
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 20 September 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.988266
This article is part of the Research Topic Oxidative Stress, Inflammation and Atherosclerosis-Related Diseases: From basic to clinical research View all 11 articles
 Wenjing Li1,2†
Wenjing Li1,2† Kehan Jin3†Jichang Luo4,5†Wenlong Xu4,5†Yujie Wu1Jia Zhou3Yilin Wang6
Kehan Jin3†Jichang Luo4,5†Wenlong Xu4,5†Yujie Wu1Jia Zhou3Yilin Wang6 Ran Xu4,5
Ran Xu4,5 Liqun Jiao4,5,7*
Liqun Jiao4,5,7* Tao Wang4,5*
Tao Wang4,5* Ge Yang1,2*
Ge Yang1,2*Atherosclerosis (AS) is a common cardiovascular disease with complex pathogenesis, in which multiple pathways and their interweaving regulatory mechanism remain unclear. The primary transcription factor NF-κB plays a critical role in AS via modulating the expression of a series of inflammatory mediators under various stimuli such as cytokines, microbial antigens, and intracellular stresses. Endoplasmic reticulum (ER) stress, caused by the disrupted synthesis and secretion of protein, links inflammation, metabolic signals, and other cellular processes via the unfolded protein response (UPR). Both NF-κB and ER stress share the intersection regarding their molecular regulation and function and are regarded as critical individual contributors to AS. In this review, we summarize the multiple interactions between NF-κB and ER stress activation, including the UPR, NLRP3 inflammasome, and reactive oxygen species (ROS) generation, which have been ignored in the pathogenesis of AS. Given the multiple links between NF-κB and ER stress, we speculate that the integrated network contributes to the understanding of molecular mechanisms of AS. This review aims to provide an insight into these interactions and their underlying roles in the progression of AS, highlighting potential pharmacological targets against the atherosclerotic inflammatory process.
The transcription factor NF-κB regulates immunity by controlling the expression of genes associated with inflammation. In mammals, five proteins belonging to the NF-κB family have been identified, NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and cRel (Table 1). NF-κB exists in the cytoplasm in the form of homodimer (e.g., p50) or heterodimer (e.g., p50/p65) as a family of structurally related proteins (1, 2). It moves into the nucleus to transcribe target genes upon activation. Highly conservative NF-κB plays critical and stable roles in the immune response or embryonic development of many species (3). Recently, some studies have found that the NF-κB signaling pathway is associated with therapy resistance in breast and ovarian cancer (4, 5). On the other hand, accumulating evidence has proved that the NF-κB signaling pathway plays a key role in the development of many inflammatory metabolic diseases such as obesity, insulin resistance, and atherosclerosis (AS) (6).

Table 1. Components and characteristics of the NF-κB signaling pathway.
The endoplasmic reticulum (ER) is an organelle responsible for protein folding. In the ER, unfolded or misfolded proteins are detected and retained until they are properly folded or degraded. Disturbance in ER protein homeostasis leads to ER stress, activating a specific signaling pathway termed the unfolded protein response (UPR). The UPR is initiated by activation of three ER membrane-bound transducers including inositol requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF6), and protein kinase-RNA like ER kinase (PERK), which alleviates ER stress and helps cells adapt to and survive from ER stress caused by various stimuli (7). However, if the ER stress cannot be resolved, the UPR initiates programmed cell death.
Atherosclerosis is a chronic inflammatory disease contributing to the main pathological basis of ischemic heart disease, myocardial infarct and stroke (8, 9). Increasing evidence has documented that both NF-κB and ER stress closely affect the course of AS, and targeting those pathways may provide new approaches for the treatments against it (10). Herein, some interesting crosstalk in the molecular signaling pathways between NF-κB and ER stress in AS has been reviewed. In this regard, it is reasonable that these links may also be related to AS, which may offer promising opportunities for new strategies against AS.
NF-κB activation is initiated from extracellular stimulation signals and is precisely regulated. NF-κB1 (p50) and NF-κB2 (p52) are produced by cleavage of precursors p105 and p100, respectively. In resting cells, NF-κB is kept in the cytosol in its inactive form by binding to IκB (inhibitor of NF-κB) molecule (11). This binding prevents its nuclear localization and transcriptional function by masking the nuclear localization sequence (NLS) at the C-terminus of Rel Homology Region (RHR) (12). RelA (p65), RelB, and cRel contain a transactivation domain (TAD) at the C-terminal end which is responsible for transcribing target genes (Table 1) (13). Thereby, NF-κB dimer consisting of at least one of these three subunits is an active transcription factor, whereas NF-κB containing only p50 and p52 suppresses transcription due to lack of TAD, despite being able to bind to DNA (14).
IκB proteins consist of three groups: the classical IκB proteins, the precursor proteins, and the atypical (nuclear) IκB proteins (14) (Table 1). All of them have an ankyrin repeat sequence (AnkR) for interaction with Rel proteins (2, 15). IκBα, IκBβ, and IκBε belong to the typical group and share the conserved two serine residues at the N-terminal whose phosphorylation regulates the ubiquitination of itself (11). IκBα is associated with dimers of p50-RelA or p50-cRel. It keeps NF-κB in the cytoplasm through an exclusive nuclear export sequence that is exposed when bound to NF-κB. In contrast, NF-κB with IκBβ can locate in the nucleus stably. IκBε and IκBα are found to be the negative feedback regulators of NF-κB back to the cytoplasm (16, 17). NF-κB precursors, p100 (IκBδ) and p105 (IκBγ), also inhibit NF-κB by assembling into high-molecular-weight complexes (18). Phosphorylation of p105 targets it for complete degradation, but it may also promote p105 to be processed into p50 in some cell types (19–21), forming p50-RelA, p50-cRel, or p50 homodimers. Atypical IκB proteins include IκBζ, BCL-3, and IκBNS (Table 1). The most distinct feature of classical IκBs is their extra functions to positively regulate NF-κB (22).
When cells are stimulated by cytokines or pathogen-associated molecular patterns (PAMPs) binding to membrane receptors, signaling cascades initiate and finally converge on the activation of the IκB kinase (IKK) complex (23). The IKK complex consists of three subunits, the catalytic subunits IKKα (IKK1) and IKKβ (IKK2), and the regulatory subunit NF-κB essential modulator (NEMO or IKKγ) (Table 1). IκBs are phosphorylated by the IKK complex, then selectively ubiquitinated by E3 ubiquitin ligase (24), and finally degraded by the proteasome, thus allowing NF-κB translocation to the nucleus. In the nucleus, it is bound to the coactivator molecule to have optimal transcriptional activity (25), leading to gene transcription of growth factors, cytokines, chemokines, adhesion molecules, and other immunoregulatory molecules (Figure 1).
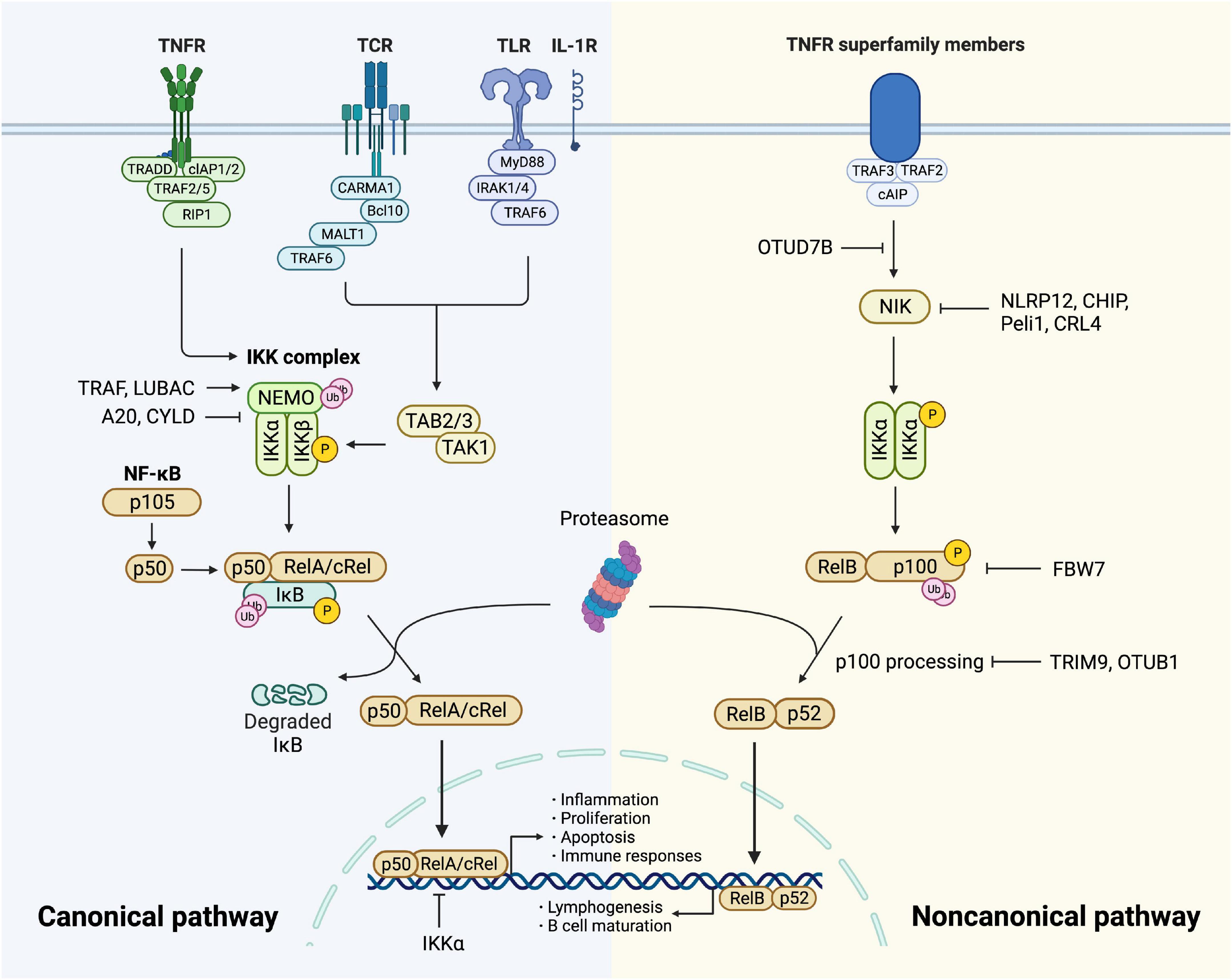
Figure 1. Canonical and non-canonical NF-κB pathway. The canonical pathway is induced via activation of receptors like TNFR, TCR, TLR, and IL-1R. When TNFR is activated by ligands, it recruits TRADD and drives the assembly of cIAP, TRAF, and RIP1 which is then recruited to NEMO and subsequent formation of IKK complex. TCR recruits CBM complex which is then ubiquitinated by TRAF6, resulting in the activation of TAK1. TLR and IL-1R recruits MyD88 and IRAK1/4, followed by TRAF6 to activate TAK and then IKK complex. TAK1 phosphorylates and activates IKK complex via phosphorylation of IKKβ. Then IκB family members phosphorylated by IKK undergo ubiquitin-dependent degradation, resulting in the release of NF-κB dimers. The canonical NF-κB pathway is regulated precisely. IKKα impedes RelA binding to DNA in nucleus. A20 and CYLD destabilize IKK complex via their deubiquitination activities. The activity of NF-κB is increased by TRAF- and LUBAC-mediated ubiquitination of NEMO. The non-canonical NF-κB pathway is initiated from the stimulation of specific TNFRs, which triggers the recruitment of TRAF3-TRAF2-cIAP and eventually results in stabilization and accumulation of NIK, which is impeded by deubiquitinase OTUD7B. Degradation of NIK is promoted by NLRP12, CHIP, Peli1, and CRL4. NIK phosphorylates and activates IKKα, triggering phosphorylation and ubiquitylation of p100. RelB and p52 generated from p100 constitute NF-κB heterodimer that conducts nuclear translocation and gene transcription. TRIM9 and OTUB1 inhibit p100 processing and FBW7 mediates p100 destruction.
Under various stimuli like cytokines, lipopolysaccharide (LPS), UV irradiation, intracellular stresses, and autoantibodies, NF-κB is activated and triggers modification signals. The activation involves two signaling pathways: the canonical and the non-canonical (alternative) pathway (26).
The canonical pathway is initiated by tumor necrosis factor receptor (TNFR), T cell receptor (TCR), Toll-like receptor (TLR), and interleukin 1 receptor (IL-1R), leading to rapid but transient NF-κB activation (23, 27). Upon TNF-α binding, TNFR1 drives the assembly of the E3 ubiquitin ligases cellular inhibitor of apoptosis (cIAP) as well as TNFR-associated factor (TRAF) 2 with the protein kinase receptor-interacting protein 1 (RIP1) (28). RIP1 is then ubiquitinated and bound to NEMO (29), forming TGF-β activated kinase 1 (TAK1)-IKK complex. TAK1 phosphorylates and activates IKKβ as well as modification signals. TCR activates NF-κB through the recruitment of CARD11/Bcl10/MALT1 (CBM) complex (30, 31), which is then ubiquitinated by recruiting TRAF6, resulting in the activation of TAK1 as well as IKK (32). TLR and IL-1R initiate signaling through recruiting myeloid differentiation primary response gene 88 (MyD88) directly (33) or indirectly (34) which induces the recruitment of IL-1 receptor-associated kinase (IRAK) 1/4, followed by TRAF6 to activate TAK complex and intracellular signaling cascades (35, 36) (Figure 1). Sequentially, variant modification signals are converged on the activation of TAK1, which activates the IKK complex via phosphorylation of IKKβ. IκB family members phosphorylated by IKK undergo ubiquitin-dependent degradation, releasing the canonical NF-κB dimers, predominantly the p50-RelA and p50-cRel (Figure 1). The regulation of the canonical NF-κB pathway occurs at different levels to maintain homeostasis. Firstly, NF-κB transcribes IκBα and IκBε genes to form negative feedback (37). NF-κB activity is also controlled at the transcriptional factor level. For example, IKKα and ubiquitin ligase complex mediate the turnover of RelA (38) and impede its binding to DNA (39). In addition, deubiquitylation of signal molecules upstream of IKK is important in the negative regulation. A20 modifies signaling molecules, especially NEMO to destabilize the IKK complex and down-regulate inflammatory response (40). Tumor suppressor protein cylindromatosis (CYLD) also inhibits the activation of IKK by a similar mechanism (41). IKK inhibitors suppress thrombosis by blocking soluble N-ethylmaleimide-sensitive factor attached protein receptor (SNARE) complex formation and platelet secretion, thus mitigating late-stage plaque development (42). Lastly, canonical NF-κB is positively regulated by ubiquitination of NEMO through TRAF and linear ubiquitin chain assembly complex (LUBAC), which is crucial for IKK activation (43) (Figure 1).
The non-canonical pathway is activated slowly and persistently compared to the canonical one. It has a central signaling component, NF-κB-inducing kinase (NIK), equivalent to TAK1 in the canonical pathway. The signaling cascade is based on the stimulation of specific TNFRs by CD40 ligand, B cell-activating factor (BAFF), and lymphotoxin-β (14). The process initiates from TRAF3-TRAF2-cIAP recruitment and ends up with NIK activation (44). NIK phosphorylates and activates IKKα (23, 45, 46), which mediates phosphorylation of p100, triggering its ubiquitylation via recruitment of the E3 ubiquitin ligase βTrCP (47–49). The processing of p100 generates p52, resulting in the nuclear translocation of p52-RelB heterodimer. Since the non-canonical activation relies on the generation of p52 from p100, the processing of p100 lies in the key position of regulation. This process is dependent on ubiquitination and phosphorylation, which are regulated by specific ubiquitin E3 ligase and NIK-IKKα axis, respectively. The former includes tripartite motif family 9 (TRIM9) which inhibits NIK-induced and β-TrCP-dependent p100 processing (50). FBW7, also an E3 ligase, exclusively interacts with glycogen synthase kinase 3β (GSK3β) phosphorylated p100 and mediates its destruction (51). OTUB1 is a deubiquitinase that stabilizes p100. As a pivotal node in the non-canonical pathway, NIK has a significant role in NF-κB regulation. Its degradation is promoted by NOD-like receptors family pyrin domain-containing (NLRP) 12 and E3 ligases, CHIP, Peli1, and CRL4 (14). Additionally, OTUD7B, an A20-like protein, deubiquitinates TRAF3 and thus negatively regulates signal-induced non-canonical NF-κB (52) (Figure 1).
