
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 29 June 2023
Sec. Heart Failure and Transplantation
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1125687
This article is part of the Research Topic Novelties in the Therapeutic Approaches for Chronic Heart Failure: Cardiovascular targets and beyond, Volume II View all 8 articles
 Zhen Hui Peh1
Zhen Hui Peh1 Adel Dihoum2
Adel Dihoum2 Dana Hutton1
Dana Hutton1 J. Simon C. Arthur3
J. Simon C. Arthur3 Graham Rena4
Graham Rena4 Faisel Khan5Chim C. Lang2
Faisel Khan5Chim C. Lang2 Ify R. Mordi2*
Ify R. Mordi2*
Heart failure with preserved ejection fraction (HFpEF) accounts for around half of all cases of heart failure and may become the dominant type of heart failure in the near future. Unlike HF with reduced ejection fraction there are few evidence-based treatment strategies available. There is a significant unmet need for new strategies to improve clinical outcomes in HFpEF patients. Inflammation is widely thought to play a key role in HFpEF pathophysiology and may represent a viable treatment target. In this review focusing predominantly on clinical studies, we will summarise the role of inflammation in HFpEF and discuss potential therapeutic strategies targeting inflammation.
Heart failure with preserved ejection fraction (HFpEF) accounts for half of all cases of Heart Failure. Broadly speaking, it is defined as symptoms and signs of HF in the presence of a left ventricular ejection fraction (LVEF) of more than 50%, usually in the presence of a cardiac structural or functional abnormality such as left ventricular hypertrophy, left atrial enlargement or diastolic dysfunction (1). At least in part due to various factors (2) such as increased life expectancy, increasing prevalence of comorbidities such as metabolic syndrome, obesity, diabetes mellitus and greater clinical recognition, the prevalence of HFpEF is increasing such that in future it is likely to be the dominant form of HF worldwide (3).
Despite intense research interest in HFpEF, there are few evidence-based treatment options. Randomized trials targeting neurohormonal and sympathetic nervous system using angiotensin converting enzyme inhibitors (ACEi), angiotensin II receptor blockers (ARBs) and beta blockers, which provide clear benefits in HF with reduced ejection fraction (1) (HFrEF), have failed to meet their primary efficacy outcome in HFpEF. The TOPCAT (4) (spironolactone) and PARAGON-HF (5) (sacubitril/valsartan) trials also failed to meet their primary outcomes, though there were perhaps some positive signals in specific groups (for example patients recruited in the Americas (6) in TOPCAT and women or those with mildly reduced ejection fraction (7) in PARAGON-HF). Until recently, the treatment of HFpEF was limited to the used of loop diuretics to relieve congestion and provide symptomatic relief, and optimization of treatment comorbidities such as hypertension and atrial fibrillation.
A recent breakthrough has been the positive outcome trials using sodium-glucose cotransporter 2 (SGLT2) inhibitors in HFpEF. EMPEROR-Preserved (8) and DELIVER (9) demonstrated reductions in cardiovascular death and heart failure hospitalization with empagliflozin and dapagliflozin respectively compared to placebo. Based on these results, SGLT2 inhibitors are likely to play a key role in the management of HFpEF in future (10). Nevertheless, with HFpEF set to the dominant form of HF worldwide yet only having one evidence-based treatment, there is undoubtedly an urgent need for additional effective therapeutic strategies.
One of the key reasons for our inability to develop successful HFpEF treatments is its pathophysiological complexity and our limited understanding of this. This even extends to our definition of HFpEF. The textbook definition of HFpEF as a form of diastolic dysfunction leading to impaired left ventricular filling and reduced cardiac output does not fully capture the complexity of HFpEF. Various definitions of HFpEF have been used in guidelines and for inclusion in clinical trials with different profiles of patients consequently included under the HFpEF umbrella (11). Detailed phenotyping studies have also consistently identified that even when using one definition of HFpEF, patients can be clustered into separate groups that have specific phenotypes and differential clinical outcomes and responses to treatment (12–14).
HFpEF involves a complex interplay between comorbidities such as hypertension, diabetes mellitus, renal failure, COPD, obesity and atrial fibrillation and cardiac structural and functional abnormalities such as LV hypertrophy, myocardial fibrosis, myocardial stiffness, endothelial dysfunction and oxidative stress (15). Different comorbidities seem to preferentially lead to different phenotypes of HFpEF although multimorbidity is common across phenotypes. The four commonest HFpEF clinical phenotypes (16) are an ageing phenotype, pulmonary hypertension phenotype, coronary artery disease phenotype and the obesity-driven cardiometabolic phenotype (Figure 1).
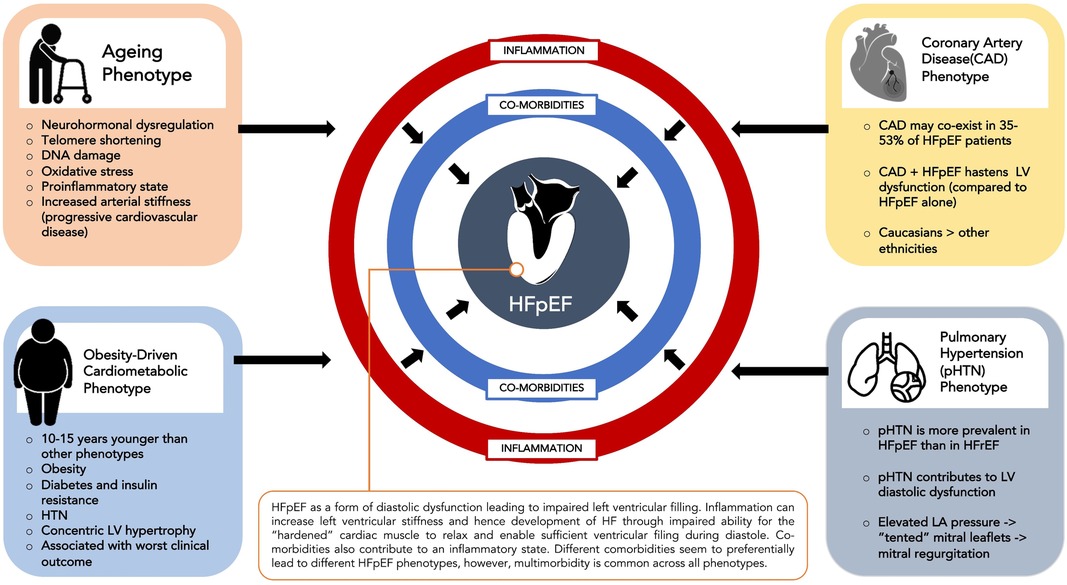
Figure 1. Common HFpEF phenotypes.
The cardiometabolic phenotype has been most concisely identified across various HFpEF cohorts. Patients with this phenotype usually have a higher prevalence of comorbidities such as hypertension, obesity, diabetes and insulin resistance and are usually 10–15 years younger (17) than patients diagnosed with other phenotypes of HFpEF. In TOPCAT, this phenotype was found to have more prevalent cardiac structural abnormalities such as concentric LVH and to have worse clinical outcomes compared to other phenotypes (18). This cardiometabolic HFpEF phenotype is consistently identified in HFpEF cohorts (12, 13, 19).
Recently, the term “metainflammation” has been used to describe the association between metabolic stress caused by conditions such as diabetes, obesity, insulin resistance and non-alcoholic fatty liver disease and chronic inflammation (20). This chronic inflammatory state can predispose to adverse cardiac remodeling such as left ventricular hypertrophy that can eventually lead to HFpEF (21). In the RELAX trial, the presence of an increased number of comorbidities such as obesity, diabetes, anaemia and chronic kidney disease in HFpEF patients was associated with higher C-reactive protein (CRP) levels (22). Further demonstrating the association between adiposity, inflammation and HFpEF, visceral adipose tissue (VAT) has been reported as an independent risk factor for hospitalization and mortality in HFpEF (21, 23), particularly in females (24). This is thought to be, at least in part, due to proinflammatory cytokines secreted by VAT that contribute to endothelial dysfunction and reduced vascular compliance (23) and subsequent concentric LV remodeling, reduced left ventricular compliance.
Regardless of the underlying phenotype, what has been consistently identified throughout these studies is that inflammation is highly prevalent in HFpEF and is associated with worse symptoms and prognosis (25). Over half of the patients in a recent HFpEF trial (RELAX) had an elevated C-reactive protein (22). Studies comparing HFpEF and HFrEF cohorts have also consistently identified that inflammation appears to be more prevalent in HFpEF compared to HFrEF, perhaps suggesting a different pathophysiology. In the BIOSTAT-CHF study, a biomarker analysis identified that there were specific biological pathways related to inflammation that were specific to HFpEF, in contrast to HFrEF (26). This finding was validated in a second cohort and replicates an earlier analysis from TIME-CHF that identified that biomarkers related to inflammation such as IL-6 and hsCRP were significantly upregulated in HFpEF compared to HFrEF (27). Levels of biomarkers such as IL-6 and CRP have also been associated with increased risk of incident HFpEF in community patients (28, 29).
As well inflammation being upregulated in HFpEF compared to HFrEF, inflammation is also associated with worse cardiac structural and functional abnormalities in HFpEF patients. A recent proteomic study of 228 HFpEF patients studied 47 proteins involved in inflammatory pathways and found that systemic inflammation was associated with an increased number of comorbidities, diastolic dysfunction and right ventricular dysfunction. Inflammation was also upregulated compared to controls (30). This was similar to other biomarker studies that have identified inflammation mediated by comorbidities as a key component of HFpEF (25).
