 Rida Raja
Rida Raja Oveena Fonseka
Oveena Fonseka Andrea-Ruiz-Velasco
Andrea-Ruiz-Velasco Wei Liu
Wei Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 02 September 2022
Sec. Cardiovascular Metabolism
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.999044
This article is part of the Research Topic Investigating Novel Mechanisms and Treatments of Metabolic Disorders Associated with Cardiovascular Disease View all 13 articles
Metabolic cardiomyopathy is a significant global financial and health challenge; however, pathophysiological mechanisms governing this entity remain poorly understood. Among the main features of metabolic cardiomyopathy, the changes to cellular lipid metabolism have been studied and targeted for the discovery of novel treatment strategies obtaining contrasting results. The endoplasmic reticulum (ER) and Golgi apparatus (GA) carry out protein modification, sorting, and secretion activities that are more commonly studied from the perspective of protein quality control; however, they also drive the maintenance of lipid homeostasis. In response to metabolic stress, ER and GA regulate the expression of genes involved in cardiac lipid biogenesis and participate in lipid droplet formation and degradation. Due to the varied roles these organelles play, this review will focus on recapitulating the alterations and crosstalk between ER, GA, and lipid metabolism in cardiac metabolic syndrome.
In the past five decades, the global prevalence of obesity, diabetes, and hypertension has reached epidemic dimensions (1, 2). These disorders, collectively termed metabolic syndrome, confer substantial morbidity and mortality of cardiovascular disease (3, 4). At the same time, they are high-risk factors for the development of cardiomyopathy (5). The metabolic syndrome encompasses a diverse set of conditions that exert chronic stress on cellular functions, such as systemic dyslipidemia, hyperglycemia, and insulin resistance. The pathophysiological changes induced in the heart by these stressors are termed metabolic cardiomyopathy. Clinically, patients with metabolic cardiomyopathy initially present diastolic dysfunction, followed by late-onset systolic dysfunction and, ultimately, heart failure (HF) (6). Along with adverse structural remodeling and increased presence of reactive oxygen species, cell death, and inflammation (7, 8), metabolic cardiomyopathy features cardiac energetic impairment (9, 10). Lipid mishandling and subsequent lipotoxicity are prominent effects of this alteration (11, 12). Efficacious therapeutic options targeting cardiac lipid derangements are lacking, therefore advanced understanding of the pathogenesis is necessary. The endoplasmic reticulum (ER) and the Golgi apparatus (GA) are essential for lipid homeostasis, hosting and processing proteins involved in triacylglycerol (TG) synthesis and lipolysis. Previous reviews have focused on the central role the mitochondria play in lipid oxidation in metabolic cardiomyopathy (13–16); however, the role of the ER and GA in lipid metabolism in the heart under metabolic stress remains poorly defined.
Fatty acids (FAs) account for approximately 70% of the energy sources required for ATP synthesis in cardiomyocytes. In circulation, FAs are commonly found as triacylglycerols (TGs) associated with lipoproteins or chylomicrons or as free FAs bound to albumin. TGs are hydrolyzed by lipoprotein lipase (LpL) Cardiomyocytes take up extracellular FAs by passive diffusion across the plasma membrane or by surface receptors such as protein cluster of differentiation 36 (CD36), fatty acid transport protein 1, and fatty acid binding protein (17). Once in the cytoplasm, FAs are shuttled into the mitochondria for β-oxidation resulting in high energy molecules required for cardiac function (18). Excessive FAs are converted back to TGs by diacylglycerol acyltransferase (DGAT) and stored in lipid droplets (LD). When required, FAs are released from LDs by adipocyte triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) (19). Lipid homeostasis also requires the participation of other organelles. For example, peroxisomes manage very long-chain fatty acid breakdown and β-oxidation, lysosomes carry out lipid catabolism, and ER and GA handle LD formation (20). It is widely accepted that in metabolic syndrome the myocardium experiences an increase in lipid consumption (9, 21), meaning that different organelles activate stress responses to manage different stages of lipid homeostasis.
Cardiac lipotoxicity develops due to a build-up of FAs and lipid intermediates that disrupt β-oxidation homeostasis. Among such intermediates are ceramides and di-acyl glycerol (DAG). In a mouse model of lipid-induced dilated cardiomyopathy by LpL cardiac-overexpression, ceramides were found to regulate substrate utilization, where inhibition of ceramide biosynthesis normalized FAs and glucose oxidation and improved survival. In addition, ceramides induce various pathological processes that promote lipotoxicity, for instance, insulin resistance, inflammation, and apoptosis (22).
Similarly, DAG induces apoptosis via mitochondrial apoptotic pathways. It also impairs insulin signaling, an effect that can be accelerated due to the reactive oxygen species production (ROS) by free FAs (19). These pathological processes associated with toxic lipid intermediates contribute to the development of metabolic cardiomyopathy; therefore, it is important to study the mechanisms available in the cell to counteract such conditions.
The ER is involved in protein quality control (PQC), calcium homeostasis, and lipid metabolism. Chronic metabolic stress can perturb ER homeostasis leading to ER stress and proteotoxicity that consist of protein misfolding and protein accumulation. Protein quality is vital to sustain cardiac function and in metabolic cardiomyopathy it has been identified as an inducer of cell death (23). ER stress triggers an adaptive signal transduction pathway known as the unfolded protein response (UPR) which neutralizes ER stress and sustains cellular function. Three transmembrane proteins maintain ER homeostasis; inositol- requiring enzyme 1α (IRE1α), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) initiate the UPR (24). Under basal conditions, these sensor proteins are associated with chaperone GRP78/Bip, rendering them inactive (25). When proteostasis is disrupted, GRP78/Bip detaches and these become active (26). IRE1α processes the transcription factor X-box binding protein 1 (XBP1), forming the transcriptionally active spliced-XBP1 (XBP1s) (25, 27). XBP1s binds to a set of UPR-target genes that upregulate organelle biosynthesis, PQC, and ER-associated degradation (ERAD) (28). PERK phosphorylates the eukaryotic translation initiating factor 2α (elf2α), attenuating protein synthesis (27, 29). In turn, elf2α also induces the translation of ATF4, which regulates the expression of genes involved in autophagy, apoptosis, antioxidant response, and the transcription factor DNA damage-inducible 34 (GADD34). GADD34 can restore protein synthesis by binding to protein phosphatase 1C (PP1C), which dephosphorylates elf2α (27). PERK upregulates the proapoptotic transcription factor CHOP/GADD153. Finally, ATF6's transcriptional activity induces the expression of ERAD and XBP1 genes (24). These transcriptional events act in a well-choreographed manner to maintain ER equilibrium.
In addition to its protein-centric role, the UPR is an essential nutrient sensing ER apparatus critical for maintaining lipid homeostasis (30, 31). Palmitate activates the PERK-eIF2α-CHOP pathway and decreases Bip expression in HepG2 liver cells. Accordingly, Bip overexpression reduced CHOP levels and attenuated palmitate-induced ER stress and apoptosis (32). Moreover, GADD34-mediated dephosphorylation of eIF2α reduces hepatosteatosis in Alb::GC transgenic mice upon high-fat diet (HFD) feeding (33). Following this, depletion of ATF4, the downstream effector of the PERK-eIF2α pathway, protects mice against hepatic steatosis and hypertriglyceridemia in response to high-fructose diet feeding (34), suggesting that the PERK pathway regulates lipogenesis in hepatocytes. Similarly, PERK depletion inhibits lipogenesis during the differentiation of mouse embryonic fibroblasts to adipocytes (35) (Figure 1).
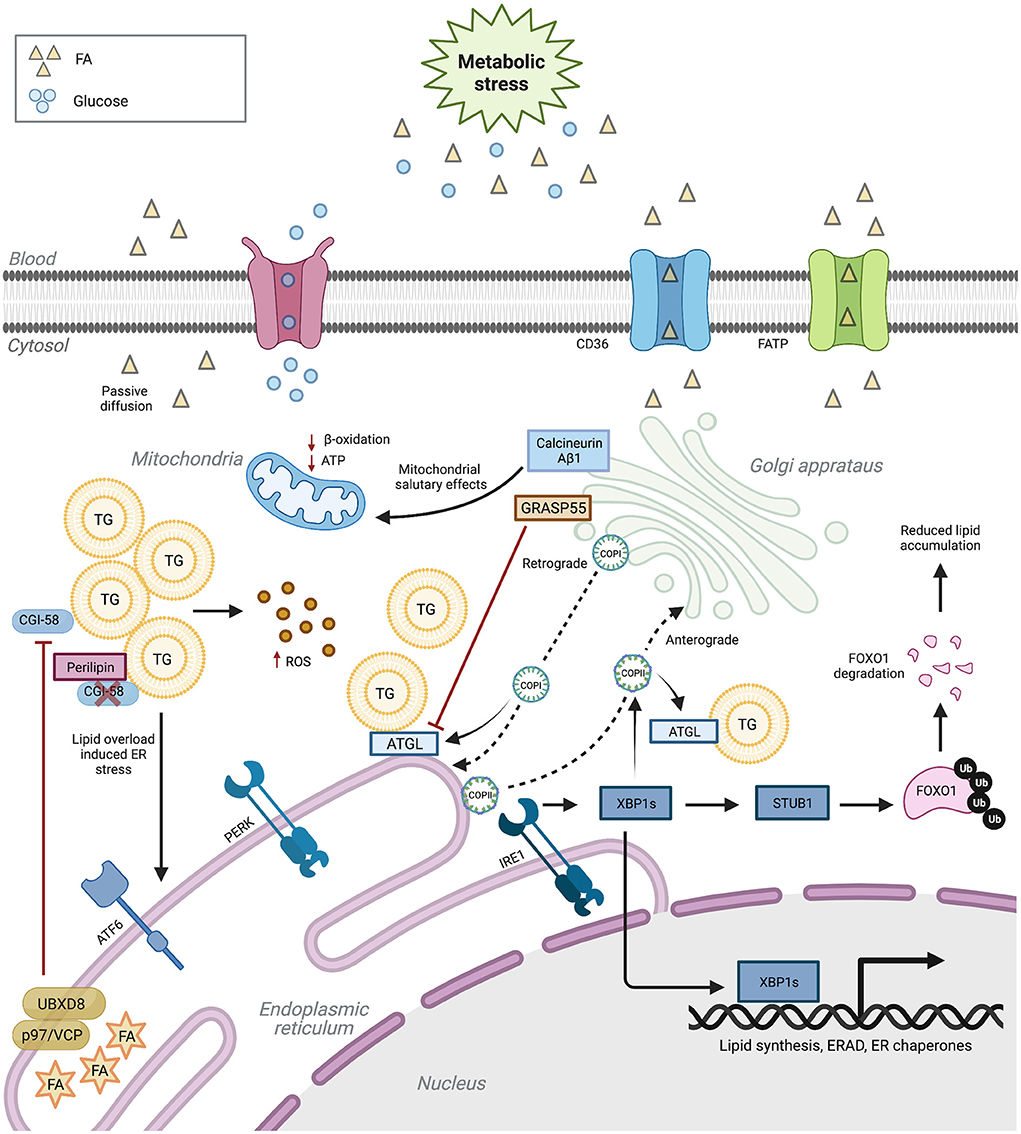
Figure 1. ER and Golgi regulation of lipid metabolism. ER and Golgi functions are paramount for lipid metabolism. Aberrant ER and Golgi function induce cellular lipid disequilibrium. In turn, lipotoxicity negatively regulates the function of these two organelles. Upon metabolic stress, ER dysfunction inhibits TG degradation, eventually leading to oxidative stress. ER stress also reduces genes participating in ER function and lipid metabolism. Impaired Golgi proteins or COPI/II pathways damage lipogenesis and lipolysis balance. CD36 (cluster of differentiation 36), FATP (fatty acid transport protein), TG (triacylglycerol), FA (fatty acid), ROS (reactive oxygen species), ATGL (adipose triglyceride lipase), XBP1s (spliced X-box binding protein 1), FoxO1 (Forkhead box protein O1), COPI (coat protein complex I) and COPII (coat protein complex II). Created with Biorender.com.
