 Keman Xu1
Keman Xu1 Mohsin Khan2
Mohsin Khan2 Jun Yu2
Jun Yu2 Nathaniel W. Snyder2
Nathaniel W. Snyder2 Sheng Wu2
Sheng Wu2 Roberto I. Vazquez-Padron3
Roberto I. Vazquez-Padron3 Hong Wang2
Hong Wang2 Xiaofeng Yang1,2*
Xiaofeng Yang1,2*- 1Departments of Cardiovascular Sciences and Biomedical Education and Data Sciences, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
- 2Departments of Cardiovascular Sciences and Biomendical Education and Data Sciences, Centers for Metabolic Disease Research, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
- 3DeWitt Daughtry Family Department of Surgery, Leonard M. Miller School of Medicine, University of Miami, Miami, FL, United States
Editorial on the Research Topic
Insights in cardiovascular therapeutics: 2021
Introduction
With the effort and support of the authors, editorial office, and editorial team, the Frontiers in Cardiovascular Medicine, Cardiovascular Therapeutics Section-Research Topic “Insights in Cardiovascular Therapeutics: 2021” has achieved great success and is attracting interest from the cardiovascular community. Here, we spotlight 12 studies published in our section that related to cell death and cardiovascular injuries, as well as some recent advances in the field that have tremendous potential in cardiovascular therapy. In addition, these highlights may serve as the foundation for some new developments in our Cardiovascular Therapeutics areas. In 2022, we will keep working to create a fantastic platform for cardiologists, translational cardiovascular scientists, and cardiovascular pharmacological scientists to share new results and data in clinical cardiology and translational cardiovascular therapeutics.
Cell death and heart diseases
Cardiovascular diseases (CVDs) are the leading cause of morbidity and mortality worldwide. An estimated 17.9 million people live with CVDs each year with no effective cures (1). Therefore, studying the pathogenesis of heart diseases and identifying potential therapies are critical. Programmed cell death is an essential but generally detrimental process in CVD development. Cardiomyocytes are terminally differentiated, have a limited division capacity, and serve vital functions. The death of cardiomyocytes affects hearts' ability to contract and causes adverse remodeling, and eventually lead to cardiac dysfunction and heart failure. Hence, cell death that leads to the loss of cardiomyocytes is a significant phase in the pathogenesis of cardiac diseases. Therefore, strongly suggesting that targeting cell death processes as a therapeutic approach to alleviate and reverse cardiomyopathy is a viable therapeutic strategy (2–4). In this editorial we will discuss a common molecular pathological theme related to research progresses in CVDs including heart failure reported by Wu et al., Liao et al., and Dash et al., atrial fibrillation reported by Lee et al. and Zheng Wang et al., refractory angina reported by Ambari et al., In-stent restenosis reported by Zhu et al., critical limb ischemia reported by Quiroz et al., protein conformational diseases reported by Zheng Song et al., mitochondrial dysfunction reported by Chen et al., and myocardial injury reported by Barbieri et al. and Cao et al.
In recent decades, new mechanisms that orchestrate various cell death pathways have been discovered, and this field continues to expand. The current well-established forms of cell death pathways include intrinsic or extrinsic apoptosis, necroptosis, pyroptosis, ferroptosis, mitochondrial permeability transition (MPT)-driven necrosis, autophagic cell death (autosis), lysosome-dependent cell death, immunogenic cell death (5), cellular senescence, parthanatos, mitotic catastrophe, neutrophil extracellular trap (NET)otic cell death, entosis (6, 7), anoikis (8), oxelptosis, and alkaliptosis (9). From a physiological point of view, cell death helps an organism develop, impacts morphogenesis and maintains homeostasis (10). However, pathological cell death is triggered when cells are subjected to various stimuli, including heart failure (11), myocardial injury, ischemia, ventricular remodeling (12), elevated troponins (13), energy production failure, oxidative damage, and imbalanced ion fluxes (14). As a result, pathological cell death does not maintain homeostasis but instead promotes disease progression.
