 Masafumi Funamoto
Masafumi Funamoto Masaki Imanishi
Masaki Imanishi Koichiro Tsuchiya2
Koichiro Tsuchiya2 Yasumasa Ikeda
Yasumasa Ikeda
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 15 March 2023
Sec. Cardiovascular Metabolism
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1133611
This article is part of the Research Topic Frontiers in Cardiovascular Medicine: Rising Stars 2022 View all 75 articles
Heart failure results from various physiological and pathological stimuli that lead to cardiac hypertrophy. This pathological process is common in several cardiovascular diseases and ultimately leads to heart failure. The development of cardiac hypertrophy and heart failure involves reprogramming of gene expression, a process that is highly dependent on epigenetic regulation. Histone acetylation is dynamically regulated by cardiac stress. Histone acetyltransferases play an important role in epigenetic remodeling in cardiac hypertrophy and heart failure. The regulation of histone acetyltransferases serves as a bridge between signal transduction and downstream gene reprogramming. Investigating the changes in histone acetyltransferases and histone modification sites in cardiac hypertrophy and heart failure will provide new therapeutic strategies to treat these diseases. This review summarizes the association of histone acetylation sites and histone acetylases with cardiac hypertrophy and heart failure, with emphasis on histone acetylation sites.
Heart failure (HF), which occurs in the late stage of most cardiovascular diseases (CVDs), has a high mortality rate and worsens the patient life expectancy. The proportion of the population affected by HF is estimated to increase annually, placing a significant burden on public health in an aging society (1). Various CVDs can lead to HF via pathological cardiac hypertrophy. Cardiac hypertrophy is an important process of cardiac remodeling that is often caused by preload, such as hypertension, or afterload, such as myocardial infarction (2, 3). Cardiac hypertrophy is a compensatory mechanism that reduces oxygen consumption, normalizes the ventricular systolic pressure, and improves the ejection function. However, long-term stress, such as hypertension, eventually leads to HF and irreversible pathological cardiac remodeling. Cardiac hypertrophy also causes fibrosis and dysfunction, leading to HF (4, 5). Various biological regulatory processes are involved in cardiac hypertrophy and remodeling. Particularly, acetylation and deacetylation of histones via epigenetic modifications have attracted researchers' attention (6, 7). Histone acetylation and deacetylation are believed to play important roles in the regulation of gene expression, leading to cardiac hypertrophy and HF under stress. Histone acetylation is important for cardiac physiology and pathophysiology (8). However, the different sites of histone acetylation are not clearly understood. This review summarizes the potential mechanisms by which the differences in histone acetylation sites in the regulation of gene expression may be involved in the etiology of cardiac hypertrophy and HF. This review provides a better understanding of the regulatory mechanisms of histone acetylation in the pathogenesis of HF and suggests new therapeutic strategies for HF.
Epigenetics is currently one of the fastest-growing areas in biological research. Epigenetics plays an important role in many cellular processes, including regulation of gene expression and transcription, cell growth and differentiation, and chromosome remodeling and inactivation (8, 9). Epigenetics refers to the regulation of gene expression via reversible chemical modifications of the DNA, histones, and even chromatin structures, without changing the DNA sequence. Epigenetic processes primarily include DNA methylation, non-coding RNAs, and histone modifications (10). The nucleosome is composed of 147 bp of DNA wrapped around an octamer containing four histone proteins: H2A, H2B, H3, and H4 (11). The linker histone protein H1 mediates interactions between adjacent nucleosomes, resulting in chromatin formation (12). Histones are characterized by their direct binding to DNA, and post-translational modifications (PTMs) of histones alter the chromatin structure and dynamically regulate the transcriptional state of genes (13). Chromatin is of two types: heterochromatin, which is closed and compressed, and euchromatin, which is relaxed and more suitable for transcription. In general, histone PTMs are controlled by “writers” that add modifier groups, “erasers” that remove modifier groups, and “readers” that specifically recognize the modification sites for downstream transcriptional activation or repression. All these PTMs are regulated by enzymes, and their enzymatic activity may be controlled by reactive cofactors, such as substrates and metabolic intermediates (14). Histone modifications include acetylation, methylation, butyrylation, propionylation, formylation, succinylation, malonylation, 2-hydroxyisobutyrylation, β-hydroxybutyrylation, glutarylation, benzoylation, and crotonylation (13, 15, 16). Among all histone modifications, acetylation and methylation are mostly shown to be involved in the regulation of CVDs, and other modifications, such as succinylation, have also been extensively studied in various diseases (15, 16). Histone acetylation is associated with the upregulation of gene transcription via various mechanisms, some of which are described below. First, PTMs of histone lysine residues alter the positive charge of the ε-amino group, which reduces nucleosome folding and affects DNA–histone or histone–histone interactions, thereby reducing the euchromatin–nucleosome interactions. However, interactions between enhancers and their corresponding promoters are strengthened (14, 17–19). Thus, PTMs of histones directly affect the structures of chromatin and nucleosomes. Second, PTMs of lysine residues on histones can serve as epigenetic landmarks directly or indirectly by the presence of transcription factors, and RNA polymerase II, which directly or indirectly mobilizes the acetylation of histone lysine residues, serves as an epigenetic landmark that is specifically recognized by transcription factors and chromatin remodeling factors. The acetylation level of lysine residues depends on the dynamic regulation of histone acetyltransferases (HATs) and histone deacetylases (HDACs) (14, 20). Acetylation sites of histones have mainly been studied for H3 and H4. Acetylation sites on histones H3 and H4 include H3K4, H3K9, H3K14, H3K18, H3K23, H3K27, H3K36, H3K56, H4K5, H4K8, H4K12, H4K16, and H4K20 (13, 21). In this review, we mainly focused on the acetylation of histones H3 and H4 and their involvement in cardiac hypertrophy and HF.
