 Myo Htet1,†
Myo Htet1,† Shunyao Lei2,†
Shunyao Lei2,† Sheetal Bajpayi1
Sheetal Bajpayi1 Asimina Zoitou2
Asimina Zoitou2 Myrsini Chamakioti3
Myrsini Chamakioti3 Emmanouil Tampakakis1,2,4*
Emmanouil Tampakakis1,2,4*
- 1Department of Medicine, Division of Cardiology, Johns Hopkins University, Baltimore, MD, United States
- 2Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD, United States
- 3School of Medicine, University of Patras, Patra, Greece
- 4Department of Genetic Medicine, Johns Hopkins University, Baltimore, MD, United States
Cardiomyopathies remain one of the leading causes of morbidity and mortality worldwide. Environmental risk factors and genetic predisposition account for most cardiomyopathy cases. As with all complex diseases, there are significant challenges in the interpretation of the molecular mechanisms underlying cardiomyopathy-associated genetic variants. Given the technical improvements and reduced costs of DNA sequence technologies, an increasing number of patients are now undergoing genetic testing, resulting in a continuously expanding list of novel mutations. However, many patients carry noncoding genetic variants, and although emerging evidence supports their contribution to cardiac disease, their role in cardiomyopathies remains largely understudied. In this review, we summarize published studies reporting on the association of different types of noncoding variants with various types of cardiomyopathies. We focus on variants within transcriptional enhancers, promoters, intronic sites, and untranslated regions that are likely associated with cardiac disease. Given the broad nature of this topic, we provide an overview of studies that are relatively recent and have sufficient evidence to support a significant degree of causality. We believe that more research with additional validation of noncoding genetic variants will provide further mechanistic insights on the development of cardiac disease, and noncoding variants will be increasingly incorporated in future genetic screening tests.
Introduction
Cardiomyopathies are disorders of the myocardium caused by genetic and environmental factors that eventually result in impaired cardiac function and heart failure (15). Depending on the specific effects in the function and morphology of the heart, and the isolated presence of arrhythmias, cardiomyopathies are divided into dilated, hypertrophic, restrictive, and arrhythmogenic (22). Dilated cardiomyopathy (DCM) is the most common cardiomyopathy affecting 1 in 250 individuals, followed by hypertrophic cardiomyopathy (HCM), which affects 1 in 500, and arrhythmogenic cardiomyopathy (ACM) encountered 1 in 5,000, while the prevalence of restrictive cardiomyopathy is even less common (40). About 30%–50% of cardiomyopathies are heritable, and the different types can have variable phenotypes, prognosis and causal mutations (16, 40, 70, 72, 81). HCM is primarily a disease of the sarcomere, as in up to 60% of patients, a pathogenic or likely pathogenic variant is detected in sarcomeric genes (22, 37). Beta-myosin heavy chain (MYH7) and myosin binding protein C3 (MYBPC3) are the most frequently affected genes, encoding for proteins of the thick sarcomeric filaments, and patients tend to exhibit disease onset in their forties. Other commonly affected genes in the thin filaments of the sarcomere are cardiac Troponin I (TNNI3) and cardiac Troponin T (TNNT2) (37). In contrast to HCM, the causative genes in DCM are functionally diverse. Titin (TTN) mutations represent 12%–25% of DCM patients and Lamin (LMNA) genetic variants represent the second most common mutations in DCM patients (40). Other genes that are associated with DCM are MYH7, TNNT2, Tropomyosin 1 (TPM1), Desmoplakin (DSP), RNA binding motif protein 20 (RBM20), and sodium voltage-gated channel alpha subunit 5 (SCN5A) (57). In arrhythmogenic cardiomyopathy (ACM), most pathogenic variants are in genes encoding desmosomal proteins such as Plakoglobin (JUP) (13, 41), DSP (48), Plakophilin-2 (PKP2), Desmoglein-2 (DSG2) and Desmocollin-2 (DSC2) (4, 51, 67). Finally, inherited restrictive cardiomyopathies are caused by mutations in sarcomeric genes such as cardiac troponin I, and less commonly by mutations in Desmin (DES) and Filamin C (FLNC) (7). It is worth noting that although cardiomyopathies are classified based on phenotypes manifested in the general population, the pathogenic mechanisms and phenotypic features among the various types of cardiomyopathies can overlap to a significant degree.
With the advancement of next generation sequencing and genome wide association studies (GWAS), our understanding of the genetic basis of cardiomyopathies has significantly improved. Multiple GWAS have identified susceptibility loci and variants associated with different types of cardiomyopathies (3, 23, 42, 68, 75). Most rare disease causal variants have been found within the coding region of the genome (81). For example, TTN coding variants usually lead to gene truncations and are viewed as the leading genetic causes in DCM patients (25). Contrarily, MYBPC3 truncating and MYH7 missense variants are the most pathogenic HCM mutations detected in next generation sequencing research studies (23). Although definitive causative genetic mutations have been identified for familial cardiomyopathies, in over half of the cases targeted genetic screening tests do not identify a contributing variant. This is because most of the current clinical genetic screening tests and earlier research studies relied heavily on whole exome sequencing (WES) or targeted sequencing of coding regions (45, 53, 54). Another explanation regarding the lack of focus in noncoding variants is that even in large meta-analyses, the power of variant detection is limited by variant frequency and penetrance, and lack of systemic interpretation. However, recent evidence from whole genome sequencing (WGS) supports a strong association between genetic variants within noncoding regions and cardiomyopathies (75). Also, emerging evidence corroborates the role of noncoding regulatory regions, where disruption of transcription factor binding sites within enhancers or promoters can alter the 3D chromatin structure and reduce target gene expression, which can be critical for disease (9, 34, 62, 64, 69, 74). Similarly, based on other studies variants within intronic or untranslated regions (UTRs) could also be involved in the pathogenesis of cardiomyopathies (6, 18, 19, 82). Furthermore, according to ClinVar, a significant percentage of non-coding variants in splice sites (∼60%) and UTRs (∼5%), are classified as pathogenic or likely pathogenic (www.ncbi.nlm.nih.gov/clinvar). In this review we will provide an overview of the role of noncoding genetic variants and their association with cardiomyopathies. We will specifically focus on variants within promoter, enhancer, untranslated, splice and intronic regions (Figure 1), where there is sufficient evidence to support a strong association with cardiac disease.
