 Nicholas D. Fried1
Nicholas D. Fried1 Joshua M. Oakes1Anna K. Whitehead1
Joshua M. Oakes1Anna K. Whitehead1 Eric Lazartigues2,3,4,5
Eric Lazartigues2,3,4,5 Xinping Yue1
Xinping Yue1 Jason D. Gardner1*
Jason D. Gardner1*- 1Department of Physiology, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 2Department of Pharmacology & Experimental Therapeutics, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 3Cardiovascular Center of Excellence, New Orleans, LA, United States
- 4Neuroscience center of Excellence, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 5Southeast Louisiana Veterans Health Care Systems, New Orleans, LA, United States
Background: The heart undergoes structural and functional changes in response to injury and hemodynamic stress known as cardiac remodeling. Cardiac remodeling often decompensates causing dysfunction and heart failure (HF). Cardiac remodeling and dysfunction are significantly associated with cigarette smoking. Although cigarette smoking has declined, the roles of nicotine and novel tobacco products (including electronic cigarettes and heat-not-burn tobacco) in cardiac remodeling are unclear. In this perspective, we present evidence demonstrating maladaptive cardiac remodeling in nicotine-exposed mice undergoing hemodynamic stress with angiotensin (Ang)-II infusion and review preclinical literature linking nicotine and novel tobacco products with cardiac remodeling and dysfunction.
Methods: Adult, male C57BL/6J mice were exposed to room air or chronic, inhaled nicotine for 8 weeks. A subset of mice was infused with Ang-II via subcutaneous osmotic mini-pumps during the final 4 weeks of exposure. Left ventricular structure and function were assessed with echocardiography.
Results: Chronic, inhaled nicotine abrogated Ang-II-induced thickening of the left ventricular posterior wall, leading to reduced relative wall thickness. Ang-II infusion was associated with increased left ventricular mass index in both air- and nicotine-exposed mice.
Conclusions: These changes suggest a phenotypic shift from concentric hypertrophy to eccentric hypertrophy in nicotine-exposed, hemodynamically-stressed mice which could drive HF pathogenesis. These findings join a growing body of animal studies demonstrating cardiac remodeling and dysfunction following nicotine and electronic cigarette exposure. Further exploration is necessary; however, clinicians and researchers should not overlook these emerging products as potential risk factors in the pathogenesis of cardiac remodeling and associated diseases including HF.
Introduction
Following injury or hemodynamic stress, the heart undergoes extensive structural and functional changes known collectively as cardiac remodeling (1). The mechanisms driving cardiac remodeling are complex and involve cellular death, inflammation, oxidative stress, modified energy and calcium homeostasis, neurohormonal activation, altered contractile machinery, fibrosis and extracellular matrix remodeling, and cardiomyocyte hypertrophy with geometric changes (2). Cardiac remodeling is concerning due to its propensity to decompensate toward cardiac dysfunction and disease states including myocardial infarction and heart failure (HF) (2, 3). Cardiac remodeling, cardiac dysfunction, and HF are significantly associated with both current and former cigarette smoking, as well as second-hand smoke exposure (4–11). Although cigarette smoking has declined worldwide (12), the cardiovascular risks of using novel tobacco products, including electronic cigarettes (13–15) and heat-not-burn tobacco (16), are poorly established.
Electronic cigarettes generate vapor by heating liquid containing nicotine, vegetable glycerin, propylene glycol, flavoring additives, and other chemicals. In contrast, heat-not-burn tobacco products generate nicotine-containing vapor by heating a cigarette-like tobacco plug to sub-combustion temperatures; this process aims to provide a similar experience to cigarette smoking while reducing exposure to toxic inhalants (17). Studies comparing nicotine emissions between electronic cigarettes, heat-not-burn tobacco products, and combustible cigarettes have been inconclusive (18–21). Clinically, differences in nicotine delivery between various products are likely insignificant due to nicotine dose titration by consumers (22).
A recent cohort study of over 5 million Korean adults found that short-term cardiovascular disease risk was reduced in patients who switched from cigarette smoking to novel tobacco product use (23). While this finding is promising from a public health perspective, patients switching from cigarette smoking to novel tobacco product use had 1.7 times greater risk of developing cardiovascular disease in comparison to patients who quit nicotine and tobacco products entirely (23). Due to concerns surrounding the shifting nicotine and tobacco consumption landscape, representatives of the American Heart Association, World Heart Federation, American College of Cardiology, and European Society of Cardiology issued a joint statement calling for further study of the cardiovascular health effects of nicotine and novel tobacco product use (24).
Despite the well-established relationship between cigarette smoking and cardiovascular pathology, the roles of nicotine and novel tobacco product use in these conditions are unclear. Epidemiological studies of novel tobacco products currently face challenges including the long time-course of cardiovascular disease pathogenesis and confounding caused by previous and current use of other tobacco products in study subjects (25). One analysis of Waves 1 through 5 (2013-2019) of the Population Assessment of Tobacco and Health (PATH) Study found reduced risk of HF, myocardial infarction, and stroke in exclusive electronic cigarette users vs. combustible tobacco users (26); a second analysis of this cohort found no differences in cardiovascular disease events between combustible tobacco users, those who transitioned to electronic cigarette use or dual-use, and those that quit tobacco products (27). Authors of both studies, however, indicate significant limitations surrounding short follow-up duration in relation to the disease endpoints (26, 27). Other clinical trials using acute endpoints, which have been limited by small sample sizes, have noted potential risk factors for cardiac remodeling and dysfunction including elevated systolic and diastolic blood pressure, vascular stiffness, endothelial dysfunction, oxidative stress, and pro-thrombotic effects with the use of electronic cigarettes containing various nicotine concentrations (14). Notably, a previously healthy 19-year-old patient developed signs and symptoms of acute cardiac dysfunction during an episode of electronic cigarette or vaping use-associated lung injury (28). Small clinical studies have also identified elevated blood pressure, vascular stiffness, oxidative stress, and acute impairment of systolic and diastolic cardiac function in users of heat-not-burn tobacco (29–31).
A growing body of preclinical literature implicates nicotine and novel tobacco products in cardiac remodeling and dysfunction, highlighting concerning connections which have not been identified in early clinical studies of these devices. In this Perspective, we discuss that mounting preclinical evidence and present original data demonstrating maladaptive cardiac remodeling driven by chronic, inhaled nicotine in a mouse model.
Materials and methods
Animals and exposure model
Adult, male C57BL/6J mice (8–12 weeks old) from Jackson Laboratory (Bar Harbor, ME) were housed in a temperature (21 C) and humidity-controlled facility under a 12-h dark/light cycle, fed standard mouse chow (iOS Teklab Extruded Rodent Diet 2019S; Envigo, Huntingdon, United Kingdom) and water ad libitum. Nicotine-exposed mice were housed in a nicotine inhalation chamber (La Jolla Alcohol Research, La Jolla, CA), while air-exposed mice were housed outside of the chamber in the same room. All procedures were approved by the Louisiana State University Health Sciences Center Institutional Animal Care and Use Committee (#3674).
Nicotine vapor was produced by bubbling air through a gas-washing bottle containing free base nicotine (Sigma-Aldrich, St. Louis, MO), as previously described (32, 33). The concentrated nicotine vapor was then diluted with air and evenly distributed to each chamber at a flow rate of 7 to 8 L/min. Flow rate was adjusted to achieve plasma cotinine levels (approximately 600 ng/mL), comparable to those observed in human combustible cigarette and novel tobacco product users (34–36). Nicotine exposure followed a 12 h on/12 h off schedule, overlapping with the dark cycle. After 4 weeks of exposure, a subset of mice was implanted with subcutaneous osmotic mini-pumps (Alzet Model 1004; Durect Corporation, Minneapolis, MN) containing angiotensin-II (Ang-II, Sigma-Aldrich) for infusion at a dose of 450 ng/kg per min for a duration of 4 weeks while maintaining either nicotine- or air-exposure.
Echocardiography and analysis
B-mode and M-mode echocardiographic assessment was performed using the Vevo 3,100 Imaging System with a 30-MHz probe (VisualSonics, Toronto, Canada) at the conclusion of the 8-week exposure period under 1–1.5% isoflurane anesthesia. Heart rate was maintained between 450 and 550 beats per min. Ultrasound images were processed using the leading-edge technique in Vevo LAB on a minimum of 3 cardiac cycles to calculate group averages. Corrected left ventricular (LV) mass was calculated as 0.8424 x [(LVID;d + LVPW;d + LVAW;d)3–LVID;d3], where LVID;d was LV internal diameter at diastole, LVPW;d was LV posterior wall thickness at diastole, and LVAW;d was LV anterior wall thickness at diastole. Systolic measurements are denoted as LVID;s and LVPW;s. LV mass index (LVMI) was calculated by dividing corrected LV mass by tibia length at 8 weeks. Relative wall thickness (RWT) was calculated as 2 x (LVPW;d / LVID;d).
Data are displayed as mean ± SEM. Findings were analyzed by two-way ANOVA using GraphPad Prism 9 (GraphPad Software, San Diego, CA). A Tukey–Kramer post hoc test for multiple comparisons between means was performed when interactions were present on two-way ANOVA. P < 0.05 was considered statistically significant.
Chronic, inhaled nicotine drives maladaptive cardiac remodeling in hemodynamically-stressed mice
Cardiac dysfunction, eventually progressing to HF, occurs as a result of poor compensation following injury (such as myocardial infarction) or hemodynamic stress (such as volume or pressure overload). Cardiac hypertrophy is one mechanism that maintains cardiac function following exposure to these stresses. The compensatory phase of cardiac hypertrophy can be described using LaPlace's Law, which states that:
In this theoretical model, parallel deposition of sarcomeres in a process called concentric hypertrophy increases LV wall thickness and decreases chamber diameter, which normalizes wall stress in response to elevated LV pressure (37). In response to increased volume, the LV walls may become thinned through series deposition of sarcomeres with dilatation of the LV chamber in a process called eccentric hypertrophy (37). Hypertrophic changes eventually become maladaptive, however, leading to impaired function and worsened pathology (37, 38). The mechanisms underlying transition from adaptive cardiac remodeling to maladaptive cardiac remodeling are poorly understood and remain an active area of research with potential ramifications in the treatment of cardiac hypertrophy and HF (39).
In this study, C57BL/6J mice were exposed to room air or inhaled nicotine for 4 weeks. A subset of mice was infused with Ang-II via osmotic mini-pumps during an additional 4-weeks exposure period. LVPW;s (1.32 ± 0.03 mm, n = 19) and LVPW;d (0.93 ± 0.02 mm, n = 19) in Air + Ang-II mice were significantly increased vs. both Air mice (LVPW;s: 1.16 ± 0.03 mm, n = 22, P < 0.001; LVPW;d: 0.75 ± 0.02 mm, n = 22, P < 0.0001) and Nicotine + Ang-II mice (LVPW;s: 1.18 ± 0.03 mm, n = 19, P < 0.01; LVPW;d: 0.83 ± 0.03 mm, n = 19, P < 0.05). There were no significant differences in LVPW;s and LVPW;d between Nicotine + Ang-II mice and Nicotine mice (LVPW;s: 1.15 ± 0.02, n = 23; LVPW;d: 0.78 ± 0.02, n = 23) (Figures 1A,B). There were also no significant differences in LVID;s and LVID;s between any of the groups (Figures 1C,D).
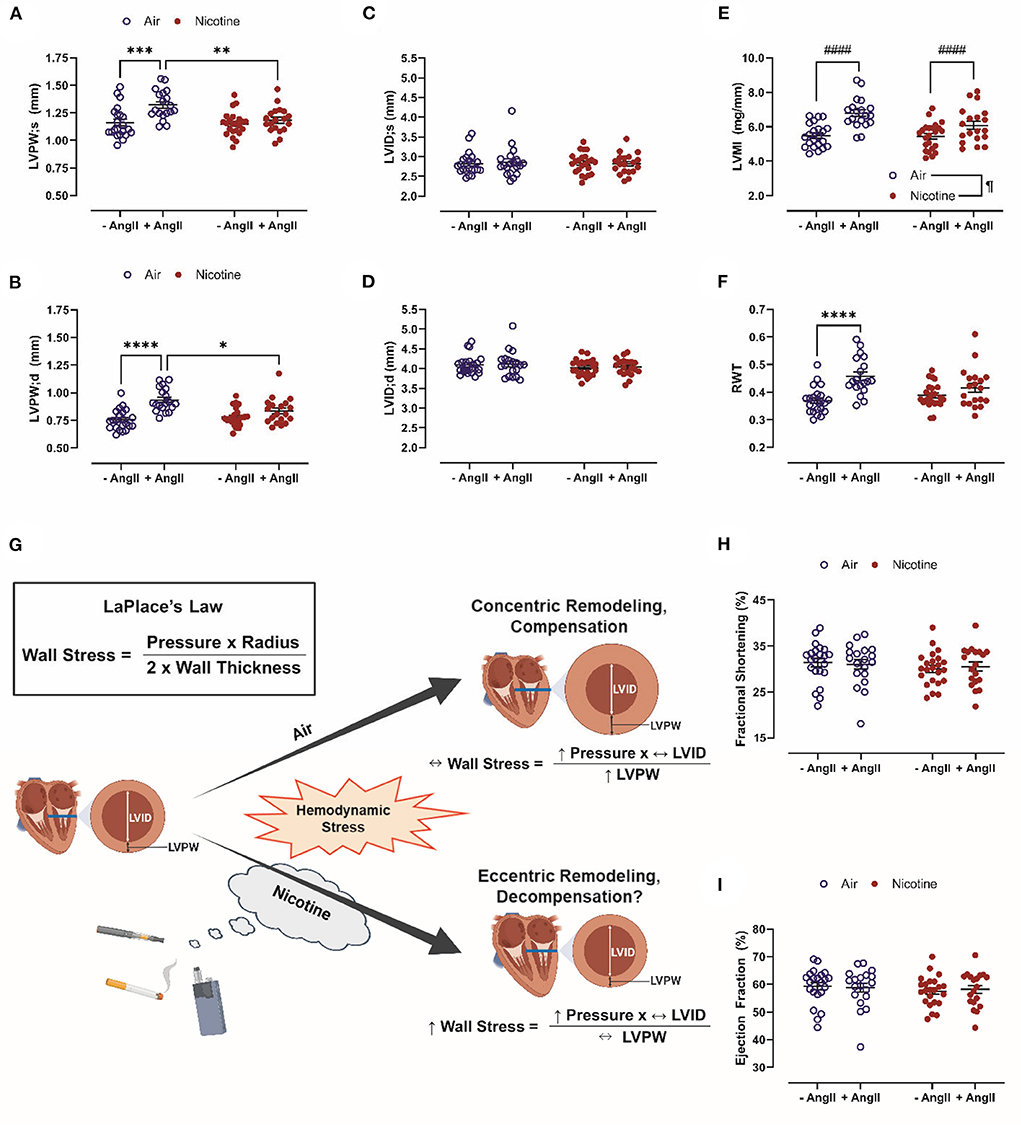
Figure 1. Chronic, inhaled nicotine drives maladaptive cardiac remodeling in mice undergoing hemodynamic stress. (A,B) Inhaled nicotine abrogates angiotensin (Ang)-II-induced left ventricular (LV) posterior wall thickening at systole and diastole (LVPW;s and ;d). (C,D) Neither Ang-II nor nicotine inhalation affect LV chamber diameter (LVID;s and ;d). (E) Ang-II increases LV mass indexed to tibia length (LVMI). (F) Ang-II increases relative wall thickness (RWT) in air-exposed mice only. (G) Inhaled nicotine exposure causes a shift from concentric to eccentric hypertrophy during hemodynamic stress which may exacerbate LV wall stress and promote decompensation based upon LaPlace's Law. (H,I) Neither Ang-II nor nicotine affect LV function measured by fractional shortening and ejection fraction. N = 19–23 per group. Data are displayed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way ANOVA with Tukey-Kramer post hoc test); #### indicates a main effect of Ang-II (P < 0.0001, two-way ANOVA); ¶indicates a main effect of nicotine (P < 0.05, two-way ANOVA). Figure 1G was created with BioRender.com.
These findings of impaired compensatory remodeling in a model of nicotine inhalation are consistent with studies using alternative nicotine exposure routes. Maternal nicotine exposure (100 μg/mL in drinking water during gestation) in rats reduces ejection fraction (EF), LVAW;s and LVPW;s in offspring via DNA methylation of cardiac-specific genes (40). Nicotine delivered by osmotic mini-pump at 6 mg/kg/day or 12 mg/kg/day in Sprague-Dawley and Long-Evans rats reduced heart length and heart weight, but did not change thickness of the LV, right ventricle (RV), or septal walls (41). Induction of pressure overload by transverse aortic constriction in ghrelin knockout mice induced elevations in heart weight indexed to tibial length, LVAW thickness, and LVPW thickness; treatment of this model with nicotine tartrate salt dissolved in the drinking water abrogated these changes (42). Rats subjected to LVAW myocardial infarction after 7-days of treatment with 1.75 mg/day of nicotine via patch were reported to have reduced LVAW thickness and increased dilatation of the LV chamber vs. mice that did not receive nicotine (43).