The IRE1α-XBP1 branch of the UPR is also a critical regulator of metabolic-stress-induced lipid disequilibrium (36). Adipocyte Xbp1s overexpression prevents obesity in HFD-fed mice and leptin-deficient ob/ob mice via increased uridine biosynthesis (37). Added to this, ectopic adipocyte Xbp1s expression promotes systemic glucose homeostasis in both lean and ob/ob mice by increasing adiponectin serum levels (38). XBP1 deletion reduces hepatic steatosis and improves insulin sensitivity in mice fed a high-fructose diet despite increased ER stress (39). In accordance, Lee et al. (40) demonstrated that XBP1 regulates lipogenesis. Finally, ATF6 modulates lipid metabolism in an XBP1-independent manner (41). ATF6α-knockout mice administered the pharmacological ER-stress inducer tunicamycin exhibit liver dysfunction and steatosis, ensued from aberrant ß-oxidation and suppression of very-low-density lipoproteins (VLDLs) formation. However, these abnormalities are obliterated by ATF6 overexpression (42). A similar account is observed in ATF6α deficient hepatocytes following a high-fat and high-sucrose diet feeding (43). During adipogenesis, ATF6α deficiency reduces the expression of key audiogenic genes needed for adipocyte differentiation (44). These studies highlight the importance of the ER stress response regulating lipid homeostasis in metabolically active peripheral tissues, such as adipose and liver. The crosstalk between such tissues and the heart is a major subject of study and their influence in the development of metabolic cardiomyopathy has been reported (45, 46). However, the amount of evidence also suggests that similar mechanisms could be present in the heart and be worth exploring for a better understanding of the disease.
ERAD is an integral part of the ER stress response. It recognizes and labels abnormal proteins in the ER directing them to the cytosol for proteasomal degradation. Cell-type-specific ERAD mouse model studies are limited. Adipocyte-specific Sel1L deficient mice are resistant to HFD-induced obesity, developing postprandial hypertriglyceridemia and displaying hepatic steatosis (47). Here, in the absence of Sel1L, LPL is retained in the ER in the form of ERAD-resistant aggregates. Similar observations were made on other LPL-expressing cells, including cardiomyocytes and macrophages (47). Hepatocyte-specific ER degradation enhancing alpha-mannosidase-like protein 3 (EDEM3) knockdown mice had increased LRP1 expression leading to enhanced VLDL uptake, thereby reducing plasma TG levels (48). Concurrently, data obtained from Gan et al. (49) showed that ischemic heart-derived small extracellular vesicles (sEVs) carrying miR-23-27-24 suppress adiponectin biosynthesis by downregulating EDEM3. This pathological communication causes adipocyte ER stress and endocrine dysfunction, which contributed to post-MI metabolic disorders.
Furthermore, Choi et al. (50) reported that depletion of the ER membrane-anchored E3 ligase gp78 stabilized DGAT2 in hepatic cells, which increased LD levels. UBXD8 is a protein that interacts with p97 during ERAD to facilitate proteasomal degradation of ubiquitinated proteins; however, UBXD8 has also emerged as a critical determinant of fatty acid (FA) metabolism and TG storage (51–54). UBXD8 acts as an unsaturated FA sensor. In FA-depleted cultured cells, UBXD8 inhibits the conversion of DAGs to TGs; this effect is reversed upon exposure to unsaturated FAs (53). Mechanistically, UBXD8 negatively regulates ATGL by promoting the dissociation of its activator CGI-58 (51). Therefore, FAs increase TG content by inactivating UBXD8. In line with this, UBXD8 depletion in murine hepatocytes leads to periportal steatosis upon HFD feeding (55). Whilst there is a vast body of literature evidencing ERAD involvement in lipid metabolism, the role of ERAD proteins in metabolic cardiomyopathy remains unexplored.
ER stress has been identified as a crucial pathophysiological driver of cardiovascular disease in preclinical and clinical studies (23). ER stress is implicated in cardiac lipotoxicity (56–58); however, the mechanisms by which ER disruptions contribute to lipotoxic alterations in cardiomyocytes are unclear. Recently, Schiattarella et al. (59) reported that cardiomyocyte-specific overexpression of Xbp1s ameliorates cardiac dysfunction and reduces myocardial steatosis in a preclinical model of HF with preserved ejection fraction. Further investigations revealed that Xbp1s promotes the ubiquitination and degradation of Forkhead box protein O1 (FoxO1) via E3 ubiquitin ligase STUB1. FoxO1 is a vital transcriptional regulator of genes involved in cellular metabolism and its transcriptional activity is increased in obese and diabetic animal models (60, 61). In cardiac muscle cells, FoxO1 orchestrates lipid accumulation; the sequential events underpining this accumulation remain unknown. Conversley, FoxO1 overexpression inhibits lipid accumulation in hepatocytes by mediating ATGL-dependent lipolysis (62). CGI-58 is an LD-associated protein that plays a vital role in TG hydrolysis by activating ATGL (63). it is decreased in failing human hearts (64). CGI-58-deficient mice display adverse cardiac remodeling accompanied by accentuated cardiac TG levels, ROS production, and inflammation when after HFD feeding (65). The chemical chaperone 4-PBA attenuated HFD-induced cardiac remodeling and dysfunction; alleviated mitochondrial dissonance, oxidative stress, and lipid accumulation in cardiac-specific CGI58- knockout mice (65). Moreover, inhibiting calpains, calcium-activated proteases, prevents lipotoxicity-associated myocardial injury in HFD-challenged mice by attenuating ER stress, thereby improving cardiac function (66). These studies suggest ER stress regulates cardiac lipid homeostasis and acts as a potential therapeutic target for metabolic cardiomyopathy.