The influence of epigenetics on disease incidence has drastically increased in the field of cancer, and in recent years, it has gradually expanded to almost all diseases, including HF (22, 23). Histone acetylation plays an important role in histone modification and affects heart diseases (22). Many histone acetylation sites have been suggested to be associated with heart diseases. Here, we discuss the relationship between histone acetylation sites and cardiac hypertrophy and HF.
Acetylation of H3K4 by p300 has been suggested to be important for the expression of the transcription factor, GATA-binding protein 4 (GATA4), in heart formation (24). Acetylation of H3K9 and H3K27 has also been suggested to be involved in the expression of GATA4, as GATA4 is involved in the development of HF and the fetal gene is reactivated during HF, suggesting that the acetylation of H3K4 may be involved in cardiac hypertrophy and HF (25, 26). Many reports have suggested that the acetylation of H3K9 by p300 causes cardiomyocyte hypertrophy (3, 4, 27–30). Furthermore, H3K9 acetylation is increased during cardiac hypertrophy and HF in TAC and Dahl rats (4, 26, 31). Acetylation of H3K9 is reduced by inhibition of the HAT activity of p300 (31, 32). Although some studies have suggested an association between H3K14 acetylation and microRNA (miR)-134–5p expression in cardiac fibroblasts, the specific role of H3K14 remains unknown due to the lack of comparisons with the normal state (33). Acetylation of H3K14 at the miR-30a-5p promoter in H9c2 cells is associated with c-Myc expression (34). However, whether H3K14 acetylation is directly involved in cardiac hypertrophy and HF remains unclear. Acetylation of H3K27 has been reported to be associated with cardiac disease, although not to the same extent as the acetylation of H3K9. Acetylation of H3K27 leads to cardiac hypertrophy and fibrosis of the heart together with bromodomain-containing 4 (35). In addition, H3K27 acetylation is associated with enhancers that are specifically activated in cardiac hypertrophy, and specific epigenetic signatures have been shown to regulate gene expression in hypertrophy by controlling promoter activity (36). Brd4 is a reader protein for H3K27 acetylation in CVDs (37, 38). Acetylation of H3K27 and H3K9 has also been shown to be a therapeutic target for CVDs (39). These acetylation sites are located in the tail domain of histones and implicated in cardiac hypertrophy and HF. Moreover, H3K122, a globular domain in the body of histones around which the DNA wraps, has also been reported to be involved in HF. In the Dahl rat model of hypertension-induced HF, acetylation of H3K9 increases during cardiac hypertrophy when the heart function is preserved, and acetylation of H3K122 in the globular domain increases during HF when the heart function is reduced. These histone acetylations are altered in the hypertrophic response gene promoter region and the recruitment of p300 remains unchanged during cardiac hypertrophy and HF. However, the binding of p300 to BRG1, a chromatin remodeling factor, increases during HF, and the recruitment of BRG1 to the promoter increases during cardiac hypertrophy. The mRNA levels of hypertrophic response genes are greatly increased in HF than in cardiac hypertrophy. This suggests that the acetylation of H3K122 in the histone body is involved in the pathogenesis of HF. Moreover, complex formation between p300 and BRG1 may be important for this acetylation of H3K122 (40).
There are no reports of any direct relationships of H3K18, H3K23, H3K36, H3K56, H4K5, H4K8, H4K12, H4K16, and H4K20 with cardiac hypertrophy and HF. However, acetylation of H3K9 and H3K27 has been reported in cardiac hypertrophy and HF. Other acetylation sites need to be investigated further based on the affected gene region and stage of the disease. Furthermore, the detailed role of these histone acetylation sites in the promoter and enhancer regions in cardiac hypertrophy and heart failure during transcription remains to be elucidated. Therefore, the relationship between histone acetylation and the expression of genes involved in cardiac hypertrophy and heart failure needs to be studied in detail.