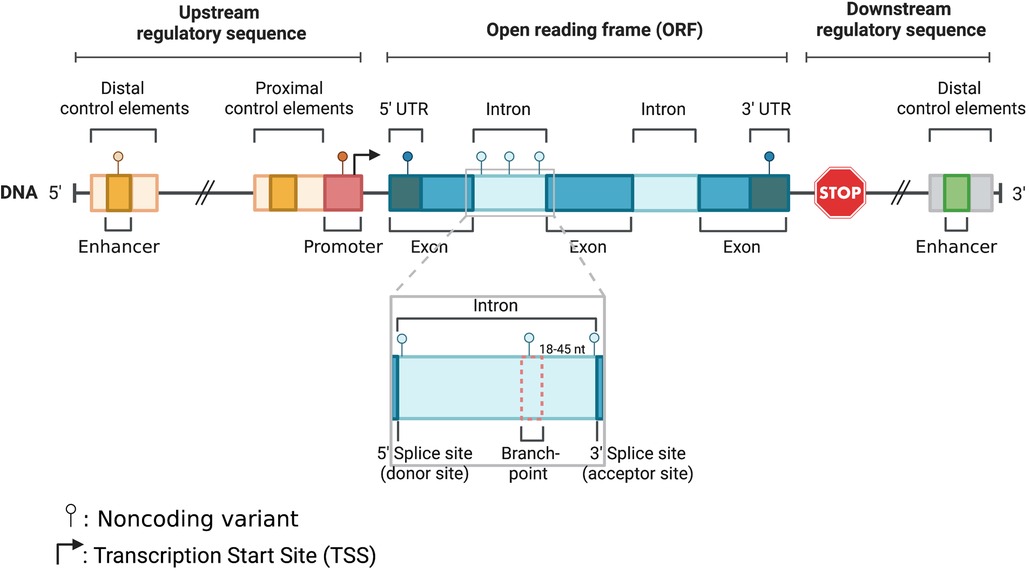
Figure 1. Schematic illustration of noncoding parts in the eukaryotic genome: enhancer, promoter, 5′ UTR, intron, and 3′ UTR.
Cardiomyopathy-associated genetic variants in enhancer and promoter regions
GWAS in heart failure patients have described a strong link with noncoding variants within transcriptional enhancer regions (3, 27, 38, 50, 60, 78) (Table 1). Enhancers are cis-regulatory DNA elements of the noncoding genome that recruit transcription factors to the promoter of target genes for temporal and tissue specific transcription regulation (10, 21, 76). Enhancers play key roles during growth and development, and many studies have shown that disease-associated variants are found within enhancers (52, 56, 74). Furthermore, thousands of cardiac specific enhancers have been described, and it is hypothesized that enhancers may have critical roles in cardiac diseases (1, 14, 24). This was also elegantly illustrated in a recent GWAS, where several regulatory variants in the promoter and enhancer regions were linked to cardiomyopathy (33).
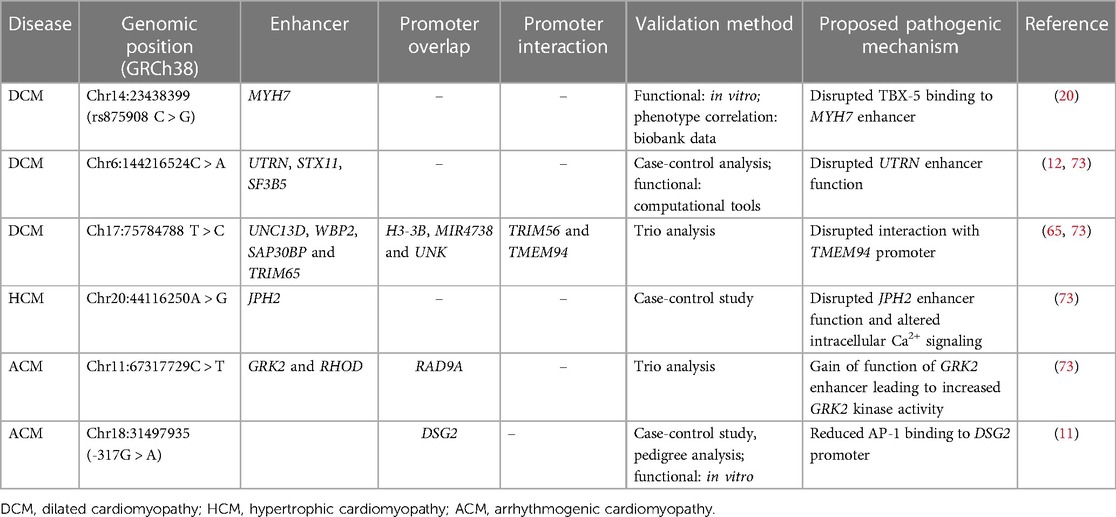
Table 1. Variants in enhancer and overlapping promoter regions of genes associated with different types of cardiomyopathies.
