
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 19 October 2022
Sec. General Cardiovascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.982871
This article is part of the Research Topic Atrial Fibrillation: Insights on mechanisms, mapping and catheter ablation View all 50 articles
 Zainu Nezami1†
Zainu Nezami1† Hannes Holm2,3*†Marcus Ohlsson1,2John Molvin2,3
Hannes Holm2,3*†Marcus Ohlsson1,2John Molvin2,3 Johan Korduner2Erasmus Bachus2
Johan Korduner2Erasmus Bachus2 Amir Zaghi2Anna Dieden2
Amir Zaghi2Anna Dieden2 Pyotr G. Platonov4
Pyotr G. Platonov4 Amra Jujic2,3
Amra Jujic2,3 Martin Magnusson2,3,5,6
Martin Magnusson2,3,5,6Background: Several studies suggest that circulating biomarkers of myocardial fibrosis are associated with worse prognosis in subjects with atrial fibrillation (AF). Here, we aimed to explore associations between fibrosis biomarkers, prevalent AF, and left atrial volume (LAV) enlargement in subjects with heart failure (HF). Additionally, we evaluated the prognostic impact of fibrotic biomarkers in HF with co-existing AF.
Materials and methods: Patients hospitalized for HF (n = 316, mean age 75 years; 30% women) were screened for AF. Seven proteins previously associated with myocardial fibrosis [metalloproteinase inhibitor 4 (TIMP-4), suppression of tumorigenicity 2 (ST-2), galectin-3 (GAL-3), growth/differentiation factor-15 (GDF-15), and matrix metalloproteinase 2, 3, and 9 (MMP-3, MMP-3, and MMP-9, respectively)] were analyzed using a proximity extension assay. Proteins with significant Bonferroni-corrected associations with mortality and re-hospitalization risk were taken forward to multivariable Cox regression analyses. Further, Bonferroni-corrected multivariable logistic regression models were used to study associations between protein plasma levels, prevalent AF, and severely enlarged left atrial volume index (LAVI ≥ 48 ml/m2).
Results: Prevalent AF was observed in 194 patients at the hospitalization of whom 178 (92%) were re-hospitalized and 111 (57%) died during the follow-up period. In multivariable logistic regression models, increased plasma levels of TIMP-4, GDF-15, and ST-2 were associated with the prevalence of AF, whereas none of the seven proteins showed any significant association with severely enlarged LAVI. Increased plasma levels of five proteins yielded significant associations with all-cause mortality in patients with co-existing AF; TIMP-4 (HR 1.33; CI95% 1.07–1.66; p = 0.010), GDF-15 (HR 1.30; CI95% 1.05–1.62; p = 0.017), GAL-3 (HR 1.29; CI95% 1.03–1.61; p = 0.029), ST-2 (HR 1.48; CI95% 1.18–1.85; p < 0.001), and MMP-3 (HR 1.33; CI95% 1.09–1.63; p = 0.006). None of the proteins showed any significant association with re-hospitalization risk.
Conclusion: In this study, we were able to demonstrate that elevated levels of three plasma proteins previously linked to myocardial fibrosis are associated with prevalent AF in a HF population. Additionally, higher levels of five plasma proteins yielded an increased risk of mortality in the HF population with or without co-existing AF.
Atrial fibrillation (AF) is the most common cardiac arrhythmia worldwide and a leading risk factor for morbidity and mortality, thus representing a high burden to affected patients and the healthcare system (1). AF often co-exists with heart failure which constitutes a challenging dilemma for clinicians since the occurrence of both conditions aggravates each other and is associated with worse prognosis (2). In this aspect, the interest in finding new pathophysiological links between AF and HF has emerged, where growing evidence suggests that myocardial fibrosis is a contributing factor for both AF and HF development (3, 4). By its contribution to cardiac remodeling, myocardial fibrosis subsequently leads to declined cardiac relaxation and contractility (5) and constitutes an atrial substrate associated with increased left atrial volume (LAV) and development of AF (6). Thus, it is well-known that each of these conditions can be either the cause or consequence of the other (7). In recent studies, several blood-based biomarkers have been suggested as markers of the fibrotic cardiac process seen in patients with AF (8). Furthermore, many of these biomarkers including transforming growth factor β 1 (TGF-β1) have also been implicated in the pathology and prognosis of HF (9). However, there is a lack of knowledge about whether biomarkers that reflect the fibrotic process within the heart are associated with the prognosis of individuals with HF and co-existing AF. The use of myocardial fibrosis biomarkers in day-to-day clinical practice can potentially bring a better understanding of the mutual pathophysiology between AF and HF. Thus, here we aim to explore if plasma levels of myocardial fibrotic proteins are associated with the prevalence of AF and enlarged LAV in a HF population as well as to explore the prognostic impact of each protein in regard to incident mortality and re-hospitalization risk in HF patients with and without AF.
The HeARt and Brain Failure inVESTigation study (HARVEST) is a prospective study undertaken in patients hospitalized for the diagnosis of HF (ICD-10: I50-) at Skane University Hospital, Sweden (10). Admission to the department of internal medicine or cardiology for the treatment of newly diagnosed or exacerbated chronic HF is the inclusion criteria for the HARVEST study. Participants who are unable to deliver informed consent are excluded. In cases of severe cognitive impairment, the relatives are informed and asked for permission on the patient’s behalf. Between 20 March 2014 and 22 January 2018, a total of 324 consecutive patients were included and underwent a clinical examination. These participants had consecutively from the study start until January 2018, been analyzed with a proximity extension assay consisting of 92 proteins. Eight patients had missing values on relevant co-variates, rendering a study population of 316 eligible participants with the complete dataset. Within the study population, 194 patients were diagnosed with co-existing AF (Figure 1). The Ethical Review Board at Lund University, Sweden has approved the study and it fulfills the Declaration of Helsinki. Written informed consent was obtained from all participants or relatives as described above.
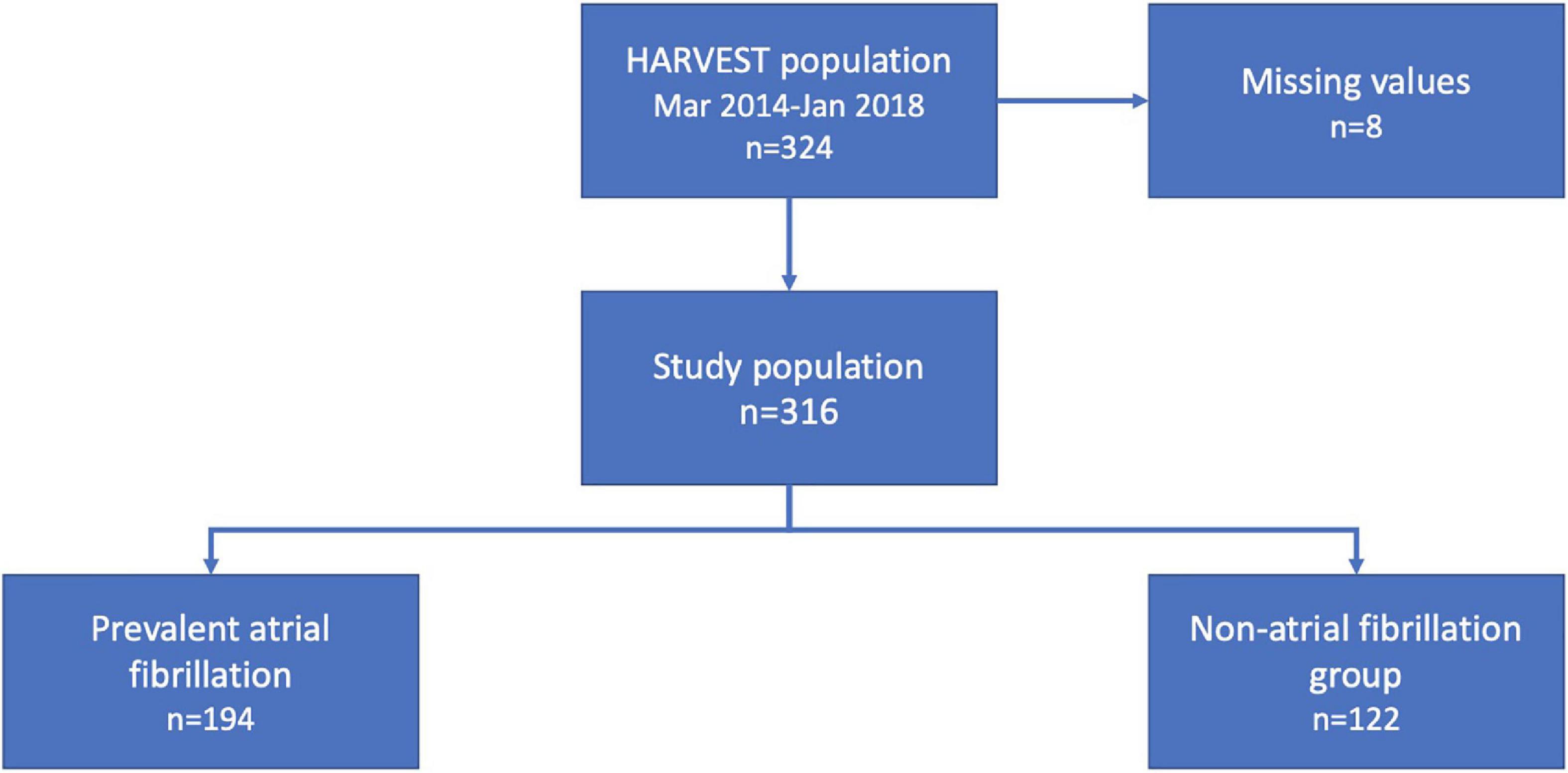
Figure 1. Flowchart of the study population.
Anthropometric measurements and blood samples were obtained after an overnight fast.
Body mass index (BMI) was calculated as kilograms per square meter, and data regarding the study participants’ medication were collected. Prevalent diabetes was defined as a self-reported diagnosis of type 2 diabetes, the use of antidiabetic medication, or fasting plasma glucose >7 mmol/L. Prevalent smoking status was self-reported as yes or no, where never and previous smokers were regarded as non-smokers, and present-day smokers were defined as smokers. Systolic and diastolic blood pressures (BPs) were measured by trained nurses using a validated automated BP monitor Boso Medicus (Bosch + Sohn GmbH u. Co. KG, Jungingen, Germany). The upper arm cuff of appropriate size was placed on the right side, and the arm was supported at the heart level. Hypertension was defined as either systolic blood pressure (SBP) ≥140 mmHg and/or diastolic blood pressure (DBP) ≥90 mmHg. Atrial fibrillation (AF) was defined as the presence of AF on an electrocardiogram at the time of hospitalization or history of AF according to the patient’s medical documentation. The use of anticoagulation medication was defined as receiving treatment with either Warfarin or novel oral anticoagulants (NOACs).
A proximity extension assay (PEA) technique using the Proseek Multiplex CVD III 96 × 96 reagents kit (Olink Bioscience, Uppsala, Sweden) was used to measure plasma levels of 92 CVD proteins (11). The PEA technique uses two oligonucleotide-labeled specific antibodies to bind to each target protein, which allows the formation of a polymerase chain reaction sequence that can then be detected and quantified. All data are presented as arbitrary units. The CVD III panel includes 92 proteins, with established or proposed associations with metabolism, inflammation, and CVD. Across all assays, the mean intra-assay and inter-assay variations were observed to be 8.1 and 11.4%, respectively. Additional information regarding the assays is available on the Olink homepage.1 Seven proteins included in the CVD III panel were selected to be part of further analyses aiming to explore the cross-sectional relationship between prevalent AF and severely enlarged LAV as well as the prognostic impact of each protein for participants with and without AF. The selected proteins have been previously associated with myocardial fibrosis, metalloproteinase inhibitor 4 (TIMP-4) (12), suppression of tumorigenicity 2 (ST-2) (13), galectin-3 (GAL-3) (14), growth/differentiation factor-15 (GDF-15) (15), and matrix metalloproteinase 2, 3, and 9 (MMP-2, MMP-3, and MMP-9, respectively) (16, 17).
Transthoracic echocardiograms were available in 237 (75%) study participants and obtained using a Philips IE33 (Philips, Andover, MA, USA) with a 1–5 MHz transducer (S5-1), or with a GE Vingmed Vivid 7 Ultrasound (GE, Vingmed Ultrasound, Horten, Norway) with a 1–4 MHz transducer (M3S). All studies were performed by experienced sonographers as a part of the clinical routine at a central echocardiographic laboratory. Measurements from the parasternal long axis, apical four- and two-chamber views were done offline using Xcelera 4.1.1 (Philips Medical Systems, Netherlands) according to the recommendations of the American Society of Echocardiography. The parasternal long-axis view was used to measure internal left and right ventricular dimensions at end-diastole. Measurements of wall thickness were obtained in a two-dimensional end-diastolic parasternal long-axis view. The Simpson method was used to calculate left ventricular volumes by manual tracing (papillary muscles included in the cavity) in two-dimensional end-diastolic and end-systolic frames defined as the largest and smallest left ventricular cavities, respectively, in apical four- and two-chamber projections. The ejection fraction (EF) was calculated automatically from end-diastolic volumes (EDV) and end-systolic volume (ESV) using the following formula: EF = (EDV-ESV)/EDV. Tricuspid annular plane systolic excursion (TAPSE) and pulsed tissue Doppler (DTI)-derived tricuspid annular systolic velocity (S’) were used to measure right ventricular systolic function. M-mode images in apical four-chamber view, with the cursor optimally aligned along the direction of the tricuspid annulus, were used to obtain TAPSE. To assess S’-wave velocity, pulsed DTI images were obtained in an apical four-chamber view on the free-wall side of the right ventricle, with the basal segment and the annulus aligned with the Doppler cursor. The LA endocardial borders were manually traced in both apical four-chamber and two-chamber views. For the assessment of left atrium (LA) volumes, the biplane area-length method was used: LA volume = (0.85 × area apical four-chamber × area apical two-chamber)/(shortest atrial length). The values were indexed to BSA and defined as left atrial volume index (LAVI). Severely enlarged atrial volume was defined as LAVI ≥ 48 ml/m2 (18).