Selected lysines are acetylated by specific biological processes, and the overall histone acetylation level is dynamically regulated by two enzyme families, HATs and HDACs (41, 42). In histone acetylation, HATs require acetyl-CoA as cofactors to catalyze the transfer of the acetyl group to the ε-amino group of the lysine side chain (43). HATs can be divided into five families: p300/CBP, Basal TF, GNAT, MYST, and NCoA (44). Importantly, changes in the formation of complexes involving HATs can alter the sites of acetylated histones. For example, complex formation between p300 and BRG1 increases during cardiac hypertrophy and HF, and the level of H3K122 acetylation in the globular domain increases (40). Furthermore, yeast Gcn5 alone acetylates free histones in vitro, but it must be combined with the Ada2 and Ada3 subunits within ADA and SAGA complexes to preferentially acetylate H2B in the nucleosome (45). This increase in histone acetylation levels and changes in histone acetylation sites are involved in the formation of HAT complexes and activation of HATs. Therefore, understanding the transcriptional regulatory mechanisms related to HATs is important for the treatment of HF. Acetylation of histones by p300 is involved in cardiac hypertrophy and heart failure (46, 47). Hypertrophic stress increases HAT activity and histone acetylation, as p300 is regulated by several proteins, including ERK1/2, Akt, and Cdk9 (3). However, for HATs such as GCN5, CBP, and PCAF, the relationship with histone acetylation is unclear, although an association between cardiac hypertrophy and heart failure has been reported (48–50).Curcumin, a natural product, inhibits histone acetylation by blocking the HAT activity of p300, thereby improving cardiomyocyte hypertrophy and HF (32). Curcumin has also been reported to inhibit heart failure with preserved ejection fraction in Dahl rats (47). Recently, curcumin-based derivatives and analogs have been shown to have beneficial effects in HF (3, 4). Anacardic and eicosapentaenoic acids improve HF by inhibiting the HAT activity of p300. Similar to curcumin, anacardic acid also inhibits the HAT activity of p300 and improves the progression of cardiac hypertrophy and HF by inhibiting the acetylation of H3K9 (31, 51). In addition, direct inhibition of p300 HAT by eicosatetraenoic acid suppresses myocardial infarction-induced HF (28). Metformin can inhibit phenylephrine-induced cardiomyocyte hypertrophy by inhibiting p300 in cultured cardiomyocytes; therefore, it can potentially be used for the treatment of patients with diabetes and HF (52). In contrast, L003 and C646, specific HAT inhibitors of p300, inhibit angiotensin-induced cardiac hypertrophy and cardiac fibrosis (53). Compounds, such as resveratrol, also improve cardiac hypertrophy and HF by activating HDACs, such as sirtuin (54). In summary, preventing histone acetylation by inhibiting the HAT activity of p300 or activating HDACs can aid in the treatment of cardiac hypertrophy and HF. Moreover, drugs targeting various epigenetic processes can inhibit the HAT activity of p300 or activate HDACs for the treatment of HF.
HDACs reverse histone acetylation by HATs and restore the original unacetylated state of histone lysine. There are four classes of HDACs, all of which are complexed with proteins that exhibit low substrate specificity (55). HDACs are classified into four major classes: HDAC class I (HDAC 1, 2, 3, and 8), HDAC class IIa (HDAC 4, 5, 7, and 9), HDAC class IIb (HDAC 6 and 10), HDAC class III (SIRT1-7), and HDAC class IV (HDAC11) (6, 56). HDACs cause chromatin enrichment by deacetylating histones, blocking access to DNA, and inhibiting transcription (57). The relationship between non-histone proteins and HDACs has been studied in cardiovascular disease. HDAC class I promotes cardiac hypertrophy and heart failure in many cases, whereas HDAC1 and HDAC2 inhibit cardioprotective and anti-hypertrophic genes (58, 59). Increased expression of HDAC8 in the heart increases p38 phosphorylation and expression, inducing cardiac hypertrophy and fibrosis (60). Trichostatin A and valproic acid have been shown to inhibit cardiac hypertrophy by inhibiting HDAC class I, making HDAC inhibition an attractive therapeutic target for heart failure (61). While HDAC class II plays a role in suppressing cardiac hypertrophy and heart failure, HDAC class IIa (HDAC 4, 5, and 9) suppresses cardiac hypertrophy by forming a complex with the transcription factor Mef2 (6). Phosphorylation of HDAC class IIa by phosphatases such as calcium/calmodulin-dependent protein kinase 2 prevents its binding to Mef2 and activates gene transcription (62). In addition, HDAC6 is involved in tubulin acetylation, myofibril stiffness and skeletal muscle wasting in cardiac disease (63–65). However, few reports on cardiac hypertrophy and heart failure associated with HDAC class IIb and IV are available, and he role of these HDACs in the heart needs to be clarified in the future. HDAC class III, also known as the SIRT protein family, plays a role in cardiac homeostasis; SIRT1 and SIRT3 promote the deacetylation of PGC-1α and reduce cardiac hypertrophy by reducing oxidative stress (66–68). SIRT2 exerts cardioprotective effect by promoting AMPK activation through deacetylation of LKB1 (69). SIRT6 inhibits cardiac hypertrophy by suppressing NFATc4 expression and activation (70). In contrast, a direct association between histone acetylation and HDACs in the heart has been reported. SIRT3 is involved in H3K27 deacetylation and regulates inflammation and fibrosis in the heart via modulation of the FOS/AP-1 pathway (71). SIRT6 binds to and represses the promoters of IGF signaling-related genes by deacetylating H3K9 through interaction with c-Jun. SIRT6 expression is downregulated in human heart failure, indicating that SIRT6 is involved in the pathogenesis of cardiac hypertrophy and heart failure (72). As mentioned above, few studies have demonstrated a direct relationship between HDACs and histone deacetylation in cardiac hypertrophy and heart failure. Therefore, the relationship between histone acetylation sites and HDACs should be studied in detail. It is also necessary to determine whether HDACs are negative or positive regulators of cardiac hypertrophy and heart failure.
