 Lijo N. Varghese
Lijo N. Varghese Daryl O. Schwenke
Daryl O. Schwenke Rajesh Katare
Rajesh Katare
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 22 March 2023
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1142575
This article is part of the Research Topic Insights in Cardiovascular Biologics and Regenerative Medicine: 2022 View all 6 articles
The global population is estimated to reach 9.8 billion by 2050, of which 2.1 billion will comprise individuals above 60 years of age. As the number of elderly is estimated to double from 2017, it is a victory of the modern healthcare system but also worrisome as ageing, and the onset of chronic disease are correlated. Among other chronic conditions, cardiovascular diseases (CVDs) are the leading cause of death in the aged population. While the underlying cause of the age-associated development of CVDs is not fully understood, studies indicate the role of non-coding RNAs such as microRNAs (miRNAs) and long noncoding RNAs (lnc-RNAs) in the development of age-associated CVDs. miRNAs and lnc-RNAs are non-coding RNAs which control gene expression at the post-transcriptional level. The expression of specific miRNAs and lnc-RNAs are reportedly dysregulated with age, leading to cardiovascular system changes and ultimately causing CVDs. Since miRNAs and lnc-RNAs play several vital roles in maintaining the normal functioning of the cardiovascular system, they are also being explored for their therapeutic potential as a treatment for CVDs. This review will first explore the pathophysiological changes associated with ageing. Next, we will review the known mechanisms underlying the development of CVD in ageing with a specific focus on miRNA and lnc-RNAs. Finally, we will discuss the therapeutic options and future challenges towards healthy cardiac ageing. With the global ageing population on the rise, this review will provide a fundamental understanding of some of the underlying molecular mechanisms of cardiac ageing.
Ageing is a natural, ineludible, and universal process for most species (1). Biological ageing is often characterised as the gradual decline in the normal physiological functioning of tissues and organs (2). Advancements in medical technology, improved sanitation facilities, availability of a nutritious diet, and better healthcare facilities have increased the global average life expectancy from 66.8 years in 2000 to 73.4 years in 2019, which is the fastest since 1950 (3, 4). This spurt in life expectancy is estimated to double the population ≥60 years of age, from 1 billion in 2017 to 2.1 billion in 2050 (5). However, increased life span is correlated with the prevalence of chronic illnesses such as cardiovascular disorders (CVDs), hypertension, diabetes, cancer, and neurodegenerative disorders (6). Among these, CVDs rank as the leading cause of death globally, with 18.6 million deaths yearly (7).
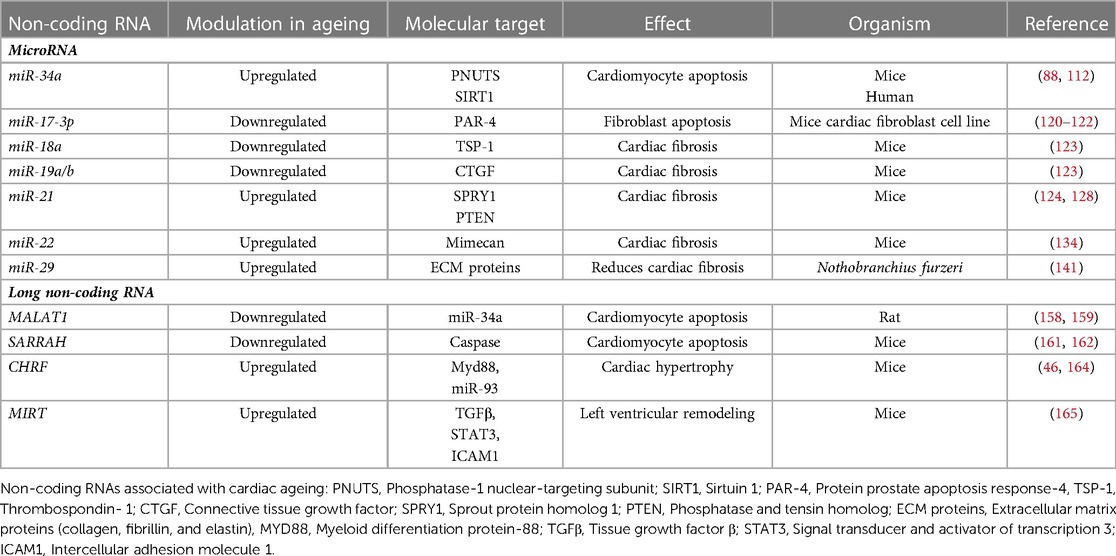
Table 1. Non-coding RNAs associated with cardiac ageing.
CVDs were earlier thought to be rampant in developed countries. However, that trend has dramatically changed due to modernisation. The numbers are steadily increasing in developing countries such as India and China, which account for most of the global population (7, 8) Among the several risk factors for CVDs, such as an unhealthy diet, lack of physical activity or exercise, smoking, and alcohol abuse, cardiac ageing is the cardinal factor in the development of CVDs (9, 10). More than 50% of individuals above 65 years of age suffer from some form of cardiac disorder. With a steady increase in the aged population, there is tremendous pressure on the healthcare system to aid the number of people affected by CVDs (11). A deeper understanding of the physiological and pathological processes in the cardiovascular system with ageing will help develop novel measures for healthy cardiac ageing and thereby reduce the burden of CVD-related deaths (12).
The cardiovascular system undergoes several pathophysiological alterations with age, which become more notable as the heart's reparative mechanisms gradually decline (13). These changes in the aged heart can differ between individuals and thus differ from the chronological age of the individual (14). Myocardial remodeling, microangiopathy, diastolic dysfunction, arrhythmia, and heart failure are some alterations that occur with cardiac ageing (11, 15) Loss of cardiomyocytes with age increases the risk for the development of cardiomyopathies, ischemic heart disease, and heart failure (16). These dead cardiomyocytes are often replaced with fibrous tissue (17). Accumulation of fibrous tissue, irrespective of cardiomyocyte apoptosis, is a hallmark of cardiac ageing which eventually impairs cardiac contractility, particularly diastolic function, commonly referred to as heart failure with preserved ejection fraction (18).
Ageing has a prominent effect not just on the cardiomyocytes but also on the pacemaker cells and valves of the heart. A gradual decrease in the number of pacemaker cells in the sinoatrial node is observed with age (19). The loss of pacemaker cells leads to a progressive decrease in the intrinsic heart rate, reducing the blood supply to vital organs (20, 21). In addition, ageing-induced thickening and calcification of the aortic valve cusps lead to aortic sclerosis, which is observed in around 20% of individuals above 65 years. Aortic sclerosis can develop into a more serious aortic valve stenosis (22). Aortic valve stenosis reduces the outflow of blood from the left ventricle, resulting in left ventricular (LV) hypertrophy as a compensatory mechanism to overcome pressure overload (23). Like the aortic valve, the mitral valve, which guards blood flow against the left atrium to the left ventricle, also shows increased deposition of collagen, lipids, and calcium with ageing. Excess deposition of calcium causes mitral annular calcification, which causes an increased risk of stroke, endocarditis, and necrosis of the calcified tissue (22).
In addition to the changes in the cardiac cells, studies have also reported impairment in vascular structure and function (24). Thickening and stiffening of the arteries are prominent features of ageing caused by increased deposition of collagen and reduced elastin. Arterial stiffness can lead to myocardial infarction (MI) due to a sustained increase in systolic pressure (15). In addition, ageing causes endothelial dysfunction in blood vessels, which can be attributed to increased inflammation and oxidative stress (25). Endothelial dysfunction is one of the first steps in the progression of atherosclerosis (26). While the exact mechanism of these ageing-induced changes is yet to be explored, they can occur due to the ageing-induced molecular changes in the cardiac cells.
Ageing-induced molecular changes in the cardiac cells form the basis of the pathophysiological changes observed in aged individuals (27). Among all the molecular changes, telomere shortening, epigenetic changes, mitochondrial dysfunction, and impaired intercellular communication are the hallmark changes observed in cardiac ageing (28, 29) (summarised in Figure 1).
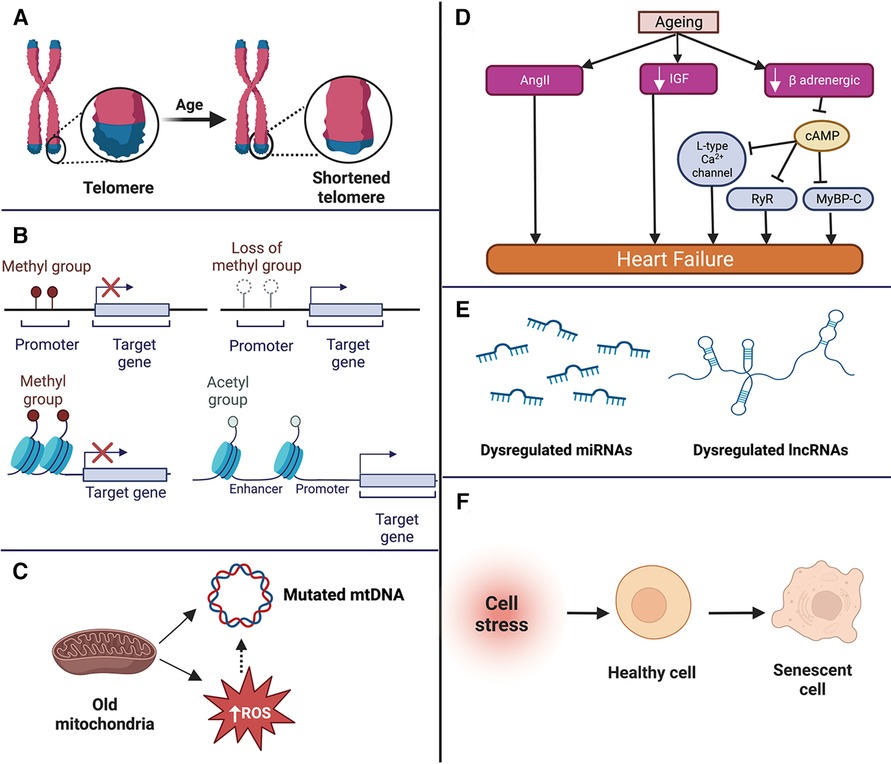
Figure 1. Summary of the molecular changes that occur in the aged heart such as telomere shortening (A), epigenetic changes (B), mitochondrial dysfunction (C), neurohormonal changes (D) dysregulation of non-coding RNAs (E), and cellular senescence (F). mtDNA, mitochondrial DNA; ROS, reactive oxygen species; miRNA, microRNA; lncRNA, long non-coding RNA; AngII, angiotensin II; IGF, insulin like growth factor; RyR, ryanodine receptor; MyBP-C, f Apoptosis of cardiomyocytes, loss of pacemaker cells, thickening and calcification of the valves, and impairment of vascular structure and function.