Upregulation of inflammation in HFpEF not only leads to worse cardiac structure and function but is also associated with worse prognosis, demonstrated using various inflammatory biomarkers. Several studies have reported an independent association between elevated C-reactive protein and increased risk of death or HF hospitalisation in HFpEF patients, including after adjustment for natriuretic peptide levels (22, 31–33). An elevated neutrophil to lymphocyte ratio, a simple measure of systemic inflammation, is also associated with adverse outcome in HFpEF (34). High levels of interleukin-6 are common in HFpEF (35) and were associated with adverse outcome. In the KaRen study of 86 HFpEF patients, higher levels of inflammation were associated with worse NYHA class (25). Elevated levels of inflammatory markers including GDF-15 and TNF receptor 1 have also been found to be independently significantly associated with a composite outcome of HF hospitalisation or all-cause mortality.
In summary, there is an abundance of evidence (albeit predominantly observational) suggesting that inflammation could play a key role in the pathophysiology and clinical outcomes of HFpEF.
The innate immune system has been proposed as a key mediator of inflammation in heart failure. Oxidative stress and hypertension cause the recruitment of inflammatory leukocytes, the most notably being neutrophils which is the most abundant leukocytes in circulation. In response to endothelial cells damaged by systemic inflammation, neutrophils secrete MPO. MPO also reduces NO availability (36). Both of these exaggerate the inflammatory response. When neutrophils die, they shed IL-6 receptors that triggers surrounding endothelial cells to recruit more monocytes and macrophage (37), further amplifying the inflammatory response.
In the context of the innate immune system, inflammasomes are also activated by tissue damage. The Nod-like receptor pyrin domain containing 3 (NLRP3) inflammasome is the best described inflammasome relating to the heart (38). NLRP3 is found in the cardiomyocytes and cardiac fibroblasts (39). The NLRP3 inflammasome is summarised in Figure 2.
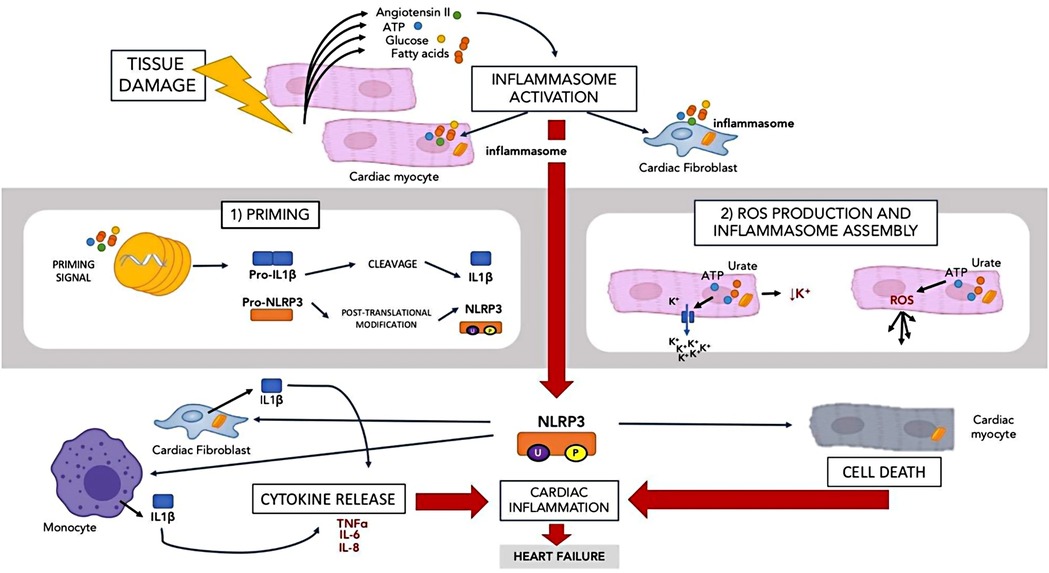
Figure 2. The innate immune system and HFpEF.
When tissue is damaged, molecules such as adenosine triphosphate, angiotensin II, fatty acids and glucose are released. The presence of these molecules activates NLRP3 in a 2-step manner (40). In the first step, there is a priming signal that leads to the transcription of IL-1 beta and NLRP3 precursors. NLRP3 precursors then undergo post translation modification via phosphorylation and ubiquitination to remain in a stable form until the second signal is activated. In the second step, the second activating signal such as ATP and urate crystal reduces in intracellular potassium and increase ROS while assembling the inflammasome. A large amount of IL-1beta is also produced by inflammatory cells such as macrophages and cardiac fibroblast (41) in response to NLRP3 activation. IL-1 beta then initiates several downstream inflammatory pathways by inducing other cytokines such as TNF, IL-6 and IL-8 (42). Activation of NLRP3 in the cardiomyocytes leads to cell death rather than IL-1beta release (43). NLRP3 activation causes IL-1 beta release by cardiac fibroblasts and cardiomyocytes death. Inhibition of NLRP3 has been shown to prevent inflammasome activation and cardiac cell death, hence reducing damage (43) and therefore may represent a potential treatment target in HFpEF.
Although much research has been focused on the innate immune system in HF, there is increasing recognition of the role of the adaptive immune system in HF pathophysiology. It is hypothesised that the adaptive immune system may play and important role in the remodeling process in response to chronic myocardial injury (44, 45). Endomyocardial biopsies of HFpEF patients show the presence of inflammatory cells and CD3+, CD11a+ and CD45+ cells, indicating underlying cardiac inflammation involving the adaptive immune system (46). Histopathological samples from HFpEF patients also demonstrate the presence of increased expression of adhesion molecules that control extravasation of T cells (46, 47). T cells may also play a role in cardiac ageing including diastolic dysfunction that is a precursor to HFpEF (48). Many of the comorbidities that are often present in HFpEF are associated with increases in circulating T cells. The metabolic changes seen in HFpEF promote pro-inflammatory T cell differentiation and lead to T cell recruitment in the heart and vasculature (49, 50). The presence of B cells may worsen cardiac injury via a few mechanisms such as the activation of T cells or the innate immune system, production of inflammatory cytokines such as IL-6, IL-1β and TNF-α or recruitment of the innate immune cells through chemokine production (51). Each of these mechanisms then further worsens the inflammatory state in the heart.
There is a large amount of evidence suggesting the role obesity and metabolic stress play in the inflammatory state of HFpEF, particularly relating to inflammation-dependent oxidative and nitrosative stress (52–56). The presence of these comorbidities increases reactive oxidative species (ROS) in the cardiac endothelial cells which further contributes to the decrease in endothelial nitric oxide (NO) (56). This results in a reduction in soluble guanylate cyclase (sGC), cyclic guanosine monophosphate (cGMP)) and cGMP protein kinase activity in cardiomyocytes which exerts a cardioprotective (57, 58) effect. The result of this is the remodeling of the left ventricle, impairing relaxation. An observation made in HFpEF is the accumulation of misfolded protein in the myocardium (59), suggesting that the unfolded protein response that mitigates stress in protein quality control is downregulated (60). In HFpEF, the pro-inflammatory molecule inducible nitric oxide synthase (iNOS) is upregulated as seen in both murine models and clinical HFpEF (20, 55).
With the burgeoning observational evidence and mechanistic studies supporting the role of inflammation in HFpEF, there has been intense interest in clinical trials of anti-inflammatory therapies in HFpEF. In this section we will summarise previously tested and potential future strategies targeting inflammation in HFpEF.
The renin-angiotensin-aldosterone system (RAAS) is central in the development of heart failure (61). When the RAAS is activated there is a reduction in blood flow to the kidney and through a cascade of different neurohormones and eventually aldosterone acts on multiple system to increase cardiac output. While initially compensatory, eventually, it acts to exacerbate heart failure. It is also involved in the inflammatory pathways of heart failure. Aldosterone encourages the expansion of adipose tissue and its transition to a proinflammatory state (62) which increases the release of proinflammatory cytokines (63). Other than mediating microvascular dysfunction and fibrosis (64), aldosterone is also found to contribute to the development of experiment HFpEF (65). Levels of aldosterone correlate to LV hypertrophy that is frequently found in HFpEF (66) and high level of aldosterone usually precede the development of metabolic syndrome (67) that also contributes to systemic inflammation. In hypertensive patients RAAS blockers do reduce inflammation (68). Hence it would seem intuitive that inhibition of the RAAS would help in the treatment of HF.
Unlike in HFrEF however, RAAS blockade in HFpEF does not improve clinical outcomes to anywhere near the same extent (69). In the TOPCAT (4) trial, spironolactone did not significantly reduce the incidence of mortality and hospitalization rates, though there were some subgroups of patients who may have benefited. Nevertheless, in a post-hoc analysis of the trial spironolactone did not cause any reduction hsCRP compared to placebo (70). Sacubitril/valsartan may be beneficial in HF with mildly reduced EF (71) and may reduce systemic inflammation in HFrEF (72), however there is no evidence as to whether this is also the case in HFpEF. Given the overall, at best neutral results of RAAS inhibitors in HFpEF it is unlikely that any anti-inflammatory effect exerted would improve clinical outcomes.
