 Yuanqiang Li
Yuanqiang Li Boyu Li
Boyu Li Wei-Dong Chen
Wei-Dong Chen Yan-Dong Wang
Yan-Dong Wang- 1State Key Laboratory of Chemical Resource Engineering, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing, China
- 2Key Laboratory of Receptors-Mediated Gene Regulation and Drug Discovery, School of Basic Medical Science, Inner Mongolia Medical University, Hohhot, China
- 3Key Laboratory of Receptors-Mediated Gene Regulation, School of Medicine, The People’s Hospital of Hebi, Henan University, Kaifeng, China
- 4Department of Gastroenterology and Hematology, The People's Hospital of Hebi, Henan, China
Cardiovascular diseases (CVDs) are the leading cause of death globally, with CVDs accounting for nearly 30% of deaths worldwide each year. G-protein-coupled receptors (GPCRs) are the most prominent family of receptors on the cell surface, and play an essential regulating cellular physiology and pathology. Some GPCR antagonists, such as β-blockers, are standard therapy for the treatment of CVDs. In addition, nearly one-third of the drugs used to treat CVDs target GPCRs. All the evidence demonstrates the crucial role of GPCRs in CVDs. Over the past decades, studies on the structure and function of GPCRs have identified many targets for the treatment of CVDs. In this review, we summarize and discuss the role of GPCRs in the function of the cardiovascular system from both vascular and heart perspectives, then analyze the complex ways in which multiple GPCRs exert regulatory functions in vascular and heart diseases. We hope to provide new ideas for the treatment of CVDs and the development of novel drugs.
Introduction
Cardiovascular diseases (CVDs) are the leading cause of death worldwide. CVDs are divided into two categories, vascular diseases and heart diseases (1). Vascular diseases include hypertension, atherosclerosis, aortic aneurysms, and vascular calcification (2), while the major components of heart diseases are ischemic heart diseases, rheumatic heart diseases, cardiomyopathy, and myocarditis (3). There are numerous options for treating CVDs, such as lipid-lowering drugs, antihypertensive drugs, antiplatelet and anticoagulant therapies. Despite the effectiveness of these approaches, there is still a long way from curing CVDs (4). Thus, it is crucial to find novel therapeutic targets and develop new drugs to treat CVDs.
G-protein-coupled receptors (GPCRs), which are the most prominent receptor family among all cell surface proteins (5), play essential roles in various human physiological and pathological processes (6). GPCRs contain seven transmembrane α-helices and are coupled with heterotrimeric GTP-binding proteins (G proteins), which are composed of Gα, Gβ, and Gγ subunits. Depending on the difference between Gα subunits, G proteins can be divided into four categories that play different roles. First of all, Gαs can generate the second messenger cyclic-3′,5′-adenosine monophosphate (cAMP) by activating adenylate cyclase, while Gαi/o exerts the opposite effect. Then Gαq/11 activates phospholipase C (PLC) to produce the second messenger inositol 1,4,5-trisphosphate (IP3). Finally, Gα12/13 can regulate downstream signals through the small GTPase Rho (7).
GPCRs are widely expressed in the cardiovascular system and play crucial roles in regulating cardiovascular function and morphology (8). β-Adrenergic receptors (βARs) and angiotensin II type 1 receptors (AT1Rs) are important GPCRs in cardiovascular function. In addition, there are many other GPCRs, such as apelin receptor (APJ), lysophosphatidic acid receptor (LPARs) and endothelin receptors (ETAR and ETBR), that play important roles in CVDs. Chronic activation by their endogenous ligands increases the workload of the heart, leading to harmful effects such as heart failure (HF) (9). For these reasons, β-blockers and angiotensin-converting enzyme inhibitors are recommended by WHO as essential medicines for patients with CVDs. Furthermore, roughly one-third of all currently used drugs in cardiovascular practice target GPCRs (10). In this review, we summarize the role of GPCRs in CVDs from both vascular diseases and heart diseases, providing new ideas for the treatment of cardiovascular diseases and the development of innovative drugs (Table 1).
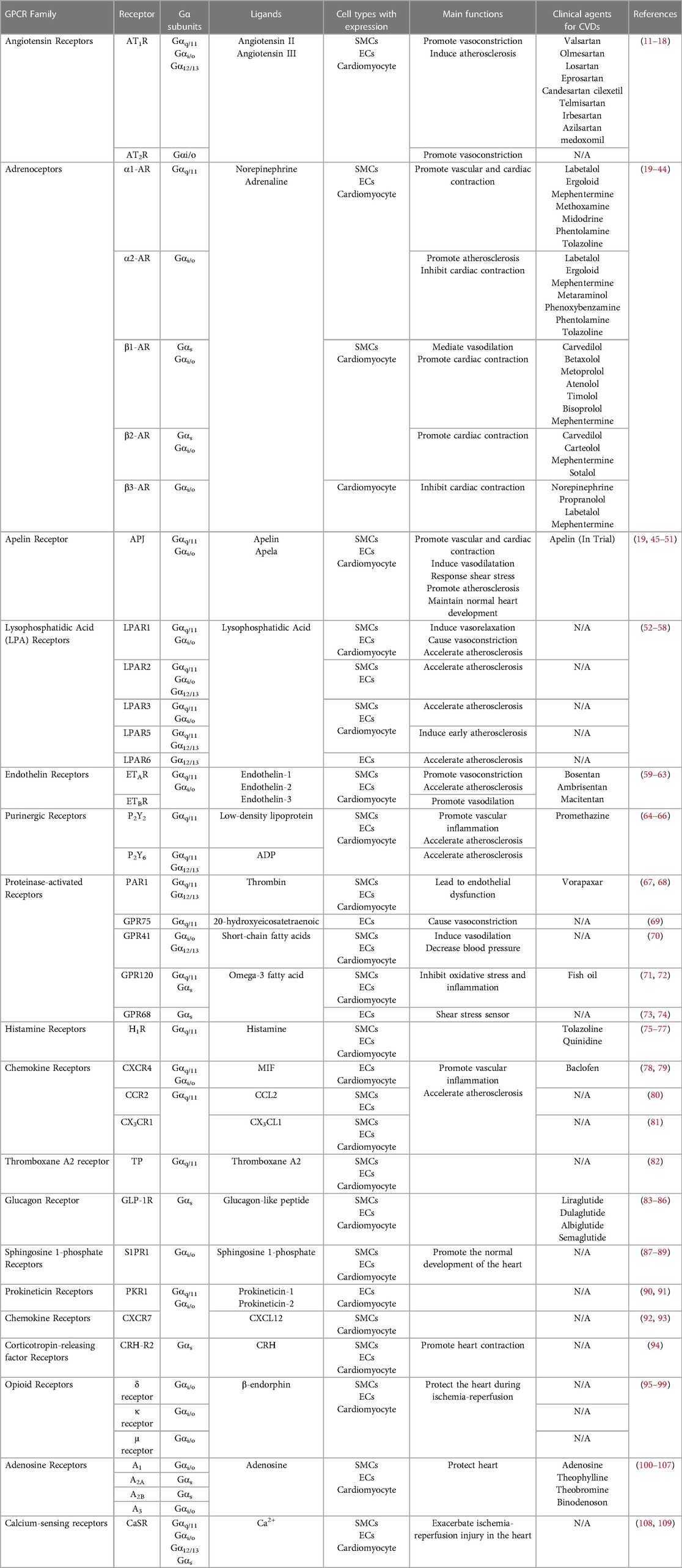
Table 1. GPCRs in cardiovascular system and cardiovascular disease.
GPCRs in vascular function and disease
GPCRs and vascular function
Vascular homeostasis is essential for maintaining the health of the body. Smooth muscle cells (SMCs) are a major structural component of the vessel wall, regulating vascular tone to maintain intravascular pressure (110). Meanwhile, endothelial cells (ECs) are critical regulators of vascular inflammation, thrombophlebitis, permeability, and vascular remodeling (111). Under normal conditions, SMCs and ECs exert a protective role, maintaining vascular stability. However, during the development of vascular disease, the dysfunction of ECs and the dedifferentiation of SMCs promote pathological changes in the vasculature, thereby accelerating the process of vascular diseases. Single-cell GPCR expression analysis demonstrates that the expression of GPCRs in ECs and SMCs is highly heterogeneous. Vascular diseases such as atherosclerosis lead to characteristic changes in the expression of GPCRs (112). Thus, in the vascular system, GPCRs are critical regulators.