There was a main effect of Ang-II (P < 0.0001) to increase LVMI in both air-exposed and nicotine-exposed mice (Figure 1E). There was also a main effect of nicotine (P < 0.05) leading to a reduction of LVMI (Figure 1E). In contrast, RWT was increased in Air + Ang-II mice (P < 0.0001 vs. Air), but not Nicotine + Ang-II mice (P = 0.416 vs. Nicotine) (Figure 1F). These changes suggest three distinct groups: absent remodeling in Air and Nicotine mice, concentric hypertrophy in Air + Ang-II mice, and eccentric hypertrophy in Nicotine + Ang-II mice (44). Concentric and eccentric hypertrophy are both clinically correlated with HF (45). HF incidence is greater in patients with eccentric hypertrophy than in patients with concentric hypertrophy (45). Additionally, concentric hypertrophy is associated with relative preservation of cardiac function (HF with preserved EF, HFpEF) vs. eccentric hypertrophy (HF with reduced EF, HFrEF) (45). Matrix metalloproteinase (MMP) activation, leading to wall thinning and LV dilatation, is thought to play a mechanistic role in eccentric hypertrophy and HFrEF (44, 46, 47). Another study using osmotic delivery of both nicotine and Ang-II in mice reported increased MMP-2 activity, increased heart weight indexed to body weight, and dilatation of the aorta providing further support for our findings indicating a shift toward eccentric hypertrophy in mice exposed to inhaled nicotine while infused with Ang-II (48). Furthermore, cigarette smoke exposure upregulated MMP-9 and inhibited adaptive remodeling in a rat model of volume overload, leading to eccentric hypertrophy and impaired ventricular function, indicating overlap in the mechanisms of nicotine- and cigarette-induced pathology (49).
These findings, when considered in the context of LaPlace's Law, suggest that nicotine exposure induces a shift from concentric to eccentric hypertrophy during hemodynamic stress which may exacerbate LV wall stress and promote cardiac decompensation (Figure 1G). It is, however, important to acknowledge that Ang-II infusion is a model of hypertension and cardiac remodeling which does not progress to HF in mice. We found no significant differences in echocardiographic measurements of cardiac function (EF and fractional shortening, FS) between Air, Air + Ang-II, Nicotine, and Nicotine+Ang-II mice (Figures 1H,I). Future studies must explore additional models of hemodynamic stress which progress to cardiac dysfunction (such as transverse aortic constriction or myocardial ischemia-reperfusion injury) to better comprehend the role of nicotine in compensatory remodeling.
Discussion
Our study and others provide evidence of impaired cardiac compensation in animal models of nicotine exposure. Nicotine is of particular interest as the primary biologically active component of novel tobacco products; however, emissions from these devices contain hundreds of additional chemicals creating complex interactions. At present, there have been no independent studies of the cardiac effects of heat-not-burn tobacco in animals. Studies conducted by researchers affiliated with Philip Morris International, a tobacco company and producer of the IQOS heat-not-burn system, reported reduced atherosclerosis, LV remodeling and dysfunction, and cardiac gene changes in female Apolipoprotein E knockout mice exposed to heat-not-burn aerosol vs. cigarette smoke (50, 51). Preclinical evidence of cardiac remodeling and dysfunction following electronic cigarette exposure is, in contrast, more robust (Table 1).
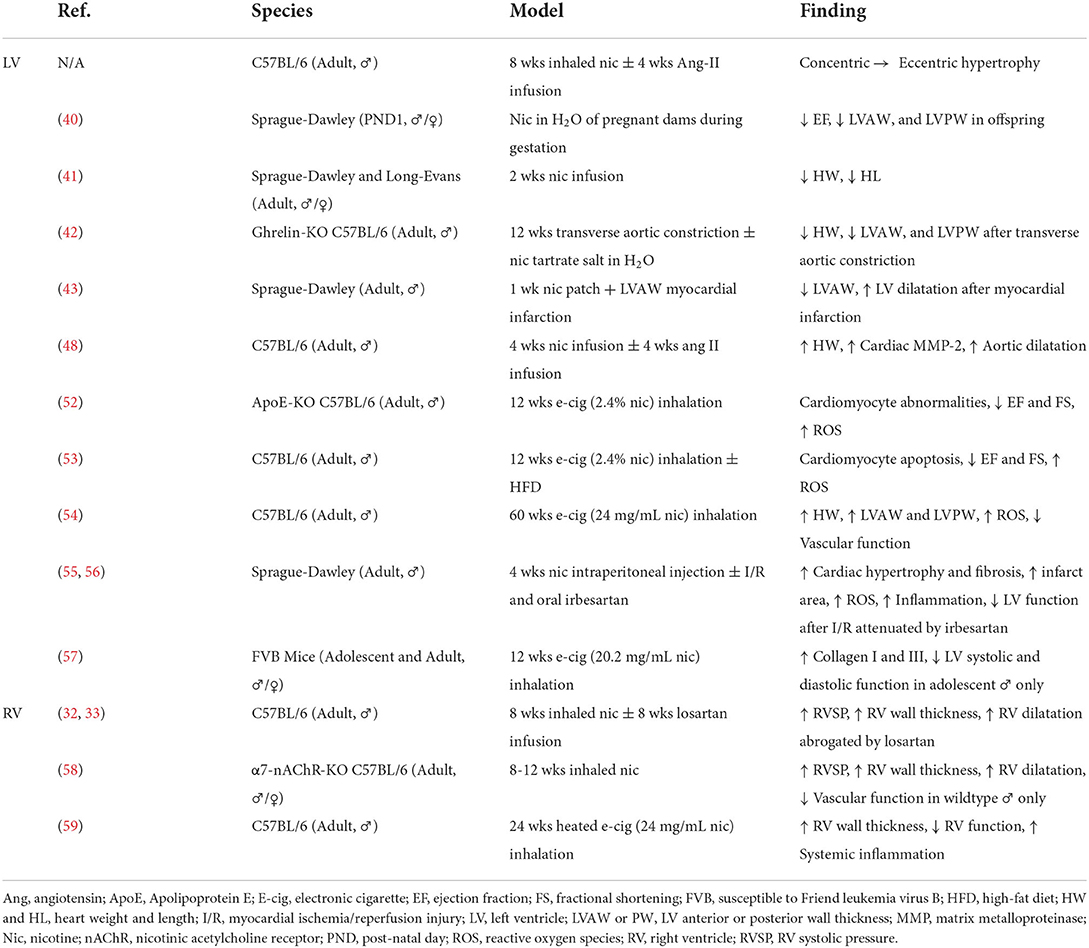
Table 1. Summary of animal studies demonstrating cardiac structural and functional changes associated with nicotine and novel tobacco products.