Beta-blockers reverse the neurohormonal effects of the sympathetic nervous system, improving symptoms and outcomes in HFrEF (73, 74). Beta-blockers also appear to reduce inflammation in HF. Hs-CRP concentration and oxidative stress in patient with chronic heart failure has been shown to be reduced by beta blocker such as bisoprolol and carvedilol (4), and in dilated cardiomyopathy, use of beta blockers is associated with a reduction in inflammatory markers such as IL-10 and TNF alpha (75).
The evidence for beta-blockers in HFpEF is weaker however (76) though there may be some reductions in mortality (77). Beta-blockers are often prescribed for other indications in patients with HFpEF such as atrial fibrillation and coronary artery disease (78). Again however, there are very few studies investigating inflammation in response to beta-blockers in HFpEF.
Sodium-Glucose co-Transporter 2 (SGLT2) Inhibitors have recently become an established part of HF care, following their initial development as type 2 diabetes treatments. They act by blocking SGLT2, a protein in the kidney that promotes the reabsorption of glomerular filtrated glucose back into the systemic circulation, contributing to about 90% of glucose reabsorption (79), causing a significant diuresis that is beneficial in HF (80). SGLT2 inhibitors cause significant reductions in HF hospitalisations and mortality in HFrEF patients with or without type 2 diabetes (81–83), and also improve outcomes in HFpEF (8, 9), meaning that for the first time we have a treatment for HFpEF that improves clinical outcomes.
The mechanisms of benefit of SGLT2 inhibitors in HF are still unclear and are a subject of intense ongoing research interest (84–86). SGLT2 inhibitors also appear to help in lowering systemic inflammation. A systematic review and meta-analysis of studies (87) in rodents showed that the use of SGLT2 inhibitors resulted in a decrease in IL-6, CRP, TNF alpha and MCP-1. SGLT2 inhibitors were also shown to significantly suppress NLRP3 inflammasome activation and in turn IL-1 beta secretion in human macrophages (88). SGLT2 inhibitors reduce epicardial fat, a source of pro-inflammatory cytokines, independently of their glycaemic effects (89, 90). In a recent trial of dapagliflozin vs. placebo in diabetic individuals with LVH, 12 months of treatment with dapagliflozin caused a significant reduction in hsCRP (91). Though is likely that the mechanisms of benefit of SGLT2 inhibitors in HF are likely to be multi-factorial, reductions in systemic inflammation may well be contributory.
Exercise Training can be a potential means of reducing systemic inflammation. Reduced exercise capacity is a significant part of the problem in patients with HFpEF, leading to reduced quality of life and worse prognosis (92). The current ESC guidelines (1) recommend exercise to improve exercise capacity, QoL and reduce HF hospitalization. The bulk of the evidence for exercise training applies to HFrEF, though there may be some benefit in HFpEF patients (93, 94).
Regular exercise helps to exert antioxidant and anti-inflammatory effects by targeting the cardiovascular system, adipose tissues and immune system (95). Upon muscle contraction, IL-6 is released by the skeletal muscle into the bloodstream. This release of IL-6 does not come with a similar release of TNF-alpha and IL-1beta, resulting in an overall anti-inflammatory effect (96). IL-6 also increases anti-inflammatory markers such as IL-1 receptor antagonist and IL-10 (97) which further exerts an inhibitory effect on TNF alpha and IL-1beta (98). Adipose tissue which serves as a source for TNF-alpha is also reduced by regular exercise (99). Lastly, NLRP3 inflammasome activation is also inhibited (100). eNOS is also upregulated by exercise resulting in increased bioavailability of NO leading to improved vascular function and reduced oxidative stress (101).
Few clinical studies have as yet evaluated the effects of exercise training on markers of inflammation. It is possible that systemic inflammation may also modify the effects of exercise in HFrEF (102). There are ongoing large studies of exercise in HFpEF (103) that will shed more light on the effects of exercise and could provide an avenue for exploring mechanisms of action.
Methotrexate is an anti-inflammatory commonly used in conditions such as rheumatoid arthritis (RA). Methotrexate use was associated with a reduction in cardiovascular events in patients with rheumatological conditions (104). A recent retrospective study (105) of 9,889 patients with RA and matched controls from the Vanderbilt University Medical center electronic health record, treatment with methotrexate was associated with lower risk of incident HFpEF. There may be a role for methotrexate in the management of inflammation in HFpEF, particularly in patients with a high inflammatory profile.
There have been no specific trials of methotrexate in HFpEF as yet. In the CIRT (106) (Cardiovascular inflammation Reduction Trial) trial, in which individuals with a history of MI or coronary artery disease who had either type 2 diabetes or metabolic syndrome were randomized to low-dose methotrexate (up to 20 mg daily) or placebo. Around 13% of participants had a history of HF. After a median of follow up of 2.3 years methotrexate did not reduce levels of IL6, Il1beta and CRP, nor did it reduce incidence of the primary endpoint of cardiovascular death, myocardial infarction or stroke. In a small randomized trial in 71 HFrEF patients 12 weeks of methotrexate treatment did cause a reduction in inflammatory markers and improved NYHA class, quality of life and increased 6-minute walk distance, without any change in LV ejection fraction.
Colchicine is an anti-inflammatory agent more commonly used to treat conditions such as gout, pericarditis and Behcet's syndrome. It exerts its anti-inflammatory effect by blocking the activation of NLRP3 inflammasome which in turn reduces the production of IL-1 beta and IL-18, inhibiting tubulin polymerization and microtubule development and thus impairing neutrophil migration. This inhibits IL-1 production by activated neutrophils and down regulates TNF alpha receptors in macrophages and endothelial cells (107).
The concept of using colchicine for chronic cardiovascular disease was tested in the COLCOT (108) trial. In this study of 4,745 individuals post-myocardial infarction randomized to colchicine or placebo there was a 23% relative risk reduction in the primary CV endpoint. Only 2% of the study population had a history of heart failure however. One small trial of colchicine in HFrEF patients found that although colchicine caused a reduction in inflammatory markers (CRP and IL-6) it did not cause any improvements in exercise capacity, symptoms or reductions in HF hospitalisation or mortality (109).
There is no clinical trial data as yet in HFpEF. A recent animal study using a murine hypertensive HFpEF model found that treatment with colchicine results reduced cardiac diastolic dysfunction, oxidative stress and fibrosis and improved exercise capacity (110). The ongoing COLpEF trial (NCT04857931) is an ongoing randomized clinical trial testing the efficacy of colchicine in reducing inflammation and improvement in left ventricular diastolic function and functional status and symptoms of patients with HFpEF and will provide further data on the role of colchicine in HFpEF.
In addition to their cholesterol-lowering properties, statins have many pleiotropic effects. These include anti-inflammatory properties—statins reduce CRP levels by 15%–30% (111). They act by inducing eNOS which improves endothelial function, inhibit adhesion molecules such as VCAM-1 and ICAM-1, reduce the effect of NFKB and disrupt T cell activation (111).
Observational studies and post-hoc subgroup analyses had suggested that statins might improve outcomes in HF patients (112–114) however large clinical randomized trials involving statins use in HFrEF failed to show any benefit (115–118). Many HFpEF patients are likely to be prescribed statins (for example for coronary artery disease)—as an example, almost 70% of patients in EMPEROR-Preserved were taking statins at baseline. Given the negative results of statins in HFrEF trials, and the widespread use of statins in HFpEF for other indications, it is unlikely that there will be a randomized trial of statin vs. placebo in HFpEF.
Other potential treatment approaches involve modulating part of the immune system in patients with heart failure to bring about a net anti-inflammatory effect. This includes the use of Prednisolone, Xanthine Oxidase Inhibitor, Pentoxifylline, IVIG or Thalidomide. Table 1 summaries some of the trials involving their use the outcomes from their use. However, most of the trials involve patients with HFrEF and outcomes have been mixed at best. Further research and trials are needed to investigate their relevancy and effect in HFpEF.
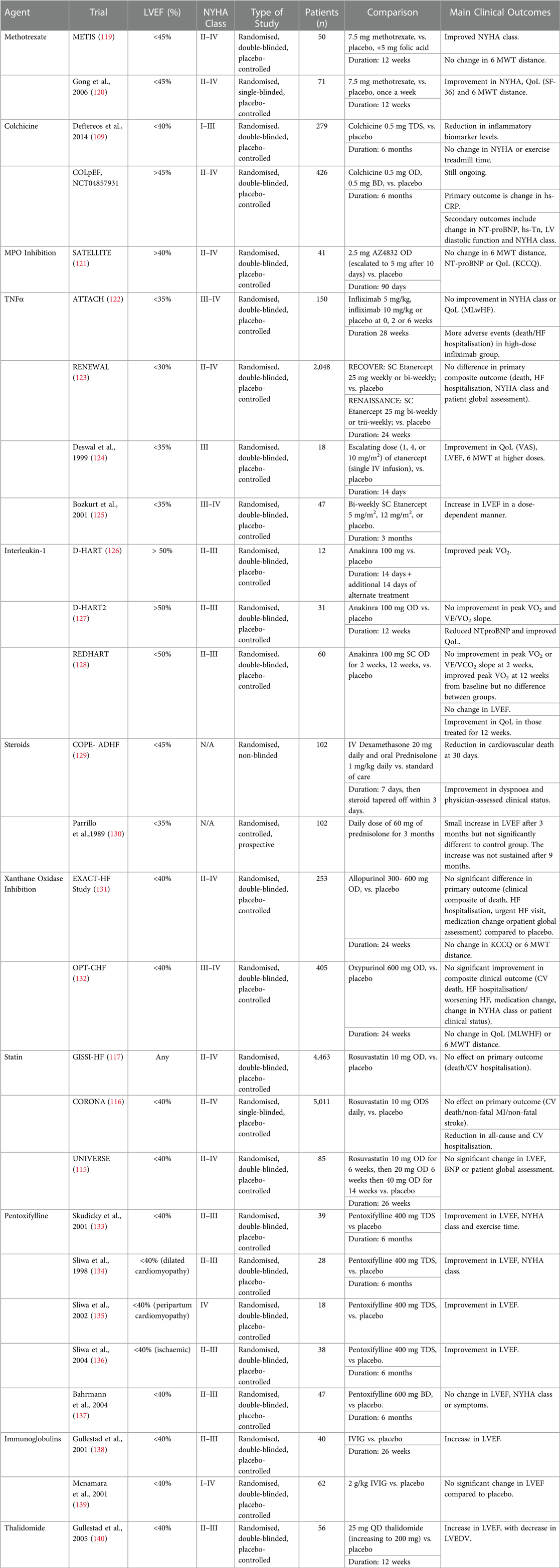
Table 1. Summary of completed and ongoing clinical trials targeting inflammation in heart failure.
