymes target protease and upon cleavage the serine protease is dynamically “trapped” in an irreversible 1:1 complex (4–6), leading to the classification of SERPINs as suicide inhibitors. The detailed structure and mechanism of SERPINs has recently been elegantly reviewed and will not be described in depth herein (7). SERPINS with a role in haemostasis include antithrombin, heparin cofactor II, protein-Z-dependent protease inhibitor, and α1-antitrypsin. This review focuses on SERPINs with a role in fibrinolysis (8, 9). Although there is some evidence that α1-antitrypsin participates in fibrinolysis its primary role is regulation of neutrophil-derived proteases (10–14). SERPINs like α2-antiplasmin (α2AP), plasminogen-activator inhibitor-1 and -2 (PAI-1 and -2), protease nexin-1 (PN-1), and C1-inhibitor (C1-INH) play crucial functions in the fibrinolytic system, by inhibiting plasmin, tissue-type plasminogen activator (tPA), and urokinase-type plasminogen activator (uPA) (Figure 1) (9, 15). Elevated levels of several SERPINs have been associated with a plethora of disease states including type 2 diabetes, obesity, metabolic syndrome, and hypertension. They have been identified as prognostic markers in several cancers including, renal, breast, lung, head and neck, colorectal, and ovarian cancers (16–19) and have been implicated in cancer progression and metastasis. These areas fall out with the scope of this review and have been described in detail elsewhere (20, 21). In this review we will focus on the biological function of the SERPINs that modulate fibrinolysis, how this is perturbed during thromboinflammatory diseases, as well as highlight existing and novel aspects to exploit for novel therapeutics.
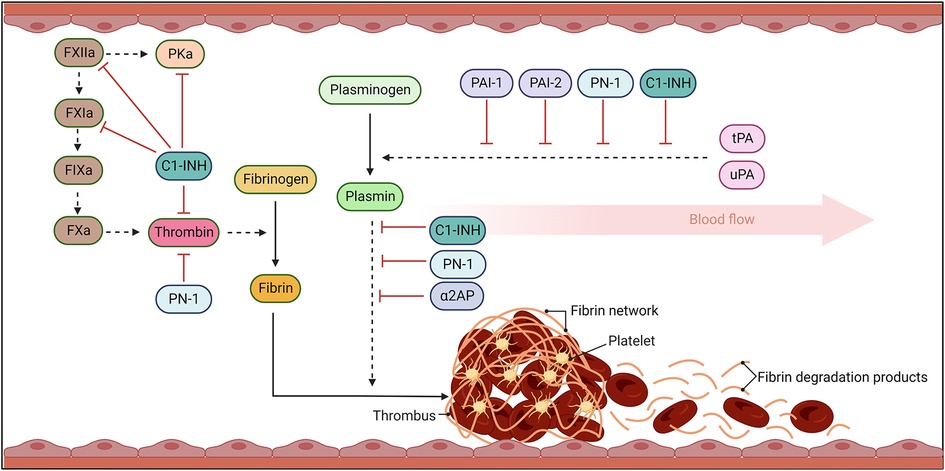
Figure 1. The fibrinolytic response. Coagulation results in thrombin cleavage of fibrinogen into fibrin. Fibrin threads become crosslinked, trapping platelets and blood cells, stabilising the clot. Once bleeding has been controlled and damage to vessel repaired, the fibrinolytic pathway acts to dissolve the fibrin network. Fibrinolysis occurs when plasminogen activators, urokinase- (uPA) and tissue-type- (tPA), cleave plasminogen to plasmin. Plasmin then degrades fibrin into fibrin degradation products (FDP) which can be efficiently cleared from the circulation. Serine protease inhibitors (SERPINs) act to inhibit the activity of these serine proteases to avoid uncontrolled activity that can lead to bleeding complications and dysregulated activity. Solid arrows signal catalytic conversion of proteins and enzymes from inactive to active states, while dashed arrows signal proteolytic activity. Blunt red arrows denote the specific inhibition of enzymes by different SERPINs.
α2-Antiplasmin—“the alpha SERPIN”
α2AP is the principal inhibitor of plasmin in vivo (22). It is an unusual SERPIN as its extensive C-terminal lysine-rich tail initially docks to the kringles of plasmin forming a non-covalent complex (23). Following plasmin cleavage of the RCL a stable covalent 1:1 plasmin-antiplasmin (PAP) complex is formed. The main plasma pool of α2AP is secreted by hepatocytes (Figure 2) (24) and circulates at 70 µg/ml, over 1,000-fold higher than PAI-1 (25). Despite being the dominant fibrinolytic SERPIN in plasma its level is surprisingly still lower than the zymogen concentration of its target enzyme, plasminogen (200 µg/ml) (26). However, the inhibitor is covalently cross-linked to fibrin through the action of the transglutaminase, activated factor XIIIa, during thrombus formation thereby localising it at the site of action and enhancing its efficacy (27, 28). Our laboratory has previously shown that cross-linking of α2AP into a thrombus is dependent on flow or shear stress (29) and others have demonstrated that compaction of clots is sufficient to promote the crosslinking reaction (30). Interestingly, despite its plethora of other substrates, the antifibrinolytic action of factor XIIIa is exclusively accounted for by cross-linking of α2AP (31) illustrating the “alpha” role of this SERPIN in modulating fibrinolysis. α2AP exists in several forms within the circulation (32). The secreted form, Met-α2AP, is cleaved by α2AP cleaving enzyme (APCE) to form Asn-α2AP (33) which cross-links thirteen times faster to fibrin, due to exposure of glutamine 2 in this truncated form (32). The C-terminus is post-translationally modified by an as yet unidentified enzyme in the circulation with downstream impact on the interaction with plasmin(ogen) (32).
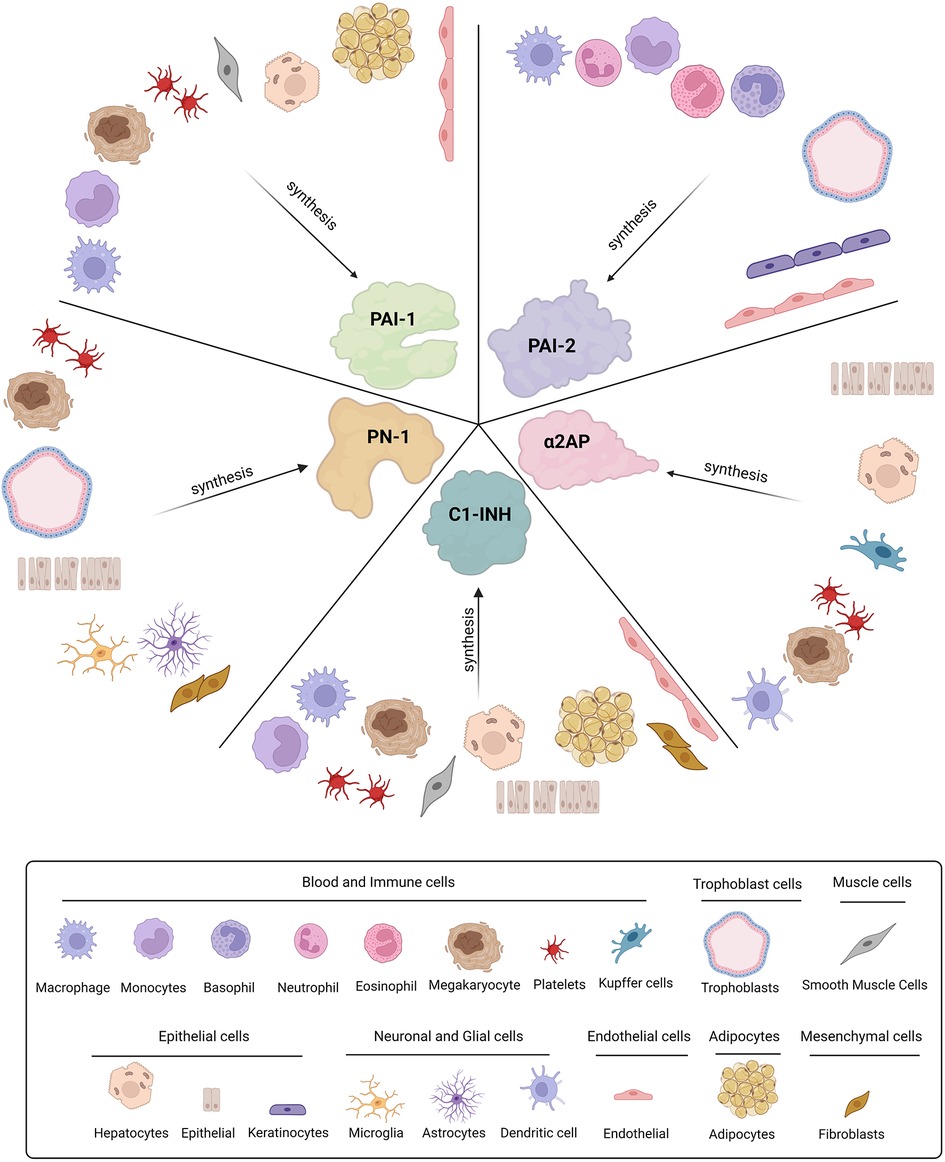
Figure 2. Cellular origins of SERPINs involved in fibrinolysis. Plasminogen activator-1 and -2 (PAI-1 and PAI-2), Protease nexin-1 (PN-1), alpha2-antiplasmin (α2AP), and C1-inhibitor (C1-INH) are all synthesised by various cell types throughout the body.
Dysregulation of α2AP disrupts the fibrinolytic balance leading to a risk of thrombosis or bleeding (34, 35). Congenital α2AP deficiencies, in which the mutation perturbs protein levels and/or function are associated with excessive or delayed bleeding following trauma and/or spontaneous rebleeding (36, 37). An acquired deficiency in α2AP can occur due to infusion of thrombolytic agents and is commonly associated with severe liver disease and acute leukaemia (36, 37). In contrast, elevated levels of α2AP are linked with diseases such as atherosclerosis thereby provoking the risk of ischemic stroke (IS) and myocardial infarction (MI) (38, 39). Experimental models of IS indicate that elevated levels of α2AP lead to a reduction in thrombus dissolution and enhanced necrosis, while neutralisation of this SERPIN during thrombolytic therapy significantly improves outcomes (40). Increased levels of α2AP are directly linked to adverse outcomes in terms of ischemic brain injury, swelling, microvascular thrombosis and mortality following cerebral thromboembolism (41). Several avenues have been pursued to neutralise the activity of α2AP, including microplasmin and plasmin infusions, neutralising antibodies, and synthetic peptides (42–44). In a mouse model of IS infusion of plasmin or immunoneutralization was shown to temporarily lower systemic α2AP levels for around 24 h (43).