Telomeres are looped DNA (TTAGGG) repeats at the ends of the chromosomes, protecting the chromosome from degradation (30). The length of telomeres reduces progressively with each cell cycle, primarily due to the inability of DNA polymerase to completely replicate the telomeres (31). When the telomeres shorten to a critical length, the cell cycle is arrested, and the cell enters a state of senescence and activates apoptosis (32). The length of the telomere is maintained by an enzyme called telomerase (31). Telomerase enzyme contains two components: Telomerase RNA component (TERC) and telomerase reverse transcriptase (TERT). TERC includes a template for telomeric DNA, which is used by TERT to synthesise new telomeric DNA repeats. Telomerase activity reduces with age (33), leading to short telomeres, and short telomeres have been linked to the progression of cancer, diabetes, and CVDs (34). In a study conducted on TERC−/− mice, the catalytic unit of the telomerase enzyme, mice displayed thinning of ventricular walls and cardiomyocyte apoptosis (35). Brouillette et al. conducted a trial with 1,500 participants to investigate the correlation between telomere length and the occurrence of coronary heart disease. They observed that the mean telomere length reduced by around 9% each year and that the participants with short telomere length had a higher risk of developing coronary heart disease than those with comparatively longer telomeres (36). Similarly, coronary endothelial cells with short telomeres exhibit impaired function and formation of plaques leading to age-dependent coronary artery disease (37, 38). Despite several studies indicating the role of short telomeres in the progression of CVDs, a large study including 7,827 participants found telomere length was not significantly associated with cardiovascular mortality (39). The reason for the inconsistency can be multi-factorial such as ethnicity of individuals, medical history, and other factors such as hypertension, smoking, and obesity, as these can attribute to telomere shortening (39, 40). Thus, using telomere length as a marker for age-associated CVD needs more conclusive evidence (41).
Epigenetic changes, including methylation of DNA, RNA methylation, and histone modifications, have been demonstrated to be associated with the development of ageing-induced CVDs (42). DNA methylation involving methylation at 5-cytosine is a highly conserved process in plants, animals, and fungi (43). DNA methylation and demethylation are essential for regulating gene expression, splicing, transposon silencing, and genomic instability (44). DNA methylation at the promoter regions inhibits the binding of transcription factors, which suppresses transcription, whereas demethylation leads to gene expression (45). Studies have identified DNA hypermethylation with age (increased DNA methylation) (44, 46). DNA hypermethylation has been observed in atherosclerotic lesions, as hypermethylation represses the expression of atheroprotective genes such as kruppel like factor 4 (KLF4) and ATP binding cassette subfamily A member 1 (ABCA1) (44). Like DNA methylation, RNA is also methylated. The most abundant modification is the methylation of adenosine in mRNA (N6-methyladenosine, M6A), which is catalysed by the enzyme methyltransferase-like 3 (METTL3) (47). N6-methyladenosine is also found in other RNAs, such as transfer RNA, ribosomal RNAs, and other non-coding RNAs (48). Even though RNA methylation profile changes with age are not elucidated, it has been linked to cardiac hypertrophy, which is frequently observed with ageing (48). Dorn et al. demonstrated cardiac hypertrophy in METTL3 overexpressing mice by 8 months of age. The mechanism by which METTL3 overexpression causes hypertrophy is still being explored. However, initial findings indicate it increases the expression of the β-myosin heavy chain (β-MHC), a critical hypertrophic protein. In vitro analysis of METTL3 inhibition attenuates cardiomyocyte hypertrophy, indicating a role in the development of cardiac hypertrophy (48).
Histones are proteins which wrap DNA to form the nucleosome. The epigenetic modifications of histones, such as methylation, acetylation, carbonylation, and phosphorylation, are associated with CVDs (49). Like DNA methylation, histone modifications repress transcription (47). Histone methylation is mediated by histone methyltransferases and demethylases, which add and remove methyl groups from histones, respectively (47). Changes in the histone methylation profile have been observed in cardiac hypertrophy, atherosclerosis, and dilated cardiomyopathy. SET and MYND domain-containing protein 1 (SMYD1) is a histone methyltransferase which represses the activity of pro-hypertrophic genes such as transforming growth factor-β-3 (TGFβ3) and natriuretic peptide A (NPPA) and its depletion is associated with hypertrophy (49). However, there is no conclusive evidence that the histone methylation pattern changes with age. Histone acetyltransferases add an acetyl group from acetyl-CoA to lysine residues, and deacetylases remove the acetyl group (47). Histone deacetylase (HDAC) is catalysed primarily by HDAC1 and HDAC2, and their global deletion has been linked to cardiac arrhythmia and dilated cardiomyopathy due to the abnormal growth of cardiomyocytes (50). Whether changes in histone acetylation lead to age-associated CVDs, is not fully understood and is an area of active research (47).
Mitochondria have a double membrane consisting of inner and outer layers. The inner layer comprises 4 complexes—Complex I to IV, which generate ATP via oxidative phosphorylation (51). With age, oxidative phosphorylation declines primarily because of the dysfunction of complexes I and IV, reducing the production of ATP in the cells (52). This dysfunction can be due to the accumulation of damage to the mitochondrial DNA (mtDNA) and excess amounts of reactive oxygen species (ROS) (53). Increased ROS accelerates atherogenesis by oxidising low-density lipoprotein, promoting endothelial cell senescence and apoptosis (54). Oxidised low-density lipoproteins increase the risk of plaque formation and subsequent atherosclerosis by reducing the availability of nitric oxide and promoting inflammation, leukocyte adhesion, and smooth muscle cell proliferation (55). Increased ROS-linked endothelial cell apoptosis also impairs blood flow to the heart and the brain due to rarefaction of the blood vessels (56). ROS is also associated with cardiomyocyte necrosis and the progression of cardiac hypertrophy (57, 58). ROS generated by monoamine oxidase (MAO), an enzyme present on the outer membrane of the mitochondria, induces cardiomyocyte necrosis by activating p53 (10, 58). ROS also induces cardiac hypertrophy through the activation of pro-hypertrophic signalling kinases and transcription factors such as mitogen-activated protein kinase (MAP-kinase) and nuclear factor kappa light chain enhancer of activated B cells (NF-κB) (59, 60). Thus, increased ROS generation due to age-dependent mitochondria dysfunction is a crucial molecular change associated with cardiac ageing and the development of CVDs (59).
Mitochondria is a unique cell organelle, as it has its DNA known as mitochondrial DNA (mtDNA). mtDNA is circular and has genes that encode proteins necessary to produce energy (61). The mtDNA accumulates mutations with age, which can also be caused due to age-impaired production of excess ROS (62). mtDNA mutations have been linked with several diseases, such as Alzheimer's disease, Parkinson's disease, CVDs, diabetes, and cancer (63). Lindroos et al. studied the effect of one of the mtDNA mutations (m.3243A > G) in 14 individuals. This mutation affects the translation of respiratory chain complex proteins by impairing the tRNA necessary for the translation (64, 65). The study revealed impaired glucose oxidation and lower stroke volume in individuals with mutation (65). Studies have also identified an association of other mtDNA mutations, such as m.3256C > T, m.12315G > A, m.13513G > A, and m.15059G > A with atherosclerosis due to impaired translation of proteins essential for oxidative phosphorylation and increased production of ROS (66). While studies suggest a strong correlation between mtDNA, ageing, and CVD, whether mtDNA is the cause of CVD remains unclear, warranting future studies to ascertain the mechanisms and its reliability as an age-associated marker of CVDs (63).
Ageing has a detrimental effect on the neuro-hormonal signalling pathways essential for the functioning of the cardiovascular system. One of the signalling pathways linked to the age-dependent progression of cardiac function decline and CVD is the renin-angiotensin-aldosterone system (RAAS) (59). RAAS regulates blood pressure by controlling fluid and electrolytes through a well-coordinated balance between the liver, kidneys, heart, lungs and blood vessels (67). In brief, RAAS is activated in the kidneys by a drop in blood pressure, leading to renin's activation. Active renin is secreted in the blood, which converts inactive angiotensinogen (produced by the liver) to angiotensin I. Angiotensin I is converted by an angiotensin-converting enzyme (primarily present in the lungs) to angiotensin II (Ang II). Ang II is the central effector molecule of the RAAS system, which acts on the adrenal gland, brain, and blood vessels to elevate blood pressure by various mechanisms (68). Ageing increases Ang II concentration, which is associated with cardiac hypertrophy, fibrosis, and increased ROS (68). Another significant signalling pathway affected by age is the insulin-like growth factor-1 (IGF), which reduces with age in humans. While the reduction in the IGF signalling pathways has been observed to improve cardiac function with age in Drosophila and mice models, the same has not been observed in humans (59). Vasan et al. examined the levels of IGF-1 in 717 elderly individuals with no known history of myocardial infarction and congestive heart failure for a mean period of 5.2 years. Of the 717 elderlies, 56 developed congestive heart failure (69).
Interestingly, all these individuals had serum IGF-1 levels below the median value—the exact reason for these contradictory findings between different species of ageing warrants further studies (59). Beta-adrenergic signalling is an important signalling pathway which regulates heart rate and contractility. There are 3 major beta-adrenergic receptors—β1, β2, and β3 receptors, which are G-coupled protein receptors. In brief, ligands (major catecholamines such as epinephrine and norepinephrine) bind to the beta receptors, which activate adenylyl cyclase and increase the level of cyclic adenosine monophosphate (cAMP). cAMP targets protein kinase A, which activates several proteins such as ryanodine receptors, L-type calcium channels and myosin binding protein-C, eventually leading to cardiomyocyte contraction (70). With age, the beta-adrenergic signalling reduces, primarily due to impaired agonist binding to the receptor and reduced β1 receptors (70, 71). A decreased beta-adrenergic response seems beneficial, especially in the aged heart, as it reduces the risk of arrhythmia, hypertrophy, and apoptosis (70, 72). However, the lower beta-adrenergic response is also associated with lowered exercise tolerance, impaired autonomic regulation and impaired arterial-ventricular load (70, 72, 73).
Cellular senescence is a complex, dynamic, and multi-step process in which a cell undergoes permanent cell cycle arrest with continued metabolic activities (74). Cellular senescence is induced as a response to stressors such as DNA damage, telomere shortening, oxidative stress, mitochondrial damage, which are a result of ageing (74, 75). A senescent cell has multiple characteristic features such as an enlarged and flattened morphology, senescence associated beta-galactosidase activity, activation of senescence associated secretory phenotype (SASP), and DNA damage response (74, 76). SASP is a prominent hallmark of a senescent cell, as it releases pro-inflammatory chemokines and cytokines, growth factors, ECM proteins, and activation of p16 and p21, which affects the neighboring cells or distant cells, if they are released in the systemic circulation (76). SASP has a deleterious effect, as it can induce senescence in the neighboring healthy cells and even 10%–15% of senescent cells in a tissue, can cause tissue degeneration (77, 78). Cellular senescence has been linked to age-related diseases such as osteoporosis, renal diseases, neurodegenerative diseases, pulmonary fibrosis, and CVDs (79).
The heart is a mosaic of different cell types such as cardiomyocytes, vascular smooth muscle cells, endothelial cells, and fibroblasts, and senescence of individual cell types can cascade to a disease (80). For example, senescent cardiomyocytes along with the phenotypical changes of a senescent cell exhibit contractile dysfunction and hypertrophic growth, which eventually leads to cardiac hypertrophy, arrhythmias, cardiac remodeling, and heart failure (81). Senescent endothelial cells have impaired production of endothelin-1 (vasoconstrictor) and nitric oxide (vasodilator) which affects vascular function. Senescent endothelial cells have been studied to develop disorders such as atherosclerosis and heart failure with preserved ejection fraction (80). Senescent fibroblasts secrete IGF-1 which promotes collagen synthesis and exacerbates cardiac fibrosis. It also induces senescence in neighboring cardiomyocytes through paracrine signalling (80). Similarly, senescent vascular smooth muscles lead to atherosclerosis and pulmonary hypertension (82).