As discussed, TNF alpha is one of the more prominent cytokines involved in inflammatory pathway of HF, associated with impaired systolic and diastolic function and adverse cardiac remodeling, and therefore has been well-studied in HF. This was supported by a small early trial that reported that TNF alpha inhibition using etanercept improved LV function at 3 months in HFrEF patients (125). Unfortunately these results were not borne out in large outcome trials. In the RENEWAL trial, etanercept, a TNF alpha receptor inhibitor did not reduce the incidence of primary outcomes of mortality or hospitalization for HF (123), while in ATTACH (122), TNF Inhibitor infliximab actually caused an increased risk of hospitalization and mortality at high doses.
These results may be partly explained by the inhibition of NFKB, a transcription regulator that is activated by TNF alpha and acts as a key effector of it. NFKB has cardioprotective effect such as reducing mitochondrial dysfunction and mitophagy, inhibiting cell death and inducing antioxidant effects (141). With the inhibiting of TNF alpha and consequently NFKB, these beneficial effects may be lost, causing HF to worsen. Furthermore, infliximab has been shown to induce apoptosis and complement mediated cell lysis (141), further contributing to cell death in the failing heart.
These trials have limited enthusiasm for targeting TNF alpha in HF, although they have only included HFrEF patients. Similar studies involving HFpEF have not been performed. There is preliminary data suggesting that anti-inflammatory therapy may be beneficial in certain subgroup of HFpEF. Unlike HFrEF where prevention of cell death in the myocardium is relevant, HFpEF is characterized by myocardial hypertrophy (142). It is conceivable that anti-TNF alpha therapy might have different effects in HFpEF. In an observational study involving the effect of anti-TNF therapy on cardiac function in rheumatoid arthritis (where HFpEF is more prevalent), it was found that anti-TNF alpha therapy was not associated with a worsening of cardiac function and in fact was associated with a 23% decrease in NT-proBNP after 6 months (143). In a Swedish registry on HF in patients with RA, patients treated with corticosteroids had a higher incidence of non-ischemic heart failure compared to patients which used biologics (144). There may still be a role for anti-TNF strategies to be tested in HFpEF.
IL-1 plays an important role in the development of systolic and diastolic dysfunction. In the context of diastolic dysfunction, IL-1 reduces calcium reuptake by sarcoplasmic reticulum through down regulation of phospholamban and SERCA, in turn affecting the initiation of cardiomyocytes relaxation (145, 146).
In the DHART trial, 2 weeks treatment with anakinra, an IL-1 receptor antagonist, reduced systemic inflammation, increased aerobic exercise tolerance and peak VO2 in patients with HFpEF (126).
Unfortunately, in the larger follow-on study DHART2 (127), despite again finding a reduction in CRP, NTproBNP and an improvement in exercise tolerance with anakinra, there was no improvement in the primary endpoint of peak VO2. The discordant results could be at least in part due to the fact that in DHART2, most of the participants were obese which in turns affects their cardiorespiratory fitness regardless of their cardiac function.
Further interest in IL-1 beta blockade as a therapeutic strategy in HF was raised by an analysis of the CANTOS (147) trial. In CANTOS, the anti-IL-1 beta monoclonal antibody canakinumab was studied in patients with a history of MI and evidence of systemic inflammation (measured by elevated hs-CRP). Participants were treated with 150 mg canakinumab or placebo every 3 months with an optimized statin regimen. Canakinumab caused a 15% decreased risk in mortality and non-fatal stroke and MI (147). In a prespecified sub analysis of the trial, canakinumab was found to reduce heart failure-related hospitalization in a dose dependent manner (148). In CANTOS, HFrEF and HFpEF was not discriminated. However, considering that many patients were older and has a history of obesity, diabetes and hypertension, this hypothesis generating result raises the possibility that IL-1 beta blockade with canakinumab might have benefit in patients with HFpEF. Further studies of IL-1 beta blockade in HFpEF are warranted.
Myeloperoxidase (MPO) is a heme containing peroxidase that is expressed mainly in neutrophils. They play a role in the development of acute and chronic vascular inflammation which is proposed as an underlying mechanism in the development of HFpEF (149). In the SATELLITE (121) (Safety and Tolerability Study of AZD4831 in Patients with Heart Failure) trail, MPO inhibitor (AZD4831) was investigated. This trial was stopped prematurely after it achieved its original aim of target engagement and satisfactory safety profile. From the trial, it showed a 69% decrease in MPO activity within the 30 days trial period and an increase in exercise capacity and wellness score, highlighting its role as a potential treatment for HFpEF. However, MPO-inhibition is a novel approach in treating HFpEF, hence further studies and trials are needed into to further out understanding and investigate the efficacy of this potential treatment.
There remain few evidence-based treatment options for HFpEF and there is an urgent need for novel therapeutic strategies. As described in Table 1 there are a number of ongoing clinical trials evaluating anti-inflammatory strategies for HFpEF. Given the heterogeneity of HFpEF, a targeted approach with detailed patient phenotyping is likely to be the most fruitful strategy. Using such an approach, patients with high levels of inflammation could be selected for anti-inflammatory therapy and could have more chance of deriving clinical benefit. This could lead to a “precision” approach where a specific cytokine is targeted. An alternative approach would be to use interventional strategies that might have multiple effects, of which an anti-inflammatory action is one. Such strategies include exercise and drugs inducing weight loss (150).
There is a plethora of observational and trial evidence supporting the role of inflammation as an important part of the pathophysiology of HF. Although the CANTOS trial showed the merit of an anti-inflammatory approach in cardiovascular disease, most other trials of anti-inflammatory therapy in HF have been neutral at best, though these have predominantly been in HFrEF. Very few clinical trials of anti-inflammatory strategies have been conducted in HFpEF populations however, and it is possible that inflammation may play a differential role in HFpEF to HFrEF. It may also be that patients could be selected for trials based on baseline levels of inflammation—those with higher levels of systemic inflammation might be more likely to benefit from anti-inflammatory therapy. Such a strategy needs to be refined as there are many potential inflammatory markers that could be used however we do not know which might translate to clinical benefit. The most widely-used is high-sensitivity C-reactive protein—in the CANTOS trial patients were eligible if their hsCRP level was ≥2 mg/l (147). Optimal levels still need to be defined. Despite the recent breakthrough of SGLT2 inhibitors in treatment of HFpEF, there is a need for further therapeutic options. Inflammation remains a viable treatment target in HFpEF and further well-designed trials are warranted.
ZP, DH and IM conceived the idea and drafted the original manuscript. SA, FK, GR and CL provided critical revision. All authors contributed to the article and approved the submitted version.