Apoptosis is the most characterized form of cell death in various cardiovascular diseases. It is characterized by a process of cellular self-destruction without inflammation (15). Although apoptosis is the most studied form of cell death, few apoptotic myocytes are observed in patients with heart failure since 80–250 myocytes are found to undergo apoptosis per 1 x 105 myocytes (2). Moreover, immunologically silent apoptosis cannot be used to explain why vasculature or myocytes injury always accompanies the excessive inflammation and immune cell infiltration during cardiac disease progression. Another five death mechanisms have been identified in heart diseases, including necroptosis, mitochondrial-mediated necrosis, pyroptosis, ferroptosis, and autophagic cell death. Among them, lytic programmed cell death, such as necroptosis and pyroptosis (16–22), has historically received the most attention. The lytic programmed cell death pathway causes cell death by making a pore on the plasma membrane. These mechanisms of cell death are associated with release damage/danger-associated molecular patterns (DAMPs) and inflammatory cytokines, which leads to inflammation (23).
Lytic programmed cell death and its role in inflammation of heart diseases
Inflammation plays an essential role in all types of cardiac diseases. The vasculature experiences inflammation as a reaction to lipid peroxidation, damage, and possibly infection. Studies in epidemiology and medicine have consistently and strongly linked the risk of cardiovascular events to inflammation (24). In contrast, the absence of inflammatory properties of apoptosis allows us to understand the importance of lytic cell death in cardiovascular diseases (25). Previous studies reported that lysophosphatidylcholine (LPC) and oxidized low-density lipoprotein (oxLDL) induce Nod-like receptor family 3 (NLRP3) and promote endothelial cell activation (26–28) in cardiac diseases (29). Further, the activation of caspase-1 canonical inflammasome pathway and caspase-4 (human)/ caspase-11 (mice) noncanonical inflammasome pathway will lead to gasdermin D cleavage and N-terminal gasdermin D protein pore formation on the plasma membrane, which could mediate endothelial pyroptosis during atherosclerosis development (30–32). In addition to pyroptosis, necroptosis, and mitochondrially mediated necrosis are the other common cell death pathways observed in heart diseases. Necroptosis is characterized by cellular enlargement, degradation of plasma membrane integrity, DAMPs release (33), and inflammation. Necroptosis could be activated when serine/threonine kinase receptor protein kinases (RIPK) 1 binds to and activates RIPK3. Then, the activated RIPK3 further activates a pseudokinase, which leads mixed lineage kinase-like domain (MLKL) phosphorylation. Phosphorylated MLKL translocates from cytosol to plasma membrane, promoting necroptotic cell death (34). Necroptosis implicated in the pathogenesis of many heart diseases. In this Research Topic, Wu et al. reported that RIPK1-RIPK3-MLKL mediated necroptosis contributes to catecholamine-induced heart failure. Moreover, necroptosis is also related to mitochondrial-mediated necrosis. RIPK1, RIPK3, and MLKL have been shown to translocate to the mitochondrial membrane during necroptosis to promote mitochondrial dysfunction, mitochondrial reactive oxygen species (mtROS) production (35–40), and cell damage (34). Chen et al. in this Research Topic demonstrated that intracellular mitochondrial transfer has been discovered in cardiovascular diseases. In pathological situations, injured cells seek recipient cells for assistance by transferring defective mitochondria; and recipient cells accept “foreign” functional mitochondria to reduce injury. Therefore, mitochondrial-targeted therapies could be a potential menthod to treat diseases. In addition to the activity of individual cell death pathways in cardiac diseases, a growing number of studies indicate crosstalk between three types of cell death of pyroptosis, apoptosis, and necroptosis, which is termed as PANoptosis. PANoptosis is a pro-inflammatory programmed cell death (PCD) pathway and has initially discovered in response to viral infections. Following infection with a virus such as influenza A virus (IAV), a master regulator of PANoptosis, Z-DNA-binding protein 1 (ZBP1) (41, 42), interacts with RIPK3 via RIP homotypic interaction motif (RHIM) domains and forms a multimeric protein complex, PANoptosome. This single multimeric complex can concurrently activate NLRP3-dependent pyroptosis, Caspase-8-dependent apoptosis, and MLKL-dependent necroptosis (43). It is believed that simultaneous activation of the three PCDs and PANoptosome formation indicate PANoptosis occurrence. PANoptosis can elicit dramatic host inflammation in response to IAV infection or severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (22), resulting in severe lung tissue damage and other lethal consequences (44). PANoptosis is not limited to virus infection but participates in other diseases including stroke, traumatic brain injury, atherosclerosis, and cancer (45). Although there is not currently much data on the involvement in PANoptosis in heart diseases, the significance of this death pathway warrants future investigation.