Exposing Apolipoprotein E knockout mice to 12 weeks of electronic cigarette vapor containing 2.4% nicotine caused extensive cellular changes in cardiomyocytes including shrunken nuclei (indicative of apoptosis), abnormal myofibrils in the cytoplasm, mitophagy, mitochondrial DNA lesions, and accumulation of lipids and reactive oxygen species (52). These cardiomyocyte changes resulted in cardiac dysfunction including reduced FS and EF (52). C567BL/6J mice fed high fat diet and exposed to 12 weeks of electronic cigarette vapor with 2.4% nicotine showed reduced FS and EF associated with cardiomyopathy-like cellular changes on electron microscopy of the ventricle (53). Cardiomyocytes from mice exposed to electronic cigarette vapor with 2.4% nicotine exhibited increased apoptosis, increased oxidative stress, and reduced AMPK phosphorylation; these in vivo and ex vivo findings were absent in mice exposed to electronic cigarette vapor with 0% nicotine (53).
In C57BL/6J mice, 60 weeks of inhalation exposure to electronic cigarettes containing 24 mg/mL of nicotine resulted in thickening of the LVAW and LVPW which were comparable to 60 weeks of exposure to 3R4F research cigarettes (54). These changes in LV chamber dimensions were associated with increased heart weight indexed to body weight, superoxide production in the cardiac tissue, and cardiovascular dysfunction (54). Treatment of Sprague-Dawley rats with irbesartan, an Ang-II type 1 receptor (AT1R) antagonist, ameliorated cardiac hypertrophy, fibrosis, reactive oxygen species accumulation, inflammation, and LV dysfunction induced by 28 days of 0.6 mg/kg of nicotine delivered by intraperitoneal injection (55). This nicotine exposure model also exacerbated LV dysfunction and infarct size in Langendorff-perfused hearts undergoing ex vivo ischemia-reperfusion injury (55, 56). Three months of exposure to electronic cigarette vapor containing 20.2 mg/mL of nicotine in male, adolescent FVB (Susceptible to the Friend leukemia virus B) mice induced LV systolic and diastolic dysfunction which were not present in adult mice or female, adolescent mice (57).
In addition to changes in the LV, nicotine and novel tobacco products induce changes in pulmonary circulation and RV remodeling. We have previously shown that chronic, inhaled nicotine exposure causes pulmonary hypertension associated with RV remodeling, elevated RV brain-type natriuretic peptide, and increased expression of angiotensin-converting enzyme in the RV, but not LV (32). These changes are abrogated by treatment with losartan, a specific AT1R antagonist (33). Nicotine-induced pulmonary hypertension, RV remodeling, and vascular dysfunction are ameliorated by α7 nicotinic cholinergic receptor knockout, and are absent in female mice (58). The pathogenesis of pulmonary hypertension induced by Sugen and hypoxia also involves α7 nicotinic cholinergic receptor-mediated cross-talk between cardiomyocytes and cardiac fibroblasts, leading to RV fibrosis (60). Treatment of cardiac fibroblasts isolated from the RV, but not the LV, with cigarette smoke extracts or 600 nM of nicotine stimulated fibroblast proliferation (61). Mice exposed to 6 months of heated electronic cigarette liquid containing 24 mg/mL of nicotine developed significant RV wall thickening and RV dysfunction associated with extensive changes in systemic inflammation (59).
This study is limited by the inclusion of a single nicotine dose and exposure duration. The dose- and time-response relationships between nicotine and maladaptive cardiac remodeling have not been established. The effect of nicotine on other cardiovascular parameters (including heart rate, skin temperature, and blood pressure), however, exhibits a flattened response with moderate doses achieving maximal effects (62). An expanding number of studies, including ours, demonstrate nicotine-associated cardiac remodeling and dysfunction using numerous dosages, exposure durations, routes of administration, and animal models. This suggests complex relationships which warrant examination in future studies.
Cigarette smoking is strongly associated with cardiac remodeling and dysfunction (4–11). Early clinical studies of novel tobacco products have shown promising reductions in cardiovascular diseases when compared to cigarette smoking (23, 26, 27). Despite this, caution must be exercised due to the prolonged disease course of cardiovascular pathology in relation to the short history of novel tobacco products. Clinical studies using acute endpoints have reported cardiovascular changes which may predispose patients to the development of cardiac remodeling and dysfunction (14). Additionally, a growing body of preclinical evidence has linked nicotine and novel tobacco product exposure with significant cardiac remodeling and dysfunction in animal models. These findings are particularly concerning due to novel tobacco product use among young, never smokers which may lead to future cigarette smoking (63). Further exploration is necessary; however, clinicians and researchers should not overlook nicotine and novel tobacco product use as potential risk factors in the pathogenesis of cardiac remodeling and associated diseases including HF.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by LSUHSC IACUC.
Author contributions
NF drafted the manuscript. EL, XY, and JG conceptualized experiments. NF and JO performed experiments and data analysis. NF and AW prepared figures. All authors edited the manuscript and approved final version.