Dilated cardiomyopathy
A study by Gacita et al. demonstrated a potential association between a variant upstream of the MYH7 enhancer (rs875908) and DCM (20). Genetic deletion of this region in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) reduced MYH7 expression and increased the alpha to beta myosin heavy chain ratio. It is predicted that this region is bound by the transcription factors GATA4 and T-box transcription factor 5 (TBX5) and that this variant likely disrupts the TBX5 binding motif. Interestingly, data from the US Northwestern biobank revealed that the same variant is associated with cardiac function in patients with heart failure. The authors also identified more than 1,700 putative enhancer variants in genes important for cardiac function such as TNNT2, Natriuretic peptide A (NPPA), Gap junction protein alpha 5 (GJA5) and Myocyte enhancer factor 2A (MEF2A) etc. (20). Recently, a study led by Vadgama and colleagues analyzed WGS data of 143 parent-offspring trios from the Genomics England 100,000 Genomes project, and found novel noncoding de novo variants in enhancer and promoter regions associated with cardiomyopathy (73). Furthermore, this study reported on a DCM patient who harbored a variant within an enhancer region which was predicted to regulate multiple genes such as Utrophin (UTRN), Syntaxin 11 (STX11), and Splicing factor 3B subunit 5 (SF3B5). Indeed, animal studies have shown that UTRN deficient mice develop DCM (12). Curiously, another DCM patient from the same cohort harbored a variant in an enhancer region that regulates multiple genes such as Unc-13 homolog D (UNC13D), WW domain binding protein 2 (WBP2), SAP30 binding protein (SAP30BP) and Tripartite motif containing 65 (TRIM65). Importantly, this enhancer region interacts with the distal promoter region of Transmembrane protein 94 (TMEM94), and biallelic TMEM94 truncating mutation is associated with congenital heart defects (65).
Hypertrophic cardiomyopathy
In the study by Vadgama et al. one HCM patient was reported to carry a variant within the enhancer of the junctophilin-2 gene (JPH2) (73). Junctophilin-2 is a major structural protein in cardiomyocytes, where it also plays a critical role in calcium handling. Heart failure is commonly associated with downregulation of JPH2, and mutations in JPH2 can result in HCM (55, 65, 77). Thus, it is possible that disrupted JPH2 can alter cytoplasmic calcium signaling leading to cardiomyopathy.
Arrhythmogenic cardiomyopathy
ACM was linked with a variant within the enhancer of G protein coupled receptor kinase 2 (GRK2) and Ras homology family member D (RHOD) (73). Furthermore, it was shown that this enhancer region overlaps with the promoter of RAD9 checkpoint clamp component A (RAD9A). Importantly, GRK2 expression is upregulated in heart failure and GRK2 inhibition improves cardiac remodeling (59). Recently, a rare noncoding variant (DSG2-317G > A) in the DSG2 promoter, was also associated with ACM. This heterozygous variant segregated in two daughters of the proband and experimental validation showed a disrupted binding site for the transcription factor AP-1 (11).
Cardiomyopathy-associated genetic variants in untranslated regions
5′ and 3′ UTRs are key mediators of post-transcriptional gene regulation. They impact mRNA processing, localization and stability (30, 44, 61). They also regulate downstream translation through elements including upstream open reading frames (ORFs), internal ribosome entry sites (IRES), m7G cap, polyadenylation signals, microRNA binding sites and secondary structures (26, 30, 39, 44). With WGS and the advancement of global RNA structure probing in vivo (46), an increasing number of UTR variants have been discovered and studied for their association with diseases (32, 63, 80) (Table 2). In addition, several UTR variants appear to increase the risk for disease because of differential microRNA binding affinity to altered alleles and subsequent changes in gene expression regulation (47, 66). Interestingly, more than 45,000 microRNA binding sites in 3′ UTRs of human genes had been discovered by 2009 (17), and these regulate nearly half of the transcriptome (58).
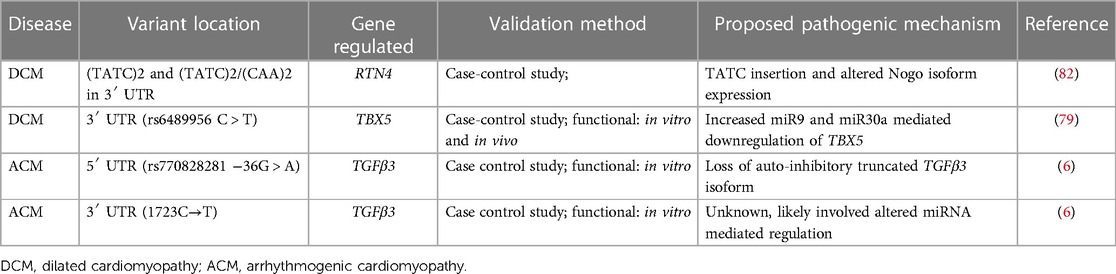
Table 2. Variants in 5′ and 3′ UTR of genes that are associated with different types of cardiomyopathies.
