 Yu-Xing Liu1,2,3†
Yu-Xing Liu1,2,3† Liang-Liang Fan
Liang-Liang Fan- 1Department of Cardiovascular Surgery, National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 2Department of Cell Biology, School of Life Sciences, Central South University, Changsha, China
- 3Hunan Key Laboratory of Animal Models for Human Disease, School of Life Sciences, Central South University, