 Yanan Zhang1,2,3,4,5,6,7
Yanan Zhang1,2,3,4,5,6,7 Jimin Wu
Jimin Wu Erdan Dong
Erdan Dong Zhanli Wang
Zhanli Wang Han Xiao
Han Xiao- 1Inner Mongolia Key Laboratory of Disease-Related Biomarkers, The Second Affiliated Hospital, Baotou Medical College, Baotou, China
- 2Department of Cardiology and Institute of Vascular Medicine, Peking University Third Hospital, Beijing, China
- 3NHC Key Laboratory of Cardiovascular Molecular Biology and Regulatory Peptides, Peking University Third Hospital, Beijing, China
- 4Key Laboratory of Molecular Cardiovascular Science, Ministry of Education, Beijing, China
- 5Beijing Key Laboratory of Cardiovascular Receptors Research, Peking University Third Hospital, Beijing, China
- 6Research Unit of Medical Science Research Management/Basic and Clinical Research of Metabolic Cardiovascular Diseases, Chinese Academy of Medical Sciences, Beijing, China
- 7Department of Clinical Laboratory, Shanghai University of Medicine & Health Sciences Affiliated Zhoupu Hospital, Shanghai, China
Toll-like receptors (TLRs) are a family of pattern recognition receptors (PRRs) that can identify pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). TLRs play an important role in the innate immune response, leading to acute and chronic inflammation. Cardiac hypertrophy, an important cardiac remodeling phenotype during cardiovascular disease, contributes to the development of heart failure. In previous decades, many studies have reported that TLR-mediated inflammation was involved in the induction of myocardium hypertrophic remodeling, suggesting that targeting TLR signaling might be an effective strategy against pathological cardiac hypertrophy. Thus, it is necessary to study the mechanisms underlying TLR functions in cardiac hypertrophy. In this review, we summarized key findings of TLR signaling in cardiac hypertrophy.
1. Introduction
Cardiac hypertrophy, characterized by an enlargement of cardiomyocyte size, is initially an adaptive response to various stimuli (Figure 1) (1). Physiological cardiac hypertrophy occurs in response to pregnancy and exercise to preserve or improve heart function without cardiac fibrosis (2). In contrast, pathological cardiac hypertrophy accompanying myocardial dysfunction and fibrosis is the cardiac response to chronic stressful conditions, such as hypertension and valvular disease (3). Pathological cardiac hypertrophy plays a causal role in the progression of heart failure. Pathological hypertrophy is associated with increased interstitial fibrosis, cell death, and cardiac dysfunction, as well as increased production of proinflammatory cytokines (3, 4). Inflammation is a characteristic feature of pathological cardiac hypertrophy (5). Toll-like receptors (TLRs), as innate immune receptors, are key factors in cardiovascular diseases (6). Insights into the precise function of TLR-mediated cardiac inflammatory signaling will aid in developing novel therapies for pathological cardiac hypertrophy.
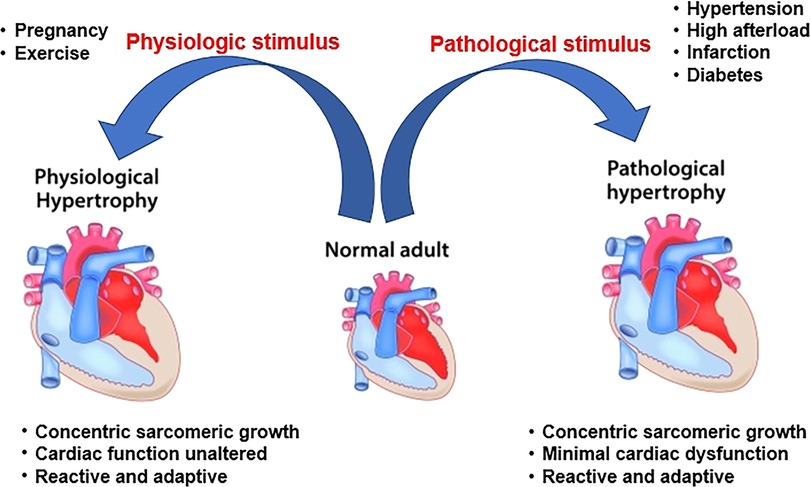
Figure 1. Phenotypes of physiological and pathological cardiac hypertrophy.
2. TLRs and downstream adaptors
TLRs have been first discovered in Drosophila melanogaster, playing a pivotal role in embryonic development and dorsal-ventral polarity (7, 8). The researchers have further found that the function of TLRs is related to innate and adaptive immunity (9). To date, 13 and 10 TLRs have been identified in mice and humans, respectively. Each TLR recognizes distinct microbial components. For example, a heterodimer of TLR2/1 or TLR2/6 recognizes lipoproteins, TLR3 responds to double-stranded RNA (dsRNA), TLR4 senses lipopolysaccharide (LPS), TLR5 binds to bacterial flagellin, TLR7/8 respond to single-stranded viral RNA (ssRNA), and TLR9 recognizes bacterial DNA containing unmethylated CpG motifs (10–15).
TLR family members usually dimerize themselves and recruit adaptor molecules with the same Toll- interleukin-1 (IL-1) receptor (TIR) domain to transmit signals. TLRs signals can be divided into myeloid differentiation factor 88 (MyD88)-dependent and MyD88-independent pathways (Figure 2). Except for TLR3, the signals of all TLR family members are conducted through the MyD88-dependent pathway, which induces the expression of proinflammatory cytokines, chemokines, and other inflammation-related molecules by activating nuclear factor-κB (NF-κB) and other transcription factors (8, 16). TLR3 signals through the MyD88-independent pathway, which includes another adaptor molecule, TIR domain-containing adaptor-inducing interferon-β (TRIF), also known as the TRIF-dependent pathway (17). TLR4 is the only TLR that triggers both MyD88- and TRIF-dependent pathways (18, 19). MyD88 and TRIF are TLR adaptor molecules, and other adaptor molecules include TIR domain-containing adaptor protein (TIRAP) and TRIF-related adaptor molecule (TRAM) (20–23).
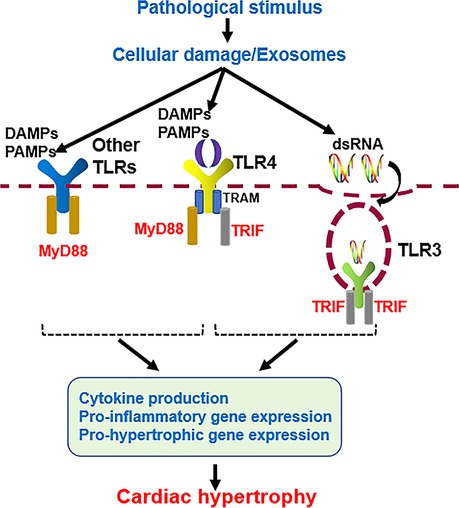
Figure 2. Cardiac hypertrophy mediated by MyD88-dependent and independent TLRs signaling pathways.
Numerous specific protein serine/threonine kinases participate in TLR signaling, such as IL-1 receptor-associated kinases (IRAKs), the transforming growth factor-β-activated kinase 1 (TAK-1), and the IκB kinase (IKK) complex. On the other hand, protein serine/threonine phosphatases, phospho-protein phosphatases (PPPs), metal-dependent protein phosphatases (PPMs), and aspartate-based phosphatases counterbalance and limit TLR signaling (24).
Different TLRs recognize specific ligands with distinct PAMPs and DAMPs, whereas all TLRs signals activate NF-κB. The excessive activation of TLR receptor signaling can also lead to autoimmune and inflammatory diseases (25). Consequently, different TLRs in a given pathological state may yield different outcomes that define the phenotype of tissue injury and organ damage.
3. Inflammation is involved in the pathogenesis of cardiac hypertrophy
Common pathophysiological mechanisms associated with cardiac hypertrophy include oxidative stress (26, 27), renin-angiotensin-aldosterone system (RAAS) (28), nervous system-activated sympathetic activity (29), pressure overload (30), and inflammation (31, 32). Inflammation is the pathological basis of myocardial hypertrophy (32). Other diseases such as hypertension (33) and ischemic injury (34) also provoke inflammatory responses, leading to cardiac hypertrophy. TLRs are widely expressed in many cells in the heart, and activating TLR-mediated inflammation signaling pathways promotes immune cell migration to the heart and cardiac fibroblast differentiation (35, 36). Therefore, TLR-induced inflammatory signaling is significant in the development of cardiac hypertrophy.
Importantly, immune cells play a pivotal role in the inflammatory response, but their cardioprotective or cardiodestructive effects differ after pressure overload. Particularly, neutrophils, dendritic cells (DCs), and mast cells demonstrate destructive functions in animal models, whereas eosinophils and natural killer T cells display cardioprotective activities (37). For example, S100 calcium-binding protein A8/A9 complex (S100a8/a9), an initial proinflammatory factor produced by neutrophils, activates the NF-κB pathway in angiotensin II (Ang II)-induced cardiac fibrosis and hypertrophy (38). Besides, TLR stimulation and DC infiltration are factors contributing to heart failure (39). Cardiac macrophages, which are highly plastic cells, are divided into two types of macrophages, including proinflammatory (M1) and anti-inflammatory (M2) phenotypes. M1 macrophages are associated with chronic inflammation, and M2 macrophages produce IL-10 and TGF-β1, which are related to tissue repair and fibrotic properties (33, 40). TLR4 activator LPS stimulates macrophages to produce mir-155 that promotes cardiac inflammation, followed by cardiac fibrosis, apoptosis, and hypertrophy (41). Overall, inflammatory cells infiltrate the heart by activating intracellular inflammatory signaling pathways, eventually contributing to cardiac hypertrophy and heart failure.
4. Toll-like receptors are associated with cardiac hypertrophy
TLRs are major components of the innate immune system that elicit cytokine and chemokine production primarily by activating the proinflammatory transcription factor NF-κB (18). Herein, we review several important TLRs associated with cardiac hypertrophy (Table 1).
