 Ding Wang
Ding Wang Xinyu Hao
Xinyu Hao Longyuan Jia1,2
Longyuan Jia1,2 Bo Jiang
Bo Jiang Shijie Xin
Shijie Xin- 1Department of Vascular Surgery, The First Affiliated Hospital of China Medical University, Shenyang, Liaoning, China
- 2Key Laboratory of Pathogenesis, Prevention and Therapeutics of Aortic Aneurysm, Shenyang, Liaoning, China
As China’s population enters the aging stage, the threat of abdominal aortic aneurysm (AAA) mainly in elderly patients is becoming more and more serious. It is of great clinical significance to study the pathogenesis of AAA and explore potential therapeutic targets. The purpose of this paper is to analyze the pathogenesis of AAA from the perspective of cellular senescence: on the basis of clear evidence of cellular senescence in aneurysm wall, we actively elucidate specific molecular and regulatory pathways, and to explore the targeted drugs related to senescence and senescent cells eliminate measures, eventually improve the health of patients with AAA and prolong the life of human beings.
Introduction
Abdominal aortic aneurysm (AAA) is locally weak and aneurysm-like dilatation of the abdominal aorta, diameter > 3 cm or more than 1.5 times the normal diameter (1). AAA is a common disease among the elderly, and the incidence of AAA increases with age. In one study, the incidence of AAA is reported to be 55 per 100,000 person-years in males aged 65–74, 112 per 100,000 person-years in males aged 75–85, and 298 per 100,000 person-years in males older than 85 (2). Most AAA patients are asymptomatic and are discovered accidentally during a physical exam or ultrasound screening. However, rupture or precursor rupture may occur when patients present with symptoms such as lumbago and abdominal pain. The in-hospital mortality rate of rupture is about 40% (3), while the out-of-hospital mortality rate can be as high as 90% (4), resulting in about 150,000–200,000 deaths globally (5), which is a serious threat to patients’ health. Currently there is no effective method to inhibit AAA progress in the treatment of clinical drug. Usually, surgical treatment (open surgery or endovascular stent repair) is the only treatment for AAA patients whose diameter is greater than 5.5 cm (greater than 5.0 cm for women) or increases by more than 1.0 cm every year (6). Patients who fail to meet the surgical indications need long-term follow-up (7), which brings heavy economic burden and psychological pressure to patients. Therefore, finding and developing potential therapeutic targets and drugs to delay aneurysm progression or prevent AAA rupture is of great clinical significance and can significantly extend human life expectancy.
The mechanism of the occurrence and development of AAA has not been fully clarified, mainly including oxidative stress, immune inflammatory response, apoptosis of vascular smooth muscle cells (VSMCs) and vascular aging (8–10). The main risk factors for AAA are age over 65, male, smoking habit and family history (11) (Figure 1). With the progress of the population aging in China, the elderly population in 2050 is expected to appear the big bang, 65 years of age or older population will reach 400 million (accounts for 26.9% of the total population), more than 80-year-old aging population will reach 150 million (12), China will become one of the countries with the highest proportion of elderly people in the world, and which will inevitably cause a series of social and economic challenges (13). Therefore, it is important to elucidate the specific mechanisms involved in the pathogenesis of age-related AAA, and to provide potential therapeutic targets for the development of novel therapeutic agents that inhibit the expansion and rupture of AAA.
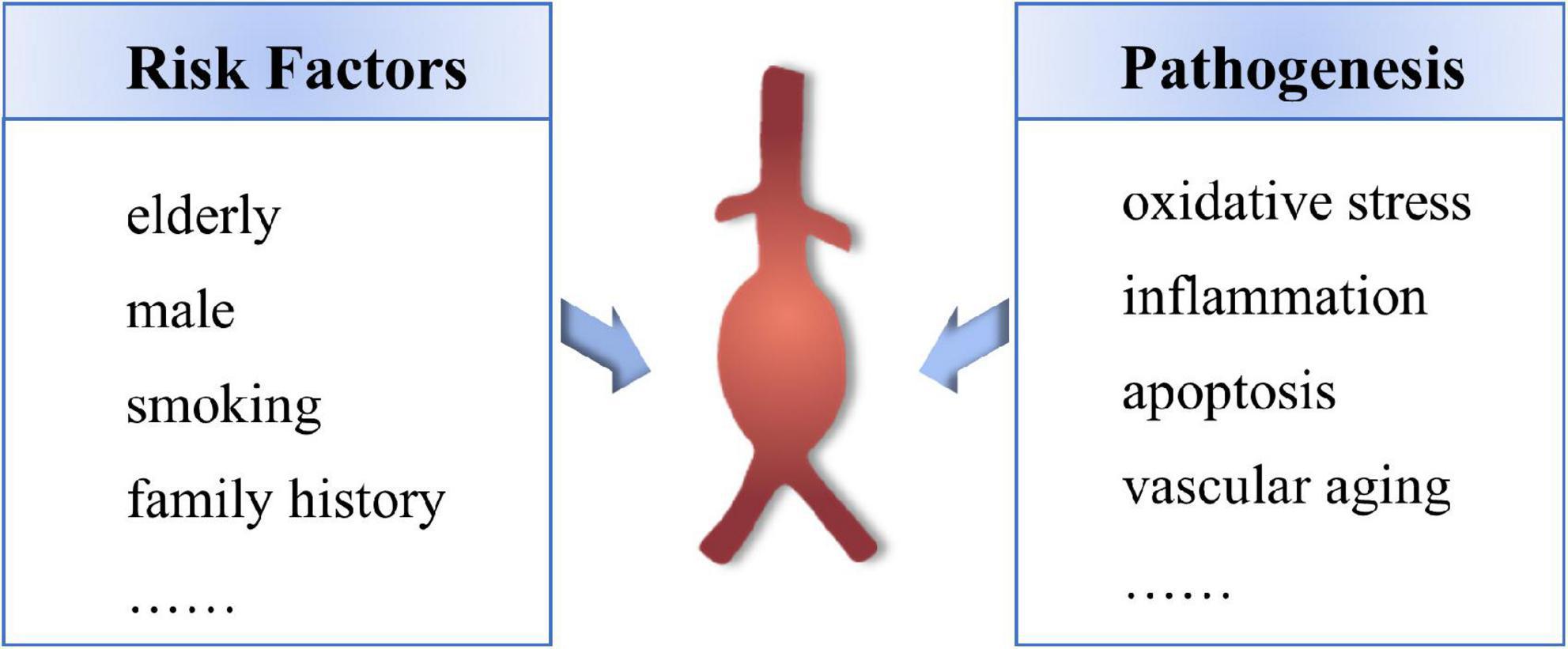
Figure 1. The remain risk factors and pathogenesis of AAA.
Hayflick and Moorhead were the first to describe “Replicative senescence (RS)” in 1961, which referred to the limited ability of human cells to proliferate (cell cycle arrest). Senescence is the irreversible loss and depletion of the cell’s ability to differentiate and divide (14). The “telomere hypothesis” emerged in the 1890s as one of the most important molecular mechanisms of RS, generating much scientific interest in the study of senescence (15). Later, it was proved that senescence may be caused by other genetic damage, such as DNA damage, chromosomal aberrations and chromatin aggregation (16, 17). Now, it is recognized that senescence also acts as a defense mechanism in response to various stresses, including telomere wear, oncogene activation, tumor suppressor gene inactivation, oxidative stress, mitochondrial dysfunction and DNA damage (radiation, chemicals, and ROS) (18, 19). This process is classified as stress-induced premature senescence (SIPS). RS is characterized by telomere shortening. Telomeres are regions thousands of bases long in DNA, covering the ends of chromosomes, and they play an essential role in maintaining the integrity and stability of DNA molecules (20). Because DNA polymerase cannot replicate the end of the DNA molecule completely each time, the DNA molecule loses 50–200 base pairs of telomeres at the 3′ end after each replication (21). Hayflick et al. observed a limit, known as the Hayflick limit, which was the maximum number of cell divisions (about 50–70) (14). When the Hayflick limit is reached, the integrity of DNA molecules is broken and the ends of chromosomes are exposed, leading to replicative senescence (22). And SIPS is triggered by telomere-independent DNA damage responses (DDR) caused by both internal and external stressors, which is often involved in the activation of cycle-dependent kinase inhibitor (CDKI) p21 and exogenous tumor suppressors (p16 and Rb) (23). CDK mediates the transition among different stages of the cell cycle, which stagnates when interrupted by DNA damage. Once stopped, the cells face three outcomes: after DNA damage is repaired, cells can re-enter the cell cycle; If not, cells die and are cleared; If still in stasis, they become senescent cells. Thus, cell cycle arrest is the first step of senescence (24).
Hallmarks of cellular senescence
Although there is no consensus on the senescence process and related mechanisms, the scientific community has proposed a senescent cell phenotype that is characterized by specific changes during cellular senescence occurred. In addition to cell cycle arrest, senescent cells undergo morphological, biochemical and functional changes, which are signs of cellular senescence (25) (Figure 2).