Potential therapeutic studies in cardiovascular diseases
Medical experts and scientists have long searched for potential cardiac disease treatments and surviving and improving patients' lives. The Frontiers in Cardiovascular Medicine -Cardiovascular Therapeutics section has provided a platform for distinguished scientists to communicate, inspire, and seek more potential therapeutic solutions (46, 47). In Table 1, we summarized 12 significant studies Wu et al., Zheng et al., Wang et al., Liao et al., Dash et al., Lee et al., Ambari et al., Zhu et al., Quiroz et al., Zheng Song et al., Chen et al., Barbieri et al., and Cao et al. on our Research Topic to illustrate the cutting-edge treatments for different cardiovascular diseases. Readers could use Table 1 as an outline to dig out their interests.
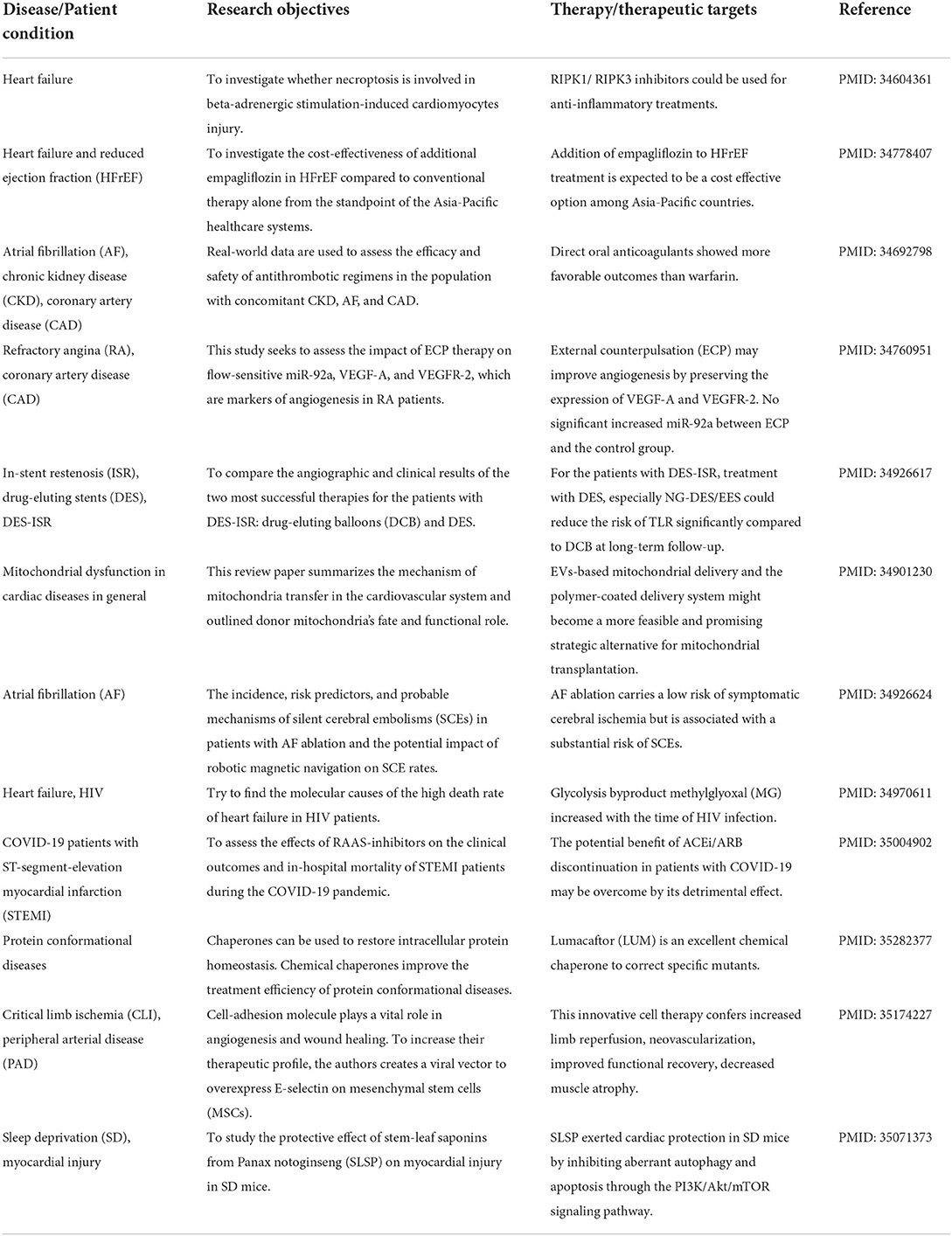
Table 1. Summary for 12 highlighted studies in Insights in cardiovascular therapeutics: 2021.