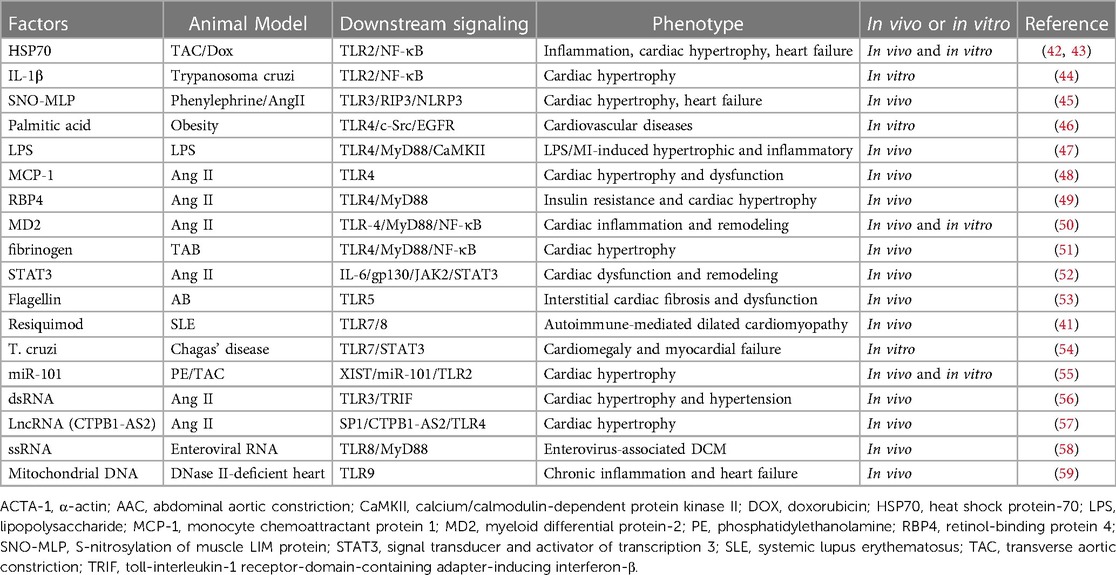
Table 1. Summary of various factors that can interact with specific TLRs, leading to cardiac hypertrophy and cardiac remodeling.
4.1. TLR2
TLR2 in complex with TLR1 or TLR6 is essential for recognizing bacterial lipoproteins and lipopeptides. After recognizing their ligands, the TLRs form stable TLR1-TLR2 or TLR2-TLR6 complexes (60). TLR2 activation has been associated with cardiovascular diseases (61, 62). Ye et al. have shown that TLR2 mediates cardiac hypertrophy and inflammation in Ang-II-treated mice through the TLR2/MyD88/NF-κB signaling pathway. Ang II significantly increased the level of the TLR2-MyD88 complex rather than that of TLR2 or MyD88 protein (63). Additionally, TLR2 activation upregulates NF-κB and inflammatory factors, such as IL-1β, which can induce cardiomyocyte hypertrophy and fibroblast and vascular endothelial cell proliferation (44). TLR2/NF-κB/IL-1β signaling is essential for activating the IGF-1/PI3K/Akt pathway and leads to adaptive cardiac hypertrophy during pressure overload (64). Besides, TLR2 is involved in renal ischemia/reperfusion (I/R)-induced cardiac hypertrophy by regulating the systemic inflammatory profile and NF-κB activation (65). A recent study has suggested that lncRNA X-inactive specific transcripts (XIST) could induce cardiac hypertrophy by targeting miR-101 and increasing TLR2 levels (55). Besides, studies have demonstrated that heat shock proteins (HSPs), such as HSP60 and HSP70, induced cardiac hypertrophy by activating NF-κB through the TLR2/MyD88-dependent pathway in Dox-induced animal models (42, 43). In contrast, HSP25 protects the heart from Dox-induced cardiotoxicity by antagonizing the binding of Dox to the TLR2 receptor (66). TLR2 deficiency in cardiac cells prevents Ang II-induced cardiac remodeling, inflammation, and dysfunction by reducing the formation of TLR2-MyD88 complexes (67). Obesity has been studied as an activator of DAMPs, which use the TLR2 signaling pathway to increase cytokine expression in heart tissue (68). Although TLR2 has been shown to induce cardiac hypertrophy, several studies have suggested that TLR2 is required for cardiac protection. TLR2-deficient mice have shown short-term advantages after myocardial I/R but promoted left ventricular dilation in the long term with reduced collagen and decorin density in the infarct scar (69). TLR2 stimulation also protects the heart from exaggerated autoimmunity in experimental autoimmune myocarditis by promoting regulatory DCs formation, which limits autoreactive T-cell responses (70). Therefore, the role of TLR2 in cardiac hypertrophy is destructive or protective, depending on the etiology and disease stage.
4.2. TLR3
TLR3 is located in the endoplasmic reticulum. Upon stimulation with dsRNA, TLR3 moves to the endosomes, where TLR3 is phosphorylated by Bruton's tyrosine kinase (BTK) and phosphorylated IRF3, triggering its downstream signaling (71, 72). Its adaptor protein for the dsRNA-induced signaling pathway is not MyD88 but TRIF (73, 74). TRIF also recruits additional proteins necessary for downstream signaling, including receptor-interacting protein 1 (RIP1), TNF receptor-associated factor 3 (TRAF3), nucleosome assembly protein 1 (NAP1), and TBK1. The TLR3/TRIF pathway then activates NF-κB and IFN regulatory factor 3 (IRF-3) (56). TLR3 deficiency in mice with Coxsackie virus B3 (CVB3) infection increases viral replication during the acute period of myocarditis. TLR3 deficiency also increases the level of cytokines related to T helper (Th) 2 response, such as IL-4, IL-10, IL-13, and TGF-β. IL-4 deficiency in mice improves heart function during acute CVB3 myocarditis, suggesting that TLR3 prevents myocarditis by reducing viral replication and IL-4 levels in the heart (75). S-nitrosylation of muscle LIM protein (MLP) induces TLR3-mediated RIP3 and nucleotide-binding oligomerization domain-like receptor pyrin domain containing 3 (NLRP3) inflammasome activation, thereby promoting the development of myocardial hypertrophy (45). Although Ang II activates both MyD88 and TRIF pathways, only the TRIF pathway is required to mediate hypertension and cardiac hypertrophy (76). A recent study has found that both TLR4-TRIF and TLR3-TRIF pathways mediate Ang II-induced cardiac hypertrophy, whereby only the TLR3-TRIF pathway is required for Ang II-induced hypertension (77).
4.3. TLR4
PAMPs and DAMPs act as exogenous or endogenous ligands for TLR4, respectively. Its co-receptor myeloid differentiation protein 2 (MD2) recognizes LPS and binds TLR4, followed by the activation of the TLR4 signaling pathway (78). Additionally, hyperthyroidism, enteroviral replication, and lifestyle-related diseases directly compromise the myocardial structure and lead to inflammation through TLR4 and downstream activation of the NLRP3 inflammasome or NF-κB-dependent pathways (16, 79, 80). For example, postnatal growth restriction (PNGR) and hyperoxia cause intestinal dysbiosis that activates pulmonary hypertension and, subsequently, promotes right ventricular hypertrophy via the TLR4/NF-κB/IL-1β pathway (81). Besides, TLR4 activation increases oxidative stress and activates MCP-1 expression, resulting in cardiac hypertrophy in Ang II-induced hypertension (48). TLR4 is the only member of the TLRs family that simultaneously activates intracellular signal transduction through two different signaling pathways, the MyD88-dependent and MyD88-independent pathways.
In the MyD88-dependent pathway, LPS binds to LPS-binding protein (LBP), and this complex then binds with CD14, transferring LPS to TLR4 and its co-receptor MD2 through hydrogen bonding on Arg-90, Glu-92, and Asp-100 (50, 82–84). Inside the cells, this CD14/TLR4/MD2 compound interacts with adaptor TIRAP, inducing IL receptor-associated kinase (IRAK) phosphorylation, MyD88 separation, and TRAF6 combination. Then, TRAF6 can activate NF-κB through TGF-β activated kinase 1 (TAK1) and MAPKs, such as JNK, extracellular-signal-regulated kinase (ERK), and p38 kinase, through mitogen-activated protein kinase ERKA 6 (MKK6), which, in turn, activates AP-1, leading to the expression of proinflammatory cytokines (51, 85–87). Several different inflammatory cytokines, including TNF-α, IL-6, and IL-1β, are induced through this signaling pathway (88). For example, retinol-binding protein 4 (RBP4) contributes to insulin resistance and heart failure by activating the TLR4/MyD88 signaling pathway (49).
Other MyD88-dependent pathways include the TLR4/MyD88/CaMK II, TLR4/MyD88/PI3K/Akt, and TLR4/MyD88/MAPK pathways, showing that TLR4/MyD88 downstream is more complicated in regulating cardiac hypertrophy. CaMK II belongs to serine/threonine kinases and plays an important role in cardiac structure remodeling and electrical activity (89). MyD88 leads to CaMK II oxidation and is essential for adverse cardiac hypertrophy and inflammation during myocardial infarction (47). The TLR4/MyD88/PI3K/Akt pathway has both adverse and protective effects on cardiac hypertrophy, probably due to the different PI3K isoforms. PI3K p110γ activates maladaptive cardiac hypertrophy, whereas PI3K p110α induces adaptive cardiac hypertrophy (90).
The MyD88-independent pathway is also named the TRIF-dependent pathway. IKKε and TBK1 are molecules downstream of TRIF, which activate NF-κB and IRF3, respectively (91, 92). NF-κB releases IκB from the binding complex, leading to NF-κB translocation from cytosol to the nucleus.
Interestingly, some molecules induce cardiac hypertrophy by multiple pathways. Nucleotide-binding oligomerization domain-2 (NOD2)-knockdown in mice increases cardiac hypertrophy and fibrosis by upregulating multiple pathways, including the TLR4/NF-κB, TLR4/MAPK, and TGF-β/Smad pathways (93). Besides, Ang II activates STAT3, which interacts with TLR4 and increases IL-6, and, in turn, promotes the second STAT3 activation, leading to an upregulated expression of genes for cardiac hypertrophy through the IL-6/glycoprotein 130 (gp130)/Janus-family tyrosine kinases 2 (JAK2) pathway (52).