Funding
This study was supported by National Heart, Lung, and Blood Institute grants R01HL135635 and R01HL135635-S1 to EL, XY, and JG.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol. (2000) 35:569–82. doi: 10.1016/S0735-1097(99)00630-0
2. Azevedo PS, Polegato BF, Minicucci MF, Paiva SAR, Zornoff LAM. Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol. (2015) 106:62–9. doi: 10.5935/abc.20160005
3. Torpy JM, Glass TJ, Glass RM. Left ventricular hypertrophy. JAMA. (2004) 292:2430. doi: 10.1001/jama.292.19.2430
4. Gopal DM, Kalogeropoulos AP, Georgiopoulou VV, Smith AL, Bauer DC, Newman AB, et al. Cigarette smoking exposure and heart failure risk in older adults: the health, aging, and body composition study. Am Heart J. (2012) 164:236–42. doi: 10.1016/j.ahj.2012.05.013
5. Skipina TM, Upadhya B, Soliman EZ. Secondhand smoke exposure is associated with prevalent heart failure: longitudinal examination of the national health and nutrition examination survey. Nicotine Tob Res. (2021) 23:1512–7. doi: 10.1093/ntr/ntab047
6. Watson M, Dardari Z, Kianoush S, Hall ME, DeFilippis AP, Keith RJ, et al. Relation between cigarette smoking and heart failure (from the multiethnic study of atherosclerosis). Am J Cardiol. (2019) 123:1972–7. doi: 10.1016/j.amjcard.2019.03.015
7. Ding N, Shah AM, Blaha MJ, Chang PP, Rosamond WD, Matsushita K. Cigarette smoking, cessation, and risk of heart failure with preserved and reduced ejection fraction. J Am Coll Cardiol. (2022) 79:2298–305. doi: 10.1016/j.jacc.2022.03.377
8. Goette A, Lendeckel U, Kuchenbecker A, Bukowska A, Peters B, Klein HU, et al. Cigarette smoking induces atrial fibrosis in humans via nicotine. Heart. (2007) 93:1056–63. doi: 10.1136/hrt.2005.087171
9. Watanabe Y, Tajiri K, Suzuki A, Nagata H, Kojima M. Influence of cigarette smoking on biventricular systolic function independent of respiratory function: a cross-sectional study. BMC Cardiovas Disord. (2020) 20:451. doi: 10.1186/s12872-020-01732-6
10. Kamimura D, Cain LR, Mentz RJ, White WB, Blaha MJ, DeFilippis AP, et al. Cigarette smoking and incident heart failure. Circulation. (2018) 137:2572–82. doi: 10.1161/CIRCULATIONAHA.117.031912
11. Skipina TM, Upadhya B, Soliman EZ. Exposure to secondhand smoke is associated with increased left ventricular mass. Tob Induc Dis. (2021) 19:1–7. doi: 10.18332/tid/136415
12. Hoffman SJ, Mammone J, Katwyk SRV, Sritharan L, Tran M, Al-Khateeb S, et al. Cigarette consumption estimates for 71 countries from 1970 to 2015: systematic collection of comparable data to facilitate quasi-experimental evaluations of national and global tobacco control interventions. BMJ. (2019) 365:l2231. doi: 10.1136/bmj.l2231
13. Tarran R, Barr RG, Benowitz NL, Bhatnagar A, Chu HW, Dalton P, et al. E-cigarettes and cardiopulmonary health. Function. (2021) 2:zqab004. doi: 10.1093/function/zqab004
14. Neczypor EW, Mears MJ, Ghosh A, Sassano MF, Gumina RJ, Wold LE, et al. E-cigarettes and cardiopulmonary health: review for clinicians. Circulation. (2022) 145:219–32. doi: 10.1161/CIRCULATIONAHA.121.056777
15. Espinoza-Derout J, Shao XM, Lao CJ, Hasan KM, Rivera JC, Jordan MC, et al. Electronic cigarette use and the risk of cardiovascular diseases. Front Cardiovas Med. (2022) 9:879726. doi: 10.3389/fcvm.2022.879726
16. Fried ND, Gardner JD. Heat-not-burn tobacco products: an emerging threat to cardiovascular health. Am J Physiol-Heart Circul Physiol. (2020) 319:H1234–9. doi: 10.1152/ajpheart.00708.2020
17. Phillip, Morris International,. IQOS Heated Tobacco Products [Internet]. Available online at: https://www.pmi.com/smoke-free-products/iqos-our-tobacco-heating-system (accessed August 10, 2022)
18. Yu SJ, Kwon MK, Choi W, Son YS. Preliminary study on the effect of using heat-not-burn tobacco products on indoor air quality. Environ Res. (2022) 212:113217. doi: 10.1016/j.envres.2022.113217
19. Cancelada L, Sleiman M, Tang X, Russell ML, Montesinos VN, Litter MI, et al. Heated tobacco products: volatile emissions and their predicted impact on indoor air quality. Environ Sci Technol. (2019) 53:7866–76. doi: 10.1021/acs.est.9b02544
20. Farsalinos KE, Yannovits N, Sarri T, Voudris V, Poulas K. Nicotine delivery to the aerosol of a heat-not-burn tobacco product: comparison with a tobacco cigarette and e-cigarettes. Nicotine Tob Res. (2018) 20:1004–9. doi: 10.1093/ntr/ntx138
21. Salman R, Talih S, El-Hage R, Haddad C, Karaoghlanian N, El-Hellani A, et al. Free-base and total nicotine, reactive oxygen species, and carbonyl emissions from IQOS, a heated tobacco product. Nicotine Tob Res. (2019) 21:1285–8. doi: 10.1093/ntr/nty235
22. Benowitz NL, St.Helen G, Liakoni E. Clinical pharmacology of electronic nicotine delivery systems (ENDS): implications for benefits and risks in the promotion of the combusted tobacco endgame. J Clin Pharmacol. (2021) 61:S18–36. doi: 10.1002/jcph.1915
23. Choi S, Lee K, Park SM. Combined associations of changes in noncombustible nicotine or tobacco product and combustible cigarette use habits with subsequent short-term cardiovascular disease risk among south korean men: a nationwide cohort study. Circulation. (2021) 144:1528–38. doi: 10.1161/CIRCULATIONAHA.121.054967