Author contributions
KX carried out literature collections, research analyses, and drafted the manuscript. MK, JY, NS, SW, RV-P, and HW provided editing input. XY supervised and edited the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Institutes of Health Grants to XY (HL132399-01A1; HL138749-01; and HL147565-01), HW (DK104116; DK113775; and HL131460), JY (HL153599), MK (HL135177), and America Heart Association Award to KX (916828).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Khan T. Cardiovascular disease. (2022). doi: 10.1002/jcla.24354. Available online at: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)#:~:text=Cardiovascular%20diseases%20(CVDs)%20are%20the,%2D%20and%20middle%2Dincome%20countries
2. Mishra PK, Adameova A, Hill JA, Baines CP, Kang PM, Downey JM, et al. Guidelines for evaluating myocardial cell death. Am J Physiol Heart Circ Physiol. (2019) 317:H891–H922. doi: 10.1152/ajpheart.00259.2019
3. Kepp O, Galluzzi L, Lipinski M, Yuan J Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov. (2011) 10:221–37. doi: 10.1038/nrd3373
4. Mery B, Guy JB, Vallard A, Espenel S, Ardail D, Rodriguez-Lafrasse C, et al. In vitro cell death determination for drug discovery: a landscape review of real issues. J Cell Death. (2017) 10:1179670717691251. doi: 10.1177/1179670717691251
5. Jiang M, Zeng J, Zhao L, Zhang M, Ma J, Guan X Zhang W. Chemotherapeutic drug-induced immunogenic cell death for nanomedicine-based cancer chemo-immunotherapy. Nanoscale. (2021) 13:17218–35. doi: 10.1039/D1NR05512G
6. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
7. Martens MD, Karch J Gordon JW. The molecular mosaic of regulated cell death in the cardiovascular system. Biochim Biophys Acta Mol Basis Dis. (2022) 1868:166297. doi: 10.1016/j.bbadis.2021.166297
8. Liao M, Qin R, Huang W, Zhu HP, Peng F, Han B Liu B. Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: a revisited perspective from molecular mechanisms to targeted therapies. J Hematol Oncol. (2022) 15:44. doi: 10.1186/s13045-022-01260-0
9. Tang D, Kang R, Berghe TV, Vandenabeele P Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–364. doi: 10.1038/s41422-019-0164-5
11. Zeglinski MR, Moghadam AR, Ande SR, Sheikholeslami K, Mokarram P, Sepehri Z, et al. Myocardial cell signaling during the transition to heart failure: cellular signaling and therapeutic approaches. Compr Physiol. (2018) 9:75–125. doi: 10.1002/cphy.c170053
12. Canty JM Jr. Myocardial injury, troponin release, and cardiomyocyte death in brief ischemia, failure, and ventricular remodeling. Am J Physiol Heart Circ Physiol. (2022) 323:H1–H15. doi: 10.1152/ajpheart.00093.2022
13. Chaulin AM. Cardiac troponins metabolism: from biochemical mechanisms to clinical practice (literature review). Int J Mol Sci. (2021) 22:10928. doi: 10.3390/ijms222010928
14. Blomgren K, Leist M Groc L. Pathological apoptosis in the developing brain. Apoptosis. (2007) 12:993–1010. doi: 10.1007/s10495-007-0754-4
15. Sauler M, Bazan IS Lee PJ. Cell death in the lung: the apoptosis-necroptosis axis. Annu Rev Physiol. (2019) 81:375–402. doi: 10.1146/annurev-physiol-020518-114320
16. Yang XF, Yin Y Wang H. Vascular inflammation and atherogenesis are activated via receptors for pamps and suppressed by regulatory t cells. Drug Discov Today Ther Strateg. (2008) 5:125–42. doi: 10.1016/j.ddstr.2008.11.003
17. Li Y-F, Xiao Huang, Xinyuan Li, Ren Gong, Ying Yin, Jun Nelson, et al. Caspase-1 mediates hyperlipidemia-weakened progenitor cell vessel repair. Front Biosci. (2015) 20:178–91. doi: 10.2741/4383