4.4. TLR5
TLR5, a transmembrane protein, is highly expressed in immune cells, cardiomyocytes, and vascular endothelial cells. TLR5 triggers inflammatory responses and promotes cardiac hypertrophy, and the deficiency of TLR5 in mice attenuates the cardiac hypertrophy and dysfunction induced by pressure overload (53). TLR5 directly interacts with spleen tyrosine kinase and activates NADPH oxidase, stimulating the p38 MAP kinase pathway in DOX-induced cardiotoxicity (94).
4.5. TLR7/8
TLR8 mediates the antiviral response by recognizing ssRNA. TLR8 is associated with the immune response to enteroviral replication and may be involved in enterovirus-associated dilated cardiomyopathy (58). Additionally, both T. cruzi trypomastigotes (extracellular form) and amastigotes (intracellular form) induce cardiomyocyte apoptosis via TLR7 signaling to activate transcription factor STAT3, which then upregulates apoptotic gene BAX and downregulates anti-apoptotic gene BCL-2 (54). Furthermore, TLR7/8 agonist resiquimod causes myocarditis and dilated cardiomyopathy, mimicking the cardiac damage induced by systemic autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis. Furthermore, the cardiac damage may be due to the systemic increase in inflammation or the direct autoimmune response toward the heart (95).
4.6. TLR9
TLR9, a receptor for unmethylated CpG-DNA, bacterial DNA, viral DNA, and fungi, was first cloned and identified in 2,000 (96–99). When TLR9 is activated by binding with its ligands, it can induce a TLR9-mediated immune response, such as an antiviral response, and the production of type I IFN through plasmacytoid DCs (100). TLR9 induces NF-κB via the MyD88-dependent pathway, where CD82 acts as an important regulator of TLR9-mediated signaling in cancer, infectious diseases, and autoimmune diseases (101). Furthermore, TLR9 triggers innate and adaptive immune responses against pathogens, such as Brucella, Streptococcus pneumoniae, Helicobacter, mouse cytomegalovirus (MCMV), herpes simplex virus (HSV) types 1 and 2, and adenovirus (102–108). Bacterial DNA could mediate neutrophil signaling by TLR9-independent and MyD88-dependent pathways (109). Mitochondrial DNA escapes from cell autophagy and leads to TLR9-mediated inflammatory responses in cardiomyocytes, followed by myocarditis and dilated cardiomyopathy (59). Inhibiting TLR9/NF-κB-mediated sterile inflammation also improves pressure overload-induced right ventricular dysfunction (110). However, TLR9 also mediates the cardiac protection of oligonucleotides or peptides. Synthetic oligonucleotides (ODNs), such as CpG-ODN C274 and 1668-thioate, attenuate ISO (isoproterenol) or I/R-induced cardiac hypertrophy by activating TLR9-mediated PI3K/AKT signaling (111, 112). Wang et al. have demonstrated that cathelicidin-related antimicrobial peptide (CRAMP) inhibited the cardiac hypertrophic response by activating the IGFR1/PI3K/AKT pathway and ameliorated cardiac oxidative stress by activating the TLR9/AMPK pathway in cardiomyocytes. TLR9 is required for the anti-oxidative effect of mCRAMP, as demonstrated by using TLR9-knockout mice. Additionally, TLR9 knockout partly reverses the antihypertrophic effect of mCRAMP, suggesting that TLR9 also contributes to protecting cardiomyocytes from hypertrophy induced by pressure overload (113).
5. Potential therapeutic approaches in cardiac hypertrophy
Several promising drugs and technologies have been developed to attenuate TLR-mediated inflammatory response and reverse cardiac hypertrophy (114). Thus, TLRs and TLR signaling medications might be potential treatment approaches in cardiac hypertrophy.
An alternative therapeutic strategy is blocking TLR upstream molecules to diminish inflammation and attenuate cardiac hypertrophy. For example, some protein molecules, such as modified citrus pectin (a specific inhibitor of galectin-3), cardiac transmembrane BAX inhibitor motif containing 1 (TMBIM1), HMGB1, EGFR, human mesenchymal stem cells, and erythropoietin (EPO), in the heart reverse pressure overload-induced cardiac hypertrophy by blocking the TLR4 signaling pathway (109, 115–117). Inhibiting some nucleic acid molecules, such as lncRNA CTPB1-AS2, lncRNA NEAT1 and miR-93, can ameliorate cardiac hypertrophy by downregulating TLR4 signaling (57–119). Silencing of protein molecule-like fatty acid-binding protein 4 protects against LPS-induced cardiomyocyte hypertrophy and apoptosis by inhibiting the TLR4/NF-κB pathway (120). Besides, CaMKIIδB silencing prevents cardiac hypertrophy independent of an inflammatory response by inhibiting the complement system and TLR2/4 NF-kB signaling (121).
In addition, TLR inhibitors can decrease cardiac hypertrophy. Some chemical compounds, such as choline and eritoran, ameliorate cardiac hypertrophy by inhibiting TLR4, which decreases inflammatory cytokines, such as IL-1β and IL-6, and increases anti-inflammatory cytokines, such as IL-10 (122, 123). Other chemical compounds, such as Triad3A (a ubiquitin E3 ligase), TAK-242, and lipopolysaccharide from Rhodobacter sphaeroides (LPS-RS), have been reported to negatively regulate the NF-κB activation pathway via the inhibition of TLR4/TLR9 or TLR4 and subsequently inhibit cardiac disease (124–127). Additionally, Ang II-induced microglia activation and oxidative stress are linked to TLR4 activation in the paraventricular nucleus (128). Inhibiting TLR4 within the paraventricular nucleus (PVN, an important cardioregulatory center in the brain) attenuates blood pressure and inflammation (129). Calcitriol infusion in the PVN ameliorates hypertensive responses and cardiac hypertrophy by decreasing TLR4-associated inflammation (130). Recombinant human relaxin (RLX) and bioactive peptides attenuated cardiac hypertrophy, inflammation, and fibrosis and appeared to involve the inhibition of TLR4 (131, 132). Interestingly, studies have reported that silencing TLR4 gene through siRNA prevents the development of diabetic cardiomyopathy in streptozotocin-induced type 1 diabetes (133).
Some TLR/MyD88 signaling inhibitors also ameliorate cardiac hypertrophy. Receptor-interacting serine/threonine-protein kinase 2 (RIP2) deficiency ameliorates cardiac hypertrophy through multiple signaling pathways that reduce TLR4/MyD88/NF-κB activation and MAPKs phosphorylation (134). In contrast, some molecules and compounds negatively regulate cardiac hypertrophy by suppressing TLR4/MyD88 signaling, which includes protein molecules such as MD1 and anti-HSP70 antibody and compounds such as Ang II type 1 receptor (AT1-R) antagonist and liver × receptors agonist (135–138). Besides, long-term oral atazanavir attenuates myocardial infarction-induced cardiac fibrosis by targeting the HMGB1/TLR9 signaling pathway (139). Pharmacologic inhibition of the MyD88 inhibitor, ST2825/IMG2005, protects against pathologic cardiac remodeling (140, 141). Moreover, other types of cardiovascular drugs, such as telmisartan, magnesium isoglycyrrhizinate (MgIG), dimethyl fumarate (DMF), and statins, including atorvastatin and simvastatin, effectively suppress the TLR-4/NF-κB signaling pathway and protect against cardiac remolding in pressure overload, chronic intermittent hypoxia, and LPS-induced conditions (46, 142–145). Traditional Chinese Medicine drugs, such as Shenqi Yangxin decoction (SQYXD), Lycium barbarum polysaccharide (LBP), arbutin, Astragaloside IV (AsIV), and Dangshen Erling decoction (DSELD) have been shown to attenuate cardiac hypertrophy by targeting the high mobility group box 1 (HMGB1)/receptor for advanced glycation end products (RAGE) and TLR4/NF-κB signaling pathway (146–150). A recent report showed that double overexpression of miR-19a and miR-20a (dOex-mIRs) in human induced pluripotent stem cell (iPS)-derived mesenchymal stem cells (MSCs) effectively preserves the left ventricular function in dilated cardiomyopathy through, at least in part, regulating TLR4/MAL/MyD88 signaling pathway (151). Nevertheless, more clinical trials and reliable measurements regarding therapeutic approaches targeting TLR signaling pathways are needed. The factors that inhibit TLR signaling-mediated cardiac hypertrophy and cardiac remodeling are listed in Table 2.
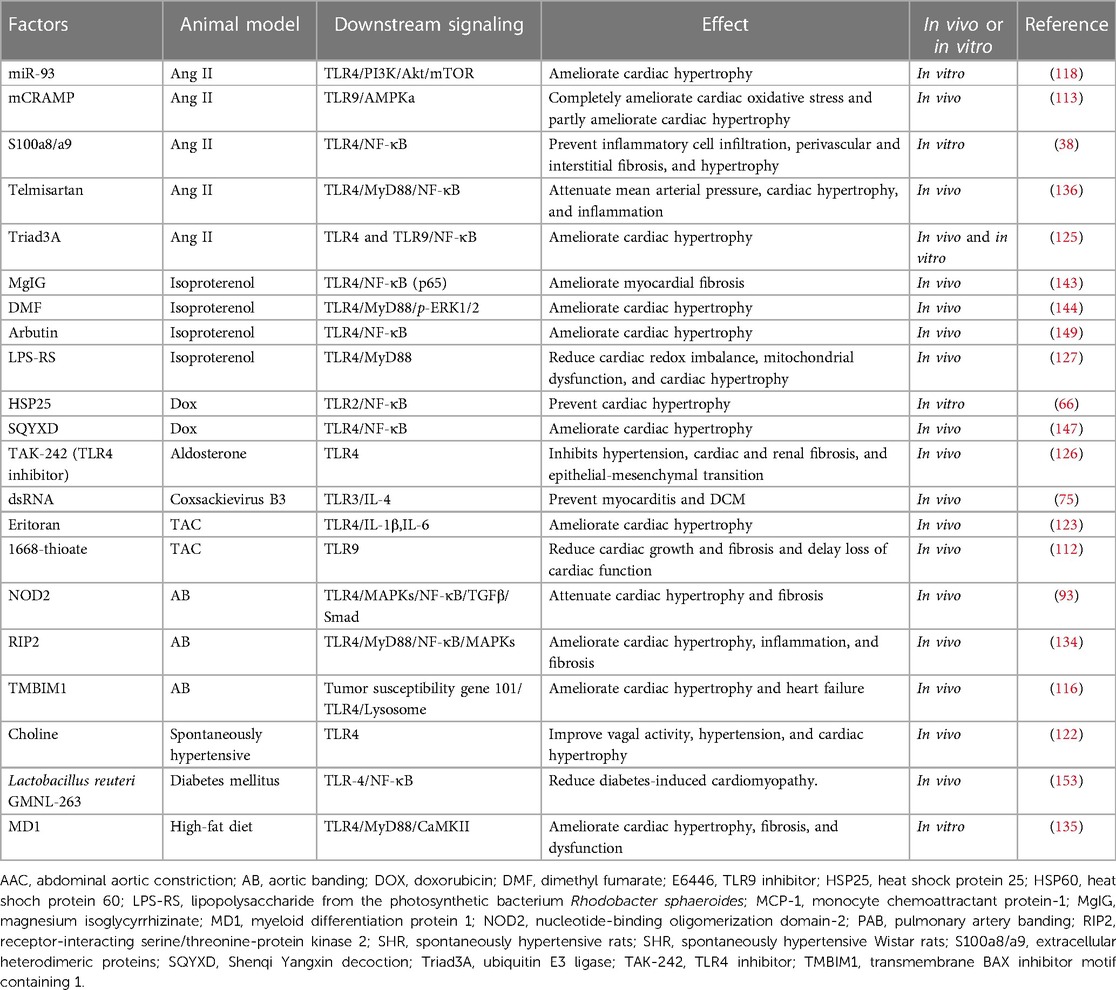
Table 2. Summary of various inhibitors that can interact with TLR signaling and ameliorate cardiac hypertrophy and cardiac remodeling.