Dilated cardiomyopathy
In a study of 159 DCM patients and 215 control subjects, Zhou et al. showed an association of DCM with TATC and CAA insertion/deletion polymorphisms in 3′ UTR of Reticulon 4 (RTN4) gene (82). The gene codes for NOGO isoforms that have been previously linked with heart failure. (TATC)2 allele and (TATC)2/(TATC)2 genotypes were reported to be associated with an increased risk for DCM. However, there are still limited insights on the functional role of this mutation as it could not be matched with any known 3′ UTR functional motifs. In the Han Chinese population, a 3′ UTR variant in the TBX5 gene, increased the risk for congenital heart disease such as atrial and ventricular septal defects by nearly two-fold (79). The mutant allele has increased binding affinity to microRNAs-9 and 30a which decreases TBX5 expression.
CTG repeat expansion in the 3′ UTR of the myotonic dystrophy protein kinase (DMPK) gene has been linked to myotonic dystrophy type 1, a neuromuscular disease that can cause cardiac conduction disorders and cardiomyopathy (8). Transcription of this expansion results in CUG repeats that fold into hairpin loops, and sequestration of the nuclear protein muscleblind like (MBNL) and heterogeneous nuclear riboprotein 1 (hnRNPH1) splicing regulators leading to aberrant alternative splicing of numerous pre-mRNAs (35).
Arrhythmogenic cardiomyopathy
Through targeted genomic DNA sequencing in a small cohort in ACM patients, Beffagna et al. described two mutations in the 5′ and 3′ UTR regions of the transforming growth factor-beta3 (TGFβ3) gene (6). They reported a 5′UTR variant (c.−36G > A) in all clinically affected individuals of the family and in 3 asymptomatic relatives. TGFβ3 has 11 upstream open reading frames (uORFs). ATG at position -142 translates to a truncated 88 amino acid peptide that has been shown to inhibit the translation of full length TGFβ3 (2). It was also hypothesized that the 5′UTR variant may result in loss of function of the inhibitory truncated peptide isoform leading to increased TGFβ signaling and fibrosis. The disease mechanism for the 3′ UTR mutation found in one patient with ACM, has not been well studied (6).
Cardiomyopathy-associated genetic variants at deep intronic sites
Deep intronic variants are defined as those located more than 20 bp away from exons, and function to introduce aberrant splicing, distort transcription regulatory motifs, alter non-coding RNA activities, etc. (31). The identification and interpretation of these variants remains challenging due to the large size of introns and lack of consensus sequences (Table 3).
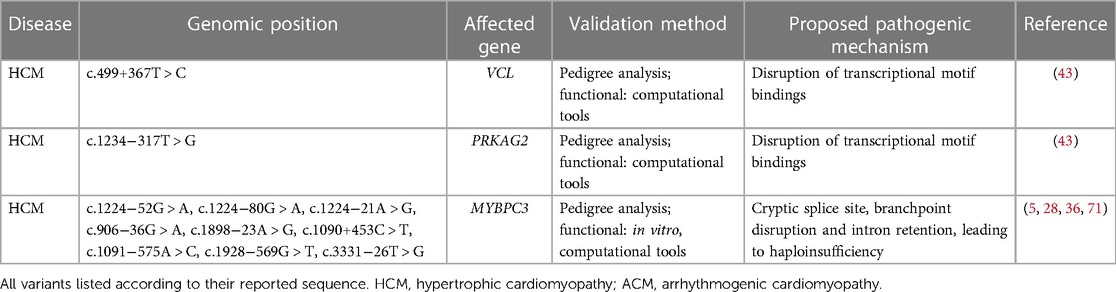
Table 3. Deep-intronic variants related to different cardiomyopathies and their proposed pathogenic mechanism.
Hypertrophic cardiomyopathy
Variants found in deep intronic regions of Vinculin (VCL) and protein kinase AMP-activated non-catalytic subunit gamma 2 (PRKAG2) were associated with HCM (43). In this study, both VCL (c.499 + 367T > C) and PRKAG2 variants (c.1234−317T > G) are predicted to be deleterious based on computational algorithms, and have a higher prevalence in patients with cardiomyopathy compared to the general population. Additionally, it has been demonstrated through pedigree analysis that the splice site mutation in MYBPC3 needs to co-exist with the VCL variant for disease manifestation. Moreover, these deep intronic variants appear to be enriched in binding sites for specific transcription factors such as FOS, JUN and EP300, and thus they may disturb the transcriptional regulation of cardiomyocytes.