18. Wang L, Fu H, Nanayakkara G, Li Y, Shao Y, Johnson C, et al. Novel extracellular and nuclear caspase-1 and inflammasomes propagate inflammation and regulate gene expression: a comprehensive database mining study. J Hematol Oncol. (2016) 9:122. doi: 10.1186/s13045-016-0351-5
19. Xi H, Zhang Y, Xu Y, Yang WY, Jiang X, Sha X, et al. Caspase-1 inflammasome activation mediates homocysteine-induced pyrop-apoptosis in endothelial cells. Circ Res. (2016) 118:1525–39. doi: 10.1161/CIRCRESAHA.116.308501
20. Li YF, Nanayakkara G, Sun Y, Li X, Wang L, Cueto R, et al. Analyses of caspase-1-regulated transcriptomes in various tissues lead to identification of novel IL-1beta-, IL-18- and sirtuin-1-independent pathways. J Hematol Oncol. (2017) 10:40. doi: 10.1186/s13045-017-0406-2
21. Fagenson AM, Xu K, Saaoud F, Nanayakkara G, Jhala NC, Liu L, et al. Liver ischemia reperfusion injury, enhanced by trained immunity, is attenuated in caspase 1/caspase 11 double gene knockout mice. Pathogens. (2020) 9:879. doi: 10.3390/pathogens9110879
22. Shao Y, Saredy J, Xu K, Sun Y, Saaoud F, Drummer CT, et al. Endothelial immunity trained by coronavirus infections, DAMP stimulations and regulated by anti-oxidant NRF2 may contribute to inflammations, myelopoiesis, COVID-19 cytokine storms and thromboembolism. Front Immunol. (2021) 12:653110. doi: 10.3389/fimmu.2021.653110
23. Bedient L, Pokharel SM, Chiok KR, Mohanty I, Beach SS, Miura TA Bose S. Lytic cell death mechanisms in human respiratory syncytial virus-infected macrophages: roles of pyroptosis and necroptosis. Viruses. (2020) 12:932. doi: 10.3390/v12090932
24. Willerson JT, Ridker PM. Inflammation as a cardiovascular risk factor. Circulation. (2004) 109:II-2–II-10. doi: 10.1161/01.CIR.0000129535.04194.38
25. Wang J, Lai B, Nanayakkara G, Yang Q, Sun Y, Lu Y, et al. Experimental data-mining analyses reveal new roles of low-intensity ultrasound in differentiating cell death regulatome in cancer and non-cancer cells via potential modulation of chromatin long-range interactions. Front Oncol. (2019) 9:600. doi: 10.3389/fonc.2019.00600
26. Drummer Ct, Saaoud F, Shao Y, Sun Y, Xu K, Lu Y, et al. Trained immunity and reactivity of macrophages and endothelial cells. Arterioscler Thromb Vasc Biol. (2021) 41:1032–46. doi: 10.1161/ATVBAHA.120.315452
27. Zhong C, Yang X, Feng Y Yu J. Trained immunity: an underlying driver of inflammatory atherosclerosis. Front Immunol. (2020) 11:284. doi: 10.3389/fimmu.2020.00284
28. Xu K, Saaoud F, Yu S, Drummer Ct, Shao Y, Sun Y, et al. Monocyte adhesion assays for detecting endothelial cell activation in vascular inflammation and atherosclerosis. Methods Mol Biol. (2022) 2419:169–82. doi: 10.1007/978-1-0716-1924-7_10
29. Lu Y, Nanayakkara G, Sun Y, Liu L, Xu K, Drummer Ct, et al. Procaspase-1 patrolled to the nucleus of proatherogenic lipid LPC-activated human aortic endothelial cells induces ROS promoter CYP1B1 and strong inflammation. Redox Biol. (2021) 47:102142. doi: 10.1016/j.redox.2021.102142
30. Yin Y, Li X, Sha X, Xi H, Li YF, Shao Y, et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol. (2015) 35:804–16. doi: 10.1161/ATVBAHA.115.305282
31. Yang Q, Nanayakkara GK, Drummer C, Sun Y, Johnson C, Cueto R, et al. Low-Intensity ultrasound-induced anti-inflammatory effects are mediated by several new mechanisms including gene induction, immunosuppressor cell promotion, and enhancement of exosome biogenesis and docking. Front Physiol. (2017) 8:818. doi: 10.3389/fphys.2017.00818