Notably, apart from those pathways mentioned above, ER stress has emerged as an important trigger upstream of NF-κB. NF-κB activation mediated by ER stress is dependent on Ca2+ efflux and subsequent production of reactive oxygen species (ROS) (13). More mechanisms and interactions will be discussed in detail later in this review.
Atherosclerosis is a common chronic inflammatory disease characterized by the accumulation of fibrin and lipids in subendothelial space, being a leading cause of cardiovascular diseases, including heart failure, stroke, and claudication (53, 54). AS dominantly occurs in the intima of middle and large-sized arteries, where endothelial cells are exposed to excessive shear stress. Vessel stenosis resulting from atherosclerotic plaque could induce CVD by abolishing blood flow. However, the dominant mechanism linking AS and CVD appears to be the vulnerability of plaque (55). Vulnerable plaque rupture exposes prothrombotic components, triggers the clotting cascade, and leads to atherothrombosis (56). Notably, inflammation is the pivotal cause of plaque progression and vulnerability.
Loss of intact endothelial functions occurs at the earliest in atherogenesis, followed by lipid accumulation and fatty streak formation under the endothelial cells. Fatty streak is a reversible lesion that can appear as early as childhood. In this process, multiple molecules mediate leukocyte adhesion, extravasation, migration, chemotaxis, activation, and the formation of foam cells from macrophages by uptake of lipids. Then the nascent plaque generally develops and forms a complex lesion with migration and proliferation of vascular smooth muscle cells (VSMCs), which secrete extracellular matrix such as collagen accumulated in the plaque (57) (Figure 2A). As plaque progresses, a necrotic core containing necrotic material, foam cells, cholesterol crystals, and lipids is formed and developed. Necrotic cores are considered to further promote inflammation, plaque rupture, and thrombosis by storing inflammatory mediators, matrix proteases, and thrombotic molecules (Figure 2B). A fissure of the fibrous cap eliminates the barrier between the tissue factor rich in the lipid core and the coagulation factors in the bloodstream, which triggers a clotting reaction and leads to thrombosis in advanced atherosclerotic lesions (58–61). Finally, the rupture of advanced plaque from the instability of the fibrous cap is primarily determined by the level of interstitial collagen (62). In addition, the disruption of fragile neovasculature in atherosclerotic plaques provides a possibility of sudden plaque progression (63) (Figure 2C).
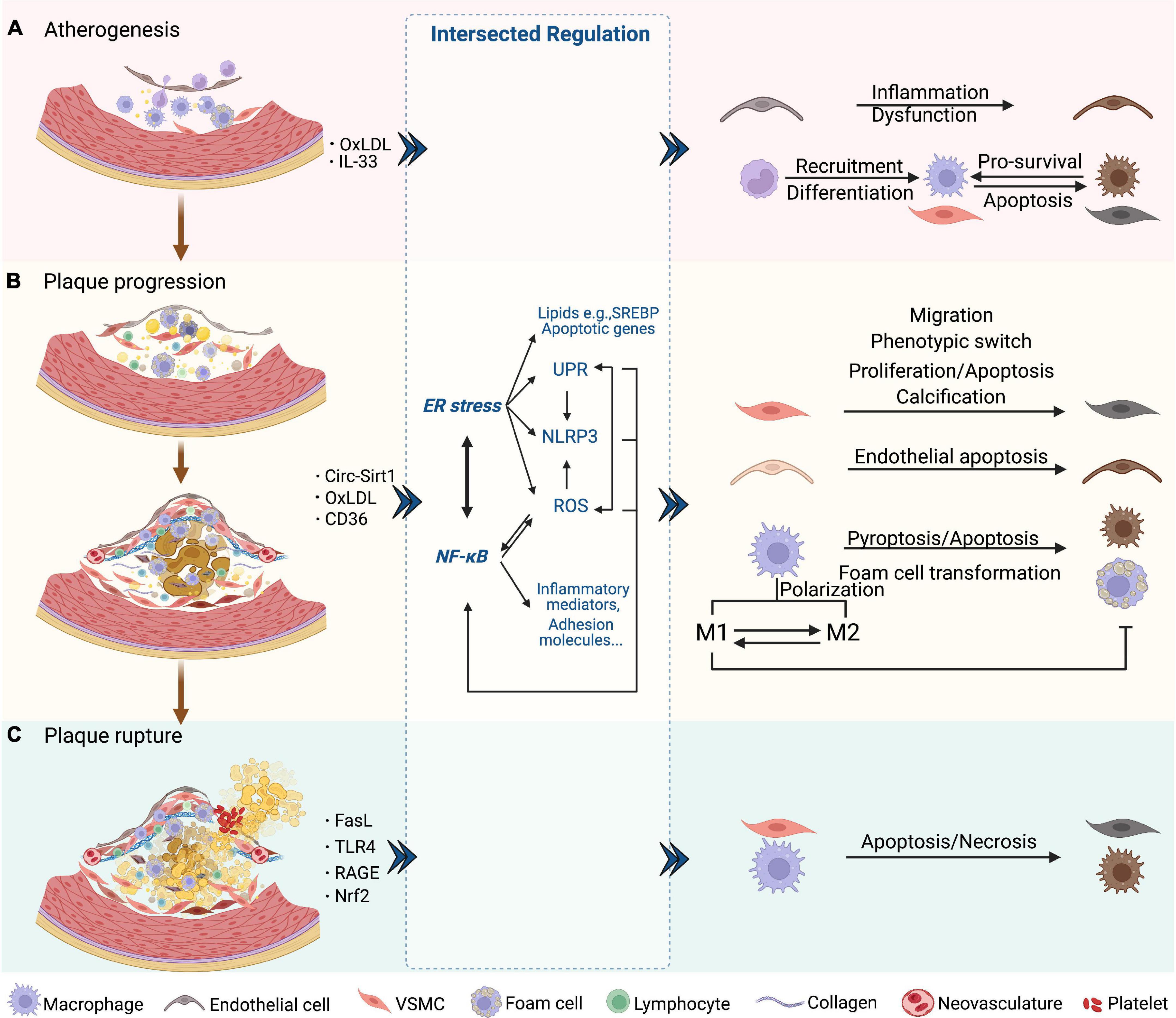
Figure 2. NF-κB and ER stress in three phases of AS. (A) Atherogenesis. Endothelial dysfunction as an initial event in atherogenesis is induced by NF-κB and downstream inflammatory mediators. The SREBP pathway is induced by ER stress and aggravates endothelial dysfunction. SREBP- and NF-κB-induced NLRP3 inflammasome contributes to atherogenesis. Chemokines induced by NF-κB attract lymphocytes and trigger endothelial inflammation. NF-κB also promotes the recruitment and differentiation of monocytes by increasing the levels of adhesion molecules and M-CSF of endothelial cells. After differentiated into macrophages, UPR markers are activated, which protects macrophages from ER stress-induced apoptosis. (B) Plaque progression. This phase is characterized by foam cell formation, VSMC migration and proliferation, ECM accumulation, and NC formation. ROS/NF-κB regulates the migration and phenotypic switch of VSMCs. Circ-Sirt1 inhibits NF-κB and thus alleviates the progression of AS. Macrophages uptake oxLDL via CD36 and this triggers the NF-κB signaling pathways, which promotes the transformation into foam cells. XBP-1 also regulates foam cell formation, endothelial apoptosis and VSMC calcification. Inhibition of ER stress promotes the formation of M1 subtype and subsequent foam cell formation. In macrophages, activated NLRP3 inflammasome causes pyroptosis and apoptosis via caspase. (C) Plaque rupture. This phase is characterized by less SMCs and collagen, and more lipids and macrophages, which could involve NF-κB-FasL pathway. Macrophages induce plaque rupture by secreting MMPs, which is regulated by TLR4/NF-κB and RAGE/NF-κB signaling. Apoptosis of macrophages and VSMCs is induced by the prolonged ER stress, including PERK and IRE-XBP1. CHOP is also a mediator of apoptosis, vascular remodeling and plaque necrosis, whose expression is promoted by UPR signaling. Nrf2, as a synergistic mediator between NF-κB and ER stress, has an athero-protective role by upregulating some antioxidant enzymes. Additionally, NLRP3 inflammasome-mediated up-regulation of MMPs predisposes plaque to rupture.
NF-κB has been regarded as a critical player in atherogenesis over the past decades partly because the genes it transcribed mediate all three phases of AS (64–66). Studies have revealed that IKK/NF-κB signaling promotes atherogenesis and that targeting NF-κB is a treatment strategy against AS and CVDs (67). Nevertheless, sufficient evidence proves that NF-κB activation leads to both protective and destructive outcomes (68). Research suggests that ER stress is associated with various lesions during AS and affects the disease course, which occurs in endothelial cells, VSMCs, and macrophages by integrating protein and lipid metabolism, cell death, and inflammatory responses (69).
Taken together, it is important to figure out how the NF-κB, ER stress-related molecules, and their functional crosstalk intervene in three stages of AS, including atherogenesis (plaque formation), plaque progression, and plaque instability.
Endothelial dysfunction, an initial factor in early atherosclerotic lesion formation, is induced by NF-κB and downstream production of inflammatory cytokines, such as IL-6 and TNF-α (70). Regenerated endothelial cells produce a large amount of NO and aggravate inflammatory response, leading to the formation of plaque (71). A recent study found that RIP1 primarily drives inflammatory cells toward activation in early atherosclerotic lesion formation in an NF-κB-dependent manner (10). Moreover, inhibiting cyclooxygenase-2 (COX-2) expression, a downstream gene of NF-κB, dramatically impedes the early evolution of AS (72). CCL20, a chemokine exerting selective attraction to lymphocytes, is upregulated by NF-κB and is strongly associated with vascular endothelial inflammation (73). At this early stage, NF-κB also participates in the production of adhesion molecules in the endothelium, including E-selectin, VCAM-1, and intercellular adhesion molecule-1 (ICAM-1), promoting the recruitment of monocytes (Figure 2A). The effects of NF-κB activation in the early stage of AS are not limited to endothelial cells but also occur in various cell types within the plaque (57). CCL20 is overexpressed in VSMCs of atherosclerotic lesions from coronary artery patients, triggers the inflammatory response, and significantly induces human lymphocyte migration (74). Besides, IL-33 upregulates the macrophage-colony stimulating factor (M-CSF) of endothelial cells through the NF-κB pathway, promoting the differentiation of monocytes (75).
Unfolded protein response activation in endothelial cells can be observed at the very beginning of AS. In athero-susceptible regions, activation of IRE1α and ATF6 is consistent with a high expression of molecular chaperones in ER. Additionally, ATF4 and CCAAT/enhancer-binding protein (CEBP) homologous protein (CHOP) mRNA are highly expressed, along with activated PERK pathway in VSMCs and macrophages at this stage (76) (Figure 2A). UPR activation aims to be a protective response to harmful stress and promotes cell survival in early atherosclerotic lesion formation. For example, UPR is a vital modulator of the sterol regulatory element binding protein (SREBP) pathway to maintain lipid homeostasis and inflammatory response, which are important contributors to atherogenesis (77–79).
NF-κB plays a considerable part in cell survival in addition to well-known pro-inflammatory functions, and the two directions may counteract each other in AS progression (80). Research has suggested that IKKβ deletion increases AS in LDLR deficient mice instead of preventing atherogenesis (68). Given the death of foam cells facilitates the necrotic core due to a defect in clearing accumulated lipids, more attention should be paid to NF-κB’s roles in limiting plaque size other than in pro-inflammation.
NF-κB activation regulates the migration and proliferation of VSMCs, whereas the detailed mechanism is still controversial (81, 82). A study by Mehrhof et al. shows that in a knock-in mouse model expressing the NF-κB super repressor, the proliferation rates of VSMCs did not differ from those in wild-type when stimulated by platelet-derived growth-factor-BB (PDGF-BB) or serum. Further study indicated that VSMC proliferation is regulated by classical mitogenic signaling pathways (MAPK and PI3K pathways) rather than NF-κB (81). These results implicate that NF-κB may essentially play a role in apoptosis and inflammatory responses in VSMCs instead of pro-survival or growth signal in the progression of AS. NF-κB-mediated phenotypic switch of VSMCs involves increased synthesis capacity and decreased contraction capacity, which is closely linked with the accumulation of extracellular matrix and plaque promotion in the progression of AS (83–85). Additionally, blocking ROS/NF-κB/mTOR/P70S6K signaling pathway prevents PDGF-BB-induced VSMC phenotypic switch, multiplication, and migration (83). Circ-Sirt1, as a non-coding RNA (ncRNA) regulator of VSMC phenotype, inhibits NF-κB translocation and binding to target DNA by directly interacting with the p65 subunit in the cytoplasm and facilitating the level of SIRT1 mRNA, respectively, which alleviates neointimal hyperplasia and the progression of AS (85) (Figure 2B). NF-κB activated by autoantibodies is also an important mediator in atherosclerotic lesion growth. 27-kDa heat shock protein (HSP27) in the blood combines with IgG anti-HSP27 auto-antibodies to form an immune complex, which has a role in anti-inflammation and anti-atherosclerosis. HSP27 immune complex activates TLR4/NF-κB signaling and increases the level of anti-inflammatory cytokine IL-10 in macrophages. Moreover, HSP27 immune complex reduces form cell formation by inhibiting oxidized low-density lipoprotein (oxLDL) binding to scavenger receptors (86). In addition, under ER stress, chaperone protein 78 kDa glucose-regulated protein (GRP78) dissociates from ER and moves to the cell surface, resulting in the generation of anti-GRP78 autoantibodies which activate NF-κB and induce the expression of adhesion molecules in human endothelial cells (87).
Generally, macrophages are divided into M1 and M2 subtypes, which have pro-inflammatory and anti-inflammatory effects, respectively. In atherosclerotic plaques, both subtypes are identified and play important roles in plaque progression (Table 2). The disruption of balance is speculated to accelerate foam cell formation and be related to plaque vulnerability (88). M2 subtype is prone to apoptosis as a result of oxLDL toxicity, leading to the accumulation of necrotic material within the plaque (89). NF-κB signaling pathway affects the transition from macrophages to foam cells and its further accumulation in the subendothelial space underlying atherosclerotic disease. In macrophages, oxLDL is taken via CD36 and other scavenger receptors and is resistant to the lysosomal enzymes (90). It signals via CD36-TLR4-TLR6 and triggers the NF-κB signaling pathway to produce proinflammatory cytokines (91). MiR-216a was found to promote telomerase activation in macrophages via the Smad3/NF-κB pathway, contributing to the transition from M2 to M1 (92). Applying fullerene derivatives inhibits the oxLDL-induced differentiation of macrophages into lipid-laden foam cells and plaque progression of apolipoprotein (Apo) E knock-out mice arteries. Mechanically, fullerene derivatives alleviate oxidative stress, inhibit CD36 receptor expression, and reduce TRAF2/NF-κB pathway activation (93).