This work has been supported by a research grant from Tenovus Scotland (T20/58).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M, et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2021) 42(36):3599–726. doi: 10.1093/eurheartj/ehab368
2. Oktay AA, Rich JD, Shah SJ. The emerging epidemic of heart failure with preserved ejection fraction. Curr Heart Fail Rep. (2013) 10(4):401–10. doi: 10.1007/s11897-013-0155-7
3. Lam CSP, Voors AA, de Boer RA, Solomon SD, van Veldhuisen DJ. Heart failure with preserved ejection fraction: from mechanisms to therapies. Eur Heart J. (2018) 39(30):2780–92. doi: 10.1093/eurheartj/ehy301
4. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. (2014) 370(15):1383–92. doi: 10.1056/NEJMoa1313731
5. Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, et al. Angiotensin-Neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. (2019) 381(17):1609–20. doi: 10.1056/NEJMoa1908655
6. Pfeffer MA, Claggett B, Assmann SF, Boineau R, Anand IS, Clausell N, et al. Regional variation in patients and outcomes in the treatment of preserved cardiac function heart failure with an aldosterone antagonist (TOPCAT) trial. Circulation. (2015) 131(1):34–42. doi: 10.1161/CIRCULATIONAHA.114.013255
7. McMurray JJV, Jackson AM, Lam CSP, Redfield MM, Anand IS, Ge J, et al. Effects of sacubitril-valsartan versus valsartan in women compared with men with heart failure and preserved ejection fraction: insights from PARAGON-HF. Circulation. (2020) 141(5):338–51. doi: 10.1161/CIRCULATIONAHA.119.044491
8. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Bohm M, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. (2021) 385(16):1451–61. doi: 10.1056/NEJMoa2107038
9. Solomon SD, McMurray JJV, Claggett B, de Boer RA, DeMets D, Hernandez AF, et al. Dapagliflozin in heart failure with mildly reduced or preserved ejection fraction. N Engl J Med. (2022) 387(12):1089–98. doi: 10.1056/NEJMoa2206286
10. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 American college of cardiology/American heart association/heart failure society of America guideline for the management of heart failure: executive summary. J Card Fail. (2022) 28(5):810–30. doi: 10.1016/j.cardfail.2022.02.009
11. Ho JE, Zern EK, Wooster L, Bailey CS, Cunningham T, Eisman AS, et al. Differential clinical profiles, exercise responses, and outcomes associated with existing HFpEF definitions. Circulation. (2019) 140(5):353–65. doi: 10.1161/CIRCULATIONAHA.118.039136
12. Segar MW, Patel KV, Ayers C, Basit M, Tang WHW, Willett D, et al. Phenomapping of patients with heart failure with preserved ejection fraction using machine learning-based unsupervised cluster analysis. Eur J Heart Fail. (2020) 22(1):148–58. doi: 10.1002/ejhf.1621
13. Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M, et al. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation. (2015) 131(3):269–79. doi: 10.1161/CIRCULATIONAHA.114.010637
14. Choy M, Liang W, He J, Fu M, Dong Y, He X, et al. Phenotypes of heart failure with preserved ejection fraction and effect of spironolactone treatment. ESC Heart Fail. (2022) 9(4):2567–75. doi: 10.1002/ehf2.13969
15. van Empel V, Brunner-La Rocca HP. Inflammation in HFpEF: key or circumstantial? Int J Cardiol. (2015) 189:259–63. doi: 10.1016/j.ijcard.2015.04.110
16. Samson R, Jaiswal A, Ennezat PV, Cassidy M, Le Jemtel TH. Clinical phenotypes in heart failure with preserved ejection fraction. J Am Heart Assoc. (2016) 5(1):e002477. doi: 10.1161/JAHA.115.002477
17. Klapholz M, Maurer M, Lowe AM, Messineo F, Meisner JS, Mitchell J, et al. Hospitalization for heart failure in the presence of a normal left ventricular ejection fraction: results of the New York heart failure registry. J Am Coll Cardiol. (2004) 43(8):1432–8. doi: 10.1016/j.jacc.2003.11.040
18. Cohen JB, Schrauben SJ, Zhao L, Basso MD, Cvijic ME, Li Z, et al. Clinical phenogroups in heart failure with preserved ejection fraction: detailed phenotypes, prognosis, and response to spironolactone. JACC Heart Fail. (2020) 8(3):172–84. doi: 10.1016/j.jchf.2019.09.009
19. Uijl A, Savarese G, Vaartjes I, Dahlstrom U, Brugts JJ, Linssen GCM, et al. Identification of distinct phenotypic clusters in heart failure with preserved ejection fraction. Eur J Heart Fail. (2021) 23(6):973–82. doi: 10.1002/ejhf.2169
20. Schiattarella GG, Rodolico D, Hill JA. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc Res. (2021) 117(2):423–34. doi: 10.1093/cvr/cvaa217
21. Schiattarella GG, Alcaide P, Condorelli G, Gillette TG, Heymans S, Jones EAV, et al. Immunometabolic mechanisms of heart failure with preserved ejection fraction. Nat Cardiovasc Res. (2022) 1(3):211–22. doi: 10.1038/s44161-022-00032-w
22. DuBrock HM, AbouEzzeddine OF, Redfield MM. High-sensitivity C-reactive protein in heart failure with preserved ejection fraction. PLoS One. (2018) 13(8):e0201836. doi: 10.1371/journal.pone.0201836
23. Rao VN, Fudim M, Mentz RJ, Michos ED, Felker GM. Regional adiposity and heart failure with preserved ejection fraction. Eur J Heart Fail. (2020) 22(9):1540–50. doi: 10.1002/ejhf.1956
24. Karlsson T, Rask-Andersen M, Pan G, Hoglund J, Wadelius C, Ek WE, et al. Contribution of genetics to visceral adiposity and its relation to cardiovascular and metabolic disease. Nat Med. (2019) 25(9):1390–5. doi: 10.1038/s41591-019-0563-7
25. Hage C, Michaelsson E, Linde C, Donal E, Daubert JC, Gan LM, et al. Inflammatory biomarkers predict heart failure severity and prognosis in patients with heart failure with preserved ejection fraction: a holistic proteomic approach. Circ Cardiovasc Genet. (2017) 10(1):e001633. doi: 10.1161/CIRCGENETICS.116.001633
26. Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, et al. Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol. (2018) 72(10):1081–90. doi: 10.1016/j.jacc.2018.06.050
27. Sanders-van Wijk S, van Empel V, Davarzani N, Maeder MT, Handschin R, Pfisterer ME, et al. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. Reduced left ventricular ejection fraction. Eur J Heart Fail. (2015) 17(10):1006–14. doi: 10.1002/ejhf.414
28. Chia YC, Kieneker LM, van Hassel G, Binnenmars SH, Nolte IM, van Zanden JJ, et al. Interleukin 6 and development of heart failure with preserved ejection fraction in the general population. J Am Heart Assoc. (2021) 10(11):e018549. doi: 10.1161/jaha.120.018549
29. Albar Z, Albakri M, Hajjari J, Karnib M, Janus SE, Al-Kindi SG. Inflammatory markers and risk of heart failure with reduced to preserved ejection fraction. Am J Cardiol. (2022) 167:68–75. doi: 10.1016/j.amjcard.2021.11.045
30. Sanders-van Wijk S, Tromp J, Beussink-Nelson L, Hage C, Svedlund S, Saraste A, et al. Proteomic evaluation of the comorbidity-inflammation paradigm in heart failure with preserved ejection fraction: results from the PROMIS-HFpEF study. Circulation. (2020) 142(21):2029–44. doi: 10.1161/CIRCULATIONAHA.120.045810
31. Koller L, Kleber M, Goliasch G, Sulzgruber P, Scharnagl H, Silbernagel G, et al. C-reactive protein predicts mortality in patients referred for coronary angiography and symptoms of heart failure with preserved ejection fraction. Eur J Heart Fail. (2014) 16(7):758–66. doi: 10.1002/ejhf.104
32. Lakhani I, Wong MV, Hung JKF, Gong M, Waleed KB, Xia Y, et al. Diagnostic and prognostic value of serum C-reactive protein in heart failure with preserved ejection fraction: a systematic review and meta-analysis. Heart Fail Rev. (2021) 26(5):1141–50. doi: 10.1007/s10741-020-09927-x
33. Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, et al. Inflammatory markers and incident heart failure risk in older adults: the health ABC (health, aging, and body composition) study. J Am Coll Cardiol. (2010) 55(19):2129–37. doi: 10.1016/j.jacc.2009.12.045
34. Curran FM, Bhalraam U, Mohan M, Singh JS, Anker SD, Dickstein K, et al. Neutrophil-to-lymphocyte ratio and outcomes in patients with new-onset or worsening heart failure with reduced and preserved ejection fraction. ESC Heart Fail. (2021) 8(4):3168–79. doi: 10.1002/ehf2.13424
35. Markousis-Mavrogenis G, Tromp J, Ouwerkerk W, Devalaraja M, Anker SD, Cleland JG, et al. The clinical significance of interleukin-6 in heart failure: results from the BIOSTAT-CHF study. Eur J Heart Fail. (2019) 21(8):965–73. doi: 10.1002/ejhf.1482
36. Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A, et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. (2002) 296(5577):2391–4. doi: 10.1126/science.1106830
37. Kratofil RM, Kubes P, Deniset JF. Monocyte conversion during inflammation and injury. Arterioscler Thromb Vasc Biol. (2017) 37(1):35–42. doi: 10.1161/ATVBAHA.116.308198