As predicted by comprehensive computational analyses (SpliceAI - prediction tool for cryptic sites, and LabBranchoR - prediction tool for branch point at the splice site (49), the study found MYBPC3 harbors four splicing-site variants (three in intron 13: c.1224−52G > A, c.1224−80G > A, and c.1224−21A > G; one in intron 9: c.906−36G > A) which result in cryptic splice sites, while one variant (c.1898v23A > G) is likely disrupting a branchpoint in intron 19 and results in nonsense mediated decay-led haploinsufficiency in HCM patients (36). Moreover, an earlier report in two South Asian HCM cohorts revealed a rare pathogenic intronic MYBPC3 variant (c.1224−52G > A) where the mutation introduces a cryptic splice acceptor site in intron 13 and 50 nucleotide inclusion, which led to altered reading frame and premature termination codon at position 438 (p.Ser408fs*31) (23). Furthermore, a different study in a French HCM patient cohort suggested that deep intronic pathogenic MYBPC3 variants account for about 6% of HCM highlighting the need for routine MYBPC3 intronic NGS (29). Moreover, WGS and transcriptomic analysis identified three other MYBPC3 deep intronic variants (c.1090 + 453C > T, c.1091−575A > C, c.1928−569G > T) in HCM patients (5, 28). RNA analyses were performed to confirm aberrant splicing through the inclusion of cryptic exons in cardiomyocytes from patient-derived induced pluripotent stem cells (iPSC-CMs) and in a myectomy sample from one affected relative of the proband (c.1928−569G > T only). In addition, the role of one MYBPC3 intronic variant (c.3331−26T > G) was found to account for a genotype-negative proband in a family with a history of HCM (71). This variant segregates with two diseased descendants of the proband and it was found in unrelated HCM patients. Through splicing assays using minigene and patient's blood, the authors confirmed that the variant leads to the retention of intron 30 and thus protein haploinsufficiency.
Conclusions
Over the last two decades, numerous novel genetic variants have been linked with different types of cardiomyopathies. However, with more information comes greater responsibility, and given the variable penetrance of genetic mutations and lack of in-depth validation studies, attributing causality to most genetic variants has been challenging. Unsurprisingly, this becomes even more complicated when assessing noncoding genetic variants. Nevertheless, analysis of noncoding variants has witnessed tremendous advancements in sequencing techniques and the booming of artificial intelligence. The transition of common methodologies from traditional WES and pedigree analysis to more advanced sequencing incorporated with in silico studies and prediction algorithms, fuels the discovery of de novo noncoding variants with a potential disease-causing or modifying role in cardiomyopathies. In this review, we provided an overview of the progress in uncovering noncoding variants and their potential pathogenic mechanisms linked with different cardiomyopathies. Given the accumulation of more genetic information and computational tools, the role of some noncoding variants in key genes can be explored further leading to a better understanding of cardiomyopathy mechanisms. Additionally, it is now more obvious that further validation of noncoding genetic variants is missing. Both in silico analyses and prediction tools are limited by the population base of rare cardiomyopathies and the oversimplification of disease mechanisms, which result in discrepancies and inaccurate classifications. This supports the development and optimization of more research protocols such as standardized high-throughput in vitro testing platforms. Moreover, patient-derived iPSCs serve as an invaluable tool in studying or modeling disease mechanisms and thus could be exploited to functionally annotate and validate the causal roles of certain noncoding variants. In addition, the rapidly evolving field of gene editing with CRISPR technologies, would further accelerate the deeper interrogation of non-coding genetic variants.
Protein coding genes comprise only a small percentage of the entire human genome and frequently their mutations cannot fully account for the observed clinical phenotypes. Noncoding genetic variants have been previously overlooked and it is gradually becoming more obvious that they have more meaningful contributions to cardiac diseases. Therefore, incorporating noncoding variants in genetic screening and demonstrating a potential association with clinical prognosis is foreseeable and could be established as part of personalized medicine in the near future.
Author contributions
MH and SL: supervised, outlined and wrote the manuscript. SB, AZ and MC: wrote parts of the manuscript. ET: supervised and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The present work is supported by NHLBI (HL-145135), AHA (CDA34660077), W.W. Smith Charitable Trust, the Magic that Matters Fund, The Johns Hopkins University Catalyst Award, and MSCRF (2023-MSCRFL-5984).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Anene-Nzelu CG, Lee MCJ, Tan WLW, Dashi A, Foo RSY. Genomic enhancers in cardiac development and disease. Nat Rev Cardiol. (2022) 19(1):7–25. doi: 10.1038/s41569-021-00597-2