Senolytics are a class of drugs which selectively induces apoptosis in senescent cells by activating the B-cell lymphoma 2 (BCL-2) family proteins, p53, phosphoinositide-3-kinases (PI3K) and other apoptotic pathways (83). Salerno et al. combined Dasatinib and Quercetin, two senolytic drugs and administered to 22–24 months old mice after acute MI, released healthy cardiac stem cells and improved cardiac remodeling and regeneration, which eventually improved LV function. Thus, removal of senescent cells by senolytics improves the overall functioning of the heart, even in aged mice (77, 83). In a recent study Cattaneo et al. reported the importance of longevity-associated BPIFB4 (LAV-BPIFB4) gene in supporting cardiac function and vascularization in ageing cardiomyopathy (84). They demonstrated reduced LAV-BPIFB4 in older hearts and that gene therapy with LAV-BPIFB4 rescued cardiac function and myocardial perfusion in aged mice by improving microvasculature density and pericyte coverage. Therefore, therapeutic modalities targeting cellular senecesnce may have therapeutic potential although long-term studies required to determine the sustainability of the effect.
The human genome consists of only 3% protein-coding genes, whereas most of the genome is transcribed to produce non-coding RNAs (nc-RNAs) (85). These nc-RNAs include a wide array of molecules such as microRNAs (miRNAs), long-noncoding RNAs (lncRNAs), small nuclear RNAs, small nucleolar RNAs, piwi-interacting RNA, circular RNA and transfer RNA etc (86).. Based on their function, these nc-RNAs can be classified into two categories: housekeeping RNAs and regulatory RNAs. Housekeeping RNAs, such as ribosomal RNA, transfer RNA, and small nuclear RNA, are present in virtually all cells and are essential for the cell's normal functioning. Regulatory RNAs such as piwi-RNA, miRNAs, and lncRNAs regulate gene expression at the transcriptional or translational levels (86, 87). Among the ncRNAs, miRNAs and lncRNAs are gaining prominence in ageing-associated diseases. The expression of miRNAs and lncRNAs dysregulate with age which leads to the onset and progression of ageing-associated diseases such as CVDs (88, 89), Alzheimer's (90), cancer (91), and diabetes (92). Furthermore, miRNAs such as miR-146a/b, -126, -34a, -22 are associated with inducing senescence in vascular smooth muscle cells, endothelial cells, cardiomyocytes, and fibroblasts, respectively (80, 93). The role of lncRNAs with respect to senescence is an active area of research, although to date only few lncRNAs that induce cellular senescence have been identified. For example, senescence associated lncRNA-1 triggers senescence in fibroblasts and lncRNA-H19 triggers senescence in cardiomyocytes (89, 94). Several studies have demonstrated the critical role of miRNAs and lncRNAs in cardiovascular system homeostasis and that their dysregulation with age leads to CVDs (95, 96). In the next section, we will focus on non-coding RNAs (miRNAs and lnc-RNAs) in the age-dependent development of CVDs.
MicroRNAs (miRNAs) are nc-RNAs that are ∼22–25 nucleotides long (97). Since their discovery in 1993 in Caenorhabditis elegans, miRNAs have been found in many organisms, including humans. It is estimated that the mammalian genome codes for more than 2000 miRNAs, and around 60% of coding genes are regulated by miRNAs (98). Several years into their discovery, the role and function of miRNAs were not fully understood. However, the discovery that two miRNAs—lin-4 and let-7 control nematode development led to a more thorough study of these small nc-RNAs (99). miRNAs inhibit protein synthesis by translational repression or mRNA degradation (100). Furthermore, each miRNA can control the expression of more than one mRNA. Hence, each mRNA can be a target for more than one miRNA (99). Thus, miRNAs control the gene expression of several proteins required for essential cellular and metabolic pathways that control cell growth, differentiation, and survival (101, 102). Concerning the cardiovascular system, several miRNAs are involved in cardiac development, differentiation, and normal function (102). Deletion or dysregulation in the expression of miRNAs leads to cardiac dysfunction and eventually death, highlighting the crucial role of miRNAs in the functioning of the cardiovascular system (102).
The biogenesis of miRNAs begins in the nucleus by the most common canonical or less common non-canonical pathway (summarised in Figure 2). In the canonical pathway, the primary transcript (pri-miRNA) transcribed by RNA-polymerase II is a several hundred nucleotides long transcript, which is processed to form a 70–100 nucleotide long precursor known as pre-miRNA (103, 104). Next, pri-miRNA is cleaved by a protein complex called the microprocessor, which comprises two enzymes—Drosha and DiGeorge syndrome critical region gene 8 (DGCR8) to form a shorter pre-miRNA. DGCR8, an RNA binding protein, binds to the pri-miRNA and Drosha cleaves it at the base of the hairpin structure, which results in a 2 nt 3′ overhang. Next, a nuclear transport receptor protein called exportin-5, along with RAN-GTPase, moves the pre-miRNA from the nucleus to the cytoplasm. Once in the cytoplasm, Dicer, an RNase III endonuclease, removes the terminal loop from the pre-miRNA to form the mature miRNA consisting of 18–22 nucleotides (104, 105). Finally, the mature miRNA duplex is loaded to RNA-induced silencing complex (RISC), a multi-protein complex consisting of Argonaute protein (Ago2). Once loaded on the RISC complex, one of the duplex strands, known as the passenger strand, is released, while the other strand, the guide strand, remains attached. The passenger strand can also be cleaved by Ago2 or C3PO endonuclease. The guide strand selection is based on the relative thermodynamic stability of the two strands. The guide strand generally has a relatively unstable 5′ end and has a uridine nucleotide in the first position (98). The loaded miRNA on the RISC (miR-RISC) binds to the complementary sequence of its target messenger RNA (mRNA). Partial complementarity between the miR-RISC and mRNA leads to translational repression, while an exact complementarity leads to mRNA cleavage and degradation (98).
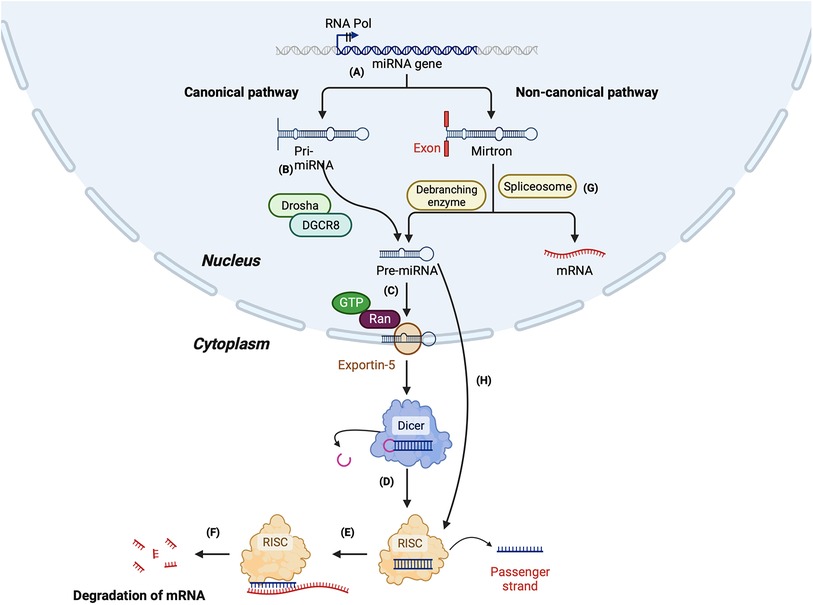
Figure 2. Summary of miRNA biogenesis via canonical and non-canonical pathway. Canonical pathway: (A) The miRNA gene is transcribed by RNA polymerase II to form a primary mi-RNA (pri-miRNA). (B) The pri-miRNA is cleaved by Drosha and Di-George syndrome critical region 8 (DGCR8) to form the precursor miRNA (pre-miRNA). (C) Pre-miRNA is transported to the cytoplasm by exportin-5. (D) In the cytoplasm, pre-miRNA is cleaved by Dicer to form a duplex mature miRNA. (E) The mature miRNA is loaded to RNA-induced silencing complex (RISC), leading to the cleavage of the passenger strand. (F) miR-RISC binds to the target mRNA leading to its degradation. Non-canonical pathway: (G) Synthesis of miRNA by Drosha/DGCR8 independent pathway: The primary miRNA is spliced by a spliceosome, forming a branched pre-miRNA. The pre-miRNA is debranched by a debranching enzyme, after which the synthesis is similar to the canonical pathway. (H) Synthesis of miRNA by Dicer-independent pathway: The pre-miRNA formed by the cleavage of pri-miRNA by Drosha/DGCR8 is exported to the cytoplasm. The pre-miRNA is not long enough to be processed by Dicer and forms the RISC.
In the non-canonical pathway of miRNA biogenesis, miRNAs can be formed either by the Drosha/DGCR8 independent pathway or the Dicer independent pathway. Mirtrons are an example of the miRNAs formed by the Drosha/DGCR8 independent pathway. The spliceosome cleaves the pri-miRNA of mirtrons, which forms a branched pre-miRNA. Next, the 3′ and 5′ ends of the pre-miRNA are ligated, forming a lariat, which is linearised by a debranching enzyme. After the debranching, the pre-miRNA can be transported to the cytoplasm by the exportin-5-RAN-GTPase complex. Once in the cytoplasm, it is cleaved by Dicer and loaded on Ago to form a mature miRNA (106, 107).
In the Dicer-independent pathway, the microprocessor complex produces the pre-miRNA as short-hairpin RNA transcripts, which are then exported to the cytoplasm. However, the exported transcripts are too short to be processed by Dicer; instead, the entire pre-miRNA duplex is loaded on the Ago complex and undergoes splicing by Ago2 to form the mature miRNA [104] (Figure 2).
While the role of miRNAs in CVD is very well established, it is only recently that researchers have started to explore the role of miRNA dysregulation in the development of age-induced CVD. Therefore, this section will review some key literature on cardiovascular-enriched microRNAs and their role in cardiac ageing (summarised in Figure 3).
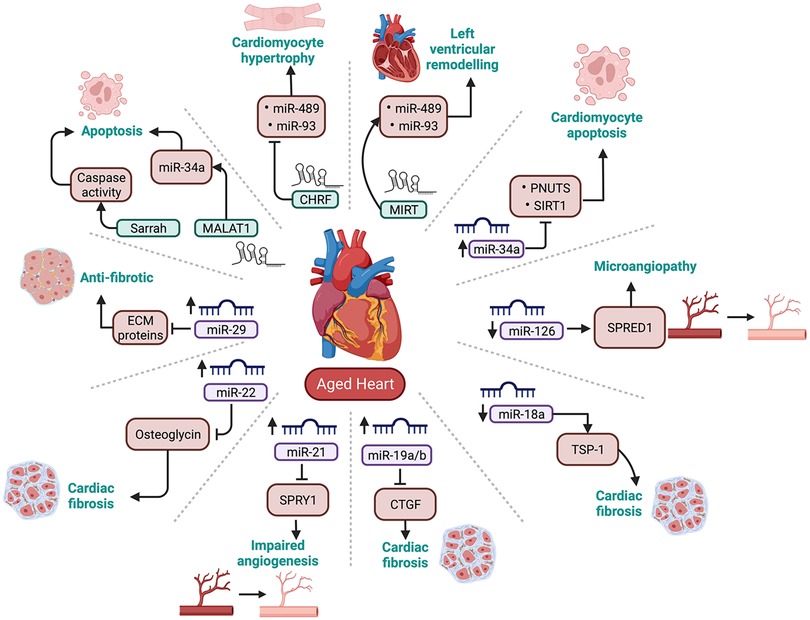
Figure 3. Summary of cardiovascular enriched miRNAs and lncRNAs that are dysregulated with ageing. PNUTS, phosphatase-1 nuclear targeting subunit; SIRT-1, sirtuin 1; TSP-1, thrombospondin-1; CTGF, connective tissue growth factor; SPRY1, sprout protein homolog 1; SPRED1, sprouty related EVH1 domain containing 1; ECM proteins, Extracellular matrix proteins.