38. Yao C, Veleva T, Scott L Jr., Cao S, Li L, Chen G, et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation. (2018) 138(20):2227–42. doi: 10.1161/CIRCULATIONAHA.118.035202
39. Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature. (2020) 588(7838):466–72. doi: 10.1038/s41586-020-2797-4
40. Sokolova M, Ranheim T, Louwe MC, Halvorsen B, Yndestad A, Aukrust P. NLRP3 Inflammasome: a novel player in metabolically induced inflammation-potential influence on the myocardium. J Cardiovasc Pharmacol. (2019) 74(4):276–84. doi: 10.1097/FJC.0000000000000704
41. Sandanger O, Ranheim T, Vinge LE, Bliksoen M, Alfsnes K, Finsen AV, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. (2013) 99(1):164–74. doi: 10.1093/cvr/cvt091
42. Chauhan D, Vande Walle L, Lamkanfi M. Therapeutic modulation of inflammasome pathways. Immunol Rev. (2020) 297(1):123–38. doi: 10.1111/imr.12908
43. Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. (2011) 108(49):19725–30. doi: 10.1073/pnas.1108586108
44. Sattler S, Fairchild P, Watt FM, Rosenthal N, Harding SE. The adaptive immune response to cardiac injury-the true roadblock to effective regenerative therapies? NPJ Regen Med. (2017) 2:19. doi: 10.1038/s41536-017-0022-3
45. Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. (2015) 15(2):117–29. doi: 10.1038/nri3800
46. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. (2011) 4(1):44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451
47. van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. (2008) 117(1):43–51. doi: 10.1161/CIRCULATIONAHA.107.728550
48. Martini E, Cremonesi M, Panico C, Carullo P, Bonfiglio CA, Serio S, et al. T cell costimulation blockade blunts age-related heart failure. Circ Res. (2020) 127(8):1115–7. doi: 10.1161/CIRCRESAHA.119.316530
49. Marelli-Berg FM, Aksentijevic D. Immunometabolic cross-talk in the inflamed heart. Cell Stress. (2019) 3(8):240–66. doi: 10.15698/cst2019.08.194
50. Guzik TJ, Skiba DS, Touyz RM, Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc Res. (2017) 113(9):1009–23. doi: 10.1093/cvr/cvx108
51. Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. (2013) 19(10):1273–80. doi: 10.1038/nm.3284
52. Paulus WJ. Unfolding discoveries in heart failure. N Engl J Med. (2020) 382(7):679–82. doi: 10.1056/NEJMcibr1913825
53. Yoon S, Kim M, Lee H, Kang G, Bedi K, Margulies KB, et al. S-nitrosylation of histone deacetylase 2 by neuronal nitric oxide synthase as a mechanism of diastolic dysfunction. Circulation. (2021) 143(19):1912–25. doi: 10.1161/CIRCULATIONAHA.119.043578
54. Schiattarella GG, Altamirano F, Kim SY, Tong D, Ferdous A, Piristine H, et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat Commun. (2021) 12(1):1684. doi: 10.1038/s41467-021-21931-9
55. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. (2019) 568(7752):351–6. doi: 10.1038/s41586-019-1100-z
56. Riehle C, Bauersachs J. Key inflammatory mechanisms underlying heart failure. Herz. (2019) 44(2):96–106. doi: 10.1007/s00059-019-4785-8
57. Fraccarollo D, Galuppo P, Motschenbacher S, Ruetten H, Schafer A, Bauersachs J. Soluble guanylyl cyclase activation improves progressive cardiac remodeling and failure after myocardial infarction. Cardioprotection over ACE inhibition. Basic Res Cardiol. (2014) 109(4):421. doi: 10.1007/s00395-014-0421-1
58. Schafer A, Galuppo P, Fraccarollo D, Vogt C, Widder JD, Pfrang J, et al. Increased cytochrome P4502E1 expression and altered hydroxyeicosatetraenoic acid formation mediate diabetic vascular dysfunction: rescue by guanylyl-cyclase activation. Diabetes. (2010) 59(8):2001–9. doi: 10.2337/db09-1668
59. Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, de Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. (2015) 36(38):2585–94. doi: 10.1093/eurheartj/ehv338
60. Wang ZV, Hill JA. Protein quality control and metabolism: bidirectional control in the heart. Cell Metab. (2015) 21(2):215–26. doi: 10.1016/j.cmet.2015.01.016
61. Orsborne C, Chaggar PS, Shaw SM, Williams SG. The renin-angiotensin-aldosterone system in heart failure for the non-specialist: the past, the present and the future. Postgrad Med J. (2017) 93(1095):29–37. doi: 10.1136/postgradmedj-2016-134045
62. Nguyen Dinh Cat A, Antunes TT, Callera GE, Sanchez A, Tsiropoulou S, Dulak-Lis MG, et al. Adipocyte-Specific mineralocorticoid receptor overexpression in mice is associated with metabolic syndrome and vascular dysfunction: role of redox-sensitive PKG-1 and rho kinase. Diabetes. (2016) 65(8):2392–403. doi: 10.2337/db15-1627
63. Kraus D, Jager J, Meier B, Fasshauer M, Klein J. Aldosterone inhibits uncoupling protein-1, induces insulin resistance, and stimulates proinflammatory adipokines in adipocytes. Horm Metab Res. (2005) 37(7):455–9. doi: 10.1055/s-2005-870240
64. Garg R, Rao AD, Baimas-George M, Hurwitz S, Foster C, Shah RV, et al. Mineralocorticoid receptor blockade improves coronary microvascular function in individuals with type 2 diabetes. Diabetes. (2015) 64(1):236–42. doi: 10.2337/db14-0670
65. Valero-Munoz M, Li S, Wilson RM, Hulsmans M, Aprahamian T, Fuster JJ, et al. Heart failure with preserved ejection fraction induces beiging in adipose tissue. Circ Heart Fail. (2016) 9(1):e002724. doi: 10.1161/CIRCHEARTFAILURE.115.002724
66. Edelmann F, Tomaschitz A, Wachter R, Gelbrich G, Knoke M, Dungen HD, et al. Serum aldosterone and its relationship to left ventricular structure and geometry in patients with preserved left ventricular ejection fraction. Eur Heart J. (2012) 33(2):203–12. doi: 10.1093/eurheartj/ehr292
67. Musani SK, Vasan RS, Bidulescu A, Liu J, Xanthakis V, Sims M, et al. Aldosterone, C-reactive protein, and plasma B-type natriuretic peptide are associated with the development of metabolic syndrome and longitudinal changes in metabolic syndrome components: findings from the Jackson heart study. Diabetes Care. (2013) 36(10):3084–92. doi: 10.2337/dc12-2562
68. Fliser D, Buchholz K, Haller H, Olmesartan EUTo, Pravastatin in I, Atherosclerosis I. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation. (2004) 110(9):1103–7. doi: 10.1161/01.CIR.0000140265.21608.8E
69. Fu M, Zhou J, Sun A, Zhang S, Zhang C, Zou Y, et al. Efficacy of ACE inhibitors in chronic heart failure with preserved ejection fraction–a meta analysis of 7 prospective clinical studies. Int J Cardiol. (2012) 155(1):33–8. doi: 10.1016/j.ijcard.2011.01.081
70. Myhre PL, Vaduganathan M, O'Meara E, Claggett BL, de Denus S, Jarolim P, et al. Mechanistic effects of spironolactone on cardiovascular and renal biomarkers in heart failure with preserved ejection fraction: a TOPCAT biorepository study. Circ Heart Fail. (2020) 13(1):e006638. doi: 10.1161/CIRCHEARTFAILURE.119.006638
71. Solomon SD, Vaduganathan M, L Claggett B, Packer M, Zile M, Swedberg K, et al. Sacubitril/valsartan across the Spectrum of ejection fraction in heart failure. Circulation. (2020) 141(5):352–61. doi: 10.1161/CIRCULATIONAHA.119.044586
72. Bolla GB, Fedele A, Faggiano A, Sala C, Santangelo G, Carugo S. Effects of sacubitril/valsartan on biomarkers of fibrosis and inflammation in patients with heart failure with reduced ejection fraction. BMC Cardiovasc Disord. (2022) 22(1):217. doi: 10.1186/s12872-022-02647-0
73. Bristow MR. Treatment of chronic heart failure with beta-adrenergic receptor antagonists: a convergence of receptor pharmacology and clinical cardiology. Circ Res. (2011) 109(10):1176–94. doi: 10.1161/CIRCRESAHA.111.245092
74. Domanski MJ, Krause-Steinrauf H, Massie BM, Deedwania P, Follmann D, Kovar D, et al. A comparative analysis of the results from 4 trials of beta-blocker therapy for heart failure: BEST, CIBIS-II, MERIT-HF, and COPERNICUS. J Card Fail. (2003) 9(5):354–63. doi: 10.1054/S1071-9164(03)00133-7
75. Ohtsuka T, Hamada M, Hiasa G, Sasaki O, Suzuki M, Hara Y, et al. Effect of beta-blockers on circulating levels of inflammatory and anti-inflammatory cytokines in patients with dilated cardiomyopathy. J Am Coll Cardiol. (2001) 37(2):412–7. doi: 10.1016/S0735-1097(00)01121-9
76. Bavishi C, Chatterjee S, Ather S, Patel D, Messerli FH. Beta-blockers in heart failure with preserved ejection fraction: a meta-analysis. Heart Fail Rev. (2015) 20(2):193–201. doi: 10.1007/s10741-014-9453-8
77. Zheng SL, Chan FT, Nabeebaccus AA, Shah AM, McDonagh T, Okonko DO, et al. Drug treatment effects on outcomes in heart failure with preserved ejection fraction: a systematic review and meta-analysis. Heart. (2018) 104(5):407–15. doi: 10.1136/heartjnl-2017-311652