While ER stress alters lipid metabolism, FAs can also induce ER stress in a bidirectional loop, creating a vicious cycle (67). Saturated fatty acids instigate lipotoxic injury in cardiomyocytes via ER stress-mediated apoptosis pathways (68–70). Cardiomyocyte-specific PPARβ/δ deletion perturbed myocardial fatty acid oxidation, induced ER stress, and lead to cardiac steatosis, hypertrophy, and congestive HF in a preclinical mouse model (71, 72). The PPARβ/δ agonist, GW501516, attenuated palmitate-induced ER-stress in AC16 cells, underscoring the therapeutic potential of PPARβ/δ for ER-stress mediated cardiac lipotoxicity (72). Moreover, CTRP9, a highly conserved paralog of adiponectin, has also been reported to regulate lipid metabolism. CTRP9-knockout mice displayed augmented ER-stressed induced apoptosis; nevertheless, recombinant CTRP9 treatment exerted anti-ER-stress-related apoptotic effects and anti-oxidative stress effects to abate lipotoxicity in neonatal rat cardiomyocytes (73).
The GA comprises a series of stacked membranes that process ER proteins and lipids. Its main functions include protein glycosylation, sorting and secretion, and phospholipid and sphingomyelin synthesis. The GA network can be divided into the cis and trans components. While the cis network is responsible for receiving cargo from the ER, the trans-Golgi network (TGN), on the other end, regulates cargo exit from the Golgi. The cargo is carried in transport vesicles formed by protein complexes that regulate their content and direction. From the ER exit sites, coat protein complex II (COPII) vesicles travel through the ER-Golgi intermediate compartment (ERGIC) to the cis-component via anterograde transport. In retrograde transport, COPI-coated vesicles recycle ER-resident proteins. Finally, cargo directed to other organelles or the plasma membrane exit the TGN in clathrin-covered vesicles (74). Like the ER, the GA can experience stress when unable to keep up with the flux of proteins and lipids, leading to the activation of the Golgi stress response. Even though this response has not been as well-characterized as the UPR in the heart, pathways such as transcription factor binding to IGHM enhancer 3 (TFE3), CAMP responsive element binding protein 3 (CREB3), and heat shock protein 47 (HSP47) have been found to regulate GA structure and functions (75).
The GA is mainly known for its role in lysosomal, transmembrane, and secretory protein glycosylation and trafficking activities. In addition to its protein processing activities, its contributions to lysosomal function and membrane dynamics are vital for lipid recycling, lysosomal lipid signaling, and membrane composition, all of which are key to lipid metabolism in the setting of metabolic disease (76–78). Presently, manifestations of direct GA involvement in lipid signaling and metabolism not associated to membrane dynamics that have not been clearly defined will be discussed. Recent observations from Fan et al. (79) demonstrated that hyperglycemia affects myocardial GA protein expression and causes fragmentation in a preclinical model of type 1 diabetes. Proofs of how this could impair lipid metabolism have been found in different animal models or cell lines. For instance, the membrane lipid transporter CD36 is processed in the GA before its transport to the plasmatic membrane. A mutation that blunted its passing through the GA resulted in ER accumulation and low lipid uptake (80).
Knocking down COPI subunits in flies (81) and Hep62 (82) cells showed they are required for lipolysis activation. They regulate LD surface proteins that inhibit lipolysis, such as Perilipins 2 and 3, and promote the formation of ER-LD bridges through which ATGL is transferred to the LDs. The deletion of oxysterol binding protein-like 2 (OSBPL2), an intracellular lipid receptor, disrupted LD localization of the subunit COPBI, leading to lipid accumulation in larger LDs and hypercholesterolemia in zebrafish (82). Furthermore, genetic or chemical interference with the ADP-ribosylation factor (ARF1)-COPI pathway at different stages gave similar results. Golgi-specific brefeldin A-resistance guanine nucleotide exchange factor 1 (GBF1) interacts directly with ATGL (83), and its deletion hindered ATGL transport to nascent LDs, inducing the lipase's proteasomal degradation (84). In addition, impeding the function of the COPII complex also disrupted lipolysis, but the effect was smaller than that of COPI. In different cells, knockdown of ARF1, a COPI complex initiator, showed that this machinery has the ability to form nano-buds from the LD membranes and target TG synthesis enzyme to the LD membrane (85), suggesting that it affects LD composition as well as degradation.
Similar to the observations above, deletion of GRASP55, another Golgi-resident protein, impaired the trafficking of ATGL and MGL to LDs, impairing lipolysis. In mice, this systemic deletion conferred resistance to HFD-induced obesity due to a decreased formation and secretion of chylomicrons and VLDLs from enterocytes resulting in low lipid uptake (86). Hepatic deletion of the initiator of COPII complex GTP-binding protein SAR1b (SAR1) in mice showed increased accumulation of TGs and cholesterol in the liver with reduced lipid plasma levels protecting them from atherosclerosis (87). In this study, Wang et al. (87) identified surfeit locus protein 4 (SURF4) as a receptor for specific VLDL secretion downstream of the COPII pathway and as a protein with a COPI sorting signal for recycling. Comparable effects were observed in liver-specific IRE1 knockout mice where COPII transport was reduced. In this case, XBP1 overexpression restored COPII gene expression and trafficking, resolving the fatty liver and hypolipidemia (88). The significance of the GA transport role in lipoprotein secretion was confirmed by a report showing eight different mutations of SARA2 (coding SAR1 protein) in patients with severe fat absorption disorders (89). These results indicate that the ER-GA transport network can be targeted for the systemic regulation of metabolic diseases.