Moreover, rather than directly targeting the TLR signaling pathway, some indirect strategies may provide additional therapeutic benefits for cardiovascular diseases. Caloric restriction is an effective therapeutic approach in the treatment of diabetes and associated cardiomyopathy by inhibition of TLR2 and TLR4 (152). Besides, probiotics Lactobacillus reuteri GMNL-263, Bifidobacterium breve CECT7263 (BFM), Lactobacillus fermentum CECT5716 (LC40), and L. coryniformis CECT5711 (K8) plus L. gasseri CECT5714 (LC9) (1:1), prevent dysbiosis, endothelial dysfunction, endotoxemia, and high blood pressure and ameliorate cardiac hypertrophy via the downregulation of their indirect target TLR4 (153–155). Apart from these, renal denervation and repetitive hyperthermia (RHT) attenuate the development of cardiac hypertrophy, at least in part by inhibiting TLR4 expression (19, 156).
6. Conclusion and future perspectives
Increased inflammatory factors and cytokines are clearly associated with cardiac hypertrophy and TLRs. In this review, we summarized comprehensive information about TLRs, such as TLR2, TLR3, TLR4, TLR5, TLR7/8, and TLR9, which are closely related to cardiac hypertrophy. TLRs interact with their ligands and co-receptors to induce the expression of numerous inflammatory factors and inflammatory cell infiltration in the heart, leading to cardiac hypertrophy and heart failure through various inflammatory signaling pathways. Reviewing the interaction between TLRs and inflammation in cardiac hypertrophy may be a research direction for the treatment of cardiovascular diseases and other inflammatory-related diseases. However, the link between TLRs and cardiac hypertrophy has not been fully explored. For example, little has been reported about the role of TLR7/8 and TLR9 in cardiac hypertrophy, especially how they mediate inflammatory signaling pathways and heart diseases. Additionally, the TLR family not only regulates inflammation but is also one of the essential mediators of the innate immune response. It is worth noting that the excessive activation of TLRs can lead to chronic inflammation and autoimmune diseases, while TLR defects can lead to cancer and allergies (157, 158). Therefore, the TLR family might play a variety of different roles in cardiovascular diseases. Still, it may need a deeper exploration of the TLR signaling pathway related to cardiac hypertrophy.
The engagement of different TLR ligands leads to unique cytokine production (159). It is likely that cross-talk within various TLR pathways is highly complex and contains many unknowns (160). Although there are many challenges in developing drugs and balancing TLR signaling, in consideration of molecular targeting therapy against TLRs and signaling molecules might be a promising approach in clinical treatment, many research centers and pharmaceutical companies are expending extensive efforts to develop TLRs modulators. Some of the TLR-based agonistic and antagonistic agents have shown to be efficacious in preclinical models and have now entered clinical trials (161, 162). Overall, these interesting findings encouraged us to set a further goal to understand the detailed mechanism of TLR-mediated inflammatory responses and cardiac hypertrophy and identify the potential targets of therapeutic interventions through TLRs' downstream and upstream signaling pathways.
Author contributions
Conceptualization, ZW and YZ; writing—original draft preparation, YZ; writing—review and editing, JW, ZW, and HX; supervision, ED. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (82030072 to HX, 82170296 to ZW). CAMS Innovation Fund for Medical Sciences (2021-I2M-5–003 to ED) and the Key Clinical Projects of Peking University Third Hospital (BYSYZD2019022 to HX).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Oldfield CJ, Duhamel TA, Dhalla NS. Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can J Physiol Pharmacol. (2020) 98:74–84. doi: 10.1139/cjpp-2019-0566
2. Wang L, Wang J, Li G, Xiao J. Non-coding RNAs in physiological cardiac hypertrophy. Adv Exp Med Biol. (2020) 1229:149–61. doi: 10.1007/978-981-15-1671-9_8
3. Samak M, Fatullayev J, Sabashnikov A, Zeriouh M, Schmack B, Farag M, et al. Cardiac hypertrophy: an introduction to molecular and cellular basis. Med Sci Monit Basic Res. (2016) 22:75–9. doi: 10.12659/MSMBR.900437
4. Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. (2010) 128:191–227. doi: 10.1016/j.pharmthera.2010.04.005
5. Besse S, Nadaud S, Balse E, Pavoine C. Early protective role of inflammation in cardiac remodeling and heart failure: focus on TNFα and resident macrophages. Cells. (2022) 11:1249. doi: 10.3390/cells11071249
6. Jaén RI, Val-Blasco A, Prieto P, Gil-Fernández M, Smani T, López-Sendón JL, et al. Innate immune receptors, key actors in cardiovascular diseases. JACC Basic Transl Sci. (2020) 5:735–49. doi: 10.1016/j.jacbts.2020.03.015
7. Imler JL, Hoffmann JA. Toll receptors in Drosophila: a family of molecules regulating development and immunity. Curr Top Microbiol Immunol. (2002) 270:63–79. doi: 10.1007/978-3-642-59430-4_4
8. Hallman M, Rämet M, Ezekowitz RA. Toll-like receptors as sensors of pathogens. Pediatr Res. (2001) 50:315–21. doi: 10.1203/00006450-200109000-00004
9. Vijay K. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int Immunopharmacol. (2018) 59:391–412. doi: 10.1016/j.intimp.2018.03.002
10. Venkataranganayaka Abhilasha K, Kedihithlu Marathe G. Bacterial lipoproteins in sepsis. Immunobiology. (2021) 226:152128. doi: 10.1016/j.imbio.2021.152128
11. Sen GC, Sarkar SN. Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growth Factor Rev. (2005) 16:1–14. doi: 10.1016/j.cytogfr.2005.01.006
12. Parra-Flores P, Espitia-Corredor J, Espinoza-Pérez C, Queirolo C, Ayala P, Brüggendieck F, et al. Toll-like receptor 4 activation prevents rat cardiac fibroblast death induced by simulated ischemia/reperfusion. Front Cardiovasc Med. (2021) 8:660197. doi: 10.3389/fcvm.2021.660197
13. Yoon SI, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, et al. Structural basis of TLR5-flagellin recognition and signaling. Science. (2012) 335:859–64. doi: 10.1126/science.1215584
14. Yu H, Wang Z, Sun G, Yu Y. Recognition of nucleic acid ligands by toll-like receptors 7/8: importance of chemical modification. Curr Med Chem. (2012) 19:1365–77. doi: 10.2174/092986712799462603
15. Krieg AM. Cpg motifs in bacterial DNA and their immune effects. Annu Rev Immunol. (2002) 20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842
16. Yao J, Li Y, Jin Y, Chen Y, Tian L, He W. Synergistic cardioptotection by tilianin and syringin in diabetic cardiomyopathy involves interaction of TLR4/NF-κB/NLRP3 and PGC1a/SIRT3 pathways. Int Immunopharmacol. (2021) 96:107728. doi: 10.1016/j.intimp.2021.107728
17. Seya T, Tatematsu M, Matsumoto M. Toward establishing an ideal adjuvant for non-inflammatory immune enhancement. Cells. (2022) 11:4006. doi: 10.3390/cells11244006
18. Lin C, Wang H, Zhang M, Mustafa S, Wang Y, Li H, et al. TLR4 Biased small molecule modulators. Pharmacol Ther. (2021) 228:107918. doi: 10.1016/j.pharmthera.2021.107918
19. Oyama J, Maeda T, Sasaki M, Higuchi Y, Node K, Makino N. Repetitive hyperthermia attenuates progression of left ventricular hypertrophy and increases telomerase activity in hypertensive rats. Am J Physiol Heart Circ Physiol. (2012) 302:H2092–101. doi: 10.1152/ajpheart.00225.2011
20. Zhang Q, Feng A, Zeng M, Zhang B, Shi J, Lv Y, et al. Chrysosplenol D protects mice against LPS-induced acute lung injury by inhibiting oxidative stress, inflammation, and apoptosis via TLR4-MAPKs/NF-κB signaling pathways. Innate Immun. (2021) 27:514–24. doi: 10.1177/17534259211051069
21. Noreen M, Arshad M. Association of TLR1, TLR2, TLR4, TLR6, and TIRAP polymorphisms with disease susceptibility. Immunol Res. (2015) 62:234–52. doi: 10.1007/s12026-015-8640-6
22. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 And CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. (2021) 78:1233–61. doi: 10.1007/s00018-020-03656-y
23. Takeda K, Akira S. TLR Signaling pathways. Semin Immunol. (2004) 16:3–9. doi: 10.1016/j.smim.2003.10.003