24. Willett J, Achenbach S, Pinto FJ, Poppas A, Elkind MSV. The tobacco endgame: eradicating a worsening epidemic a joint opinion from the American heart association, world heart federation, American college of cardiology, and the european society of cardiology. J Am Coll Cardiol. (2021) 78:77–81. doi: 10.1016/j.jacc.2021.04.005
25. Benowitz NL, Liakoni E. Tobacco use disorder and cardiovascular health. Addiction. (2022) 117:1128–38. doi: 10.1111/add.15703
26. Berlowitz JB, Xie W, Harlow AF, Hamburg NM, Blaha MJ, Bhatnagar A, et al. E-cigarette use and risk of cardiovascular disease: a longitudinal analysis of the PATH study (2013–2019). Circulation. (2022) 145:1557–9. doi: 10.1161/CIRCULATIONAHA.121.057369
27. Mahoney MC, Rivard C, Kimmel HL, Hammad HT, Sharma E, Halenar MJ, et al. Cardiovascular outcomes among combustible-tobacco and electronic nicotine delivery system (ENDS) users in waves 1 through 5 of the population assessment of tobacco and health (PATH) study, 2013–2019. Int J Environ Res Public Health. (2022) 19:4137. doi: 10.3390/ijerph19074137
28. Amirahmadi R, Childress J, Patel S, Wagner LA. Electric cigarette-related lung injury and cardiovascular insult. BMJ Case Rep CP. (2021) 14:e238352. doi: 10.1136/bcr-2020-238352
29. Hwang JH Ryu DH, Park SW. Heated tobacco products: cigarette complements, not substitutes. Drug Alcohol Depend. (2019) 204:107576. doi: 10.1016/j.drugalcdep.2019.107576
30. Ioakeimidis N, Emmanouil E, Terentes-Printzios D, Dima I, Aznaouridis K, Tousoulis D, et al. Acute effect of heat-not-burn versus standard cigarette smoking on arterial stiffness and wave reflections in young smokers. Eur J Prev Cardiolog. (2020) 2047487320918365. doi: 10.1177/2047487320918365
31. Yaman B, Akpinar O, Kemal HS, Cerit L, Yüksek Ü, Söylemez N, et al. Comparison of IQOS (heated tobacco) and cigarette smoking on cardiac functions by two-dimensional speckle tracking echocardiography. Toxicol Appl Pharmacol. (2021) 423:115575. doi: 10.1016/j.taap.2021.115575
32. Oakes JM, Xu J, Morris TM, Fried ND, Pearson CS, Lobell TD, et al. Effects of chronic nicotine inhalation on systemic and pulmonary blood pressure and right ventricular remodeling in mice. Hypertension. (2020) 75:1305–14. doi: 10.1161/HYPERTENSIONAHA.119.14608
33. Fried ND, Morris TM, Whitehead A, Lazartigues E, Yue X, Gardner JD. Angiotensin II type 1 receptor mediates pulmonary hypertension and right ventricular remodeling induced by inhaled nicotine. Am J Physiol-Heart Circul Physiol. (2021) 320:H1526–34. doi: 10.1152/ajpheart.00883.2020
34. Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology. (2007) 190:269–319. doi: 10.1007/s00213-006-0441-0
35. Marsot A, Simon N. Nicotine and cotinine levels with electronic cigarette: a review. Int J Toxicol. (2016) 35:179–85. doi: 10.1177/1091581815618935
36. Schirone L, Loffredo L, Carnevale R, Battaglia S, Marti R, Pizzolo S, et al. Sex-related differences in oxidative, platelet, and vascular function in chronic users of heat-not-burn vs. traditional combustion cigarettes. Antioxidants. (2022) 11:1237. doi: 10.3390/antiox11071237
37. Nadruz W. Myocardial remodeling in hypertension. J Hum Hypertens. (2015) 29:1–6. doi: 10.1038/jhh.2014.36
38. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. (2018) 15:387–407. doi: 10.1038/s41569-018-0007-y
39. Ren Z, Yu P, Li D, Li Z, Liao Y, Wang Y, et al. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation. (2020) 141:1704–19. doi: 10.1161/CIRCULATIONAHA.119.043053
40. Jiang XY, Feng YL, Ye LT, Li XH, Feng J, Zhang MZ, et al. Inhibition of gata4 and Tbx5 by nicotine-mediated DNA methylation in myocardial differentiation. Stem Cell Rep. (2017) 8:290–304. doi: 10.1016/j.stemcr.2016.12.016
41. Elliott BM, Faraday MM, Grunberg NE. Effects of nicotine on heart dimensions and blood volume in male and female rats. Nicotine Tob Res. (2003) 5:341–8. doi: 10.1080/1462220031000094114
42. Mao Y, Tokudome T, Kishimoto I, Otani K, Nishimura H, Yamaguchi O, et al. Endogenous ghrelin attenuates pressure overload–induced cardiac hypertrophy via a cholinergic anti-inflammatory pathway. Hypertension. (2015) 65:1238–44. doi: 10.1161/HYPERTENSIONAHA.114.04864
43. Villarreal FJ, Hong D, Omens J. Nicotine-modified postinfarction left ventricular remodeling. Am J Physiol-Heart Circul Physiol. (1999) 276:H1103–6. doi: 10.1152/ajpheart.1999.276.3.H1103
44. Nauta JF, Hummel YM, Tromp J, Ouwerkerk W, van der Meer P, Jin X, et al. Concentric vs. eccentric remodelling in heart failure with reduced ejection fraction: clinical characteristics, pathophysiology and response to treatment. Eur J Heart Fail. (2020) 22:1147–55. doi: 10.1002/ejhf.1632
45. Velagaleti RS, Gona P, Pencina MJ, Aragam J, Wang TJ, Levy D, et al. Left ventricular hypertrophy patterns and incidence of heart failure with preserved versus reduced ejection fraction. Am J Cardiol. (2014) 113:117–22. doi: 10.1016/j.amjcard.2013.09.028
46. Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. (2010) 122:2727–35. doi: 10.1161/CIRCULATIONAHA.110.942268