The primary endpoints were all-cause mortality and the first post-discharge hospitalization. All-cause mortality was defined as death by any cause and was retrieved from the National Board of Health and Welfare’s Cause of Death Register. Re-hospitalizations defined as the first of any re-admission to the hospital were retrieved from electronic medical charts (Melior, Siemens Health Services, Solna, Sweden). All subjects were followed from study inclusion until 31 December 2020.
Group differences in continuous variables between study participants with or without prevalent AF were compared using a one-way ANOVA test, whereas categorical variables were compared using Pearson’s chi-square test. The variables are presented as means (standard deviation (SD)) and medians (25–75 interquartile range). All analyses were performed using SPSS Windows version 25.0 and R version 4.0.4, and a p-value of < 0.05 was considered statistically significant.
In 316 subjects with a complete dataset on all co-variates, unadjusted logistic regressions were carried out exploring associations between the seven proteins and prevalence of AF. Proteins that presented with significant Bonferroni-corrected associations with prevalent AF were further adjusted for age and sex (Model 1). Proteins with significant associations with prevalent AF in Model 1 were further adjusted according to Model 2 [prevalent diabetes, current smoking, BMI, systolic blood pressure (SBP), New York heart association classification (NYHA class), anticoagulation treatment, and prior HF]. To analyze the associations of the seven myocardial fibrosis proteins with the severely enlarged atrial volume defined as LAVI ≥48 ml/m2, the same statistical procedure as described above was performed. Pearson’s correlation analysis was performed to explore correlations between the levels of proteins in the whole population.
In the next set of analyses, associations between the seven proteins and (1) mortality and (2) re-hospitalization risk were explored using unadjusted Cox regression analyses in three groups; all patients, subjects with and without prevalent AF. Associations that were Bonferroni-corrected significantly associated with (1) mortality and/or (2) re-hospitalization risk were further adjusted according to Model 1 (age and sex). Associations that were significant in Model 1 were further adjusted according to Model 2 (prevalent diabetes, current smoking, BMI, SBP, NYHA class, prior HF, and anticoagulation treatment). In Cox regression models including all patients, prevalent AF was added as a confounder in Model 2.
The study population had a mean age of 75 (±12) years and was predominantly male participants (70%; n = 220) (Table 1). More than half of the study population had known AF diagnosed by the time of index admission, i.e., prevalent AF (61%; n = 194). Of these, 83% (n = 161) had at baseline ongoing treatment with an anticoagulant agent. Patients with co-existing AF were more likely to be older, and have lower BMI and lower SBP compared to individuals without AF. The prevalence of diabetes, current smoking, or level of NYHA class at admission did not differ between the groups (Table 1). Except for GAL-3 and MMP-9, patients with AF had higher levels of proteins associated with myocardial fibrosis as compared to those without co-existing AF. Correlations between the proteins are shown in Supplementary Table 1.
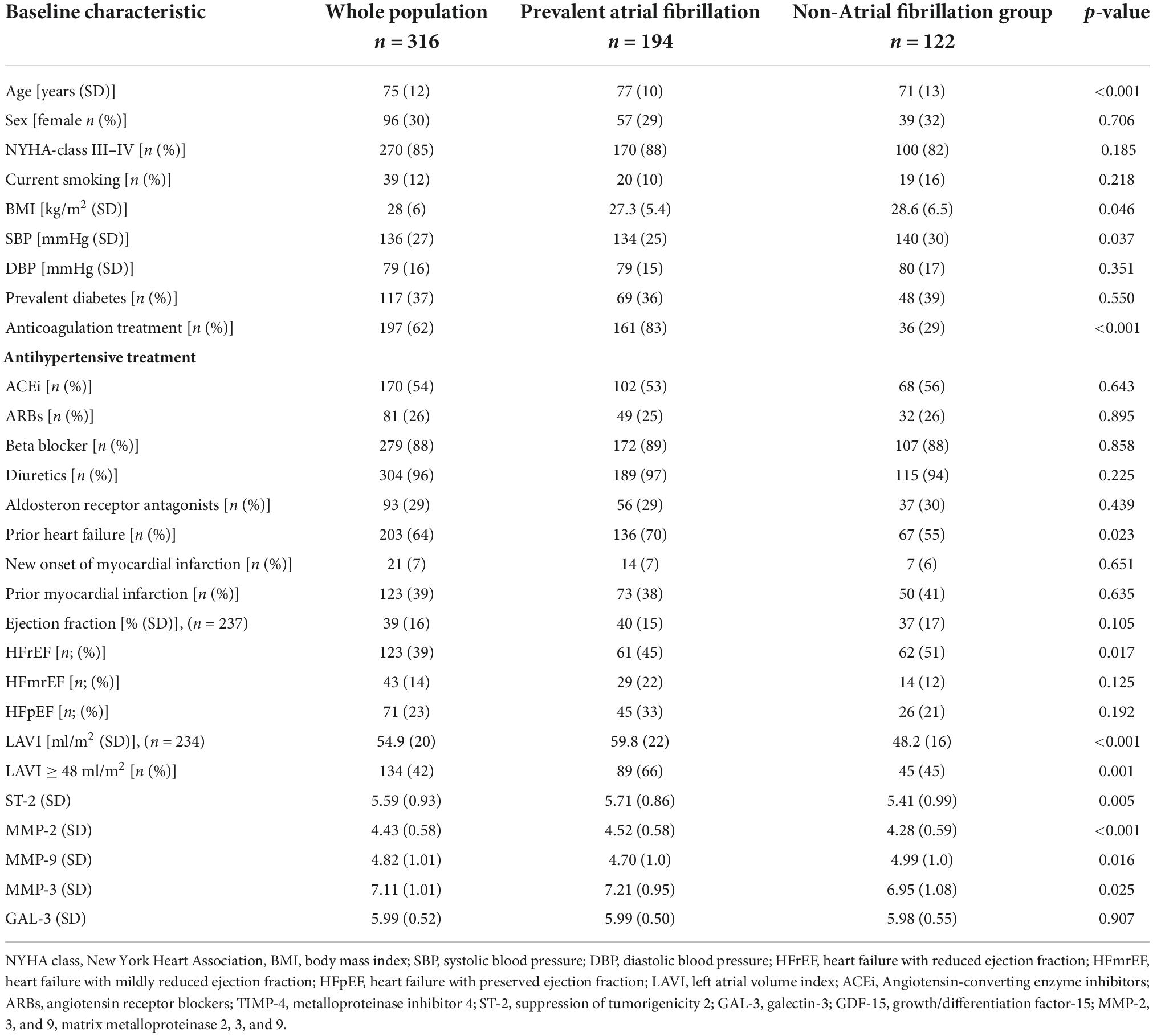
Table 1. Characteristics of study participants (n = 316) at baseline stratified according to the prevalence of atrial fibrillation.