The miR-34 family consists of 3 miRNAs, miR-34a, -34b and -34c. Among these, miR-34a has been well-characterised and demonstrated to play a significant role in age-related CVD. The gene for miR-34a is located in chromosome 1p36.22 (108). Studies have reported that activation of miR-34a following cardiac injury leads to increased apoptosis of cardiomyocytes (109, 110). Boon et al. observed an upregulation of miR-34a with age in mice cardiomyocytes by inhibiting its direct target phosphatase-1 nuclear-targeting subunit (PNUTS). In support of this, the expression of PNUTS was downregulated in aged mouse hearts (88). PNUTS interacts with telomere repeat factor (TRF2), which protects chromosome ends by enforcing a T-loop structure. In a healthy cell, DNA damage promotes PNUTS translocation to, and repair of, double-stranded DNA breaks during the G2-M checkpoint (111). However, this effect is lost in the aged heart due to the upregulation of miR-34a. Notably, the researchers also demonstrated that forced PNUTS overexpression reduced age- and miR-34a-related markers of DNA damage and protected cardiomyocytes from apoptosis by preventing telomere shortening and promoting the DNA damage response (88).
In addition to apoptosis, miR-34a targets another pro-survival gene, Sirtuin1 (SIRT1) (112). SIRT1 is a deacetylase enzyme which deacetylates p53, a protein essential for cell cycle and survival. With the repression of SIRT1, p53 is acetylated, leading to an increase in pro-apoptotic proteins like p21 and p53 upregulated modulator of apoptosis (PUMA), eventually leading to apoptosis (113). p53 has a binding site located on the MiR-34a gene. Thus, activation of p53 can result in increased transcription of miR-34a, which leads to repression of SIRT1 and further acetylation of p53. Thus, miR-34a, SIRT1, and p53 have a strong positive feedback loop (113). Interestingly, a recent study from our laboratory showed a marked upregulation of miR-34a in type 2 diabetic hearts from the early stages of the disease (114). Diabetes is a disease which accelerates the ageing of the cells by promoting activation of the pro-senescence signalling cascade (115). Importantly, in vitro inhibition of miR-34a markedly reduced high glucose-induced deleterious effects in cardiomyocytes. This evidence suggests that targeting miR-34a could be beneficial for the healthy ageing of the heart.
The miR-17-92 cluster is transcribed from the miR-17-92 cluster host gene (MIR17HG) located in chromosome 13q31.3. The primary transcript is ∼0.8 kb long polycistron which is processed to form seven mature miRNAs, miR-17-3p, -17-5p, -18a, -19a, -19b, -20a and -92a. The miR-17-92 cluster is also referred to as oncomir-1, as its overexpression has been demonstrated to be associated with cancer (116, 117). The miR-17-92 cluster plays a protective role in the heart, with its reduced expression linked to left ventricular hypertrophy, arrhythmias, and heart failure (118, 119). Among all the miRNAs within the cluster, miR-17-3p, -18a, and -19a/b have been shown to have a role in cardiac ageing. The expression of miR-17-3p is downregulated in aged mice hearts, which was associated with the upregulation of its target protein prostate apoptosis response-4 (PAR-4) (120). PAR-4 is a pro-apoptotic protein, and its upregulation negatively affects the expression of anti-apoptotic proteins CCAAT enhancer binding protein beta (CEBPB) and focal adhesion kinase (FAK). Reduced expression of CEBPB and FAK enhances cellular senescence and apoptosis in mouse cardiac fibroblast cells (121, 122).
miR-18a and miR-19a/b are also downregulated in aged mice cardiomyocytes and are associated with an increase in their target proteins, thrombospondin- 1 (TSP-1) and connective tissue growth factor (CTGF), respectively. The increase in TSP-1 and CTGF causes an increase in collagen 1A1 and collagen 3A1 levels leading to the development of cardiac fibrosis (123). This evidence suggests that the members of the miR-17-92 play an essential role in normal cardiac functioning, and their regulation can reduce apoptosis and fibrosis in the aged heart.
miR-21 is a highly conserved miRNA encoded by the MIR21 gene, located on chromosome 17q23.2 (124, 125). miR-21 is expressed in cardiac fibroblasts, endothelial cells and vascular smooth muscle cells. Conditions such as cardiac hypertrophy and heart failure upregulate the expression of miR-21. Of note, ageing is associated with both these conditions (126, 127). Therefore, miR-21 upregulation may be associated with cardiac ageing. Sprout protein homolog 1 (SPRY1), a direct target of miR-21, is an inhibitor of the extracellular signal-regulated kinase–mitogen-activated protein (ERK-MAP) kinase. ERK-MAP kinase activates the pathways associated with increased interstitial fibrosis and cardiac hypertrophy (124). Therefore, inhibition of SPRY1 following upregulation of miR-21 may increase fibrosis in the aged heart. Similar to the expression of miR-21 in fibroblasts, the expression of miR-21 is upregulated in the vascular smooth muscles, which inhibits the expression of its other target protein phosphatase and tensin homolog (PTEN). Inhibition of PTEN activates the Phosphatidylinositol-3-kinase and its downstream molecule serine/threonine kinase B (PI3K/AKT) signalling pathway significantly regulates cell proliferation and survival. Activating the PI3K/AKT pathway facilitates the increased proliferation of smooth muscle cells and the increased formation of atherosclerotic plaques (128) Contrary to its role on fibroblasts, miR-21 exhibits pro-angiogenic effects on endothelial cells. Zhang et al. found that the level of miR-21 increased with age in mice, peaking around 18 months, after which it decreased (129). Another study showed that the downregulation of miR-21 was associated with impaired angiogenesis and a reduction in the renewal capacity of endothelial cells (130). Even though several studies have reported the role of miR-21 in CVDs, dysregulation of miR-21 may not be specific to cardiac ageing. This is likely due to the ubiquitous expression of miR-21 in several other tissues. Therefore, dysregulation in the levels of miR-21 cannot be solely associated with cardiac dysfunction (124), especially because it is well-known that miRNAs can transport between cells and in circulation to the distant organ. Thus, a more detailed study is required to ascertain the role of miR-21 in the aged heart (131).
miR-22 is an oncogenic miRNA, transcribed from the MIR22 gene located on chromosome 17p13.3 (132). Recently, miR-22 has been identified to have a role in senescence, apoptosis, and angiogenesis (133). To understand the role of miR-22 in cardiac ageing, Jazbutye et al. determined the expression of miR-22 in 4, 24, and 76-week-old mice. They observed an upregulation of miR-22 in fibroblasts with age. Using luciferase assay, they confirmed osteoglycin (mimecan) as the target protein. Interestingly, osteoglycin was downregulated with age (134, 135). Osteoglycin has a protective effect on the heart by reducing collagen production, which eventually reduces fibrosis. This is supported by increased cardiac fibrosis due to the activation of fibroblasts following the downregulation of osteoglycin (134). Like several other miRNAs, studies have demonstrated increased expression of circulating miR-22 in the plasma of patients with acute MI (136) or pancreatic cancer (137). This suggests a possibility for using miR-22 as a potential biomarker for the early diagnosis of ageing-induced cardiac fibrosis.
miR-29 comprises miR-29a, -29b1, -29b2, -29c, are highly conserved miRNAs. The genes encoding miR-29 family are located on chromosome 7q32.3 (miR-29a and -29b1) and 1q32.2 (miR-29b2 and -29c (138, 139). miR-29 regulates the expression of several proteins such as collagen, fibrillin, and elastin that form the extracellular matrix (ECM) (140). The expression of miR-29 family, especially miR-29a and -29b1 is upregulated with age (140, 141). A study by Heid et al. on aged Nothobranchius furzeri hearts observed the upregulation of miR-29a due to increased oxidative stress, a key molecular changes that occur with ageing. Interestingly, upregulation of miR-29a has a cardioprotective effect, as it decreases age-dependent deposition of ECM proteins such as collagens 1A1, 1A2, 11A1, 15A1 and elastin (141, 142). Since increase in collagen deposition is a hallmark sign of cardiac fibrosis, miR-29a acts as anti-fibrotic miRNA (143). While reduced ECM proteins is beneficial against cardiac fibrosis, it has been linked to formation of aneurysm (140, 144). Therefore, upregulation of miR-29 and subsequent reduction in ECM proteins can disrupt the integrity of the vascular wall and can lead to aortic aneurysm (140). Hence a balanced and tissue specific expression levels of miR-29a may be critical. Interestingly, the age-dependent upregulation in the expression of miR-29 and its protective effect was not confined to the heart, but studies have demonstrated these changes in brain as well (145). Although interesting, further studies are required to highlight the functional effects of age-dependent upregulation of miR-29.
The role of miRNAs as regulators of age-associated cardiovascular dysfunction is an exciting area of research. These small noncoding molecules have been linked to several pathologies, such as left ventricular hypertrophy, atherosclerosis, hypertension, MI, and arrhythmia (146).
As discussed above, several studies have established the role of dysregulation of miR-34a, -17-92 cluster, -21, -22, and -29 is a significant factor in the development of age-associated CVDs. However, further investigations are required to accurately determine the role of miRNAs in ageing-induced CVD. For example, it remains unknown whether the miRNAs' dysregulation is cell-specific or occurs in all the tissues. Further, the mechanism and factors that cause dysregulation of these miRNAs with age remain unknown (147). Furthermore, it is vital to determine the role of other well-established cardiovascular enriched and cardiac-specific miRNAs such as miR-1, -208, 133, and -206 in cardiac ageing (148) (summarised in Table 1).
The human genome codes for approximately 16,000 lncRNAs. They are generally more than 200 nucleotides long (149). Unlike miRNAs that repress mRNAs' translation, lncRNAs can upregulate or downregulate gene expression (149). lncRNAs are gaining prominence in cardiovascular studies, with several lncRNAs having been identified and demonstrated to play an essential role in cardiovascular homeostasis. For example, Braveheart (Bvht) is necessary for the differentiation of cardiac stem cells into cardiomyocytes, while FOXF1 adjacent non-coding developmental regulatory RNA (Fendrr) aids in the development of ventricles (150). Similarly, they are also demonstrated to play a role in the development of CVD. lncRNAs such as myosin heavy chain associated RNA transcript (Mhrt), cardiac hypertrophy related factor (Chrf), and HOX antisense intergenic RNA (Hotair) are associated with cardiac hypertrophy. In contrast, cardiac hypertrophy associated with epigenetic regulator (Chaer), maternally expressed gene 3 (Meg3), the non-coding repressor of NFAT (Nron), is associated with heart failure (89). In addition, recent studies have identified a role for Meg3, autophagy promoting factor (Apf), and myocardial infarction-associated transcript (MIRT) in myocardial infarction (89). Despite being extensively studied for their role in several cardiac disorders, the role of lncRNAs in the age-associated development of CVDs is still in its infancy (151). We will next review the biogenesis of lncRNAs and some of the known functions of lncRNAs in association with cardiac ageing.
The biogenesis of lncRNA is different from that of miRNAs but is similar to the formation of mRNAs. LncRNAs are transcribed by RNA polymerase II (89). Most of the lncRNA has a polyadenylated 3′ end and is capped with methyl-guanosine at the 5′ end (152). They undergo alternative splicing and modifications to form lncRNA (153). Based on the cellular fate and function of the specific lncRNA, it can either be localised in the nucleus or transported to the cytoplasm. LncRNAs are exported to the cytoplasm by the nuclear RNA export factor 1 (NXF1) (154). Once in the cytoplasm, according to their function, they are associated with ribosomes located in the mitochondria or exosomes. In general, the biogenesis of lncRNA varies significantly based on the function of the lncRNA (154).