Significantly elevated levels of circulating α2AP have been identified as a key component in the pathophysiology of pulmonary arterial hypertension (PAH), contributing to poorer outcomes and higher mortality (45, 46). Reduced interactions between prekallikrein, factor XI (FXI) or α2AP, with small high-density lipoprotein subclasses correlate with higher mortality in PAH patients (47). A significant relationship has also been observed between α2AP and obstructive sleep apnoea (OSA), cardiomyopathy, congestive heart failure, and atrial fibrillation (48). Interestingly, increased levels of both α2AP and PAI-1 correlate with augmented prothrombotic activity in patients with OSA (49). These data suggest that inhibition of these SERPINs may be potential therapeutic option to down-regulate thrombotic complications in OSA. Interestingly, a significant decrease in α2AP activity has been observed in arterial hypertension, reflecting a shift to a hyperfibrinolytic state (50). The implication of these findings is not year clear and requires further exploration.
Therapeutic targeting of α2AP is an attractive strategy which could mitigate the bleeding risk associated with conventional anticoagulant and thrombolytic therapy. A phase 1 clinical trial in patients with acute lower extremity arterial or bypass occlusion indicated that catheter-delivered plasmin at high doses significantly reduced systemic α2AP by 39% resulting in >50% clearance of the thrombus in 79% of patients (51). This interesting study indicates that consumption of endogenous α2AP is a safe and effective way of promoting thrombolysis in vivo. A novel monoclonal antibody that inactivates α2AP, TS23 (42, 52), has been used in phase 1 clinical trials in healthy individuals, but outcomes are not yet available. A randomised phase 2 clinical trial was started earlier this year to evaluate the efficacy of TS23 in treating acute pulmonary embolism (53), with results expected in 2024. These developments are promising and signify considerable interest in targeting this SERPIN for a variety of thrombotic and cardiovascular conditions and one would hope that new drugs and compounds directed toward α2AP may be on the horizon in the not-too-distant future.
Plasminogen activator inhibitor-1—“the premier PAI”
PAI-1 is a 45 kDa glycoprotein and the “premier” inhibitor of plasminogen activators (PAs) (54). PAI-1 is highly expressed in many tissues, including placenta, gallbladder, liver, lung, and kidney as well as various cell-types such as platelets, syncytiotrophoblasts, adipocytes, smooth muscle cells (SMCs), endothelial cells, and fibroblasts (Figure 2) (55–59). Levels of PAI-1 in plasma are relativity low (20 ng/ml) (60) with the main circulating pool found within the α-granules of platelets (61). PAI-1 is expressed by megakaryocytes, the precursor cells of platelets (62, 63) and is packaged into α-granules during biogenesis. However, mRNA can be found within platelets where the inhibitor is synthesised to a limited degree in its active form (62). Unlike other SERPINs, PAI-1 can spontaneously transition from its unstable active form (half-life ∼1 h) (64), to a more thermodynamically stable latent form, extending the half-life to ∼2–4 h (65). Plasma PAI-1 levels are known to oscillate in a circadian rhythm, peaking in the morning (66, 67) thereby reducing fibrinolytic potential (68). This diurnal variation in PAI-1 has been postulated to explain the morning peak in adverse cardiovascular events (69, 70). Circulating levels of PAI-1 are influenced by the 4G/5G genetic polymorphism in the promoter region (71). Elevated levels of PAI-1 in the morning are most apparent in homozygotes for 4G and intermediate for heterozygotes (72, 73).
Deficiency in PAI-1 is associated with hyperfibrinolysis, which can be either congenital or acquired. A complete PAI-1 deficiency is characterised by a mild-moderate bleeding diathesis most commonly associated with trauma or surgery (74). Despite equal prevalence in males and females, PAI-1 deficiency is usually diagnosed earlier in females due to menorrhagia and postpartum haemorrhage (75). Acquired deficiency in PAI-1 is a common consequence of cirrhosis of the liver and observed clinically as increased levels of circulating D-dimers and fibrin degradation products (FDPs) (76–78). Conversely, elevated PAI-1 incites a hypofibrinolytic state, which has been linked to increased risk of venous and arterial thrombosis (79, 80).
Elevated levels of PAI-1 have been correlated with IS (81–83). Similarly increased levels of inflammatory markers and fibrinolytic inhibitors, including PAI-1, were recorded in intracranial large artery atherosclerosis (ILA), a major cause of IS (81, 84). Importantly, PAI-1 and C-reactive protein could predict ILA progression independent of other biomarkers assessed; these data highlight the unique relationship between pro-inflammatory and hypofibrinolytic states (81). Elevated plasma levels of TNF-α, PAI-1, and tPA, markers of immuno-inflammatory activation and endothelial dysfunction, correlate with stroke diagnosis (85). The “sterile” thromboinflammatory state observed in stroke patients is observed in other conditions such as venous thrombosis, diabetes and autoimmune diseases (86). A retrospective observational clinical trial is currently investigating differences in factor VIII, tPA, and PAI-1 as biomarkers for the occurrence of intracranial haemorrhage in patients with acute IS, pre- and post-treatment with alteplase (87). A population-based case-control study demonstrated that elevated levels of PAI-1 are predictive of venous thrombosis (88). Interestingly, a recent study investigating deep vein thrombosis (DVT) in patients following total hip arthroplasty, found that PAI-1 expression was a postoperative biomarker (89). Similarly, PAI-1, fibrinogen, and D-dimers were predictive of postoperative DVT following surgery for lower limb trauma (90). Collectively these studies highlight the prognostic power of PAI-1 in a variety of thromboinflammatory conditions, however, this would require further work to establish and standardise normal ranges.
Attenuated fibrinolysis is a significant factor in obesity-associated thrombosis, attributed to an increase in PAI-1 levels (91–93). Elevated gene expression and plasma tPA are also observed in obesity which is suggested to be a compensatory mechanism to overcome PAI-1, despite this, hypofibrinolysis persists (94, 95). Adipocytes are proposed as the principal source of PAI-1 with enhanced synthesis primarily driven by the proinflammatory cytokines TNF-α and TGF-β1 (93, 96, 97). Interestingly, in healthy individuals there was no arterio-venous differences in PAI-1 antigen or activity across an adipose tissue bed (98). An interesting study on glycosylation of PAI-1 suggested that in lean individuals the primary source of PAI-1 is platelets while in obese subjects the glycosylation pattern of plasma PAI-1 mimicked that of adipose tissue, suggesting these cells contribute to circulating levels (99). Recently hepatocytes have been described as an alternative source of increased PAI-1 and tPA in obesity due to alterations in the hepatic PAI-1/tPA gene regulatory pathways (95). Further research into the various cellular sources of plasma PAI-1 could be fruitful for future targeting of this SERPIN in various cardiometabolic diseases.
The most frequent cause of MI is rupture of atherosclerotic plaque, leading to occlusive thrombus formation. Many cells that express PAI-1, such as platelets, macrophages, and SMCs reside within the plaque milieu (100–102). Atherosclerotic progression is associated with platelet activation and PAI-1 secretion thereby promoting thrombus resistance to lysis (103–106). Recurrence of MI has also been linked to increased plasma levels of PAI-1 (107, 108). It was suggested that this causality may arise due to the multifaceted role of PAI-1 in aging, fibrinolysis, and endothelial injury (109, 110), with elevated levels provoking a prothrombotic and profibrotic state, thereby impairing clot breakdown and driving endothelial dysfunction (111). The onset of acute MI follows diurnal oscillations, with most cases occurring in the morning between 6:00 am and noon (112). Misalignment between the internal and external clock, for instance during daylight saving time, have been associated with increased risk of MI (113). A recent study on acute MI determined a correlation between tPA and PAI-1 levels with highest levels recorded in the morning (114), in agreement with earlier studies (66–68, 70). However, treatment with the anti-platelet drug clopidogrel gave rise to elevated levels of PAI-1 in those acute MI patients resistant to clopidogrel, but not clopidogrel-sensitive patients (114). These interesting data suggest that increased evening PAI-1 may relate to platelet activation and underscore the diurnal changes that occur both naturally in this SERPIN and in response to drugs. Several clinical trials are currently investigating PAI-1 in different thromboembolic conditions. Eplerenone, a selective aldosterone receptor antagonist, was shown to attenuate the morbidity and mortality of patients with acute MI (115), and is being analysed to reduce PAI-1 levels in aldosterone-induced MI in a randomised control study (116). A first-in-class orally available PAI-1 inhibitor for the treatment of fibrosis and fibroproliferative disorders is currently in phase 1 clinical trials (117). Metformin, a type-2 diabetes treatment, is currently being investigated in a phase 4 non-randomised trial for its use in PAI-1 deficient individuals to prevent/stabilise cardiac fibrosis (118).
During the COVID-19 pandemic thromboembolic complications were quickly recognised as a consequence of severe disease (119, 120). Reduced fibrinolytic capacity, observed as prolonged clot lysis, increased clot strength, and elevated platelet counts, were observed in 70% of COVID-19 patients (121). This has been attributed to variations in fibrinolytic factors such as PAI-1 (>2-fold), tPA (>1.6-fold), and thrombin activatable fibrinolysis inhibitor (TAFI) (>1.7-fold) in critically ill COVID-19 patients (122). Dysregulation of haemostasis in patients with severe COVID-19 is linked to an aberrant hyperinflammatory state triggered by the cytokine storm (123). We demonstrated elevated PAI-1 in a cohort of hospitalised COVID-19 patients that supressed plasmin generation and tPA-mediated lysis thereby driving the suboptimal fibrinolytic response (124). Interestingly, a direct association between PAI-1 and hypofibrinolysis was also observed during the SARS-CoV outbreak of 2002 (125). A double-blind randomised phase 2 clinical trial of a novel PAI-1 inhibitor (TM5614) to evaluate its efficacy in treating severe COVID-19 (126) has unfortunately been suspended due to challenges in patient recruitment and drug manufacturing issues. Similarly, a case-control prospective clinical trial to assess the predictive power of PAI-1 in the development of severe COVID-19 was initiated but has not been updated suggesting issues with recruitment (127).
These studies and observations indicate that PAI-1 is a powerful player in defining the physiological and pathophysiological response. A diverse portfolio of PAI-1 inhibitors, including antibodies, nanobodies, antibody fragments, and peptides, have been reported in the literature but we are yet to see a drug traverse the pipeline for clinical use (128–130).
Plasminogen activator inhibitor-2—“the incognito PAI”
PAI-2 is expressed in multiple cells, such as monocytes, granulocytes, trophoblasts, epithelial and endothelial cells (Figure 2) (131–133). Elevated PAI-2 is observed in pregnancy and is related to increased cellular expression by the trophoblastic epithelium of the placenta (134). PAI-2 lacks a signal peptide for release (135), and therefore its function is presumed to be intracellular, as reflected by the negligible levels in plasma. Nonetheless, this “incognito” SERPIN has the capacity to inhibit many extracellular proteases, despite reduced efficacy toward tPA and uPA compared to PAI-1 (136, 137). Studies related to PAI-2 in pathophysiological states are limited, however there are reports to suggest that PAI-2 can modulate venous thrombus (138), coronary artery disease (139, 140), and cerebral artery occlusion (83). Upregulation of PAI-2 has also been observed in patients with acute respiratory distress syndrome (ARDS) (141). Interestingly, PAI-2 expression is markedly increased after exposure to inflammatory stimuli such as TNF-α, suggesting a role for this SERPIN in inflammation (131, 132) as well as other processes such as apoptosis (142), infection (143) and cancer (144).