GPCRs regulate blood pressure by modulating the dynamic balance of vasoconstriction and relaxation (113, 114). Gαq/11-coupled GPCRs and Gα12/13-coupled GPCRs cause vasoconstriction via Ca2+ and RhoA, respectively. Conversely, Gαs-coupled GPCRs can generate cAMP, and then promote blood vessel relaxation (115, 116). One typical example is that angiotensin II (AngII) binds to and activates angiotensin receptors (AT1R and AT2R), causing the smooth muscle to contract (11). And α1-adrenergic receptor (α1-AR), mainly expressed in SMCs, has a similar function (117). The APJ is highly expressed in cardiovascular tissues, and the apelin/APJ system is vital for regulating vascular tone (45, 46). Apelin/APJ system can inhibit the BKCa signaling pathway (118), increase the phosphorylation of MLC (119), or cooperate with α1-AR to promote vasoconstriction (19). However, the apelin/APJ system can induce vasodilatation by stimulating the release of nitric oxide (NO) (47, 120). This different regulation depends on the type of blood vessels and pathological condition (45). Besides, many other GPCRs are implicated in the regulation of vasoconstriction and relaxation. LPA stimulates LPA receptor 1 (LPAR1), then activates PLC and releases NO to induce vasorelaxation. In addition, activation of LPAR1 can also produce thromboxane A2 (TxA2), which can bind to prostaglandin receptors, leading to vasoconstriction (52–54). The endothelin system includes two GPCRs: endothelin receptor A (ETAR) and B (ETBR). Endothelin 1 (ET-1) can promote vasoconstriction by activating ETAR or promote vasodilation by activating ETBR (59). Recent studies have shown that the binding of the orphan receptor GPR75 to 20-hydroxyeicosatetraenoic acid (20-HETE) activates the Gαq/11 protein, which causes vasoconstriction (69). And short-chain fatty acids (SCFAs) can activate GPR41 to induce vasodilation (70). In conclusion, GPCRs are critical regulators of vascular tone.
Normal vascular endothelial function is highly crucial for vascular homeostasis. Endothelial dysfunction leads to the destruction of cell connections, vascular leakage, tissue edema, and organ failure (111, 121). Vascular endothelial dysfunction is caused by inflammation, and GPCRs play an essential role in this process (122). Multiple GPCRs agonists, including thrombin, histamine, and prostaglandin E2, stimulate robust p38 autophosphorylation to promote endothelial inflammatory responses (123). Purinergic GPCRs (P2Ys) are widely expressed in the cardiovascular system. P2Y1, P2Y2, P2Y4, and P2Y6 can promote vascular inflammation and reduce endothelial barrier function through the Gαq-PLC pathway (124). Protease-activated receptors (PARs) are specific GPCRs that can be cleaved by serine proteases thrombin or trypsin and then regulate downstream signaling pathways. The sustained activation of PAR1 promotes the disruption of endothelial junction proteins, increases endothelial permeability and plasma extravasation, and leads to endothelial dysfunction (67). In contrast, activation of GPR120, a recently identified omega-3 fatty acid receptor, inhibits oxidative stress and inflammation by suppressing the production of reactive oxygen species (ROS) and the expression of pro-inflammatory cytokines. It also can protect vascular endothelial function by preventing monocyte attachment to endothelial cells (71).
The flow of blood causes shear stress, which is a mechanical stimulus. Mechanical stimuli can be sensed by cells and converted into biochemical signals to inspire diverse cellular functions (125, 126). Some GPCRs are initial sensors of mechanical stimulation, they can be activated by shear stress to regulate downstream signals. For instance, GPR68, a mechanosensor expressed in ECs, is significantly responsive to shear stress and is required for ECs' shear stress sensitivity. After the absence of GPR68 in mice, the vasodilation response brought about by the increase in blood flow was disrupted, suggesting that GPR68 is involved in flow-mediated vasodilation and remodeling (73, 74). Similar to GPR68, the H1 histamine receptor (H1R) is mechanosensitive Gαq/11 coupled GPCR highly expressed in ECs. Shear stress activates H1R in an agonist-independent manner, leading to vasodilation (75). APJ is another GPCR that can be activated by mechanical stimulation. Flow-induced signaling through APJ is crucial for cell morphology, endothelial elasticity, and cellular adhesion. Deleting APJ not only impairs the elasticity and cell adhesion of ECs but also alters the remodeling of actin filaments and the distribution of vinculin particles (48).
In summary, GPCRs play an important role in regulating vascular tension, maintaining vascular endothelial barrier function, and sensing blood flow shear stress.
GPCRs and atherosclerosis
Atherosclerosis is a progressive disease. The accumulation of lipids, fibers, or cell debris on the arterial intima interferes normal function of blood vessels and impedes blood flow. In severe cases, it can lead to myocardial infarction or stroke (127). Atherosclerosis can be seen as a response to injury and is a chronic inflammatory disease of blood vessels. Endothelial dysfunction caused by vascular inflammation initiates the process of atherosclerosis. In the presence of inflammatory factors, SMCs migrate to the vascular intima and then proliferate, resulting in atherosclerotic plaques (128, 129). Endothelial dysfunction and the proliferation and migration of SMCs are the fundamental factors of atherosclerosis. Many GPCRs play an essential role in vascular endothelial dysfunction, therefore significantly influencing on the atherosclerotic process.
The role of P2Ys in atherosclerosis can be anticipated due to their role in endothelial dysfunction (130). A typical example is P2Y2. The ATP released by oxidized low-density lipoprotein (LDL) activates P2Y2, therefore promoting vascular inflammation, ensuring penetration and adhesion of monocytes, and accelerating the process of atherosclerosis (64). In addition, P2Y6 is upregulated in atherosclerotic lesions, suggesting that it may also promote atherosclerosis. Due to reduced vascular inflammation, P2Y6-deficient mice have a slowed atherosclerotic process (65). LPA accumulates in atherosclerosis, and the expression of LPAR1-LPAR6 in human arterial plaques and normal arteries is significantly different, suggesting that LPARs may play a role in atherosclerosis (131, 132). Activation of LPAR1 and LPAR3 can promote the expression of hypoxia-inducible factor 1 subunit alpha (HIF-1α), then upregulate C-X-C motif chemokine ligand 1 (CXCL1) in cells, thereby accelerating atherosclerosis (133). LPAR6 can induce actin stress crack formation through the RhoA/ROCK pathway to increase endothelial permeability and advance the occurrence of atherosclerosis (55). Furthermore, LPAR5 activates the TGFBR1, which stimulates the glycosaminoglycan (GAG) chain elongation, resulting in the early pathogenesis of atherosclerosis (56). The interaction between chemokines and their GPCR-type receptors (CRKs) is an element that promotes atherosclerosis. CXC-motif chemokine receptor-4 (CXCR4) (78), CC chemokine receptor 2 (CCR2) (80), and C-X3-C Motif Chemokine Receptor 1 (CX3CR1) (81) accelerate atherosclerosis by promoting vascular inflammation.
The proliferation and migration of SMCs accelerate the occurrence of atherosclerosis, and GPCRs also regulate this process. LPARs play a vital role in the dedifferentiation, proliferation, and migration of SMCs (52). LPA promotes the dedifferentiation of SMCs through LPAR3 (57) and promotes SMC proliferation and migration through LPAR1 and LAPR2 (58). And in-depth studies suggest that LPA may accelerate the atherosclerosis process by activating Gαq/11-coupled GPCRs to promote the proliferation and migration of SMCs (134, 135). Yes-associated protein (YAP) signaling pathway is a crucial regulator of the proliferation and migration of SMCs (136). Depending on the difference of the coupled G protein, GPCRs have different effects on the regulation of YAP. Briefly, Gαi/o, Gαq/11, and Gα12/13 can activate YAP, while Gαs exerts an inhibitory effect (137). For instance, the thromboxane A2 receptor (TP) (82), AT1R (138), and ETAR (60) can activate YAP, then promote the proliferation and migration of SMCs. Activation of APJ by apelin promotes the proliferation of SMCs, while the knockout of APJ reduces the production of ROS and the formation of atherosclerosis (49). By promoting the activity of growth factor receptors EGFR and HGFR, the α2-adrenergic receptor (α2-AR) promotes the proliferation of SMCs cells (20). Furthermore, the Glucagon-like peptide 1 receptor (GLP-1R) is located in the nucleus of rat SMCs, and artificially keeping it in the cytoplasm can promote the proliferation of SMCs (83). Together, these studies show that GPCRs are of profound significance to the occurrence and development of atherosclerosis, and the development of drugs targeting GPCRs to treat atherosclerosis is very necessary.
GPCRs in heart function and disease
GPCRs and heart function
The heart principal function is to pump blood to the circulation of the various organs and systems of the human body to achieve the purpose of oxygen supply and nutrient exchange. The normal development of the heart is of great importance. Abnormal heart development leads to heart malformations and congenital heart disease, affecting human life and health (139, 140). After years of research, a large number of pathways and processes that play a regulatory role in the development of the heart have come to light. Among them, GPCRs are regulators that cannot be neglected (90). Sphingosine 1-phosphate (S1P) is a lipid with biological activity, and the activation of its receptors S1P receptors (S1PRs) is essential in the normal development of the heart (87). Mice with global loss of S1PR1 will die 12.5–14.5 days post-coitus due to cardiovascular defects (88). In addition, knocking out S1PR1 in mouse cardiomyocytes will affect standard ventricular compaction, septation, and embryo survival, indicating that S1PR1 in cardiomyocytes is required for the normal development of the heart (89). The apelin-APJ system is an essential regulator of the cardiovascular system. Loss of APJ leads to abnormal development of myocardial progenitor and defects in heart development (50, 51). Apela is an endogenous ligand of APJ newly discovered in recent years (141), and the absence of apela gene also leads to early deformation of heart development (142, 143). Many other GPCRs are also important in heart development. Prokineticin receptor-1 (PKR1) (90), C-X-C motif chemokine receptor 7 (CXCR7) (92, 93), 5-hydroxytryptamine receptor 2B (5-HT2B) (144), and atypical chemokine receptor (ACKR) (145) are critical to the development of the heart, and the absence of either of them leads to incomplete heart development and thus death in mice.