Histone acetylation and HAT are therapeutic targets, and their proper regulation is a promising therapeutic strategy for HF; however, some problems should be mentioned. Currently, only a limited number of histone acetylation sites have been implicated in HF (Figure 1). Therefore, future studies should investigate other histone acetylation sites throughout the genome. As the sites of histone acetylation are not similar in all gene regions, the overall map of histone acetylation sites in HF and the related HAT-centered transcriptional regulatory mechanisms need to be clarified. Since histone acetylation is regulated by the balance between HATs and HDACs, it is necessary to develop therapeutic agents targeting HDACs (9, 73). Although histone acetylation has been associated with gene expression during heart failure, there are also genes whose expression is altered independently of histone acetylation (9, 36, 40). Histone acetylation is associated with other epigenetic mechanisms such as histone methylation and DNA methylation (35). Therefore, the relationship between histone acetylation and other epigenetic mechanisms in cardiac hypertrophy and heart failure should be further investigated. In addition, post-translational modifications of histones, such as acetylation, in patients with heart failure have rarely been examined. Increased HAT activation of p300 has been reported in patients with heart failure (74); however, the association between histone acetylation and the development of heart failure in humans needs to be clarified.
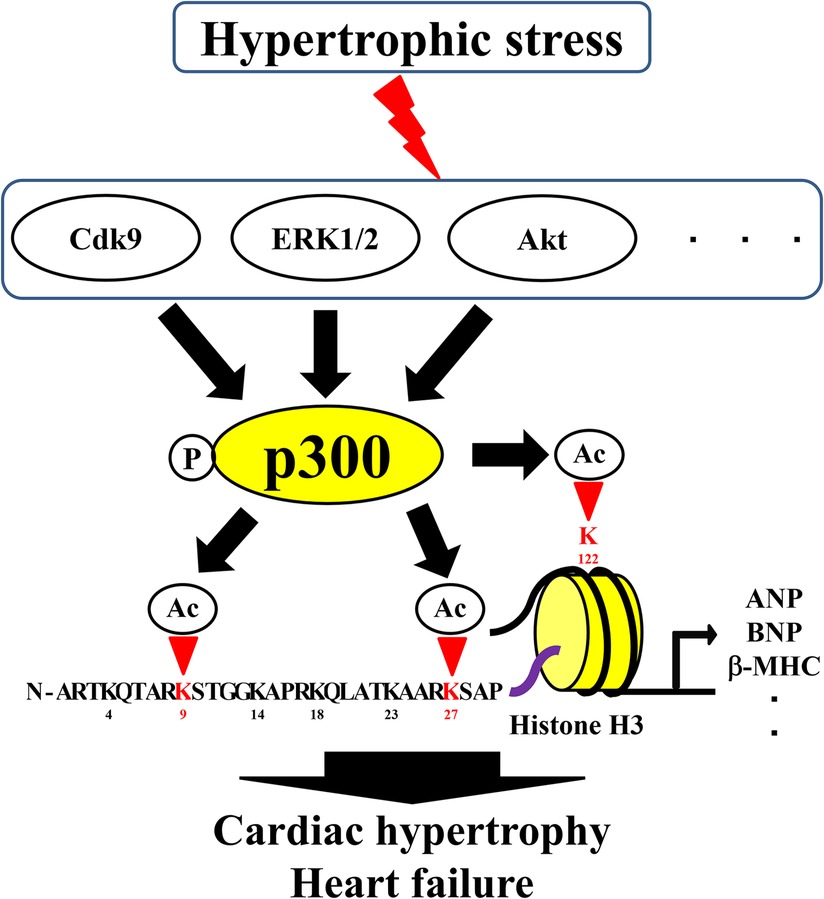
Figure 1. Roles of histone acetylation sites in cardiac hypertrophy and heart failure. Ac, acetylation; HATs, histone acetyltransferases; P, phosphorylation.
In summary, epigenetic regulation by histone modifiers is critical in the pathogenesis of cardiac hypertrophy. Identifying the molecular mechanisms underlying the roles of histone modifiers in normal and disease states is essential for the development of novel “epigenetic” drugs to treat cardiac hypertrophy. Furthermore, as different histone acetylation sites have different effects on transcription, identifying time-specific acetylation sites in disease states is also important for the development of novel therapeutic agents for HF.