78. Solomon SD, Rizkala AR, Lefkowitz MP, Shi VC, Gong J, Anavekar N, et al. Baseline characteristics of patients with heart failure and preserved ejection fraction in the PARAGON-HF trial. Circ Heart Fail. (2018) 11(7):e004962. doi: 10.1161/CIRCHEARTFAILURE.118.004962
79. Shubrook JH, Bokaie BB, Adkins SE. Empagliflozin in the treatment of type 2 diabetes: evidence to date. Drug Des Devel Ther. (2015) 9:5793–803. doi: 10.2147/DDDT.S69926
80. Mordi NA, Mordi IR, Singh JS, McCrimmon RJ, Struthers AD, Lang CC. Renal and cardiovascular effects of SGLT2 inhibition in combination with loop diuretics in patients with type 2 diabetes and chronic heart failure: the RECEDE-CHF trial. Circulation. (2020) 142(18):1713–24. doi: 10.1161/CIRCULATIONAHA.120.048739
81. Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. (2020) 383(15):1413–24. doi: 10.1056/NEJMoa2022190
82. McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. (2019) 381(21):1995–2008. doi: 10.1056/NEJMoa1911303
83. Bhatt DL, Szarek M, Steg PG, Cannon CP, Leiter LA, McGuire DK, et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. (2021) 384(2):117–28. doi: 10.1056/NEJMoa2030183
84. Fonseca-Correa JI, Correa-Rotter R. Sodium-glucose cotransporter 2 inhibitors mechanisms of action: a review. Front Med (Lausanne). (2021) 8:777861. doi: 10.3389/fmed.2021.777861
85. Wilcox CS. Antihypertensive and renal mechanisms of SGLT2 (sodium-glucose linked transporter 2) inhibitors. Hypertension. (2020) 75(4):894–901. doi: 10.1161/HYPERTENSIONAHA.119.11684
86. Mordi IR, Lang CC. Glucose-lowering and metabolic effects of SGLT2 inhibitors. Heart Fail Clin. (2022) 18(4):529–38. doi: 10.1016/j.hfc.2022.03.004
87. Theofilis P, Sagris M, Oikonomou E, Antonopoulos AS, Siasos G, Tsioufis K, et al. The impact of SGLT2 inhibitors on inflammation: a systematic review and meta-analysis of studies in rodents. Int Immunopharmacol. (2022) 111:109080. doi: 10.1016/j.intimp.2022.109080
88. Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 Inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. (2020) 11(1):2127. doi: 10.1038/s41467-020-15983-6
89. Requena-Ibanez JA, Santos-Gallego CG, Rodriguez-Cordero A, Vargas-Delgado AP, Mancini D, Sartori S, et al. Mechanistic insights of empagliflozin in nondiabetic patients with HFrEF: from the EMPA-TROPISM study. JACC Heart Fail. (2021) 9(8):578–89. doi: 10.1016/j.jchf.2021.04.014
90. Sato T, Aizawa Y, Yuasa S, Kishi S, Fuse K, Fujita S, et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc Diabetol. (2018) 17(1):6. doi: 10.1186/s12933-017-0658-8
91. Brown AJM, Gandy S, McCrimmon R, Houston JG, Struthers AD, Lang CC. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: the DAPA-LVH trial. Eur Heart J. (2020) 41(36):3421–32. doi: 10.1093/eurheartj/ehaa419
92. Nayor M, Houstis NE, Namasivayam M, Rouvina J, Hardin C, Shah RV, et al. Impaired exercise tolerance in heart failure with preserved ejection fraction: quantification of multiorgan system reserve capacity. JACC Heart Fail. (2020) 8(8):605–17. doi: 10.1016/j.jchf.2020.03.008
93. Long L, Mordi IR, Bridges C, Sagar VA, Davies EJ, Coats AJ, et al. Exercise-based cardiac rehabilitation for adults with heart failure. Cochrane Database Syst Rev. (2019) 1:CD003331. doi: 10.1002/14651858.cd003331.pub5
94. Pandey A, Parashar A, Kumbhani D, Agarwal S, Garg J, Kitzman D, et al. Exercise training in patients with heart failure and preserved ejection fraction: meta-analysis of randomized control trials. Circ Heart Fail. (2015) 8(1):33–40. doi: 10.1161/CIRCHEARTFAILURE.114.001615
95. Sallam N, Laher I. Exercise modulates oxidative stress and inflammation in aging and cardiovascular diseases. Oxid Med Cell Longev. (2016) 2016:7239639. doi: 10.1155/2016/7239639
96. Pedersen BK, Febbraio MA. Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev. (2008) 88(4):1379–406. doi: 10.1152/physrev.90100.2007
97. Steensberg A, Fischer CP, Keller C, Moller K, Pedersen BK. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am J Physiol Endocrinol Metab. (2003) 285(2):E433–7. doi: 10.1152/ajpendo.00074.2003
98. Schneider CP, Schwacha MG, Chaudry IH. The role of interleukin-10 in the regulation of the systemic inflammatory response following trauma-hemorrhage. Biochim Biophys Acta. (2004) 1689(1):22–32. doi: 10.1016/j.bbadis.2004.01.003
99. Ross R, Dagnone D, Jones PJ, Smith H, Paddags A, Hudson R, et al. Reduction in obesity and related comorbid conditions after diet-induced weight loss or exercise-induced weight loss in men. A randomized, controlled trial. Ann Intern Med. (2000) 133(2):92–103. doi: 10.7326/0003-4819-133-2-200007180-00008
100. Butts B, Butler J, Dunbar SB, Corwin E, Gary RA. Effects of exercise on ASC methylation and IL-1 cytokines in heart failure. Med Sci Sports Exerc. (2018) 50(9):1757–66. doi: 10.1249/MSS.0000000000001641
101. Sessa WC, Pritchard K, Seyedi N, Wang J, Hintze TH. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ Res. (1994) 74(2):349–53. doi: 10.1161/01.RES.74.2.349
102. Fernandes-Silva MM, Guimaraes GV, Rigaud VO, Lofrano-Alves MS, Castro RE, de Barros Cruz LG, et al. Inflammatory biomarkers and effect of exercise on functional capacity in patients with heart failure: insights from a randomized clinical trial. Eur J Prev Cardiol. (2017) 24(8):808–17. doi: 10.1177/2047487317690458
103. Lang CC, Smith K, Wingham J, Eyre V, Greaves CJ, Warren FC, et al. A randomised controlled trial of a facilitated home-based rehabilitation intervention in patients with heart failure with preserved ejection fraction and their caregivers: the REACH-HFpEF pilot study. BMJ Open. (2018) 8(4):e019649. doi: 10.1136/bmjopen-2017-019649
104. Balanescu AR, Bojinca VC, Bojinca M, Donisan T, Balanescu SM. Cardiovascular effects of methotrexate in immune-mediated inflammatory diseases. Exp Ther Med. (2019) 17(2):1024–9. doi: 10.3892/etm.2018.6992
105. Ahlers MJ, Lowery BD, Farber-Eger E, Wang TJ, Bradham W, Ormseth MJ, et al. Heart failure risk associated with rheumatoid arthritis-related chronic inflammation. J Am Heart Assoc. (2020) 9(10):e014661. doi: 10.1161/JAHA.119.014661
106. Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, et al. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. (2019) 380(8):752–62. doi: 10.1056/NEJMoa1809798
107. Deftereos S, Giannopoulos G, Papoutsidakis N, Panagopoulou V, Kossyvakis C, Raisakis K, et al. Colchicine and the heart: pushing the envelope. J Am Coll Cardiol. (2013) 62(20):1817–25. doi: 10.1016/j.jacc.2013.08.726
108. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381(26):2497–505. doi: 10.1056/NEJMoa1912388
109. Deftereos S, Giannopoulos G, Panagopoulou V, Bouras G, Raisakis K, Kossyvakis C, et al. Anti-inflammatory treatment with colchicine in stable chronic heart failure: a prospective, randomized study. JACC Heart Fail. (2014) 2(2):131–7. doi: 10.1016/j.jchf.2013.11.006
110. Shen S, Duan J, Hu J, Qi Y, Kang L, Wang K, et al. Colchicine alleviates inflammation and improves diastolic dysfunction in heart failure rats with preserved ejection fraction. Eur J Pharmacol. (2022) 929:175126. doi: 10.1016/j.ejphar.2022.175126
111. Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. (2005) 4(12):977–87. doi: 10.1038/nrd1901
112. Khush KK, Waters DD, Bittner V, Deedwania PC, Kastelein JJ, Lewis SJ, et al. Effect of high-dose atorvastatin on hospitalizations for heart failure: subgroup analysis of the treating to new targets (TNT) study. Circulation. (2007) 115(5):576–83. doi: 10.1161/CIRCULATIONAHA.106.625574
113. Horwich TB, MacLellan WR, Fonarow GC. Statin therapy is associated with improved survival in ischemic and non-ischemic heart failure. J Am Coll Cardiol. (2004) 43(4):642–8. doi: 10.1016/j.jacc.2003.07.049
114. Marume K, Takashio S, Nagai T, Tsujita K, Saito Y, Yoshikawa T, et al. Effect of statins on mortality in heart failure with preserved ejection fraction without coronary artery disease- report from the JASPER study. Circ J. (2019) 83(2):357–67. doi: 10.1253/circj.CJ-18-0639
115. Krum H, Ashton E, Reid C, Kalff V, Rogers J, Amarena J, et al. Double-blind, randomized, placebo-controlled study of high-dose HMG CoA reductase inhibitor therapy on ventricular remodeling, pro-inflammatory cytokines and neurohormonal parameters in patients with chronic systolic heart failure. J Card Fail. (2007) 13(1):1–7. doi: 10.1016/j.cardfail.2006.09.008
116. Kjekshus J, Apetrei E, Barrios V, Bohm M, Cleland JG, Cornel JH, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med. (2007) 357(22):2248–61. doi: 10.1056/NEJMoa0706201