An instrumental study into the role of PAI-2 in fibrinolysis demonstrated that venous thrombi from PAI-2 deficient mice harboured 12-fold higher levels of active uPA (138). There was also pronounced improvement in thrombus resolution in both PAI-1 and PAI-2 deficient mice; this is likely linked to the overexpression of uPA and the observed infiltration of inflammatory cells, such as neutrophils and macrophages (138, 145, 146). Interestingly, enhanced monocyte-derived uPA activity (150-fold) has been associated with reductions in PAI-1 and PAI-2, 1.6- and 2.1-fold respectively, and more efficient dissolution of thrombi (146–148). In contrast, formation of thrombi was only impaired by PAI-1 deficiency (138), indicating a complex interplay of these two SERPINs in venous thrombus formation and resolution.
PAI-1 and PAI-2 have been investigated in IS, in a model of middle cerebral artery occlusion (MCAO) (83). They found that 24 h post-stroke, levels of PAI-2 and PAI-1 mRNA were significantly overexpressed, 19- and 237-fold, respectively. PAI-1 deficient mice had a 31% decrease in brain infarct volume 24 h post-MCAO, however this was not observed in PAI-2 deficient mice, suggesting this SERPIN has limited impact on secondary brain damage post-stroke (83). The role of PAI-1 in brain injury is well established but given the limited studies on PAI-2 the significance of this SERPIN in this setting remains unclear (109, 149, 150).
Clearly, we currently lack an understanding of the relevance of PAI-2 in both the intracellular and extracellular space. The distinct elevation of plasma PAI-2 in pregnancy and the relationship with inflammation demands further attention and underscores are limited knowledge. PAI-2 has been presumed to function intracellularly yet the limited studies available indicate that it can “escape” to the circulation which may be promoted under certain pathophysiological conditions and/or environmental settings.
Protease nexin-1—“the binary SERPIN”
PN-1 was originally identified in glial cells in the 1970s (151) and has subsequently been found in a variety of cells including epithelial, fibroblasts, glial cells, and trophoblasts (Figure 2) (152, 153). Within the SERPIN superfamily PN-1 is the phylogenetically closest relative to PAI-1, sharing both structural and functional homologies (154, 155). PN-1 levels are negligible in plasma but, like its phylogenetic cousin PAI-1, it is found at high concentrations within platelet α-granules (156). Activated platelets secrete PN-1 in response to agonist stimulation which attenuates plasmin generation and activity (156). PN-1 demonstrates “binary” function as it targets the primary serine proteases of the coagulation and fibrinolytic cascade, thrombin and plasmin, as well as other enzymes within these pathways including factor XI (FXI), tPA and uPA (156–158).
In the last two decades the focus on PN-1 has intensified with various links to haemophilia (159) and fibrosis (160) reported. Cardiac fibrosis is defined as an excessive accumulation of extracellular matrix (ECM) proteins (161) promoting myocardial stiffness and altered systolic function (162). Elevated PN-1 was recorded both in vivo and in vitro in mouse models of fibrosis (163), with mRNA expression increased 5.5- and 1.9-fold in the plasma and myocardium, respectively. This increase is hypothesised to be attributed to PN-1 inhibition of uPA in the extracellular space (164). Aspirin attenuates PN-1 levels in cardiac fibrosis due to repressed phosphorylation of Erk1/2-PN-1 and blockage of the MAPK-Erk1/2-PN-1 pathway in cardiac fibroblasts, suggesting utility as a drug target or marker of disease progression (163, 165). There are currently no drugs with antifibrotic activity approved for clinical use (166), although PAI-1 has been highlighted (117, 118). Given the close phylogenetic connection between these SERPINs further work into the role of PN-1 in this setting could be of value.
PN-1 inhibits thrombin generation and activity in vitro, more efficiently than other SERPINs, with an inhibitory constant (Ki) of 1.41 × 10−6 M−1s−1 (167) compared to Ki of 1.08 × 10−4 M−1s−1 for antithrombin (168). In PN-1 deficient mice thrombus formation was augmented, as characterised both in ex vivo collagen-induced flow models and in vivo FeCl3 induced injury (169). Neutralising antibodies to PN-1 enhance thrombin activity in mild to moderate haemophilia A and mild haemophilia B patients highlighting its potential as a treatment for haemophilia activity (159, 170). These studies reveal that the role of PN-1 in thrombin inhibition could be exploited as a future pharmacological intervention. Nonetheless, the capacity of this “binary SERPIN” to also inhibit plasmin and uPA in vivo must be teased out before development of therapeutic strategies.
C1-inhibtor—“the bountiful SERPIN”
C1-INH is the most abundant protease inhibitor in plasma circulating at a massive 250 µg/ml (171), perhaps reflecting the diversity and number of protease targets. It was conventionally considered an inhibitor of the complement pathway (C1r and C1s) and the contact system (FXIIa, FXIa, and kallikrein), however, it has also been shown to target enzymes in the fibrinolytic pathway, including plasmin, tPA, and uPA (15, 172). C1-INH is primarily produced by the liver, but is expressed in other cell types, such as monocytes/macrophages, endothelial cells, SMCs and fibroblasts (Figure 2) (173–176). Platelet α-granules also harbour C1-INH although this accounts for only 0.08% of the total circulating pool (177, 178).
A deficiency or impaired function of C1-INH is associated with angioedema, both hereditary (HAE) and acquired (179, 180). Acute attacks in HAE patients (C1-INH-HAE) are characterised by swelling of deep tissue and mucosa, as a consequence of local vascular leakage and an increase in bradykinin (181). Interestingly, the most common sites of swelling in these patients are in areas of high fibrinolytic activity, such as the lips, mouth, tongue, eyelids and genitalia (182, 183). A recent study investigating the risk of comorbidities in HAE found increased risk of hypertension, arterial and venous embolism, in line with C1-INH's multifunctional role in various pathways (184, 185). Antifibrinolytic drugs, such as tranexamic acid can reduce the severity of HAE attacks (186, 187), however the efficacy of this treatment is exceptionally variable (188, 189). There are many studies, both interventional and observational, investigating C1-INH deficiency, but the majority focus on the link with angioedema (190–193). Interestingly, angioedema is a common side-effect of thrombolytic therapies (194, 195) suggesting further linkage between these pathways, however, C1-INH function in the fibrinolytic pathway has largely been overlooked and understudied (171, 196). To add to the complexity while C1-INH directly inhibits both plasmin and tPA, it can itself be inactivated by plasmin cleavage (171). Additionally, there have now been several studies to indicate an increase in various prothrombotic plasma proteins, including factor XIIa, prothrombin, D-dimer and thrombin generation in C1-INH deficient patients (181, 197–199). Replacement therapies with human C1-INH reduce circulating levels of these markers thus lowering thrombotic risk (200, 201). C1-INH deficient mice also show increased activation of the coagulation pathway and venous thrombosis (202). Interestingly, in the general population low levels of C1-INH correlate with an increased risk of venous thrombosis (203).
C1-INH has been investigated in COVID-19 disease, partly due to its similarities in disease progression as HAE, including immune activation driving inflammation, endothelial dysfunction, and altered fibrinolysis (204, 205). Elevated C1-INH was identified in patients with severe, but not non-severe COVID-19 (206, 207). Current randomised clinical trials are studying C1-INH in relation to COVID-19 to reduce disease progression (208) and its use as a co-therapy with icatibant, a competitive antagonist to bradykinin, on the pulmonary manifestations of COVID-19 disease (209).
C1-INH is critical in the inhibition of multiple enzymes/factors in the contact, complement, and fibrinolytic systems (172). Research in this field is divided on whether there are thrombotic risks associated with C1-INH deficiency, accentuating an opportunity to bridge gaps in our knowledge. The “bountiful” array of protease targets of this SERPIN in the inflammatory, haemostatic and complement pathways has directed attention toward its value as a therapeutic and predictive biomarker, with ongoing clinical studies highlighting its potential in various thromboinflammatory conditions.
Concluding remarks
SERPINs play multi-functional roles targeting serine proteases in the complement, coagulation and fibrinolytic pathways, orchestrating a fine balance between promotion and inhibition of these pathways. In the last few decades, many studies have helped decipher the behaviour of α2AP, PAI-1, PAI-2, PN-1, and C1-INH in thrombotic and inflammatory diseases. Primarily these SERPINs have been suggested to have predictive powers, although their precise role in different disease settings has not been fully disentangled and additional studies would be necessary to standardise normal ranges. Nevertheless, elevated levels of these SERPINs are related to the incidence, severity, and prognosis of various thromboembolic diseases, including ischemic stroke, myocardial infarction, along with pathogenic infections, such as COVID-19 and sepsis, and inflammation, such as angioedema. Evidently, the power and regulatory capacity of PN-1 and PAI-2 are understudied thereby currently precluding their viability as therapeutics, but yet leave the door open to future research in the field. However, there is currently significant interest in targeting α2AP, PAI-1, and C1-INH in a variety of thromboembolic and inflammatory conditions. Understanding the complex interplay of SERPINs in regulating the fibrinolytic response is imperative to define their critical role in maintaining haemostasis and will provide clues as to the intertwined relationship between the fibrinolytic, inflammatory and complement systems. Despite the current state of knowledge and rational for various SERPINs as therapeutics, there are currently an exceptionally limited repertoire of drugs with regulatory approval to target these inhibitors. With the current interest in thromboinflammation it is a prime opportunity to propel these “super” SERPINs into the limelight to understand their biological function and utility as therapeutic targets.
Author contributions
SJH researched the data, wrote, and revised the article. CSW wrote and edited the article. NJM conceived, wrote, and edited the article. All authors contributed to the article and approved the submitted version.
Funding
SH, CW and NM are supported by an NC3Rs-British Heart Foundation Studentship (NC/W001810/1) and an Animal Free Research UK grant (170_Mutch_ABE_CT). CW and NM are supported by the British Heart Foundation (PG/20/17/35050).