MF: wrote the article. MI, KT, and YI: edited and reviewed the article. All authors read and agreed to the article's final version for publication. All authors contributed to the article and approved the submitted version.
This work was supported by JSPS KAKENHI Grant Number JP22K16106.
This work was supported by JSPS KAKENHI Grant Number JP22K16106. We would like to thank Editage (www.editage.com) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The handling editor TM declared a past co-authorship with the authors MF and YI.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics-2022 update: a report from the American heart association. Circulation. (2022) 145:E153–639. doi: 10.1161/CIR.0000000000001052
2. Maillet M, Van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. (2013) 14:38–48. doi: 10.1038/NRM3495
3. Funamoto M, Sunagawa Y, Gempei M, Shimizu K, Katanasaka Y, Shimizu S, et al. Pyrazole-Curcumin suppresses cardiomyocyte hypertrophy by disrupting the CDK9/CyclinT1 Complex. Pharmaceutics. (2022) 14:1269. doi: 10.3390/PHARMACEUTICS14061269
4. Shimizu K, Sunagawa Y, Funamoto M, Wakabayashi H, Genpei M, Miyazaki Y, et al. The synthetic curcumin analogue GO-Y030 effectively suppresses the development of pressure overload-induced heart failure in mice. Sci Rep. (2020) 10:7172. doi: 10.1038/s41598-020-64207-w
5. Butler J, Packer M, Filippatos G, Ferreira JP, Zeller C, Schnee J, et al. Effect of empagliflozin in patients with heart failure across the spectrum of left ventricular ejection fraction. Eur Heart J. (2022) 43:416. doi: 10.1093/EURHEARTJ/EHAB798
6. Han Y, Nie J, Wang DW, Ni L. Mechanism of histone deacetylases in cardiac hypertrophy and its therapeutic inhibitors. Front Cardiovasc Med. (2022) 9:931475. doi: 10.3389/FCVM.2022.931475
7. Yan K, Wang K, Li P. The role of post-translational modifications in cardiac hypertrophy. J Cell Mol Med. (2019) 23:3795–807. doi: 10.1111/JCMM.14330
8. Greco CM, Condorelli G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol. (2015) 12:488–97. doi: 10.1038/NRCARDIO.2015.71
9. Papait R, Condorelli G. Epigenetics in heart failure. Ann N Y Acad Sci. (2010) 1188:159–64. doi: 10.1111/J.1749-6632.2009.05096.X
10. Yin H, Xie Y, Gu P, Li W, Zhang Y, Yao Y, et al. The emerging role of epigenetic regulation in the progression of silicosis. Clin Epigenetics. (2022) 14:169. doi: 10.1186/S13148-022-01391-8
11. Furumatsu T, Ozaki T. Epigenetic regulation in chondrogenesis. Acta Med Okayama. (2010) 64:155–61. doi: 10.18926/AMO/40007
12. Duffney LJ, Valdez P, Tremblay MW, Cao X, Montgomery S, McConkie-Rosell A, et al. Epigenetics and autism spectrum disorder: a report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am J Med Genet B Neuropsychiatr Genet. (2018) 177:426–33. doi: 10.1002/AJMG.B.32631
13. Cavalieri V. The expanding constellation of histone post-translational modifications in the epigenetic landscape. Genes. (2021) 12:1596. doi: 10.3390/GENES12101596
14. Huang Z, Song S, Zhang X, Zeng L, Sun A, Ge J. Metabolic substrates, histone modifications, and heart failure. Biochim Biophys Acta Gene Regul Mech. (2023) 1866:194898. doi: 10.1016/J.BBAGRM.2022.194898
15. Ntorla A, Burgoyne JR. The regulation and function of histone crotonylation. Front Cell Dev Biol. (2021) 9:624914. doi: 10.3389/FCELL.2021.624914
16. Amamoto Y, Aoi Y, Nagashima N, Suto H, Yoshidome D, Arimura Y, et al. Synthetic posttranslational modifications: chemical catalyst-driven regioselective histone acylation of native chromatin. J Am Chem Soc. (2017) 139:7568–76. doi: 10.1021/JACS.7B02138/SUPPL_FILE/JA7B02138_SI_002.PDF
17. Shimko JC, North JA, Bruns AN, Poirier MG, Ottesen JJ. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J Mol Biol. (2011) 408:187–204. doi: 10.1016/J.JMB.2011.01.003
18. Tropberger P, Pott S, Keller C, Kamieniarz-Gdula K, Caron M, Richter F, et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell. (2013) 152:859–72. doi: 10.1016/J.CELL.2013.01.032
19. Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, et al. Histone fold modifications control nucleosome unwrapping and disassembly. Proc Natl Acad Sci U S A. (2011) 108:12711–6. doi: 10.1073/PNAS.1106264108