In multivariable logistic regression models (Model 2), increased plasma levels of TIMP-4 (OR 1.58; CI95% 1.15–2.18; p = 0.005), ST-2 (OR 1.42; CI95% 1.06–1.91; p = 0.020), and GDF-15 (OR 1.40; CI95% 1.01–1.94; p = 0.046) were associated with prevalent AF (Table 2). In a sub-analysis stratified according to gender, increased plasma levels of TIMP-4 and ST-2 were associated with prevalent AF in male participants (Supplementary Table 2). In female participants, none of the proteins showed any significant association with prevalent AF. In multivariable logistic regression models (Model 2), none of the proteins were associated with severely enlarged LAVI (≥48 ml/m2) (Supplementary Table 3).
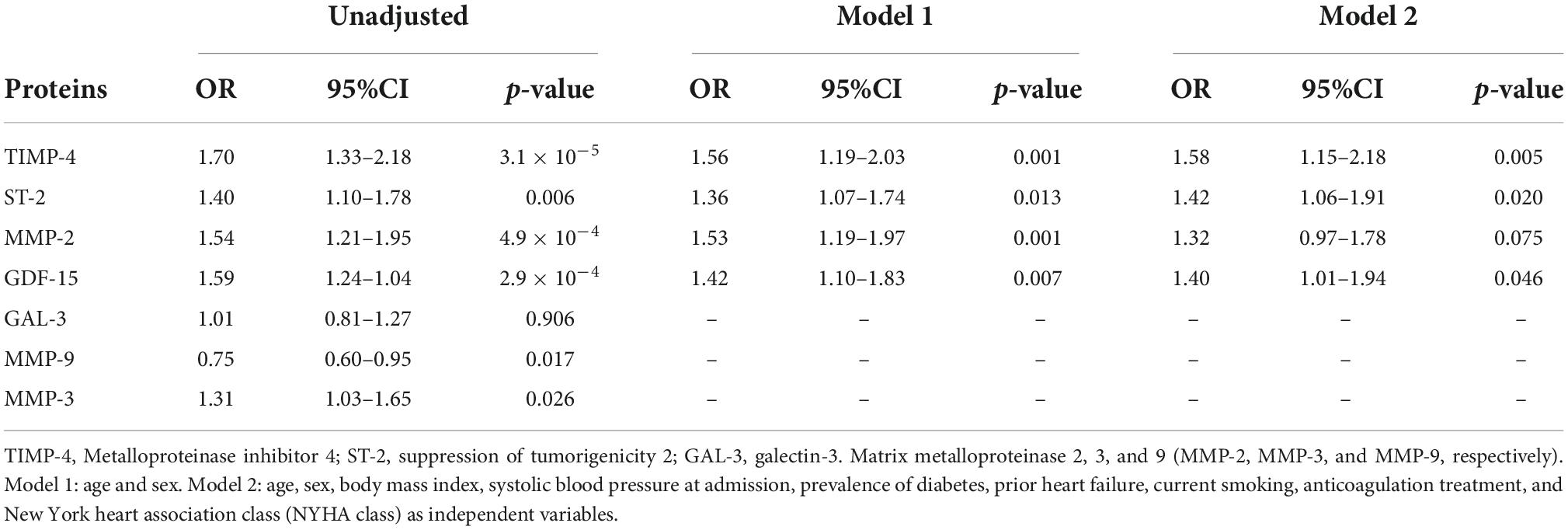
Table 2. Logistic regression analysis examining proteins association with prevalent atrial fibrillation.
During the follow-up period (March 2014 to January 2018), a total of 277 (88%) were re-hospitalized and 177 (56%) patients died. The median follow-up time to hospital admission and death was 136 [interquartile range (IQR) 533] and 1,180 (IQR 1178) days, respectively. Of these cases, 178 (64%) and 111 (63%) had AF, respectively. In the whole population, increased levels of six proteins were associated with mortality; TIMP-4, GDF-15, GAL-3, ST-2, MMP-2, and MMP-3. For re-hospitalization risk within the whole population, increased levels of TIMP-4 remained significant in the fully adjusted model (Supplementary Table 4). In multivariable Cox regression models (Model 2), increased plasma levels of five proteins yielded significant associations with increased risk of mortality for study participants with AF; TIMP-4, GDF-15, GAL-3, ST-2, and MMP-3 (Table 3). None of the proteins were found to be significantly associated with re-hospitalization risk (Table 3). For participants without AF, multivariable Cox regression models (Model 2) showed that increased levels of TIMP-4, ST-2, MMP-2, MMP-3, and GDF-15 yielded significant associations with mortality whereas only higher levels of GDF-15 were significantly associated with re-hospitalization (Table 4).
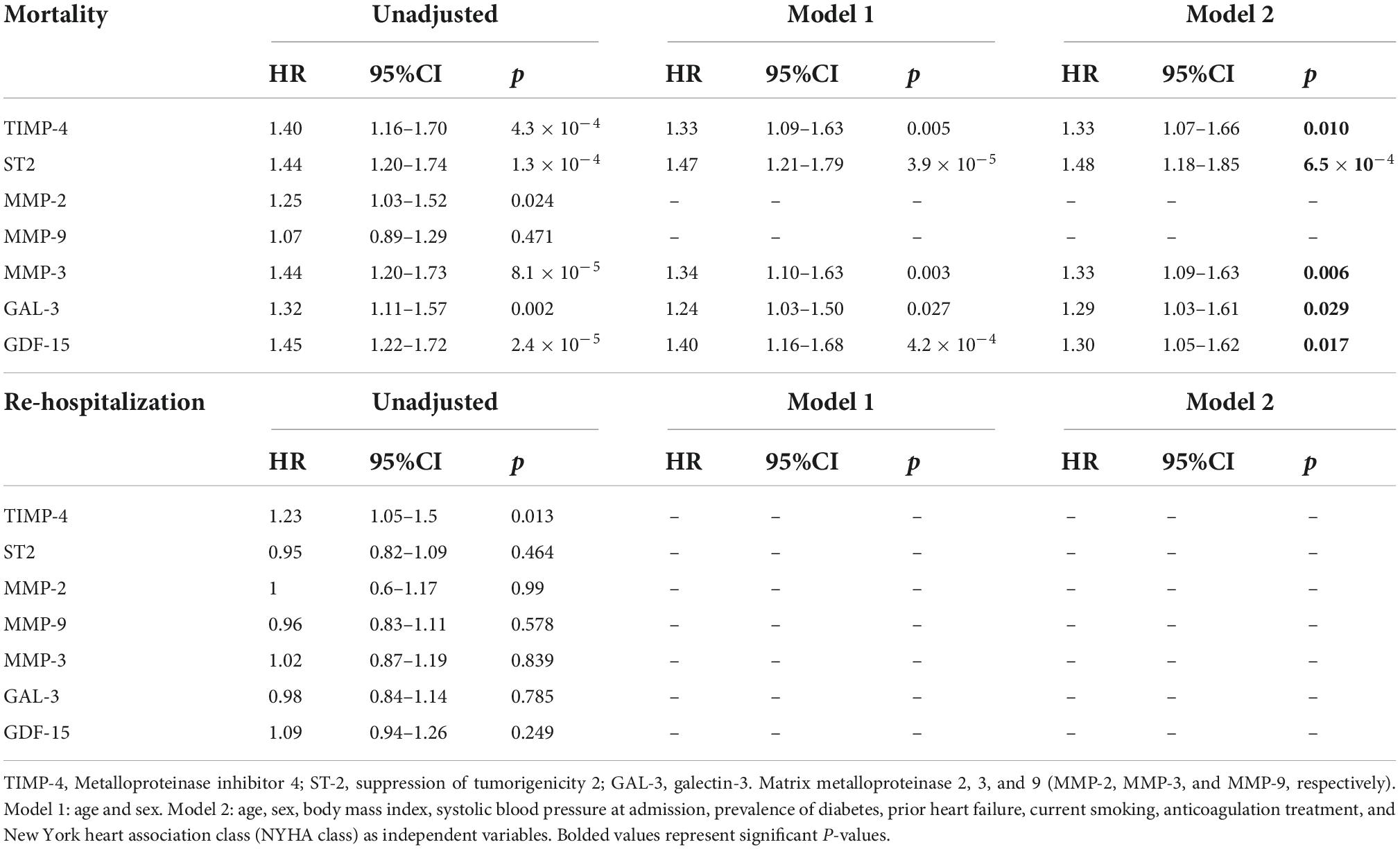
Table 3. Cox regression analyses display associations between myocardial fibrosis biomarkers and mortality and re-hospitalization in patients with prevalent AF.