Acknowledgments
Figures were created with BioRender.com and exported under a paid subscription.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Carrell RW, Pemberton PA, Boswell DR. The serpins: evolution and adaptation in a family of protease inhibitors. Cold Spring Harb Symp Quant Biol. (1987) 52:527–35. doi: 10.1101/SQB.1987.052.01.060
2. Huntington JA, Read RJ, Carrell RW. Structure of a serpin–protease complex shows inhibition by deformation. Nature. (2000) 407(6806):923–6. doi: 10.1038/35038119
3. Sim RB, Laich A. Serine proteases of the complement system. Biochem Soc Trans. (2000) 28(5):545–50. doi: 10.1042/bst0280545
4. Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, et al. An overview of the serpin superfamily. Genome Biol. (2006) 7(5):216. doi: 10.1186/gb-2006-7-5-216
5. Heit C, Jackson BC, McAndrews M, Wright MW, Thompson DC, Silverman GA, et al. Update of the human and mouse SERPINgene superfamily. Hum Genomics. (2013) 7(1):22. doi: 10.1186/1479-7364-7-22
6. Gooptu B, Lomas DA. Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu Rev Biochem. (2009) 78(1):147–76. doi: 10.1146/annurev.biochem.78.082107.133320
7. Sanrattana W, Maas C, de Maat S. SERPINs-from trap to treatment. Front Med. (2019) 6:25. doi: 10.3389/fmed.2019.00025
8. Rau JC, Beaulieu LM, Huntington JA, Church FC. Serpins in thrombosis, hemostasis and fibrinolysis. J Thromb Haemostasis. (2007) 5:102–15 doi: 10.1111/j.1538-7836.2007.02516.x
9. Yaron JR, Zhang L, Guo Q, Haydel SE, Lucas AR. Fibrinolytic serine proteases, therapeutic serpins and inflammation: fire dancers and firestorms. Front Cardiovasc Med. (2021) 8:648947. doi: 10.3389/fcvm.2021.648947
10. Basil N, Ekström M, Piitulainen E, Lindberg A, Rönmark E, Jehpsson L, et al. Severe alpha-1-antitrypsin deficiency increases the risk of venous thromboembolism. J Thromb Haemostasis. (2021) 19(6):1519–25. doi: 10.1111/jth.15302
11. Manderstedt E, Halldén C, Lind-Halldén C, Elf J, Svensson PJ, Engström G, et al. Thrombotic risk determined by rare and common SERPINA1 variants in a population-based cohort study. J Thromb Haemost. (2022) 20(6):1421–7. doi: 10.1111/jth.15696
12. Riis J, Nordestgaard BG, Afzal S. α1-Antitrypsin Z allele and risk of venous thromboembolism in the general population. J Thromb Haemostasis. (2022) 20(1):115–25. doi: 10.1111/jth.15556
13. Grover SP, Mackman N. Anticoagulant SERPINs: endogenous regulators of hemostasis and thrombosis. Front Cardiovasc Med. (2022) 9:878199. doi: 10.3389/fcvm.2022.878199
14. Janciauskiene S, Wrenger S, Immenschuh S, Olejnicka B, Greulich T, Welte T, et al. The multifaceted effects of Alpha1-antitrypsin on neutrophil functions. Front Pharmacol. (2018) 9:341. doi: 10.3389/fphar.2018.00341
15. Mutch NJ. Chapter 23: the role of platelets in fibrinolysis. In: Michelson AD, editors. Platelets. 3rd ed. Aberdeen: Academic Press (2013). p. 469–85.
16. Kuhn W, Schmalfeldt B, Reuning U, Pache L, Berger U, Ulm K, et al. Prognostic significance of urokinase (uPA) and its inhibitor PAI-1 for survival in advanced ovarian carcinoma stage FIGO IIIc. Br J Cancer. (1999) 79(11-12):1746–51. doi: 10.1038/sj.bjc.6690278
17. Nielsen HJ, Christensen IJ, Sørensen S, Moesgaard F, Brünner N. Preoperative plasma plasminogen activator inhibitor type-1 and serum C-reactive protein levels in patients with colorectal cancer. Ann Surg Oncol. (2000) 7(8):617–23. doi: 10.1007/BF02725342
18. Castelló R, Landete JM, España F, Vázquez C, Fuster C, Almenar SM, et al. Expression of plasminogen activator inhibitors type 1 and type 3 and urokinase plasminogen activator protein and mRNA in breast cancer. Thromb Res. (2007) 120(5):753–62. doi: 10.1016/j.thromres.2006.12.016
19. Pavón MA, Arroyo-Solera I, Téllez-Gabriel M, León X, Virós D, López M, et al. Enhanced cell migration and apoptosis resistance may underlie the association between high SERPINE1 expression and poor outcome in head and neck carcinoma patients. Oncotarget. (2015) 6(30):29016–33. doi: 10.18632/oncotarget.5032
20. Pączek S, Mroczko B. The role of selected serpins in gastrointestinal (GI) malignancies. J Clin Med. (2022) 11(20):6225. doi: 10.3390/jcm11206225
21. Kubala MH, DeClerck YA. The plasminogen activator inhibitor-1 paradox in cancer: a mechanistic understanding. Cancer Metastasis Rev. (2019) 38(3):483–92. doi: 10.1007/s10555-019-09806-4
22. Wiman B, Collen D. On the mechanism of the reaction between human alpha 2-antiplasmin and plasmin. J Biol Chem. (1979) 254(18):9291–7. doi: 10.1016/S0021-9258(19)86843-6
23. Lu BG, Sofian T, Law RH, Coughlin PB, Horvath AJ. Contribution of conserved lysine residues in the alpha2-antiplasmin C terminus to plasmin binding and inhibition. J Biol Chem. (2011) 286(28):24544–52. doi: 10.1074/jbc.M111.229013
24. Fair DS, Plow EF. Synthesis and secretion of the fibrinolytic components, including alpha 2-antiplasmin, by a human hepatoma cell line. J Lab Clin Med. (1983) 101(3):372–84.6186756
25. Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. (2015) 29(1):17–24. doi: 10.1016/j.blre.2014.09.003
26. Mutch NJ. Regulation of fibrinolysis by platelets. Platelets. (2019) 4:417–31. doi: 10.1016/B978-0-12-813456-6.00023-0
27. Sakata Y, Aoki N. Cross-linking of alpha 2-plasmin inhibitor to fibrin by fibrin-stabilizing factor. J Clin Invest. (1980) 65(2):290–7. doi: 10.1172/JCI109671
28. Sakata Y, Aoki N. Significance of cross-linking of alpha 2-plasmin inhibitor to fibrin in inhibition of fibrinolysis and in hemostasis. J Clin Invest. (1982) 69(3):536–42. doi: 10.1172/JCI110479
29. Mutch NJ, Koikkalainen JS, Fraser SR, Duthie KM, Griffin M, Mitchell J, et al. Model thrombi formed under flow reveal the role of factor XIII-mediated cross-linking in resistance to fibrinolysis. J Thromb Haemost. (2010) 8(9):2017–24. doi: 10.1111/j.1538-7836.2010.03963.x
30. Rijken DC, Abdul S, Malfliet JJ, Leebeek FW, Uitte de Willige S. Compaction of fibrin clots reveals the antifibrinolytic effect of factor XIII. J Thromb Haemost. (2016) 14(7):1453–61. doi: 10.1111/jth.13354
31. Fraser SR, Booth NA, Mutch NJ. The antifibrinolytic function of factor XIII is exclusively expressed through α₂-antiplasmin cross-linking. Blood. (2011) 117(23):6371–4. doi: 10.1182/blood-2011-02-333203
32. Abdul S, Leebeek FW, Rijken DC, Uitte de Willige S. Natural heterogeneity of alpha2-antiplasmin: functional and clinical consequences. Blood. (2016) 127(5):538–45. doi: 10.1182/blood-2015-09-670117
33. Lee KN, Jackson KW, Christiansen VJ, Chung KH, McKee PA. A novel plasma proteinase potentiates alpha2-antiplasmin inhibition of fibrin digestion. Blood. (2004) 103(10):3783–8. doi: 10.1182/blood-2003-12-4240
34. Pretorius E, Vlok M, Venter C, Bezuidenhout JA, Laubscher GJ, Steenkamp J, et al. Persistent clotting protein pathology in long COVID/post-acute sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol. (2021) 20(1):172. doi: 10.1186/s12933-021-01359-7
35. Carpenter SL, Mathew P. α2-Antiplasmin and its deficiency: fibrinolysis out of balance. Haemophilia. (2008) 14(6):1250–4. doi: 10.1111/j.1365-2516.2008.01766.x
36. Reed GL, Houng AK, Singh S, Wang D. Alpha2-antiplasmin: new insights and opportunities for ischemic stroke. Semin Thromb Hemost. (2017) 43(2):191–9. doi: 10.1055/s-0036-1585077
37. Singh S, Saleem S, Reed GL. Alpha2-antiplasmin: the devil you don't know in cerebrovascular and cardiovascular disease. Front Cardiovasc Med. (2020) 7:608899. doi: 10.3389/fcvm.2020.608899
38. The ARIC Investigators. The atherosclerosis risk in communit (ARIC) study: design and objectives. Am J Epidemiol. (1989) 129(4):687–702. doi: 10.1093/oxfordjournals.aje.a115184
39. Suri MFK, Yamagishi K, Aleksic N, Hannan PJ, Folsom AR. Novel hemostatic factor levels and risk of ischemic stroke: the atherosclerosis risk in communities (ARIC) study. Cerebrovascular Diseases. (2010) 29(5):497–502. doi: 10.1159/000297966
40. Houng AK, Wang D, Reed GL. Reversing the deleterious effects of α2-antiplasmin on tissue plasminogen activator therapy improves outcomes in experimental ischemic stroke. Exp Neurol. (2014) 255:56–62. doi: 10.1016/j.expneurol.2014.02.009
41. Reed GL, Houng AK, Wang D. Microvascular thrombosis, fibrinolysis, ischemic injury, and death after cerebral thromboembolism are affected by levels of circulating α2-antiplasmin. Arterioscler Thromb Vasc Biol. (2014) 34(12):2586–93. doi: 10.1161/ATVBAHA.114.304530
42. ClinicalTrials.gov. Translational sciences I. Study of safety and biomarker efficacy of TS23 in healthy volunteer. Identifier NCT03001544.