24. Seumen CHT, Grimm TM, Hauck CR. Protein phosphatases in TLR signaling. Cell Commun Signal. (2021) 19:45. doi: 10.1186/s12964-021-00722-1
25. Chen JQ, Szodoray P, Zeher M. Toll-like receptor pathways in autoimmune diseases. Clin Rev Allergy Immunol. (2016) 50:1–17. doi: 10.1007/s12016-015-8473-z
26. Ramachandra CJA, Cong S, Chan X, Yap EP, Yu F, Hausenloy DJ. Oxidative stress in cardiac hypertrophy: from molecular mechanisms to novel therapeutic targets. Free Radic Biol Med. (2021) 166:297–312. doi: 10.1016/j.freeradbiomed.2021.02.040
27. Maulik SK, Kumar S. Oxidative stress and cardiac hypertrophy: a review. Toxicol Mech Methods. (2012) 22:359–66. doi: 10.3109/15376516.2012.666650
28. Raman VK, Lee YA, Lindpaintner K. The cardiac renin-angiotensin-aldosterone system and hypertensive cardiac hypertrophy. Am J Cardiol. (1995) 76:18D–23D. doi: 10.1016/s0002-9149(99)80487-1
29. Ghosh SS, Krieg RJ, Sica DA, Wang R, Fakhry I, Gehr T. Cardiac hypertrophy in neonatal nephrectomized rats: the role of the sympathetic nervous system. Pediatr Nephrol. (2009) 24:367–77. doi: 10.1007/s00467-008-0978-8
30. Frohlich ED, Susic D. Pressure overload. Heart Fail Clin. (2012) 8:21–32. doi: 10.1016/j.hfc.2011.08.005
31. Bacmeister L, Schwarzl M, Warnke S, Stoffers B, Blankenberg S, Westermann D, et al. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol. (2019) 114:19. doi: 10.1007/s00395-019-0722-5
32. Xu L, Brink M. mTOR, cardiomyocytes and inflammation in cardiac hypertrophy. Biochim Biophys Acta. (2016) 1863:1894–903. doi: 10.1016/j.bbamcr.2016.01.003
33. Fujiu K, Wang J, Nagai R. Cardioprotective function of cardiac macrophages. Cardiovasc Res. (2014) 102:232–9. doi: 10.1093/cvr/cvu059
34. Gounis MJ, van der Marel K, Marosfoi M, Mazzanti ML, Clarençon F, Chueh JY, et al. Imaging inflammation in cerebrovascular disease. Stroke. (2015) 46:2991–7. doi: 10.1161/STROKEAHA.115.008229
35. Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. (2011) 8:292–300. doi: 10.1038/nrcardio.2011.38
36. Song L, Zhang Z, Qiu Z, Jiang T. Serum metabonomic study of patients with acute coronary syndrome using ultra-performance liquid chromatography orbitrap mass spectrometer. Front Cardiovasc Med. (2021) 8:637621. doi: 10.3389/fcvm.2021.637621
37. Liu X, Shi GP, Guo J. Innate immune cells in pressure overload-induced cardiac hypertrophy and remodeling. Front Cell Dev Biol. (2021) 9:659666. doi: 10.3389/fcell.2021.659666
38. Wu Y, Li Y, Zhang C, A X, Wang Y, Cui W, et al. S100a8/a9 released by CD11b + Gr1 + neutrophils activates cardiac fibroblasts to initiate angiotensin II-induced cardiac inflammation and injury. Hypertension. (2014) 63:1241–50. doi: 10.1161/HYPERTENSIONAHA.113.02843
39. Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med. (2003) 9:1484–90. doi: 10.1038/nm960
40. Yang D, Liu HQ, Liu FY, Tang N, Guo Z, Ma SQ, et al. Critical roles of macrophages in pressure overload-induced cardiac remodeling. J Mol Med (Berl). (2021) 99:33–46. doi: 10.1007/s00109-020-02002-w
41. Zahid A, Ismail H, Li B, Jin T. Molecular and structural basis of DNA sensors in antiviral innate immunity. Front Immunol. (2020) 11:613039. doi: 10.3389/fimmu.2020.613039
42. Ferenčić A, Cuculić D, Stemberga V, Šešo B, Arbanas S, Jakovac H. Left ventricular hypertrophy is associated with overexpression of HSP60, TLR2, and TLR4 in the myocardium. Scand J Clin Lab Invest. (2020) 80:236–46. doi: 10.1080/00365513.2020.1725977
43. Liu P, Bao HY, Jin CC, Zhou JC, Hua F, Li K, et al. Targeting extracellular heat shock protein 70 ameliorates doxorubicin-induced heart failure through resolution of toll-like receptor 2-mediated myocardial inflammation. J Am Heart Assoc. (2019) 8:e012338. doi: 10.1161/JAHA.119.012338
44. Petersen CA, Krumholz KA, Burleigh BA. Toll-like receptor 2 regulates interleukin-1beta-dependent cardiomyocyte hypertrophy triggered by Trypanosoma cruzi. Infect Immun. (2005) 73:6974–80. doi: 10.1128/IAI.73.10.6974-6980.2005
45. Tang X, Pan L, Zhao S, Dai F, Chao M, Jiang H, et al. SNO-MLP (S-nitrosylation of muscle LIM protein) facilitates myocardial hypertrophy through TLR3 (toll-like receptor 3)-mediated RIP3 (receptor-interacting protein kinase 3) and NLRP3 (NOD-like receptor pyrin domain containing 3) inflammasome activation. Circulation. (2020) 141:984–1000. doi: 10.1161/CIRCULATIONAHA.119.042336
46. Li W, Fang Q, Zhong P, Chen L, Wang L, Zhang Y, et al. EGFR Inhibition blocks palmitic acid-induced inflammation in cardiomyocytes and prevents hyperlipidemia-induced cardiac injury in mice. Sci Rep. (2016) 6:24580. doi: 10.1038/srep24580
47. Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. Myd88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol. (2012) 52:1135–44. doi: 10.1016/j.yjmcc.2012.01.021
48. Matsuda S, Umemoto S, Yoshimura K, Itoh S, Murata T, Fukai T, et al. Angiotensin II activates MCP-1 and induces cardiac hypertrophy and dysfunction via toll-like receptor 4. J Atheroscler Thromb. (2015) 22:833–44. doi: 10.5551/jat.27292
49. Gao W, Wang H, Zhang L, Cao Y, Bao JZ, Liu ZX, et al. Retinol-binding protein 4 induces cardiomyocyte hypertrophy by activating TLR4/MyD88 pathway. Endocrinology. (2016) 157:2282–93. doi: 10.1210/en.2015-2022
50. Han J, Zou C, Mei L, Zhang Y, Qian Y, You S, et al. MD2 Mediates angiotensin II-induced cardiac inflammation and remodeling via directly binding to ang II and activating TLR4/NF-κB signaling pathway. Basic Res Cardiol. (2017) 112:9. doi: 10.1007/s00395-016-0599-5
51. Li T, Wang Y, Liu C, Hu Y, Wu M, Li J, et al. MyD88-dependent nuclear factor-kappaB activation is involved in fibrinogen-induced hypertrophic response of cardiomyocytes. J Hypertens. (2009) 27:1084–93. doi: 10.1097/HJH.0b013e3283293c93
52. Han J, Ye S, Zou C, Chen T, Wang J, Li J, et al. Angiotensin II causes biphasic STAT3 activation through TLR4 to initiate cardiac remodeling. Hypertension. (2018) 72:1301–11. doi: 10.1161/HYPERTENSIONAHA.118.11860
53. Liu Y, Hu ZF, Liao HH, Liu W, Liu J, Ma ZG, et al. Toll-like receptor 5 deficiency attenuates interstitial cardiac fibrosis and dysfunction induced by pressure overload by inhibiting inflammation and the endothelial-mesenchymal transition. Biochim Biophys Acta. (2015) 1852:2456–66. doi: 10.1016/j.bbadis.2015.08.013
54. Stahl P, Ruppert V, Meyer T, Schmidt J, Campos MA, Gazzinelli RT, et al. Trypomastigotes and amastigotes of Trypanosoma cruzi induce apoptosis and STAT3 activation in cardiomyocytes in vitro. Apoptosis. (2013) 18:653–63. doi: 10.1007/s10495-013-0822-x
55. Xiao L, Gu Y, Sun Y, Chen J, Wang X, Zhang Y, et al. The long noncoding RNA XIST regulates cardiac hypertrophy by targeting miR-101. J Cell Physiol. (2019) 234:13680–92. doi: 10.1002/jcp.28047
56. Funami K, Sasai M, Ohba Y, Oshiumi H, Seya T, Matsumoto M. Spatiotemporal mobilization of toll/IL-1 receptor domain-containing adaptor molecule-1 in response to dsRNA. J Immunol. (2007) 179:6867–72. doi: 10.4049/jimmunol.179.10.6867
57. Luo X, He S, Hu Y, Liu J, Chen X. Sp1-induced LncRNA CTBP1-AS2 is a novel regulator in cardiomyocyte hypertrophy by interacting with FUS to stabilize TLR4. Cardiovasc Pathol. (2019) 42:21–9. doi: 10.1016/j.carpath.2019.04.005
58. Satoh M, Akatsu T, Ishikawa Y, Minami Y, Takahashi Y, Nakamura M. Association between toll-like receptor 8 expression and adverse clinical outcomes in patients with enterovirus-associated dilated cardiomyopathy. Am Heart J. (2007) 154:581–8. doi: 10.1016/j.ahj.2007.05.010
59. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. (2012) 485:251–5. doi: 10.1038/nature10992
60. Jin MS, Lee JO. Structures of the toll-like receptor family and its ligand complexes. Immunity. (2008) 29:182–91. doi: 10.1016/j.immuni.2008.07.007
61. Wang JW, Fontes MSC, Wang X, Chong SY, Kessler EL, Zhang YN, et al. Leukocytic toll-like receptor 2 deficiency preserves cardiac function and reduces fibrosis in sustained pressure overload. Sci Rep. (2017) 7:9193. doi: 10.1038/s41598-017-09451-3
62. Lu X, Gong S, Wang X, Hu N, Pu D, Zhang J, et al. Celastrol exerts cardioprotective effect in rheumatoid arthritis by inhibiting TLR2/HMGB1 signaling pathway-mediated autophagy. Int Arch Allergy Immunol. (2021) 182:1245–54. doi: 10.1159/000517185