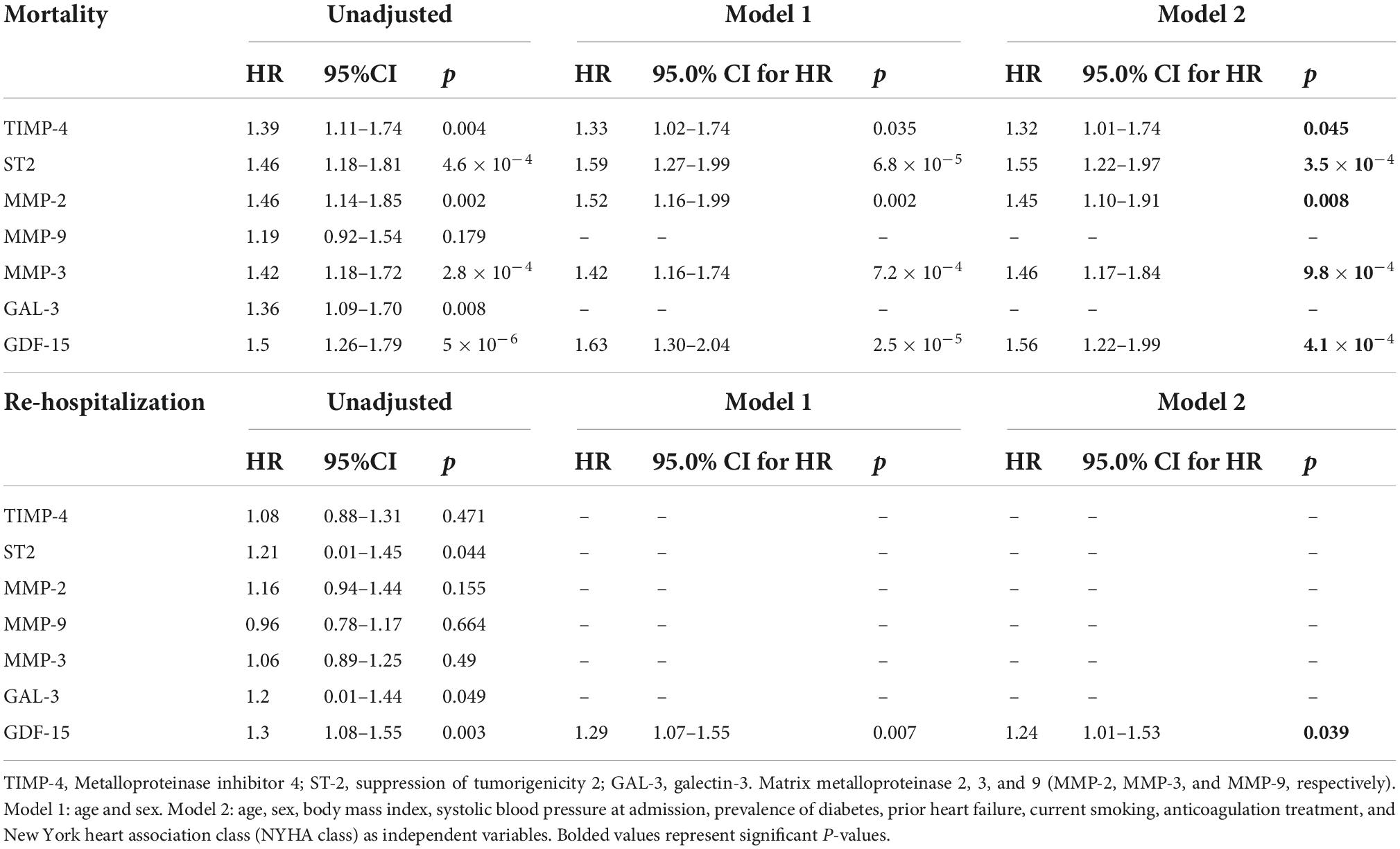
Table 4. Cox regression analyses displaying the associations between myocardial fibrosis biomarkers and mortality and re-hospitalization for participants without prevalent AF.
In this study, we were able to demonstrate that elevated levels of three plasma proteins previously linked to myocardial fibrosis are associated with prevalent AF in a HF population. Additionally, higher levels of five plasma proteins previously linked to myocardial fibrosis yielded an increased risk of mortality in the HF population with or without co-existing AF.
The co-existence between atrial fibrillation (AF) and heart failure has been well-established over the last decade and together they are responsible for substantial morbidity and burden on the healthcare system (1). Despite sharing common precipitating factors such as older age, hypertension, diabetes, obesity, and ischemic and non-ischemic cardiac disease, there are still limited data providing insights into the causal relationship between AF and HF. Hence, a better understanding of the pathophysiological properties of these conditions is crucial for sustainable treatment in this challenging patient population. The above-mentioned risk factors principally lead to cellular and extracellular myocardial changes such as electrophysiological and neurohormonal changes within the heart (19, 20). Furthermore, AF or HF may facilitate the progression and development of each other in several ways such as inflammatory cardiac (myocarditis and pericarditis) and non-cardiac (infections and inflammatory bowel diseases) conditions, whereas ischemic heart diseases contribute to AF/HF development due to subclinical inflammatory appearances (21–23). Evidence from large HF trials has shown that the prevalence of AF increases with the severity of HF symptoms (24) ranging from 10% in HF with NYHA classes I–II to 50% in HF with NYHA class IV. Correspondingly, studies of AF have revealed a high burden of concomitant HF with a prevalence between 21 and 68% (25). The most unfavorable hemodynamic effect linked with AF results mainly from the loss of atrial systole, ventricular chronotropic ineffectiveness, and irregular ventricular rate leading to shortened diastolic phase (26). In addition to atrial contractility loss, AF further contributes to dysfunctional ventricular filling and reduced stroke volume. The impaired atrial systolic function can precipitate HF in situations of dysfunctional ventricular filling that often accompany AF, such as mitral stenosis, ischemic heart disease, and diastolic dysfunction (27). This property together with the suboptimal electromechanical function and activation of the neurohumoral system due to reduced cardiac output creates an environment causing the deterioration of AF and HF simultaneously (28).