The heart's contraction is a complex process involving action potentials, contractile proteins, and excitation-contraction coupling, which has been thoroughly reviewed by predecessors (146–150). Heart contractility is extremely important to the pumping function of the heart, and the decline of contractility can lead to HF, which results in sudden death (147). GPCRs expressed in the human myocardium have both positive and negative regulatory effects on heart contractility. AR family mainly includes five receptors, α1, α2, β1, β2, and β3 (117). These five receptors are all expressed in the heart, and the regulatory role of βARs in the heart is crucial. β1-AR accounts for about 80% of the βARs in the heart, followed by β2-AR, accounting for 15%–18%, and the remaining β3-AR (10). Activation of β1-AR or β2-AR will activate Gαs protein and promote the production of cAMP. Then cAMP acts on protein kinase A (PKA), thereby causing heart contraction (21, 151). Conversely, activation of β3-AR promotes cardiac relaxation through the release of NO (22). Besides, α1-AR (23) and α2-AR (24) perform functions that promote or inhibit cardiac contraction, respectively. In addition, using corticotropin-releasing hormone (CRH) to activate CRH receptor 2 (CRH-R2) in mice can promote heart contraction through a variety of signaling pathways, including adenylate cyclase, PKC, and PKA (94). And the binding of myosuppressin (MS) to its receptor can decrease heart contractility to a great extent (152). In summary, the strategic role of GPCRs in heart development and contraction is evident.
GPCRs and cardiac ischemia-reperfusion injury
Ischemic heart disease occupies an essential position in all types of heart disease, and its fatality rate has reached almost half of all CVDs, and it is the leading cause of death around the world (3). The ischemia-reperfusion (IR) process is a pathological phenomenon, which refers to first restricting the blood supply to the organ, then restoring the perfusion and corresponding oxygen supply (153). Heart IR will cause many deaths of cardiomyocytes and induce severe autoimmune responses, which may lead to long-term cardiac dysfunction (153, 154). Therefore, effective interventions to limit IR injury (IRI) are critical to protecting the heart. GPCRs have been proven to play a significant role in inhibiting IRI and protecting the heart. The opioid receptor (OR) family is a cardioprotective system, and opioid preconditioning has shown a strong protective effect on IRI (155). δ opioid receptors (DOR) and κ opioid receptors (KOR) are expressed in human cardiomyocytes, while the expression of μ opioid receptors (MOR) is dependent on species (95). During IR, ORs are vital determinants of ischemia and hypoxia tolerance; opioid levels are upregulated in heart ischemic, which leads to the activation of ORs and induces cardioprotective responses (96). ORs preconditioning effects activate a series of downstream signal pathways through the Gαi/o-PKC pathway to protect mitochondrial function, inhibit cell death signals, and achieve the purpose of protecting the heart (96–99). Adenosine receptors are another GPCR family that can protect the heart in IR. Studies have shown that the four subtypes of adenosine receptors, A1, A2A, A2B, and A3, have beneficial effects in protecting the heart (156). Activating A1 and A3 adenosine receptors before ischemia can initiate the ischemic preconditioning response, improve the ischemia tolerance of the heart, and avoid heart damage (100, 101). At the same time, the A2 adenosine receptors protect the heart during reperfusion, and the synergistic effect of A2A and A2B may play a non-negligible role in avoiding reperfusion injury (102, 103). S1P is released in the ischemic damaged heart and then binds to S1PRs to protect the heart from IRI by the Gα12/13-RhoA-protein kinase D (PKD) pathway (157, 158). However, some GPCRs can exacerbate the IRI of the heart, and the most prominent example is the calcium-sensing receptor (CaSR). CasR is widely expressed throughout the body, and its primary function is to maintain a constant concentration of extracellular ionized Ca2+ (108). Research has shown that the activation of CaSR by IR induces mitochondrial apoptosis, which promotes cardiomyocyte apoptosis, causing heart damage (109). In short, GPCRs have a critical role in the positive and negative regulation of IRI. Targeting GPCRs to prevent heart damage, alleviate heart disease, and avoid HF is a promising treatment.
Conclusion
Since CVDs are the leading cause of death globally, it is essential to find therapeutic targets for CVDs and develop drugs to treat CVDs. Because of their signal transduction function, GPCRs play a critical role in the occurrence and development of CVDs (Figure 1). In terms of vascular function and disease, GPCRs can receive a variety of extracellular stimuli, including their ligands or mechanical stress, regulate vascular tension and endothelial function, then positively or negatively adjust vascular diseases such as hypertension and atherosclerosis. There are significant gender differences in the occurrence of CVDs, with a higher incidence of CVDs in men compared to women (159). Multiple GPCRs play an integral role in this phenomenon, the most prominent is the G protein-coupled estrogen receptor (GPER), whose activation by estrogen is a key factor in female-specific cardiovascular protection (160). In addition, the orphan receptor GPR37L1 is also involved in the sex differences in CVDs. Mice lacking GPR37L1 exhibited female-specific increases in systolic, diastolic and mean arterial pressure. However, the gender issues on GPCR functions in CVDs are still not clear and further studies are still needed (161–163). In addition, GPCRs regulate the development and function of the heart, and further participate in heart diseases as a target for treatment. Finally, an understanding of the roles of these GPCRs in the cardiovascular system and CVDs will provide new insights into GPCRs and new ideas for fully exploiting the enormous treasure trove of GPCRs.
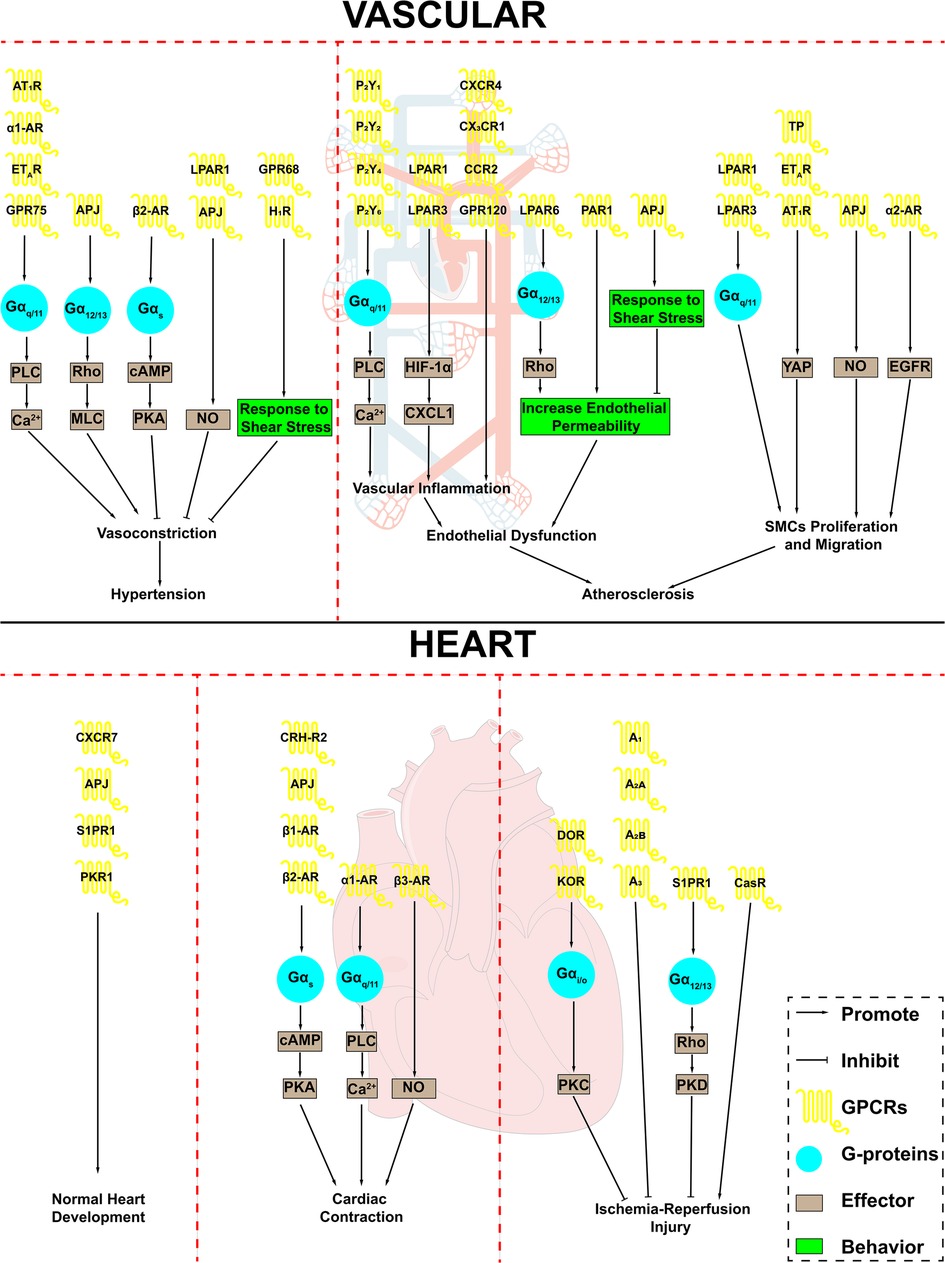
Figure 1. GPCRs in cardiovascular system and cardiovascular disease. GPCRs play an important role in the cardiovascular system and are involved in a variety of cardiovascular disease processes. AT1R, angiotensin II type 1 receptor; α1-AR, α1-adrenergic receptor; α2-AR, α2-adrenergic receptor; β1-AR, β1-adrenergic receptor; β2-AR, β2-adrenergic receptor; β3-AR, β3-adrenergic receptor; ETAR, endothelin receptor A; APJ, apelin receptor; LPAR1, lysophosphatidic acid receptor 1; LPAR3, lysophosphatidic acid receptor 3; LPAR6, lysophosphatidic acid receptor 6; H1R, H1 histamine receptor; P2Y1, purinergic receptor P2Y1; P2Y2, purinergic receptor P2Y2; P2Y4, purinergic receptor P2Y4; P2Y6, purinergic receptor P2Y6; CXCR4, CXC-motif chemokine receptor-4; CCR2, combining CC chemokine receptor 2; PAR1, Protease-activated receptor 1; TP, thromboxane A2 receptor; S1PR1, Sphingosine 1-phosphate receptor 1; PKR1, Prokineticin receptor-1; CRH-R2, corticotropin-releasing hormone receptor 2; DOR, δ opioid receptor; KOR, κ opioid receptor; A1, adenosine A1 receptor; A2A, adenosine A2a receptor; A2B, adenosine A2b receptor; A3, adenosine A3 receptor; CasR, calcium-sensing receptor; PLC, phospholipase C; MLC, myosin light chain; cAMP, cyclic-3′,5′-adenosine monophosphate; PKA, protein kinase A; NO, nitric oxide; HIF-1α, hypoxia inducible factor 1 subunit alpha; CXCL1, C-X-C motif chemokine ligand 1; YAP, Yes-associated protein; EGFR, epidermal growth factor receptor; SMCs, smooth muscle cells.