MALAT1 is a highly conserved lncRNA first identified in the metastasis of early-stage non-small cell lung cancer (155). MALAT1 encoded by the MALAT1 gene located on chromosome 11q.13 (156) maintains basal endothelial cell migration (157), suggesting its crucial role in cardiovascular homeostasis. A recent study by Ruan et. al established a direct link between MALAT1 and miR-34a, where MALAT1 downregulates the expression of miR-34a. As discussed above, miR-34a is upregulated with age which can be attributed to the downregulation of MALAT1 (88, 158). In contrast, Li et al. demonstrated enhanced expression of MALAT1 in cardiac fibroblasts using a rat model of hypertension, which promoted the expression of collagen I and fibronectin leading to myocardial fibrosis (159). Since hypertension is a commonly associated cardiovascular comorbidity with ageing (160) it will be interesting to determine if MALAT1 has differential roles depending on the circumstances and disease conditions.
Sarrah is an anti-apoptotic lncRNA which enhances cardiomyocyte survival and contractility. The expression of Sarrah is downregulated with ageing (161). A downregulation or inhibition of Sarrah activates the caspase activity, thus leading to cardiomyocyte apoptosis (162). In vivo overexpression of Sarrah forms a triple helix in the promoter regions and activates transcription of pro-survival genes such as NRF2 and GPC6, PDE3A, ITPR2, PARP8, and SSBP2 (161). Besides its role as an anti-apoptotic lncRNA, Sarrah is also associated with angiogenesis, as it stimulates an increase in VEGF signalling (161). To date, there is no direct evidence for the role of Sarrah on cardiac ageing. However, due to its functional role in cardiomyocyte apoptosis, which is increased with cardiac ageing, it is logical that Sarrah will have a role in cardiac ageing, requiring further investigations.
CHRF has been studied to be upregulated in ang-II-induced hypertrophy (163). There are two mechanisms by which CHRF expression causes hypertrophy. The first is by repressing the activity of miR-489, an anti-hypertrophic miRNA (46). CHRF acts as a sponge and inhibits the activity of miR-489, which increases the expression of one of its target proteins, the myeloid differentiation protein (Myd88). Myd88 induces hypertrophy by the NF-κB pathway (46). CHRF also inhibits miR-93, an anti-hypertrophic miRNA. Inhibition of miR-93 increases the expression of Akt3, its target protein, which induces cardiomyocyte hypertrophy (164). The change in the expression of CHRF concerning ageing has not been studied yet, but as discussed above, angII signalling increased with age-induced hypertrophy. Thus, the expression of CHRF might increase with age and can augment hypertrophy in the aged heart (46, 163).
MIRT1 and MIRT2 are upregulated in acute myocardial infarction models (163). In addition, MIRT1 and MIRT2 upregulation increased the expression of several genes, such as TGFβ, stat3, and Icam1, all known to be associated with left ventricular remodeling (165). Left ventricular remodeling occurs after MI, in which the LV architecture is altered to distribute pressure overload after MI and an increase in cardiomyocyte hypertrophy (166). Thus, MIRT1 and MIRT2 can play a critical role in LV remodeling after MI, and their regulation can help prevent LV remodeling (165).
LncRNAs and their role in the progression of CVD is relatively new and is less explored than miRNAs (167). LncRNAs have been associated with MI, cardiac hypertrophy, LV remodeling, arrhythmias and other processes (167). Unlike miRNAs, lncRNAs are not highly conserved, and since most of the studies are conducted on mice models or in vitro, whether the lncRNAs will have the same function in humans has yet to be fully understood (168). The research on lncRNAs is still in its infancy, and the discovery of new lncRNAs associated with CVDs is rising (summarised in Table 1).
Nc-RNAs such as miRNAs and lncRNAs control gene expression and thus play a critical role in the cell's normal functioning. MiRNAs and lncRNAs are released in the blood, bound in exosomes, microvesicles, or RNA-binding proteins (166). The tissue-specific expression of ncRNAs, dysregulation of their expression in diseases and their release in bodily fluids such as blood and saliva makes them ideal as a potential biomarker for early diagnosis of the disease (169, 170). MiRNAs as therapeutic molecules have been delivered using viral vectors to treat arrhythmia, atherosclerosis, cardiac hypertrophy, fibrosis, and angiogenesis, exhibiting beneficial effects (169). MGN-1374, an antisense oligonucleotide (ASO) therapeutic molecule which inhibits miR-15 and miR-195, which are involved in cardiac ischemic injury and cardiomyopathy, is currently being studied as a potential and novel treatment of acute myocardial infarction (171). MRG-110 is a synthetic molecule being tested to promote angiogenesis by inhibiting anti-angiogenic miR-92a and is currently in phase 1 clinical trials (172). An ASO developed by Haya therapeutics that inhibits the expression of pro-fibrotic lncRNA Wisp2 super-enhancer–associated RNA (Wisper) is also currently in clinical trials (170). LncRNA therapeutics for CVDs are still nascent and restricted to murine models (173). Hence, further studies are required to translate these findings to the clinic.
While our understanding of miRNAs and lncRNAs has been increasing significantly, and several miRNA-based drugs are in clinical trials, we need a better understanding of their mechanisms to identify them as biomarkers for diagnosing CVD. A significant challenge is the varying results in miRNA levels with specific cardiac pathologies (166). For example, some studies indicate the expression of miR-146 increases with age which causes inflammation and oxidative stress, but some studies indicate the expression decreases with age (95). This discrepancy can be attributed to differences in miRNA quantification methods, sample collection and normalisation of sampling parameters, and associated comorbidities such as diabetes and hypertension (174, 175). Different techniques of miRNA quantification, such as qRT-PCR, stem-loop RT-PCR, digital droplet PCR, and RNAseq, can produce different results, as per the method's sensitivity (174). In most studies cited, small nucleolar RNA U6 (snRNA U6) is used as an internal control, but even snRNA U6 may change in some conditions. Hence it is not a reliable internal control (174). Thus, the variability arising from different sampling methods can be reduced by establishing a standardised operating procedure and statistical analysis tools for quantification (166). Other challenges in developing nc-RNA-based therapeutics are RNA instability, difficult delivery of the molecules across the cell membrane, toxicity of exogenous nucleic acids and off-target effects (169, 170). An example of miR-34a can explain the off-target effects of nc-RNA modulation. As discussed above, miR-34a levels increase with age, leading to increased apoptosis of cardiomyocytes. Inhibiting miR-34a to lower apoptosis of cardiomyocytes can be detrimental, as miR-34a is also a tumour suppressor miRNA, and low levels of miR-34a have been associated with carcinomas (176). Inhibiting miR-34a can thus increase the incidence of cancer. Thus, it is essential to consider the function of each nc-RNA in different conditions before modulating its expression. One possibility to overcome this could be to develop a cell-specific delivery strategy. Another way could be the development of transient modulation of the miRNAs rather than a permanent upregulation or knockdown. Despite the challenges, it is beyond doubt that nc-RNAs have the potential as a therapeutic molecule for the treatment of CVDs. Further, combining nc-RNAs based therapeutics with known biomarkers such as cardiac troponins can be more beneficial for diagnosing and treating CVDs (146, 169).
In conclusion, ageing is a dominant risk factor in the progression of CVDs, as even those individuals leading a healthy lifestyle can also develop CVDs as they age. Since the human heart cannot regenerate, it is more susceptible to diseases. Thus, general screening of cardiac functioning tests such as lipid profile and electrocardiogram beginning from a young age can help identify any dysfunction before the disease progresses. Since the dysregulation of miRNAs and lncRNAs occurs with age, tests to screen the levels of miRNAs can be crucial to understanding cardiac functioning. The emergence of miRNAs and lncRNAs in the normal functioning of the cardiovascular system makes them ideal as novel therapeutic targets to prevent the onset of the disease.
LNJ wrote the manuscript and made figures; DS cricitically revised the manuscript; RK designed and conceptualised the idea, wrote the manuscript and made critical revisions to the draft and the figures. All authors contributed to the article and approved the submitted version.
RK received a project grant from MBIE, NZ (UOOX2205) that supports part of his research time to write this review. LNJ is supported by the PhD scholarship from the Government of Maharastra, India and PhD consumables fund from the Department of Physiology, University of Otago, New Zealand.
The handling editor [PM] declared a past collaboration with the authors [DS, RK].
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bulterijs S, Hull RS, Bjork VC, Roy AG. It is time to classify biological aging as a disease. Front Genet. (2015) 6:205. doi: 10.3389/fgene.2015.00205
2. Maffei VJ, Kim S, Blanchard ET, Luo M, Jazwinski SM, Taylor CM, et al. Biological aging and the human gut Microbiota. J Gerontol A Biol Sci Med Sci. (2017) 72:1474–82. doi: 10.1093/gerona/glx042
3. Moga Rogoz AT, Sart G, Bayar Y, Gavriletea MD. Impact of economic freedom and educational attainment on life expectancy: evidence from the new EU member states. Front Public Health. (2022) 10:907138. doi: 10.3389/fpubh.2022.907138
4. Durrani H. Healthcare and healthcare systems: inspiring progress and future prospects. Mhealth. (2016) 2:3. doi: 10.3978/j.issn.2306-9740.2016.01.03
5. Rudnicka E, Napierala P, Podfigurna A, Meczekalski B, Smolarczyk R, Grymowicz M. The world health organization (WHO) approach to healthy ageing. Maturitas. (2020) 139:6–11. doi: 10.1016/j.maturitas.2020.05.018
6. Squier TC. Oxidative stress and protein aggregation during biological aging. Exp Gerontol. (2001) 36:1539–50. doi: 10.1016/S0531-5565(01)00139-5
7. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
8. Gaziano TA. Cardiovascular disease in the developing world and its cost-effective management. Circulation. (2005) 112:3547–53. doi: 10.1161/CIRCULATIONAHA.105.591792
9. Dai X, Hummel SL, Salazar JB, Taffet GE, Zieman S, Schwartz JB. Cardiovascular physiology in the older adults. J Geriatr Cardiol. (2015) 12:196–201. doi: 10.11909/j.issn.1671-5411.2015.03.015
10. Ciumarnean L, Milaciu MV, Negrean V, Orasan OH, Vesa SC, Salagean O, et al. Cardiovascular risk factors and physical activity for the prevention of cardiovascular diseases in the elderly. Int J Environ Res Public Health. (2021) 19:207. doi: 10.3390/ijerph19010207
11. Yan M, Sun S, Xu K, Huang X, Dou L, Pang J, et al. Cardiac aging: from basic research to therapeutics. Oxid Med Cell Longev. (2021) 2021:9570325. doi: 10.1155/2021/9570325
12. Guerville F, De Souto Barreto P, Ader I, Andrieu S, Casteilla L, Dray C, et al. Revisiting the hallmarks of aging to identify markers of biological age. J Prev Alzheimers Dis. (2020) 7:56–64. doi: 10.14283/jpad.2019.50
13. Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin. (2012) 8:143–64. doi: 10.1016/j.hfc.2011.08.011
14. Cheitlin MD. Cardiovascular physiology-changes with aging. Am J Geriatr Cardiol. (2003) 12:9–13. doi: 10.1111/j.1076-7460.2003.01751.x
15. Singam NSV, Fine C, Fleg JL. Cardiac changes associated with vascular aging. Clin Cardiol. (2020) 43:92–8. doi: 10.1002/clc.23313
16. Kim NH, Kang PM. Apoptosis in cardiovascular diseases: mechanism and clinical implications. Korean Circ J. (2010) 40:299–305. doi: 10.4070/kcj.2010.40.7.299
17. Ribera-Casado JM. Ageing and the cardiovascular system. Z Gerontol Geriatr. (1999) 32:412–9. doi: 10.1007/s003910050138
19. Pugh KG, Wei JY. Clinical implications of physiological changes in the aging heart. Drugs Aging. (2001) 18:263–76. doi: 10.2165/00002512-200118040-00004
20. Peters CH, Sharpe EJ, Proenza C. Cardiac pacemaker activity and aging. Annu Rev Physiol. (2020) 82:21–43. doi: 10.1146/annurev-physiol-021119-034453
21. Behar J, Yaniv Y. Age-related pacemaker deterioration is due to impaired intracellular and membrane mechanisms: insights from numerical modeling. J Gen Physiol. (2017) 149:935–49. doi: 10.1085/jgp.201711792
22. Jagdish Butany MSA, Feinde C. Age-related morphological changes in cardiac valves. Geriatrics and Aging. (2003) 6:49–54.