20. Jing Y, Li X, Liu Z, Li XD. Roles of negatively charged histone lysine acylations in regulating nucleosome structure and dynamics. Front Mol Biosci. (2022) 9:899013. doi: 10.3389/FMOLB.2022.899013
21. Shvedunova M, Akhtar A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol. (2022) 23:329–49. doi: 10.1038/S41580-021-00441-Y
22. Qin J, Guo N, Tong J, Wang Z. Function of histone methylation and acetylation modifiers in cardiac hypertrophy. J Mol Cell Cardiol. (2021) 159:120–9. doi: 10.1016/J.YJMCC.2021.06.011
23. Napoli C, Bontempo P, Palmieri V, Coscioni E, Maiello C, Donatelli F, et al. Epigenetic therapies for heart failure: current insights and future potential. Vasc Health Risk Manag. (2021) 17:247–54. doi: 10.2147/VHRM.S287082
24. Zhou W, Jiang D, Tian J, Liu L, Lu T, Huang X, et al. Acetylation of H3K4, H3K9, and H3K27 mediated by p300 regulates the expression of GATA4 in cardiocytes. Genes Dis. (2018) 6:318–25. doi: 10.1016/J.GENDIS.2018.10.002
25. Sunagawa Y, Katanasaka Y, Wada H, Hasegawa K, Morimotoa T. Functional analysis of GATA4 complex, a cardiac hypertrophy-response transcriptional factor, using a proteomics approach. Yakugaku Zasshi. (2016) 136:151–6. doi: 10.1248/YAKUSHI.15-00226-2
26. Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. (2008) 118:868–78. doi: 10.1172/JCI33160
27. Sunagawa Y, Funamoto M, Sono S, Shimizu K, Shimizu S, Genpei M, et al. Curcumin and its demethoxy derivatives possess p300 HAT inhibitory activity and suppress hypertrophic responses in cardiomyocytes. J Pharmacol Sci. (2018) 136:212–7. doi: 10.1016/j.jphs.2017.12.013
28. Sunagawa Y, Katayama A, Funamoto M, Shimizu K, Shimizu S, Sari N, et al. The polyunsaturated fatty acids, EPA and DHA, ameliorate myocardial infarction-induced heart failure by inhibiting p300-HAT activity in rats. J Nutr Biochem. (2022) 106:109031. doi: 10.1016/J.JNUTBIO.2022.109031
29. Katagiri T, Sunagawa Y, Maekawa T, Funamoto M, Shimizu S, Shimizu K, et al. Ecklonia stolonifera okamura extract suppresses myocardial infarction-induced left ventricular systolic dysfunction by inhibiting p300-HAT activity. Nutrients. (2022) 14:580. doi: 10.3390/NU14030580
30. Peng B, Peng C, Luo X, Wu S, Mao Q, Zhang H, et al. JNK signaling-dependent regulation of histone acetylation are involved in anacardic acid alleviates cardiomyocyte hypertrophy induced by phenylephrine. PLoS One. (2021) 16:e0261388. doi: 10.1371/JOURNAL.PONE.0261388
31. Li S, Peng B, Luo X, Sun H, Peng C. Anacardic acid attenuates pressure-overload cardiac hypertrophy through inhibiting histone acetylases. J Cell Mol Med. (2019) 23:2744–52. doi: 10.1111/JCMM.14181
32. Morimoto T, Sunagawa Y, Fujita M, Hasegawa K. Novel heart failure therapy targeting transcriptional pathway in cardiomyocytes by a natural compound, curcumin. Circ J. (2010) 74:1060–6. doi: 10.1253/circj.cj-09-1012
33. Hao S, Sui X, Wang J, Zhang J, Pei Y, Guo L, et al. Secretory products from epicardial adipose tissue induce adverse myocardial remodeling after myocardial infarction by promoting reactive oxygen species accumulation. Cell Death Dis. (2021) 12:848. doi: 10.1038/S41419-021-04111-X
34. Xu L, Zhang H, Wang Y, Guo W, Gu L, Yang A, et al. H3k14 hyperacetylation-mediated c-Myc binding to the miR-30a-5p gene promoter under hypoxia postconditioning protects senescent cardiomyocytes from hypoxia/reoxygenation injury. Mol Med Rep. (2021) 23:468. doi: 10.3892/MMR.2021.12107
35. McKinsey TA, Vondriska TM, Wang Y. Epigenomic regulation of heart failure: integrating histone marks, long noncoding RNAs, and chromatin architecture. F1000Res. (2018) 7:F1000 Faculty Rev-1713. doi: 10.12688/F1000RESEARCH.15797.1
36. Papait R, Cattaneo P, Kunderfranco P, Greco C, Carullo P, Guffanti A, et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci U S A. (2013) 110:20164–9. doi: 10.1073/PNAS.1315155110/-/DCSUPPLEMENTAL/SD01.XLSX
37. Mohammed SA, Albiero M, Ambrosini S, Gorica E, Karsai G, Caravaggi CM, et al. The BET protein inhibitor apabetalone rescues diabetes-induced impairment of angiogenic response by epigenetic regulation of thrombospondin-1. Antioxid Redox Signal. (2022) 36:667–84. doi: 10.1089/ARS.2021.0127