Author contributions
YL and BL: writing—original draft preparation; Y-DW and W-DC: writing—revision, review and editing, supervision and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Foundation of China (grant no. 81970551) and National Key R&D Program of China (2021YFC2103900) to Y-DW, the National Natural Science Foundation of China (grant no. 81970726), Henan Provincial Natural Science Foundation (grant no.182300410323), Program for Science & Technology Innovation Talents in Universities of Henan Province (HASTIT, grant no. 13HASTIT024), Plan for Scientific Innovation Talent of Henan Province, and National College Student Innovation and Entrepreneurship Training Program (grant no. 202010475032) to W-DC.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yu Y, Su X, Qin Q, Hou Y, Zhang X, Zhang H, et al. Yes-associated protein and transcriptional coactivator with pdz-binding motif as new targets in cardiovascular diseases. Pharmacol Res. (2020) 159:105009. doi: 10.1016/j.phrs.2020.105009
2. Ladich E, Yahagi K, Romero ME, Virmani R. Vascular diseases: aortitis, aortic aneurysms, and vascular calcification. Cardiovasc Pathol. (2016) 25(5):432–41. doi: 10.1016/j.carpath.2016.07.002
3. Joseph P, Leong D, McKee M, Anand SS, Schwalm JD, Teo K, et al. Reducing the global burden of cardiovascular disease, part 1: the epidemiology and risk factors. Circ Res. (2017) 121(6):677–94. doi: 10.1161/circresaha.117.308903
4. Flora GD, Nayak MK. A brief review of cardiovascular diseases, associated risk factors and current treatment regimes. Curr Pharm Des. (2019) 25(38):4063–84. doi: 10.2174/1381612825666190925163827
5. Lin HH. G-protein-coupled receptors and their (bio) chemical significance win 2012 nobel prize in chemistry. Biomed J. (2013) 36(3):118–24. doi: 10.4103/2319-4170.113233
6. Lefkowitz RJ. A brief history of G-protein coupled receptors (nobel lecture). Angew Chem Int Ed Engl. (2013) 52(25):6366–78. doi: 10.1002/anie.201301924
7. Moreira IS. Structural features of the G-protein/gpcr interactions. Biochim Biophys Acta. (2014) 1840(1):16–33. doi: 10.1016/j.bbagen.2013.08.027
8. Wang J, Gareri C, Rockman HA. G-protein-coupled receptors in heart disease. Circ Res. (2018) 123(6):716–35. doi: 10.1161/circresaha.118.311403
9. Woo AY, Xiao RP. Β-adrenergic receptor subtype signaling in heart: from bench to bedside. Acta Pharmacol Sin. (2012) 33(3):335–41. doi: 10.1038/aps.2011.201
10. Capote LA, Mendez Perez R, Lymperopoulos A. Gpcr signaling and cardiac function. Eur J Pharmacol. (2015) 763(Pt B):143–8. doi: 10.1016/j.ejphar.2015.05.019
11. Touyz RM. The role of angiotensin ii in regulating vascular structural and functional changes in hypertension. Curr Hypertens Rep. (2003) 5(2):155–64. doi: 10.1007/s11906-003-0073-2
12. Azizi M, Menard J, Bissery A, Guyenne TT, Bura-Riviere A, Vaidyanathan S, et al. Pharmacologic demonstration of the synergistic effects of a combination of the renin inhibitor aliskiren and the At1 receptor antagonist valsartan on the angiotensin ii-renin feedback interruption. J Am Soc Nephrol. (2004) 15(12):3126–33. doi: 10.1097/01.ASN.0000146686.35541.29
13. Koike H, Sada T, Mizuno M. In vitro and in vivo pharmacology of olmesartan medoxomil, an angiotensin ii type At1 receptor antagonist. J Hypertens Suppl. (2001) 19(1):S3–S14. doi: 10.1097/00004872-200106001-00002
14. Dickstein K, Timmermans P, Segal R. Losartan: a selective angiotensin ii type 1 (At1) receptor antagonist for the treatment of heart failure. Expert Opin Investig Drugs. (1998) 7(11):1897–914. doi: 10.1517/13543784.7.11.1897
15. van den Meiracker AH, Admiraal PJ, Janssen JA, Kroodsma JM, de Ronde WA, Boomsma F, et al. Hemodynamic and biochemical effects of the At1 receptor antagonist irbesartan in hypertension. Hypertension. (1995) 25(1):22–9. doi: 10.1161/01.hyp.25.1.22
16. Vauquelin G, Fierens F, Van Liefde I. Long-lasting angiotensin type 1 receptor binding and protection by candesartan: comparison with other biphenyl-tetrazole sartans. J Hypertens Suppl. (2006) 24(1):S23–S30. doi: 10.1097/01.hjh.0000220403.61493.18
17. Karlberg BE, Lins LE, Hermansson K. Efficacy and safety of telmisartan, a selective At1 receptor antagonist, compared with enalapril in elderly patients with primary hypertension. Tees study group. J Hypertens. (1999) 17(2):293–302. doi: 10.1097/00004872-199917020-00015
18. Jones JD, Jackson SH, Agboton C, Martin TS. Azilsartan medoxomil (edarbi): the eighth angiotensin ii receptor blocker. P T. (2011) 36(10):634–40.22346296
19. Nagano K, Kwon C, Ishida J, Hashimoto T, Kim JD, Kishikawa N, et al. Cooperative action of apj and Α1a-adrenergic receptor in vascular smooth muscle cells induces vasoconstriction. J Biochem. (2019) 166(5):383–92. doi: 10.1093/jb/mvz071
20. Huhtinen A, Hongisto V, Laiho A, Löyttyniemi E, Pijnenburg D, Scheinin M. Gene expression profiles and signaling mechanisms in Α(2b)-adrenoceptor-evoked proliferation of vascular smooth muscle cells. BMC Syst Biol. (2017) 11(1):65. doi: 10.1186/s12918-017-0439-8
21. Lymperopoulos A, Bathgate A. Arrestins in the cardiovascular system. Prog Mol Biol Transl Sci. (2013) 118:297–334. doi: 10.1016/b978-0-12-394440-5.00012-7
22. Moens AL, Yang R, Watts VL, Barouch LA. Beta 3-adrenoreceptor regulation of nitric oxide in the cardiovascular system. J Mol Cell Cardiol. (2010) 48(6):1088–95. doi: 10.1016/j.yjmcc.2010.02.011
23. Shannon R, Chaudhry M. Effect of alpha1-adrenergic receptors in cardiac pathophysiology. Am Heart J. (2006) 152(5):842–50. doi: 10.1016/j.ahj.2006.05.017
24. Lymperopoulos A, Rengo G, Koch WJ. Adrenal adrenoceptors in heart failure: fine-tuning cardiac stimulation. Trends Mol Med. (2007) 13(12):503–11. doi: 10.1016/j.molmed.2007.10.005
25. Cohen ML, Berkowitz BA. Decreased vascular relaxation in hypertension. J Pharmacol Exp Ther. (1976) 196(2):396–406.176346
26. Bernstein JS, Ebert TJ, Stowe DF, Schmeling WT, Nelson MA, Woods MP. Partial attenuation of hemodynamic responses to rapid sequence induction and intubation with labetalol. J Clin Anesth. (1989) 1(6):444–51. doi: 10.1016/0952-8180(89)90009-3
27. Korneyev AY, Cincotta AH. Identification of hepatic, non-monoamine, dihydroergocryptine binding sites with significant gender differences. Life Sci. (1996) 58(12):241–8. doi: 10.1016/0024-3205(96)00053-7
28. Mohta M, Janani SS, Sethi AK, Agarwal D, Tyagi A. Comparison of phenylephrine hydrochloride and mephentermine sulphate for prevention of post spinal hypotension. Anaesthesia. (2010) 65(12):1200–5. doi: 10.1111/j.1365-2044.2010.06559.x
29. Zeng A, Yuan B, Wang C, Yang G, He L. Frontal analysis of cell-membrane chromatography for determination of drug-alpha(1d) adrenergic receptor affinity. J Chromatogr B Analyt Technol Biomed Life Sci. (2009) 877(20–21):1833–7. doi: 10.1016/j.jchromb.2009.05.021
30. Altenbach RJ, Khilevich A, Meyer MD, Buckner SA, Milicic I, Daza AV, et al. N-[3-(1h-Imidazol-4-ylmethyl)phenyl]ethanesulfonamide (Abt-866, 1),(1) a novel alpha(1)-adrenoceptor ligand with an enhanced in vitro and in vivo profile relative to phenylpropanolamine and midodrine. J Med Chem. (2002) 45(20):4395–7. doi: 10.1021/jm025550h