32. Xu K, Shao Y, Saaoud F, Gillespie A, Drummer Ct, Liu L, et al. Novel knowledge-based transcriptomic profiling of lipid lysophosphatidylinositol-induced endothelial cell activation. Front Cardiovasc Med. (2021) 8:773473. doi: 10.3389/fcvm.2021.773473
33. Wang X, Li YF, Nanayakkara G, Shao Y, Liang B, Cole L, et al. Lysophospholipid receptors, as novel conditional danger receptors and homeostatic receptors modulate inflammation-novel paradigm and therapeutic potential. J Cardiovasc Transl Res. (2016) 9:343–59. doi: 10.1007/s12265-016-9700-6
34. Guo X, Chen Y Liu Q. Necroptosis in heart disease: molecular mechanisms and therapeutic implications. J Mol Cell Cardiol. (2022) 169:74–83. doi: 10.1016/j.yjmcc.2022.05.006
35. Li X, Fang P, Li Y, Kuo YM, Andrews AJ, Nanayakkara G, et al. Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arterioscler Thromb Vasc Biol. (2016) 36:1090–100. doi: 10.1161/ATVBAHA.115.306964
36. Li X, Shao Y, Sha X, Fang P, Kuo YM, Andrews AJ, et al. IL-35 (Interleukin-35) suppresses endothelial cell activation by inhibiting mitochondrial reactive oxygen species-mediated site-specific acetylation of H3K14 (Histone 3 Lysine 14). Arterioscler Thromb Vasc Biol. (2018) 38:599–609. doi: 10.1161/ATVBAHA.117.310626
37. Sun Y, Lu Y, Saredy J, Wang X, Drummer Iv C, Shao Y, et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. (2020) 37:101696. doi: 10.1016/j.redox.2020.101696
38. Li X, Fang P, Yang WY, Chan K, Lavallee M, Xu K, et al. Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Can J Physiol Pharmacol. (2017) 95:247–52. doi: 10.1139/cjpp-2016-0515
39. Cheng J, Nanayakkara G, Shao Y, Cueto R, Wang L, Yang WY, et al. Mitochondrial proton leak plays a critical role in pathogenesis of cardiovascular diseases. Adv Exp Med Biol. (2017) 982:359–70. doi: 10.1007/978-3-319-55330-6_20
40. Nanayakkara GK, Wang H Yang X. Proton leak regulates mitochondrial reactive oxygen species generation in endothelial cell activation and inflammation-a novel concept. Arch Biochem Biophys. (2019) 662:68–74. doi: 10.1016/j.abb.2018.12.002
41. Karki R, Lee S, Mall R, Pandian N, Wang Y, Sharma BR, et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci Immunol. (2022) 2022:eabo6294. doi: 10.1126/sciimmunol.abo6294
42. Yuan F, Cai J, Wu J, Tang Y, Zhao K, Liang F, et al. Z-DNA binding protein 1 promotes heatstroke-induced cell death. Science. (2022) 376:609–15. doi: 10.1126/science.abg5251
43. Christgen S, Place DE Kanneganti TD. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res. (2020) 30:315–27. doi: 10.1038/s41422-020-0295-8
44. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. (2021) 184:149–168 e17. doi: 10.1016/j.cell.2020.11.025
45. Samir P, Malireddi RKS Kanneganti TD. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. (2020) 10:238. doi: 10.3389/fcimb.2020.00238
46. Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A Kelly GL. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer. (2022) 22:45–64. doi: 10.1038/s41568-021-00407-4
Keywords: cell death, cardiovascular injuries, novel targets, cardiovascular therapeutics, inflammation
Citation: Xu K, Khan M, Yu J, Snyder NW, Wu S, Vazquez-Padron RI, Wang H and Yang X (2022) Editorial: Insights in cardiovascular therapeutics: 2021 – cell death, cardiovascular injuries, and novel targets of cardiovascular therapeutics. Front. Cardiovasc. Med. 9:981544. doi: 10.3389/fcvm.2022.981544
Received: 29 June 2022; Accepted: 11 July 2022;
Published: 26 July 2022.
Edited and reviewed by: Masanori Aikawa, Harvard Medical School, United States
Copyright © 2022 Xu, Khan, Yu, Snyder, Wu, Vazquez-Padron, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Yang, eGZ5YW5nQHRlbXBsZS5lZHU=