Increasing evidence suggests myocardial fibrosis as a crucial contributor to the cardiac remodeling seen in patients with AF (29). Myocardial fibrosis is histologically defined by a dispersed disposition of excess fibrous tissue in relation to the total mass of cardiomyocytes within the myocardial interstitium (8, 30). Replacement fibrosis is frequently initiated by cardiomyocyte death, which causes inflammatory responses and release of cytokines, chemokines, and oxidative stress, whereas in reactive fibrosis, various stimuli such as ischemia, metabolic injury, or pressure overload are responsible for the accumulation of fibrotic tissue in the absence of cell death (8, 31–33). Additionally, different cell types are involved with the fibrotic response, either directly by myofibroblast (producing fibrous tissue) or indirectly by macrophages, lymphocytes, cardiomyocytes, and mast cells (secreting profibrotic mediators). Significant accumulation of fibrotic tissue within the heart will eventually lead to left ventricular (LV) dysfunction and risk of arrhythmia (34, 35).
Although the golden standard method to assess myocardial fibrosis is through tissue biopsy, increasing evidence has shown promising links between circulating biomarkers and myocardial fibrosis. Recently Lopez et al. considered 28 plasma molecules as potential candidates for myocardial fibrosis (8). To date, only procollagen types I and III have been proven to be associated with histologically verified myocardial fibrosis (8).
Matrix metalloproteinases (MMPs) belong to zinc-dependent proteolytic enzymes and, together with tissue metalloproteinase inhibitors (TIMPs), regulate extracellular matrix (ECM) (16, 36). The balance between the synthesis and degradation process is maintained by ECM homeostasis (16). A misbalance toward MMPs results in increased fragmentation of ECM proteins, while a misbalance toward TIMP results in the protection of ECM proteins (37). High levels of both MMP-2 and TIMP-4 were demonstrated in this study, which most likely represents the counterbalance between the two proteins. Usually, profibrotic stimuli, such as proinflammatory cytokines or intensification of oxidative stress, cause a disruption in favor of the ECM protein synthesis process causing excessive fibrosis (38). Increased TIMP is suggested to be involved in ECM deposition or fibrotic processes, while reduced levels or loss of TIMP prolong the ECM degradation (39). Thus, in the normal state, TIMP can directly inhibit ECM degradation or MMP activation. Reports confirm that AF is mediated by increased activity of MMPs which has also been shown to predict HF development (40). The current study could not find any associations between AF, LAVI, and MMP-2, even though the associations were estimated as borderline significant. The lack of balance between TIMPs and MMPs could potentially be the major component of the heart remodeling process, and an area with a clear need for more evidence.
Galectin-3 is a galactoside-binding lectin involved in important regulatory processes such as inflammation, fibrosis, adhesion, and immunity (41). Activated macrophages secrete galectin leading to the proliferation of fibroblasts, collagen disposition following cardiac fibrosis, and myocyte disruption and potentially predispose AF (42, 43). The ARIC study (n = 8,436) with a median follow-up time of 16 years stated that elevated plasma galectin-3 is associated with an increased risk of incident AF (44). After adjustment for AF risk factors, participants with galectin-3 levels above the 90th percentile had a significantly higher risk of incident AF. Furthermore, studies have shown that elevated galectin-3 levels are associated with a more advanced AF, accompanied by severe comorbidities and worse outcomes (45). Even though levels of Gal-3 did not differ between participants with or without co-existent AF, higher levels of Gal-3 were associated with all-cause mortality only in participants with AF.
ST-2 belongs to the interleukin-1 receptor family and is released from the myocardium and vascular endothelial cells in response to pressure and hemodynamic volume overload (46). From the pathophysiological point of view, increased hemodynamic load with subsequent atrial stretch is a mechanism of AF pathophysiology and is responsible for the release of brain natriuretic peptide. Additionally, atrial stretch may lead to increased levels of ST-2. ST-2 actions within the cardiovascular system have raised questions about whether it could potentially become a novel biomarker of cardiac remodeling, myocardial infarction, HF, and AF (47). Chen et al. also pointed out that increased ST-2 levels in an AF population could be associated with an increase in the heart rate and atrial pressure compared to patients with sinus rhythm. Based on these findings, the significant association between elevated levels of ST-2 and prevalent AF demonstrated in the current study is not surprising.
Growth differentiation factor-15 (GDF-15) is a member of the transforming growth factor b cytokine superfamily and is described as a stress-responsive cytokine. Based on current evidence, it may be involved in inflammation, coagulation, oxidative stress, endothelial dysfunction, and homeostasis (48). The PARADIGM-HF trial has shown that higher levels of GDF-15 were associated with mortality and cardiovascular events in patients with HF with reduced ejection fraction (49). A two-sample Mendelian randomization study by Wang et al. was carried out using five independent large genome-wide association studies to investigate the causal association between circulating GDF-15 levels and prevalent CVD. This study provided genetic evidence, suggesting that circulating GDF-15 levels are significantly linked to the increased risk of AF, coronary artery disease, myocardial infarction, and cardioembolic stroke (50). The ARISTOTLE trail has also pinpointed that increased concentration of GDF-15 is associated with mortality, major bleeding, and stroke in a population with prevalent AF (51). Thus, GDF-15 is increased in several conditions which might explain our findings that higher GDF-15 is strongly associated with mortality also in those without prevalent AF (p = 4.1 × 10–4) and is the only protein biomarker associated with re-hospitalization.
As this is an observational study, no conclusions about causality can be drawn. Further, increased levels of several proteins were associated with mortality for both study sub-populations, prevalent AF vs. no AF. This might be explained by the fact that the current cohort consists of HF patients with a high burden of CV risk factors which might have affected the levels of the included proteins. Additionally, the association between higher levels of the proteins and higher mortality risk might be an adverse effect of severe HF since the vast majority belong to NYHA classes III–IV. Another limitation is the lack of complete data on echocardiographic variables, which might have affected the power needed to explore associations between proteins and left atrial volume. Blood samples were obtained after an overnight fast. It is, therefore, possible that the initial medical treatment (including furosemide) for decompensated HF might have influenced the plasma levels of biomarkers included in the CVD III panel. The collected data origin from a single regional hospital and the included participants are mainly of Caucasian descent. Hence, the applicability for other HF populations is limited.
In this study, we were able to demonstrate that elevated levels of five plasma proteins previously associated with myocardial fibrosis were associated with increased mortality in a HF population with and without co-existing AF. Additionally, higher levels of four fibrosis-associated proteins were significantly associated with prevalent atrial fibrillation. Considering our findings, one may argue that we still lack an understanding of the accurate pathophysiological mechanism taking part in HF with co-existing AF. To establish this, further studies are warranted.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Ethical Review Board at Lund University. The patients/participants provided their written informed consent to participate in this study.
ZN and HH contributed equally to writing the manuscript. AZ, AJ, AD, EB, JK, JM, MO, MM, and PP contributed equally with statistical help, data interpretation, and literature research. All authors contributed to the article and approved the submitted version.