31. Koshimizu TA, Tsujimoto G, Hirasawa A, Kitagawa Y, Tanoue A. Carvedilol selectively inhibits oscillatory intracellular calcium changes evoked by human alpha1d- and alpha1b-adrenergic receptors. Cardiovasc Res. (2004) 63(4):662–72. doi: 10.1016/j.cardiores.2004.05.014
32. Sengupta JN, Hamada A, Miller DD, Patil PN. Interaction of enantiomers of hydroxy tolazoline with adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol. (1987) 335(4):391–6. doi: 10.1007/BF00165553
33. Song W, Zhang Y, Xia L, Liu G. Effects of some mexiletine derivatives on alpha 1-adrenoceptors. Yao Xue Xue Bao. (1998) 33(2):102–5.11938943
34. Tatsuta M, Iishi H, Baba M, Yano H, Sakai N, Uehara H, et al. Alpha1-adrenoceptor stimulation enhances experimental gastric carcinogenesis induced by N-methyl-N'-nitro-N-nitrosoguanidine in wistar rats. Int J Cancer. (1998) 77(3):467–9. doi: 10.1002/(sici)1097-0215(19980729)77:3%3C467::aid-ijc25%3E3.0.co;2-3
35. Frang H, Cockcroft V, Karskela T, Scheinin M, Marjamaki A. Phenoxybenzamine binding reveals the helical orientation of the third transmembrane domain of adrenergic receptors. J Biol Chem. (2001) 276(33):31279–84. doi: 10.1074/jbc.M104167200
36. Rudoy CA, Van Bockstaele EJ. Betaxolol, a selective beta(1)-adrenergic receptor antagonist, diminishes anxiety-like behavior during early withdrawal from chronic cocaine administration in rats. Prog Neuropsychopharmacol Biol Psychiatry. (2007) 31(5):1119–29. doi: 10.1016/j.pnpbp.2007.04.005
37. Johnson JA, Zineh I, Puckett BJ, McGorray SP, Yarandi HN, Pauly DF. Beta 1-adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin Pharmacol Ther. (2003) 74(1):44–52. doi: 10.1016/S0009-9236(03)00068-7
38. Horinouchi T, Morishima S, Tanaka T, Suzuki F, Tanaka Y, Koike K, et al. Different changes of plasma membrane beta-adrenoceptors in rat heart after chronic administration of propranolol, atenolol and bevantolol. Life Sci. (2007) 81(5):399–404. doi: 10.1016/j.lfs.2007.06.003
39. Wang T, Kaumann AJ, Brown MJ. (–)-Timolol is a more potent antagonist of the positive inotropic effects of (–)-adrenaline than of those of (–)-noradrenaline in human atrium. Br J Clin Pharmacol. (1996) 42(2):217–23. doi: 10.1046/j.1365-2125.1996.39412.x
40. Lipworth BJ, Irvine NA, McDevitt DG. A dose-ranging study to evaluate the beta 1-adrenoceptor selectivity of bisoprolol. Eur J Clin Pharmacol. (1991) 40(2):135–9. doi: 10.1007/BF00280067
41. Nichols AJ, Gellai M, Ruffolo RR Jr. Studies on the mechanism of arterial vasodilation produced by the novel antihypertensive agent, carvedilol. Fundam Clin Pharmacol. (1991) 5(1):25–38. doi: 10.1111/j.1472-8206.1991.tb00698.x
42. Doggrell SA. The effects of (+/−)-, (+)-, and (−)-atenolol, sotalol, and amosulalol on the rat left atria and portal vein. Chirality. (1993) 5(1):8–14. doi: 10.1002/chir.530050103
43. Imming P, Sinning C, Drugs MA. Their targets and the nature and number of drug targets. Nat Rev Drug Discov. (2006) 5(10):821–34. doi: 10.1038/nrd2132
44. Berg T, Piercey BW, Jensen J. Role of beta1-3-adrenoceptors in blood pressure control at rest and during tyramine-induced norepinephrine release in spontaneously hypertensive rats. Hypertension. (2010) 55(5):1224–30. doi: 10.1161/HYPERTENSIONAHA.109.149286
45. Rikitake Y. The apelin/apj system in the regulation of vascular tone: friend or foe? J Biochem. (2021) 169(4):383–6. doi: 10.1093/jb/mvaa129
46. Kidoya H, Takakura N. Biology of the apelin-apj axis in vascular formation. J Biochem. (2012) 152(2):125–31. doi: 10.1093/jb/mvs071
47. Maguire JJ, Kleinz MJ, Pitkin SL, Davenport AP. [Pyr1]Apelin-13 identified as the predominant apelin isoform in the human heart: vasoactive mechanisms and inotropic action in disease. Hypertension. (2009) 54(3):598–604. doi: 10.1161/hypertensionaha.109.134619
48. Strohbach A, Pennewitz M, Glaubitz M, Palankar R, Groß S, Lorenz F, et al. The apelin receptor influences biomechanical and morphological properties of endothelial cells. J Cell Physiol. (2018) 233(8):6250–61. doi: 10.1002/jcp.26496
49. Hashimoto T, Kihara M, Imai N, Yoshida S, Shimoyamada H, Yasuzaki H, et al. Requirement of apelin-apelin receptor system for oxidative stress-linked atherosclerosis. Am J Pathol. (2007) 171(5):1705–12. doi: 10.2353/ajpath.2007.070471
50. Charo DN, Ho M, Fajardo G, Kawana M, Kundu RK, Sheikh AY, et al. Endogenous regulation of cardiovascular function by apelin-apj. Am J Physiol Heart Circ Physiol. (2009) 297(5):H1904–13. doi: 10.1152/ajpheart.00686.2009
51. Paskaradevan S, Scott IC. The aplnr gpcr regulates myocardial progenitor development via a novel cell-non-autonomous, gα(I/O) protein-independent pathway. Biol Open. (2012) 1(3):275–85. doi: 10.1242/bio.2012380
52. Zhou Y, Little PJ, Ta HT, Xu S, Kamato D. Lysophosphatidic acid and its receptors: pharmacology and therapeutic potential in atherosclerosis and vascular disease. Pharmacol Ther. (2019) 204:107404. doi: 10.1016/j.pharmthera.2019.107404
53. Ruisanchez É, Dancs P, Kerék M, Németh T, Faragó B, Balogh A, et al. Lysophosphatidic acid induces vasodilation mediated by Lpa1 receptors, phospholipase C, and endothelial nitric oxide synthase. FASEB J. (2014) 28(2):880–90. doi: 10.1096/fj.13-234997
54. Dancs PT, Ruisanchez É, Balogh A, Panta CR, Miklós Z, Nüsing RM, et al. Lpa(1) receptor-mediated thromboxane a(2) release is responsible for lysophosphatidic acid-induced vascular smooth muscle contraction. FASEB J. (2017) 31(4):1547–55. doi: 10.1096/fj.201600735R
55. Yukiura H, Kano K, Kise R, Inoue A, Aoki J. Lpp3 localizes Lpa6 signalling to non-contact sites in endothelial cells. J Cell Sci. (2015) 128(21):3871–7. doi: 10.1242/jcs.172098
56. Zhou Y, Little PJ, Cao Y, Ta HT, Kamato D. Lysophosphatidic acid receptor 5 transactivation of Tgfbr1 stimulates the mrna expression of proteoglycan synthesizing genes Xylt1 and Chst3. Biochim Biophys Acta Mol Cell Res. (2020) 1867(12):118848. doi: 10.1016/j.bbamcr.2020.118848
57. Zhou Z, Niu J, Zhang Z. The role of lysophosphatidic acid receptors in phenotypic modulation of vascular smooth muscle cells. Mol Biol Rep. (2010) 37(6):2675–86. doi: 10.1007/s11033-009-9798-6
58. Panchatcharam M, Miriyala S, Yang F, Rojas M, End C, Vallant C, et al. Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ Res. (2008) 103(6):662–70. doi: 10.1161/circresaha.108.180778
59. Wolf P, Beck-Sickinger AG. The ring size of monocyclic et-1 controls selectivity and signaling efficiency at both endothelin receptor subtypes. J Pept Sci. (2021) 27(7):e3325. doi: 10.1002/psc.3325
60. Wang Z, Liu P, Zhou X, Wang T, Feng X, Sun YP, et al. Endothelin promotes colorectal tumorigenesis by activating yap/taz. Cancer Res. (2017) 77(9):2413–23. doi: 10.1158/0008-5472.Can-16-3229
61. Bolli MH, Boss C, Binkert C, Buchmann S, Bur D, Hess P, et al. The discovery of N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-n'-propylsulfamide (macitentan), an orally active, potent dual endothelin receptor antagonist. J Med Chem. (2012) 55(17):7849–61. doi: 10.1021/jm3009103
62. Kiowski W, Sutsch G, Oechslin E, Bertel O. Hemodynamic effects of bosentan in patients with chronic heart failure. Heart Fail Rev. (2001) 6(4):325–34. doi: 10.1023/a:1011460426786
63. Casserly B, Klinger JR. Ambrisentan for the treatment of pulmonary arterial hypertension. Drug Des Devel Ther. (2009) 2:265–80. doi: 10.2147/dddt.s3057