2. Arrick BA, Lee AL, Grendell RL, Derynck R. Inhibition of translation of transforming growth factor-beta 3 mRNA by its 5′ untranslated region. Mol Cell Biol. (1991) 11(9):4306–13. doi: 10.1128/mcb.11.9.4306-4313.1991
3. Arvanitis M, Tampakakis E, Zhang Y, Wang W, Auton A, Dutta D, et al. Genome-wide association and multi-omic analyses reveal ACTN2 as a gene linked to heart failure. Nat Commun. (2020) 11(1):1122. doi: 10.1038/s41467-020-14843-7
4. Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, et al. DSG2 Mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet. (2006) 79(1):136–42. doi: 10.1086/504393
5. Bagnall RD, Ingles J, Dinger ME, Cowley MJ, Ross SB, Minoche AE, et al. Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. (2018) 72(4):419–29. doi: 10.1016/j.jacc.2018.04.078
6. Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. (2005) 65(2):366–73. doi: 10.1016/j.cardiores.2004.10.005
7. Brodehl A, Gerull B. Genetic insights into primary restrictive cardiomyopathy. J Clin Med. (2022) 11(8):1–25. doi: 10.3390/jcm11082094
8. Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. (1992) 68(4):799–808. doi: 10.1016/0092-8674(92)90154-5
9. Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. (2001) 106(6):709–21. doi: 10.1016/s0092-8674(01)00493-7
10. Catarino RR, Stark A. Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev. (2018) 32(3-4):202–23. doi: 10.1101/gad.310367.117
11. Christensen AH, Andersen CB, Wassilew K, Svendsen JH, Bundgaard H, Brand SM, et al. Rare non-coding desmoglein-2 variant contributes to arrhythmogenic right ventricular cardiomyopathy. J Mol Cell Cardiol. (2019) 131:164–70. doi: 10.1016/j.yjmcc.2019.04.029
12. Chun JL, O'Brien R, Berry SE. Cardiac dysfunction and pathology in the dystrophin and utrophin-deficient mouse during development of dilated cardiomyopathy. Neuromuscul Disord. (2012) 22(4):368–79. doi: 10.1016/j.nmd.2011.07.003
13. Coonar AS, Protonotarios N, Tsatsopoulou A, Needham EW, Houlston RS, Cliff S, et al. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation. (1998) 97(20):2049–58. doi: 10.1161/01.cir.97.20.2049
14. Dickel DE, Barozzi I, Zhu Y, Fukuda-Yuzawa Y, Osterwalder M, Mannion BJ, et al. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat Commun. (2016) 7:12923. doi: 10.1038/ncomms12923
15. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. (2008) 29(2):270–6. doi: 10.1093/eurheartj/ehm342
16. Elliott PM. Classification of cardiomyopathies: evolution or revolution? J Am Coll Cardiol. (2013) 62(22):2073–4. doi: 10.1016/j.jacc.2013.10.008
17. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. (2009) 19(1):92–105. doi: 10.1101/gr.082701.108
18. Friedrich FW, Dilanian G, Khattar P, Juhr D, Gueneau L, Charron P, et al. A novel genetic variant in the transcription factor Islet-1 exerts gain of function on myocyte enhancer factor 2C promoter activity. Eur J Heart Fail. (2013) 15(3):267–76. doi: 10.1093/eurjhf/hfs178
19. Frisso G, Detta N, Coppola P, Mazzaccara C, Pricolo MR, D’Onofrio A, et al. Functional studies and in silico analyses to evaluate non-coding variants in inherited cardiomyopathies. Int J Mol Sci. (2016) 17(11):1–13. doi: 10.3390/ijms17111883
20. Gacita AM, Fullenkamp DE, Ohiri J, Pottinger T, Puckelwartz MJ, Nobrega MA, et al. Genetic variation in enhancers modifies cardiomyopathy gene expression and progression. Circulation. (2021) 143(13):1302–16. doi: 10.1161/circulationaha.120.050432
21. Gao T, Qian J. Enhanceratlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. (2020) 48(D1):D58–D64. doi: 10.1093/nar/gkz980
23. Harper AR, Bowman M, Hayesmoore JBG, Sage H, Salatino S, Blair E, et al. Reevaluation of the south Asian MYBPC3(Δ25 bp) intronic deletion in hypertrophic cardiomyopathy. Circ Genom Precis Med. (2020) 13(3):e002783. doi: 10.1161/circgen.119.002783
24. He A, Kong SW, Ma Q, Pu WT. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc Natl Acad Sci U S A. (2011) 108(14):5632–7. doi: 10.1073/pnas.1016959108
25. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. (2012) 366(7):619–28. doi: 10.1056/NEJMoa1110186
26. Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. (2016) 352(6292):1413–6. doi: 10.1126/science.aad9868
27. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. (2013) 155(4):934–47. doi: 10.1016/j.cell.2013.09.053
28. Holliday M, Singer ES, Ross SB, Lim S, Lal S, Ingles J, et al. Transcriptome sequencing of patients with hypertrophic cardiomyopathy reveals novel splice-altering variants in MYBPC3. Circ Genom Precis Med. (2021) 14(2):e003202. doi: 10.1161/circgen.120.003202
29. Janin A, Chanavat V, Rollat-Farnier PA, Bardel C, Nguyen K, Chevalier P, et al. Whole MYBPC3 NGS sequencing as a molecular strategy to improve the efficiency of molecular diagnosis of patients with hypertrophic cardiomyopathy. Hum Mutat. (2020) 41(2):465–75. doi: 10.1002/humu.23944