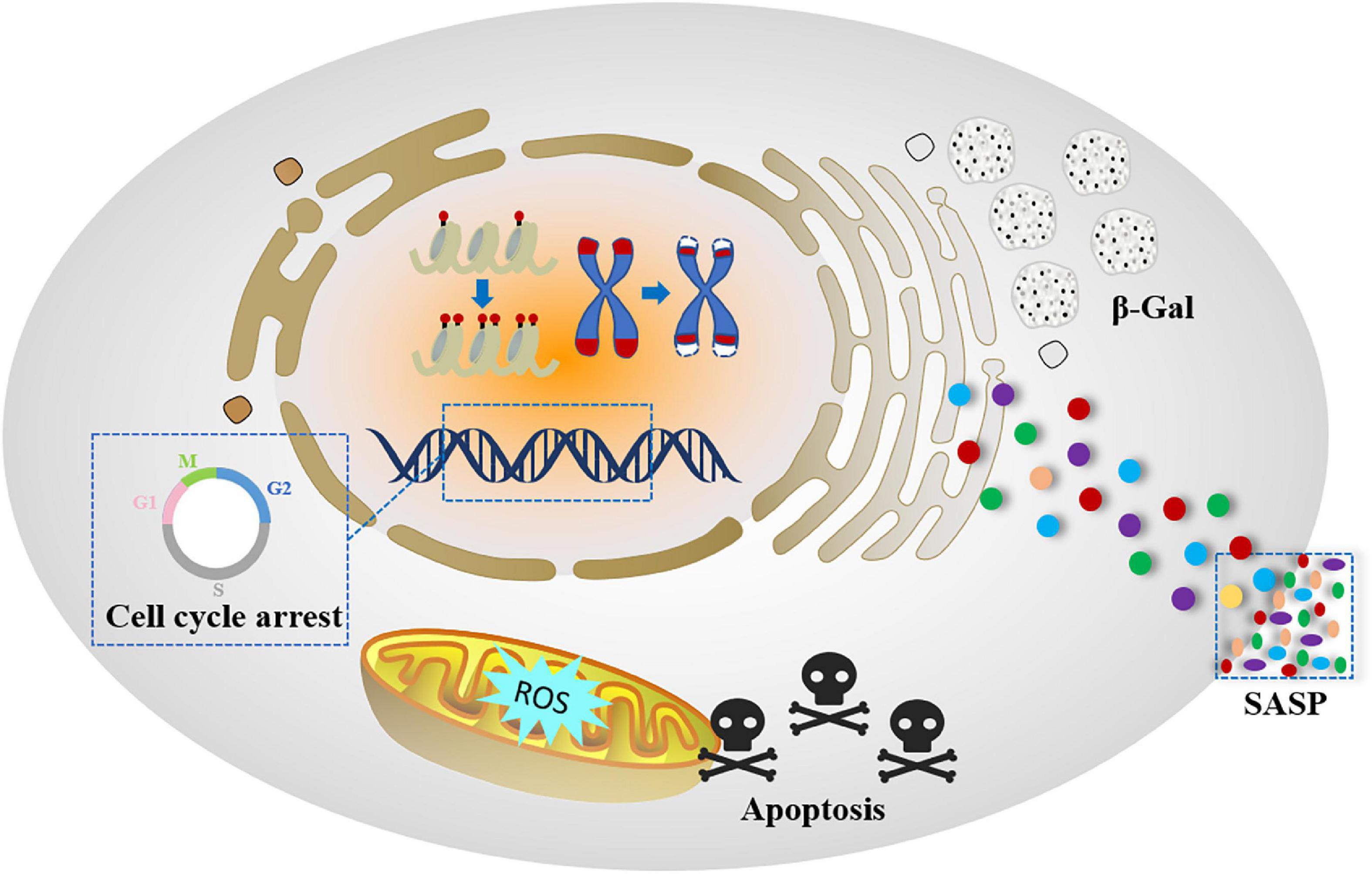
Figure 2. Pattern diagram of senescent cell hallmarks.
Morphogenesis and chromatin remodeling
Senescent cells undergo significant morphological and structural changes, including enlargement, flattening, nuclear enlargement, multinucleation (26) and changing the composition of plasma membrane (27, 28). These seemingly simple changes help establish and maintain the senescent state of cells.
Senescent cells have also been observed with significant chromatin related changes, most notably the formation of senescence associated heterochromia lesions (SAHF), which were first described by Lowe et al. (29). SAHF, as a DNase-resistant and DAPI-dense subnuclear cell structure, enriches in histone modification (H3K9me) associated with transcription inhibition (30). The formation of SAHF leads to transcriptional inhibition of E2F target genes, which triggers permanent cell cycle arrest (31). The emergence and function of SAHF depend on an effective INK4A-Rb pathway, since INK4A inactivation prevents SAHF formation and Rb is recruited into proliferation-related genes to inhibit them (32). However, recent research results challenged some of earlier explanations. It has been reported that cellular senescence can be robustly established in the absence of SAHF. One study found that telomere shortening, ionizing radiation, and prolonged exposure to hydroxyurea or etoposide also can trigger senescence without the distinct formation or the typical heterochromatin markers of SAHF (33). Surprisingly, SAHF formation was observed under similar conditions but with different cell lines, which may be related to INK4A induction (31).
Senescence-associated-β galactosidase (SA-β-Gal)
Detection of SA-β-Gal activity is the most commonly used senescence discrimination method (27). Endogenous β-Gal is the product of GLB1 gene encoding (34), and the transcription process is negatively regulated by the NOTCH1 signaling pathway (35). The lysosome content in senescent cells increases significantly, leading to a significant increase in the endogenous β-Gal activity of lysosomes (34). Lysosomal β-galactosidase activity is usually most active at pH 4.0, but which can also be detected at pH 6.0 in senescent cells (36). Therefore, SA-β-Gal activity can reflect lysosome β -galactosidase activity, thus revealing the increase of lysosome content in senescent cells. Although these histochemical and immunohistochemical staining methods are widely performed, SA-β-Gal activity is not only present in senescent cells (37). Studies have found that cells lacking GLB1 gene undergo unimpeded senescence (38). In addition, osteoclasts and macrophages themselves have higher levels of β-Gal activity (39, 40). Therefore, SA-β-Gal activity assay combined with other markers may be more reliable for determining cellular senescence.
Senescence-associated secretory phenotype
Senescent cells influence surrounding environment and communicate with their neighbors by producing a complex mixture of secreted factors that change the behavior of both themselves and nearby non-senescent cells (41–43).
The attribute of senescent cells secreting pro-inflammatory cytokines, chemokines, angiogenic factors, and proteases is referred to as senescence-associated secretory phenotype (SASP) (44). SASP can have positive or negative effects on the organism. When it is excessively elevated or persistent, SASP causes local and potentially systemic inflammation, destroys tissue structure, and stimulates adjacent cells to senescence. In contrast, local or transient SASP stimulation may promote recovery of tissue damage (44). Cytokines IL-6 and IL-8 in SASP enhance the growth stagnation of senescent cells, which is detrimental (45). MMPs, the component of SASP, can limit fibrosis after liver injury or during skin wound healing, which is beneficial (46, 47). SASP enhances and spreads senescence through autocrine and paracrine. IL-1α, TGF-β and IL-6 promote senescence in a cellular autonomous manner, while many other SASP factors act in a non-cellular autonomous manner, thus altering the behavior of neighboring cells (48, 49).
SASP is highly heterogeneous and regulated at multiple levels (50). Transcriptional level regulation: SASP is mainly regulated by activation of nuclear factor kappa-B (NF-κB) and CCAAT/enhancer binding protein β (C/EBPβ) transcription factors (47). Janus kinase (JAK), transcription activator (STAT) pathways and NOTCH signaling are also involved in regulating SASP expression (51). Epigenetic regulation: Bromine domain and extra-terminal (BET) family protein BRD4 recruited the super enhancer element adjacent to SASP gene, resulting in the remodeling of the enhancer (52). Decreased histone H3K9me2 (53) and histone deacetylase silencing information regulator 2 related enzyme 1 (SIRT1) (54), increased expression of histone variant macroH2A1 (55) in the promoter region of key SASP factors are involved in the regulation of SASP. Histone variant H2AJ accumulated in senescent cells is also involved in the induction of SASP (56). Other epigenetic mediators, such as histone lysine-n-methyltransferase 2A (MLL1 and HMGB2), also promote SASP production by keeping their gene loci open and active (57, 58). Post-transcriptional regulation: Rapamycin (mTOR) is a key regulator of protein translation in senescent cells. MTOR mainly regulates SASP through two mechanisms. On the one hand, mTOR promotes IL-1α translation and activates NF-κB and C/EBPβ (59). On the other hand, mTOR indirectly inhibits the RNA-binding protein ZFP36L1, preventing it from degrading the mRNA encoding SASP factor (60).
P53/p21 and p16/Rb pathways
The p53/p21 signaling pathway is activated in response to DNA damage caused by oncogenic factor mutations, telomere wear or oxidative stress, thereby inducing cellular senescence (61, 62). The activation of p53 is regulated by post-translational modifications, including ubiquitination, methylation, phosphorylation and acetylation (63). Mouse double minute 2 (MDM2) is an E3 ubiquitin ligase that plays an important role in p53 degradation as a negative regulator (64). Activated p53 regulates the expression of numerous anti-proliferation genes and participates in cellular senescence (65, 66). In addition, p53 is an important tumor suppressor protein that regulates p21 transcription process (67). P21 is a cyclin kinase (CDK) inhibitor that inactivates all CDKs, thereby inhibiting cell cycle progression. Specifically, p21 inhibits the CDK complex activity by interacting with two cyclin-binding mods (Cy1 and Cy2), resulting in the removal of phosphorylation of the Rb protein family and subsequent binding to E2F to form the DREAM complex, eventually which leads to cell cycle stagnation (68, 69). P21 also promotes senescence by down-regulating mitotic gene cyclin E2 and up-regulating senescence-related gene fibronectin-1 (70). However, p21 can also be activated in a p53-independent manner, induced by pathway such as TGF-β and by using Sp1 as a main transcription factor (71–73).
P14 (ARF), p15 and p16 are three tumor suppressor proteins encoded by INK4/ARF gene (74). ARF regulates the stability of p53 by binding to MDM2, whereas p53 regulates the expression of ARF through a negative feedback mechanism (75, 76). P16 directly binds to CDK4/6 and blocks the formation of CDK4/6 complex, thereby inhibiting Rb phosphorylation and facilitating E2F target gene expression (77). Rb family is one of the main targets of CDK complex, and its most significant function is to bind and inactivate E2F complex and prevent transcription of E2F target genes. Dephosphorylated pRb binds to E2F to form the Rb-E2F complex that inhibits transcription of genes involved in cell cycle progression by binding to the promoter region of E2F target genes (78). This inhibitory mechanism is eliminated by CDK2-mediated Rb hyperphosphorylation, which releases E2F and promotes S-phase gene transcription and cell cycle progression (79). In addition, p16 has also been reported to induce cellular senescence in a manner independent of p53 (80).