64. Eun SY, Park SW, Lee JH, Chang KC, Kim HJ. P2y(2)R activation by nucleotides released from oxldl-treated endothelial cells (ecs) mediates the interaction between ecs and immune cells through rage expression and reactive oxygen species production. Free Radic Biol Med. (2014) 69:157–66. doi: 10.1016/j.freeradbiomed.2014.01.022
65. Stachon P, Peikert A, Michel NA, Hergeth S, Marchini T, Wolf D, et al. P2y6 deficiency limits vascular inflammation and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2014) 34(10):2237–45. doi: 10.1161/atvbaha.114.303585
66. Cantisani C, Ricci S, Grieco T, Paolino G, Faina V, Silvestri E, et al. Topical promethazine side effects: our experience and review of the literature. Biomed Res Int. (2013) 2013:151509. doi: 10.1155/2013/151509
67. Peng S, Grace MS, Gondin AB, Retamal JS, Dill L, Darby W, et al. The transient receptor potential vanilloid 4 (Trpv4) ion channel mediates protease activated receptor 1 (Par1)-induced vascular hyperpermeability. Lab Invest. (2020) 100(8):1057–67. doi: 10.1038/s41374-020-0430-7
68. Bonaca MP, Morrow DA. Sch 530348: a novel oral thrombin receptor antagonist. Future Cardiol. (2009) 5(5):435–42. doi: 10.2217/fca.09.27
69. Garcia V, Gilani A, Shkolnik B, Pandey V, Zhang FF, Dakarapu R, et al. 20-Hete signals through G-protein-coupled receptor Gpr75 (G(Q)) to affect vascular function and trigger hypertension. Circ Res. (2017) 120(11):1776–88. doi: 10.1161/circresaha.116.310525
70. Natarajan N, Hori D, Flavahan S, Steppan J, Flavahan NA, Berkowitz DE, et al. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol Genomics. (2016) 48(11):826–34. doi: 10.1152/physiolgenomics.00089.2016
71. Jiang T, Jiang D, You D, Zhang L, Liu L, Zhao Q. Agonism of Gpr120 prevents ox-ldl-induced attachment of monocytes to endothelial cells. Chem Biol Interact. (2020) 316:108916. doi: 10.1016/j.cbi.2019.108916
72. Visioli F, Giordano E, Nicod NM, Davalos A. Molecular targets of Omega 3 and conjugated linoleic fatty acids—“micromanaging” cellular response. Front Physiol. (2012) 3:42. doi: 10.3389/fphys.2012.00042
73. Iliff AJ, Xu XZS. A mechanosensitive gpcr that detects the bloody force. Cell. (2018) 173(3):542–4. doi: 10.1016/j.cell.2018.04.001
74. Xu J, Mathur J, Vessières E, Hammack S, Nonomura K, Favre J, et al. Gpr68 senses flow and is essential for vascular physiology. Cell. (2018) 173(3):762–75.e16. doi: 10.1016/j.cell.2018.03.076
75. Erdogmus S, Storch U, Danner L, Becker J, Winter M, Ziegler N, et al. Helix 8 is the essential structural motif of mechanosensitive gpcrs. Nat Commun. (2019) 10(1):5784. doi: 10.1038/s41467-019-13722-0
76. Roll C, Hanssler L. Interaction of tolazoline and cimetidine in persistent fetal circulation of the newborn infant. Monatsschr Kinderheilkd. (1993) 141(4):297–9.8487790
77. Vitali Serdoz L, Rittger H, Furlanello F, Bastian D. Quinidine-a legacy within the modern era of antiarrhythmic therapy. Pharmacol Res. (2019) 144:257–63. doi: 10.1016/j.phrs.2019.04.028
78. Kontos C, El Bounkari O, Krammer C, Sinitski D, Hille K, Zan C, et al. Designed Cxcr4 mimic acts as a soluble chemokine receptor that blocks atherogenic inflammation by agonist-specific targeting. Nat Commun. (2020) 11(1):5981. doi: 10.1038/s41467-020-19764-z
79. Guyon A, Kussrow A, Olmsted IR, Sandoz G, Bornhop DJ, Nahon JL. Baclofen and other gabab receptor agents are allosteric modulators of the Cxcl12 chemokine receptor Cxcr4. J Neurosci. (2013) 33(28):11643–54. doi: 10.1523/JNEUROSCI.6070-11.2013
80. Taylor BC, Lee CT, Amaro RE. Structural basis for ligand modulation of the Ccr2 conformational landscape. Proc Natl Acad Sci U S A. (2019) 116(17):8131–6. doi: 10.1073/pnas.1814131116
81. Low S, Wu H, Jerath K, Tibolla A, Fogal B, Conrad R, et al. Vhh antibody targeting the chemokine receptor Cx3cr1 inhibits progression of atherosclerosis. MAbs. (2020) 12(1):1709322. doi: 10.1080/19420862.2019.1709322
82. Feng X, Liu P, Zhou X, Li MT, Li FL, Wang Z, et al. Thromboxane A2 activates yap/taz protein to induce vascular smooth muscle cell proliferation and migration. J Biol Chem. (2016) 291(36):18947–58. doi: 10.1074/jbc.M116.739722
83. Zhang X, Fan S, Zhang L, Shi Y. Glucagon-like peptide-1 receptor undergoes importin-Α-dependent nuclear localization in rat aortic smooth muscle cells. FEBS Lett. (2020) 594(10):1506–16. doi: 10.1002/1873-3468.13751
84. Bock T, Pakkenberg B, Buschard K. The endocrine pancreas in non-diabetic rats after short-term and long-term treatment with the long-acting glp-1 derivative Nn2211. APMIS. (2003) 111(12):1117–24. doi: 10.1111/j.1600-0463.2003.apm1111207.x
85. Lorenz M, Lawson F, Owens D, Raccah D, Roy-Duval C, Lehmann A, et al. Differential effects of glucagon-like peptide-1 receptor agonists on heart rate. Cardiovasc Diabetol. (2017) 16(1):6. doi: 10.1186/s12933-016-0490-6
86. Htike ZZ, Zaccardi F, Papamargaritis D, Webb DR, Khunti K, Davies MJ. Efficacy and safety of glucagon-like peptide-1 receptor agonists in type 2 diabetes: a systematic review and mixed-treatment comparison analysis. Diabetes Obes Metab. (2017) 19(4):524–36. doi: 10.1111/dom.12849
87. Sanchez T, Hla T. Structural and functional characteristics of S1p receptors. J Cell Biochem. (2004) 92(5):913–22. doi: 10.1002/jcb.20127
88. Gaengel K, Niaudet C, Hagikura K, Laviña B, Muhl L, Hofmann JJ, et al. The sphingosine-1-phosphate receptor S1pr1 restricts sprouting angiogenesis by regulating the interplay between ve-cadherin and Vegfr2. Dev Cell. (2012) 23(3):587–99. doi: 10.1016/j.devcel.2012.08.005
89. Clay H, Wilsbacher LD, Wilson SJ, Duong DN, McDonald M, Lam I, et al. Sphingosine 1-phosphate receptor-1 in cardiomyocytes is required for normal cardiac development. Dev Biol. (2016) 418(1):157–65. doi: 10.1016/j.ydbio.2016.06.024
90. Nebigil CG, Désaubry L. The role of gpcr signaling in cardiac epithelial to mesenchymal transformation (emt). Trends Cardiovasc Med. (2019) 29(4):200–4. doi: 10.1016/j.tcm.2018.08.007
91. Urayama K, Guilini C, Turkeri G, Takir S, Kurose H, Messaddeq N, et al. Prokineticin receptor-1 induces neovascularization and epicardial-derived progenitor cell differentiation. Arterioscler Thromb Vasc Biol. (2008) 28(5):841–9. doi: 10.1161/atvbaha.108.162404
92. Sierro F, Biben C, Martínez-Muñoz L, Mellado M, Ransohoff RM, Li M, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second Cxcl12/sdf-1 receptor, Cxcr7. Proc Natl Acad Sci U S A. (2007) 104(37):14759–64. doi: 10.1073/pnas.0702229104
93. Klein KR, Karpinich NO, Espenschied ST, Willcockson HH, Dunworth WP, Hoopes SL, et al. Decoy receptor Cxcr7 modulates adrenomedullin-mediated cardiac and lymphatic vascular development. Dev Cell. (2014) 30(5):528–40. doi: 10.1016/j.devcel.2014.07.012
94. Walczewska J, Dzieza-Grudnik A, Siga O, Grodzicki T. The role of urocortins in the cardiovascular system. J Physiol Pharmacol. (2014) 65(6):753–66.25554979
95. Wang JW, Xue ZY, Wu AS. Mechanistic insights into Δ-opioid-induced cardioprotection: involvement of caveolin translocation to the mitochondria. Life Sci. (2020) 247:116942. doi: 10.1016/j.lfs.2019.116942
96. Headrick JP, See Hoe LE, Du Toit EF, Peart JN. Opioid receptors and cardioprotection—‘opioidergic conditioning’ of the heart. Br J Pharmacol. (2015) 172(8):2026–50. doi: 10.1111/bph.13042
97. Cohen MV, Philipp S, Krieg T, Cui L, Kuno A, Solodushko V, et al. Preconditioning-mimetics bradykinin and dadle activate Pi3-kinase through divergent pathways. J Mol Cell Cardiol. (2007) 42(4):842–51. doi: 10.1016/j.yjmcc.2007.01.004
98. Lishmanov AY, Maslov LN, Lasukova TV, Crawford D, Wong TM. Activation of kappa-opioid receptor as a method for prevention of ischemic and reperfusion arrhythmias: role of protein kinase C and K(atp) channels. Bull Exp Biol Med. (2007) 143(2):187–90. doi: 10.1007/s10517-007-0046-6
99. Shen H, Ben Q, Zhang Y, Jiang G, Yao C, Shi D, et al. Role of Δ2 opioid receptor in cardioprotection against hypoxia-reoxygenation injury. J Cardiovasc Pharmacol. (2012) 60(3):253–61. doi: 10.1097/FJC.0b013e31825e215f
100. Thornton JD, Liu GS, Olsson RA, Downey JM. Intravenous pretreatment with A1-selective adenosine analogues protects the heart against infarction. Circulation. (1992) 85(2):659–65. doi: 10.1161/01.cir.85.2.659
101. Maddock HL, Gardner NM, Khandoudi N, Bril A, Broadley KJ. Protection from myocardial stunning by ischaemia and hypoxia with the adenosine A3 receptor agonist, ib-meca. Eur J Pharmacol. (2003) 477(3):235–45. doi: 10.1016/j.ejphar.2003.08.024
102. Busse H, Bitzinger D, Höcherl K, Seyfried T, Gruber M, Graf BM, et al. Adenosine A2a and A2b receptor substantially attenuate ischemia/reperfusion injury in septic rat hearts. Cardiovasc Drugs Ther. (2016) 30(6):551–8. doi: 10.1007/s10557-016-6693-y
103. Guerrero A. A2a adenosine receptor agonists and their potential therapeutic applications. An update. Curr Med Chem. (2018) 25(30):3597–612. doi: 10.2174/0929867325666180313110254
104. Funakoshi H, Zacharia LC, Tang Z, Zhang J, Lee LL, Good JC, et al. A1 adenosine receptor upregulation accompanies decreasing myocardial adenosine levels in mice with left ventricular dysfunction. Circulation. (2007) 115(17):2307–15. doi: 10.1161/CIRCULATIONAHA.107.694596
105. Daly JW, Jacobson KA, Ukena D. Adenosine receptors: development of selective agonists and antagonists. Prog Clin Biol Res. (1987) 230:41–63.3588607
106. Udelson JE, Heller GV, Wackers FJ, Chai A, Hinchman D, Coleman PS, et al. Randomized, controlled dose-ranging study of the selective adenosine A2a receptor agonist binodenoson for pharmacological stress as an adjunct to myocardial perfusion imaging. Circulation. (2004) 109(4):457–64. doi: 10.1161/01.CIR.0000114523.03312.7D
107. Rodriguez-Rodriguez P, Ragusky K, Phuthong S, Ruvira S, Ramiro-Cortijo D, Canas S, et al. Vasoactive properties of a cocoa shell extract: mechanism of action and effect on endothelial dysfunction in aged rats. Antioxidants (Basel). (2022) 11(2):429–46. doi: 10.3390/antiox11020429
108. Paquot F, Huart J, Defraigne JO, Krzesinski JM, Jouret F. Implications of the calcium-sensing receptor in ischemia/reperfusion. Acta Cardiol. (2017) 72(2):125–31. doi: 10.1080/00015385.2017.1291136
109. Zheng H, Liu J, Liu C, Lu F, Zhao Y, Jin Z, et al. Calcium-sensing receptor activating phosphorylation of pkcδ translocation on mitochondria to induce cardiomyocyte apoptosis during ischemia/reperfusion. Mol Cell Biochem. (2011) 358(1–2):335–43. doi: 10.1007/s11010-011-0984-1
110. Wang G, Jacquet L, Karamariti E, Xu Q. Origin and differentiation of vascular smooth muscle cells. J Physiol. (2015) 593(14):3013–30. doi: 10.1113/jp270033
111. Flavahan NA. In development-a new paradigm for understanding vascular disease. J Cardiovasc Pharmacol. (2017) 69(5):248–63. doi: 10.1097/FJC.0000000000000480
112. Kaur H, Carvalho J, Looso M, Singh P, Chennupati R, Preussner J, et al. Single-cell profiling reveals heterogeneity and functional patterning of gpcr expression in the vascular system. Nat Commun. (2017) 8:15700. doi: 10.1038/ncomms15700
113. Kuo IY, Ehrlich BE. Signaling in muscle contraction. Cold Spring Harb Perspect Biol. (2015) 7(2):a006023. doi: 10.1101/cshperspect.a006023
114. Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol. (2011) 3(9):a004549. doi: 10.1101/cshperspect.a004549
115. Nash CA, Nelson CP, Mistry R, Moeller-Olsen C, Christofidou E, Challiss RAJ, et al. Differential regulation of Β2-adrenoceptor and adenosine A2b receptor signalling by grk and arrestin proteins in arterial smooth muscle. Cell Signal. (2018) 51:86–98. doi: 10.1016/j.cellsig.2018.07.013
116. Brinks HL, Eckhart AD. Regulation of gpcr signaling in hypertension. Biochim Biophys Acta. (2010) 1802(12):1268–75. doi: 10.1016/j.bbadis.2010.01.005
117. Motiejunaite J, Amar L, Vidal-Petiot E. Adrenergic receptors and cardiovascular effects of catecholamines. Ann Endocrinol (Paris). (2021) 82(3–4):193–7. doi: 10.1016/j.ando.2020.03.012
118. Modgil A, Guo L, O'Rourke ST, Sun C. Apelin-13 inhibits large-conductance Ca2+-activated K+ channels in cerebral artery smooth muscle cells via a Pi3-kinase dependent mechanism. PLoS One. (2013) 8(12):e83051. doi: 10.1371/journal.pone.0083051
119. Hashimoto T, Kihara M, Ishida J, Imai N, Yoshida S, Toya Y, et al. Apelin stimulates myosin light chain phosphorylation in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2006) 26(6):1267–72. doi: 10.1161/01.Atv.0000218841.39828.91
120. Ishida J, Hashimoto T, Hashimoto Y, Nishiwaki S, Iguchi T, Harada S, et al. Regulatory roles for apj, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo*. J Biol Chem. (2004) 279(25):26274–9. doi: 10.1074/jbc.M404149200
121. Weis SM. Vascular permeability in cardiovascular disease and cancer. Curr Opin Hematol. (2008) 15(3):243–9. doi: 10.1097/MOH.0b013e3282f97d86
122. Birch CA, Molinar-Inglis O, Trejo J. Subcellular hot spots of gpcr signaling promote vascular inflammation. Curr Opin Endocr Metab Res. (2021) 16:37–42. doi: 10.1016/j.coemr.2020.07.011
123. Grimsey NJ, Lin Y, Narala R, Rada CC, Mejia-Pena H, Trejo J. G protein-coupled receptors activate P38 mapk via a non-canonical Tab1-Tab2- and Tab1-Tab3-dependent pathway in endothelial cells. J Biol Chem. (2019) 294(15):5867–78. doi: 10.1074/jbc.RA119.007495
124. Wettschureck N, Strilic B, Offermanns S. Passing the vascular barrier: endothelial signaling processes controlling extravasation. Physiol Rev. (2019) 99(3):1467–525. doi: 10.1152/physrev.00037.2018
125. Wolfenson H, Yang B, Sheetz MP. Steps in mechanotransduction pathways that control cell morphology. Annu Rev Physiol. (2019) 81:585–605. doi: 10.1146/annurev-physiol-021317-121245
126. Yamashiro Y, Yanagisawa H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin Sci (Lond). (2020) 134(17):2399–418. doi: 10.1042/cs20190488
128. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16(12):727–44. doi: 10.1038/s41569-019-0227-9
129. Theodorou K, Boon RA. Endothelial cell metabolism in atherosclerosis. Front Cell Dev Biol. (2018) 6:82. doi: 10.3389/fcell.2018.00082
130. Strassheim D, Verin A, Batori R, Nijmeh H, Burns N, Kovacs-Kasa A, et al. P2y purinergic receptors, endothelial dysfunction, and cardiovascular diseases. Int J Mol Sci. (2020) 21(18):6855–876. doi: 10.3390/ijms21186855
131. Siess W, Zangl KJ, Essler M, Bauer M, Brandl R, Corrinth C, et al. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc Natl Acad Sci U S A. (1999) 96(12):6931–6. doi: 10.1073/pnas.96.12.6931