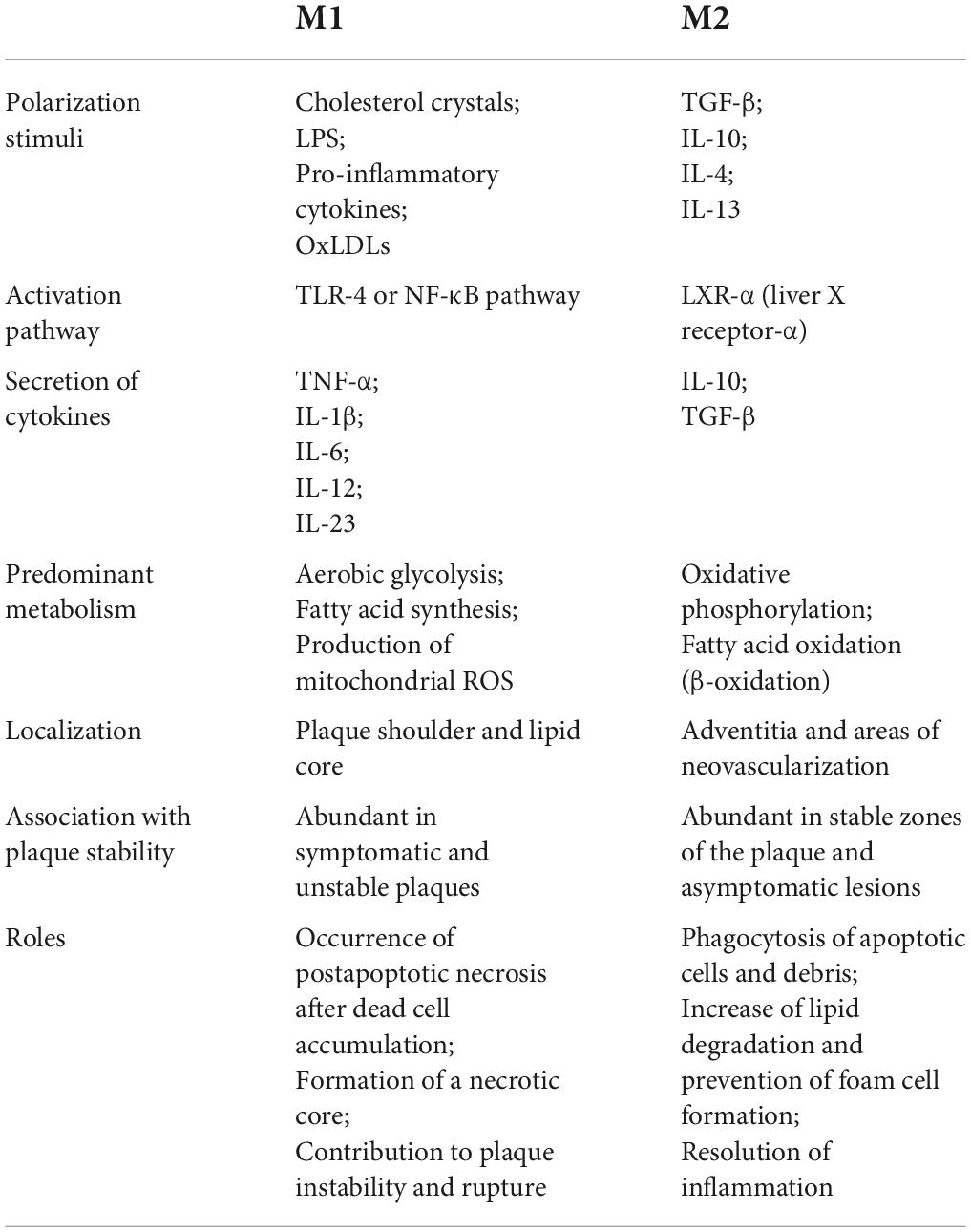
Table 2. Differences between M1 and M2 macrophages in atherosclerosis.
Endoplasmic reticulum stress is also a pivotal mechanism regulating plaque progression. Spliced X-box binding protein-1 (XBP-1), a molecule downstream of IRE1 and ATF6, modulates many aspects involved in AS progression, such as macrophage apoptosis, foam cell formation, and IL-8 and TNF-α production. Uncontrolled activation and excessive expression of splicing XBP-1 contribute to endothelial apoptosis and eventually AS evolution, as discovered in the branches and plaques of arteries in ApoE knock-out mice, which may also be related to induction of VSMC calcification (94, 95) (Figure 2B). ER stress is also considered to have an important role in macrophage differentiation. Inhibition of ER stress affects lipid metabolism characterized by an increase in cholesterol efflux, which shifts the M2 subtype to M1 and reduces foam cell formation (96). These studies imply that inhibition of ER stress, which promotes transition toward M1, may decrease foam cell formation, inhibit macrophage apoptosis, and block plaque development.
During the last decades, people have been trying to understand the pathophysiology of atherosclerosis, though the precise mechanisms underlying plaque destabilization still remain unclear. In this phase, studies have suggested that macrophages secrete proteases, especially matrix metalloproteinase-9 (MMP-9), to destroy elastin, fibrin, and other matrix proteins that the tension of the fibrous cap comes from, making macrophages an important player in plaque destabilization (97). Several studies support that downregulation of MMP-9 expression in macrophages is mediated by suppressing TLR4/NF-κB signaling, which is associated with attenuation of plaque vulnerability (98, 99). Receptor for advanced glycation end products (RAGE) is a key factor for plaque destabilization in diabetes mellitus, where its downregulation may suppress atherosclerotic plaque development, an effect mediated by NF-κB inhibition (100, 101). Statistical analysis of atherosclerotic lesions from carotid arteries revealed colocalized NF-κB activation and FasL overexpression, and a similar result was also found in peripheral blood mononuclear cells (PBMCs), indicating the NF-κB/FasL pathway may contribute to plaque vulnerability (102) (Figure 2C).
Advanced atheroma provides environmental and molecular bases that trigger ER stress and the UPR. ER-resident molecular chaperone, GRP78/94, and HSP47 are predominantly localized to the VSMC-rich fibrous cap of advanced plaques, suggesting activation of the UPR in VSMCs (103). On the other hand, under prolonged and enhanced ER stress, the activated PERK pathway promotes the level of death effector, and IRE1α/XBP-1 may activate the apoptosis signaling pathway in macrophages and VSMCs at this stage (104, 105). Thin-cap atheroma and ruptured plaques display abundant dead macrophages and VSMCs featuring strongly activated PERK/CHOP which is a mediator of apoptosis on chronic ER stress and a contributor to vascular remodeling and plaque necrosis (106–108) (Figure 2C). The effects of ER stress on the advanced plaque in macrophages are further demonstrated in AS-prone mice lacking CHOP, which shows blockage of macrophage apoptosis and inhibition of necrotic core formation (107, 109, 110).
Various pathological factors which activate NF-κB, such as ROS, lipids, TLR ligands, and some cytokines (e.g., TNF-α and IL-1), disrupt ER homeostasis and activate the UPR, leading to the situation called ER stress (111). Of note, this relationship is not likely one-sided. There are several potential avenues through which ER function also affects inflammatory signaling. And their interplay constitutes the pathological basis of many inflammatory and metabolic diseases, including AS (112–114). The ER stress is initiated with the dissociation of chaperone proteins such as GRP78/Bip and GRP94 with the ER stress sensor proteins (IRE1α, PERK, and ATF6), which leads to UPR activation. Chaperones also directly participate in subsequential UPR and NF-κB signaling. ATF6 and IRE1α pathways promote the transcription of the ER chaperones, which is necessary for the alleviation of the misfolded proteins to restore homeostasis (115). GRP78 is a member of the chaperone HSP70 family which is closely relevant to the endothelial dysfunction in the development of AS, with a fundamental role in protecting protein stabilization and also in anti-inflammation (116). Note that HSP70s suppress the expression of inflammatory cytokines via inhibiting the NF-κB. HSP70s stabilize the IκB complex through its binding and block IKK kinase activity and further NF-κB mediated transcription (117, 118).
Three branches of UPR (IRE1α, PERK, and ATF6) of ER stress have been reported to have crosstalk with many inflammation-related signaling, including the NF-κB pathway. For example, activated IRE1α and recruited TRAF2 activate JNK, inducing the production of IL-6 and TNF-α by phosphorylation of AP-1 and consequent NF-κB activation. ER stress induces TRAIL receptor activation which leads to apoptosis through the FADD/caspase-8 pathway, or alternative production of inflammatory cytokines through NF-κB activation (119–121). However, ER stress can also lead to inhibition of inflammation. The ER E3 ubiquitin ligase TRIM13 ubiquitylates the IKK regulatory subunit NEMO, blocking the degradation of IκBα, which consequently inhibits NF-κB translocation into the nucleus (122). Hence, it makes sense to unravel the exact molecular mechanisms of ER-stress-induced inflammation. Here we focus on how ER stress intersects with NF-κB through various inflammatory signaling pathways to form this integrated network (Figure 3).
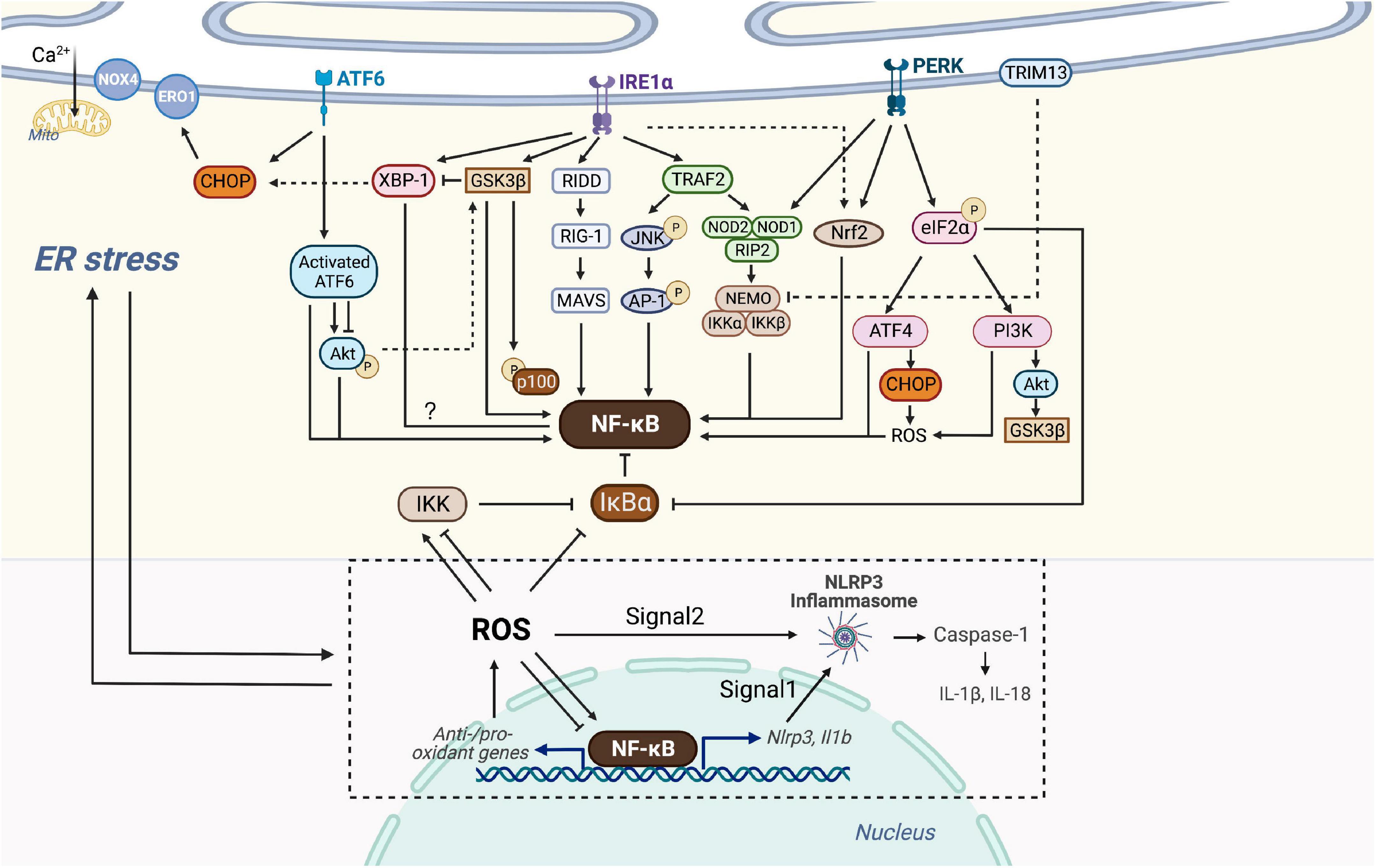
Figure 3. Crosstalk of NF-κB and ER stress. Three branches of UPR (IRE1α, PERK, and ATF6) of ER stress are able to intersect with NF-κB. Activated IRE1α recruits TRAF2, which activates JNK and then AP-1 or associates with IKK probably via NOD1/2 and RIP2. IRE1α is also linked with the RIDD/RIG-I/MAVS pathway and GSK3β to activate NF-κB. IRE1α oligomerization increases XBP-1 expression which might be associated with decreased NF-κB expression, but GSK3β activation inhibits IRE1α-dependent XBP-1 splicing. PERK branch can induce NF-κB activation essentially by translation attenuation of the free IκBα mediated by phosphorylated eIF2α. Additionally, PERK-eIF2α could also contributes to inflammation via ATF4 or PI3K-Akt pathway. Both of NOD1 and Nrf2 could be activated by PERK and IRE1, but Nrf2 has both positive and negative effects on NF-κB, dependent on cellular circumstances. Through the ATF6 branch transient phosphorylation of Akt activates NF-κB, whereas ATF6 activation could inhibit Akt-GSK3β and enhance NF-κB signaling. Additionally, ER E3 ubiquitin ligase, TRIM13 ubiquitylates NEMO and prevents nuclear translocation of NF-κB. CHOP could be activated by all three branches of UPR, causing ROS-mediated ER stress and NF-κB inhibition or activation. ER stress-induced NLRP3 inflammasome is dependent on NF-κB and UPR activation. Signal 1 of NLRP3 inflammasome activation is transcriptional upregulation of NLRP3 along with pro-IL-1β provided by NF-κB. Signal 2 is a posttranscriptional modification which can be provided by ROS. NF-κB controls the levels of ROS by regulating anti-oxidant and pro-oxidant genes, and ROS in turn inhibits or enhances the DNA binding activity of NF-κB itself, depending on modifications of NF-κB. ROS also regulates the IKK complex and phosphorylates IκBα. ROS produced by Nox4 transduces ER stress signals to the UPR to maintain homeostasis, whereas ROS produced by ERO1 or mitochondrial damage leads to cell death. ROS, NF-κB, NLRP3 inflammasome and the production of IL-1β and IL-18, in turn, trigger chronic ER stress.
Several signal cascades have been discovered in the NF-κB activation via IRE1α kinase activity. Activated IRE1α kinase recruits TRAF2, which associates with IKK and degrades IκBα to activate NF-κB (123). It is confirmed in endothelial cells that LPS induces ER stress and overproduction of IL-6 and MCP-1 through IRE1α/NF-κB pathway, resulting in endothelial dysfunction (124). Moreover, Keestra et al. found that Brucella abortus infection triggered ER stress and induced inflammation and IL-6 production in a TRAF2, nucleotide-binding oligomerization domain-containing protein (NOD) 1/2, and RIP2-dependent manner, providing a novel connection between ER stress and NF-κB activation (125). IRE1α is also linked with the RIDD/RIG-I pathway upon encountering viral RNAs, which induces an inflammatory response through MAVS and downstream NF-κB (126). In addition, IRE1α oligomerization generates spliced XBP-1 mRNAs that are translated into potent transcription factors (127). Increased XBP-1 expression contributes to the secretion of myeloperoxidases, TNF-α, IL-6, and IL-1β, and is negatively correlated with NF-κB expression in the colon (128). Also, ER stress-induced IRE1α activation mediates GSK3β activation and subsequent IL-1β gene expression (129). XBP1s K60/77R mutation, preventing the ubiquitination and proteasome-degradation of XBP1s, mimics the constitutive activation of IRE1α elevated, and results in the elevated GSK3β phosphorylation (130). In vivo and in vitro studies have confirmed that GSK-3β activation is involved in NF-κB activation, suggesting crosstalk between ER stress and NF-κB through IRE1α/GSK3β pathway (131, 132).
Interestingly, GSK3β activation inhibits IRE1α-dependent XBP-1 splicing, and they differentially regulate proinflammatory cytokine gene expression, indicating complex signaling crosstalk in inflammatory pathways (Figure 3).
Protein kinase-RNA like ER kinase branch can induce NF-κB activation essentially by translation attenuation, including the free IκBα, mediated by phosphorylated eIF2α. Zhang et al. observed that anti-dsDNA antibodies activate NF-κB and upregulate various inflammatory cytokines through PERK-eIF2α-ATF4 (133). Besides, a recent study has shown that thapsigargin-induced PERK activation along with the inositol triphosphate receptor (IP3R)-mediated calcium flux makes cells more responsive to Salmonella typhimurium through the NOD1-stimulated NF-κB activation and subsequent inflammatory response (134). Nuclear erythroid-related factor 2 (Nrf2), a transcription factor mainly activated by PERK and IRE1, also plays a pivotal role in the crosstalk between UPR and NF-κB. Studies on the linkage between Nrf2 and autophagy have shown that Nrf2 activates IKK and subsequent NF-κB by enhancing the expression of p62, which explains NF-κB-dependent autophagy activation (135–137). Complex interrelation indicates that Nrf2 influences NF-κB both positively and negatively due to various circumstances. For instance, studies on Nrf2 knock-out mouse embryo fibroblasts have shown increased activity of IKKβ and degradation of IκBα (138). Moreover, the increase of Nrf2 activity in patients with lupus nephritis prevents p65 activation by accumulating glutathione. Increased heme oxygenase-1 (HO-1), a product of the Nrf2 target gene, inhibits adhesion molecules such as E-selectin and vascular cell adhesion molecule-1 (VCAM-1) expressed in endothelial cells via NF-κB downregulation (139). Additional experiments have implicated the PERK-eIF2α signaling as a contributor to inflammation via the JNK and PI3K-Akt pathway, but the detailed interaction with NF-κB has not been well defined (140) (Figure 3).
Since Nrf2 serves as a platform of interrelation between NF-κB and ER stress (Figure 3), special attention has been paid to this transcription factor to better define its possible contribution to oxidative stress of the vulnerable plaque (141) (Figure 2C). The expansion of the necrotic core and the disruption of the plaque are largely determined by the accelerated number of apoptotic cells and phagocytic clearance defect. Nrf2 not only upregulates the expression of different antioxidant enzymes but also regulates mitochondrial ROS production through NADPH oxidase (Nox) activity. Though most studies have demonstrated the protective roles of Nrf2 against AS, several studies have revealed that it might play antagonistic roles, both preventing and enhancing AS. Studies found that laminar blood flow stimulates the anti-atherogenic activation of Nrf2, whereas oscillatory blood flow promotes the opposite effect (142). Nrf2 in bone marrow-derived cells promotes plaque progression in ApoE knock-out mice (143), while early AS is aggravated in LDLR knock-out mice with Nrf2-deficient macrophages (144). The positive atherogenic role of Nrf2 appears to be implemented by IL-1 release and by promoting foam cell formation through the expression of the CD36 scavenger receptor (145, 146).
As one of the UPR branches, ATF6 also plays a nonnegligible role during ER stress and in its crosstalk with NF-κB. However, Yamazaki et al. have confirmed that subtilase cytotoxin-triggered rapid cleavage of molecular chaperone GRP78/BiP (78-kD glucose-regulated protein/immunoglobulin heavy chain binding protein in pre-B cells) leads to Akt phosphorylation mediated by ATF6, contributing to downstream NF-κB activation (147). Recently, another study reported that the decrease of ATF6 expression induced by miR-149 might attenuate inflammation and apoptosis through NF-κB and Akt signaling cascades (148). In addition, in vitro study showed that ATF6 activation induced by chemical agents inhibits Akt/GSK3β and increases NF-κB activity, thus improving the pro-inflammatory effect of TLR4 in ER-stressed macrophages (149). Despite representing unique signaling cascades, ample evidence has indicated that the UPR and NF-κB may converge on nuclear transcription factors, such as ATF3/4/6α, CHOP, and XBP-1 (150) (Figure 3). Taken together, the UPR has crosstalk with NF-κB at various levels, which offers perspectives on the adjustment of cellular stress responses and therapeutic application in the future.
The NLRP3 inflammasome is a multi-protein complex that recognizes PAMPs or damage-related molecular patterns (DAMPs) and activates the protease caspase-1, leading to pyroptosis and the formation of mature IL-1β and IL-18 to mediate the inflammatory response (151). NLRP3 inflammasome connects lipid metabolism and inflammation because it is activated by crystalline cholesterol and oxLDL in plaques of AS, making it a possible player in the development of AS. In general, transcription and modification signals of the NLRP3 are necessary for its function. The former is provided by the binding of LPS to TLR4, resulting in NF-κB activation and consequent transcription of NLRP3 and IL-1β precursor (152). The modification signals occur after transcription, one of which is BRCA1/BRCA2-containing complex subunit 3 (BRCC3)-mediated deubiquitination (153–155). Though the exact process remains unanswered, it is considered that the activation of the NLRP3 inflammasome is possibly associated with factors such as K+ outflow, ROS, Ca2+ flux, and lysosomal rupture, all of which can provide modification signal (156). Notably, these mechanisms contribute to signal one by activating NF-κB through ROS production. Hu et al. demonstrated that in LPS-induced endometritis in mice, NLRP3 inflammasome is activated via ER stress-associated pathway, along with increased NF-κB and ROS (157). In LPS-induced liver injury, NF-κB and the NLRP3 inflammasome activation along with cytokine production such as TNF-α, IL-1β, and IL-18, in turn, contribute to chronic ER stress to form negative feedback (158). A recent study has observed that the ER stress-induced NLRP3 inflammasome is dependent on NF-κB activation and pro-inflammatory cytokine secretion, which is linked to the pathogenesis of atrial fibrillation and can be potentially targeted in cardiac tissue (159). Nevertheless, evidence has revealed that UPR is not indispensable for inflammasome activation (160). Since UPR is involved in NF-κB activation and ROS production, which are related to the activation of the NLRP3 inflammasome, these controversial results call for further insight into UPR pathways as inflammasome mediators (Figure 3).
Atherosclerosis has been considered an inflammatory and lipid metabolic condition, and since the NLRP3 inflammasome is activated by lipids such as crystalline cholesterol and oxLDL, it presumably combines different pathological bases of AS. The NLRP3 inflammasome and subsequent caspase-1 activation cause pyroptosis in macrophages after uptake of oxLDL and might contribute to the progression of atheroma (161, 162). On the other hand, the NLRP3 inflammasome induces macrophage apoptosis via caspase-8 activation (163), though to what extent this pro-apoptotic function protects against AS development is still unanswered (Figure 2B). IL-1β and IL-18 produced by the NLRP3 inflammasome increase the expression of many endothelial molecules such as MCP-1, VCAM-1, and IL-8, involving inflammatory cell adhesion, chemotaxis, recruitment, and activation (164). Moreover, the NLRP3 inflammasome promotes plaque instability and subsequent thrombogenesis (165). Blocking NLRP3 signaling reduces the production of pro-inflammatory cytokines in ApoE knock-out mice and contributes to plaque stabilization by reducing macrophages and lipids as well as increasing SMCs and collagen (166).
Although numerous studies have reported the impact of NLRP3 inflammasome on the progression of AS, evidence has suggested it is not as important as we have thought. In vivo NLRP3 inflammasome is not critically implicated in AS progression, infiltration by macrophages, and stability of plaques (167). Research also supported that NLRP1 is more likely to be a critical factor for the initiation of endothelium inflammation (168). In addition, JNK1 and apoptosis signal-regulating kinase 1 (ASK1) contribute to inflammasome activation and caspase-8-mediated macrophage apoptosis, though whether this JNK1/ASK1/caspase-8-dependent apoptosis is directly mediated by NLRP3 inflammasome is uncertain (155). The identified pro-apoptotic activity of NLRP3 inflammasome might produce an anti-atherogenic effect, which could partly explain its controversial functions in AS.
The relationship between NF-κB and ROS is not one-sided. ROS is a key route linking the two events. Firstly, ROS activates or inactivates the IKK complex in different cell types (169). Often ROS alternatively phosphorylate IκBα, which may result in the release and activation of NF-κB (169, 170). Also, ROS may inhibit or enhance the DNA binding affinity of NF-κB itself, depending on different forms of modification in NF-κB heterodimers (171, 172). Another manner in which ROS interacts with NF-κB is the crosstalk between JNK and NF-κB, preventing persistent JNK activation and promoting cell survival (173).
As to the interactions between ER stress and ROS, it is proved that ROS plays both positive and negative roles during ER stress and in determining cell fate (174). Upon being produced by Nox4, an ER-resident oxygen-sensing enzyme, ROS acts as a signaling intermediate to transduce ER stress-related signals to the UPR, resulting in the correction of the unsteady state. However, ROS as a pro-inflammatory stimulus can further exacerbate inflammation after the UPR activation (111). On the other hand, if ER stress persists, delayed expression of the transcription factor CHOP leads to induction of ER oxidase 1 (ERO1) to produce ROS. Meanwhile, mitochondria exaggerate ROS production stimulated by the Ca2+ released from ER. Both contribute to a secondary increase in ROS, generally leading to cell death. Therefore, ROS lies both upstream and downstream of the UPR, making the network composed of ER stress, ROS, and NF-κB more complex than we have imagined.
Substantial evidence indicates that ROS is a central factor through which ER stress functions cooperatively with NF-κB in inflammation and other cellular processes. Li et al. observed that recombinant Treponema pallidum protein regulates the ROS/NF-κB pathway through ER stress. PERK induces the activation of the NF-κB and JNK pathways, leading to the production of IL-1β, IL-6, and IL-8 by macrophages (175). In another study, NF-κB signaling is activated by phosphoinositol 3-kinase δ (PI3Kδ) through ER-associated ROS and RIDD-RIG-I activation, which may induce severe airway inflammation and hyperresponsiveness (176). In human lung cancer cells, it is observed that a CHOP activator induces necrotic cell death via ROS-mediated ER stress induction and unusual NF-κB inhibition (177) (Figure 3).
The contribution of ROS to AS has been well investigated. ROS causes endothelial dysfunction (178), atherogenesis (179), and LDL oxidation (180). OxLDL has pro-inflammatory effects and participates in the phenotype switching and apoptosis of macrophages and VSMC in the AS progression (181, 182). ROS is positively related to atherosclerotic risk factors, such as diabetes and hypertension, etc. In vivo studies of the animal model have also shown that anti-oxidant treatments delay or prevent AS (183), suggesting the aggravating role of ROS in AS. A recent study has demonstrated that nicotine-induced autophagy and subsequent phenotypic transition of VSMCs accelerate AS, which is partly mediated by the nAChRs/ROS/NF-κB signaling pathway (184). In addition, in cultured VSMCs, chicoric acid impeded PDGF-BB-induced VSMC phenotypic alteration, proliferation, and migration mechanistically by blocking ROS/NF-κB/mTOR/P70S6K pathway (83) (Figure 2B). However, the diverse effects of ROS have been reported in AS. Nox4 is a major ROS-producing NADPH oxidase and is widely expressed in VSMCs. Its endothelial-specific overexpression increases ROS level, promotes aging, and makes cells susceptible to apoptosis, resulting in aggravated AS lesions in animals (185–187). Of note, it is also found in several mice models that Nox4 knock-out promotes initial plaque formation (188). Unlike Nox4, another NADPH oxidase Nox2 overexpressing leads to atherogenic rather than protective consequences (189), highlighting the controversial roles of Nox-dependent ROS in AS. The crosstalk between ER stress and ROS may be pivotal to understanding the controversial effect of ROS. Nox4 but not Nox2 selectively phosphorylates eIF2α, the downstream PERK arm of UPR, thus providing a direct route for integrating ROS and ER stress. In addition, Nox4 is central to a signaling feedback loop of Rho/Ras GTPase and ER stress. RhoA activation occurs on ER surface in response to UPR and further promotes Nox4-dependent ROS production (190). Nox4-generated oxygen inactivates ER calcium transporter SERCA (Sarcoplasmic Reticulum Ca2+ ATPases) and causes calcium-calmodulin-dependent activation of RasGRF1/2, which further mediates the UPR activation (191). Thus, ROS is more than a marker of oxidative stress, but plays two opposite roles in ER stress (restoration of homeostasis or apoptosis) and involves inflammation and cell growth. These data emphasize the controversial effects of ROS and careful considerations in Nox inhibitor development aiming to reduce ROS levels. It is challenging for Nox4 inhibitor development to retain the ER stress inhibition activity and the athero-protective function of Nox4. Given the diverse signaling roles served by Nox4, more specific Nox inhibitors targeting Nox1 and Nox2 while excluding Nox4 could be an optimal treatment strategy (174).
Innovation of prevention and treatment strategies against AS is still a pressing mission given being the leading cause of mortality and morbidity in developed and developing countries. Despite various interactions between ER stress and NF-κB, whether and to what extent these mediator molecules play a role in AS remains unanswered. Conceptually, several existing pharmacological targeting on UPR, ROS, NLRP3 inflammasome or other crossroads between ER stress and NF-κB could potentially influence both of them and impede the progression of AS. Herein, we focus on NF-κB inhibitors, UPR inhibitors, ROS-interfering molecules, natural compounds, and some ncRNAs with anti-atherogenic protective effects, targeting ER stress and/or NF-κB, which are attractive potential therapeutic strategies for AS (Table 3).