43. Nagai N, De Mol M, Van Hoef B, Verstreken M, Ds C. Depletion of circulating α2-antiplasmin by intravenous plasmin or immunoneutralization reduces focal cerebral ischemic injury in the absence of arterial recanalization. Blood. (2001) 97(10):3086–92. doi: 10.1182/blood.V97.10.3086
44. Briquez PS, Lorentz KM, Larsson HM, Frey P, Hubbell JA. Human kunitz-type protease inhibitor engineered for enhanced matrix retention extends longevity of fibrin biomaterials. Biomaterials. (2017) 135:1–9. doi: 10.1016/j.biomaterials.2017.04.048
45. Welsh CH, Hassell KL, Badesch DB, Kressin DC, Marlar RA. Coagulation and fibrinolytic profiles in patients with severe pulmonary hypertension. Chest. (1996) 110(3):710–7. doi: 10.1378/chest.110.3.710
46. Lu M, Blaine KP, Cullinane A, Hall C, Dulau-Florea A, Sun J, et al. Pulmonary arterial hypertension patients display normal kinetics of clot formation using thrombelastography. Pulm Circ. (2021) 11(3):20458940211022204. doi: 10.1177/20458940211022204
47. Harbaum L, Ghataorhe P, Wharton J, Jiménez B, Howard LSG, Gibbs JSR, et al. Reduced plasma levels of small HDL particles transporting fibrinolytic proteins in pulmonary arterial hypertension. Thorax. (2019) 74(4):380–9. doi: 10.1136/thoraxjnl-2018-212144
48. Gilat H, Vinker S, Buda I, Soudry E, Shani M, Bachar G. Obstructive sleep apnea and cardiovascular comorbidities: a large epidemiologic study. Medicine. (2014) 93(9):e45. doi: 10.1097/MD.0000000000000045
49. Zakrzewski M, Zakrzewska E, Kiciński P, Przybylska-Kuć S, Dybała A, Myśliński W, et al. Evaluation of fibrinolytic inhibitors: alpha-2-antiplasmin and plasminogen activator inhibitor 1 in patients with obstructive sleep apnoea. PLoS One. (2016) 11(11):e0166725. doi: 10.1371/journal.pone.0166725
50. Tomczykowska M, Bielak J, Bodys A. Evaluation of platelet activation, plasma antithrombin III and alpha2-antiplasmin activities in hypertensive patients. Ann Univ Mariae Curie Sklodowska Med. (2003) 58(1):15–20.15314952
51. Marder VJ, Comerota AJ, Shlansky-Goldberg RD, Davis JP, Deng C, Hanna K, et al. Safety of catheter-delivered plasmin in patients with acute lower extremity arterial or bypass graft occlusion: phase I results. J Thromb Haemost. (2012) 10(6):985–91. doi: 10.1111/j.1538-7836.2012.04728.x
52. Singh S, Houng AK, Reed GL. Venous stasis-induced fibrinolysis prevents thrombosis in mice: role of α2-antiplasmin. Blood. (2019) 134(12):970–8. doi: 10.1182/blood.2019000049
53. ClinicalTrials.gov. Translational sciences I. Novel α2-antiplasmin inactivation for lysis of intravascular thrombi (NAIL-IT) trial. Identifier NCT0540854.
54. Kruithof EK, Tran-Thang C, Ransijn A, Bachmann F. Demonstration of a fast-acting inhibitor of plasminogen activators in human plasma. Blood. (1984) 64(4):907–13. doi: 10.1182/blood.V64.4.907.907
55. Handt S, Jerome W, Tietze L, Hantgan R. Plasminogen activator inhibitor-1 secretion of endothelial cells increases fibrinolytic resistance of an in vitro fibrin clot: evidence for a key role of endothelial cells in thrombolytic resistance. Blood. (1996) 87(10):4204–13. doi: 10.1182/blood.V87.10.4204.bloodjournal87104204
56. Loskutoff DJ, Samad F. The adipocyte and hemostatic balance in obesity. Arterioscler Thromb Vasc Biol. (1998) 18(1):1–6. doi: 10.1161/01.ATV.18.1.1
57. Floridon C, Nielsen O, Hølund B, Sweep F, Sunde L, Thomsen SG, et al. Does plasminogen activator inhibitor-1 (PAI-1) control trophoblast invasion? A study of fetal and maternal tissue in intrauterine, tubal and molar pregnancies. Placenta. (2000) 21(8):754–62. doi: 10.1053/plac.2000.0573
58. Garg N, Goyal N, Strawn TL, Wu J, Mann KM, Lawrence DA, et al. Plasminogen activator inhibitor-1 and vitronectin expression level and stoichiometry regulate vascular smooth muscle cell migration through physiological collagen matrices. J Thromb Haemostasis. (2010) 8(8):1847–54. doi: 10.1111/j.1538-7836.2010.03907.x
59. Galgoczi E, Jeney F, Gazdag A, Erdei A, Katko M, Nagy DM, et al. Cell density-dependent stimulation of PAI-1 and hyaluronan synthesis by TGF-β in orbital fibroblasts. J Endocrinol. (2016) 229(2):187–96. doi: 10.1530/JOE-15-0524
60. Fay WP, Murphy JG, Owen WG. High concentrations of active plasminogen activator inhibitor-1 in porcine coronary artery thrombi. Arterioscler Thromb Vasc Biol. (1996) 16(10):1277–84. doi: 10.1161/01.ATV.16.10.1277
61. Booth NA, Simpson AJ, Croll A, Bennett B, MacGregor IR. Plasminogen activator inhibitor (PAI-1) in plasma and platelets. Br J Haematol. (1988) 70(3):327–33. doi: 10.1111/j.1365-2141.1988.tb02490.x
62. Morrow GB, Mutch NJ. Past, present, and future perspectives of plasminogen activator inhibitor 1 (PAI-1). Semin Thromb Hemost. (2022) 49:303–13. doi: 10.1055/s-0042-1758791
63. Brogren H, Karlsson L, Andersson M, Wang L, Erlinge D, Jern S. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood. (2004) 104(13):3943–8. doi: 10.1182/blood-2004-04-1439
64. Lawrence DA, Olson ST, Palaniappan S, Ginsburg D. Engineering plasminogen activator inhibitor 1 mutants with increased functional stability. Biochemistry. (1994) 33(12):3643–8. doi: 10.1021/bi00178a022
65. Vaughan DE, Declerck PJ, Van Houtte E, De Mol M, Collen D. Studies of recombinant plasminogen activator inhibitor-1 in rabbits. Pharmacokinetics and evidence for reactivation of latent plasminogen activator inhibitor-1 in vivo. Circ Res. (1990) 67(5):1281–6. doi: 10.1161/01.RES.67.5.1281
66. Scheer FA, Shea SA. Human circadian system causes a morning peak in prothrombotic plasminogen activator inhibitor-1 (PAI-1) independent of the sleep/wake cycle. Blood. (2014) 123(4):590–3. doi: 10.1182/blood-2013-07-517060
67. Kluft C, Jie AF, Rijken DC, Verheijen JH. Daytime fluctuations in blood of tissue-type plasminogen activator (t-PA) and its fast-acting inhibitor (PAI-1). Thromb Haemostasis. (1988) 59(2):329–32. doi: 10.1055/s-0038-1642781
68. Angleton P, Chandler WL, Schmer G. Diurnal variation of tissue-type plasminogen activator and its rapid inhibitor (PAI-1). Circulation. (1989) 79(1):101–6. doi: 10.1161/01.CIR.79.1.101
69. Tofler GH, Brezinski D, Schafer AI, Czeisler CA, Rutherford JD, Willich SN, et al. Concurrent morning increase in platelet aggregability and the risk of myocardial infarction and sudden cardiac death. N Engl J Med. (1987) 316(24):1514–8. doi: 10.1056/NEJM198706113162405
70. Andreotti F, Davies GJ, Hackett DR, Khan MI, De Bart AC, Aber VR, et al. Major circadian fluctuations in fibrinolytic factors and possible relevance to time of onset of myocardial infarction, sudden cardiac death and stroke. Am J Cardiol. (1988) 62(9):635–7. doi: 10.1016/0002-9149(88)90669-8
71. Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. (2000) 342(24):1792–801. doi: 10.1056/NEJM200006153422406
72. van der Bom JG, Bots ML, Haverkate F, Kluft C, Grobbee DE. The 4G5G polymorphism in the gene for PAI-1 and the circadian oscillation of plasma PAI-1. Blood. (2003) 101(5):1841–4. doi: 10.1182/blood-2002-07-2181
73. Asselbergs FW, Pattin K, Snieder H, Hillege HL, van Gilst WH, Moore JH. Genetic architecture of tissue-type plasminogen activator and plasminogen activator inhibitor-1. Semin Thromb Hemost. (2008) 34(6):562–8. doi: 10.1055/s-0028-1103367
74. Fay WP, Parker AC, Condrey LR, Shapiro AD. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. (1997) 90(1):204–8. doi: 10.1182/blood.V90.1.204.204_204_208
75. Heiman M GS, Khan SS, Vaughan DE, Shapiro AD. Complete plasminogen activator inhibitor 1 deficiency. Seattle (WA): University of Washington, Seattle: GeneReviews (2017).
76. Violi F, Ferro D, Quintarelli C, Musca A, Balsano F, Cordova C, et al. Hyperfibrinolysis resulting from clotting activation in patients with different degrees of cirrhosis. Hepatology. (1993) 17(1):78–83. doi: 10.1002/hep.1840170115
77. Matsuura R, Soma M, Maeda Y, Kasakura S. Studies on the mechanism of hyperfibrinolysis in liver cirrhosis–changes of plasma t-PA, PAI-1 and active PAI-1 levels in liver cirrhosis. Rinsho Byori. (1995) 43(12):1256–60.8569037
78. Arteel GE. New role of plasminogen activator inhibitor-1 in alcohol-induced liver injury. J Gastroenterol Hepatol. (2008) 23(Suppl 1):S54–9. doi: 10.1111/j.1440-1746.2007.05285.x
79. Vaughan DE. PAI-1 and atherothrombosis. J Thromb Haemostasis. (2005) 3(8):1879–83. doi: 10.1111/j.1538-7836.2005.01420.x
80. Meltzer ME, Lisman T, Doggen CJM, de Groot PG, Rosendaal FR. Synergistic effects of hypofibrinolysis and genetic and acquired risk factors on the risk of a first venous thrombosis. PLoS Med. (2008) 5(5):e97. doi: 10.1371/journal.pmed.0050097
81. Arenillas JF, ÁLvarez-SabíN J, Molina CA, ChacóN P, FernáNdez-Cadenas I, Ribó M, et al. Progression of symptomatic intracranial large artery atherosclerosis is associated with a proinflammatory state and impaired fibrinolysis. Stroke. (2008) 39(5):1456–63. doi: 10.1161/STROKEAHA.107.498600
82. Cesari M, Pahor M, Incalzi RA. REVIEW: plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc Ther. (2010) 28(5):e72–91. doi: 10.1111/j.1755-5922.2010.00171.x
83. Griemert EV, Recarte Pelz K, Engelhard K, Schäfer MK, Thal SC. PAI-1 but not PAI-2 gene deficiency attenuates ischemic brain injury after experimental stroke. Transl Stroke Res. (2019) 10(4):372–80. doi: 10.1007/s12975-018-0644-9
84. Sacco RL, Kargman DE, Gu Q, Zamanillo MC. Race-Ethnicity and determinants of intracranial atherosclerotic cerebral infarction. Stroke. (1995) 26(1):14–20. doi: 10.1161/01.STR.26.1.14
85. Tuttolomondo A, Pinto A, Corrao S, Di Raimondo D, Fernandez P, Di Sciacca R, et al. Immuno-inflammatory and thrombotic/fibrinolytic variables associated with acute ischemic stroke diagnosis. Atherosclerosis. (2009) 203(2):503–8. doi: 10.1016/j.atherosclerosis.2008.06.030
86. Parikh NS, Merkler AE, Iadecola C. Inflammation, autoimmunity, infection, and stroke: epidemiology and lessons from therapeutic intervention. Stroke. (2020) 51(3):711–8. doi: 10.1161/STROKEAHA.119.024157
87. ClinicalTrials.gov, Zhujiang H, Nanfang Hospital of Southern Medical U, First Affiliated Hospital of Jinan U. Relationships among FVIII, t-PA/PAI-1, and MMP-9 levels and intracranial hemorrhage complications after thrombolysis with alteplase in patients with acute ischemic stroke: protocol for a multicenter retrospective study. Identifier NCT03733223.