117. Tavazzi L, Maggioni AP, Marchioli R, Barlera S, Franzosi MG, Latini R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. (2008) 372(9645):1231–9. doi: 10.1016/S0140-6736(08)61240-4
118. Alehagen U, Benson L, Edner M, Dahlstrom U, Lund LH. Association between use of statins and mortality in patients with heart failure and ejection fraction of ≥50. Circ Heart Fail. (2015) 8(5):862–70. doi: 10.1161/CIRCHEARTFAILURE.115.002143
119. Moreira DM, Vieira JL, Gottschall CA. The effects of METhotrexate therapy on the physical capacity of patients with ISchemic heart failure: a randomized double-blind, placebo-controlled trial (METIS trial). J Card Fail. (2009) 15(10):828–34. doi: 10.1016/j.cardfail.2009.06.439
120. Gong K, Zhang Z, Sun X, Zhang X, Li A, Yan J, et al. The nonspecific anti-inflammatory therapy with methotrexate for patients with chronic heart failure. Am Heart J. (2006) 151(1):62–8. doi: 10.1016/j.ahj.2005.02.040
121. Lam CSP, Lund LH, Shah SJ, Voors AA, Erlinge D, Saraste A, et al. Myeloperoxidase inhibition in heart failure with preserved or mildly reduced ejection fraction: SATELLITE trial results. J Card Fail. (2023) S1071-9164(23)00142-2. doi: 10.1016/j.cardfail.2023.04.003. [Epub ahead of print]
122. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Anti TNFTACHFI. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF therapy against congestive heart failure (ATTACH) trial. Circulation. (2003) 107(25):3133–40. doi: 10.1161/01.CIR.0000077913.60364.D2
123. Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the randomized etanercept worldwide evaluation (RENEWAL). Circulation. (2004) 109(13):1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2
124. Deswal A, Bozkurt B, Seta Y, Parilti-Eiswirth S, Hayes FA, Blosch C, et al. Safety and efficacy of a soluble P75 tumor necrosis factor receptor (enbrel, etanercept) in patients with advanced heart failure. Circulation. (1999) 99(25):3224–6. doi: 10.1161/01.CIR.99.25.3224
125. Bozkurt B, Torre-Amione G, Warren MS, Whitmore J, Soran OZ, Feldman AM, et al. Results of targeted anti-tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation. (2001) 103(8):1044–7. doi: 10.1161/01.CIR.103.8.1044
126. Van Tassell BW, Arena R, Biondi-Zoccai G, Canada JM, Oddi C, Abouzaki NA, et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol. (2014) 113(2):321–7. doi: 10.1016/j.amjcard.2013.08.047
127. Van Tassell BW, Trankle CR, Canada JM, Carbone S, Buckley L, Kadariya D, et al. IL-1 blockade in patients with heart failure with preserved ejection fraction. Circ Heart Fail. (2018) 11(8):e005036. doi: 10.1161/CIRCHEARTFAILURE.118.005036
128. Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, et al. Interleukin-1 blockade in recently decompensated systolic heart failure: results from REDHART (recently decompensated heart failure anakinra response trial). Circ Heart Fail. (2017) 10(11):e004373. doi: 10.1161/CIRCHEARTFAILURE.117.004373
129. Liu C, Liu K, Group C-AS. Cardiac outcome prevention effectiveness of glucocorticoids in acute decompensated heart failure: cOPE-ADHF study. J Cardiovasc Pharmacol. (2014) 63(4):333–8. doi: 10.1097/FJC.0000000000000048
130. Parrillo JE, Cunnion RE, Epstein SE, Parker MM, Suffredini AF, Brenner M, et al. A prospective, randomized, controlled trial of prednisone for dilated cardiomyopathy. N Engl J Med. (1989) 321(16):1061–8. doi: 10.1056/NEJM198910193211601
131. Givertz MM, Anstrom KJ, Redfield MM, Deswal A, Haddad H, Butler J, et al. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: the xanthine oxidase inhibition for hyperuricemic heart failure patients (EXACT-HF) study. Circulation. (2015) 131(20):1763–71. doi: 10.1161/CIRCULATIONAHA.114.014536
132. Hare JM, Mangal B, Brown J, Fisher C Jr., Freudenberger R, Colucci WS, et al. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol. (2008) 51(24):2301–9. doi: 10.1016/j.jacc.2008.01.068
133. Skudicky D, Bergemann A, Sliwa K, Candy G, Sareli P. Beneficial effects of pentoxifylline in patients with idiopathic dilated cardiomyopathy treated with angiotensin-converting enzyme inhibitors and carvedilol: results of a randomized study. Circulation. (2001) 103(8):1083–8. doi: 10.1161/01.CIR.103.8.1083
134. Sliwa K, Skudicky D, Candy G, Wisenbaugh T, Sareli P. Randomised investigation of effects of pentoxifylline on left-ventricular performance in idiopathic dilated cardiomyopathy. Lancet. (1998) 351(9109):1091–3. doi: 10.1016/S0140-6736(97)09338-0
135. Sliwa K, Woodiwiss A, Candy G, Badenhorst D, Libhaber C, Norton G, et al. Effects of pentoxifylline on cytokine profiles and left ventricular performance in patients with decompensated congestive heart failure secondary to idiopathic dilated cardiomyopathy. Am J Cardiol. (2002) 90(10):1118–22. doi: 10.1016/S0002-9149(02)02779-0
136. Sliwa K, Woodiwiss A, Kone VN, Candy G, Badenhorst D, Norton G, et al. Therapy of ischemic cardiomyopathy with the immunomodulating agent pentoxifylline: results of a randomized study. Circulation. (2004) 109(6):750–5. doi: 10.1161/01.CIR.0000112568.48837.60
137. Bahrmann P, Hengst UM, Richartz BM, Figulla HR. Pentoxifylline in ischemic, hypertensive and idiopathic-dilated cardiomyopathy: effects on left-ventricular function, inflammatory cytokines and symptoms. Eur J Heart Fail. (2004) 6(2):195–201. doi: 10.1016/j.ejheart.2003.09.005
138. Gullestad L, Aass H, Fjeld JG, Wikeby L, Andreassen AK, Ihlen H, et al. Immunomodulating therapy with intravenous immunoglobulin in patients with chronic heart failure. Circulation. (2001) 103(2):220–5. doi: 10.1161/01.CIR.103.2.220
139. McNamara DM, Holubkov R, Starling RC, Dec GW, Loh E, Torre-Amione G, et al. Controlled trial of intravenous immune globulin in recent-onset dilated cardiomyopathy. Circulation. (2001) 103(18):2254–9. doi: 10.1161/01.CIR.103.18.2254
140. Gullestad L, Ueland T, Fjeld JG, Holt E, Gundersen T, Breivik K, et al. Effect of thalidomide on cardiac remodeling in chronic heart failure: results of a double-blind, placebo-controlled study. Circulation. (2005) 112(22):3408–14. doi: 10.1161/CIRCULATIONAHA.105.564971
141. Hori M, Yamaguchi O. Is tumor necrosis factor-alpha friend or foe for chronic heart failure? Circ Res. (2013) 113(5):492–4. doi: 10.1161/CIRCRESAHA.113.302024
142. van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, et al. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. (2006) 113(16):1966–73. doi: 10.1161/CIRCULATIONAHA.105.587519
143. Baniaamam M, Handoko ML, Agca R, Heslinga SC, Konings TC, van Halm VP, et al. The effect of anti-TNF therapy on cardiac function in rheumatoid arthritis: an observational study. J Clin Med. (2020) 9(10):3145. doi: 10.3390/jcm9103145
144. Mantel A, Holmqvist M, Andersson DC, Lund LH, Askling J. Association between rheumatoid arthritis and risk of ischemic and nonischemic heart failure. J Am Coll Cardiol. (2017) 69(10):1275–85. doi: 10.1016/j.jacc.2016.12.033
145. McTiernan CF, Lemster BH, Frye C, Brooks S, Combes A, Feldman AM. Interleukin-1 beta inhibits phospholamban gene expression in cultured cardiomyocytes. Circ Res. (1997) 81(4):493–503. doi: 10.1161/01.RES.81.4.493
146. Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin-1 in heart disease. Circulation. (2013) 128(17):1910–23. doi: 10.1161/CIRCULATIONAHA.113.003199
147. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
148. Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-Inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. (2019) 139(10):1289–99. doi: 10.1161/CIRCULATIONAHA.118.038010
149. Hage C, Michaelsson E, Kull B, Miliotis T, Svedlund S, Linde C, et al. Myeloperoxidase and related biomarkers are suggestive footprints of endothelial microvascular inflammation in HFpEF patients. ESC Heart Fail. (2020) 7(4):1534–46. doi: 10.1002/ehf2.12700
Keywords: inflammation, heart failure, HFpEF, interleukins, cardiometabolic
Citation: Peh ZH, Dihoum A, Hutton D, Arthur JSC, Rena G, Khan F, Lang CC and Mordi IR (2023) Inflammation as a therapeutic target in heart failure with preserved ejection fraction. Front. Cardiovasc. Med. 10:1125687. doi: 10.3389/fcvm.2023.1125687
Received: 16 December 2022; Accepted: 15 June 2023;
Published: 29 June 2023.
Edited by:
Massimo Iacoviello, University of Foggia, ItalyReviewed by:
Bibhiti Das, University of Mississippi Medical Center, United States© 2023 Peh, Dihoum, Hutton, Arthur, Rena, Khan, Lang and Mordi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ify R. Mordi aS5tb3JkaUBkdW5kZWUuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.