63. Ye S, Lin K, Wu G, Xu MJ, Shan P, Huang W, et al. Toll-like receptor 2 signaling deficiency in cardiac cells ameliorates ang II-induced cardiac inflammation and remodeling. Transl Res. (2021) 233:62–76. doi: 10.1016/j.trsl.2021.02.011
64. Higashikuni Y, Tanaka K, Kato M, Nureki O, Hirata Y, Nagai R, et al. Toll-like receptor-2 mediates adaptive cardiac hypertrophy in response to pressure overload through interleukin-1β upregulation via nuclear factor κB activation. J Am Heart Assoc. (2013) 2:e000267. doi: 10.1161/JAHA.113.000267
65. Trentin-Sonoda M, da Silva RC, Kmit FV, Abrahão MV, Monnerat Cahli G, Brasil GV, et al. Knockout of toll-like receptors 2 and 4 prevents renal ischemia-reperfusion-induced cardiac hypertrophy in mice. PLoS One. (2015) 10:e0139350. doi: 10.1371/journal.pone.0139350
66. Krishnamurthy K, Kanagasabai R, Druhan LJ, Ilangovan G. Heat shock protein 25-enriched plasma transfusion preconditions the heart against doxorubicin-induced dilated cardiomyopathy in mice. J Pharmacol Exp Ther. (2012) 341:829–39. doi: 10.1124/jpet.112.192245
67. Bualeong T, Kebir S, Hof D, Goelz L, Graewe M, Ehrentraut SF, et al. Tlr2 deficiency does not limit the development of left ventricular hypertrophy in a model of transverse aortic constriction induced pressure overload. J Negat Results Biomed. (2016) 15:9. doi: 10.1186/s12952-016-0050-3
68. Gaisler-Silva F, Junho CVC, Fredo-da-Costa I, Christoffolete MA, Carneiro-Ramos MS. Diet-induced obesity differentially modulates cardiac inflammatory status in the C57 and FVB mouse strains. Curr Mol Med. (2022) 22:365–73. doi: 10.2174/1566524021666210603163613
69. Mersmann J, Habeck K, Latsch K, Zimmermann R, Jacoby C, Fischer JW, et al. Left ventricular dilation in toll-like receptor 2 deficient mice after myocardial ischemia/reperfusion through defective scar formation. Basic Res Cardiol. (2011) 106:89–98. doi: 10.1007/s00395-010-0127-y
70. Kania G, Siegert S, Behnke S, Prados-Rosales R, Casadevall A, Lüscher TF, et al. Innate signaling promotes formation of regulatory nitric oxide-producing dendritic cells limiting T-cell expansion in experimental autoimmune myocarditis. Circulation. (2013) 127:2285–94. doi: 10.1161/CIRCULATIONAHA.112.000434
71. Schröder M, Bowie AG. TLR3 In antiviral immunity: key player or bystander? Trends Immunol. (2005) 26:462–8. doi: 10.1016/j.it.2005.07.002
72. Lee KG, Xu S, Kang ZH, Huo J, Huang M, Liu D, et al. Bruton's tyrosine kinase phosphorylates toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci U S A. (2012) 109:5791–6. doi: 10.1073/pnas.1119238109
73. Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly (I:c). Adv Drug Deliv Rev. (2008) 60:805–12. doi: 10.1016/j.addr.2007.11.005
74. Gorbea C, Makar KA, Pauschinger M, Pratt G, Bersola JL, Varela J, et al. A role for toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J Biol Chem. (2010) 285:23208–23. doi: 10.1074/jbc.M109.047464
75. Abston ED, Coronado MJ, Bucek A, Onyimba JA, Brandt JE, Frisancho JA, et al. TLR3 Deficiency induces chronic inflammatory cardiomyopathy in resistant mice following coxsackievirus B3 infection: role for IL-4. Am J Physiol Regul Integr Comp Physiol. (2013) 304:R267–77. doi: 10.1152/ajpregu.00516.2011
76. Singh MV, Cicha MZ, Meyerholz DK, Chapleau MW, Abboud FM. Dual activation of TRIF and MyD88 adaptor proteins by angiotensin II evokes opposing effects on pressure, cardiac hypertrophy, and inflammatory gene expression. Hypertension. (2015) 66:647–56. doi: 10.1161/HYPERTENSIONAHA.115.06011
77. Singh MV, Cicha MZ, Nunez S, Meyerholz DK, Chapleau MW, Abboud FM. Angiotensin II-induced hypertension and cardiac hypertrophy are differentially mediated by TLR3- and TLR4-dependent pathways. Am J Physiol Heart Circ Physiol. (2019) 316:H1027–38. doi: 10.1152/ajpheart.00697.2018
78. Mishra V, Pathak C. Structural insights into pharmacophore-assisted in silico identification of protein-protein interaction inhibitors for inhibition of human toll-like receptor 4—myeloid differentiation factor-2 (hTLR4-MD-2) complex. J Biomol Struct Dyn. (2019) 37:1968–91. doi: 10.1080/07391102.2018.1474804
79. Satoh M, Nakamura M, Akatsu T, Shimoda Y, Segawa I, Hiramori K. Toll-like receptor 4 is expressed with enteroviral replication in myocardium from patients with dilated cardiomyopathy. Lab Invest. (2004) 84:173–81. doi: 10.1038/labinvest.3700031
80. Teixeira RB, Barboza TE, DE Araujo CC, Siqueira R, DE Castro AL, Bonetto JHP, et al. Decreased PGC1- α levels and increased apoptotic protein signaling are associated with the maladaptive cardiac hypertrophy in hyperthyroidism. J Biosci. (2018) 43:887–95. doi: 10.1007/s12038-018-9816-8
81. Wedgwood S, Gerard K, Halloran K, Hanhauser A, Monacelli S, Warford C, et al. Intestinal dysbiosis and the developing lung: the role of toll-like receptor 4 in the gut-lung axis. Front Immunol. (2020) 11:357. doi: 10.3389/fimmu.2020.00357
82. Sumneang N, Apaijai N, Chattipakorn SC, Chattipakorn N. Myeloid differentiation factor 2 in the heart: bench to bedside evidence for potential clinical benefits? Pharmacol Res. (2021) 163:105239. doi: 10.1016/j.phrs.2020.105239
83. da Silva Correia J, Ulevitch RJ. MD-2 and TLR4 N-linked glycosylations are important for a functional lipopolysaccharide receptor. J Biol Chem. (2002) 277:1845–54. doi: 10.1074/jbc.M109910200
84. Płóciennikowska A, Hromada-Judycka A, Borzęcka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. (2015) 72:557–81. doi: 10.1007/s00018-014-1762-5
85. Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys Res Commun. (2008) 368:94–9. doi: 10.1016/j.bbrc.2008.01.061
86. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM Couples endocytosis of toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. (2008) 9:361–8. doi: 10.1038/ni1569
87. Jakus PB, Kalman N, Antus C, Radnai B, Tucsek Z, Gallyas F Jr, et al. TRAF6 Is functional in inhibition of TLR4-mediated NF-κB activation by resveratrol. J Nutr Biochem. (2013) 24:819–23. doi: 10.1016/j.jnutbio.2012.04.017
88. Zhang Q, Sun W, Li T, Liu F. Polarization behavior of bone macrophage as well as associated osteoimmunity in glucocorticoid-induced osteonecrosis of the femoral head. J Inflamm Res. (2023) 16:879–94. doi: 10.2147/JIR.S401968
89. Zhong P, Quan D, Peng J, Xiong X, Liu Y, Kong B, et al. Role of CaMKII in free fatty acid/hyperlipidemia-induced cardiac remodeling both in vitro and in vivo. J Mol Cell Cardiol. (2017) 109:1–16. doi: 10.1016/j.yjmcc.2017.06.010
90. Aoyagi T, Matsui T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr Pharm Des. (2011) 17:1818–24. doi: 10.2174/138161211796390976
91. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. (2003) 301:640–3. doi: 10.1126/science.1087262
92. Frantz S, Ertl G, Bauersachs J. Toll-like receptor signaling in the ischemic heart. Front Biosci. (2008) 13:5772–9. doi: 10.2741/3114
93. Zong J, Salim M, Zhou H, Bian ZY, Dai J, Yuan Y, et al. NOD2 Deletion promotes cardiac hypertrophy and fibrosis induced by pressure overload. Lab Invest. (2013) 93:1128–36. doi: 10.1038/labinvest.2013.99
94. Ma ZG, Kong CY, Wu HM, Song P, Zhang X, Yuan YP, et al. Toll-like receptor 5 deficiency diminishes doxorubicin-induced acute cardiotoxicity in mice. Theranostics. (2020) 10:11013–25. doi: 10.7150/thno.47516
95. Hasham MG, Baxan N, Stuckey DJ, Branca J, Perkins B, Dent O, et al. Systemic autoimmunity induced by the TLR7/8 agonist resiquimod causes myocarditis and dilated cardiomyopathy in a new mouse model of autoimmune heart disease. Dis Model Mech. (2017) 10:259–70. doi: 10.1242/dmm.027409
96. Kumagai Y, Takeuchi O, Akira S. TLR9 As a key receptor for the recognition of DNA. Adv Drug Deliv Rev. (2008) 60:795–804. doi: 10.1016/j.addr.2007.12.004
97. Jin JW, Tang SQ, Rong MZ, Zhang MQ. Synergistic effect of dual targeting vaccine adjuvant with aminated β-glucan and CpG-oligodeoxynucleotides for both humoral and cellular immune responses. Acta Biomater. (2018) 78:211–23. doi: 10.1016/j.actbio.2018.08.002
98. Jin Y, Zhuang Y, Dong X, Liu M. Development of CpG oligodeoxynucleotide TLR9 agonists in anti-cancer therapy. Expert Rev Anticancer Ther. (2021) 21:841–51. doi: 10.1080/14737140.2021.1915136