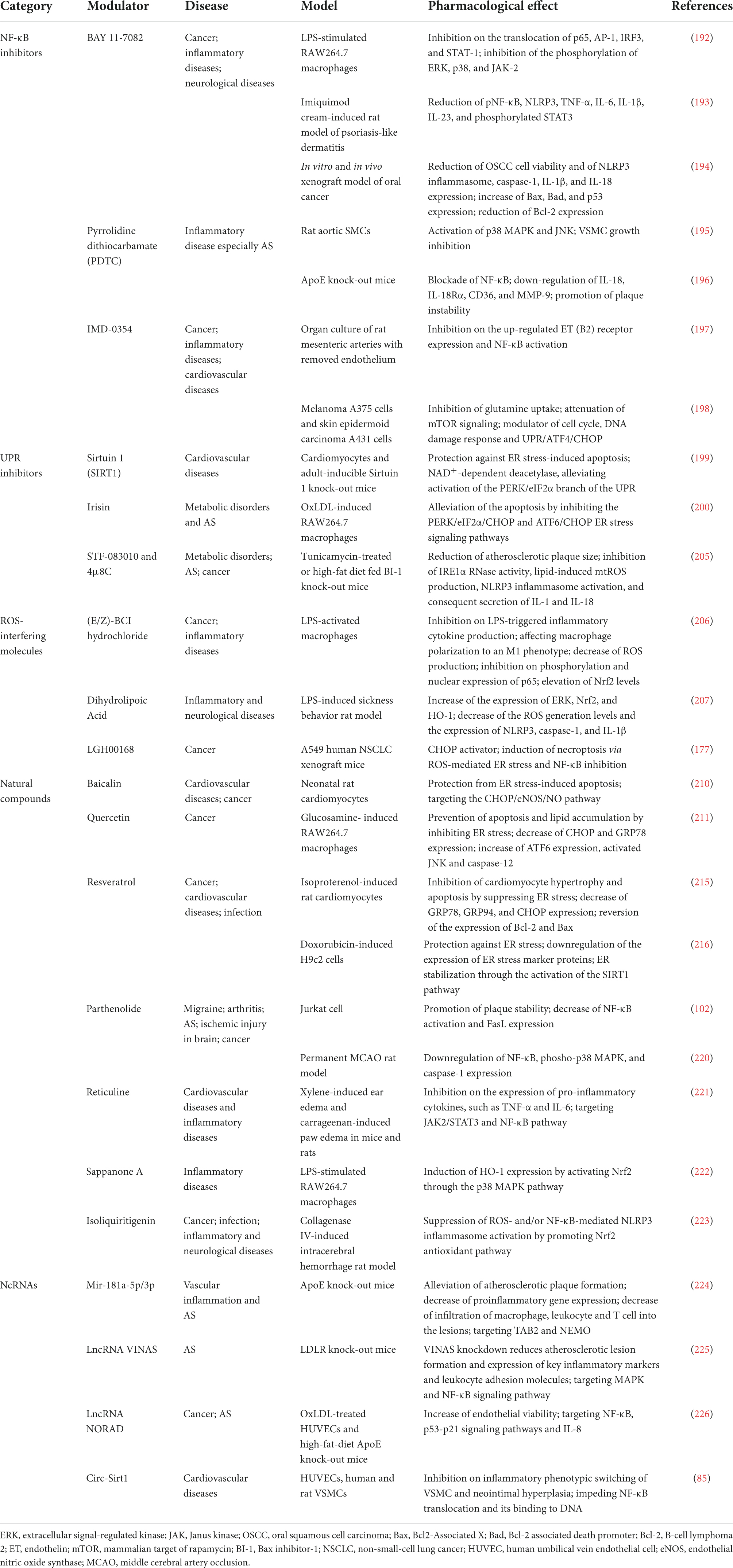
Table 3. NF-κB and/or ER stress modulators in experimental atherosclerosis and associated disease models.
BAY 11-7082 (BAY) inhibits IKK-mediated phosphorylation of IκBα, resulting in decreased NF-κB and decreased expression of adhesion molecules. In addition, BAY also suppresses the translocation and activation of AP-1, interferon regulatory factor-3 (IRF-3), and signal transducer and activator of transcription-1 (STAT-1) by inhibiting the phosphorylation or activation of ERK, p38, and JAK-2 (192). BAY is also an inhibitor of NLRP3 inflammasome and a modulator of apoptosis pathways shown in the management of psoriasis-like dermatitis and oral cancer (193, 194). These suggest that BAY could serve as a lead compound in developing potent anti-inflammatory drugs with multiple targets in inflammatory responses.
Pyrrolidine dithiocarbamate (PDTC), another NF-κB inhibitor, leads to PDTC-dependent VSMC growth inhibition by inducing marked activation of p38 MAPK and JNK (195). In addition, PDTC blocks IL-18 signaling in ApoE knock-out mice, thus reducing inflammation and restoring plaque instability (196). A better understanding of the molecular mechanisms of PDTC provides a theoretical basis for clinical applications of antioxidants in AS.
IMD-0354 is an IKKβ inhibitor known to exert anti-inflammatory, antitumor, and radioprotective effects. The NF-κB activation induced by TNF-α and associated up-regulation of endothelin B2 receptor could be effectively suppressed by IMD-0354 in VSMCs (197). Additionally, IMD-0354 is confirmed as a potent inhibitor of glutamine uptake that concomitantly attenuates mTOR signaling, but not IKK-NF-κB signaling, suppresses the growth of melanoma cells, and induces autophagy and apoptosis. Affected genes and molecules are implicated in ROS/UPR signaling, including ATF4 and CHOP (198). IMD-0354 has been applied in phase I clinical trials for atopic dermatitis and choroidal neovascularization, though its cardiovascular protective effect has not been verified in clinical trials.
Blockage of NF-κB alone might be insufficient for AS mitigation. Combination with NF-κB inhibitors and lipid-regulating drugs such as statins could be a feasible scheme. Considering that persistent NF-κB inhibition could cause immune deficiency, future NF-κB inhibitors for AS treatment should only be used as adjuvant and intermittent medicine. In a word, the diversity of NF-κB modification signals makes it a long way to apply NF-κB inhibitors in anti-atherosclerotic therapy.
Given the associations mentioned above between the UPR and NF-κB, the new functions of UPR inhibitors deserve to be reconsidered. Three representative molecules are listed in Table 3, with a special focus on their influences on PERK/eIF2α, ROS production, and NLRP3 inflammasome activation. Sirtuin-1 (SIRT1), an NAD+-dependent deacetylase, protects cardiomyocytes from ER stress-induced apoptosis by attenuating PERK/eIF2α pathway activation (199). A myokine, irisin, inhibits the PERK/eIF2α/CHOP and ATF6/CHOP pathways and alleviates the apoptosis of macrophages induced by oxLDL (200). Mouse models have shown that irisin promotes endothelial cell proliferation and significantly reduces AS in mice by upregulating the expression of miRNA126-5p (201). In the last decade, abundant clinical studies on the protective functions of irisin in the cardiovascular system have made breakthroughs. A recent cohort study has indicated low serum irisin levels as biomarkers of subclinical AS (202). However, existing studies mainly focus on serum irisin level increase after beneficial interventions such as simvastatin or Omega-3 fatty acids, and direct clinical evidence is necessary before irisin application (203, 204). Still, irisin has a promising preventive and therapeutic prospect for AS. In mouse models, small molecules STF-083010 and 4μ8C have shown a role in reducing atherosclerotic plaque size by inhibiting IRE1α RNase activity, lipid-induced mtROS production, and NLRP3 inflammasome activation (205).
Although people already have much knowledge of UPR and its roles in the development of AS, clinical trials evaluating UPR inhibitors are still scanty. Considering that adaptive UPR is important for the recovery of ER homeostasis, UPR inhibition is possibly only an incidental anti-atherogenic mechanism for potential UPR inhibitor drugs. For clinical use, specific inhibition of critical interaction between NF-κB and ER stress in one checkpoint of UPR branches could be an optimal strategy.
Many molecules present with anti-oxidant activities are promising anti-atherogenic drugs. (E/Z)-BCI hydrochloride (BCI), a small molecule inhibitor of dual-specificity phosphatase 6 (DUSP6), activates the Nrf2 signaling pathway and inhibits NF-κB activity, alleviating inflammatory response and decreasing ROS production in LPS-activated macrophages (206). Dihydrolipoic acid exhibits strong antioxidant activities in many conditions, especially neuroinflammation and provides protection via Nrf2/HO-1/ROS/NLRP3 signaling cascade in LPS-induced behavioral deficits in rats (207). Novel CHOP activator LGH00168 inhibits the NF-κB pathway and induces ROS-mediated ER stress, leading to necroptosis in A549 human lung cancer cells (177).
Reactive oxygen species is an identified risk factor for cardiovascular diseases. The activation of UPR branches, especially IRE1α and PERK, leads to the abrogation of ER stress-generated ROS, thus alleviating endothelial dysfunction. As discussed later, many natural compounds work by mediating ROS generation. Physical exercise is regarded as a supplement to pharmacotherapy for cardiovascular diseases by reducing ER stress and ROS (208, 209). In conclusion, numerous pathways upstream of ROS make interventions on ROS one of the most prospective strategies in extensive clinical settings more than AS. One limitation of the clinical application of ROS-interfering small molecules is toxicity.
Baicalin is a primary active substance from the Scutellaria root and attenuates ER stress-related apoptosis in vivo mediated by CHOP/eNOS signaling pathway (210). Baicalin is a marketed drug in China for the treatment of hepatitis, but more convincing clinical outcomes are required to evaluate its efficacy in treating AS. Quercetin existing in the pericarp, flower, leaf, and seed of various plants has an effect on maintaining ER protein homeostasis probably by increasing ATF6 and reducing CHOP and GRP78 in glucosamine-induced macrophages (211). Quercetin has been applied in Phase 2/3 clinical trials on coronary artery disease, venous thromboembolism, hypertension, and heart failure, and assessed as disease improvement effects (212–214). Resveratrol found in red wine attenuates cardiomyocyte hypertrophy and apoptosis in isoproterenol-induced rat cardiomyocytes, characterized by a low level of GRP78, GRP94, and CHOP, and by a reversed level of Bcl-2 and Bax (215). Resveratrol also alleviates doxorubicin-induced cardiocyte apoptosis of rats by relieving ER stress-related inflammatory response and activating SIRT1 signaling (216). A series of clinical studies have shown that dietary resveratrol improves endothelial function and exerts a beneficial effect on AS (217–219). Parthenolide is demonstrated to be an anti-inflammatory mediator and an NF-κB inhibitor, which has a potential application in cardiovascular and cerebrovascular diseases. Studies have demonstrated that the NF-κB/FasL signaling contributing to plaque rupture could be inhibited by parthenolide (102). Furthermore, the neuroprotective effect of parthenolide is characterized by the downregulation of NF-κB, phospho-p38 MAPK, and caspase-1 (220). Reticuline has anti-inflammation roles in CVDs by targeting the JAK2/STAT3 and NF-κB pathway, though the specific mechanisms are still unknown and further verification in atherosclerotic models is required (221). Sappanone A increases the level of HO-1 mediated by p38/Nrf2 signaling and suppresses LPS-induced NF-κB activation by modulating the p65 subunit, indicating its anti-inflammatory effect (222). Isoliquiritigenin from Glycyrrhiza glabra could reduce early neuronal degeneration after intracerebral hemorrhage, involving the NLRP3 inflammasome regulated by ROS and/or NF-κB through inducing Nrf2-mediated antioxidant activity (223).
The health effects of natural compounds in humans are limited by their purity and poor bioavailability, as they are extracted from plants and rapidly metabolized and excreted. Nevertheless, due to their easy availability from daily meals, diet change could be a simple and beneficial intervention. We can assume that natural compounds have a very high application value in AS prevention and treatment as well as improvement of general health conditions.
NcRNAs have received most and more attention over the last decades for their involvement in the progression of AS. Research has identified two microRNAs, miR-181a-5p and miR-181a-3p, cooperatively recede endothelium inflammation through blockade of the NF-κB signaling pathway by post-transcriptional regulation of TAB2 and NEMO expression, respectively (224). Long ncRNA (lncRNA) VINAS is highly expressed in intimal AS lesions and promotes vascular inflammation by a possible mechanism involving MAPK and NF-κB signaling pathways. Knockdown of lncRNA VINAS decreases the expression of adhesion molecules such as E-selectin, VCAM-1, and ICAM-1 and inflammatory molecules such as MCP-1, TNF-α, IL-1β, and COX-2 (225). LncRNA NORAD (non-coding RNA activated by DNA damage) knockdown aggravates oxidative stress, increases phosphorylated IκBα level and NF-κB nuclear translocation, and directly promotes IL-8 transcription in AS model. Therefore, lncRNA NORAD has a role in attenuating endothelial cell injury and alleviating AS (226). In contrast, ncRNA circ-Sirt1 directly binds to NF-κB and inhibits its translocation (85).
A number of RNA therapeutics have been in clinical phase II or III for various diseases, but lncRNAs are not among them. Moreover, up to now, few RNA therapies have been explored for cardiovascular diseases. The application of ncRNA therapeutics in AS requires overcoming many challenges, including immunogenicity, lack of specificity, and delivery difficulty.
As NF-κB and ER stress are involved in many human physiological processes, such as immunity and cancer, there are certain limitations to be overcome before therapeutically targeting them in AS. Also, new drug development is limited by the complexity of intrinsic pathways and crosstalk with other pathways. Therefore, the unexpected effects should be considered with caution when evaluating the safety of NF-κB and ER stress as targets for treatment. In this regard, it is significant to further explore more specific and effective crosstalk inhibitors and/or enhancers for atherogenesis, while leaving the normal physiological functions unaffected. On the other hand, these crossover effects also mean that a single successful drug may have utility in multiple diseases.
Indeed, currently available studies provide only a theoretical prospect of targeting interactions between NF-κB and ER stress against AS, and more convincing experiments are required to come closer to the production of an effective NF-κB targeting anti-atherogenic drug. Nevertheless, a broader and deeper understanding of NF-κB signaling and recognition of the potential direct or indirect links between these divergent pathogenic processes may eventually define the value of targeting their crosstalk as a clinical application to AS.
WL, KJ, JL, and WX contributed to the conception, reviewed for important intellectual content, and wrote the majority of the text and created the figures. YJW, JZ, YLW, and RX provided some text. LJ, TW, and GY edited the manuscript. All authors read and approved the final manuscript.