88. Meltzer ME, Lisman T, de Groot PG, Meijers JC, le Cessie S, Doggen CJ, et al. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood. (2010) 116(1):113–21. doi: 10.1182/blood-2010-02-267740
89. Tang J, Zhu W, Mei X, Zhang Z. Plasminogen activator inhibitor-1: a risk factor for deep vein thrombosis after total hip arthroplasty. J Orthop Surg Res. (2018) 13(1):8. doi: 10.1186/s13018-018-0716-2
90. Cheng J, Fu Z, Zhu J, Zhou L, Song W. The predictive value of plasminogen activator inhibitor-1, fibrinogen, and D-dimer for deep venous thrombosis following surgery for traumatic lower limb fracture. Ann Palliat Med. (2020) 9(5):3385–92. doi: 10.21037/apm-20-1604
91. Blokhin IO, Lentz SR. Mechanisms of thrombosis in obesity. Curr Opin Hematol. (2013) 20(5):437–44. doi: 10.1097/MOH.0b013e3283634443
92. Lijnen HR. Role of fibrinolysis in obesity and thrombosis. Thromb Res. (2009) 123(Suppl 4):S46–9. doi: 10.1016/S0049-3848(09)70143-4
93. Skurk T, Hauner H. Obesity and impaired fibrinolysis: role of adipose production of plasminogen activator inhibitor-1. Int J Obes Relat Metab Disord. (2004) 28(11):1357–64. doi: 10.1038/sj.ijo.0802778
94. Zheng Z, Nakamura K, Gershbaum S, Tabas I. The markedly increased PAI1 in obesity induces a compensatory increase of hepatocyte tpa expression by activating a LRP1-CREB1 pathway. Blood. (2019) 134:3625. doi: 10.1182/blood-2019-129271
95. Zheng Z, Nakamura K, Gershbaum S, Wang X, Thomas S, Bessler M, et al. Interacting hepatic PAI-1/tPA gene regulatory pathways influence impaired fibrinolysis severity in obesity. J Clin Invest. (2020) 130(8):4348–59. doi: 10.1172/JCI135919
96. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. (2011) 11(2):85–97. doi: 10.1038/nri2921
97. Eriksson P, Reynisdottir S, Lönnqvist F, Stemme V, Hamsten A, Arner P. Adipose tissue secretion of plasminogen activator inhibitor-1 in non-obese and obese individuals. Diabetologia. (1998) 41(1):65–71. doi: 10.1007/s001250050868
98. Yudkin JS, Coppack SW, Bulmer K, Rawesh A, Mohamed-Ali V. Lack of evidence for secretion of plasminogen activator inhibitor-1 by human subcutaneous adipose tissue in vivo. Thromb Res. (1999) 96(1):1–9. doi: 10.1016/S0049-3848(99)00061-4
99. Brogren H, Sihlbom C, Wallmark K, Lonn M, Deinum J, Karlsson L, et al. Heterogeneous glycosylation patterns of human PAI-1 may reveal its cellular origin. Thromb Res. (2008) 122(2):271–81. doi: 10.1016/j.thromres.2008.04.008
100. DeBakey ME, Lawrie GM, Glaeser DH. Patterns of atherosclerosis and their surgical significance. Ann Surg. (1985) 201(2):115–31. doi: 10.1097/00000658-198502000-00001
101. Rylander AC Jr, Lindgren A, Deinum J, Bergström GM, Böttcher G, Kalies I, et al. Fibrinolysis inhibitors in plaque stability: a morphological association of PAI-1 and TAFI in advanced carotid plaque. J Thromb Haemost. (2017) 15(4):758–69. doi: 10.1111/jth.13641
102. Sillen M, Declerck PJ. A narrative review on plasminogen activator inhibitor-1 and its (patho)physiological role: to target or not to target? Int J Mol Sci. (2021) 22(5):2721. doi: 10.3390/ijms22052721
103. Badimon L, Padró T, Vilahur G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur Heart J Acute Cardiovasc Care. (2012) 1(1):60–74. doi: 10.1177/2048872612441582
104. Huebner BR, Moore EE, Moore HB, Stettler GR, Nunns GR, Lawson P, et al. Thrombin provokes degranulation of platelet α-granules leading to the release of active plasminogen activator inhibitor-1 (PAI-1). Shock. (2018) 50(6):671–6. doi: 10.1097/SHK.0000000000001089
105. Morrow G, Whyte C, Mutch N. Functional plasminogen activator inhibitor 1 is retained on the activated platelet membrane following platelet activation. Haematologica. (2019) 105(12):2824–33. doi: 10.3324/haematol.2019.230367
106. Napolitano F, Montuori N. Role of plasminogen activation system in platelet pathophysiology: emerging concepts for translational applications. Int J Mol Sci. (2022) 23(11):4303–11. doi: 10.3390/ijms23116065
107. Hamsten A, de Faire U, Walldius G, Dahlen G, Szamosi A, Landou C, et al. Plasminogen activator inhibitor in plasma: risk factor for recurrent myocardial infarction. Lancet. (1987) 2(8549):3–9. doi: 10.1016/S0140-6736(87)93050-9
108. Tofler GH, Massaro J, O'Donnell CJ, Wilson PWF, Vasan RS, Sutherland PA, et al. Plasminogen activator inhibitor and the risk of cardiovascular disease: the framingham heart study. Thromb Res. (2016) 140:30–5. doi: 10.1016/j.thromres.2016.02.002
109. Condron M, Rowell S, Dewey E, Anderson T, Lealiiee L, Farrell D, et al. The procoagulant molecule plasminogen activator inhibitor-1 is associated with injury severity and shock in patients with and without traumatic brain injury. J Trauma Acute Care Surg. (2018) 85(5):888–93. doi: 10.1097/TA.0000000000002040
110. Eren M, Boe A, Klyachko E, Vaughan D. Role of plasminogen activator inhibitor-1 in senescence and aging. Semin Thromb Hemost. (2014) 40(06):645–51. doi: 10.1055/s-0034-1387883
111. Shimizu T, Uematsu M, Yoshizaki T, Obata J-E, Nakamura T, Fujioka D, et al. Myocardial production of plasminogen activator inhibitor-1 is associated with coronary endothelial and ventricular dysfunction after acute myocardial infarction. J Atheroscler Thromb. (2016) 23(5):557–66. doi: 10.5551/jat.32300
112. Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation. (1989) 79(4):733–43. doi: 10.1161/01.CIR.79.4.733
113. Janszky I, Ljung R. Shifts to and from daylight saving time and incidence of myocardial infarction. N Engl J Med. (2008) 359(18):1966–8. doi: 10.1056/NEJMc0807104
114. Boinska J, Koziński M, Kasprzak M, Ziołkowski M, Kubica J, Rość D. Diurnal oscillations of fibrinolytic parameters in patients with acute myocardial infarction and their relation to platelet reactivity: preliminary insights. J Clin Med. (2022) 11(23):1–11. doi: 10.3390/jcm11237105
115. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. (2003) 348(14):1309–21. doi: 10.1056/NEJMoa030207
116. ClinicalTrials.gov, Vanderbilt University Medical C, National Center for Research R. Diurnal variation of plasminogen activator inhibitor-1. Identifier NCT00515021.
118. ClinicalTrials.gov, Indiana H, Thrombosis Center I. Metformin use in cardiac fibrosis in PAI-1 deficiency. Identifier NCT05317806.
119. Llitjos JF, Leclerc M, Chochois C, Monsallier JM, Ramakers M, Auvray M, et al. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost. (2020) 18(7):1743–6. doi: 10.1111/jth.14869
120. Kunutsor SK, Laukkanen JA. Incidence of venous and arterial thromboembolic complications in COVID-19: a systematic review and meta-analysis. Thromb Res. (2020) 196:27–30. doi: 10.1016/j.thromres.2020.08.022
121. Bachler M, Bösch J, Stürzel DP, Hell T, Giebl A, Ströhle M, et al. Impaired fibrinolysis in critically ill COVID-19 patients. Br J Anaesth. (2021) 126(3):590–8. doi: 10.1016/j.bja.2020.12.010
122. Nougier C, Benoit R, Simon M, Desmurs-Clavel H, Marcotte G, Argaud L, et al. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J Thromb Haemostasis. (2020) 18(9):2215–9. doi: 10.1111/jth.15016
123. Darif D, Hammi I, Kihel A, El Idrissi Saik I, Guessous F, Akarid K. The pro-inflammatory cytokines in COVID-19 pathogenesis: what goes wrong? Microb Pathog. (2021) 153:104799. doi: 10.1016/j.micpath.2021.104799
124. Whyte CS, Simpson M, Morrow GB, Wallace CA, Mentzer AJ, Knight JC, et al. The suboptimal fibrinolytic response in COVID-19 is dictated by high PAI-1. J Thromb Haemostasis. (2022) 20:2394–406. doi: 10.1111/jth.15806
125. Prabhakaran P, Ware LB, White KE, Cross MT, Matthay MA, Olman MA. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2003) 285(1):L20–8. doi: 10.1152/ajplung.00312.2002
126. ClinicalTrials.gov, Northwestern U. Study to antagonize plasminogen activator inhibitor-1 in severe COVID-19. Identifier NCT04634799.
127. ClinicalTrials.gov, Saglik Bilimleri U. PAI-1 levels and predicting Covid-19 patients’ prognosis. Identifier NCT04997928.