99. da Silva-Junior EB, Firmino-Cruz L, Guimarães-de-Oliveira JC, De-Medeiros JVR, de Oliveira Nascimento D, Freire-de-Lima M, et al. The role of toll-like receptor 9 in a murine model of Cryptococcus gattii infection. Sci Rep. (2021) 11:1407. doi: 10.1038/s41598-021-80959-5
100. Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. (2003) 198:513–20. doi: 10.1084/jem.20030162
101. Khan NS, Lukason DP, Feliu M, Ward RA, Lord AK, Reedy JL, et al. CD82 Controls CpG-dependent TLR9 signaling. FASEB J. (2019) 33:12500–14. doi: 10.1096/fj.201901547R
102. Peng Y, Bai W, Wang Z, Yu H. TLR9/NF-kB Pathway regulates Brucella CpG DNA-mediated cytokine response in human peripheral blood mononuclear cells. Iran J Immunol. (2021) 18:268–78. doi: 10.22034/IJI.2021.84578.1665
103. Gomes MT, Campos PC, de Almeida LA, Oliveira FS, Costa MM, Marim FM, et al. The role of innate immune signals in immunity to Brucella abortus. Front Cell Infect Microbiol. (2012) 2:130. doi: 10.3389/fcimb.2012.00130
104. Lee KS, Scanga CA, Bachelder EM, Chen Q, Snapper CM. TLR2 Synergizes with both TLR4 and TLR9 for induction of the MyD88-dependent splenic cytokine and chemokine response to Streptococcus pneumoniae. Cell Immunol. (2007) 245:103–10. doi: 10.1016/j.cellimm.2007.04.003
105. Varga MG, Piazuelo MB, Romero-Gallo J, Delgado AG, Suarez G, Whitaker ME, et al. TLR9 Activation suppresses inflammation in response to Helicobacter pylori infection. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G852–8. doi: 10.1152/ajpgi.00175.2016
106. Puttur F, Francozo M, Solmaz G, Bueno C, Lindenberg M, Gohmert M, et al. Conventional dendritic cells confer protection against mouse cytomegalovirus infection via TLR9 and MyD88 signaling. Cell Rep. (2016) 17:1113–27. doi: 10.1016/j.celrep.2016.09.055
107. Hochrein H, Schlatter B, O'Keeffe M, Wagner C, Schmitz F, Schiemann M, et al. Herpes simplex virus type-1 induces IFN-alpha production via toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A. (2004) 101:11416–21. doi: 10.1073/pnas.0403555101
108. Zang J, Zheng MH, Cao XL, Zhang YZ, Zhang YF, Gao XY, et al. Adenovirus infection promotes the formation of glioma stem cells from glioblastoma cells through the TLR9/NEAT1/STAT3 pathway. Cell Commun Signal. (2020) 18:135. doi: 10.1186/s12964-020-00598-7
109. Li Y, Zhou WW, Sun JH, Yang HX, Xu GR, Zhang Y, et al. Modified citrus pectin prevents isoproterenol-induced cardiac hypertrophy associated with p38 signalling and TLR4/JAK/STAT3 pathway. Biomed Pharmacother. (2021) 143:112178. doi: 10.1016/j.biopha.2021.112178
110. Yoshida K, Abe K, Ishikawa M, Saku K, Shinoda-Sakamoto M, Ishikawa T, et al. Inhibition of TLR9-NF-κB-mediated sterile inflammation improves pressure overload-induced right ventricular dysfunction in rats. Cardiovasc Res. (2019) 115:658–68. doi: 10.1093/cvr/cvy209
111. Yang L, Cai X, Liu J, Jia Z, Jiao J, Zhang J, et al. CpG-ODN attenuates pathological cardiac hypertrophy and heart failure by activation of PI3Kα-akt signaling. PLoS One. (2013) 8:e62373. doi: 10.1371/journal.pone.0062373
112. Duerr GD, Wu S, Schneider ML, Marggraf V, Weisheit CK, Velten M, et al. Cpg postconditioning after reperfused myocardial infarction is associated with modulated inflammation, less apoptosis, and better left ventricular function. Am J Physiol Heart Circ Physiol. (2020) 319:H995–1007. doi: 10.1152/ajpheart.00269.2020
113. Wang X, Chen L, Zhao X, Xiao L, Yi S, Kong Y, et al. A cathelicidin-related antimicrobial peptide suppresses cardiac hypertrophy induced by pressure overload by regulating IGFR1/PI3K/AKT and TLR9/AMPKα. Cell Death Dis. (2020) 11:96. doi: 10.1038/s41419-020-2296-4
114. Xiao Z, Kong B, Yang H, Dai C, Fang J, Qin T, et al. Key player in cardiac hypertrophy, emphasizing the role of toll-like receptor 4. Front Cardiovasc Med. (2020) 7:579036. doi: 10.3389/fcvm.2020.579036
115. Liu F, Wen Y, Kang J, Wei C, Wang M, Zheng Z, et al. Regulation of TLR4 expression mediates the attenuating effect of erythropoietin on inflammation and myocardial fibrosis in rat heart. Int J Mol Med. (2018) 42:1436–44. doi: 10.3892/ijmm.2018.3707
116. Deng KQ, Zhao GN, Wang Z, Fang J, Jiang Z, Gong J, et al. Targeting transmembrane BAX inhibitor motif containing 1 alleviates pathological cardiac hypertrophy. Circulation. (2018) 137:1486–504. doi: 10.1161/CIRCULATIONAHA.117.031659
117. Zhang L, Yang X, Jiang G, Yu Y, Wu J, Su Y, et al. HMGB1 Enhances mechanical stress-induced cardiomyocyte hypertrophy in vitro via the RAGE/ERK1/2 signaling pathway. Int J Mol Med. (2019) 44:885–92. doi: 10.3892/ijmm.2019.4276
118. Li Y, Wang J, Sun L, Zhu S. LncRNA myocardial infarction-associated transcript (MIAT) contributed to cardiac hypertrophy by regulating TLR4 via miR-93. Eur J Pharmacol. (2018) 818:508–17. doi: 10.1016/j.ejphar.2017.11.031
119. Wang SM, Liu GQ, Xian HB, Si JL, Qi SX, Yu YP. LncRNA NEAT1 alleviates sepsis-induced myocardial injury by regulating the TLR2/NF-κB signaling pathway. Eur Rev Med Pharmacol Sci. (2019) 23:4898–907. doi: 10.26355/eurrev_201906_18078
120. Sun F, Chen G, Yang Y, Lei M. Fatty acid-binding protein 4 silencing protects against lipopolysaccharide-induced cardiomyocyte hypertrophy and apoptosis by inhibiting the toll-like receptor 4-nuclear factor-κB pathway. J Int Med Res. (2021) 49:300060521998233. doi: 10.1177/0300060521998233
121. Cruz Junho CV, Trentin-Sonoda M, Alvim JM, Gaisler-Silva F, Carneiro-Ramos MS. Ca2+/calmodulin-dependent kinase II delta B is essential for cardiomyocyte hypertrophy and complement gene expression after LPS and HSP60 stimulation in vitro. Braz J Med Biol Res. (2019) 52:e8732. doi: 10.1590/1414-431X20198732
122. Liu L, Lu Y, Bi X, Xu M, Yu X, Xue R, et al. Choline ameliorates cardiovascular damage by improving vagal activity and inhibiting the inflammatory response in spontaneously hypertensive rats. Sci Rep. (2017) 7:42553. doi: 10.1038/srep42553
123. Ehrentraut H, Weber C, Ehrentraut S, Schwederski M, Boehm O, Knuefermann P, et al. The toll-like receptor 4-antagonist eritoran reduces murine cardiac hypertrophy. Eur J Heart Fail. (2011) 13:602–10. doi: 10.1093/eurjhf/hfr035
124. Mian MOR, He Y, Bertagnolli M, Mai-Vo TA, Fernandes RO, Boudreau F, et al. TLR (Toll-like receptor) 4 antagonism prevents left ventricular hypertrophy and dysfunction caused by neonatal hyperoxia exposure in rats. Hypertension. (2019) 74:843–53. doi: 10.1161/HYPERTENSIONAHA.119.13022
125. Lu X, He Y, Tang C, Wang X, Que L, Zhu G, et al. Triad3A attenuates pathological cardiac hypertrophy involving the augmentation of ubiquitination-mediated degradation of TLR4 and TLR9. Basic Res Cardiol. (2020) 115:19. doi: 10.1007/s00395-020-0779-1
126. Zhang Y, Peng W, Ao X, Dai H, Yuan L, Huang X, et al. TAK-242, a toll-like receptor 4 antagonist, protects against aldosterone-induced cardiac and renal injury. PLoS One. (2015) 10:e0142456. doi: 10.1371/journal.pone.0142456
127. Katare PB, Bagul PK, Dinda AK, Banerjee SK. Toll-like receptor 4 inhibition improves oxidative stress and mitochondrial health in isoproterenol-induced cardiac hypertrophy in rats. Front Immunol. (2017) 8:719. doi: 10.3389/fimmu.2017.00719
128. Winklewski PJ, Radkowski M, Demkow U. Neuroinflammatory mechanisms of hypertension: potential therapeutic implications. Curr Opin Nephrol Hypertens. (2016) 25:410–6. doi: 10.1097/MNH.0000000000000250
129. Dange RB, Agarwal D, Teruyama R, Francis J. Toll-like receptor 4 inhibition within the paraventricular nucleus attenuates blood pressure and inflammatory response in a genetic model of hypertension. J Neuroinflammation. (2015) 12:31. doi: 10.1186/s12974-015-0242-7
130. Xu ML, Yu XJ, Zhao JQ, Du Y, Xia WJ, Su Q, et al. Calcitriol ameliorated autonomic dysfunction and hypertension by down-regulating inflammation and oxidative stress in the paraventricular nucleus of SHR. Toxicol Appl Pharmacol. (2020) 394:114950. doi: 10.1016/j.taap.2020.114950
131. Tapia Cáceres F, Gaspari TA, Hossain MA, Samuel CS. Relaxin inhibits the cardiac myofibroblast NLRP3 inflammasome as part of its anti-fibrotic actions via the angiotensin type 2 and ATP (P2X7) receptors. Int J Mol Sci. (2022) 23:7074. doi: 10.3390/ijms23137074
132. Huang CY, Nithiyanantham S, Liao JY, Lin WT. Bioactive peptides attenuate cardiac hypertrophy and fibrosis in spontaneously hypertensive rat hearts. J Food Drug Anal. (2020) 28:94–102. doi: 10.1016/j.jfda.2019.11.002