30. Jia L, Mao Y, Ji Q, Dersh D, Yewdell JW, Qian SB. Decoding mRNA translatability and stability from the 5′ UTR. Nat Struct Mol Biol. (2020) 27(9):814–21. doi: 10.1038/s41594-020-0465-x
31. Jung H, Lee KS, Choi JK. Comprehensive characterisation of intronic mis-splicing mutations in human cancers. Oncogene. (2021) 40(7):1347–61. doi: 10.1038/s41388-020-01614-3
32. Lee DSM, Park J, Kromer A, Baras A, Rader DJ, Ritchie MD, et al. Disrupting upstream translation in mRNAs is associated with human disease. Nat Commun. (2021) 12(1):1515. doi: 10.1038/s41467-021-21812-1
33. Lesurf R, Said A, Akinrinade O, Breckpot J, Delfosse K, Liu T, et al. Whole genome sequencing delineates regulatory, copy number, and cryptic splice variants in early onset cardiomyopathy. NPJ Genom Med. (2022) 7(1):18. doi: 10.1038/s41525-022-00288-y
34. Liu CF, Abnousi A, Bazeley P, Ni Y, Morley M, Moravec CS, et al. Global analysis of histone modifications and long-range chromatin interactions revealed the differential cistrome changes and novel transcriptional players in human dilated cardiomyopathy. J Mol Cell Cardiol. (2020) 145:30–42. doi: 10.1016/j.yjmcc.2020.06.001
35. Llamusí B, Artero R. Molecular effects of the CTG repeats in mutant dystrophia myotonica protein kinase gene. Curr Genomics. (2008) 9(8):509–16. doi: 10.2174/138920208786847944
36. Lopes LR, Barbosa P, Torrado M, Quinn E, Merino A, Ochoa JP, et al. Cryptic splice-altering variants in MYBPC3 are a prevalent cause of hypertrophic cardiomyopathy. Circ Genom Precis Med. (2020) 13(3):e002905. doi: 10.1161/circgen.120.002905
37. Marian AJ. Clinical interpretation and management of genetic variants. JACC Basic Transl Sci. (2020) 5(10):1029–42. doi: 10.1016/j.jacbts.2020.05.013
38. May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, et al. Large-scale discovery of enhancers from human heart tissue. Nat Genet. (2011) 44(1):89–93. doi: 10.1038/ng.1006
39. Mazumder B, Seshadri V, Fox PL. Translational control by the 3′-UTR: the ends specify the means. Trends Biochem Sci. (2003) 28(2):91–8. doi: 10.1016/s0968-0004(03)00002-1
40. McKenna WJ, Judge DP. Epidemiology of the inherited cardiomyopathies. Nat Rev Cardiol. (2021) 18(1):22–36. doi: 10.1038/s41569-020-0428-2
41. McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. (2000) 355(9221):2119–24. doi: 10.1016/s0140-6736(00)02379-5
42. Meder B, Rühle F, Weis T, Homuth G, Keller A, Franke J, et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J. (2014) 35(16):1069–77. doi: 10.1093/eurheartj/eht251
43. Mendes de Almeida R, Tavares J, Martins S, Carvalho T, Enguita FJ, Brito D, et al. Whole gene sequencing identifies deep-intronic variants with potential functional impact in patients with hypertrophic cardiomyopathy. PLoS One. (2017) 12(8):e0182946. doi: 10.1371/journal.pone.0182946
44. Mignone F., Gissi C., Liuni S., Pesole G. (2002). Untranslated regions of mRNAs. Genome Biol, 3(3), Reviews0004. doi: 10.1186/gb-2002-3-3-reviews0004
45. Minoche AE, Horvat C, Johnson R, Gayevskiy V, Morton SU, Drew AP, et al. Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Genet Med. (2019) 21(3):650–62. doi: 10.1038/s41436-018-0084-7
46. Mortimer SA, Kidwell MA, Doudna JA. Insights into RNA structure and function from genome-wide studies. Nat Rev Genet. (2014) 15(7):469–79. doi: 10.1038/nrg3681
47. Moszyńska A, Gebert M, Collawn JF, Bartoszewski R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. (2017) 7(4):1–13. doi: 10.1098/rsob.170019
48. Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. (2000) 9(18):2761–6. doi: 10.1093/hmg/9.18.2761
49. Paggi JM, Bejerano G. A sequence-based, deep learning model accurately predicts RNA splicing branchpoints. Rna. (2018) 24(12):1647–58. doi: 10.1261/rna.066290.118
50. Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. (2013) 110(44):17921–6. doi: 10.1073/pnas.1317023110
51. Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. (2006) 113(9):1171–9. doi: 10.1161/circulationaha.105.583674
52. Plank JL, Dean A. Enhancer function: mechanistic and genome-wide insights come together. Mol Cell. (2014) 55(1):5–14. doi: 10.1016/j.molcel.2014.06.015
53. Pua CJ, Bhalshankar J, Miao K, Walsh R, John S, Lim SQ, et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J Cardiovasc Transl Res. (2016) 9(1):3–11. doi: 10.1007/s12265-016-9673-5
54. Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. (2014) 16(8):601–8. doi: 10.1038/gim.2013.204
55. Reynolds JO, Quick AP, Wang Q, Beavers DL, Philippen LE, Showell J, et al. Junctophilin-2 gene therapy rescues heart failure by normalizing RyR2-mediated Ca(2+) release. Int J Cardiol. (2016) 225:371–80. doi: 10.1016/j.ijcard.2016.10.021
56. Rickels R, Shilatifard A. Enhancer logic and mechanics in development and disease. Trends Cell Biol. (2018) 28(8):608–30. doi: 10.1016/j.tcb.2018.04.003
57. Rosenbaum AN, Agre KE, Pereira NL. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol. (2020) 17(5):286–97. doi: 10.1038/s41569-019-0284-0
58. Salman OF, El-Rayess HM, Abi Khalil C, Nemer G, Refaat MM. Inherited cardiomyopathies and the role of mutations in non-coding regions of the genome. Front Cardiovasc Med. (2018) 5:77. doi: 10.3389/fcvm.2018.00077