Apoptosis resistance
Apoptosis is a kind of spontaneous and orderly death controlled by genes, which helps to maintain homeostasis (81, 82). Various pro-apoptotic signals initially stimulate different signaling pathways and eventually converge into a common mechanism: up-regulating the expression of cysteine proteases, activating caspase and executioner caspase, and which ultimately leads to degradation of cell components (83). This mechanism is negatively regulated by a variety of genes, the most common of which is the Bcl-2 family (84). Studies have shown that anti-apoptotic factors Bcl-W, Bcl-XL and Bcl-2 of the Bcl-2 family are significantly upregulated in senescence induced cells, and these proteins can successfully induce cellular senescence after being removed (85, 86). However, there are still great differences in apoptosis resistance among different cell types (87).
Metabolic reprogramming
Senescent cells undergo a series of metabolic changes related to increased oxidative stress and oxidative protein accumulation, impaired protein homeostasis and specific metabolic pathways (88). Increased metabolic activity is considered to be an adaptive change, and senescent cells need metabolic reprogramming to meet their nutritional and energy requirements in the senescent state, such as increased SASP secretion, oxidative stress and endoplasmic reticulum stress (87). In addition, studies have also found that autophagy in senescent cells is combined with protein synthesis to overcome the amino acid shortage (89). Meanwhile, the combination of metabolomics and proteomics has revealed that senescent cells can flexibly use different metabolic pathways to improve survival and support function. Specifically, the tricarboxylic acid (TCA) cycle, pentose phosphate and nucleotide pathways are upregulated in senescent cells, while the fatty acid (FA) pathway is downregulated (90).
Relationship between cellular senescence and abdominal aortic aneurysm
AAA is a degenerative cardiovascular disease, partly characterized by local weakness of the abdominal aorta, which may be an objective manifestation of changes in the internal microenvironment of local cellular components and extracellular matrix components, namely, the result of vascular remodeling. In the process of vascular remodeling, the most significant feature is the loss of VSMCs and infiltration of immune inflammation, which is very consistent with the two typical characteristics of senescent cells, the reduced proliferation ability and SASP, suggesting that cellular senescence may play an important role in the pathogenesis of AAA. It can’t be ignored that AAA is more common in elderly patients, and elderly patients are the main population of aging. Below, we will elaborate on the evidence and mechanisms of local and circulatory senescence in patients with AAA (Table 1).

Table 1. Evidence of cell senescence in the aneurysm wall.
Evidence and mechanisms of cellular senescence in abdominal aortic aneurysm
A large number of studies have found that the formation and progress of AAA is closely related to cellular senescence. Liao et al. obtained VSMCs derived from paired AAA and adjacent (non-aneurysmal) inferior mesenteric artery (IMA) using explants and observed their differences during successive passages in culture. The results showed that VSMCs derived from AAA were larger and rounder in appearance than VSMCs derived from IMA, with significantly reduced proliferation ability and limited in vitro life (91). This is the first report of signs of senescence in AAA medial VSMCs, which lays the foundation for us to explore the correlation between the two. VSMCs senescence characterization has also been confirmed in human end-stage aneurysm tissues and age-matched saphenous vein tissues (92). Subsequent study has shown that, compared with the control group, the expression of telomerase in endothelial cells of AAA patients is significantly reduced. The association persisted after adjusting for age, sex, coronary artery disease, hypercholesterolemia, hypertension and smoking (93). The finding further suggests that cellular senescence is closely related to AAA, and we have reason to believe that cellular senescence is a key link in the pathogenesis of AAA. Meanwhile, it has been reported that circulating leukocyte DNA content can predict vascular telomere content, which can serve as an accurate proxy for human vascular age (94, 95). In recent years, it has been found that the mesenchymal stem cells (MSC) of aneurysm wall have senescence state, which promotes the occurrence and development of AAA. Compared with adipose-derived mesenchymal stem cells from healthy donors (H-ASC), adipose-derived mesenchymal stem cells from patients with AAA (AAA-ASC) were characterized by enhanced senescence, such as increased SA-β-Gal activity and decreased proliferation and migration. In addition, AAA-ASC showed decreased cell function with mitochondrial kinetic disorder, production of reactive oxygen species (ROS) and decreased mitochondrial membrane potential (96). Similar studies have found that vascular-resident mesenchymal stromal cells (AAA-MSC) and circulating endothelial progenitor cells (EPCs) in patients with AAA have the characteristics of senescent cells and damage of vascular repair ability of MSC (97, 98). Besides, another study has found that pretreatment of elderly mice with oral anti-aging agents (dasatinib + quercetin) can reduce the abundance of senescent cells in the arterial wall and surrounding tissues, and inhibit the severity of Ang II-induced AAA (99). Further elucidation that cellular senescence supports the occurrence and development of AAA, and reversal of cellular senescence is helpful to explore a new strategy to restore AAA therapy.
VSMCs play a key role in maintaining vascular system development and normal vascular homeostasis (100–102). Under physiological conditions, VSMCs are located in the vascular media, and their main functions are to regulate vasoconstriction, blood pressure, arterial tension diameter and flow distribution (103). VSMCs have differentiated plasticity in response to abnormal environmental stimuli, and tend to transform from a contractile phenotype to a synthetic phenotype and secrete pathogenic factors, such as ROS, inflammatory cytokines and MMPs (104). Importantly, oxidative stress and the secretion of inflammatory cytokines are also important features of cellular senescence. The phenotypic transformation process has been fully verified and is closely related to the occurrence and development of AAA (105). In terms of mechanism, cellular senescence is also an important pathological process in the formation of AAA. In addition, aging has become an independent risk factor for cardiovascular diseases (CVDs), so how cellular senescence is involved in CVDs has been the focus of researchers for the past several decades (106). Signs of VSMCs senescence have been found in aging vascular walls, represented by reduced proliferation rate and phenotypic transformation (107). Although the current understanding of VSMCs senescence is not fully adequate, it is necessary to decipher the correlation between senescence-related molecules and AAA progression in the search for new therapies.
SIRT1-related senescence and abdominal aortic aneurysm
The SIRT protein family is a group of histone deacetyltransferases with nicotinamide adenine dinucleotide (NAD)+ dependence, including seven homologous genes of SIRT1-SIRT7 (108). SIRT1 is the most well-studied member of the SIRT protein family and has been proven to regulate senescence and age-related diseases (109, 110). A Study has shown that endogenous SIRT1 expression in VSMCs of elderly donors is significantly reduced compared with that of young donors. The loss of SIRT1 expression leads to cellular senescence in VSMCs, which is related to cell proliferation and migration ability, even VSMCs from young donors showed cellular senescence after si-SIRT1 knockout (111). These data suggest that down-regulation of SIRT1 motivates to VSMCs senescence. It is found that the expression and activity of SIRT1 are significantly reduced in human AAA samples. VSMC-specific knockout of SIRT1 accelerates Ang II-induced AAA formation and rupture and AAA-related pathological changes, while VSMC-specific overexpression of SIRT1 inhibits Ang II-induced AAA formation and progression. Mechanistically, SIRT1 inhibits p21-dependent VSMCs senescence by blocking Ang II-induced binding of NF-κB to monocyte chemotactic protein-1 promoter (MCP-1) (112). In addition, the role of phosphodiesterase 1C (PDE1C) in VSMCs senescence in vitro and in vivo depends on SIRT1. Mechanism studies further demonstrate that inhibiting cAMP by PDE1C stimulates SIRT1 activation, which leads to subsequent upregulation of SIRT1 expression. It is also found that the pharmacological inhibition of PDE1C significantly slows down the progression of AAA (113). Importantly, SIRT1 activators show good effects in regulating lipid metabolism, inhibiting inflammation and protecting CVDs (110).
Non-coding RNAs play an important role in regulating the formation of AAA and the senescence of VSMCs (114–116). Upregulation of miR-199a-5p, enhanced expression of p-p53 and p21, and decreased expression of SIRT1 were observed in plasma and Ang II-treated VSMCs of patients with AAA. Bioinformatics prediction of TargetScan database and analysis of dual luciferase reporter genes showed that the 3′-UTR of SIRT1 had a potential binding site for miR-199a-5p, indicating that SIRT1 was a potential target of miR-199a-5p. Mechanically speaking, Ang II treatment significantly increased the level of miR-199a-5p, thereby promoting VSMCs senescence to participate in the formation of AAA by inhibiting the expression of SIRT1 (117). During the continuous culture of human aortic VSMCs in vitro, an increase in miR-34a level was observed accompanied by down-regulation of SIRT1. Surprisingly, even in young human aortic VSMCs, miR-34a overexpression and SIRT1 inhibition may induce cellular senescence. However, cellular senescence induced by overexpression of miR-34a in VSMCs was saved by transfection of SIRT1 protein, implying that the pro-senescence effect of miR-34a depended on SIRT1 participation (118). In addition, a research has confirmed that miR-34a induces senescence in endothelial cells, which is also achieved by targeted inhibition of SIRT1 (119). Meanwhile, other study has found that miR-188-5p with senescence induction in aneurysm wall co-locates with CD68 and CD3, suggesting that miR-188-5p may regulate the senescence of macrophages and T cells to involve in the occurrence and development of AAA (120). Therefore, different cell types may have different mechanisms of senescence regulation. In addition to miRNAs, long non-coding RNAs (lncRNAs) are also participated in AAA lesions and VSMCs senescence. Both ANRIL and SIRT1 were down-regulated and miR-181a expression was up-regulated in dual-channel-triggered senescent VSMCs. However, the overexpression of ANRIL prominently restrained senescence and promoted VSMCs viability via p53/p21 regulatory pathway. Mechanically, ANRIL overexpression inhibited miR-181a expression, which decreased the 3′-UTR inhibitory targeted binding of miR-181a to SIRT1 and increased SIRT1 expression (120). In the IgE-induced VSMCs senescence model, lincRNA-p21 was found to be upregulated, thereby enhancing p21 expression without altering p53 expression. Rescue experiments also proved that IgE induced VSMCs senescence and aggravated the development of AAA by upregulation of lincRNAp21/p21 pathway (121). The above studies provide more new potential targets for the treatment of AAA.