Finally, in addition to its role in protein and LD processing, the GA is involved in metabolic and cell death signaling. In the heart, activating an alternative isoform of calcineurin presenting a new Golgi-localization signal was found to regulate metabolism through AKT phosphorylation, reducing mitochondrial dysfunction and preventing myocardial remodeling following transaortic constriction (90). AKT signaling is impaired in the hearts of obese mice (91), and this pathway inhibits cardiac lipolysis (92). Furthermore, GA stress pathways have been found to promote apoptosis, particularly in neurological diseases, by modulating caspase cleavage and calcium influx (93). In Hela cells, the ferroptosis pathway was also activated by inducers of GA stress (94). Ferroptosis is an iron-dependent cell death process driven by lipid peroxidation, making these observations relevant for the study of GA and metabolic stress. There is clear evidence that the GA regulates lipid metabolism and that it responds to stress in relevant metabolically active tissues. However, the specific links involved in metabolic cardiomyopathy, particularly taking place in the myocardium, need further study to identify better potential therapeutic targets.
The ER and GA regulation of lipid metabolism are complex and largely unexplored. Both organelles have a clear role in sustaining protein synthesis and processing during metabolic stress conditions, which is essential for preventing HF. However, their part goes beyond protein quality control; they are deeply involved in different stages of lipid metabolism, from synthesis to storage and catabolism, by processing lipids and governing lipid droplet dynamics. Moreover, their sensing abilities trigger signaling pathways that coordinate their own functional capacity and that of other organelles. In metabolic cardiomyopathy, these tasks become even more critical due to the altered lipid profile state and the high lipotoxicity risks. Therefore, it follows that further studying and targeting ER and GA homeostasis for the treatment of metabolic cardiomyopathy could provide opportunities for the prevention of HF.
RR, OF, and AR-V collected references, drafted, and proofread the manuscript. RR and HG generated the figure. AR-V and WL designed the manuscript. All authors contributed to the article and approved the submitted version.
This work as supported by grants FS/15/16/31477, FS/18/73/33973, PG/19/66/34600, and FS/19/70/34650 to WL from the British Heart Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lee IM, Shiroma EJ, Lobelo F, Puska P, Blair SN, Katzmarzyk PT, et al. Effect of physical inactivity on major non-communicable diseases worldwide: an analysis of burden of disease and life expectancy. Lancet. (2012) 380:219–29. doi: 10.1016/S0140-6736(12)61031-9
2. Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet. (2012) 380:2224–60. doi: 10.1016/S0140-6736(12)61766-8
3. Bozkurt B, Aguilar D, Deswal A, Dunbar SB, Francis GS, Horwich T, et al. Contributory risk and management of comorbidities of hypertension, obesity, diabetes mellitus, hyperlipidemia, and metabolic syndrome in chronic heart failure: a scientific statement from the American heart association. Circulation. (2016) 134:e535–e78. doi: 10.1161/CIR.0000000000000450
4. Low Wang CC, Hess CN, Hiatt WR, Goldfine AB. Clinical update: cardiovascular disease in diabetes mellitus: atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus - mechanisms, management, and clinical considerations. Circulation. (2016) 133:2459–502. doi: 10.1161/CIRCULATIONAHA.116.022194
5. Jia G, Demarco V, Sowers Jr. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. (2016) 12:144–53. doi: 10.1038/nrendo.2015.216
6. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. (2020) 17:585–607. doi: 10.1038/s41569-020-0339-2
7. Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev. (2013) 18:149–66. doi: 10.1007/s10741-012-9313-3
8. Wong C, Marwick TH. Obesity cardiomyopathy: diagnosis and therapeutic implications. Nat Clin Pract Cardiovasc Med. (2007) 4:480–90. doi: 10.1038/ncpcardio0964
9. Liu W, Ruiz-Velasco A, Wang S, Khan S, Zi M, Jungmann A, et al. Metabolic stress-induced cardiomyopathy is caused by mitochondrial dysfunction due to attenuated Erk5 signaling. Nat Commun. (2017) 8:494. doi: 10.1038/s41467-017-00664-8
10. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. (2005) 146:5341–9. doi: 10.1210/en.2005-0938
11. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. (2014) 57:660–71. doi: 10.1007/s00125-014-3171-6
12. Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. (2008) 88:389–419. doi: 10.1152/physrev.00017.2007
13. Nguyen BY, Ruiz-Velasco A, Bui T, Collins L, Wang X, Liu W. Mitochondrial function in the heart: the insight into mechanisms and therapeutic potentials. Br J Pharmacol. (2019) 176:4302–18. doi: 10.1111/bph.14431
14. Bugger H, Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res. (2010) 88:229–40. doi: 10.1093/cvr/cvq239
15. Galloway CA, Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal. (2015) 22:1545–62. doi: 10.1089/ars.2015.6293
16. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. (2014) 114:524–37. doi: 10.1161/CIRCRESAHA.114.300559
17. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
18. Fillmore N MJ, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol. (2014) 171:2080–90. doi: 10.1111/bph.12475
19. Drosatos K, Schulze PC. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep. (2013) 10:109–21. doi: 10.1007/s11897-013-0133-0
20. Gordaliza-Alaguero I, Canto C, Zorzano A. Metabolic implications of organelle-mitochondria communication. EMBO Rep. (2019) 20:e47928. doi: 10.15252/embr.201947928
21. Jia AG, Hill RM, Sowers RJ. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
22. D'Souza K, Nzirorera C, Kienesberger PC. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim Biophys Acta. (2016) 1861:1513–24. doi: 10.1016/j.bbalip.2016.02.016
23. Kaur N, Raja R, Ruiz-Velasco A, Liu W. Cellular protein quality control in diabetic cardiomyopathy: from bench to bedside. Front Cardiovasc Med. (2020) 7:5309. doi: 10.3389/fcvm.2020.585309
24. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. (2012) 13:89–102. doi: 10.1038/nrm3270
25. Wang S, Binder P, Fang Q, Wang Z, Xiao W, Liu W, et al. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. Br J Pharmacol. (2018) 175:1293–304. doi: 10.1111/bph.13888
26. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. (2000) 2:326–32. doi: 10.1038/35014014
27. Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. (2013) 12:703–19. doi: 10.1038/nrd3976
28. Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Mol Cell. (2018) 69:169–81. doi: 10.1016/j.molcel.2017.06.017