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 82171303 and 91954201) and the Beijing Municipal Science & Technology Commission (Grant No. 5202022).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. (1998) 16:225–60. doi: 10.1146/annurev.immunol.16.1.225
2. Baldwin ASJr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. (1996) 14:649–83. doi: 10.1146/annurev.immunol.14.1.649
3. Williams LM, Gilmore TD. Looking down on NF-κB. Mol Cell Biol. (2020) 40:e104–20. doi: 10.1128/mcb.00104-20
4. Tian M, Tian D, Qiao X, Li J, Zhang L. Modulation of Myb-induced NF-kB -STAT3 signaling and resulting cisplatin resistance in ovarian cancer by dietary factors. J Cell Physiol. (2019) 234:21126–34. doi: 10.1002/jcp.28715
5. Khongthong P, Roseweir AK, Edwards J. The NF-KB pathway and endocrine therapy resistance in breast cancer. Endocr Relat Cancer. (2019) 26:R369–80. doi: 10.1530/erc-19-0087
6. Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. (2011) 13:11–22. doi: 10.1016/j.cmet.2010.12.008
7. Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. (2013) 12:703–19. doi: 10.1038/nrd3976
9. DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. (2012) 246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x
10. Karunakaran D, Nguyen MA, Geoffrion M, Vreeken D, Lister Z, Cheng HS, et al. RIPK1 expression associates with inflammation in early atherosclerosis in humans and can be therapeutically silenced to reduce NF-κB activation and atherogenesis in mice. Circulation. (2021) 143:163–77. doi: 10.1161/circulationaha.118.038379
11. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
12. Henkel T, Zabel U, van Zee K, Müller JM, Fanning E, Baeuerle PA. Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-kappa B subunit. Cell. (1992) 68:1121–33. doi: 10.1016/0092-8674(92)90083-o
13. Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. (2014) 8:213. doi: 10.3389/fncel.2014.00213
14. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. (2020) 5:209. doi: 10.1038/s41392-020-00312-6
15. Tam WF, Sen R. IkappaB family members function by different mechanisms. J Biol Chem. (2001) 276:7701–4. doi: 10.1074/jbc.C000916200
16. Kearns JD, Basak S, Werner SL, Huang CS, Hoffmann A. IkappaBepsilon provides negative feedback to control NF-kappaB oscillations, signaling dynamics, and inflammatory gene expression. J Cell Biol. (2006) 173:659–64. doi: 10.1083/jcb.200510155
17. Arenzana-Seisdedos F, Thompson J, Rodriguez MS, Bachelerie F, Thomas D, Hay RT. Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF-kappa B. Mol Cell Biol. (1995) 15:2689–96. doi: 10.1128/mcb.15.5.2689
18. Savinova OV, Hoffmann A, Ghosh G. The Nfkb1 and Nfkb2 proteins p105 and p100 function as the core of high-molecular-weight heterogeneous complexes. Mol Cell. (2009) 34:591–602. doi: 10.1016/j.molcel.2009.04.033
19. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. (2009) 27:693–733. doi: 10.1146/annurev.immunol.021908.132641
20. Yang HT, Papoutsopoulou S, Belich M, Brender C, Janzen J, Gantke T, et al. Coordinate regulation of TPL-2 and NF-κB signaling in macrophages by NF-κB1 p105. Mol Cell Biol. (2012) 32:3438–51. doi: 10.1128/mcb.00564-12
21. Sriskantharajah S, Belich MP, Papoutsopoulou S, Janzen J, Tybulewicz V, Seddon B, et al. Proteolysis of NF-kappaB1 p105 is essential for T cell antigen receptor-induced proliferation. Nat Immunol. (2009) 10:38–47. doi: 10.1038/ni.1685
22. Schuster M, Annemann M, Plaza-Sirvent C, Schmitz I. Atypical IκB proteins - nuclear modulators of NF-κB signaling. Cell Commun Signal. (2013) 11:23. doi: 10.1186/1478-811x-11-23
23. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
24. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. (2002) 105:1135–43. doi: 10.1161/hc0902.104353
25. Abraham E. NF-kappaB activation. Crit Care Med. (2000) 28(Suppl. 4):N100–4. doi: 10.1097/00003246-200004001-00012
26. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. (2009) 1:a000034. doi: 10.1101/cshperspect.a000034
27. Hu H, Sun SC. Ubiquitin signaling in immune responses. Cell Res. (2016) 26:457–83. doi: 10.1038/cr.2016.40
28. Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. (1995) 81:495–504. doi: 10.1016/0092-8674(95)90070-5
29. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. (2006) 22:245–57. doi: 10.1016/j.molcel.2006.03.026
30. Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, et al. PKC-beta controls I kappa B kinase lipid raft recruitment and activation in response to BCR signaling. Nat Immunol. (2002) 3:780–6. doi: 10.1038/ni823
31. Bertin J, Wang L, Guo Y, Jacobson MD, Poyet JL, Srinivasula SM, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. (2001) 276:11877–82. doi: 10.1074/jbc.M010512200
32. Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. (2004) 14:289–301. doi: 10.1016/s1097-2765(04)00236-9
33. Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. (1997) 7:837–47. doi: 10.1016/s1074-7613(00)80402-1
34. Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R. TLR-4, IL-1R and TNF-R signaling to NF-kappaB: variations on a common theme. Cell Mol Life Sci. (2008) 65:2964–78. doi: 10.1007/s00018-008-8064-8
35. Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J Biol Chem. (2007) 282:4102–12. doi: 10.1074/jbc.M609503200
36. Jiang Z, Ninomiya-Tsuji J, Qian Y, Matsumoto K, Li X. Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol Cell Biol. (2002) 22:7158–67. doi: 10.1128/mcb.22.20.7158-7167.2002
37. Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. (1993) 259:1912–5. doi: 10.1126/science.8096091
38. Li Q, Lu Q, Bottero V, Estepa G, Morrison L, Mercurio F, et al. Enhanced NF-kappaB activation and cellular function in macrophages lacking IkappaB kinase 1 (IKK1). Proc Natl Acad Sci U S A. (2005) 102:12425–30. doi: 10.1073/pnas.0505997102
39. Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. (2007) 8:584–91. doi: 10.1038/ni1464
40. Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. (2004) 5:1052–60. doi: 10.1038/ni1110
41. Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. (2003) 424:797–801. doi: 10.1038/nature01811
42. Karim ZA, Zhang J, Banerjee M, Chicka MC, Al Hawas R, Hamilton TR, et al. IκB kinase phosphorylation of SNAP-23 controls platelet secretion. Blood. (2013) 121:4567–74. doi: 10.1182/blood-2012-11-470468
43. Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. (2012) 246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x
44. Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. (2008) 9:1371–8. doi: 10.1038/ni.1676
45. Dixit V, Mak TW. NF-kappaB signaling. Many roads lead to madrid. Cell. (2002) 111:615–9. doi: 10.1016/s0092-8674(02)01166-2
46. Huxford T, Ghosh G. A structural guide to proteins of the NF-kappaB signaling module. Cold Spring Harb Perspect Biol. (2009) 1:a000075. doi: 10.1101/cshperspect.a000075
47. Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. (2001) 7:401–9. doi: 10.1016/s1097-2765(01)00187-3
48. Fong A, Sun SC. Genetic evidence for the essential role of beta-transducin repeat-containing protein in the inducible processing of NF-kappa B2/p100. J Biol Chem. (2002) 277:22111–4. doi: 10.1074/jbc.C200151200
49. Liang C, Zhang M, Sun SC. Beta-TrCP binding and processing of NF-kappaB2/p100 involve its phosphorylation at serines 866 and 870. Cell Signal. (2006) 18:1309–17. doi: 10.1016/j.cellsig.2005.10.011
50. Sun M, Li S, Yu K, Xiang J, Li F. An E3 ubiquitin ligase TRIM9 is involved in WSSV infection via interaction with β-TrCP. Dev Comp Immunol. (2019) 97:57–63. doi: 10.1016/j.dci.2019.03.014
51. Arabi A, Ullah K, Branca RM, Johansson J, Bandarra D, Haneklaus M, et al. Proteomic screen reveals Fbw7 as a modulator of the NF-κB pathway. Nat Commun. (2012) 3:976. doi: 10.1038/ncomms1975
52. Hu H, Brittain GC, Chang JH, Puebla-Osorio N, Jin J, Zal A, et al. OTUD7B controls non-canonical NF-κB activation through deubiquitination of TRAF3. Nature. (2013) 494:371–4. doi: 10.1038/nature11831
53. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. (2011) 12:204–12. doi: 10.1038/ni.2001
54. Libby P. The changing landscape of atherosclerosis. Nature. (2021) 592:524–33. doi: 10.1038/s41586-021-03392-8
55. Frostegård J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. (2013) 11:117. doi: 10.1186/1741-7015-11-117
56. Shah PK. Mechanisms of plaque vulnerability and rupture. J Am Coll Cardiol. (2003) 41:15s–22s. doi: 10.1016/s0735-1097(02)02834-6
57. Pateras I, Giaginis C, Tsigris C, Patsouris E, Theocharis S. NF-κB signaling at the crossroads of inflammation and atherogenesis: searching for new therapeutic links. Expert Opin Ther Targets. (2014) 18:1089–101. doi: 10.1517/14728222.2014.938051
58. Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. (2011) 14:575–85. doi: 10.1016/j.cmet.2011.07.015
59. Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. (2005) 25:1256–61. doi: 10.1161/01.ATV.0000166517.18801.a7
60. Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. (2009) 86:1089–95. doi: 10.1189/jlb.0209115
61. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. (2015) 209:13–22. doi: 10.1083/jcb.201412052
62. Jeziorska M, Woolley DE. Local neovascularization and cellular composition within vulnerable regions of atherosclerotic plaques of human carotid arteries. J Pathol. (1999) 188:189–96. doi: 10.1002/(sici)1096-9896(199906)188:23.0.Co;2-n
63. Brogi E, Winkles JA, Underwood R, Clinton SK, Alberts GF, Libby P. Distinct patterns of expression of fibroblast growth factors and their receptors in human atheroma and nonatherosclerotic arteries. Association of acidic FGF with plaque microvessels and macrophages. J Clin Invest. (1993) 92:2408–18. doi: 10.1172/jci116847
64. Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. (2001) 107:255–64. doi: 10.1172/jci10373
65. Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, et al. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci U S A. (2004) 101:5634–9. doi: 10.1073/pnas.0401060101
66. de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. (2005) 25:904–14. doi: 10.1161/01.Atv.0000160340.72641.87
67. Wolfrum S, Teupser D, Tan M, Chen KY, Breslow JL. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-kappaB target genes. Proc Natl Acad Sci U S A. (2007) 104:18601–6. doi: 10.1073/pnas.0709011104
68. Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, et al. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. (2003) 112:1176–85. doi: 10.1172/jci18580
69. Yang S, Wu M, Li X, Zhao R, Zhao Y, Liu L, et al. Role of endoplasmic reticulum stress in atherosclerosis and its potential as a therapeutic target. Oxid Med Cell Longev. (2020) 2020:9270107. doi: 10.1155/2020/9270107
70. Battson ML, Lee DM, Gentile CL. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am J Physiol Heart Circ Physiol. (2017) 312:H355–67. doi: 10.1152/ajpheart.00437.2016
71. Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol (Oxf). (2017) 219:22–96. doi: 10.1111/apha.12646
72. Burleigh ME, Babaev VR, Yancey PG, Major AS, McCaleb JL, Oates JA, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in ApoE-deficient and C57BL/6 mice. J Mol Cell Cardiol. (2005) 39:443–52. doi: 10.1016/j.yjmcc.2005.06.011
73. Elnabawi YA, Garshick MS, Tawil M, Barrett TJ, Fisher EA, Lo Sicco K, et al. CCL20 in psoriasis: a potential biomarker of disease severity, inflammation, and impaired vascular health. J Am Acad Dermatol. (2021) 84:913–20. doi: 10.1016/j.jaad.2020.10.094
74. Calvayrac O, Rodríguez-Calvo R, Alonso J, Orbe J, Martín-Ventura JL, Guadall A, et al. CCL20 is increased in hypercholesterolemic subjects and is upregulated by LDL in vascular smooth muscle cells: role of NF-κB. Arterioscler Thromb Vasc Biol. (2011) 31:2733–41. doi: 10.1161/atvbaha.111.235721
75. Montanari E, Stojkovic S, Kaun C, Lemberger CE, de Martin R, Rauscher S, et al. Interleukin-33 stimulates GM-CSF and M-CSF production by human endothelial cells. Thromb Haemost. (2016) 116:317–27. doi: 10.1160/th15-12-0917
76. Civelek M, Manduchi E, Riley RJ, Stoeckert CJ Jr., Davies PF. Coronary artery endothelial transcriptome in vivo: identification of endoplasmic reticulum stress and enhanced reactive oxygen species by gene connectivity network analysis. Circ Cardiovasc Genet. (2011) 4:243–52. doi: 10.1161/circgenetics.110.958926
77. Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. (2000) 6:1355–64. doi: 10.1016/s1097-2765(00)00133-7
78. Bobrovnikova-Marjon E, Hatzivassiliou G, Grigoriadou C, Romero M, Cavener DR, Thompson CB, et al. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc Natl Acad Sci U S A. (2008) 105:16314–9. doi: 10.1073/pnas.0808517105
79. Ning J, Hong T, Ward A, Pi J, Liu Z, Liu HY, et al. Constitutive role for IRE1α-XBP1 signaling pathway in the insulin-mediated hepatic lipogenic program. Endocrinology. (2011) 152:2247–55. doi: 10.1210/en.2010-1036
80. Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res. (2011) 108:1122–32. doi: 10.1161/circresaha.110.226928
81. Mehrhof FB, Schmidt-Ullrich R, Dietz R, Scheidereit C. Regulation of vascular smooth muscle cell proliferation: role of NF-kappaB revisited. Circ Res. (2005) 96:958–64. doi: 10.1161/01.Res.0000166924.31219.49
82. Yang D, Sun C, Zhang J, Lin S, Zhao L, Wang L, et al. Proliferation of vascular smooth muscle cells under inflammation is regulated by NF-κB p65/microRNA-17/RB pathway activation. Int J Mol Med. (2018) 41:43–50. doi: 10.3892/ijmm.2017.3212
83. Lu QB, Wan MY, Wang PY, Zhang CX, Xu DY, Liao X, et al. Chicoric acid prevents PDGF-BB-induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFκB/mTOR/P70S6K signaling cascade. Redox Biol. (2018) 14:656–68. doi: 10.1016/j.redox.2017.11.012
84. Monaco C, Paleolog E. Nuclear factor kappaB: a potential therapeutic target in atherosclerosis and thrombosis. Cardiovasc Res. (2004) 61:671–82. doi: 10.1016/j.cardiores.2003.11.038
85. Kong P, Yu Y, Wang L, Dou YQ, Zhang XH, Cui Y, et al. circ-Sirt1 controls NF-κB activation via sequence-specific interaction and enhancement of SIRT1 expression by binding to miR-132/212 in vascular smooth muscle cells. Nucleic Acids Res. (2019) 47:3580–93. doi: 10.1093/nar/gkz141
86. Shi C, Deng J, Chiu M, Chen YX, O’Brien ER. Heat shock protein 27 immune complex altered signaling and transport (ICAST): novel mechanisms of attenuating inflammation. Faseb J. (2020) 34:14287–301. doi: 10.1096/fj.202001389RR
87. Crane ED, Al-Hashimi AA, Chen J, Lynn EG, Won KD, Lhoták S, et al. Anti-GRP78 autoantibodies induce endothelial cell activation and accelerate the development of atherosclerotic lesions. JCI Insight. (2018) 3:e99363. doi: 10.1172/jci.insight.99363
88. Xie Z, Wang X, Liu X, Du H, Sun C, Shao X, et al. Adipose-derived exosomes exert proatherogenic effects by regulating macrophage foam cell formation and polarization. J Am Heart Assoc. (2018) 7:e007442. doi: 10.1161/jaha.117.007442