Therefore, it can be seen that SIRT1 is a key factor in regulating cellular senescence, and it must be the most promising protective regulator for AAA. It was found that the protective effect of calorie restriction (CR) on AAA development was eliminated in VSMC-specific SIRT1 knockout mice (122). In addition, licorice chaltone A (LA), an ingredient in licorice, prevented Ang II-induced AAA formation in apoE–/– mice by upregulating SIRT1 in VSMCs (123). Resveratrol (RSV), a natural activator of SIRT1, significantly alleviated Ang II-induced VSMCs senescence (124). A selective activator of SIRT1, SRT2104, was found to improve serum lipid metabolism parameters including total cholesterol, low-density lipoprotein and triglycerides in CVDs (125), and also attenuated serum pro-inflammatory cytokine levels and coagulation activation induced by intravenous LPS in healthy subjects (126). Senescent mechanisms and intervention measures for AAA patients based on SIRT1 are listed in Figure 3.
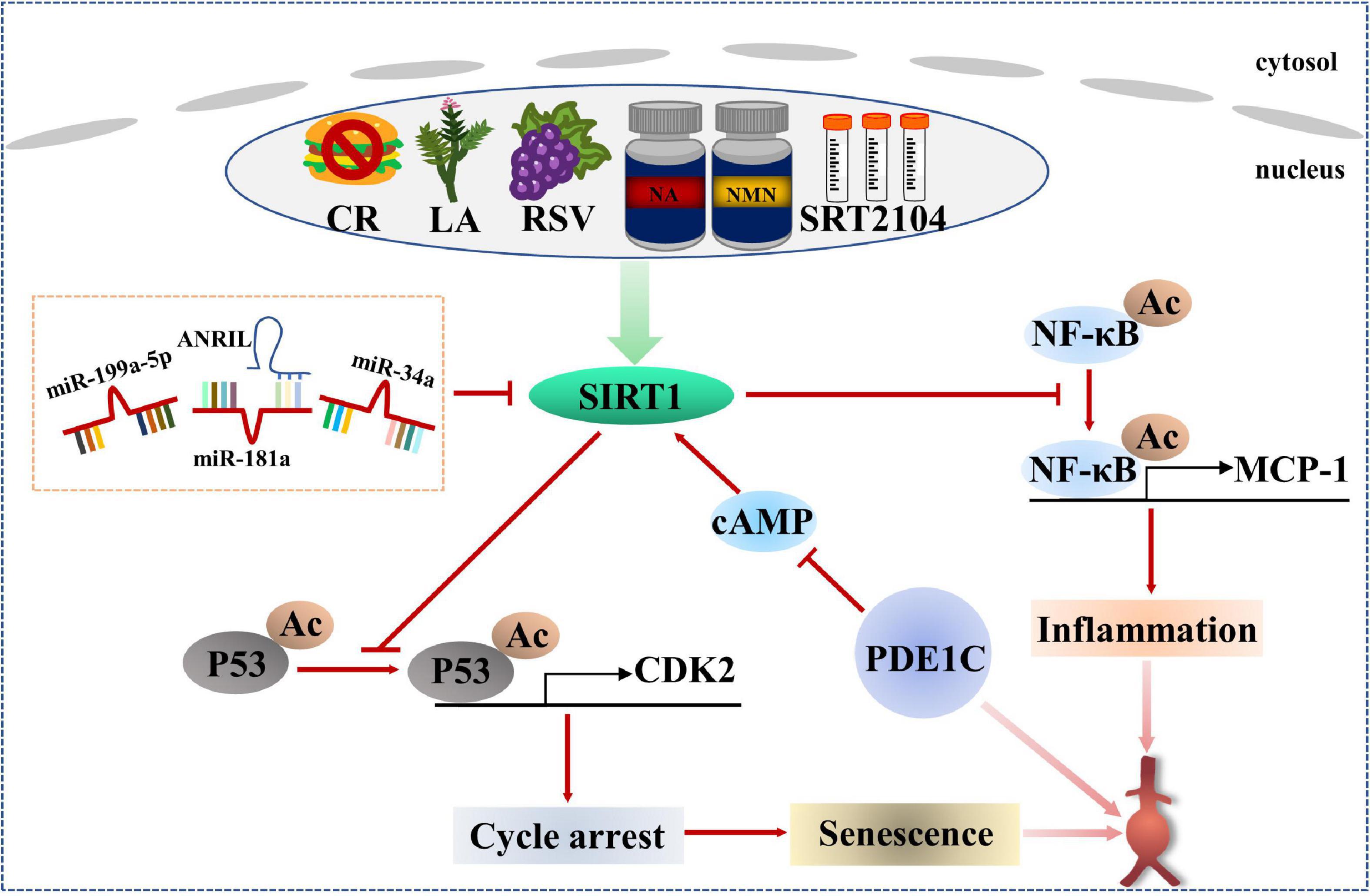
Figure 3. Regulation mechanism and intervention between SIRT1-related senescence and AAA.
Mitochondrial dysfunction-related senescence and abdominal aortic aneurysm
Mitochondrial dysfunction is an important cause of cellular senescence, which is different from senescence caused by genotoxic stress. Mitochondria oxidize NADH to NAD+ to maintain their normal function, but when mitochondria are dysfunctional, the ratio of NAD+ to NADH is out of whack, leading to the activation of adenosine monophosphate activated protein kinase (AMPK) and p53, which triggers mitochondrial dysfunction related senescence (miDAS) (127). Mitochondria are the main source of ROS, and mitochondrial dysfunction may contribute to abnormal ROS production (128). Specifically, elevated ROS levels impair the lifespan of vascular cells during cellular senescence and have been demonstrated in human brain aneurysms. Dysregulation of mitochondrial dynamics is also a key factor in cellular senescence, which is mainly regulated by fission and fusion processes and is closely related to cardiovascular diseases (129, 130). Fortunately, studies have confirmed that targeted inhibition of Drp1, a key protein of mitochondrial fission, and enhancement of Mfn1, a key protein of mitochondrial fusion, can inhibit the production of ROS and the senescence of VSMCs to maintain mitochondrial integrity, which is a potential treatment method to inhibit the formation and progression of AAA (131, 132). In addition, CR is a non-pharmacological intervention that regulates mitochondrial function to prevent AAA formation. Mechanistically, p53 knockout almost completely blocks the protective effect of CR by inhibiting the activity of cytochrome C oxidase assembly protein 2 (SCO2)-dependent mitochondrial complex IV. On the contrary, SCO2 overexpression restores the beneficial effect of CR on antagonizing Ang II-induced expression of AAA-related molecules and ROS production in VSMCs (133).
MiDAS is characterized by imbalance of NAD+/NADH and mitochondrial fusion/fission. It is noteworthy that SIRT1 is an NAD+ dependent histone deacetylase, so SIRT1 is closely related to NAD+/NADH (108). The therapies that improve mitochondrial function by exogenous supplementation of their precursors, such as nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), have been demonstrated to be effective in the treatment of CVDs in vivo experiments (134, 135). The natural compound resveratrol not only acts as an activator of SIRT1, but also acts as an antioxidant, helping to remove excessive ROS and reducing mitochondrial damage to protect cellular senescence and AAA (136, 137). In addition, mdivi1, an inhibitor of Drp1, can also effectively maintain mitochondrial integrity and reduce vascular senescence (132, 138). Intervention targets and methods related to MiDAS for the occurrence and progression of AAA are listed in Figure 4.