128. Zhou X, Hendrickx ML, Hassanzadeh-Ghassabeh G, Muyldermans S, Declerck PJ. Generation and in vitro characterisation of inhibitory nanobodies towards plasminogen activator inhibitor 1. Thromb Haemost. (2016) 116(6):1032–40. doi: 10.1160/TH16-04-0306
129. Vousden KA, Lundqvist T, Popovic B, Naiman B, Carruthers AM, Newton P, et al. Discovery and characterisation of an antibody that selectively modulates the inhibitory activity of plasminogen activator inhibitor-1. Sci Rep. (2019) 9(1):1605. doi: 10.1038/s41598-019-38842-x
130. Sillen M, Weeks SD, Zhou X, Komissarov AA, Florova G, Idell S, et al. Molecular mechanism of two nanobodies that inhibit PAI-1 activity reveals a modulation at distinct stages of the PAI-1/plasminogen activator interaction. J Thromb Haemostasis. (2020) 18(3):681–92. doi: 10.1111/jth.14716
131. Cater JH, Mañucat-Tan NB, Georgiou DK, Zhao G, Buhimschi IA, Wyatt AR, et al. A novel role for plasminogen activator inhibitor type-2 as a hypochlorite-resistant serine protease inhibitor and holdase chaperone. Cells. (2022) 11(7):1152. doi: 10.3390/cells11071152
132. Boncela J, Przygodzka P, Papiewska-Pajak I, Wyroba E, Cierniewski CS. Association of plasminogen activator inhibitor type 2 (PAI-2) with proteasome within endothelial cells activated with inflammatory stimuli. J Biol Chem. (2011) 286(50):43164–71. doi: 10.1074/jbc.M111.245647
133. Ritchie H, Robbie LA, Kinghorn S, Exley R, Booth NA. Monocyte plasminogen activator inhibitor 2 (PAI-2) inhibits u-PA-mediated fibrin clot lysis and is cross-linked to fibrin. Thromb Haemostasis. (1999) 81(1):96–103. doi: 10.1055/s-0037-1614425
134. Astedt B, Lindoff C, Lecander I. Significance of the plasminogen activator inhibitor of placental type (PAI-2) in pregnancy. Semin Thromb Hemost. (1998) 24(5):431–5. doi: 10.1055/s-2007-996035
135. Bachmann F. The enigma PAI-2. Gene expression, evolutionary and functional aspects. Thromb Haemost. (1995) 74(1):172–9. doi: 10.1055/s-0038-1642672
136. Dougherty KM, Pearson JM, Yang AY, Westrick RJ, Baker MS, Ginsburg D. The plasminogen activator inhibitor-2 gene is not required for normal murine development or survival. Proc Natl Acad Sci USA. (1999) 96(2):686–91. doi: 10.1073/pnas.96.2.686
137. Thorsen S, Philips M, Selmer J, Lecander I, Astedt B. Kinetics of inhibition of tissue-type and urokinase-type plasminogen activator by plasminogen-activator inhibitor type 1 and type 2. Eur J Biochem. (1988) 175(1):33–9. doi: 10.1111/j.1432-1033.1988.tb14162.x
138. Siefert SA, Chabasse C, Mukhopadhyay S, Hoofnagle MH, Strickland DK, Sarkar R, et al. Enhanced venous thrombus resolution in plasminogen activator inhibitor type-2 deficient mice. J Thromb Haemostasis. (2014) 12(10):1706–16. doi: 10.1111/jth.12657
139. Corsetti JP, Salzman P, Ryan D, Moss AJ, Zareba W, Sparks CE. Plasminogen activator inhibitor-2 polymorphism associates with recurrent coronary event risk in patients with high HDL and C-reactive protein levels. PLoS One. (2013) 8(7):e68920. doi: 10.1371/journal.pone.0068920
140. Li X, Luo JY, Zhang L, Yang YN, Xie X, Liu F, et al. Variant of PAI-2 gene is associated with coronary artery disease and recurrent coronary event risk in Chinese han population. Lipids Health Dis. (2015) 14:148. doi: 10.1186/s12944-015-0150-y
141. Shetty S, Idell S. Fibrinolysis | plasminogen activator and plasmin. In: Laurent GJ, Shapiro SD, editors. Encyclopedia of respiratory medicine. Oxford: Academic Press (2006). p. 205–10.
142. Kumar S, Baglioni C. Protection from tumor necrosis factor-mediated cytolysis by overexpression of plasminogen activator inhibitor type-2. J Biol Chem. (1991) 266(31):20960–4. doi: 10.1016/S0021-9258(18)54804-3
143. Shafren DR, Gardner J, Mann VH, Antalis TM, Suhrbier A. Picornavirus receptor down-regulation by plasminogen activator inhibitor type 2. J Virol. (1999) 73(9):7193–8. doi: 10.1128/JVI.73.9.7193-7198.1999
144. Mueller BM, Yu YB, Laug WE. Overexpression of plasminogen activator inhibitor 2 in human melanoma cells inhibits spontaneous metastasis in scid/scid mice. Proc Natl Acad Sci U S A. (1995) 92(1):205–9. doi: 10.1073/pnas.92.1.205
145. Gossage JA, Humphries J, Modarai B, Burnand KG, Smith A. Adenoviral urokinase-type plasminogen activator (uPA) gene transfer enhances venous thrombus resolution. J Vasc Surg. (2006) 44(5):1085–90. doi: 10.1016/j.jvs.2006.07.020
146. Humphries J, Gossage JA, Modarai B, Burnand KG, Sisson TH, Murdoch C, et al. Monocyte urokinase-type plasminogen activator up-regulation reduces thrombus size in a model of venous thrombosis. J Vasc Surg. (2009) 50(5):1127–34. doi: 10.1016/j.jvs.2009.06.047
147. Northeast AD, Soo KS, Bobrow LG, Gaffney PJ, Burnand KG. The tissue plasminogen activator and urokinase response in vivo during natural resolution of venous thrombus. J Vasc Surg. (1995) 22(5):573–9. doi: 10.1016/S0741-5214(95)70041-2
148. Ali T, Humphries J, Burnand K, Sawyer B, Bursill C, Channon K, et al. Monocyte recruitment in venous thrombus resolution. J Vasc Surg. (2006) 43(3):601–8. doi: 10.1016/j.jvs.2005.10.073
149. Griemert EV, Schwarzmaier SM, Hummel R, Gölz C, Yang D, Neuhaus W, et al. Plasminogen activator inhibitor-1 augments damage by impairing fibrinolysis after traumatic brain injury. Ann Neurol. (2019) 85(5):667–80. doi: 10.1002/ana.25458
150. Dietzmann K, von Bossanyi P, Krause D, Wittig H, Mawrin C, Kirches E. Expression of the plasminogen activator system and the inhibitors PAI-1 and PAI-2 in posttraumatic lesions of the CNS and brain injuries following dramatic circulatory arrests: an immunohistochemical study. Pathol Res Pract. (2000) 196(1):15–21. doi: 10.1016/S0344-0338(00)80017-5
151. Barde YA, Lindsay RM, Monard D, Thoenen H. New factor released by cultured glioma cells supporting survival and growth of sensory neurones. Nature. (1978) 274(5673):818. doi: 10.1038/274818a0
152. Mansilla S, Boulaftali Y, Venisse L, Arocas VR, Meilhac O, Michel J-B, et al. Macrophages and platelets are the Major source of protease nexin-1 in human atherosclerotic plaque. Arterioscler Thromb Vasc Biol. (2008) 28(10):1844–50. doi: 10.1161/ATVBAHA.108.171389
153. Santoro A, Conde J, Scotece M, Abella V, Lois A, Lopez V, et al. SERPINE2 inhibits IL-1α-induced MMP-13 expression in human chondrocytes: involvement of ERK/NF-κB/AP-1 pathways. PLoS One. (2015) 10(8):e0135979. doi: 10.1371/journal.pone.0135979
154. Sommer J, Gloor SM, Rovelli GF, Hofsteenge J, Nick H, Meier R, et al. cDNA sequence coding for a rat glia-derived nexin and its homology to members of the serpin superfamily. Biochemistry. (1987) 26(20):6407–10. doi: 10.1021/bi00394a016
155. Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. (2000) 10(12):1845–64. doi: 10.1101/gr.147800
156. Boulaftali Y, Ho-Tin-Noe B, Pena A, Loyau S, Venisse L, Francois D, et al. Platelet protease nexin-1, a serpin that strongly influences fibrinolysis and thrombolysis. Circulation. (2011) 123(12):1326–34. doi: 10.1161/CIRCULATIONAHA.110.000885
157. Yang Y, Xin X, Fu X, Xu D. Expression pattern of human SERPINE2 in a variety of human tumors. Oncol Lett. (2018) 15:4523–30. doi: 10.3892/ol.2018.7819
158. Madjene C, Boutigny A, Bouton MC, Arocas V, Richard B. Protease nexin-1 in the cardiovascular system: wherefore art thou? Front Cardiovasc Med. (2021) 8:652852. doi: 10.3389/fcvm.2021.652852
159. Kawecki C, Aymonnier K, Ferrière S, Vénisse L, Arocas V, Boulaftali Y, et al. Development and characterization of single-domain antibodies neutralizing protease nexin-1 as tools to increase thrombin generation. J Thromb Haemostasis. (2020) 18:2155–68. doi: 10.1111/jth.14940
160. Selbonne S, Madjene C, Salmon B, Boulaftali Y, Bouton M-C, Arocas V. Protease nexin-1 deficiency increases mouse hindlimb neovascularisation following ischemia and accelerates femoral artery perfusion. Sci Rep. (2021) 11(1):13412. doi: 10.1038/s41598-021-92794-9
161. Hinderer S, Schenke-Layland K. Cardiac fibrosis – a short review of causes and therapeutic strategies. Adv Drug Delivery Rev. (2019) 146:77–82. doi: 10.1016/j.addr.2019.05.011
162. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. (2014) 71(4):549–74. doi: 10.1007/s00018-013-1349-6
163. Li X, Zhao D, Guo Z, Li T, Qili M, Xu B, et al. Overexpression of SerpinE2/protease nexin-1 contribute to pathological cardiac fibrosis via increasing collagen deposition. Sci Rep. (2016) 6(1):37635. doi: 10.1038/srep37635
164. Li C, Zhu HY, Bai WD, Su LL, Liu JQ, Cai WX, et al. MiR-10a and miR-181c regulate collagen type I generation in hypertrophic scars by targeting PAI-1 and uPA. FEBS Lett. (2015) 589(3):380–9. doi: 10.1016/j.febslet.2014.12.024
165. Li X, Wang G, QiLi M, Liang H, Li T, E X, et al. Aspirin reduces cardiac interstitial fibrosis by inhibiting Erk1/2-Serpine2 and P-akt signalling pathways. Cell Physiol Biochem. (2018) 45(5):1955–65. doi: 10.1159/000487972
166. Morfino P, Aimo A, Castiglione V, Gálvez-Montón C, Emdin M, Bayes-Genis A. Treatment of cardiac fibrosis: from neuro-hormonal inhibitors to CAR-T cell therapy. Heart Fail Rev. (2022) 28:555–69. doi: 10.1007/s10741-022-10279-x