29. Claudio H. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. (2012) 13:89.
30. Basseri S, Austin RC. Endoplasmic reticulum stress and lipid metabolism: mechanisms and therapeutic potential. Biochem Res Int. (2012) 2012:841362. doi: 10.1155/2012/841362
31. Han J, Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res. (2016) 57:1329–38. doi: 10.1194/jlr.R067595
32. Gu X, Li K, Laybutt DR, He ML, Zhao HL, Chan JC, et al. Bip overexpression, but not CHOP inhibition, attenuates fatty-acid-induced endoplasmic reticulum stress and apoptosis in HepG2 liver cells. Life Sci. (2010) 87:724–32. doi: 10.1016/j.lfs.2010.10.012
33. Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. (2008) 7:520–32. doi: 10.1016/j.cmet.2008.04.011
34. Xiao F, Deng J, Guo Y, Niu Y, Yuan F, Yu J, et al. BTG1 ameliorates liver steatosis by decreasing stearoyl-CoA desaturase 1 (SCD1) abundance and altering hepatic lipid metabolism. Sci Signal. (2016) 9:ra50. doi: 10.1126/scisignal.aad8581
35. Bobrovnikova-Marjon E, Hatzivassiliou G, Grigoriadou C, Romero M, Cavener DR, Thompson CB, et al. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc Natl Acad Sci USA. (2008) 105:16314–9. doi: 10.1073/pnas.0808517105
36. Sha H, He Y, Chen H, Wang C, Zenno A, Shi H, et al. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. (2009) 9:556–64. doi: 10.1016/j.cmet.2009.04.009
37. Deng Y, Wang ZV, Gordillo R, Zhu Y, Ali A, Zhang C, et al. Adipocyte Xbp1s overexpression drives uridine production and reduces obesity. Mol Metab. (2018) 11:1–17. doi: 10.1016/j.molmet.2018.02.013
38. Sha H, Yang L, Liu M, Xia S, Liu Y, Liu F, et al. Adipocyte spliced form of X-box-binding protein 1 promotes adiponectin multimerization and systemic glucose homeostasis. Diabetes. (2014) 63:867–79. doi: 10.2337/db13-1067
39. Jurczak MJ, Lee AH, Jornayvaz FR, Lee HY, Birkenfeld AL, Guigni BA, et al. Dissociation of inositol-requiring enzyme (IRE1alpha)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J Biol Chem. (2012) 287:2558–67. doi: 10.1074/jbc.M111.316760
40. Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. (2008) 320:1492–6. doi: 10.1126/science.1158042
41. Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci. (2009) 122(Pt 10):1626–36. doi: 10.1242/jcs.045625
42. Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, et al. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. (2010) 21:2975–86. doi: 10.1091/mbc.e09-02-0133
43. Chen X, Zhang F, Gong Q, Cui A, Zhuo S, Hu Z, et al. Hepatic ATF6 increases fatty acid oxidation to attenuate hepatic steatosis in mice through peroxisome proliferator-activated receptor alpha. Diabetes. (2016) 65:1904–15. doi: 10.2337/db15-1637
44. Lowe CE, Dennis RJ, Obi U, O'Rahilly S, Rochford JJ. Investigating the involvement of the ATF6alpha pathway of the unfolded protein response in adipogenesis. Int J Obes. (2012) 36:1248–51. doi: 10.1038/ijo.2011.233
45. Kim JB. Dynamic cross talk between metabolic organs in obesity and metabolic diseases. Exp Mol Med. (2016) 48:e214. doi: 10.1038/emm.2015.119
46. Jahng JW, Song E, Sweeney G. Crosstalk between the heart and peripheral organs in heart failure. Exp Mol Med. (2016) 48:e217. doi: 10.1038/emm.2016.20
47. Sha H, Sun S, Francisco AB, Ehrhardt N, Xue Z, Liu L, et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab. (2014) 20:458–70. doi: 10.1016/j.cmet.2014.06.015
48. Xu YX, Peloso GM, Nagai TH, Mizoguchi T, Deik A, Bullock K, et al. EDEM3 modulates plasma triglyceride level through its regulation of LRP1 expression. iScience. (2020) 23:100973. doi: 10.1016/j.isci.2020.100973
49. Gan L, Liu D, Xie D, Bond Lau W, Liu J, Christopher TA, et al. Ischemic Heart-Derived Small Extracellular Vesicles Impair Adipocyte Function. Circ Res. (2022) 130:48–66. doi: 10.1161/CIRCRESAHA.121.320157
50. Choi K, Kim H, Kang H, Lee SY, Lee SJ, Back SH, et al. Regulation of diacylglycerol acyltransferase 2 protein stability by gp78-associated endoplasmic-reticulum-associated degradation. FEBS J. (2014) 281:3048–60. doi: 10.1111/febs.12841
51. Olzmann JA, Richter CM, Kopito RR. Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc Natl Acad Sci USA. (2013) 110:1345–50. doi: 10.1073/pnas.1213738110
52. Suzuki M, Otsuka T, Ohsaki Y, Cheng J, Taniguchi T, Hashimoto H, et al. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol Biol Cell. (2012) 23:800–10. doi: 10.1091/mbc.e11-11-0950
53. Lee JN, Kim H, Yao H, Chen Y, Weng K, Ye J. Identification of Ubxd8 protein as a sensor for unsaturated fatty acids and regulator of triglyceride synthesis. Proc Natl Acad Sci U S A. (2010) 107:21424–9. doi: 10.1073/pnas.1011859107