89. Isa SA, Ruffino JS, Ahluwalia M, Thomas AW, Morris K, Webb R. M2 macrophages exhibit higher sensitivity to oxLDL-induced lipotoxicity than other monocyte/macrophage subtypes. Lipids Health Dis. (2011) 10:229. doi: 10.1186/1476-511x-10-229
90. Yamada Y, Doi T, Hamakubo T, Kodama T. Scavenger receptor family proteins: roles for atherosclerosis, host defence and disorders of the central nervous system. Cell Mol Life Sci. (1998) 54:628–40. doi: 10.1007/s000180050191
91. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
92. Yang S, Li J, Chen Y, Zhang S, Feng C, Hou Z, et al. MicroRNA-216a promotes M1 macrophages polarization and atherosclerosis progression by activating telomerase via the Smad3/NF-κB pathway. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:1772–81. doi: 10.1016/j.bbadis.2018.06.016
93. Plotkin JD, Elias MG, Dellinger AL, Kepley CL. NF-κB inhibitors that prevent foam cell formation and atherosclerotic plaque accumulation. Nanomedicine. (2017) 13:2037–48. doi: 10.1016/j.nano.2017.04.013
94. Sage AP, Nus M, Bagchi Chakraborty J, Tsiantoulas D, Newland SA, Finigan AJ, et al. X-Box binding protein-1 dependent plasma cell responses limit the development of atherosclerosis. Circ Res. (2017) 121:270–81. doi: 10.1161/circresaha.117.310884
95. Zeng L, Zampetaki A, Margariti A, Pepe AE, Alam S, Martin D, et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc Natl Acad Sci U S A. (2009) 106:8326–31. doi: 10.1073/pnas.0903197106
96. Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, et al. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem. (2012) 287:11629–41. doi: 10.1074/jbc.M111.338673
97. Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. (2006) 116:59–69. doi: 10.1172/jci25074
98. Wang N, Zhang X, Ma Z, Niu J, Ma S, Wenjie W, et al. Combination of tanshinone IIA and astragaloside IV attenuate atherosclerotic plaque vulnerability in ApoE(−/−) mice by activating PI3K/AKT signaling and suppressing TRL4/NF-κB signaling. Biomed Pharmacother. (2020) 123:109729. doi: 10.1016/j.biopha.2019.109729
99. Zhang M, Xue Y, Chen H, Meng L, Chen B, Gong H, et al. Resveratrol inhibits MMP3 and MMP9 expression and secretion by suppressing TLR4/NF-κB/STAT3 activation in Ox-LDL-Treated HUVECs. Oxid Med Cell Longev. (2019) 2019:9013169. doi: 10.1155/2019/9013169
100. Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ Jr., Chow WS, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. (1998) 4:1025–31. doi: 10.1038/2012
101. Cuccurullo C, Iezzi A, Fazia ML, De Cesare D, Di Francesco A, Muraro R, et al. Suppression of RAGE as a basis of simvastatin-dependent plaque stabilization in type 2 diabetes. Arterioscler Thromb Vasc Biol. (2006) 26:2716–23. doi: 10.1161/01.Atv.0000249630.02085.12
102. Martín-Ventura JL, Blanco-Colio LM, Muñoz-García B, Gómez-Hernández A, Arribas A, Ortega L, et al. NF-kappaB activation and Fas ligand overexpression in blood and plaques of patients with carotid atherosclerosis: potential implication in plaque instability. Stroke. (2004) 35:458–63. doi: 10.1161/01.Str.0000114876.51656.7a
103. Zhou J, Werstuck GH, Lhoták S, de Koning AB, Sood SK, Hossain GS, et al. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation. (2004) 110:207–13. doi: 10.1161/01.Cir.0000134487.51510.97
104. Wu R, Zhang QH, Lu YJ, Ren K, Yi GH. Involvement of the IRE1α-XBP1 pathway and XBP1s-dependent transcriptional reprogramming in metabolic diseases. DNA Cell Biol. (2015) 34:6–18. doi: 10.1089/dna.2014.2552
105. Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. (2011) 31:2792–7. doi: 10.1161/atvbaha.111.224881
106. Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. (2007) 116:1226–33. doi: 10.1161/circulationaha.106.682054
107. Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. Cell Metab. (2009) 9:474–81. doi: 10.1016/j.cmet.2009.03.003
108. Gao J, Ishigaki Y, Yamada T, Kondo K, Yamaguchi S, Imai J, et al. Involvement of endoplasmic stress protein C/EBP homologous protein in arteriosclerosis acceleration with augmented biological stress responses. Circulation. (2011) 124:830–9. doi: 10.1161/circulationaha.110.014050
109. Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, et al. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. (2010) 121:792–803. doi: 10.1161/circulationaha.109.900928
110. Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. (2009) 15:1383–91. doi: 10.1038/nm.2067
111. Mogilenko DA, Haas JT, L’Homme L, Fleury S, Quemener S, Levavasseur M, et al. Metabolic and innate immune cues merge into a specific inflammatory response via the UPR. Cell. (2019) 177:1201–16.e19. doi: 10.1016/j.cell.2019.03.018
112. Burgos-Morón E, Abad-Jiménez Z, Marañón AM, Iannantuoni F, Escribano-López I, López-Domènech S, et al. Relationship between oxidative stress, ER stress, and inflammation in Type 2 diabetes: the battle continues. J Clin Med. (2019) 8:1385. doi: 10.3390/jcm8091385
113. Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. (2017) 168:692–706. doi: 10.1016/j.cell.2016.12.004
114. Inagi R. Glycative stress and glyoxalase in kidney disease and aging. Biochem Soc Trans. (2014) 42:457–60. doi: 10.1042/bst20140007
115. Zhou AX, Tabas I. The UPR in atherosclerosis. Semin Immunopathol. (2013) 35:321–32. doi: 10.1007/s00281-013-0372-x
116. Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. (2019) 20:665–80. doi: 10.1038/s41580-019-0133-3
117. Chen HW, Kuo HT, Wang SJ, Lu TS, Yang RC. In vivo heat shock protein assembles with septic liver NF-kappaB/I-kappaB complex regulating NF-kappaB activity. Shock. (2005) 24:232–8. doi: 10.1097/01.shk.0000174020.87439.f2
118. Kim I, Shin HM, Baek W. Heat-shock response is associated with decreased production of interleukin-6 in murine aortic vascular smooth muscle cells. Naunyn Schmiedebergs Arch Pharmacol. (2005) 371:27–33. doi: 10.1007/s00210-004-1007-5
119. Stöhr D, Jeltsch A, Rehm M. TRAIL receptor signaling: from the basics of canonical signal transduction toward its entanglement with ER stress and the unfolded protein response. Int Rev Cell Mol Biol. (2020) 351:57–99. doi: 10.1016/bs.ircmb.2020.02.002
120. Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. (2000) 2:241–3. doi: 10.1038/35008667
121. Sullivan GP, O’Connor H, Henry CM, Davidovich P, Clancy DM, Albert ML, et al. TRAIL receptors serve as stress-associated molecular patterns to promote ER-Stress-Induced inflammation. Dev Cell. (2020) 52:714–30.e5. doi: 10.1016/j.devcel.2020.01.031
122. Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol. (2021) 18:499–521. doi: 10.1038/s41569-021-00511-w
123. Shen CH, Tung SY, Huang WS, Lu CC, Lee KC, Hsieh YY, et al. Exploring the effects of tert-butylhydroperoxide induced liver injury using proteomic approach. Toxicology. (2014) 316:61–70. doi: 10.1016/j.tox.2013.12.007
124. Chen J, Zhang M, Zhu M, Gu J, Song J, Cui L, et al. Paeoniflorin prevents endoplasmic reticulum stress-associated inflammation in lipopolysaccharide-stimulated human umbilical vein endothelial cells via the IRE1α/NF-κB signaling pathway. Food Funct. (2018) 9:2386–97. doi: 10.1039/c7fo01406f
125. Keestra-Gounder AM, Byndloss MX, Seyffert N, Young BM, Chávez-Arroyo A, Tsai AY, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature. (2016) 532:394–7. doi: 10.1038/nature17631
126. Lencer WI, DeLuca H, Grey MJ, Cho JA. Innate immunity at mucosal surfaces: the IRE1-RIDD-RIG-I pathway. Trends Immunol. (2015) 36:401–9. doi: 10.1016/j.it.2015.05.006
127. Tam AB, Koong AC, Niwa M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. (2014) 9:850–8. doi: 10.1016/j.celrep.2014.09.016
128. Zhang H, Song G, Zhang Z, Song H, Tang X, Deng A, et al. Colitis is effectively ameliorated by (±)-8-Acetonyl-dihydrocoptisine via the XBP1-NF-κB pathway. Front Pharmacol. (2017) 8:619. doi: 10.3389/fphar.2017.00619
129. Kim S, Joe Y, Kim HJ, Kim YS, Jeong SO, Pae HO, et al. Endoplasmic reticulum stress-induced IRE1α activation mediates cross-talk of GSK-3β and XBP-1 to regulate inflammatory cytokine production. J Immunol. (2015) 194:4498–506. doi: 10.4049/jimmunol.1401399
130. Sun H, Wei G, Liu H, Xiao D, Huang J, Lu J, et al. Inhibition of XBP1s ubiquitination enhances its protein stability and improves glucose homeostasis. Metabolism. (2020) 105:154046. doi: 10.1016/j.metabol.2019.154046
131. Schwabe RF, Brenner DA. Role of glycogen synthase kinase-3 in TNF-alpha-induced NF-kappaB activation and apoptosis in hepatocytes. Am J Physiol Gastrointest Liver Physiol. (2002) 283:G204–11. doi: 10.1152/ajpgi.00016.2002
132. Demarchi F, Bertoli C, Sandy P, Schneider C. Glycogen synthase kinase-3 beta regulates NF-kappa B1/p105 stability. J Biol Chem. (2003) 278:39583–90. doi: 10.1074/jbc.M305676200
133. Zhang H, Zhao C, Wang S, Huang Y, Wang H, Zhao J, et al. Anti-dsDNA antibodies induce inflammation via endoplasmic reticulum stress in human mesangial cells. J Transl Med. (2015) 13:178. doi: 10.1186/s12967-015-0536-7
134. Mendez JM, Kolora LD, Lemon JS, Dupree SL, Keestra-Gounder AM. Activation of the endoplasmic reticulum stress response impacts the NOD1 signaling pathway. Infect Immun. (2019) 87:e826–818. doi: 10.1128/iai.00826-18
135. Stêpkowski TM, Kruszewski MK. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic Biol Med. (2011) 50:1186–95. doi: 10.1016/j.freeradbiomed.2011.01.033
136. Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. (2010) 285:22576–91. doi: 10.1074/jbc.M110.118976
137. Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. (2008) 13:343–54. doi: 10.1016/j.ccr.2008.02.001
138. Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. (2006) 116:984–95. doi: 10.1172/jci25790
139. Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, et al. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol. (2004) 172:3553–63. doi: 10.4049/jimmunol.172.6.3553
140. Li J, Holbrook NJ. Elevated gadd153/chop expression and enhanced c-Jun N-terminal protein kinase activation sensitizes aged cells to ER stress. Exp Gerontol. (2004) 39:735–44. doi: 10.1016/j.exger.2004.02.008
141. Alonso-Piñeiro JA, Gonzalez-Rovira A, Sánchez-Gomar I, Moreno JA, Durán-Ruiz MC. Nrf2 and heme oxygenase-1 involvement in atherosclerosis related oxidative stress. Antioxidants (Basel). (2021) 10:1463. doi: 10.3390/antiox10091463
142. Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K, et al. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem. (2005) 280:27244–50. doi: 10.1074/jbc.M502551200
143. Harada N, Ito K, Hosoya T, Mimura J, Maruyama A, Noguchi N, et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic Biol Med. (2012) 53:2256–62. doi: 10.1016/j.freeradbiomed.2012.10.001
144. Ruotsalainen AK, Inkala M, Partanen ME, Lappalainen JP, Kansanen E, Mäkinen PI, et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc Res. (2013) 98:107–15. doi: 10.1093/cvr/cvt008
145. Freigang S, Ampenberger F, Spohn G, Heer S, Shamshiev AT, Kisielow J, et al. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol. (2011) 41:2040–51. doi: 10.1002/eji.201041316
146. Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. (2004) 94:609–16. doi: 10.1161/01.Res.0000119171.44657.45
147. Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol. (2009) 183:1480–7. doi: 10.4049/jimmunol.0900017
148. Chen Z, Liu Y, Yang L, Liu P, Zhang Y, Wang X. MiR-149 attenuates endoplasmic reticulum stress-induced inflammation and apoptosis in nonalcoholic fatty liver disease by negatively targeting ATF6 pathway. Immunol Lett. (2020) 222:40–8. doi: 10.1016/j.imlet.2020.03.003
149. Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, et al. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am J Transplant. (2014) 14:1552–61. doi: 10.1111/ajt.12711
150. Schmitz ML, Shaban MS, Albert BV, Gökçen A, Kracht M. The crosstalk of endoplasmic reticulum (ER) stress pathways with NF-κB: complex mechanisms relevant for cancer, inflammation and infection. Biomedicines. (2018) 6:58. doi: 10.3390/biomedicines6020058
151. Kim YG, Kim SM, Kim KP, Lee SH, Moon JY. The role of inflammasome-dependent and inflammasome-independent NLRP3 in the kidney. Cells. (2019) 8:1389. doi: 10.3390/cells8111389
152. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
153. Awad F, Assrawi E, Louvrier C, Jumeau C, Georgin-Lavialle S, Grateau G, et al. Inflammasome biology, molecular pathology and therapeutic implications. Pharmacol Ther. (2018) 187:133–49. doi: 10.1016/j.pharmthera.2018.02.011
154. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
155. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: its regulation and involvement in atherosclerosis. J Cell Physiol. (2018) 233:2116–32. doi: 10.1002/jcp.25930
156. Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. (2021) 18:1141–60. doi: 10.1038/s41423-021-00670-3
157. Hu X, Li D, Wang J, Guo J, Li Y, Cao Y, et al. Melatonin inhibits endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation in lipopolysaccharide-induced endometritis in mice. Int Immunopharmacol. (2018) 64:101–9. doi: 10.1016/j.intimp.2018.08.028
158. Du RH, Tan J, Yan N, Wang L, Qiao C, Ding JH, et al. Kir6.2 knockout aggravates lipopolysaccharide-induced mouse liver injury via enhancing NLRP3 inflammasome activation. J Gastroenterol. (2014) 49:727–36. doi: 10.1007/s00535-013-0823-0
159. Yarmohammadi F, Hayes AW, Karimi G. Possible protective effect of resolvin D1 on inflammation in atrial fibrillation: involvement of ER stress mediated the NLRP3 inflammasome pathway. Naunyn Schmiedebergs Arch Pharmacol. (2021) 394:1613–9. doi: 10.1007/s00210-021-02115-0
160. Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, et al. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. (2012) 3:e261. doi: 10.1038/cddis.2011.132
161. Zeng W, Wu D, Sun Y, Suo Y, Yu Q, Zeng M, et al. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci Rep. (2021) 11:19305. doi: 10.1038/s41598-021-98437-3
162. Sharma BR, Kanneganti TD. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol. (2021) 22:550–9. doi: 10.1038/s41590-021-00886-5
163. Van Hauwermeiren F, Van Opdenbosch N, Van Gorp H, de Vasconcelos N, van Loo G, Vandenabeele P, et al. Bacillus anthracis induces NLRP3 inflammasome activation and caspase-8-mediated apoptosis of macrophages to promote lethal anthrax. Proc Natl Acad Sci U S A. (2022) 119:e2116415119. doi: 10.1073/pnas.2116415119
164. Wan Z, Fan Y, Liu X, Xue J, Han Z, Zhu C, et al. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes Metab Syndr Obes. (2019) 12:1931–42. doi: 10.2147/dmso.S222053
165. Burger F, Baptista D, Roth A, da Silva RF, Montecucco F, Mach F, et al. NLRP3 inflammasome activation controls vascular smooth muscle cells phenotypic switch in atherosclerosis. Int J Mol Sci. (2021) 23:340. doi: 10.3390/ijms23010340