This work was supported by the HARVEST-Malmö study, supported by grants from the Medical Faculty of Lund University, Skane University Hospital, the Crafoord Foundation, the Ernhold Lundstroms Research Foundation, the Region Skane, the Hulda and Conrad Mossfelt Foundation, the Southwest Skanes Diabetes Foundation, the Kockska Foundation, the Research Funds of Region Skåne, the Swedish Heart and Lung Foundation, the Wallenberg Center for Molecular Medicine, Lund University, and Lund University.
The Knut and Alice Wallenberg Foundation was acknowledged for generous support. We thank the research nurses Hjördis Jernhed and Dina Chatziapostolou for their valuable contributions.
Author EB was employed by AstraZeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.982871/full#supplementary-material
1. Hindricks G, Potpara T, Dagres N, Arbelo E, Bax JJ, Blomstrom-Lundqvist C, et al. 2020 ESC guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European association for cardio-thoracic surgery (EACTS): the task force for the diagnosis and management of atrial fibrillation of the European society of cardiology (ESC) developed with the special contribution of the European heart rhythm association (EHRA) of the ESC. Eur Heart J. (2021) 42:373–498. doi: 10.1093/eurheartj/ehab648
2. Anter E, Jessup M, Callans DJ. Atrial fibrillation and heart failure: treatment considerations for a dual epidemic. Circulation. (2009) 119:2516–25. doi: 10.1161/CIRCULATIONAHA.108.821306
3. Dzeshka MS, Lip GY, Snezhitskiy V, Shantsila E. Cardiac fibrosis in patients with atrial fibrillation: mechanisms and clinical implications. J Am Coll Cardiol. (2015) 66:943–59. doi: 10.1016/j.jacc.2015.06.1313
4. Webber M, Jackson SP, Moon JC, Captur G. Myocardial fibrosis in heart failure: anti-fibrotic therapies and the role of cardiovascular magnetic resonance in drug trials. Cardiol Ther. (2020) 9:363–76. doi: 10.1007/s40119-020-00199-y
5. Hinderer S, Schenke-Layland K. Cardiac fibrosis – a short review of causes and therapeutic strategies. Adv Drug Deliv Rev. (2019) 146:77–82. doi: 10.1016/j.addr.2019.05.011
6. Nattel S, Harada M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol. (2014) 63:2335–45. doi: 10.1016/j.jacc.2014.02.555
7. Prabhu S, Voskoboinik A, Kaye DM, Kistler PM. Atrial fibrillation and heart failure – cause or effect? Heart Lung Circ. (2017) 26:967–74. doi: 10.1016/j.hlc.2017.05.117
8. Lopez B, Gonzalez A, Ravassa S, Beaumont J, Moreno MU, San Jose G, et al. Circulating biomarkers of myocardial fibrosis: the need for a reappraisal. J Am Coll Cardiol. (2015) 65:2449–56. doi: 10.1016/j.jacc.2015.04.026
9. Topf A, Mirna M, Ohnewein B, Jirak P, Kopp K, Fejzic D, et al. The diagnostic and therapeutic value of multimarker analysis in heart failure. An approach to biomarker-targeted therapy. Front Cardiovasc Med. (2020) 7:579567. doi: 10.3389/fcvm.2020.579567
10. Christensson A, Grubb A, Molvin J, Holm H, Gransbo K, Tasevska-Dinevska G, et al. The shrunken pore syndrome is associated with declined right ventricular systolic function in a heart failure population – the HARVEST study. Scand J Clin Lab Investig. (2016) 76:568–74. doi: 10.1080/00365513.2016.1223338
11. Assarsson E, Lundberg M, Holmquist G, Bjorkesten J, Thorsen SB, Ekman D, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. (2014) 9:e95192. doi: 10.1371/journal.pone.0095192
12. Takawale A, Zhang P, Patel VB, Wang X, Oudit G, Kassiri Z. Tissue inhibitor of matrix metalloproteinase-1 promotes myocardial fibrosis by mediating CD63-integrin beta1 interaction. Hypertension. (2017) 69:1092–103. doi: 10.1161/HYPERTENSIONAHA.117.09045
13. Bayes-Genis A, de Antonio M, Vila J, Penafiel J, Galan A, Barallat J, et al. Head-to-head comparison of 2 myocardial fibrosis biomarkers for long-term heart failure risk stratification: ST2 versus galectin-3. J Am Coll Cardiol. (2014) 63:158–66. doi: 10.1016/j.jacc.2013.07.087
14. Vergaro G, Del Franco A, Giannoni A, Prontera C, Ripoli A, Barison A, et al. Galectin-3 and myocardial fibrosis in nonischemic dilated cardiomyopathy. Int J Cardiol. (2015) 184:96–100. doi: 10.1016/j.ijcard.2015.02.008
15. Lok SI, Winkens B, Goldschmeding R, van Geffen AJ, Nous FM, van Kuik J, et al. Circulating growth differentiation factor-15 correlates with myocardial fibrosis in patients with non-ischaemic dilated cardiomyopathy and decreases rapidly after left ventricular assist device support. Eur J Heart Fail. (2012) 14:1249–56. doi: 10.1093/eurjhf/hfs120
16. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. (2012) 5:15. doi: 10.1186/1755-1536-5-15
17. Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiology. (2013) 28:391–403. doi: 10.1152/physiol.00029.2013
18. Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American society of echocardiography and the European association of cardiovascular imaging. J Am Soc Echocardiogr. (2015) 28:1–39.e14. doi: 10.1016/j.echo.2014.10.003
19. Tarone G, Balligand JL, Bauersachs J, Clerk A, De Windt L, Heymans S, et al. Targeting myocardial remodelling to develop novel therapies for heart failure: a position paper from the working group on myocardial function of the European society of cardiology. Eur J Heart Fail. (2014) 16:494–508. doi: 10.1002/ejhf.62
20. Hartupee J, Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol. (2017) 14:30–8. doi: 10.1038/nrcardio.2016.163
21. Boos CJ, Anderson RA, Lip GY. Is atrial fibrillation an inflammatory disorder? Eur Heart J. (2006) 27:136–49. doi: 10.1093/eurheartj/ehi645
22. Zhang H, Li J, Chen X, Wu N, Xie W, Tang H, et al. Association of systemic inflammation score with atrial fibrillation: a case-control study with propensity score matching. Heart Lung Circ. (2018) 27:489–96. doi: 10.1016/j.hlc.2017.04.007
23. Zhou X, Dudley SC Jr. Evidence for inflammation as a driver of atrial fibrillation. Front Cardiovasc Med. (2020) 7:62. doi: 10.3389/fcvm.2020.00062
24. Maisel WH, Stevenson LW. Atrial fibrillation in heart failure: epidemiology, pathophysiology, and rationale for therapy. Am J Cardiol. (2003) 91:2D–8D. doi: 10.1016/S0002-9149(02)03373-8