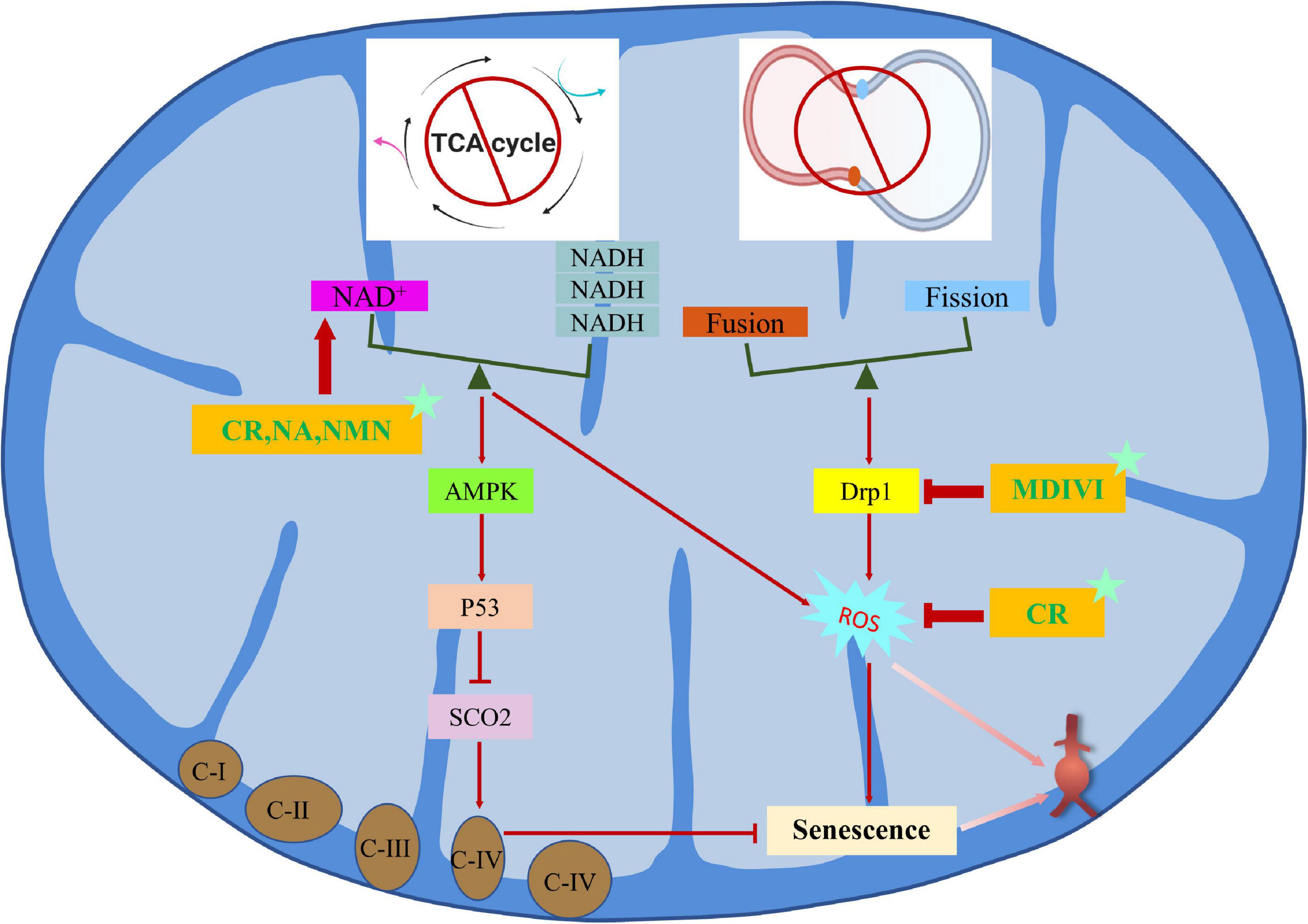
Figure 4. Regulation mechanism and intervention between mitochondrial dysfunction-related senescence and AAA.
Autophagy-related senescence and abdominal aortic aneurysm
Autophagy plays an important role through a complex network of autophagy related proteins (ATG) involved in the induction and formation of autophagosomes (139). The key step of autophagosome evolution is to transform LC3-I into lipidized LC3-II (140). Cellular senescence under normal and pathological conditions is often closely related to the reduction of autophagy (141). Aortic autophagy decreases with age, leading to age-related dysfunction of endothelial cells (ECs) and arterial calcification (142). Defective autophagy has been shown to promote senescence in VSMCs. It was reported that a large amount of SQSTM1/p62 was accumulated in the VSMCs of ATG7 knockout mice, which accelerated the occurrence of SIPS, such as cell enlargement, nuclear hyperplasia, cell cycle arrest and increased SA-β-Gal activity (143). Importantly, trehalose or spermidine can slow age-related aortic pulse wave velocity and reduce the accumulation of oxidative stress and advanced glycation end-products (AGEs) in arterial wall (144). At present, there are few mechanisms about how autophagy regulates the senescence of aneurysm wall cells, but the veil behind autophagy will be revealed with the further research.
Other
The nucleoli are sites where ribosomal DNA (rDNA) transcription (mediated by RNA polymerase I or Pol I) and ribosomal biogenesis occur. Studies have shown that inhibition of rDNA transcription in eukaryotic cells triggers a conserved cellular stress response called nucleolar stress (also known as ribosomal stress) (145, 146). Nucleolar stress induces activation of p53 pathway, thus promoting cellular senescence (147). In human AAA tissue, increased nucleolar stress in the medial cells is accompanied by localized DNA damage responses in the nucleolar septum. In vitro, nucleolar stress induces atypical DNA damage responses, mediating phosphorylation of p53 and senescence of VSMCs (148). However, clearance of rDNA transcription disturbance and nucleolar stress can contribute to recovery and prevention of AAA (148–150).
In addition, studies have found that both exogenous PM2.5 and endogenous intestinal microbiota metabolite trimethylamine N-oxide (TMAO) can mediate VSMCs senescence and promote the formation of AAA (151, 152). Other researchers found that treatment with soluble recombinant human ApoER2 or hydroxyurea, a DNA synthesis inhibitor, can inhibit PCSK9-induced VSMCs senescence (153). Meanwhile, a study have shown that inhibition of VSMCs senescence by targeting the MKL1/p38/MAPK pathway can be a potentially effective method for the treatment of AAA (154).
At the same time, a study found that excessive mechanical stress induces the continuous release of ADP and promotes VSMCs senescence through P2RY12-dependent Ras activation, which leads to excessive inflammation and degeneration and ultimately accelerates the formation and progression of thoracic aortic aneurysm (155). Another study reported that the reduced number and impaired function of circulating EPCs in patients with intracranial aneurysms may contribute to the pathophysiological process of aneurysm formation (156). In addition, a study has confirmed that cellular senescence is closely related to the formation process of retinal microaneurysms in the elderly (157).
Conclusion
With the discovery of the physiological and pathological effects of senescence, many researchers have focused on the exploration and development of therapeutic methods targeting senescent cells. On the one hand, we have made good achievements in pre-clinical basic research, such as the research and development of senescent cell scavenger and SASP inhibitor(dasatinib + quercetin), which can effectively improve the continuous deterioration of senescence and even reverse senescence. Of course, on the other hand, we still need to do a lot of more difficult work, such as the search for specific markers of senescence cells, the heterogeneity and dynamics of senescence, the combined application of anti-senescence and pro-senescence therapy, and the precise change of SASP. However, as biological approaches and drug discovery technologies continue to advance, the underlying mechanisms that regulate senescence will come to light, offering promising transformational opportunities for the development of new treatments that minimize the harmful consequences of senescence and bring great benefits to elderly patients with AAA.
Although a large amount of evidence has been found to prove that cellular senescence is closely related to AAA, there are still some limitations in this paper. 1. Most of this paper discusses the evidence of VSMCs senescence in the vascular wall, but lacks the description of ECs, macrophages and other vascular cells. 2. The evidence chain between cellular senescence and AAA has not been fully elucidated in some of the included literature.
In this paper, we conclude that the reduction of SIRT1 molecular activity, on the one hand, leads to cell cycle arrest through p53/CDK2 signaling, and on the other hand, induces inflammation through the activation of NF-κB signaling pathway, both of which jointly promote aging and eventually form AAA. Therefore, designing drugs targeted SIRT1 may be effective in preventing AAA formation and progression.
Author contributions
DW and SX devised the research plan. DW wrote the original draft. DW, XH, and LJ drew the picture. YJ, BJ, and SX modified and polished the manuscript. All authors contributed to the article and approved the submitted version.
Funding
SX was supported by National Natural Science Foundation of China (No. 81974049).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AAA, abdominal aortic aneurysm; AMPK, adenosine monophosphate activated protein kinase; AGE, advanced glycation end-product; BET, bromine domain and extra-terminal; CDKI, cycle-dependent kinase inhibitor; C/EBP β , CCAAT/enhancer binding protein β; CVD, cardiovascular disease; DDR, DNA damage responses; EC, endothelial cell; EPC, endothelial progenitor cell; FA, fatty acid; JAK, janus kinase; LA, licorice chaltone A; MCP-1, monocyte chemotactic protein-1; MDM2, mouse double minute 2; miDAS, mitochondrial dysfunction related senescence; MSC, mesenchymal stem cell; NAD+, nicotinamide adenine dinucleotide; NF- κ B, nuclear factor kappa-B; NMN, nicotinamide mononucleotide; NR, nicotinamide riboside; rDNA, ribosomal DNA; ROS, reactive oxygen species; SIPS, stress-induced premature senescence; SAHF, senescence associated heterochromia lesions; SASP, senescence-associated secretory phenotype; SIRT1, silencing information regulator 2 related enzyme 1; STAT, signal transduction and transcription activator; TMAO, trimethylamine N-oxide; TVA, tricarboxylic acid; VSMC, vascular smooth muscle cell.
References
1. Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat Rev Cardiol. (2019) 16:225–42. doi: 10.1038/s41569-018-0114-9
2. Howard DPJ, Banerjee A, Fairhead JF, Handa A, Silver LE, Rothwell PM, et al. Population-Based study of incidence of acute abdominal aortic aneurysms with projected impact of screening strategy. JAHA. (2015) 4:e001926. doi: 10.1161/JAHA.115.001926
3. Kühnl A, Erk A, Trenner M, Salvermoser M, Schmid V, Eckstein H-H. Incidence, treatment and mortality in patients with abdominal aortic aneurysms. Dtsch Arztebl Int. (2017) 114:391–8. doi: 10.3238/arztebl.2017.0391
4. Kent KC. Clinical practice. Abdominal aortic aneurysms. N Engl J Med. (2014) 371:2101–8. doi: 10.1056/NEJMcp1401430
5. GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (London, England). (2015) 385:117–71. doi: 10.1016/S0140-6736(14)61682-2
6. Golledge J, Moxon JV, Singh TP, Bown MJ, Mani K, Wanhainen A. Lack of an effective drug therapy for abdominal aortic aneurysm. J Intern Med. (2020) 288:6–22. doi: 10.1111/joim.12958
7. Cooper MA, Upchurch GR. The society of vascular surgery practice guidelines on the care of patients with abdominal aortic aneurysms. JAMA Surg. (2019) 154:553–4. doi: 10.1001/jamasurg.2019.0566
8. Sánchez-Infantes D, Nus M, Navas-Madroñal M, Fité J, Pérez B, Barros-Membrilla AJ, et al. Oxidative stress and inflammatory markers in abdominal aortic aneurysm. Antioxidants. (2021) 10:602. doi: 10.3390/antiox10040602
9. Rombouts KB, Merrienboer TAR, Ket JCF, Bogunovic N, Velden J, Yeung KK. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur J Clin Invest. (2021) 52:e13697. doi: 10.1111/eci.13697
10. Wu J-Q, Wang W, Zheng Y-H. [Role of vascular aging in the pathogenesis of abdominal aortic aneurysm and potential therapeutic targets]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. (2021) 43:962–8.