59. Schlegel P, Reinkober J, Meinhardt E, Tscheschner H, Gao E, Schumacher SM, et al. G protein-coupled receptor kinase 2 promotes cardiac hypertrophy. PLoS One. (2017) 12(7):e0182110. doi: 10.1371/journal.pone.0182110
60. Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. (2011) 43(4):333–8. doi: 10.1038/ng.784
61. Singer RH. RNA Zipcodes for cytoplasmic addresses. Curr Biol. (1993) 3(10):719–21. doi: 10.1016/0960-9822(93)90079-4
62. Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum Mol Genet. (2012) 21(14):3255–63. doi: 10.1093/hmg/dds165
63. Soukarieh O, Meguerditchian C, Proust C, Aïssi D, Eyries M, Goyenvalle A, et al. Common and rare 5′UTR variants altering upstream open Reading frames in cardiovascular genomics. Front Cardiovasc Med. (2022) 9:841032. doi: 10.3389/fcvm.2022.841032
64. Spurrell CH, Barozzi I, Kosicki M, Mannion BJ, Blow MJ, Fukuda-Yuzawa Y, et al. Genome-wide fetalization of enhancer architecture in heart disease. Cell Rep. (2022) 40(12):111400. doi: 10.1016/j.celrep.2022.111400
65. Stephen J, Maddirevula S, Nampoothiri S, Burke JD, Herzog M, Shukla A, et al. Bi-allelic TMEM94 truncating variants are associated with neurodevelopmental delay, congenital heart defects, and distinct facial dysmorphism. Am J Hum Genet. (2018) 103(6):948–67. doi: 10.1016/j.ajhg.2018.11.001
66. Steri M, Idda ML, Whalen MB, Orrù V. Genetic variants in mRNA untranslated regions. Wiley Interdiscip Rev RNA. (2018) 9(4):e1474. doi: 10.1002/wrna.1474
67. Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. (2006) 79(5):978–84. doi: 10.1086/509122
68. Tadros R, Francis C, Xu X, Vermeer AMC, Harper AR, Huurman R, et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet. (2021) 53(2):128–34. doi: 10.1038/s41588-020-00762-2
69. Tan WLW, Anene-Nzelu CG, Wong E, Lee CJM, Tan HS, Tang SJ, et al. Epigenomes of human hearts reveal new genetic variants relevant for cardiac disease and phenotype. Circ Res. (2020) 127(6):761–77. doi: 10.1161/circresaha.120.317254
70. Tobita T, Nomura S, Fujita T, Morita H, Asano Y, Onoue K, et al. Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci Rep. (2018) 8(1):1998. doi: 10.1038/s41598-018-20114-9
71. Torrado M, Maneiro E, Lamounier Junior A, Fernández-Burriel M, Sánchez Giralt S, Martínez-Carapeto A, et al. Identification of an elusive spliceogenic MYBPC3 variant in an otherwise genotype-negative hypertrophic cardiomyopathy pedigree. Sci Rep. (2022) 12(1):7284. doi: 10.1038/s41598-022-11159-y
72. Towbin JA. Inherited cardiomyopathies. Circ J. (2014) 78(10):2347–56. doi: 10.1253/circj.cj-14-0893
73. Vadgama N, Ameen M, Sundaram L, Gaddam S, Gifford C, Nasir J, et al. De novo and inherited variants in coding and regulatory regions in genetic cardiomyopathies. Hum Genomics. (2022) 16(1):55. doi: 10.1186/s40246-022-00420-0
74. van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. (2012) 122(7):2519–30. doi: 10.1172/jci62613
75. Villar D, Frost S, Deloukas P, Tinker A. The contribution of non-coding regulatory elements to cardiovascular disease. Open Biol. (2020) 10(7):200088. doi: 10.1098/rsob.200088
76. Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. (2009) 461(7261):199–205. doi: 10.1038/nature08451
77. Walsh R, Offerhaus JA, Tadros R, Bezzina CR. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. (2022) 19(3):151–67. doi: 10.1038/s41569-021-00608-2
78. Wamstad JA, Wang X, Demuren OO, Boyer LA. Distal enhancers: new insights into heart development and disease. Trends Cell Biol. (2014) 24(5):294–302. doi: 10.1016/j.tcb.2013.10.008
79. Wang F, Liu D, Zhang RR, Yu LW, Zhao JY, Yang XY, et al. A TBX5 3′UTR variant increases the risk of congenital heart disease in the Han Chinese population. Cell Discov. (2017) 3:17026. doi: 10.1038/celldisc.2017.26
80. Whiffin N, Karczewski KJ, Zhang X, Chothani S, Smith MJ, Evans DG, et al. Characterising the loss-of-function impact of 5′ untranslated region variants in 15,708 individuals. Nat Commun. (2020) 11(1):2523. doi: 10.1038/s41467-019-10717-9
81. Yamada T, Nomura S. Recent findings related to cardiomyopathy and genetics. Int J Mol Sci. (2021) 22:22. doi: 10.3390/ijms222212522
Keywords: cardiomyopathy, noncoding variants, enhancers, promoters, untranslated regions, intronic variants
Citation: Htet M, Lei S, Bajpayi S, Zoitou A, Chamakioti M and Tampakakis E (2023) The role of noncoding genetic variants in cardiomyopathy. Front. Cardiovasc. Med. 10:1116925. doi: 10.3389/fcvm.2023.1116925
Received: 6 December 2022; Accepted: 4 May 2023;
Published: 22 May 2023.
Edited by:
John Lynn Jefferies, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Paul Cheng, Stanford Healthcare, United StatesRoddy Walsh, Academic Medical Center, Netherlands
Elena V. Zaklyazminskaya, Russian National Research Center of Surgery named after B.V. Petrovsky, Russia
© 2023 Htet, Lei, Bajpayi, Zoitou, Chamakioti and Tampakakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emmanouil Tampakakis ZXRhbXBhazFAamhtaS5lZHU=
†These authors have contributed equally to this work