23. Carabello BA. How does the heart respond to aortic stenosis: let me count the ways. Circ Cardiovasc Imaging. (2013) 6:858–60. doi: 10.1161/CIRCIMAGING.113.001242
24. Xu X, Wang B, Ren C, Hu J, Greenberg DA, Chen T, et al. Age-related impairment of vascular structure and functions. Aging Dis. (2017) 8:590–610. doi: 10.14336/AD.2017.0430
25. Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci (Lond). (2011) 120:357–75. doi: 10.1042/CS20100476
26. Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR Jr., Lerman A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. (2000) 101:948–54. doi: 10.1161/01.CIR.101.9.948
27. North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res. (2012) 110:1097–108. doi: 10.1161/CIRCRESAHA.111.246876
28. Wagner JUG, Dimmeler S. Cellular cross-talks in the diseased and aging heart. J Mol Cell Cardiol. (2020) 138:136–46. doi: 10.1016/j.yjmcc.2019.11.152
29. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
30. Rizvi S, Raza ST, Mahdi F. Telomere length variations in aging and age-related diseases. Curr Aging Sci. (2014) 7:161–7. doi: 10.2174/1874609808666150122153151
31. Koliada AK, Krasnenkov DS, Vaiserman AM. Telomeric aging: mitotic clock or stress indicator? Front Genet. (2015) 6:82. doi: 10.3389/fgene.2015.00082
32. Artandi SE, Attardi LD. Pathways connecting telomeres and p53 in senescence, apoptosis, and cancer. Biochem Biophys Res Commun. (2005) 331:881–90. doi: 10.1016/j.bbrc.2005.03.211
33. Razgonova MP, Zakharenko AM, Golokhvast KS, Thanasoula M, Sarandi E, Nikolouzakis K, et al. Telomerase and telomeres in aging theory and chronographic aging theory (review). Mol Med Rep. (2020) 22:1679–94. doi: 10.3892/mmr.2020.11274
34. Xu C, Wang Z, Su X, Da M, Yang Z, Duan W, et al. Association between leucocyte telomere length and cardiovascular disease in a large general population in the United States. Sci Rep. (2020) 10:80. doi: 10.1038/s41598-019-57050-1
35. Yeh JK, Wang CY. Telomeres and telomerase in cardiovascular diseases. Genes (Basel). (2016) 7:35. doi: 10.3390/genes7090058
36. Brouilette SW, Moore JS, McMahon AD, Thompson JR, Ford I, Shepherd J, et al. Telomere length, risk of coronary heart disease, and statin treatment in the west of Scotland primary prevention study: a nested case-control study. Lancet. (2007) 369:107–14. doi: 10.1016/S0140-6736(07)60071-3
37. Ogami M, Ikura Y, Ohsawa M, Matsuo T, Kayo S, Yoshimi N, et al. Telomere shortening in human coronary artery diseases. Arterioscler Thromb Vasc Biol. (2004) 24:546–50. doi: 10.1161/01.ATV.0000117200.46938.e7
38. Scheller Madrid A, Rode L, Nordestgaard BG, Bojesen SE. Short telomere length and ischemic heart disease: observational and genetic studies in 290 022 individuals. Clin Chem. (2016) 62:1140–9. doi: 10.1373/clinchem.2016.258566
39. Huang YQ, Lo K, Feng YQ, Zhang B. The association of mean telomere length with all-cause, cerebrovascular and cardiovascular mortality. Biosci Rep. (2019) 39. doi: 10.1042/BSR20192306
40. Hoffmann J, Richardson G, Haendeler J, Altschmied J, Andres V, Spyridopoulos I. Telomerase as a therapeutic target in cardiovascular disease. Arterioscler Thromb Vasc Biol. (2021) 41:1047–61. doi: 10.1161/ATVBAHA.120.315695
41. Pusceddu I, Herrmann W, Kleber ME, Scharnagl H, Marz W, Herrmann M. Telomere length, vitamin B12 and mortality in persons undergoing coronary angiography: the ludwigshafen risk and cardiovascular health study. Aging (Albany NY). (2019) 11:7083–97. doi: 10.18632/aging.102238
42. Herman AB, Occean JR, Sen P. Epigenetic dysregulation in cardiovascular aging and disease. J Cardiovasc Aging. (2021) 1:1–10. doi: 10.20517/jca.2021.16
43. Smith ZD, Meissner A. DNA Methylation: roles in mammalian development. Nat Rev Genet. (2013) 14:204–20. doi: 10.1038/nrg3354
44. Zhang W, Song M, Qu J, Liu GH. Epigenetic modifications in cardiovascular aging and diseases. Circ Res. (2018) 123:773–86. doi: 10.1161/CIRCRESAHA.118.312497
45. Jiang YZ, Jimenez JM, Ou K, McCormick ME, Zhang LD, Davies PF. Hemodynamic disturbed flow induces differential DNA methylation of endothelial kruppel-like factor 4 promoter in vitro and in vivo. Circ Res. (2014) 115:32–43. doi: 10.1161/CIRCRESAHA.115.303883
46. Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y, et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. (2014) 114:1377–88. doi: 10.1161/CIRCRESAHA.114.302476
47. Pagiatakis C, Musolino E, Gornati R, Bernardini G, Papait R. Epigenetics of aging and disease: a brief overview. Aging Clin Exp Res. (2021) 33:737–45. doi: 10.1007/s40520-019-01430-0
48. Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, et al. The N(6)-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation. (2019) 139:533–45. doi: 10.1161/CIRCULATIONAHA.118.036146
49. Yang Y, Luan Y, Yuan RX, Luan Y. Histone methylation related therapeutic challenge in cardiovascular diseases. Front Cardiovasc Med. (2021) 8:710053. doi: 10.3389/fcvm.2021.710053
50. Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. (2007) 21:1790–802. doi: 10.1101/gad.1563807
51. Sharma LK, Lu J, Bai Y. Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr Med Chem. (2009) 16:1266–77. doi: 10.2174/092986709787846578
52. Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol-Cell Ph. (2007) 292:C670–86. doi: 10.1152/ajpcell.00213.2006
53. Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. (2005) 19:419–21. doi: 10.1096/fj.04-2622fje
54. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. (2018) 50:121–7. doi: 10.1080/07853890.2017.1417631
55. Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. (2007) 100:460–73. doi: 10.1161/01.RES.0000258450.44413.96
56. Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. (2012) 110:1109–24. doi: 10.1161/CIRCRESAHA.111.246140
57. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. (2019) 51:1–13. doi: 10.1038/s12276-019-0355-7
58. Villeneuve C, Guilbeau-Frugier C, Sicard P, Lairez O, Ordener C, Duparc T, et al. p53-PGC-1alpha pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxid Redox Signal. (2013) 18:5–18. doi: 10.1089/ars.2011.4373
59. Dai DF, Chen T, Johnson SC, Szeto H, Rabinovitch PS. Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. (2012) 16:1492–526. doi: 10.1089/ars.2011.4179
60. Maulik SK, Kumar S. Oxidative stress and cardiac hypertrophy: a review. Toxicol Mech Methods. (2012) 22:359–66. doi: 10.3109/15376516.2012.666650
61. Poznyak AV, Ivanova EA, Sobenin IA, Yet SF, Orekhov AN. The role of mitochondria in cardiovascular diseases. Biology (Basel). (2020) 9:137. doi: 10.3390/biology9060137
62. Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. (2018) 592:728–42. doi: 10.1002/1873-3468.12956
63. Chocron ES, Munkacsy E, Pickering AM. Cause or casualty: the role of mitochondrial DNA in aging and age-associated disease. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:285–97. doi: 10.1016/j.bbadis.2018.09.035
64. Kirino Y, Yasukawa T, Ohta S, Akira S, Ishihara K, Watanabe K, et al. Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc Natl Acad Sci U S A. (2004) 101:15070–5. doi: 10.1073/pnas.0405173101
65. Lindroos MM, Parkka JP, Taittonen MT, Iozzo P, Karppa M, Hassinen IE, et al. Myocardial glucose uptake in patients with the m.3243A > G mutation in mitochondrial DNA. J Inherit Metab Dis. (2016) 39:67–74. doi: 10.1007/s10545-015-9865-1
66. Mitrofanov KY, Zhelankin AV, Shiganova GM, Sazonova MA, Bobryshev YV, Postnov AY, et al. Analysis of mitochondrial DNA heteroplasmic mutations A1555G, C3256T, T3336C, capital ES, Cyrillic5178capital A, cyrillic, G12315A, G13513A, G14459A, G14846capital A, cyrillic and G15059A in CHD patients with the history of myocardial infarction. Exp Mol Pathol. (2016) 100:87–91. doi: 10.1016/j.yexmp.2015.12.003
67. Kelly TN, Raj D, Rahman M, Kretzler M, Kallem RR, Ricardo AC, et al. The role of renin-angiotensin-aldosterone system genes in the progression of chronic kidney disease: findings from the chronic renal insufficiency cohort (CRIC) study. Nephrol Dial Transplant. (2015) 30:1711–8. doi: 10.1093/ndt/gfv125
68. Ames MK, Atkins CE, Pitt B. The renin-angiotensin-aldosterone system and its suppression. J Vet Intern Med. (2019) 33:363–82. doi: 10.1111/jvim.15454
69. Vasan RS, Sullivan LM, D'Agostino RB, Roubenoff R, Harris T, Sawyer DB, et al. Serum insulin-like growth factor I and risk for heart failure in elderly individuals without a previous myocardial infarction: the framingham heart study. Ann Intern Med. (2003) 139:642–8. doi: 10.7326/0003-4819-139-8-200310210-00007
70. Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. (2003) 93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA
71. White M, Roden R, Minobe W, Khan MF, Larrabee P, Wollmering M, et al. Age-related changes in beta-adrenergic neuroeffector systems in the human heart. Circulation. (1994) 90:1225–38. doi: 10.1161/01.CIR.90.3.1225
72. Ferrara N, Komici K, Corbi G, Pagano G, Furgi G, Rengo C, et al. beta-adrenergic receptor responsiveness in aging heart and clinical implications. Front Physiol. (2014) 4:396. doi: 10.3389/fphys.2013.00396
73. de Lucia C, Eguchi A, Koch WJ. New insights in cardiac beta-adrenergic signaling during heart failure and aging. Front Pharmacol. (2018) 9:904. doi: 10.3389/fphar.2018.00904
74. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. (2021) 9:645593. doi: 10.3389/fcell.2021.645593
75. Hu C, Zhang X, Teng T, Ma ZG, Tang QZ. Cellular senescence in cardiovascular diseases: a systematic review. Aging Dis. (2022) 13:103–28. doi: 10.14336/AD.2021.0927
76. Cianflone E, Torella M, Biamonte F, De Angelis A, Urbanek K, Costanzo FS, et al. Targeting cardiac stem cell senescence to treat cardiac aging and disease. Cells. (2020) 9:1558. doi: 10.3390/cells9061558