25. Gomez-Outes A, Terleira-Fernandez AI, Calvo-Rojas G, Suarez-Gea ML, Vargas-Castrillon E. Dabigatran, rivaroxaban, or apixaban versus warfarin in patients with nonvalvular atrial fibrillation: a systematic review and meta-analysis of subgroups. Thrombosis. (2013) 2013:640723. doi: 10.1155/2013/640723
26. Ling LH, Kistler PM, Kalman JM, Schilling RJ, Hunter RJ. Comorbidity of atrial fibrillation and heart failure. Nat Rev Cardiol. (2016) 13:131–47. doi: 10.1038/nrcardio.2015.191
27. Meisner JS, Keren G, Pajaro OE, Mani A, Strom JA, Frater RW, et al. Atrial contribution to ventricular filling in mitral stenosis. Circulation. (1991) 84:1469–80. doi: 10.1161/01.CIR.84.4.1469
28. Olsson LG, Swedberg K, Ducharme A, Granger CB, Michelson EL, McMurray JJ, et al. Atrial fibrillation and risk of clinical events in chronic heart failure with and without left ventricular systolic dysfunction: results from the candesartan in heart failure-assessment of reduction in mortality and morbidity (CHARM) program. J Am Coll Cardiol. (2006) 47:1997–2004. doi: 10.1016/j.jacc.2006.01.060
29. Platonov PG, Mitrofanova LB, Orshanskaya V, Ho SY. Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J Am Coll Cardiol. (2011) 58:2225–32. doi: 10.1016/j.jacc.2011.05.061
30. Gonzalez A, Schelbert EB, Diez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. (2018) 71:1696–706. doi: 10.1016/j.jacc.2018.02.021
31. Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, et al. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation. (1994) 89:151–63. doi: 10.1161/01.CIR.89.1.151
32. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. (2014) 71:549–74. doi: 10.1007/s00018-013-1349-6
33. Weber KT, Sun Y, Guarda E. Structural remodeling in hypertensive heart disease and the role of hormones. Hypertension. (1994) 23(6 Pt 2):869–77. doi: 10.1161/01.HYP.23.6.869
34. Nguyen MN, Kiriazis H, Gao XM, Du XJ. Cardiac fibrosis and arrhythmogenesis. Compr Physiol. (2017) 7:1009–49. doi: 10.1002/cphy.c160046
35. Villari B, Campbell SE, Hess OM, Mall G, Vassalli G, Weber KT, et al. Influence of collagen network on left ventricular systolic and diastolic function in aortic valve disease. J Am Coll Cardiol. (1993) 22:1477–84. doi: 10.1016/0735-1097(93)90560-n
36. Kakkar R, Lee RT. Intramyocardial fibroblast myocyte communication. Circ Res. (2010) 106:47–57. doi: 10.1161/CIRCRESAHA.109.207456
37. Sygitowicz G, Maciejak-Jastrzebska A, Sitkiewicz D. A review of the molecular mechanisms underlying cardiac fibrosis and atrial fibrillation. J Clin Med. (2021) 10:4430. doi: 10.3390/jcm10194430
38. Spinale FG. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res. (2002) 90:520–30. doi: 10.1161/01.RES.0000013290.12884.A3
39. Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. (2015) 44-46:247–54. doi: 10.1016/j.matbio.2015.03.005
40. McManus DD, Shaikh AY, Abhishek F, Vasan RS. Atrial fibrillation and heart failure parallels: lessons for atrial fibrillation prevention. Crit Pathw Cardiol. (2011) 10:46–51. doi: 10.1097/HPC.0b013e31820e1a4b
41. de Boer RA, Yu L, van Veldhuisen DJ. Galectin-3 in cardiac remodeling and heart failure. Curr Heart Fail Rep. (2010) 7:1–8. doi: 10.1007/s11897-010-0004-x
42. Jalife J. Mechanisms of persistent atrial fibrillation. Curr Opin Cardiol. (2014) 29:20–7. doi: 10.1097/HCO.0000000000000027
43. Lippi G, Cervellin G, Sanchis-Gomar F. Galectin-3 in atrial fibrillation: simple bystander, player or both? Clin Biochem. (2015) 48:818–22.
44. Fashanu OE, Norby FL, Aguilar D, Ballantyne CM, Hoogeveen RC, Chen LY, et al. Galectin-3 and incidence of atrial fibrillation: the atherosclerosis risk in communities (ARIC) study. Am Heart J. (2017) 192:19–25.
45. Clementy N, Piver E, Bisson A, Andre C, Bernard A, Pierre B, et al. Galectin-3 in atrial fibrillation: mechanisms and therapeutic implications. Int J Mol Sci. (2018) 19:976. doi: 10.3390/ijms19040976
46. Bartunek J, Delrue L, Van Durme F, Muller O, Casselman F, De Wiest B, et al. Nonmyocardial production of ST2 protein in human hypertrophy and failure is related to diastolic load. J Am Coll Cardiol. (2008) 52:2166–74.
47. Chen C, Qu X, Gao Z, Zheng G, Wang Y, Chen X, et al. Soluble ST2 in patients with nonvalvular atrial fibrillation and prediction of heart failure. Int Heart J. (2018) 59:58–63. doi: 10.1536/ihj.16-520
48. Wollert KC, Kempf T. Growth differentiation factor 15 in heart failure: an update. Curr Heart Fail Rep. (2012) 9:337–45. doi: 10.1007/s11897-012-0113-9
49. Bouabdallaoui N, Claggett B, Zile MR, McMurray JJV, O’Meara E, Packer M, et al. Growth differentiation factor-15 is not modified by sacubitril/valsartan and is an independent marker of risk in patients with heart failure and reduced ejection fraction: the PARADIGM-HF trial. Eur J Heart Fail. (2018) 20:1701–9. doi: 10.1002/ejhf.1301
50. Wang Z, Yang F, Ma M, Bao Q, Shen J, Ye F, et al. The impact of growth differentiation factor 15 on the risk of cardiovascular diseases: two-sample Mendelian randomization study. BMC Cardiovasc Disord. (2020) 20:462. doi: 10.1186/s12872-020-01744-2
51. Wallentin L, Hijazi Z, Andersson U, Alexander JH, De Caterina R, Hanna M, et al. Growth differentiation factor 15, a marker of oxidative stress and inflammation, for risk assessment in patients with atrial fibrillation: insights from the apixaban for reduction in stroke and other thromboembolic events in atrial fibrillation (ARISTOTLE) trial. Circulation. (2014) 130:1847–58. doi: 10.1161/CIRCULATIONAHA.114.011204
Keywords: heart failure, biomarkers, fibrosis, echocardiography, atrial fibrillation
Citation: Nezami Z, Holm H, Ohlsson M, Molvin J, Korduner J, Bachus E, Zaghi A, Dieden A, Platonov PG, Jujic A and Magnusson M (2022) The impact of myocardial fibrosis biomarkers in a heart failure population with atrial fibrillation—The HARVEST-Malmö study. Front. Cardiovasc. Med. 9:982871. doi: 10.3389/fcvm.2022.982871
Received: 30 June 2022; Accepted: 30 September 2022;
Published: 19 October 2022.
Edited by:
Isabel Moscoso, Health Research Institute of Santiago de Compostela (IDIS), SpainReviewed by:
Jawed Fareed, Loyola University Medical Center, United StatesCopyright © 2022 Nezami, Holm, Ohlsson, Molvin, Korduner, Bachus, Zaghi, Dieden, Platonov, Jujic and Magnusson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannes Holm, aGFubmVzLmhvbG1AbWVkLmx1LnNl
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.