11. Sakalihasan N, Michel J-B, Katsargyris A, Kuivaniemi H, Defraigne J-O, Nchimi A, et al. Abdominal aortic aneurysms. Nat Rev Dis Primers. (2018) 4:34. doi: 10.1038/s41572-018-0030-7
12. Zeng Y. Towards deeper research and better policy for healthy aging –using the unique data of chinese longitudinal healthy longevity survey. China Economic J. (2012) 5:131–49. doi: 10.1080/17538963.2013.764677
13. Fang EF, Scheibye-Knudsen M, Jahn HJ, Li J, Ling L, Guo H, et al. A research agenda for aging in China in the 21st century. Ageing Res Rev. (2015) 24:197–205. doi: 10.1016/j.arr.2015.08.003
14. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. (1965) 37:614–36. doi: 10.1016/0014-4827(65)90211-9
15. Greider CW. Telomeres and senescence: The history, the experiment, the future. Curr Biol. (1998) 8:R178–81. doi: 10.1016/S0960-9822(98)70105-8
16. von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. (2002) 27:339–44. doi: 10.1016/S0968-0004(02)02110-2
17. von Zglinicki T, Saretzki G, Ladhoff J, d’Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev. (2005) 126:111–7. doi: 10.1016/j.mad.2004.09.034
18. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. (2014) 28:99–114. doi: 10.1101/gad.235184.113
19. Liao Z, Chua D, Tan NS. Reactive oxygen species: a volatile driver of field cancerization and metastasis. Mol Cancer. (2019) 18:65. doi: 10.1186/s12943-019-0961-y
20. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet. (2019) 20:299–309. doi: 10.1038/s41576-019-0099-1
22. Jacobs JJL. Loss of telomere protection: consequences and opportunities. Front Oncol. (2013) 3:88. doi: 10.3389/fonc.2013.00088
23. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
24. Centner AM, Bhide PG, Salazar G. Nicotine in Senescence and Atherosclerosis. Cells. (2020) 9:1035. doi: 10.3390/cells9041035
25. Bernadotte A, Mikhelson VM, Spivak IM. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany NY). (2016) 8:3–11. doi: 10.18632/aging.100871
26. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. (2007) 8:729–40. doi: 10.1038/nrm2233
27. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol. (2018) 28:436–53. doi: 10.1016/j.tcb.2018.02.001
28. Ramos I, Stamatakis K, Oeste CL, Pérez-Sala D. Vimentin as a multifaceted player and potential therapeutic target in viral infections. IJMS. (2020) 21:4675. doi: 10.3390/ijms21134675
29. Narita M, Nuñez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-Mediated heterochromatin formation and silencing of e2f target genes during cellular senescence. Cell. (2003) 113:703–16. doi: 10.1016/s0092-8674(03)00401-x
30. Zhang R, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol. (2007) 27:2343–58. doi: 10.1128/MCB.02019-06
31. Ye X, Zerlanko B, Zhang R, Somaiah N, Lipinski M, Salomoni P, et al. Definition of pRB- and p53-dependent and -independent steps in HIRA/ASF1a-mediated formation of senescence-associated heterochromatin foci. Mol Cell Biol. (2007) 27:2452–65. doi: 10.1128/MCB.01592-06
32. Sulli G, Di Micco R, d’Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. (2012) 12:709–20. doi: 10.1038/nrc3344
33. Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol. (2011) 13:292–302. doi: 10.1038/ncb2170
34. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell. (2006) 5:187–95. doi: 10.1111/j.1474-9726.2006.00199.x
35. Hoare M, Ito Y, Kang T-W, Weekes MP, Matheson NJ, Patten DA, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. (2016) 18:979–92. doi: 10.1038/ncb3397
36. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. (1995) 92:9363–7. doi: 10.1073/pnas.92.20.9363
37. Noren Hooten N, Evans MK. Techniques to induce and quantify cellular senescence. J Vis Exp. (2017) 55533.
38. Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. (2000) 113(Pt 20):3613–22. doi: 10.1242/jcs.113.20.3613
39. Bursuker I, Rhodes JM, Goldman R. Beta-galactosidase–an indicator of the maturational stage of mouse and human mononuclear phagocytes. J Cell Physiol. (1982) 112:385–90. doi: 10.1002/jcp.1041120312
40. Odgren PR, MacKay CA, Mason-Savas A, Yang M, Mailhot G, Birnbaum MJ. False-positive beta-galactosidase staining in osteoclasts by endogenous enzyme: studies in neonatal and month-old wild-type mice. Connect Tissue Res. (2006) 47:229–34.
41. Sun Y, Coppé J-P, Lam EW-F. Cellular senescence: the sought or the unwanted? Trends Mol Med. (2018) 24:871–85. doi: 10.1016/j.molmed.2018.08.002
42. Lopes-Paciencia S, Saint-Germain E, Rowell M-C, Ruiz AF, Kalegari P, Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine. (2019) 117:15–22. doi: 10.1016/j.cyto.2019.01.013
43. Mohamad Kamal NS, Safuan S, Shamsuddin S, Foroozandeh P. Aging of the cells: Insight into cellular senescence and detection Methods. Eur J Cell Biol. (2020) 99:151108. doi: 10.1016/j.ejcb.2020.151108
44. Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The Senescence-Associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol Mech Dis. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
45. Ortiz-Montero P, Londoño-Vallejo A, Vernot J-P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. (2017) 15:17. doi: 10.1186/s12964-017-0172-3
46. Jun J-I, Lau LF. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY). (2010) 2:627–31. doi: 10.18632/aging.100201
47. Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-Induced senescence relayed by an interleukin-dependent inflammatory network. Cell. (2008) 133:1019–31. doi: 10.1016/j.cell.2008.03.039
48. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell. (2008) 133:1006–18. doi: 10.1016/j.cell.2008.03.038
49. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. (2013) 15:978–90. doi: 10.1038/ncb2784
50. Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol. (2017) 27:2652–60. doi: 10.1016/j.cub.2017.07.033
51. Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in pten -deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. (2014) 9:75–89. doi: 10.1016/j.celrep.2014.08.044
52. Tasdemir N, Banito A, Roe J-S, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. (2016) 6:612–29. doi: 10.1158/2159-8290.CD-16-0217
53. Takahashi A, Imai Y, Yamakoshi K, Kuninaka S, Ohtani N, Yoshimoto S, et al. DNA Damage Signaling Triggers Degradation of Histone Methyltransferases through APC/CCdh1 in Senescent Cells. Mol Cell. (2012) 45:123–31. doi: 10.1016/j.molcel.2011.10.018
54. Hayakawa T, Iwai M, Aoki S, Takimoto K, Maruyama M, Maruyama W, et al. SIRT1 Suppresses the Senescence-Associated Secretory Phenotype through Epigenetic Gene Regulation. PLoS One. (2015) 10:e0116480. doi: 10.1371/journal.pone.0116480
55. Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ. MacroH2A1 and ATM Play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol Cell. (2015) 59:719–31. doi: 10.1016/j.molcel.2015.07.011
56. Contrepois K, Coudereau C, Benayoun BA, Schuler N, Roux P-F, Bischof O, et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat Commun. (2017) 8:14995. doi: 10.1038/ncomms14995
57. Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. (2016) 30:321–36. doi: 10.1101/gad.271882.115
58. Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol. (2016) 215:325–34. doi: 10.1083/jcb.201608026