77. Lewis-McDougall FC, Ruchaya PJ, Domenjo-Vila E, Shin Teoh T, Prata L, Cottle BJ, et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell. (2019) 18:e12931. doi: 10.1111/acel.12931
78. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. (2012) 11:345–9. doi: 10.1111/j.1474-9726.2012.00795.x
79. Kaur J, Farr JN. Cellular senescence in age-related disorders. Transl Res. (2020) 226:96–104. doi: 10.1016/j.trsl.2020.06.007
80. Chen MS, Lee RT, Garbern JC. Senescence mechanisms and targets in the heart. Cardiovasc Res. (2022) 118:1173–87. doi: 10.1093/cvr/cvab161
81. Tang X, Li PH, Chen HZ. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front Endocrinol (Lausanne). (2020) 11:280. doi: 10.3389/fendo.2020.00280
82. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. (2012) 111:245–59. doi: 10.1161/CIRCRESAHA.111.261388
83. Salerno N, Marino F, Scalise M, Salerno L, Molinaro C, Filardo A, et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech Ageing Dev. (2022) 208:111740. doi: 10.1016/j.mad.2022.111740
84. Cattaneo M, Beltrami AP, Thomas AC, Spinetti G, Alvino V, Avolio E, et al. The longevity-associated BPIFB4 gene supports cardiac function and vascularization in aging cardiomyopathy. Cardiovasc Res. (2023):cvad008. doi: 10.1093/cvr/cvad008. [Epub ahead of print]36635236
85. Marttila S, Chatsirisupachai K, Palmer D, de Magalhaes JP. Ageing-associated changes in the expression of lncRNAs in human tissues reflect a transcriptional modulation in ageing pathways. Mech Ageing Dev. (2020) 185:111177. doi: 10.1016/j.mad.2019.111177
86. Zhang P, Wu W, Chen Q, Chen M. Non-Coding RNAs and their integrated networks. J Integr Bioinform. (2019) 16:20190027. doi: 10.1515/jib-2019-0027
87. Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. (2014) 157:77–94. doi: 10.1016/j.cell.2014.03.008
88. Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, et al. MicroRNA-34a regulates cardiac ageing and function. Nature. (2013) 495:107–10. doi: 10.1038/nature11919
89. Lozano-Vidal N, Bink DI, Boon RA. Long noncoding RNA in cardiac aging and disease. J Mol Cell Biol. (2019) 11:860–7. doi: 10.1093/jmcb/mjz046
90. Pereira Fernandes D, Bitar M, Jacobs FMJ, Barry G. Long non-coding RNAs in neuronal aging. Noncoding RNA. (2018) 4:12. doi: 10.3390/ncrna4020012
91. Kondo Y, Shinjo K, Katsushima K. Long non-coding RNAs as an epigenetic regulator in human cancers. Cancer Sci. (2017) 108:1927–33. doi: 10.1111/cas.13342
92. He X, Ou C, Xiao Y, Han Q, Li H, Zhou S. LncRNAs: key players and novel insights into diabetes mellitus. Oncotarget. (2017) 8:71325–41. doi: 10.18632/oncotarget.19921
93. Colpani O, Spinetti G. MicroRNAs orchestrating senescence of endothelial and vascular smooth muscle cells. Vasc Biol. (2019) 1:H75–81. doi: 10.1530/VB-19-0017
94. Zhuang Y, Li T, Xiao H, Wu J, Su S, Dong X, et al. LncRNA-H19 drives cardiomyocyte senescence by targeting miR-19a/socs1/p53 axis. Front Pharmacol. (2021) 12:631835. doi: 10.3389/fphar.2021.631835
95. de Lucia C, Komici K, Borghetti G, Femminella GD, Bencivenga L, Cannavo A, et al. microRNA in cardiovascular aging and age-related cardiovascular diseases. Front Med (Lausanne). (2017) 4:74. doi: 10.3389/fmed.2017.00074
96. Jin L, Song Q, Zhang W, Geng B, Cai J. Roles of long noncoding RNAs in aging and aging complications. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:1763–71. doi: 10.1016/j.bbadis.2018.09.021
97. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. (2014) 15:509–24. doi: 10.1038/nrm3838
98. Li WJ, Wang Y, Liu R, Kasinski AL, Shen H, Slack FJ, et al. MicroRNA-34a: potent tumor suppressor, cancer stem cell inhibitor, and potential anticancer therapeutic. Front Cell Dev Biol. (2021) 9:640587. doi: 10.3389/fcell.2021.640587
99. Cai Y, Yu X, Hu S, Yu J. A brief review on the mechanisms of miRNA regulation. Genomics Proteomics Bioinformatics. (2009) 7:147–54. doi: 10.1016/S1672-0229(08)60044-3
100. Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. (2007) 23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406
101. Li M, Marin-Muller C, Bharadwaj U, Chow KH, Yao Q, Chen C. MicroRNAs: control and loss of control in human physiology and disease. World J Surg. (2009) 33:667–84. doi: 10.1007/s00268-008-9836-x
102. Ouyang Z, Wei K. miRNA in cardiac development and regeneration. Cell Regen. (2021) 10:14. doi: 10.1186/s13619-021-00077-5
103. Titov II, Vorozheykin PS. Comparing miRNA structure of mirtrons and non-mirtrons. BMC Genomics. (2018) 19:114. doi: 10.1186/s12864-018-4473-8
104. O'Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). (2018) 9:402. doi: 10.3389/fendo.2018.00402
105. Bhaskaran M, Mohan M. MicroRNAs: history, biogenesis, and their evolving role in animal development and disease. Vet Pathol. (2014) 51:759–74. doi: 10.1177/0300985813502820
106. Westholm JO, Lai EC. Mirtrons: microRNA biogenesis via splicing. Biochimie. (2011) 93:1897–904. doi: 10.1016/j.biochi.2011.06.017
107. Havens MA, Reich AA, Duelli DM, Hastings ML. Biogenesis of mammalian microRNAs by a non-canonical processing pathway. Nucleic Acids Res. (2012) 40:4626–40. doi: 10.1093/nar/gks026
108. Hua CC, Liu XM, Liang LR, Wang LF, Zhong JC. Targeting the microRNA-34a as a novel therapeutic strategy for cardiovascular diseases. Front Cardiovasc Med. (2021) 8:784044. doi: 10.3389/fcvm.2021.784044
109. Pan J, Zhou L, Lin C, Xue W, Chen P, Lin J. MicroRNA-34a promotes ischemia-induced cardiomyocytes apoptosis through targeting Notch1. Evid Based Complement Alternat Med. (2022) 2022:1388415. doi: 10.1155/2022/1388415
110. Wang ZC, Wang ZZ, Ma HJ, Wang CC, Wang HT. Attenuation of the hypoxia-induced miR-34a protects cardiomyocytes through maintenance of glucose metabolism. Biochem Biophys Res Commun. (2018) 498:375–81. doi: 10.1016/j.bbrc.2017.06.030
111. Landsverk HB, Mora-Bermudez F, Landsverk OJ, Hasvold G, Naderi S, Bakke O, et al. The protein phosphatase 1 regulator PNUTS is a new component of the DNA damage response. EMBO Rep. (2010) 11:868–75. doi: 10.1038/embor.2010.134
112. Rezk NA, Lashin MB, Sabbah NA. MiRNA 34-a regulate SIRT-1 and foxo-1 expression in endometriosis. Noncoding RNA Res. (2021) 6:35–41. doi: 10.1016/j.ncrna.2021.02.002
113. Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. (2008) 105:13421–6. doi: 10.1073/pnas.0801613105
114. Fomison-Nurse I, Saw EEL, Gandhi S, Munasinghe PE, Van Hout I, Williams MJA, et al. Diabetes induces the activation of pro-ageing miR-34a in the heart, but has differential effects on cardiomyocytes and cardiac progenitor cells. Cell Death Differ. (2018) 25:1336–49. doi: 10.1038/s41418-017-0047-6
115. Burton DGA, Faragher RGA. Obesity and type-2 diabetes as inducers of premature cellular senescence and ageing. Biogerontology. (2018) 19:447–59. doi: 10.1007/s10522-018-9763-7
116. Fuziwara CS, Kimura ET. Insights into regulation of the miR-17-92 cluster of miRNAs in cancer. Front Med (Lausanne). (2015) 2:64. doi: 10.3389/fmed.2015.00064
117. Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. (2013) 20:1603–14. doi: 10.1038/cdd.2013.125
118. Gu H, Liu Z, Zhou L. Roles of miR-17-92 cluster in cardiovascular development and common diseases. Biomed Res Int. (2017) 2017:9102909. doi: 10.1155/2017/9102909
119. Danielson LS, Park DS, Rotllan N, Chamorro-Jorganes A, Guijarro MV, Fernandez-Hernando C, et al. Cardiovascular dysregulation of miR-17-92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J. (2013) 27:1460–7. doi: 10.1096/fj.12-221994
120. Wang H, Bei Y, Shi J, Xiao J, Kong X. Non-Coding RNAs in cardiac aging. Cell Physiol Biochem. (2015) 36:1679–87. doi: 10.1159/000430141
121. Du WW, Li X, Li T, Li H, Khorshidi A, Liu F, et al. The microRNA miR-17-3p inhibits mouse cardiac fibroblast senescence by targeting Par4. J Cell Sci. (2015) 128:293–304. doi: 10.1242/jcs.158360
122. Lu Q, Rounds S. Focal adhesion kinase and endothelial cell apoptosis. Microvasc Res. (2012) 83:56–63. doi: 10.1016/j.mvr.2011.05.003
123. van Almen GC, Verhesen W, van Leeuwen RE, van de Vrie M, Eurlings C, Schellings MW, et al. MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell. (2011) 10:769–79. doi: 10.1111/j.1474-9726.2011.00714.x
124. Surina S, Fontanella RA, Scisciola L, Marfella R, Paolisso G, Barbieri M. miR-21 in human cardiomyopathies. Front Cardiovasc Med. (2021) 8:767064. doi: 10.3389/fcvm.2021.767064
125. Hebbar N, Wang C, Rangnekar VM. Mechanisms of apoptosis by the tumor suppressor par-4. J Cell Physiol. (2012) 227:3715–21. doi: 10.1002/jcp.24098
126. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. (2008) 456:980–4. doi: 10.1038/nature07511
127. Triposkiadis F, Xanthopoulos A, Butler J. Cardiovascular aging and heart failure: jACC review topic of the week. J Am Coll Cardiol. (2019) 74:804–13. doi: 10.1016/j.jacc.2019.06.053
128. Zhang M, Li C, Zhao L, Liu G, Yang G. MiR-21 facilitates the proliferation of smooth muscle cells and atherosclerotic plaque formation by suppressing PTEN to activate AKT. Int J Clin Exp Med. (2019) 12:3316–24. doi: 1940-5901/IJCEM0074006
129. Zhang X, Azhar G, Wei JY. The expression of microRNA and microRNA clusters in the aging heart. PLoS One. (2012) 7:e34688. doi: 10.1371/journal.pone.0034688
130. Zhu S, Deng S, Ma Q, Zhang T, Jia C, Zhuo D, et al. MicroRNA-10A* and MicroRNA-21 modulate endothelial progenitor cell senescence via suppressing high-mobility group A2. Circ Res. (2013) 112:152–64. doi: 10.1161/CIRCRESAHA.112.280016
131. Jenike AE, Halushka MK. miR-21: a non-specific biomarker of all maladies. Biomark Res. (2021) 9:18. doi: 10.1186/s40364-021-00272-1