133. Zhang Y, Li Y, Huang X, Zhang F, Tang L, Xu S, et al. Systemic delivery of siRNA specific for silencing TLR4 gene expression reduces diabetic cardiomyopathy in a mouse model of streptozotocin-induced type 1 diabetes. Diabetes Ther. (2020) 11:1161–73. doi: 10.1007/s13300-020-00802-4
134. Zhao CH, Ma X, Guo HY, Li P, Liu HY. RIP2 Deficiency attenuates cardiac hypertrophy, inflammation and fibrosis in pressure overload induced mice. Biochem Biophys Res Commun. (2017) 493:1151–8. doi: 10.1016/j.bbrc.2017.07.035
135. Shuai W, Kong B, Fu H, Shen C, Huang H. Loss of MD1 increases vulnerability to ventricular arrhythmia in diet-induced obesity mice via enhanced activation of the TLR4/MyD88/CaMKII signaling pathway. Nutr Metab Cardiovasc Dis. (2019) 29:991–8. doi: 10.1016/j.numecd.2019.06.004
136. Li HB, Li X, Huo CJ, Su Q, Guo J, Yuan ZY, et al. TLR4/MyD88/NF-κB Signaling and PPAR-γ within the paraventricular nucleus are involved in the effects of telmisartan in hypertension. Toxicol Appl Pharmacol. (2016) 305:93–102. doi: 10.1016/j.taap.2016.06.014
137. Wu S, Yin R, Ernest R, Li Y, Zhelyabovska O, Luo J, et al. Liver X receptors are negative regulators of cardiac hypertrophy via suppressing NF-kappaB signalling. Cardiovasc Res. (2009) 84:119–26. doi: 10.1093/cvr/cvp180
138. Cai WF, Zhang XW, Yan HM, Ma YG, Wang XX, Yan J, et al. Intracellular or extracellular heat shock protein 70 differentially regulates cardiac remodelling in pressure overload mice. Cardiovasc Res. (2010) 88:140–9. doi: 10.1093/cvr/cvq182
139. Zhang G, Zhang X, Li D, Tian J, Jiang W. Long-term oral atazanavir attenuates myocardial infarction-induced cardiac fibrosis. Eur J Pharmacol. (2018) 828:97–102. doi: 10.1016/j.ejphar.2018.03.041
140. Van Tassell BW, Seropian IM, Toldo S, Salloum FN, Smithson L, Varma A, et al. Pharmacologic inhibition of myeloid differentiation factor 88 (MyD88) prevents left ventricular dilation and hypertrophy after experimental acute myocardial infarction in the mouse. J Cardiovasc Pharmacol. (2010) 55:385–90. doi: 10.1097/FJC.0b013e3181d3da24
141. Ha T, Hua F, Li Y, Ma J, Gao X, Kelley J, et al. Blockade of MyD88 attenuates cardiac hypertrophy and decreases cardiac myocyte apoptosis in pressure overload-induced cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol. (2006) 290:H985–94. doi: 10.1152/ajpheart.00720.2005
142. Yuan X, Deng Y, Guo X, Shang J, Zhu D, Liu H. Atorvastatin attenuates myocardial remodeling induced by chronic intermittent hypoxia in rats: partly involvement of TLR-4/MYD88 pathway. Biochem Biophys Res Commun. (2014) 446:292–7. doi: 10.1016/j.bbrc.2014.02.091
143. Ma D, Zhang J, Zhang Y, Zhang X, Han X, Song T, et al. Inhibition of myocardial hypertrophy by magnesium isoglycyrrhizinate through the TLR4/NF-κB signaling pathway in mice. Int Immunopharmacol. (2018) 55:237–44. doi: 10.1016/j.intimp.2017.12.019
144. Ahmed AA, Ahmed AAE, El Morsy EM, Nofal S. Dimethyl fumarate interferes with MyD88-dependent toll-like receptor signalling pathway in isoproterenol-induced cardiac hypertrophy model. J Pharm Pharmacol. (2018) 70:1521–30. doi: 10.1111/jphp.13000
145. Venardos N, Deng XS, Yao Q, Weyant MJ, Reece TB, Meng X, et al. Simvastatin reduces the TLR4-induced inflammatory response in human aortic valve interstitial cells. J Surg Res. (2018) 230:101–9. doi: 10.1016/j.jss.2018.04.054
146. Yang J, Wang HX, Zhang YJ, Yang YH, Lu ML, Zhang J, et al. Astragaloside IV attenuates inflammatory cytokines by inhibiting TLR4/NF-кB signaling pathway in isoproterenol-induced myocardial hypertrophy. J Ethnopharmacol. (2013) 150:1062–70. doi: 10.1016/j.jep.2013.10.017
147. Shen L, Xing Q, Lu S, Zhou Y, Zhou C, Li L. Effect of shenqi yangxin decoction on high mobility group box 1 and inflammatory signal pathway in a rat model of dilated cardiomyopathy. J Tradit Chin Med. (2018) 38:862–71. doi: 10.1016/S0254-6272(18)30985-3
148. Liu Q, Han Q, Lu M, Wang H, Tang F. Lycium barbarum polysaccharide attenuates cardiac hypertrophy, inhibits calpain-1 expression and inhibits NF-κB activation in streptozotocin-induced diabetic rats. Exp Ther Med. (2019) 18:509–16. doi: 10.3892/etm.2019.7612
149. Nalban N, Sangaraju R, Alavala S, Mir SM, Jerald MK, Sistla R. Arbutin attenuates isoproterenol-induced cardiac hypertrophy by inhibiting TLR-4/NF-κB pathway in mice. Cardiovasc Toxicol. (2020) 20:235–48. doi: 10.1007/s12012-019-09548-3
150. Zhong Y, Chen L, Li M, Chen L, Qian Y, Chen C, et al. Dangshen erling decoction ameliorates myocardial hypertrophy via inhibiting myocardial inflammation. Front Pharmacol. (2022) 12:725186. doi: 10.3389/fphar.2021.725186
151. Sheu JJ, Chai HT, Sung PH, Chiang JY, Huang TH, Shao PL, et al. Double overexpression of miR-19a and miR-20a in induced pluripotent stem cell-derived mesenchymal stem cells effectively preserves the left ventricular function in dilated cardiomyopathic rat. Stem Cell Res Ther. (2021) 12:371. doi: 10.1186/s13287-021-02440-4
152. Cohen K, Waldman M, Abraham NG, Laniado-Schwartzman M, Gurfield D, Aravot D, et al. Caloric restriction ameliorates cardiomyopathy in animal model of diabetes. Exp Cell Res. (2017) 350:147–53. doi: 10.1016/j.yexcr.2016.11.016
153. Chiang CJ, Tsai BC, Lu TL, Chao YP, Day CH, Ho TJ, et al. Diabetes-induced cardiomyopathy is ameliorated by heat-killed Lactobacillus reuteri GMNL-263 in diabetic rats via the repression of the toll-like receptor 4 pathway. Eur J Nutr. (2021) 60:3211–23. doi: 10.1007/s00394-020-02474-z
154. Gómez-Guzmán M, Toral M, Romero M, Jiménez R, Galindo P, Sánchez M, et al. Antihypertensive effects of probiotics Lactobacillus strains in spontaneously hypertensive rats. Mol Nutr Food Res. (2015) 59:2326–36. doi: 10.1002/mnfr.201500290
155. Robles-Vera I, Toral M, de la Visitación N, Sánchez M, Gómez-Guzmán M, Romero M, et al. Probiotics prevent dysbiosis and the rise in blood pressure in genetic hypertension: role of short-chain fatty acids. Mol Nutr Food Res. (2020) 64:e1900616. doi: 10.1002/mnfr.201900616
156. Jiang W, Tan L, Guo Y, Li X, Tang X, Yang K. Effect of renal denervation procedure on left ventricular hypertrophy of hypertensive rats and its mechanisms. Acta Cir Bras. (2012) 27:815–20. doi: 10.1590/s0102-86502012001100012
157. Soraci L, Gambuzza ME, Biscetti L, Laganà P, Lo Russo C, Buda A, et al. Toll-like receptors and NLRP3 inflammasome-dependent pathways in Parkinson's Disease: mechanisms and therapeutic implications. J Neurol. (2023) 270:1346–60. doi: 10.1007/s00415-022-11491-3
158. Bezhaeva T, Karper J, Quax PHA, de Vries MR. The intriguing role of TLR accessory molecules in cardiovascular health and disease. Front Cardiovasc Med. (2022) 9:820962. doi: 10.3389/fcvm.2022.820962
159. Tan RS, Ho B, Leung BP, Ding JL. TLR cross-talk confers specificity to innate immunity. Int Rev Immunol. (2014) 33:443–53. doi: 10.3109/08830185.2014.921164
160. Sallustio F, Picerno A, Tatullo M, Rampino A, Rengo C, Valletta A, et al. Toll-like receptors in stem/progenitor cells. Handb Exp Pharmacol. (2022) 276:175–212. doi: 10.1007/164_2021_539
161. Anwar MA, Shah M, Kim J, Choi S. Recent clinical trends in toll-like receptor targeting therapeutics. Med Res Rev. (2019) 39:1053–90. doi: 10.1002/med.21553
Keywords: cardiac hypertrophy, toll-like receptor, innate immune, inflammation, signaling pathway
Citation: Zhang Y, Wu J, Dong E, Wang Z and Xiao H (2023) Toll-like receptors in cardiac hypertrophy. Front. Cardiovasc. Med. 10:1143583. doi: 10.3389/fcvm.2023.1143583
Received: 13 January 2023; Accepted: 24 March 2023;
Published: 11 April 2023.
Edited by:
Junjie Xiao, Shanghai University, ChinaReviewed by:
Shijun Wang, Fudan University, ChinaLiming Yang, Harbin Medical University, China
Yichao Zhao, Shanghai Jiao Tong University, China
Zhen Guo, Washington University in St. Louis, United States
© 2023 Zhang, Wu, Dong, Wang and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhanli Wang d2FuZy56aGFubGlAaG90bWFpbC5jb20= Han Xiao eGlhb2hhbkBiam11LmVkdS5jbg==
Specialty Section: This article was submitted to General Cardiovascular Medicine, a section of the journal Frontiers in Cardiovascular Medicine