38. Fang M, Luo J, Zhu X, Wu Y, Li X. BRD4 Silencing protects angiotensin II-induced cardiac hypertrophy by inhibiting TLR4/NF- κ B and activating Nrf2-HO-1 pathways. Cardiol Res Pract. (2022) 2022:8372707. doi: 10.1155/2022/8372707
39. Kashyap S, Mukker A, Gupta D, Datta PK, Rappaport J, Jacobson JM, et al. Antiretroviral drugs regulate epigenetic modification of cardiac cells through modulation of H3K9 and H3K27 acetylation. Front Cardiovasc Med. (2021) 8:634774. doi: 10.3389/FCVM.2021.634774
40. Funamoto M, Sunagawa Y, Katanasaka Y, Shimizu K, Miyazaki Y, Sari N, et al. Histone acetylation domains are differentially induced during development of heart failure in dahl salt-sensitive rats. Int J Mol Sci. (2021) 22:1771. doi: 10.3390/ijms22041771
41. Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. (2007) 100:1512–21. doi: 10.1161/01.RES.0000267723.65696.4A
42. Wang X, Hayes JJ. Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol Cell Biol. (2008) 28:227–36. doi: 10.1128/MCB.01245-07
43. Berndsen CE, Denu JM. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr Opin Struct Biol. (2008) 18:682–9. doi: 10.1016/J.SBI.2008.11.004
44. Li P, Ge J, Li H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat Rev Cardiol. (2019) 17:96–115. doi: 10.1038/s41569-019-0235-9
45. Grant PA, Duggan L, Côté J, Roberts SM, Brownell JE, Candau R, et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. (1997) 11:1640–50. doi: 10.1101/GAD.11.13.1640
46. Miyamoto S, Kawamura T, Morimoto T, Ono K, Wada H, Kawase Y, et al. Histone acetyltransferase activity of p300 is required for the promotion of left ventricular remodeling after myocardial infarction in adult mice in vivo. Circulation. (2006) 113:679–90. doi: 10.1161/CIRCULATIONAHA.105.585182
47. Sunagawa Y, Funamoto M, Shimizu K, Shimizu S, Sari N, Katanasaka Y, et al. Curcumin, an inhibitor of p300-HAT activity, suppresses the development of hypertension-induced left ventricular hypertrophy with preserved ejection fraction in dahl rats. Nutrients. (2021) 13:2608. doi: 10.3390/NU13082608
48. Mao Q, Wu S, Peng C, Peng B, Luo X, Huang L, et al. Interactions between the ERK1/2 signaling pathway and PCAF play a key role in PE-induced cardiomyocyte hypertrophy. Mol Med Rep. (2021) 24:636. doi: 10.3892/MMR.2021.12275
49. Li J, Yan C, Wang Y, Chen C, Yu H, Liu D, et al. GCN5-mediated Regulation of pathological cardiac hypertrophy via activation of the TAK1-JNK/p38 signaling pathway. Cell Death Dis. (2022) 13:421. doi: 10.1038/S41419-022-04881-Y
50. Zhou H, Li N, Yuan Y, Jin YG, Guo H, Deng W, et al. Activating transcription factor 3 in cardiovascular diseases: a potential therapeutic target. Basic Res Cardiol. (2018) 113:37. doi: 10.1007/S00395-018-0698-6
51. Peng C, Luo X, Li S, Sun H. Phenylephrine-induced cardiac hypertrophy is attenuated by a histone acetylase inhibitor anacardic acid in mice. Mol Biosyst. (2017) 13:714–24. doi: 10.1039/C6MB00692B
52. Sunagawa Y, Shimizu K, Katayama A, Funamoto M, Shimizu K, Nurmila S, et al. Metformin suppresses phenylephrine-induced hypertrophic responses by inhibiting p300-HAT activity in cardiomyocytes. J Pharmacol Sci. (2021) 147:169–75. doi: 10.1016/J.JPHS.2021.07.001
53. Rai R, Sun T, Ramirez V, Lux E, Eren M, Vaughan DE, et al. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J Cell Mol Med. (2019) 23:3026. doi: 10.1111/JCMM.14162
54. Zhang L, Chen J, Yan L, He Q, Xie H, Chen M. Resveratrol ameliorates cardiac remodeling in a murine model of heart failure with preserved ejection fraction. Front Pharmacol. (2021) 12:646240. doi: 10.3389/FPHAR.2021.646240
55. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. (2014) 6:a018713. doi: 10.1101/CSHPERSPECT.A018713
56. Chen H, Xie C, Chen Q, Zhuang S. HDAC11, An emerging therapeutic target for metabolic disorders. Front Endocrinol. (2022) 13:989305. doi: 10.3389/FENDO.2022.989305
57. Weeks KL, Avkiran M. Roles and post-translational regulation of cardiac class IIa histone deacetylase isoforms. J Physiol. (2015) 593:1785–97. doi: 10.1113/JPHYSIOL.2014.282442