132. Jiang X, Hu C, Arnovitz S, Bugno J, Yu M, Zuo Z, et al. miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia. Nat Commun. (2016) 7:11452. doi: 10.1038/ncomms11452
133. Wang J, Li Y, Ding M, Zhang H, Xu X, Tang J. Molecular mechanisms and clinical applications of miR-22 in regulating malignant progression in human cancer (review). Int J Oncol. (2017) 50:345–55. doi: 10.3892/ijo.2016.3811
134. Jazbutyte V, Fiedler J, Kneitz S, Galuppo P, Just A, Holzmann A, et al. MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr). (2013) 35:747–62. doi: 10.1007/s11357-012-9407-9
135. Deckx S, Heggermont W, Carai P, Rienks M, Dresselaers T, Himmelreich U, et al. Osteoglycin prevents the development of age-related diastolic dysfunction during pressure overload by reducing cardiac fibrosis and inflammation. Matrix Biol. (2018) 66:110–24. doi: 10.1016/j.matbio.2017.09.002
136. Li H, Zhang P, Li F, Yuan G, Wang X, Zhang A, et al. Plasma miR-22-5p, miR-132-5p, and miR-150-3p are associated with acute myocardial infarction. Biomed Res Int. (2019) 2019:5012648. doi: 10.1155/2019/5012648
137. Hussein NA, Kholy ZA, Anwar MM, Ahmad MA, Ahmad SM. Plasma miR-22-3p, miR-642b-3p and miR-885-5p as diagnostic biomarkers for pancreatic cancer. J Cancer Res Clin Oncol. (2017) 143:83–93. doi: 10.1007/s00432-016-2248-7
138. Kriegel AJ, Liu Y, Fang Y, Ding X, Liang M. The miR-29 family: genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol Genomics. (2012) 44:237–44. doi: 10.1152/physiolgenomics.00141.2011
139. Horita M, Farquharson C, Stephen LA. The role of miR-29 family in disease. J Cell Biochem. (2021) 122:696–715. doi: 10.1002/jcb.29896
140. Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, et al. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res. (2011) 109:1115–9. doi: 10.1161/CIRCRESAHA.111.255737
141. Heid J, Cencioni C, Ripa R, Baumgart M, Atlante S, Milano G, et al. Age-dependent increase of oxidative stress regulates microRNA-29 family preserving cardiac health. Sci Rep. (2017) 7:16839. doi: 10.1038/s41598-017-16829-w
142. Querejeta R, Lopez B, Gonzalez A, Sanchez E, Larman M, Martinez Ubago JL, et al. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation. (2004) 110:1263–8. doi: 10.1161/01.CIR.0000140973.60992.9A
143. Hinderer S, Schenke-Layland K. Cardiac fibrosis—a short review of causes and therapeutic strategies. Adv Drug Deliv Rev. (2019) 146:77–82. doi: 10.1016/j.addr.2019.05.011
144. Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, et al. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest. (2012) 122:497–506. doi: 10.1172/JCI61598
145. Ripa R, Dolfi L, Terrigno M, Pandolfini L, Savino A, Arcucci V, et al. MicroRNA miR-29 controls a compensatory response to limit neuronal iron accumulation during adult life and aging. BMC Biol. (2017) 15:9. doi: 10.1186/s12915-017-0354-x
146. Romaine SP, Tomaszewski M, Condorelli G, Samani NJ. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart. (2015) 101:921–8. doi: 10.1136/heartjnl-2013-305402
147. Smith-Vikos T, Slack FJ. MicroRNAs and their roles in aging. J Cell Sci. (2012) 125:7–17. doi: 10.1242/jcs.099200
148. Winbanks CE, Ooi JY, Nguyen SS, McMullen JR, Bernardo BC. MicroRNAs differentially regulated in cardiac and skeletal muscle in health and disease: potential drug targets? Clin Exp Pharmacol Physiol. (2014) 41:727–37. doi: 10.1111/1440-1681.12281
149. Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang CP, et al. Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol. (2015) 12:415–25. doi: 10.1038/nrcardio.2015.55
150. Boon RA, Jae N, Holdt L, Dimmeler S. Long noncoding RNAs: from clinical genetics to therapeutic targets? J Am Coll Cardiol. (2016) 67:1214–26. doi: 10.1016/j.jacc.2015.12.051
151. Greco S, Salgado Somoza A, Devaux Y, Martelli F. Long noncoding RNAs and cardiac disease. Antioxid Redox Signal. (2018) 29:880–901. doi: 10.1089/ars.2017.7126
152. Liu Y, Ding W, Yu W, Zhang Y, Ao X, Wang J. Long non-coding RNAs: biogenesis, functions, and clinical significance in gastric cancer. Mol Ther Oncolytics. (2021) 23:458–76. doi: 10.1016/j.omto.2021.11.005
153. Correia CCM, Rodrigues LF, de Avila Pelozin BR, Oliveira EM, Fernandes T. Long non-coding RNAs in cardiovascular diseases: potential function as biomarkers and therapeutic targets of exercise training. Noncoding RNA. (2021) 7:65. doi: 10.3390/ncrna7040065
154. Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. (2021) 22:96–118. doi: 10.1038/s41580-020-00315-9
155. Wang JZ, Xiang JJ, Wu LG, Bai YS, Chen ZW, Yin XQ, et al. A genetic variant in long non-coding RNA MALAT1 associated with survival outcome among patients with advanced lung adenocarcinoma: a survival cohort analysis. BMC Cancer. (2017) 17:167. doi: 10.1186/s12885-017-3151-6
156. Zhang X, Hamblin MH, Yin KJ. The long noncoding RNA Malat1: its physiological and pathophysiological functions. RNA Biol. (2017) 14:1705–14. doi: 10.1080/15476286.2017.1358347
157. Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. (2014) 114:1389–97. doi: 10.1161/CIRCRESAHA.114.303265
158. Ruan L, Mendhe B, Parker E, Kent A, Isales CM, Hill WD, et al. Long non-coding RNA MALAT1 is depleted with age in skeletal muscle in vivo and MALAT1 silencing increases expression of TGF-beta1 in vitro. Front Physiol. (2021) 12:742004. doi: 10.3389/fphys.2021.742004
159. Li D, Zhang C, Li J, Che J, Yang X, Xian Y, et al. Long non-coding RNA MALAT1 promotes cardiac remodeling in hypertensive rats by inhibiting the transcription of MyoD. Aging (Albany NY). (2019) 11:8792–809. doi: 10.18632/aging.102265
160. Pinto E. Blood pressure and ageing. Postgrad Med J. (2007) 83:109–14. doi: 10.1136/pgmj.2006.048371
161. Trembinski DJ, Bink DI, Theodorou K, Sommer J, Fischer A, van Bergen A, et al. Aging-regulated anti-apoptotic long non-coding RNA sarrah augments recovery from acute myocardial infarction. Nat Commun. (2020) 11:2039. doi: 10.1038/s41467-020-15995-2
162. Santos F, Correia M, Nobrega-Pereira S, Bernardes de Jesus B. Age-Related pathways in cardiac regeneration: a role for lncRNAs? Front Physiol. (2020) 11:583191. doi: 10.3389/fphys.2020.583191
163. Greco S, Gorospe M, Martelli F. Noncoding RNA in age-related cardiovascular diseases. J Mol Cell Cardiol. (2015) 83:142–55. doi: 10.1016/j.yjmcc.2015.01.011
164. Wo Y, Guo J, Li P, Yang H, Wo J. Long non-coding RNA CHRF facilitates cardiac hypertrophy through regulating Akt3 via miR-93. Cardiovasc Pathol. (2018) 35:29–36. doi: 10.1016/j.carpath.2018.04.003
165. Zangrando J, Zhang L, Vausort M, Maskali F, Marie PY, Wagner DR, et al. Identification of candidate long non-coding RNAs in response to myocardial infarction. BMC Genomics. (2014) 15:460. doi: 10.1186/1471-2164-15-460
166. Zhou SS, Jin JP, Wang JQ, Zhang ZG, Freedman JH, Zheng Y, et al. miRNAS in cardiovascular diseases: potential biomarkers, therapeutic targets and challenges. Acta Pharmacol Sin. (2018) 39:1073–84. doi: 10.1038/aps.2018.30
167. Ratti M, Lampis A, Ghidini M, Salati M, Mirchev MB, Valeri N, et al. MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) as new tools for cancer therapy: first steps from bench to bedside. Target Oncol. (2020) 15:261–78. doi: 10.1007/s11523-020-00717-x
168. Fang Y, Xu Y, Wang R, Hu L, Guo D, Xue F, et al. Recent advances on the roles of LncRNAs in cardiovascular disease. J Cell Mol Med. (2020) 24:12246–57. doi: 10.1111/jcmm.15880
169. Cakmak HA, Demir M. MicroRNA and cardiovascular diseases. Balkan Med J. (2020) 37:60–71. doi: 10.4274/balkanmedj.galenos.2020.2020.1.94
170. Halasz H, Carpenter S. Challenges and future directions for LncRNAs and inflammation. Adv Exp Med Biol. (2022) 1363:179–83. doi: 10.1007/978-3-030-92034-0_10
171. Chakraborty C, Sharma AR, Sharma G, Lee SS. Therapeutic advances of miRNAs: a preclinical and clinical update. J Adv Res. (2021) 28:127–38. doi: 10.1016/j.jare.2020.08.012
172. Hanna J, Hossain GS, Kocerha J. The potential for microRNA therapeutics and clinical research. Front Genet. (2019) 10:478. doi: 10.3389/fgene.2019.00478
173. Das S, Shah R, Dimmeler S, Freedman JE, Holley C, Lee JM, et al. Noncoding RNAs in cardiovascular disease: current knowledge, tools and technologies for investigation, and future directions: a scientific statement from the American heart association. Circ Genom Precis Med. (2020) 13:e000062. doi: 10.1161/HCG.0000000000000062
174. Tijsen AJ, Pinto YM, Creemers EE. Circulating microRNAs as diagnostic biomarkers for cardiovascular diseases. Am J Physiol Heart Circ Physiol. (2012) 303:H1085–95. doi: 10.1152/ajpheart.00191.2012
175. Navickas R, Gal D, Laucevicius A, Taparauskaite A, Zdanyte M, Holvoet P. Identifying circulating microRNAs as biomarkers of cardiovascular disease: a systematic review. Cardiovasc Res. (2016) 111:322–37. doi: 10.1093/cvr/cvw174
Keywords: cardiac ageing, cardiovascular disease, molecular changes, non-coding RNA, microRNA, long non-coding RNA
Citation: Varghese LN, Schwenke DO and Katare R (2023) Role of noncoding RNAs in cardiac ageing. Front. Cardiovasc. Med. 10:1142575. doi: 10.3389/fcvm.2023.1142575
Received: 11 January 2023; Accepted: 6 March 2023;
Published: 22 March 2023.
Edited by:
Paolo Madeddu, University of Bristol, United KingdomReviewed by:
Carlo Gaetano, Scientific Clinical Institute Maugeri (ICS Maugeri), Italy© 2023 Varghese, Schwenke and Katare. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajesh Katare cmFqZXNoLmthdGFyZUBvdGFnby5hYy5ueg==
Specialty Section: This article was submitted to Cardiovascular Biologics and Regenerative Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.