54. Wang CW, Lee SC. The ubiquitin-like (UBX)-domain-containing protein Ubx2/Ubxd8 regulates lipid droplet homeostasis. J Cell Sci. (2012) 125(Pt 12):2930–9. doi: 10.1242/jcs.100230
55. Imai N, Suzuki M, Hayashi K, Ishigami M, Hirooka Y, Abe T, et al. Hepatocyte-specific depletion of UBXD8 induces periportal steatosis in mice fed a high-fat diet. PLoS ONE. (2015) 10:e0127114. doi: 10.1371/journal.pone.0127114
56. Bosma M, Dapito DH, Drosatos-Tampakaki Z, Huiping-Son N, Huang LS, Kersten S, et al. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim Biophys Acta. (2014) 1841:1648–55. doi: 10.1016/j.bbalip.2014.09.012
57. Park SY, Cho YR, Finck BN, Kim HJ, Higashimori T, Hong EG, et al. Cardiac-specific overexpression of peroxisome proliferator-activated receptor-alpha causes insulin resistance in heart and liver. Diabetes. (2005) 54:2514–24. doi: 10.2337/diabetes.54.9.2514
58. Ljubkovic M, Gressette M, Bulat C, Cavar M, Bakovic D, Fabijanic D, et al. Disturbed fatty acid oxidation, endoplasmic reticulum stress, and apoptosis in left ventricle of patients with type 2 diabetes. Diabetes. (2019) 68:1924–33. doi: 10.2337/db19-0423
59. Schiattarella GG, Altamirano F, Kim SY, Tong D, Ferdous A, Piristine H, et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat Commun. (2021) 12:1684. doi: 10.1038/s41467-021-21931-9
60. Kim JJ Li P, Huntley J, Chang JP, Arden KC, Olefsky JM. FoxO1 haploinsufficiency protects against high-fat diet-induced insulin resistance with enhanced peroxisome proliferator-activated receptor gamma activation in adipose tissue. Diabetes. (2009) 58:1275–82. doi: 10.2337/db08-1001
61. Behl Y, Krothapalli P, Desta T, Roy S, Graves DT. FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes. (2009) 58:917–25. doi: 10.2337/db08-0537
62. Zhao N, Tan H, Wang L, Han L, Cheng Y, Feng Y, et al. Palmitate induces fat accumulation via repressing FoxO1-mediated ATGL-dependent lipolysis in HepG2 hepatocytes. PLoS ONE. (2021) 16:e0243938. doi: 10.1371/journal.pone.0243938
63. Lu X, Yang X, Liu J. Differential control of ATGL-mediated lipid droplet degradation by CGI-58 and G0S2. Cell Cycle. (2010) 9:2719–25. doi: 10.4161/cc.9.14.12181
64. Jebessa ZH, Shanmukha KD, Dewenter M, Lehmann LH, Xu C, Schreiter F, et al. The lipid droplet-associated protein ABHD5 protects the heart through proteolysis of HDAC4. Nat Metab. (2019) 1:1157–67. doi: 10.1038/s42255-019-0138-4
65. Xie X, Tie YF, Lai S, Zhang YL Li HH, Liu Y. Cardiac-specific CGI-58 deficiency activates the ER stress pathway to promote heart failure in mice. Cell Death Dis. (2021) 12:1003. doi: 10.1038/s41419-021-04282-7
66. Li S, Zhang L, Ni R, Cao T, Zheng D, Xiong S, et al. Disruption of calpain reduces lipotoxicity-induced cardiac injury by preventing endoplasmic reticulum stress. Biochim Biophys Acta. (2016) 1862:2023–33. doi: 10.1016/j.bbadis.2016.08.005
67. Otoda T, Takamura T, Misu H, Ota T, Murata S, Hayashi H, et al. Proteasome dysfunction mediates obesity-induced endoplasmic reticulum stress and insulin resistance in the liver. Diabetes. (2013) 62:811–24. doi: 10.2337/db11-1652
68. Akoumi A, Haffar T, Mousterji M, Kiss RS, Bousette N. Palmitate mediated diacylglycerol accumulation causes endoplasmic reticulum stress, Plin2 degradation, and cell death in H9C2 cardiomyoblasts. Exp Cell Res. (2017) 354:85–94. doi: 10.1016/j.yexcr.2017.03.032
69. Zou L, Li X, Wu N, Jia P, Liu C, Jia D. Palmitate induces myocardial lipotoxic injury via the endoplasmic reticulum stressmediated apoptosis pathway. Mol Med Rep. (2017) 16:6934–9. doi: 10.3892/mmr.2017.7404
70. Park M, Sabetski A, Kwan Chan Y, Turdi S, Sweeney G. Palmitate induces ER stress and autophagy in H9c2 cells: implications for apoptosis and adiponectin resistance. J Cell Physiol. (2015) 230:630–9. doi: 10.1002/jcp.24781
71. Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. (2004) 10:1245–50. doi: 10.1038/nm1116
72. Palomer X, Capdevila-Busquets E, Botteri G, Salvado L, Barroso E, Davidson MM, et al. PPARbeta/delta attenuates palmitate-induced endoplasmic reticulum stress and induces autophagic markers in human cardiac cells. Int J Cardiol. (2014) 174:110–8. doi: 10.1016/j.ijcard.2014.03.176
73. Zuo A, Zhao X, Li T, Li J, Lei S, Chen J, et al. CTRP9 knockout exaggerates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy through inhibiting the LKB1/AMPK pathway. J Cell Mol Med. (2020) 24:2635–47. doi: 10.1111/jcmm.14982