132. Aldi S, Matic LP, Hamm G, van Keulen D, Tempel D, Holmstrøm K, et al. Integrated human evaluation of the lysophosphatidic acid pathway as a novel therapeutic target in atherosclerosis. Mol Ther Methods Clin Dev. (2018) 10:17–28. doi: 10.1016/j.omtm.2018.05.003
133. Akhtar S, Hartmann P, Karshovska E, Rinderknecht FA, Subramanian P, Gremse F, et al. Endothelial hypoxia-inducible factor-1α promotes atherosclerosis and monocyte recruitment by upregulating microrna-19a. Hypertension. (2015) 66(6):1220–6. doi: 10.1161/hypertensionaha.115.05886
134. Kim J, Keys JR, Eckhart AD. Vascular smooth muscle migration and proliferation in response to lysophosphatidic acid (lpa) is mediated by lpa receptors coupling to gq. Cell Signal. (2006) 18(10):1695–701. doi: 10.1016/j.cellsig.2006.01.009
135. Liu Y, Chen F, Ji L, Zhang L, Xu YJ, Dhalla NS. Role of lysophosphatidic acid in vascular smooth muscle cell proliferation. Can J Physiol Pharmacol. (2020) 98(2):103–10. doi: 10.1139/cjpp-2019-0264
136. Wang X, Hu G, Gao X, Wang Y, Zhang W, Harmon EY, et al. The induction of yes-associated protein expression after arterial injury is crucial for smooth muscle phenotypic modulation and neointima formation. Arterioscler Thromb Vasc Biol. (2012) 32(11):2662–9. doi: 10.1161/atvbaha.112.254730
137. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the hippo-yap pathway by G-protein-coupled receptor signaling. Cell. (2012) 150(4):780–91. doi: 10.1016/j.cell.2012.06.037
138. Lin M, Yuan W, Su Z, Lin C, Huang T, Chen Y, et al. Yes-associated protein mediates angiotensin ii-induced vascular smooth muscle cell phenotypic modulation and hypertensive vascular remodelling. Cell Prolif. (2018) 51(6):e12517. doi: 10.1111/cpr.12517
139. Courchaine K, Rykiel G, Rugonyi S. Influence of blood flow on cardiac development. Prog Biophys Mol Biol. (2018) 137:95–110. doi: 10.1016/j.pbiomolbio.2018.05.005
140. Buijtendijk MFJ, Barnett P, van den Hoff MJB. Development of the human heart. Am J Med Genet C Semin Med Genet. (2020) 184(1):7–22. doi: 10.1002/ajmg.c.31778
141. Perjés Á, Kilpiö T, Ulvila J, Magga J, Alakoski T, Szabó Z, et al. Characterization of apela, a novel endogenous ligand of apelin receptor, in the adult heart. Basic Res Cardiol. (2016) 111(1):2. doi: 10.1007/s00395-015-0521-6
142. Chng SC, Ho L, Tian J, Reversade B. Elabela: a hormone essential for heart development signals via the apelin receptor. Dev Cell. (2013) 27(6):672–80. doi: 10.1016/j.devcel.2013.11.002
143. Papangeli I, Chun HJ. A tale of two elabela null mice. Trends Endocrinol Metab. (2017) 28(11):759–60. doi: 10.1016/j.tem.2017.09.004
144. Nebigil CG, Choi DS, Dierich A, Hickel P, Le Meur M, Messaddeq N, et al. Serotonin 2b receptor is required for heart development. Proc Natl Acad Sci U S A. (2000) 97(17):9508–13. doi: 10.1073/pnas.97.17.9508
145. Quinn KE, Mackie DI, Caron KM. Emerging roles of atypical chemokine receptor 3 (Ackr3) in normal development and physiology. Cytokine. (2018) 109:17–23. doi: 10.1016/j.cyto.2018.02.024
146. Spinale FG. Assessment of cardiac function–basic principles and approaches. Compr Physiol. (2015) 5(4):1911–46. doi: 10.1002/cphy.c140054
147. MacLeod KT. Recent advances in understanding cardiac contractility in health and disease. F1000Res. (2016) 5:1770–83. doi: 10.12688/f1000research.8661.1
148. Gordan R, Gwathmey JK, Xie LH. Autonomic and endocrine control of cardiovascular function. World J Cardiol. (2015) 7(4):204–14. doi: 10.4330/wjc.v7.i4.204
149. Vikhorev PG, Vikhoreva NN. Cardiomyopathies and related changes in contractility of human heart muscle. Int J Mol Sci. (2018) 19(8):2234–53. doi: 10.3390/ijms19082234
150. Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. (2017) 121(2):181–95. doi: 10.1161/circresaha.117.310230
151. Lefkowitz RJ, Rockman HA, Koch WJ. Catecholamines, cardiac beta-adrenergic receptors, and heart failure. Circulation. (2000) 101(14):1634–7. doi: 10.1161/01.cir.101.14.1634
152. Leander M, Bass C, Marchetti K, Maynard BF, Wulff JP, Ons S, et al. Cardiac contractility structure-activity relationship and ligand-receptor interactions; the discovery of unique and novel molecular switches in myosuppressin signaling. PLoS One. (2015) 10(3):e0120492. doi: 10.1371/journal.pone.0120492
153. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. (2011) 17(11):1391–401. doi: 10.1038/nm.2507
154. Lefort C, Benoist L, Chadet S, Piollet M, Heraud A, Babuty D, et al. Stimulation of P2y11 receptor modulates cardiac fibroblasts secretome toward immunomodulatory and protective roles after hypoxia/reoxygenation injury. J Mol Cell Cardiol. (2018) 121:212–22. doi: 10.1016/j.yjmcc.2018.07.245
155. Schultz JE, Hsu AK, Gross GJ. Morphine mimics the cardioprotective effect of ischemic preconditioning via a glibenclamide-sensitive mechanism in the rat heart. Circ Res. (1996) 78(6):1100–4. doi: 10.1161/01.res.78.6.1100
156. Ashton KJ, Peart JN, Morrison RR, Matherne GP, Blackburn MR, Headrick JP. Genetic modulation of adenosine receptor function and adenosine handling in murine hearts: insights and issues. J Mol Cell Cardiol. (2007) 42(4):693–705. doi: 10.1016/j.yjmcc.2006.12.012
157. Xiang SY, Ouyang K, Yung BS, Miyamoto S, Smrcka AV, Chen J, et al. Plcε, Pkd1, and Ssh1l transduce rhoa signaling to protect mitochondria from oxidative stress in the heart. Sci Signal. (2013) 6(306):ra108. doi: 10.1126/scisignal.2004405
158. Yung BS, Brand CS, Xiang SY, Gray CB, Means CK, Rosen H, et al. Selective coupling of the S1p(3) receptor subtype to S1p-mediated rhoa activation and cardioprotection. J Mol Cell Cardiol. (2017) 103:1–10. doi: 10.1016/j.yjmcc.2016.12.008
159. Pilote L, Dasgupta K, Guru V, Humphries KH, McGrath J, Norris C, et al. A comprehensive view of sex-specific issues related to cardiovascular disease. CMAJ. (2007) 176(6):S1–S44. doi: 10.1503/cmaj.051455
160. Mouat MA, Coleman JLJ, Smith NJ. Gpcrs in context: sexual dimorphism in the cardiovascular system. Br J Pharmacol. (2018) 175(21):4047–59. doi: 10.1111/bph.14160
161. Coleman JLJ, Mouat MA, Wu J, Jancovski N, Bassi JK, Chan AY, et al. Orphan receptor Gpr37l1 contributes to the sexual dimorphism of central cardiovascular control. Biol Sex Differ. (2018) 9(1):14. doi: 10.1186/s13293-018-0173-y
162. Mouat MA, Coleman JLJ, Wu J, Dos Remedios CG, Feneley MP, Graham RM, et al. Involvement of Gpr37l1 in murine blood pressure regulation and human cardiac disease pathophysiology. Am J Physiol Heart Circ Physiol. (2021) 321(4):H807–H17. doi: 10.1152/ajpheart.00198.2021
Keywords: G-protein-coupled receptors, cardiovascular diseases, vascular tone, ischemia-reperfusion, heart function
Citation: Li Y, Li B, Chen W-D and Wang Y-D (2023) Role of G-protein coupled receptors in cardiovascular diseases. Front. Cardiovasc. Med. 10:1130312. doi: 10.3389/fcvm.2023.1130312
Received: 23 December 2022; Accepted: 9 May 2023;
Published: 5 June 2023.
Edited by:
Phung N. Thai, University of California, United StatesReviewed by:
Jinan Wang, University of Kansas, United StatesEugenia Gurevich, Vanderbilt University, United States
© 2023 Li, Li, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Dong Chen d2RjaGVuNjY2QDE2My5jb20= Yan-Dong Wang eWR3YW5nQG1haWwuYnVjdC5lZHUuY24=