167. Wallace A, Rovelli G, Hofsteenge J, Stone SR. Effect of heparin on the glia-derived-nexin-thrombin interaction. Biochem J. (1989) 257(1):191–6. doi: 10.1042/bj2570191
168. Hofsteenge J, Taguchi H, Stone SR. Effect of thrombomodulin on the kinetics of the interaction of thrombin with substrates and inhibitors. Biochem J. (1986) 237(1):243–51. doi: 10.1042/bj2370243
169. Boulaftali Y, Adam F, Venisse L, Ollivier V, Richard B, Taieb S, et al. Anticoagulant and antithrombotic properties of platelet protease nexin-1. Blood. (2010) 115(1):97–106. doi: 10.1182/blood-2009-04-217240
170. Aymonnier K, Kawecki C, Venisse L, Boulaftali Y, Christophe OD, Lenting PJ, et al. Targeting protease nexin-1, a natural anticoagulant serpin, to control bleeding and improve hemostasis in hemophilia. Blood. (2019) 134(19):1632–44. doi: 10.1182/blood.2019000281
171. Kajdácsi E, Jandrasics Z, Veszeli N, Makó V, Koncz A, Gulyás D, et al. Patterns of C1-inhibitor/plasma serine protease complexes in healthy humans and in hereditary angioedema patients. Front Immunol. (2020) 11:794. doi: 10.3389/fimmu.2020.00794
172. Maas C, Oschatz C, Rennéo T. The plasma contact system 2.0. Semin Thromb Hemost. (2011) 37(4):375–81. doi: 10.1055/s-0031-1276586
173. Schmaier AH, Murray SC, Heda GD, Farber A, Kuo A, McCrae K, et al. Synthesis and expression of C1 inhibitor by human umbilical vein endothelial cells. J Biol Chem. (1989) 264(30):18173–9. doi: 10.1016/S0021-9258(19)84693-8
174. Lappin DF, McPhaden AR, Yap PL, Carter PE, Birnie GD, Fothergill JE, et al. Monocyte C1-inhibitor synthesis in patients with C1-inhibitor deficiency. Eur J Clin Invest. (1989) 19(1):45–52. doi: 10.1111/j.1365-2362.1989.tb00194.x
175. Zuraw BL, Lotz M. Regulation of the hepatic synthesis of C1 inhibitor by the hepatocyte stimulating factors interleukin 6 and interferon gamma. J Biol Chem. (1990) 265(21):12664–70. doi: 10.1016/S0021-9258(19)38395-4
176. Kramer J, Katz Y, Rosen FS, Davis AE 3rd, Strunk RC. Synthesis of C1 inhibitor in fibroblasts from patients with type I and type II hereditary angioneurotic edema. J Clin Invest. (1991) 87(5):1614–20. doi: 10.1172/JCI115175
177. Schmaier AH, Smith PM, Colman RW. Platelet C1- inhibitor. A secreted alpha-granule protein. J Clin Invest. (1985) 75(1):242–50. doi: 10.1172/JCI111680
178. Schmaier AH, Amenta S, Xiong T, Heda GD, Gewirtz AM. Expression of platelet C1 inhibitor. Blood. (1993) 82(2):465–74. doi: 10.1182/blood.V82.2.465.465
179. Georgy MS, Pongracic JA. Chapter 22: hereditary and acquired angioedema. Allergy Asthma Proc. (2012) 33(Suppl 1):73–6. doi: 10.2500/aap.2012.33.3555
180. Napolitano F, Montuori N. The role of the plasminogen activation system in angioedema: novel insights on the pathogenesis. J Clin Med. (2021) 10(3):518. doi: 10.3390/jcm10030518
181. Csuka D, Veszeli N, Imreh É, Zotter Z, Skopál J, Prohászka Z, et al. Comprehensive study into the activation of the plasma enzyme systems during attacks of hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis. (2015) 10(1):132. doi: 10.1186/s13023-015-0351-5
182. Nilsson T, Back O. Elevated plasmin-α2-antiplasmin complex levels in hereditary angioedema: evidence for the in vivo efficiency of the intrinsic fibrinolytic system. Thromb Res. (1985) 40(6):817–21. doi: 10.1016/0049-3848(85)90318-4
183. Kaplan AP. Angioedema. World Allergy Organ J. (2008) 1(6):103–13. doi: 10.1097/WOX.0b013e31817aecbe
184. Björkman LS, Persson B, Aronsson D, Skattum L, Nordenfelt P, Egesten A. Comorbidities in hereditary angioedema—a population-based cohort study. Clin Transl Allergy. (2022) 12(3):e12135. doi: 10.1002/clt2.12135
185. Grover SP, Sundler Björkman L, Egesten A, Moll S, Mackman N. Hereditary angioedema is associated with an increased risk of venous thromboembolism. J Thromb Haemostasis. (2022) 20(11):2703–6. doi: 10.1111/jth.15870
186. Hamilton AG, Bosley AR, Bowen DJ. Laryngeal oedema due to hereditary angioedema. Anaesthesia. (1977) 32(3):265–7. doi: 10.1111/j.1365-2044.1977.tb11606.x
187. Cai J, Ribkoff J, Olson S, Raghunathan V, Al-Samkari H, DeLoughery TG, et al. The many roles of tranexamic acid: an overview of the clinical indications for TXA in medical and surgical patients. Eur J Haematol. (2020) 104(2):79–87. doi: 10.1111/ejh.13348
188. Horiuchi T, Hide M, Yamashita K, Ohsawa I. The use of tranexamic acid for on-demand and prophylactic treatment of hereditary angioedema—a systematic review. J Cutan Immunol Allergy. (2018) 1(4):126–38. doi: 10.1002/cia2.12029
189. Maurer M, Magerl M, Ansotegui I, Aygören-Pürsün E, Betschel S, Bork K, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2017 revision and update. Allergy. (2018) 73(8):1575–96. doi: 10.1111/all.13384
190. ClinicalTrials.gov, Shire, Takeda. C1 esterase inhibitor (C1INH-nf) for the prevention of acute hereditary angioedema (HAE) attacks. Identifier NCT01005888.
191. ClinicalTrials.gov, Shire, Takeda. Subcutaneous CINRYZE with recombinant human hyaluronidase for prevention of angioedema attacks. Identifier NCT01756157.
192. ClinicalTrials.gov, Shire, Takeda. Safety and efficacy study of CINRYZE for prevention of angioedema attacks in children ages 6-11 with hereditary angioedema. Identifier NCT02052141.
193. ClinicalTrials.gov, HAEGR. F. Global registry to gather data on natural history of patients with hereditary angioedema type I and II. Identifier NCT03828279.
194. Fröhlich K, Macha K, Gerner ST, Bobinger T, Schmidt M, Dörfler A, et al. Angioedema in stroke patients with thrombolysis. Stroke. (2019) 50(7):1682–7. doi: 10.1161/STROKEAHA.119.025260
195. Khalid M, Kanaa M, Alkawaleet Y, Ayub MT. Angioedema: a life-threatening complication of tissue plasminogen activator. Cureus. (2018) 10(3):e2392. doi: 10.7759/cureus.2392
196. Maas C. Plasminflammation-an emerging pathway to bradykinin production. Front Immunol. (2019) 10:2046. doi: 10.3389/fimmu.2019.02046
197. Nielsen EW, Morrissey J, Olsen JO, Osterud B. Factor VIIa in patients with C1-inhibitor deficiency. Thromb Haemost. (1995) 74(4):1103–6. doi: 10.1055/s-0038-1649888
198. Joseph K, Tholanikunnel TE, Kaplan AP. Treatment of episodes of hereditary angioedema with C1 inhibitor: serial assessment of observed abnormalities of the plasma bradykinin-forming pathway and fibrinolysis. Ann Allergy Asthma Immunol. (2010) 104(1):50–4. doi: 10.1016/j.anai.2009.11.014
199. van Geffen M, Cugno M, Lap P, Loof A, Cicardi M, van Heerde W. Alterations of coagulation and fibrinolysis in patients with angioedema due to C1-inhibitor deficiency. Clin Exp Immunol. (2012) 167(3):472–8. doi: 10.1111/j.1365-2249.2011.04541.x
200. Farkas H, Kőhalmi KV, Veszeli N, Zotter Z, Várnai K, Varga L. Risk of thromboembolism in patients with hereditary angioedema treated with plasma-derived C1-inhibitor. Allergy Asthma Proc. (2016) 37(2):164–70. doi: 10.2500/aap.2016.37.3933
201. Reshef A, Zanichelli A, Longhurst H, Relan A, Hack CE. Elevated D-dimers in attacks of hereditary angioedema are not associated with increased thrombotic risk. Allergy. (2015) 70(5):506–13. doi: 10.1111/all.12587
202. Grover SP, Kawano T, Wan J, Tanratana P, Polai Z, Shim YJ, et al. C1 inhibitor deficiency enhances contact pathway mediated activation of coagulation and venous thrombosis. Blood. (2023) doi: 10.1182/blood.2022018849. Online ahead of print.
203. Jensen SB, Hindberg K, Solomon T, Smith EN, Lapek JD Jr, Gonzalez DJ, et al. Discovery of novel plasma biomarkers for future incident venous thromboembolism by untargeted synchronous precursor selection mass spectrometry proteomics. J Thromb Haemost. (2018) 16(9):1763–74. doi: 10.1111/jth.14220
204. Adesanya TMA, Campbell CM, Cheng L, Ogbogu PU, Kahwash R. C1 esterase inhibition: targeting multiple systems in COVID-19. J Clin Immunol. (2021) 41(4):729–32. doi: 10.1007/s10875-021-00972-1
205. Thomson TM, Toscano-Guerra E, Casis E, Paciucci R. C1 esterase inhibitor and the contact system in COVID-19. Br J Haematol. (2020) 190(4):520–4. doi: 10.1111/bjh.16938
206. Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. (2020) 182(1):59–72.e15. doi: 10.1016/j.cell.2020.05.032
207. Hausburg MA, Banton KL, Roshon M, Bar-Or D. Clinically distinct COVID-19 cases share notably similar immune response progression: a follow-up analysis. Heliyon. (2021) 7(1):e05877. doi: 10.1016/j.heliyon.2020.e05877
Keywords: SERPIN, thromboinflammation, plasminogen activator-inhibitor-1, α2-antiplasmin, protease nexin-1, C1-inhibitor
Citation: Humphreys SJ, Whyte CS and Mutch NJ (2023) “Super” SERPINs—A stabilizing force against fibrinolysis in thromboinflammatory conditions. Front. Cardiovasc. Med. 10:1146833. doi: 10.3389/fcvm.2023.1146833
Received: 17 January 2023; Accepted: 31 March 2023;
Published: 19 April 2023.
Edited by:
Laurent Mosnier, The Scripps Research Institute, United StatesReviewed by:
Evi X. Stavrou, Case Western Reserve University, United StatesSteven Philip Grover, University of North Carolina at Chapel Hill, United States
© 2023 Humphreys, Whyte and Mutch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola J. Mutch bi5qLm11dGNoQGFiZG4uYWMudWs=
Specialty Section: This article was submitted to Thrombosis, a section of the journal Frontiers in Cardiovascular Medicine