166. Zheng F, Xing S, Gong Z, Mu W, Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. (2014) 2014:507208. doi: 10.1155/2014/507208
167. Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, et al. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. (2011) 2:e137. doi: 10.1038/cddis.2011.18
168. Bleda S, de Haro J, Varela C, Esparza L, Ferruelo A, Acin F. NLRP1 inflammasome, and not NLRP3, is the key in the shift to proinflammatory state on endothelial cells in peripheral arterial disease. Int J Cardiol. (2014) 172:e282–4. doi: 10.1016/j.ijcard.2013.12.201
169. Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J Biol Chem. (2003) 278:24233–41. doi: 10.1074/jbc.M212389200
170. Schoonbroodt S, Ferreira V, Best-Belpomme M, Boelaert JR, Legrand-Poels S, Korner M, et al. Crucial role of the amino-terminal tyrosine residue 42 and the carboxyl-terminal PEST domain of I kappa B alpha in NF-kappa B activation by an oxidative stress. J Immunol. (2000) 164:4292–300. doi: 10.4049/jimmunol.164.8.4292
171. Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A. (1991) 88:4328–32. doi: 10.1073/pnas.88.10.4328
172. Liu J, Yoshida Y, Yamashita U. DNA-binding activity of NF-kappaB and phosphorylation of p65 are induced by N-acetylcysteine through phosphatidylinositol (PI) 3-kinase. Mol Immunol. (2008) 45:3984–9. doi: 10.1016/j.molimm.2008.06.012
173. Morgan MJ, Kim YS, Liu ZG. TNFalpha and reactive oxygen species in necrotic cell death. Cell Res. (2008) 18:343–9. doi: 10.1038/cr.2008.31
174. Ochoa CD, Wu RF, Terada LS. ROS signaling and ER stress in cardiovascular disease. Mol Aspects Med. (2018) 63:18–29. doi: 10.1016/j.mam.2018.03.002
175. Li W, Zhou X, Cai J, Zhao F, Cao T, Ning L, et al. Recombinant Treponema pallidum protein Tp0768 promotes proinflammatory cytokine secretion of macrophages through ER stress and ROS/NF-κB pathway. Appl Microbiol Biotechnol. (2021) 105:353–66. doi: 10.1007/s00253-020-11018-8
176. Kim HK, Lee GH, Bhattarai KR, Junjappa RP, Lee HY, Handigund M, et al. PI3Kδ contributes to ER stress-associated asthma through ER-redox disturbances: the involvement of the RIDD-RIG-I-NF-κB axis. Exp Mol Med. (2018) 50:e444. doi: 10.1038/emm.2017.270
177. Ma YM, Peng YM, Zhu QH, Gao AH, Chao B, He QJ, et al. Novel CHOP activator LGH00168 induces necroptosis in A549 human lung cancer cells via ROS-mediated ER stress and NF-κB inhibition. Acta Pharmacol Sin. (2016) 37:1381–90. doi: 10.1038/aps.2016.61
178. Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. (2018) 100:1–19. doi: 10.1016/j.vph.2017.05.005
179. Ungvari Z, Tarantini S, Kiss T, Wren JD, Giles CB, Griffin CT, et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nature Reviews Cardiology. (2018) 15:555–65. doi: 10.1038/s41569-018-0030-z
180. Lara-Guzmán OJ, Gil-Izquierdo A, Medina S, Osorio E, Alvarez-Quintero R, Zuluaga N, et al. Oxidized LDL triggers changes in oxidative stress and inflammatory biomarkers in human macrophages. Redox Biol. (2018) 15:1–11. doi: 10.1016/j.redox.2017.11.017
181. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. (2018) 122:877–902. doi: 10.1161/circresaha.117.311401
182. Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res. (2006) 99:924–32. doi: 10.1161/01.RES.0000248212.86638.e9
183. Niki E. Do free radicals play causal role in atherosclerosis? Low density lipoprotein oxidation and vitamin E revisited. J Clin Biochem Nutr. (2011) 48:3–7. doi: 10.3164/jcbn.11-007FR
184. Wang Z, Liu B, Zhu J, Wang D, Wang Y. Nicotine-mediated autophagy of vascular smooth muscle cell accelerates atherosclerosis via nAChRs/ROS/NF-κB signaling pathway. Atherosclerosis. (2019) 284:1–10. doi: 10.1016/j.atherosclerosis.2019.02.008
185. Lozhkin A, Vendrov AE, Pan H, Wickline SA, Madamanchi NR, Runge MS. NADPH oxidase 4 regulates vascular inflammation in aging and atherosclerosis. J Mol Cell Cardiol. (2017) 102:10–21. doi: 10.1016/j.yjmcc.2016.12.004
186. Hu P, Wu X, Khandelwal AR, Yu W, Xu Z, Chen L, et al. Endothelial Nox4-based NADPH oxidase regulates atherosclerosis via soluble epoxide hydrolase. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1382–91. doi: 10.1016/j.bbadis.2017.02.004
187. Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, et al. Endothelial NADPH oxidase 4 protects ApoE-/- mice from atherosclerotic lesions. Free Radic Biol Med. (2015) 89:1–7. doi: 10.1016/j.freeradbiomed.2015.07.004
188. Gray SP, Di Marco E, Kennedy K, Chew P, Okabe J, El-Osta A, et al. Reactive oxygen species can provide atheroprotection via NOX4-dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol. (2016) 36:295–307. doi: 10.1161/atvbaha.115.307012
189. Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. (2010) 191:1113–25. doi: 10.1083/jcb.201006121
190. Ewald CY, Hourihan JM, Bland MS, Obieglo C, Katic I, Moronetti Mazzeo LE, et al. NADPH oxidase-mediated redox signaling promotes oxidative stress resistance and longevity through memo-1 in C. elegans. Elife. (2017) 6:e19493. doi: 10.7554/eLife.19493
191. Wu RF, Liao C, Hatoum H, Fu G, Ochoa CD, Terada LS. RasGRF couples Nox4-dependent endoplasmic reticulum signaling to Ras. Arteriosclerosis Thrombosis Vasc Biol. (2017) 37:98–107. doi: 10.1161/ATVBAHA.116.307922
192. Lee J, Rhee MH, Kim E, Cho JY. BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediators Inflamm. (2012) 2012:416036. doi: 10.1155/2012/416036
193. Irrera N, Vaccaro M, Bitto A, Pallio G, Pizzino G, Lentini M, et al. BAY 11-7082 inhibits the NF-κB and NLRP3 inflammasome pathways and protects against IMQ-induced psoriasis. Clin Sci (Lond). (2017) 131:487–98. doi: 10.1042/cs20160645
194. Scuderi SA, Casili G, Basilotta R, Lanza M, Filippone A, Raciti G, et al. NLRP3 inflammasome inhibitor BAY-117082 reduces oral squamous cell carcinoma progression. Int J Mol Sci. (2021) 22:11108. doi: 10.3390/ijms222011108
195. Moon SK, Jung SY, Choi YH, Lee YC, Patterson C, Kim CH. PDTC, metal chelating compound, induces G1 phase cell cycle arrest in vascular smooth muscle cells through inducing p21Cip1 expression: involvement of p38 mitogen activated protein kinase. J Cell Physiol. (2004) 198:310–23. doi: 10.1002/jcp.10728
196. Bhat OM, Kumar PU, Giridharan NV, Kaul D, Kumar MJ, Dhawan V. Interleukin-18-induced atherosclerosis involves CD36 and NF-κB crosstalk in Apo E-/- mice. J Cardiol. (2015) 66:28–35. doi: 10.1016/j.jjcc.2014.10.012
197. Zhang W, Li XJ, Zeng X, Shen DY, Liu CQ, Zhang HJ, et al. Activation of nuclear factor-κB pathway is responsible for tumor necrosis factor-α-induced up-regulation of endothelin B2 receptor expression in vascular smooth muscle cells in vitro. Toxicol Lett. (2012) 209:107–12. doi: 10.1016/j.toxlet.2011.12.005
198. Feng Y, Pathria G, Heynen-Genel S, Jackson M, James B, Yin J, et al. Identification and characterization of IMD-0354 as a glutamine carrier protein inhibitor in melanoma. Mol Cancer Ther. (2021) 20:816–32. doi: 10.1158/1535-7163.Mct-20-0354
199. Prola A, Pires Da Silva J, Guilbert A, Lecru L, Piquereau J, Ribeiro M, et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2α deacetylation. Cell Death Differ. (2017) 24:343–56. doi: 10.1038/cdd.2016.138
200. Zheng G, Li H, Zhang T, Yang L, Yao S, Chen S, et al. Irisin protects macrophages from oxidized low density lipoprotein-induced apoptosis by inhibiting the endoplasmic reticulum stress pathway. Saudi J Biol Sci. (2018) 25:849–57. doi: 10.1016/j.sjbs.2017.08.018
201. Zhang Y, Song H, Zhang Y, Wu F, Mu Q, Jiang M, et al. Irisin inhibits atherosclerosis by promoting endothelial proliferation through microRNA126-5p. J Am Heart Assoc. (2016) 5:e004031. doi: 10.1161/jaha.116.004031
202. Remuzgo-Martínez S, Rueda-Gotor J, Pulito-Cueto V, López-Mejías R, Corrales A, Lera-Gómez L, et al. Irisin as a novel biomarker of subclinical atherosclerosis, cardiovascular risk and severe disease in axial spondyloarthritis. Front Immunol. (2022) 13:894171. doi: 10.3389/fimmu.2022.894171
203. Gouni-Berthold I, Berthold HK, Huh JY, Berman R, Spenrath N, Krone W, et al. Effects of lipid-lowering drugs on irisin in human subjects in vivo and in human skeletal muscle cells ex vivo. PLoS One. (2013) 8:e72858. doi: 10.1371/journal.pone.0072858
204. Agh F, Mohammadzadeh Honarvar N, Djalali M, Nematipour E, Gholamhoseini S, Zarei M, et al. Omega-3 fatty acid could increase one of myokines in male patients with coronary artery disease: a randomized, double-blind, placebo-controlled trial. Arch Iran Med. (2017) 20: 28–33.
205. Lebeaupin C, Vallée D, Rousseau D, Patouraux S, Bonnafous S, Adam G, et al. Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice. Hepatology. (2018) 68:515–32. doi: 10.1002/hep.29847
206. Zhang F, Tang B, Zhang Z, Xu D, Ma G. DUSP6 Inhibitor (E/Z)-BCI hydrochloride attenuates lipopolysaccharide-induced inflammatory responses in murine macrophage cells via activating the Nrf2 signaling axis and inhibiting the NF-κB pathway. Inflammation. (2019) 42:672–81. doi: 10.1007/s10753-018-0924-2
207. Bian H, Wang G, Huang J, Liang L, Zheng Y, Wei Y, et al. Dihydrolipoic acid protects against lipopolysaccharide-induced behavioral deficits and neuroinflammation via regulation of Nrf2/HO-1/NLRP3 signaling in rat. J Neuroinflamm. (2020) 17:166. doi: 10.1186/s12974-020-01836-y
208. Wu NN, Tian H, Chen P, Wang D, Ren J, Zhang Y. Physical exercise and selective autophagy: benefit and risk on cardiovascular health. Cells. (2019) 8:1436. doi: 10.3390/cells8111436
209. Chang P, Zhang X, Zhang M, Li G, Hu L, Zhao H, et al. Swimming exercise inhibits myocardial ER stress in the hearts of aged mice by enhancing cGMP-PKG signaling. Mol Med Rep. (2020) 21:549–56. doi: 10.3892/mmr.2019.10864
210. Shen M, Wang L, Yang G, Gao L, Wang B, Guo X, et al. Baicalin protects the cardiomyocytes from ER stress-induced apoptosis: inhibition of CHOP through induction of endothelial nitric oxide synthase. PLoS One. (2014) 9:e88389. doi: 10.1371/journal.pone.0088389
211. Cai X, Bao L, Dai X, Ding Y, Zhang Z, Li Y. Quercetin protects RAW264.7 macrophages from glucosamine-induced apoptosis and lipid accumulation via the endoplasmic reticulum stress pathway. Mol Med Rep. (2015) 12:7545–53. doi: 10.3892/mmr.2015.4340
212. Zwicker JI, Schlechter BL, Stopa JD, Liebman HA, Aggarwal A, Puligandla M, et al. Targeting protein disulfide isomerase with the flavonoid isoquercetin to improve hypercoagulability in advanced cancer. JCI Insight. (2019) 4:e125851. doi: 10.1172/jci.insight.125851
213. Dagher O, Mury P, Noly PE, Fortier A, Lettre G, Thorin E, et al. Design of a randomized placebo-controlled trial to evaluate the anti-inflammatory and senolytic effects of quercetin in patients undergoing coronary artery bypass graft surgery. Front Cardiovasc Med. (2021) 8:741542. doi: 10.3389/fcvm.2021.741542
214. Kar P, Laight D, Rooprai HK, Shaw KM, Cummings M. Effects of grape seed extract in Type 2 diabetic subjects at high cardiovascular risk: a double blind randomized placebo controlled trial examining metabolic markers, vascular tone, inflammation, oxidative stress and insulin sensitivity. Diabet Med. (2009) 26:526–31. doi: 10.1111/j.1464-5491.2009.02727.x
215. Lin Y, Zhu J, Zhang X, Wang J, Xiao W, Li B, et al. Inhibition of cardiomyocytes hypertrophy by resveratrol is associated with amelioration of endoplasmic reticulum stress. Cell Physiol Biochem. (2016) 39:780–9. doi: 10.1159/000447788
216. Lou Y, Wang Z, Xu Y, Zhou P, Cao J, Li Y, et al. Resveratrol prevents doxorubicin-induced cardiotoxicity in H9c2 cells through the inhibition of endoplasmic reticulum stress and the activation of the Sirt1 pathway. Int J Mol Med. (2015) 36:873–80. doi: 10.3892/ijmm.2015.2291
217. van der Made SM, Plat J, Mensink RP. Trans-resveratrol supplementation and endothelial function during the fasting and postprandial phase: a randomized placebo-controlled trial in overweight and slightly obese participants. Nutrients. (2017) 9:596. doi: 10.3390/nu9060596
218. Marques B, Trindade M, Aquino JCF, Cunha AR, Gismondi RO, Neves MF, et al. Beneficial effects of acute trans-resveratrol supplementation in treated hypertensive patients with endothelial dysfunction. Clin Exp Hypertens. (2018) 40:218–23. doi: 10.1080/10641963.2017.1288741
219. Pecoraro L, Zoller T, Atkinson RL, Nisi F, Antoniazzi F, Cavarzere P, et al. Supportive treatment of vascular dysfunction in pediatric subjects with obesity: the OBELIX study. Nutr Diabetes. (2022) 12:2. doi: 10.1038/s41387-021-00180-1
220. Dong L, Qiao H, Zhang X, Zhang X, Wang C, Wang L, et al. Parthenolide is neuroprotective in rat experimental stroke model: downregulating NF-κB, phospho-p38MAPK, and caspase-1 and ameliorating BBB permeability. Mediators Inflamm. (2013) 2013:370804. doi: 10.1155/2013/370804
221. Yang X, Gao X, Cao Y, Guo Q, Li S, Zhu Z, et al. Anti-inflammatory effects of boldine and reticuline isolated from litsea cubeba through JAK2/STAT3 and NF-κB signaling pathways. Planta Med. (2018) 84:20–5. doi: 10.1055/s-0043-113447
222. Lee S, Choi SY, Choo YY, Kim O, Tran PT, Dao CT, et al. Sappanone A exhibits anti-inflammatory effects via modulation of Nrf2 and NF-κB. Int Immunopharmacol. (2015) 28:328–36. doi: 10.1016/j.intimp.2015.06.015
223. Zeng J, Chen Y, Ding R, Feng L, Fu Z, Yang S, et al. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J Neuroinflamm. (2017) 14:119. doi: 10.1186/s12974-017-0895-5
224. Su Y, Yuan J, Zhang F, Lei Q, Zhang T, Li K, et al. MicroRNA-181a-5p and microRNA-181a-3p cooperatively restrict vascular inflammation and atherosclerosis. Cell Death Dis. (2019) 10:365. doi: 10.1038/s41419-019-1599-9
225. Simion V, Zhou H, Pierce JB, Yang D, Haemmig S, Tesmenitsky Y, et al. LncRNA VINAS regulates atherosclerosis by modulating NF-κB and MAPK signaling. JCI Insight. (2020) 5:e140627. doi: 10.1172/jci.insight.140627
Keywords: NF-κB, endoplasmic reticulum stress, atherosclerosis, unfolded protein response, NLRP3 inflammasome, reactive oxygen species
Citation: Li W, Jin K, Luo J, Xu W, Wu Y, Zhou J, Wang Y, Xu R, Jiao L, Wang T and Yang G (2022) NF-κB and its crosstalk with endoplasmic reticulum stress in atherosclerosis. Front. Cardiovasc. Med. 9:988266. doi: 10.3389/fcvm.2022.988266
Received: 07 July 2022; Accepted: 30 August 2022;
Published: 20 September 2022.
Edited by:
Mark Slevin, Manchester Metropolitan University, United KingdomReviewed by:
Preetha Shridas, University of Kentucky, United StatesCopyright © 2022 Li, Jin, Luo, Xu, Wu, Zhou, Wang, Xu, Jiao, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liqun Jiao, bGlxdW5qaWFvQHNpbmEuY24=; Tao Wang, d2FuZ3Rhb19kckBzaW5hLmNvbQ==; Ge Yang, R2UueWFuZ0BpYS5hYy5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.