74. De Matteis MA, Luini A. Exiting the Golgi complex. Nat Rev Mol Cell Biol. (2008) 9:273–84. doi: 10.1038/nrm2378
75. Sasaki K, Yoshida H. Golgi stress response and organelle zones. FEBS Lett. (2019) 593:2330–40. doi: 10.1002/1873-3468.13554
76. Xu J, Huang X. Lipid metabolism at membrane contacts: dynamics and functions beyond lipid homeostasis. Front Cell Dev Biol. (2020) 8:615856. doi: 10.3389/fcell.2020.615856
77. Thelen AM, Zoncu R. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol. (2017) 27:833–50. doi: 10.1016/j.tcb.2017.07.006
78. Bankaitis VA, Garcia-Mata R, Mousley CJ. Golgi membrane dynamics and lipid metabolism. Curr Biol. (2012) 22:R414–24. doi: 10.1016/j.cub.2012.03.004
79. Fan YQ, Wang YH, Shao YY, Shao XT, Hu J, Xu CQ, et al. [Exogenous spermine alleviates myocardial injury induced by hyperglycemia by inhibiting golgi stress]. Zhongguo Ying Yong Sheng Li Xue Za Zhi. (2019) 35:385–9. doi: 10.12047/j.cjap.5795.2019.082
80. Thorne RF, Ralston KJ, de Bock CE, Mhaidat NM, Zhang XD, Boyd AW, et al. Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochim Biophys Acta. (2010) 1803:1298–307. doi: 10.1016/j.bbamcr.2010.07.002
81. Beller M, Sztalryd C, Southall N, Bell M, Jackle H, Auld DS, et al. COPI complex is a regulator of lipid homeostasis. PLoS Biol. (2008) 6:e292. doi: 10.1371/journal.pbio.0060292
82. Wang T, Wei Q, Liang L, Tang X, Yao J, Lu Y, et al. OSBPL2 Is Required for the BInding of COPB1 to ATGL and the regulation of lipid droplet lipolysis. iScience. (2020) 23:101252. doi: 10.1016/j.isci.2020.101252
83. Ellong EN, Soni KG, Bui QT, Sougrat R, Golinelli-Cohen MP, Jackson CL. Interaction between the triglyceride lipase ATGL and the Arf1 activator GBF1. PLoS One. (2011) 6:e21889. doi: 10.1371/journal.pone.0021889
84. Soni KG, Mardones GA, Sougrat R, Smirnova E, Jackson CL, Bonifacino JS. Coatomer-dependent protein delivery to lipid droplets. J Cell Sci. (2009) 122(Pt 11):1834-41. doi: 10.1242/jcs.045849
85. Wilfling F, Thiam AR, Olarte MJ, Wang J, Beck R, Gould TJ, et al. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife. (2014) 3:e01607. doi: 10.7554/eLife.01607
86. Kim J, Kim H, Noh SH, Jang DG, Park SY, Min D, et al. Grasp55(-/-) mice display impaired fat absorption and resistance to high-fat diet-induced obesity. Nat Commun. (2020) 11:1418. doi: 10.1038/s41467-020-14912-x
87. Wang X, Wang H, Xu B, Huang D, Nie C, Pu L, et al. Receptor-mediated ER export of lipoproteins controls lipid homeostasis in mice and humans. Cell Metab. (2021) 33:350–66. doi: 10.1016/j.cmet.2020.10.020
88. Liu L, Cai J, Wang H, Liang X, Zhou Q, Ding C, et al. Coupling of COPII vesicle trafficking to nutrient availability by the IRE1alpha-XBP1s axis. Proc Natl Acad Sci USA. (2019) 116:11776–85. doi: 10.1073/pnas.1814480116
89. Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, et al. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet. (2003) 34:29–31. doi: 10.1038/ng1145
90. Padron-Barthe L, Villalba-Orero M, Gomez-Salinero JM, Acin-Perez R, Cogliati S, Lopez-Olaneta M, et al. Activation of serine one-carbon metabolism by calcineurin abeta1 reduces myocardial hypertrophy and improves ventricular function. J Am Coll Cardiol. (2018) 71:654–67. doi: 10.1016/j.jacc.2017.11.067
91. Zhang L, Ussher JR, Oka T, Cadete VJ, Wagg C, Lopaschuk GD. Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res. (2011) 89:148–56. doi: 10.1093/cvr/cvq266
92. Zheng P, Xie Z, Yuan Y, Sui W, Wang C, Gao X, et al. Plin5 alleviates myocardial ischaemia/reperfusion injury by reducing oxidative stress through inhibiting the lipolysis of lipid droplets. Sci Rep. (2017) 7:42574. doi: 10.1038/srep42574
93. He Q, Liu H, Deng S, Chen X, Li D, Jiang X, et al. The golgi apparatus may be a potential therapeutic target for apoptosis-related neurological diseases. Front Cell Dev Biol. (2020) 8:830. doi: 10.3389/fcell.2020.00830
Keywords: metabolic syndrome, metabolic cardiomyopathy, cardiac lipid metabolism, endoplasmic reticulum, Golgi apparatus
Citation: Raja R, Fonseka O, Ganenthiran H, Ruiz-Velasco A and Liu W (2022) The multifaceted roles of ER and Golgi in metabolic cardiomyopathy. Front. Cardiovasc. Med. 9:999044. doi: 10.3389/fcvm.2022.999044
Received: 20 July 2022; Accepted: 15 August 2022;
Published: 02 September 2022.
Edited by:
Xiaoqiang Tang, Sichuan University, ChinaCopyright © 2022 Raja, Fonseka, Ganenthiran, Ruiz-Velasco and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Liu, d2VpLmxpdUBtYW5jaGVzdGVyLmFjLnVr; Andrea-Ruiz-Velasco, YW5kcmVhLnJ1aXpAbWFuY2hlc3Rlci5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.