47. Parente JM, Blascke de. Mello MM, Silva PHL da, Omoto ACM, Pernomian L, Oliveira IS de, et al. MMP inhibition attenuates hypertensive eccentric cardiac hypertrophy and dysfunction by preserving troponin I and dystrophin. Biochem Pharmacol. (2021) 193:114744. doi: 10.1016/j.bcp.2021.114744
48. Colombo ES, Davis J, Makvandi M, Aragon M, Lucas SN, Paffett ML, et al. Effects of nicotine on cardiovascular remodeling in a mouse model of systemic hypertension. Cardiovasc Toxicol. (2013) 13:364–9. doi: 10.1007/s12012-013-9217-z
49. Bradley JM, Nguyen JB, Fournett AC, Gardner JD. Cigarette smoke exacerbates ventricular remodeling and dysfunction in the volume overloaded heart. Microsc Microanal. (2012) 18:91–8. doi: 10.1017/S1431927611012207
50. Szostak J, Titz B, Schlage WK, Guedj E, Sewer A, Phillips B, et al. Structural, functional, and molecular impact on the cardiovascular system in ApoE-/- mice exposed to aerosol from candidate modified risk tobacco products, Carbon Heated Tobacco Product 12 and Tobacco Heating System 22, compared with cigarette smoke. Chemico-Biol Interact. (2020) 315:108887. doi: 10.1016/j.cbi.2019.108887
51. Szostak J, Boué S, Talikka M, Guedj E, Martin F, Phillips B, et al. Aerosol from Tobacco Heating System 22 has reduced impact on mouse heart gene expression compared with cigarette smoke. Food Chem Toxicol. (2017) 101:157–67. doi: 10.1016/j.fct.2017.01.013
52. Espinoza-Derout J, Hasan KM, Shao XM, Jordan MC, Sims C, Lee DL, et al. Chronic intermittent electronic cigarette exposure induces cardiac dysfunction and atherosclerosis in apolipoprotein-E knockout mice. Am J Physiol-Heart Circul Physiol. (2019) 317:H445–59. doi: 10.1152/ajpheart.00738.2018
53. Hasan KM, Friedman TC, Parveen M, Espinoza-Derout J, Bautista F, Razipour MM, et al. Electronic cigarettes cause alteration in cardiac structure and function in diet-induced obese mice. PLoS ONE. (2020) 15:e0239671. doi: 10.1371/journal.pone.0239671
54. El-Mahdy MA, Mahgoup EM, Ewees MG, Eid MS, Abdelghany TM, Zweier JL. Long-term electronic cigarette exposure induces cardiovascular dysfunction similar to tobacco cigarettes: role of nicotine and exposure duration. Am J Physiol-Heart Circul Physiol. (2021) 320:H2112–29. doi: 10.1152/ajpheart.00997.2020
55. Ramalingam A, Budin SB, Fauzi NM, Ritchie RH, Zainalabidin S. Angiotensin II type I receptor antagonism attenuates nicotine-induced cardiac remodeling, dysfunction, and aggravation of myocardial ischemia-reperfusion injury in rats. Front Pharmacol. (2019) 10:1493. doi: 10.3389/fphar.2019.01493
56. Ramalingam A, Budin SB, Mohd Fauzi N, Ritchie RH, Zainalabidin S. Targeting mitochondrial reactive oxygen species-mediated oxidative stress attenuates nicotine-induced cardiac remodeling and dysfunction. Sci Rep. (2021) 11:13845. doi: 10.1038/s41598-021-93234-4
57. Neczypor EW, Saldaña TA, Mears MJ, Aslaner DM, Escobar YNH, Gorr MW, et al. E-cigarette aerosol reduces left ventricular function in adolescent mice. Circulation. (2022) 145:868–70. doi: 10.1161/CIRCULATIONAHA.121.057613
58. Whitehead AK, Fried ND Li Z, Neelamegam K, Pearson CS, LaPenna KB, et al. Alpha7 nicotinic acetylcholine receptor mediates chronic nicotine inhalation-induced cardiopulmonary dysfunction. Clin Sci. (2022) 136:973–87. doi: 10.1042/CS20220083
59. Getiye Y, Peterson MR, Phillips BD, Carrillo D, Bisha B, He G. E-cigarette exposure with or without heating the e-liquid induces differential remodeling in the lungs and right heart of mice. J Mol Cell Cardiol. (2022) 168:83–95. doi: 10.1016/j.yjmcc.2022.04.014
60. Vang A, Bos D da SG, Fernandez-Nicolas A, Zhang P, Morrison AR, Mancini TJ, et al. α7 Nicotinic acetylcholine receptor mediates right ventricular fibrosis and diastolic dysfunction in pulmonary hypertension. JCI Insight. (2021) 12:e142945. doi: 10.1172/jci.insight.142945
61. Vang A, Clements RT, Chichger H, Kue N, Allawzi A, O'Connell K, et al. Effect of α7 nicotinic acetylcholine receptor activation on cardiac fibroblasts: a mechanism underlying RV fibrosis associated with cigarette smoke exposure. Am J Physiol-Lung Cel Mol Physiol. (2017) 312:L748–59. doi: 10.1152/ajplung.00393.2016
62. Benowitz NL, Jacob III P, Herrera B. Nicotine intake and dose response when smoking reduced–nicotine content cigarettes. Clin Pharmacol Ther. (2006) 80:703–14. doi: 10.1016/j.clpt.2006.09.007
Keywords: nicotine, electronic cigarettes (E-cigarettes), heat-not-burn (HNB), cardiac remodeling, novel tobacco products
Citation: Fried ND, Oakes JM, Whitehead AK, Lazartigues E, Yue X and Gardner JD (2022) Nicotine and novel tobacco products drive adverse cardiac remodeling and dysfunction in preclinical studies. Front. Cardiovasc. Med. 9:993617. doi: 10.3389/fcvm.2022.993617
Received: 13 July 2022; Accepted: 20 September 2022;
Published: 06 October 2022.
Edited by:
Inna P. Gladysheva, University of Arizona, United StatesReviewed by:
Xuesi Wang, Capital Medical University, ChinaCopyright © 2022 Fried, Oakes, Whitehead, Lazartigues, Yue and Gardner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason D. Gardner, amdhcmRuQGxzdWhzYy5lZHU=