58. Bush EW, McKinsey TA. Targeting histone deacetylases for heart failure. Expert Opin Ther Targets. (2009) 13:767–84. doi: 10.1517/14728220902939161
59. Bush EW, McKinsey TA. Protein acetylation in the cardiorenal axis: the promise of histone deacetylase inhibitors. Circ Res. (2010) 106:272–84. doi: 10.1161/CIRCRESAHA.109.209338
60. Zhao T, Kee HJ, Bai L, Kim MK, Kee SJ, Jeong MH. Selective HDAC8 inhibition attenuates isoproterenol-induced cardiac hypertrophy and fibrosis via p38 MAPK pathway. Front Pharmacol. (2021) 12:677757. doi: 10.3389/FPHAR.2021.677757
61. Mathiyalagan P, Keating ST, Du XJ, El-Osta A. Chromatin modifications remodel cardiac gene expression. Cardiovasc Res. (2014) 103:7–16. doi: 10.1093/CVR/CVU122
62. Hohl M, Wagner M, Reil JC, Müller SA, Tauchnitz M, Zimmer AM, et al. HDAC4 Controls histone methylation in response to elevated cardiac load. J Clin Invest. (2013) 123:1359–70. doi: 10.1172/JCI61084
63. Lin YH, Major JL, Liebner T, Hourani Z, Travers JG, Wennersten SA, et al. HDAC6 Modulates myofibril stiffness and diastolic function of the heart. J Clin Invest. (2022) 132:e148333. doi: 10.1172/JCI148333
64. Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, et al. HDAC6 Contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Hear Circ Physiol. (2014) 307:H252. doi: 10.1152/AJPHEART.00149.2014
65. Sanbe A, Inomata Y, Matsushita N, Sawa Y, Hino C, Yamazaki H, et al. Modification of cardiac disease by transgenically altered histone deacetylase 6. Biochem Biophys Res Commun. (2022) 631:48–54. doi: 10.1016/J.BBRC.2022.09.055
66. Guo L, Yin A, Zhang Q, Zhong T, O’Rourke ST, Sun C. Angiotensin-(1-7) attenuates angiotensin II-induced cardiac hypertrophy via a Sirt3-dependent mechanism. Am J Physiol Heart Circ Physiol. (2017) 312:H980–91. doi: 10.1152/AJPHEART.00768.2016
67. Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with PPARα to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. (2011) 90:276–84. doi: 10.1093/CVR/CVQ376
68. Yu L, Gong B, Duan W, Fan C, Zhang J, Li Z, et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: role of AMPK-PGC-1α-SIRT3 signaling. Sci Rep. (2017) 7:41337. doi: 10.1038/SREP41337
69. Tang X, Chen XF, Wang NY, Wang XM, Liang ST, Zheng W, et al. SIRT2 Acts as a cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation. (2017) 136:2051–67. doi: 10.1161/CIRCULATIONAHA.117.028728
70. Li Z, Zhang X, Guo Z, Zhong Y, Wang P, Li J, et al. SIRT6 Suppresses NFATc4 expression and activation in cardiomyocyte hypertrophy. Front Pharmacol. (2019) 9:1519. doi: 10.3389/FPHAR.2018.01519
71. Palomer X, Román-Azcona MS, Pizarro-Delgado J, Planavila A, Villarroya F, Valenzuela-Alcaraz B, et al. SIRT3-mediated Inhibition of FOS through histone H3 deacetylation prevents cardiac fibrosis and inflammation. Signal Transduct Target Ther. (2020) 5:1519. doi: 10.1038/S41392-020-0114-1
72. Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, et al. The sirtuin SIRT6 blocks IGF-akt signaling and development of cardiac hypertrophy by targeting c-jun. Nat Med. (2012) 18:1643–50. doi: 10.1038/nm.2961
73. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. (2002) 110:479. doi: 10.1016/S0092-8674(02)00861-9
Keywords: epigenetics, histone post-translational modification, cardiac hypertrophy, heart failure, acetylation
Citation: Funamoto M, Imanishi M, Tsuchiya K and Ikeda Y (2023) Roles of histone acetylation sites in cardiac hypertrophy and heart failure. Front. Cardiovasc. Med. 10:1133611. doi: 10.3389/fcvm.2023.1133611
Received: 29 December 2022; Accepted: 24 February 2023;
Published: 15 March 2023.
Edited by:
Tatsuya Morimoto, University of Shizuoka, JapanReviewed by:
Shafeeq Ahmed Mohammed, University of Zurich, Switzerland© 2023 Funamoto, Imanishi, Tsuchiya and Ikeda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masafumi Funamoto ZnVuYW1vdG9AdG9rdXNoaW1hLXUuYWMuanA= Yasumasa Ikeda eWFzdWlrZUB0b2t1c2hpbWEtdS5hYy5qcA==
Specialty Section: This article was submitted to Cardiovascular Metabolism, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.