59. Laberge R-M, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. (2015) 17:1049–61.
60. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. (2015) 17:1205–17. doi: 10.1038/ncb3225
61. Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. (2016) 6:a026070. doi: 10.1101/cshperspect.a026070
62. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. (2021) 9:645593. doi: 10.3389/fcell.2021.645593
63. Kruse J-P, Gu W. Modes of p53 Regulation. Cell. (2009) 137:609–22. doi: 10.1016/j.cell.2009.04.050
64. Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. (2010) 20:299–309. doi: 10.1016/j.tcb.2010.01.009
65. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspect Biol. (2010) 2:a001008. doi: 10.1101/cshperspect.a001008
66. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. (2017) 170:1062–78. doi: 10.1016/j.cell.2017.08.028
67. Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair. (2016) 42:63–71. doi: 10.1016/j.dnarep.2016.04.008
68. Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Therapeut. (2017) 173:83–105. doi: 10.1016/j.pharmthera.2017.02.008
69. Wiedemeyer WR. Resistance Mechanisms to Cyclin-Dependent Kinase Inhibitors. In: Yarden Y, Elkabets M editors. Resistance to Anti-Cancer Therapeutics Targeting Receptor Tyrosine Kinases and Downstream Pathways. Cham: Springer International Publishing (2018). p. 181–210. doi: 10.1007/978-3-319-67932-7_8
70. Ferrándiz N, Caraballo JM, García-Gutierrez L, Devgan V, Rodriguez-Paredes M, Lafita MC, et al. p21 as a Transcriptional Co-Repressor of S-Phase and Mitotic Control Genes. PLoS One. (2012) 7:e37759. doi: 10.1371/journal.pone.0037759
71. Huang W, Tan D, Wang X, Han S, Tan J, Zhao Y, et al. Histone deacetylase 3 represses p15INK4b and p21WAF1/cip1 transcription by interacting with Sp1. Biochem Biophys Res Commun. (2006) 339:165–71. doi: 10.1016/j.bbrc.2005.11.010
72. Jung Y-S, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. (2010) 22:1003–12. doi: 10.1016/j.cellsig.2010.01.013
73. Koo B-H, Kim Y, Je Cho Y, Kim D-S. Distinct roles of transforming growth factor-β signaling and transforming growth factor-β receptor inhibitor SB431542 in the regulation of p21 expression. Eur J Pharmacol. (2015) 764:413–23. doi: 10.1016/j.ejphar.2015.07.032
74. Gil J, Peters G. Regulation of the INK4b–ARF–INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. (2006) 7:667–77. doi: 10.1038/nrm1987
75. Kim WY, Sharpless NE. The Regulation of INK4/ARF in Cancer and Aging. Cell. (2006) 127:265–75. doi: 10.1016/j.cell.2006.10.003
76. Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15INK4B tumor suppressor gene. Oncogene. (2011) 30:1956–62. doi: 10.1038/onc.2010.568
77. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. (1993) 366:704–7. doi: 10.1038/366704a0
78. Fischer M, Müller GA. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit Rev Biochem Mol Biol. (2017) 52:638–62. doi: 10.1080/10409238.2017.1360836
79. Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, et al. Exit from G1 and S Phase of the Cell Cycle Is Regulated by Repressor Complexes Containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. (2000) 101:79–89. doi: 10.1016/S0092-8674(00)80625-X
80. Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. (2012) 130:1715–25. doi: 10.1002/ijc.27316
81. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. (1972) 26:239–57. doi: 10.1038/bjc.1972.33
82. D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. (2019) 43:582–92. doi: 10.1002/cbin.11137
83. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. (1998) 281:1312–6. doi: 10.1126/science.281.5381.1312
84. Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. (1998) 281:1322–6. doi: 10.1126/science.281.5381.1322
85. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. (2016) 7:11190. doi: 10.1038/ncomms11190
86. Chang J, Wang Y, Shao L, Laberge R-M, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. (2016) 22:78–83. doi: 10.1038/nm.4010
87. Frasca D, Saada YB, Garcia D, Friguet B. Effects of cellular senescence on metabolic pathways in non-immune and immune cells. Mech Ageing Dev. (2021) 194:111428. doi: 10.1016/j.mad.2020.111428
88. Hamon M-P, Ahmed EK, Baraibar MA, Friguet B. Proteome oxidative modifications and impairment of specific metabolic pathways during cellular senescence and aging. Proteomics. (2020) 20:e1800421. doi: 10.1002/pmic.201800421
89. Narita M, Young ARJ, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial Coupling of mTOR and Autophagy Augments Secretory Phenotypes. Science. (2011) 332:966–70. doi: 10.1126/science.1205407
90. Wu M, Ye H, Shao C, Zheng X, Li Q, Wang L, et al. Metabolomics-Proteomics combined approach identifies differential metabolism-associated molecular events between senescence and apoptosis. J Proteome Res. (2017) 16:2250–61. doi: 10.1021/acs.jproteome.7b00111
91. Liao S, Curci JA, Kelley BJ, Sicard GA, Thompson RW. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery. J Surg Res. (2000) 92:85–95. doi: 10.1006/jsre.2000.5878
92. Riches K, Angelini TG, Mudhar GS, Kaye J, Clark E, Bailey MA, et al. Exploring smooth muscle phenotype and function in a bioreactor model of abdominal aortic aneurysm. J Transl Med. (2013) 11:208. doi: 10.1186/1479-5876-11-208
93. Dimitroulis D, Katsargyris A, Klonaris C, Avgerinos ED, Fragou-Plemenou M, Kouraklis G, et al. Telomerase expression on aortic wall endothelial cells is attenuated in abdominal aortic aneurysms compared to healthy nonaneurysmal aortas. J Vascular Surg. (2011) 54:1778–83. doi: 10.1016/j.jvs.2011.06.079
94. Wilson WRW, Herbert KE, Mistry Y, Stevens SE, Patel HR, Hastings RA, et al. Blood leucocyte telomere DNA content predicts vascular telomere DNA content in humans with and without vascular disease. Eur Heart J. (2008) 29:2689–94. doi: 10.1093/eurheartj/ehn386
95. Atturu G, Brouilette S, Samani NJ, London NJM, Sayers RD, Bown MJ. Short Leukocyte Telomere Length is Associated with Abdominal Aortic Aneurysm (AAA). Eur J Vascular Endovasc Surg. (2010) 39:559–64. doi: 10.1016/j.ejvs.2010.01.013
96. Huang X, Zhang H, Liang X, Hong Y, Mao M, Han Q, et al. Adipose-Derived Mesenchymal Stem Cells Isolated from Patients with Abdominal Aortic Aneurysm Exhibit Senescence Phenomena. Oxid Med Cell Longev. (2019) 2019:1–12. doi: 10.1155/2019/1305049
97. Teti G, Chiarini F, Mazzotti E, Ruggeri A, Carano F, Falconi M. Cellular senescence in vascular wall mesenchymal stromal cells, a possible contribution to the development of aortic aneurysm. Mech Ageing Dev. (2021) 197:111515. doi: 10.1016/j.mad.2021.111515
98. Sung S-H, Wu T-C, Chen J-S, Chen Y-H, Huang P-H, Lin S-J, et al. Reduced number and impaired function of circulating endothelial progenitor cells in patients with abdominal aortic aneurysm. Int J Cardiol. (2013) 168:1070–7. doi: 10.1016/j.ijcard.2012.11.002
99. Parvizi M, Franchi F, Arendt BK, Ebtehaj S, Rodriguez-Porcel M, Lanza IR. Senolytic agents lessen the severity of abdominal aortic aneurysm in aged mice. Exp Gerontol. (2021) 151:111416. doi: 10.1016/j.exger.2021.111416
100. Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. (1995) 75:487–517. doi: 10.1152/physrev.1995.75.3.487
101. Thompson RW, Liao S, Curci JA. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron Artery Dis. (1997) 8:623–31. doi: 10.1097/00019501-199710000-00005
102. Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension. (2001) 38:581–7. doi: 10.1161/hy09t1.096249
103. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. (2004) 84:767–801. doi: 10.1152/physrev.00041.2003
104. Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, Morgan KG. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol Rev. (2016) 68:476–532. doi: 10.1124/pr.115.010652
105. Gurung R, Choong AM, Woo CC, Foo R, Sorokin V. Genetic and epigenetic mechanisms underlying vascular smooth muscle cell phenotypic modulation in abdominal aortic aneurysm. IJMS. (2020) 21:6334. doi: 10.3390/ijms21176334
106. Owens WA, Walaszczyk A, Spyridopoulos I, Dookun E, Richardson GD. Senescence and senolytics in cardiovascular disease: Promise and potential pitfalls. Mech Ageing Dev. (2021) 198:111540. doi: 10.1016/j.mad.2021.111540
107. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
108. Finkel T, Deng C-X, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. (2009) 460:587–91. doi: 10.1038/nature08197
109. Ding Y-N, Tang X, Chen H-Z, Liu D-P. Epigenetic regulation of vascular aging and age-related vascular diseases. Adv Exp Med Biol. (2018) 1086:55–75. doi: 10.1007/978-981-13-1117-8_4
110. Wang F, Chen H-Z. Histone Deacetylase SIRT1, Smooth Muscle Cell Function, and Vascular Diseases. Front Pharmacol. (2020) 11:537519. doi: 10.3389/fphar.2020.537519
111. Thompson AM, Wagner R, Rzucidlo EM. Age-related loss of SirT1 expression results in dysregulated human vascular smooth muscle cell function. Am J Physiol Heart Circ Physiol. (2014) 307:H533–41. doi: 10.1152/ajpheart.00871.2013
112. Chen H-Z, Wang F, Gao P, Pei J-F, Liu Y, Xu T-T, et al. Age-Associated Sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ Res. (2016) 119:1076–88. doi: 10.1161/CIRCRESAHA.116.308895
113. Zhang C, Zhao H, Cai Y, Xiong J, Mohan A, Lou D, et al. Cyclic nucleotide phosphodiesterase 1C contributes to abdominal aortic aneurysm. Proc Natl Acad Sci USA. (2021) 118:e2107898118. doi: 10.1073/pnas.2107898118
114. Abdelmohsen K, Panda A, Kang M-J, Xu J, Selimyan R, Yoon J-H, et al. Senescence-associated lncRNAs: senescence-associated long noncoding RNAs. Aging Cell. (2013) 12:890–900. doi: 10.1111/acel.12115
115. Bielak-Zmijewska A, Wnuk M, Przybylska D, Grabowska W, Lewinska A, Alster O, et al. A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology. (2014) 15:47–64. doi: 10.1007/s10522-013-9477-9
116. Raucci A, Macrì F, Castiglione S, Badi I, Vinci MC, Zuccolo E. MicroRNA-34a: the bad guy in age-related vascular diseases. Cell Mol Life Sci. (2021) 78:7355–78. doi: 10.1007/s00018-021-03979-4
117. Tao W, Hong Y, He H, Han Q, Mao M, Hu B, et al. MicroRNA-199a-5p aggravates angiotensin II–induced vascular smooth muscle cell senescence by targeting Sirtuin-1 in abdominal aortic aneurysm. J Cell Mol Med. (2021) 25:6056–69. doi: 10.1111/jcmm.16485
118. Badi I, Burba I, Ruggeri C, Zeni F, Bertolotti M, Scopece A, et al. MicroRNA-34a induces vascular smooth muscle cells senescence by sirt1 downregulation and promotes the expression of age-associated pro-inflammatory secretory factors. GERONA. (2015) 70:1304–11. doi: 10.1093/gerona/glu180
119. Ito T, Yagi S, Yamakuchi M. MicroRNA-34a regulation of endothelial senescence. Biochem Biophys Res Commun. (2010) 398:735–40. doi: 10.1016/j.bbrc.2010.07.012
120. Tan P, Guo Y-H, Zhan J-K, Long L-M, Xu M-L, Ye L, et al. LncRNA-ANRIL inhibits cell senescence of vascular smooth muscle cells by regulating miR-181a/Sirt1. Biochem Cell Biol. (2019) 97:571–80. doi: 10.1139/bcb-2018-0126
121. Guo W, Gao R, Zhang W, Ge W, Ren M, Li B, et al. IgE Aggravates the Senescence of Smooth Muscle Cells in Abdominal Aortic Aneurysm by Upregulating LincRNA-p21. Aging Dis. (2019) 10:699. doi: 10.14336/AD.2018.1128
122. Liu Y, Wang T-T, Zhang R, Fu W-Y, Wang X, Wang F, et al. Calorie restriction protects against experimental abdominal aortic aneurysms in mice. J Exp Med. (2016) 213:2473–88. doi: 10.1084/jem.20151794
123. Hou X, Yang S, Zheng Y. Licochalcone A attenuates abdominal aortic aneurysm induced by angiotensin II via regulating the miR-181b/SIRT1/HO-1 signaling. J Cell Physiol. (2019) 234:7560–8. doi: 10.1002/jcp.27517
124. Kim EN, Kim MY, Lim JH, Kim Y, Shin SJ, Park CW, et al. The protective effect of resveratrol on vascular aging by modulation of the renin-angiotensin system. Atherosclerosis. (2018) 270:123–31. doi: 10.1016/j.atherosclerosis.2018.01.043
125. Venkatasubramanian S, Noh RM, Daga S, Langrish JP, Joshi NV, Mills NL, et al. Cardiovascular effects of a novel SIRT1 activator, SRT2104, in otherwise healthy cigarette smokers. J Am Heart Assoc. (2013) 2:e000042. doi: 10.1161/JAHA.113.000042
126. van der Meer AJ, Scicluna BP, Moerland PD, Lin J, Jacobson EW, Vlasuk GP, et al. The Selective Sirtuin 1 Activator SRT2104 reduces endotoxin-induced cytokine release and coagulation activation in humans. Crit Care Med. (2015) 43:e199–202. doi: 10.1097/CCM.0000000000000949
127. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. (2016) 23:303–14.
128. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
129. Vásquez-Trincado C, García-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease: Mitochondria and cardiovascular disease. J Physiol. (2016) 594:509–25. doi: 10.1113/JP271301
130. Anderson GR, Wardell SE, Cakir M, Yip C, Ahn Y, Ali M, et al. Dysregulation of mitochondrial dynamics proteins are a targetable feature of human tumors. Nat Commun. (2018) 9:1677. doi: 10.1038/s41467-018-04033-x
131. Ma D, Zheng B, Liu H, Zhao Y, Liu X, Zhang X, et al. Klf5 down-regulation induces vascular senescence through eIF5a depletion and mitochondrial fission. PLoS Biol. (2020) 18:e3000808. doi: 10.1371/journal.pbio.3000808
132. Cooper HA, Cicalese S, Preston KJ, Kawai T, Okuno K, Choi ET, et al. Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovasc Res. (2021) 117:971–82.
133. Gao P, Zhang H, Zhang Q, Fang X, Wu H, Wang M, et al. Caloric Restriction Exacerbates Angiotensin II–Induced Abdominal Aortic Aneurysm in the Absence of p53. Hypertension. (2019) 73:547–60. doi: 10.1161/HYPERTENSIONAHA.118.12086
134. Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol. (2016) 17:679–90. doi: 10.1038/nrm.2016.93
135. Kane AE, Sinclair DA. Sirtuins and NAD+ in the development and treatment of metabolic and cardiovascular diseases. Circ Res. (2018) 123:868–85. doi: 10.1161/CIRCRESAHA.118.312498
136. Bilen A, Mercantepe F, Tümkaya L, Yilmaz A, Batcik Ş. The hepatoprotective potential of resveratrol in an experimental model of ruptured abdominal aortic aneurysm via oxidative stress and apoptosis. J Biochem Mol Toxicol. (2021) 35:e22836. doi: 10.1002/jbt.22836
137. Moran CS, Biros E, Krishna SM, Wang Y, Tikellis C, Morton SK, et al. Resveratrol Inhibits Growth of Experimental Abdominal Aortic Aneurysm Associated With Upregulation of Angiotensin-Converting Enzyme 2. Arterioscler Thromb Vasc Biol. (2017) 37:2195–203. doi: 10.1161/ATVBAHA.117.310129
138. Uchikado Y, Ikeda Y, Sasaki Y, Iwabayashi M, Akasaki Y, Ohishi M. Association of Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 With Angiotensin II Type 1 Receptor Impacts Mitochondrial Quality Control, Offering Promise for the Treatment of Vascular Senescence. Front Cardiovasc Med. (2021) 8:788655. doi: 10.3389/fcvm.2021.788655
139. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. (2010) 466:68–76.
140. Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. (2010) 6:764–76. doi: 10.4161/auto.6.6.12709
141. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. (2015) 349:aaa5612.
142. Salabei JK, Hill BG. Implications of autophagy for vascular smooth muscle cell function and plasticity. Free Radical Biol Med. (2013) 65:693–703.
143. Grootaert MO, da Costa Martins PA, Bitsch N, Pintelon I, De Meyer GR, Martinet W, et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy. (2015) 11:2014–32. doi: 10.1080/15548627.2015.1096485
144. LaRocca TJ, Gioscia-Ryan RA, Hearon CM, Seals DR. The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev. (2013) 134:314–20. doi: 10.1016/j.mad.2013.04.004
145. Boulon S, Westman BJ, Hutten S, Boisvert F-M, Lamond AI. The nucleolus under stress. Mol Cell. (2010) 40:216–27.
146. Yang K, Yang J, Yi J. Nucleolar Stress: hallmarks, sensing mechanism and diseases. Cell Stress. (2018) 2:125–40. doi: 10.15698/cst2018.06.139
147. Yang L, Song T, Chen L, Soliman H, Chen J. Nucleolar repression facilitates initiation and maintenance of senescence. Cell Cycle. (2015) 14:3613–23. doi: 10.1080/15384101.2015.1100777
148. Zhang W, Cheng W, Parlato R, Guo X, Cui X, Dai C, et al. Nucleolar stress induces a senescence-like phenotype in smooth muscle cells and promotes development of vascular degeneration. Aging (Albany NY). (2020) 12:22174–98. doi: 10.18632/aging.104094
149. Bi X, Ye Q, Li D, Peng Q, Wang Z, Wu X, et al. Inhibition of nucleolar stress response by Sirt1: A potential mechanism of acetylation-independent regulation of p53 accumulation. Aging Cell. (2019) 18:e12900. doi: 10.1111/acel.12900
150. Pinho M, Macedo JC, Logarinho E, Pereira PS. NOL12 repression induces nucleolar stress-driven cellular senescence and is associated with normative aging. Mol Cell Biol. (2019) 39:e99–19. doi: 10.1128/MCB.00099-19
151. Jun X, Jin G, Fu C, Jinxuan Z, Xueling L, Jiaxin H, et al. PM2.5 promotes abdominal aortic aneurysm formation in angiotensin II-infused apoe-/- mice. Biomed Pharmacother. (2018) 104:550–7. doi: 10.1016/j.biopha.2018.04.107
152. Hu J, Xu J, Shen S, Zhang W, Chen H, Sun X, et al. Trimethylamine N-oxide promotes abdominal aortic aneurysm formation by aggravating aortic smooth muscle cell senescence in mice. J Cardiovasc Transl Res. (2022). doi: 10.1007/s12265-022-10211-6 [Epub ahead of print].
153. Guo Y, Tang Z, Yan B, Yin H, Tai S, Peng J, et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) Triggers Vascular Smooth Muscle Cell Senescence and Apoptosis: Implication of Its Direct Role in Degenerative Vascular Disease. Arterioscler Thromb Vasc Biol. (2022) 42:67–86. doi: 10.1161/ATVBAHA.121.316902
154. Gao P, Gao P, Zhao J, Shan S, Luo W, Slivano OJ, et al. MKL1 cooperates with p38MAPK to promote vascular senescence, inflammation, and abdominal aortic aneurysm. Redox Biol. (2021) 41:101903. doi: 10.1016/j.redox.2021.101903
155. Zhang W-M, Liu Y, Li T-T, Piao C-M, Liu O, Liu J-L, et al. Sustained activation of ADP/P2ry12 signaling induces SMC senescence contributing to thoracic aortic aneurysm/dissection. J Mol Cell Cardiol. (2016) 99:76–86.
156. Wei H, Mao Q, Liu L, Xu Y, Chen J, Jiang R, et al. Changes and function of circulating endothelial progenitor cells in patients with cerebral aneurysm. J Neurosci Res. (2011) 89:1822–8. doi: 10.1002/jnr.22696
Keywords: pathogenesis, cellular senescence, therapeutics, longevity, abdominal aortic aneurysm
Citation: Wang D, Hao X, Jia L, Jing Y, Jiang B and Xin S (2022) Cellular senescence and abdominal aortic aneurysm: From pathogenesis to therapeutics. Front. Cardiovasc. Med. 9:999465. doi: 10.3389/fcvm.2022.999465
Received: 21 July 2022; Accepted: 15 August 2022;
Published: 14 September 2022.
Edited by:
Ding Yuan, Sichuan University, ChinaReviewed by:
Ziheng Wu, The First Affiliated Hospital of Zhejiang University, ChinaWei Guo, Chinese PLA General Hospital, China
Copyright © 2022 Wang, Hao, Jia, Jing, Jiang and Xin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shijie Xin, sjxin@cmu.edu.cn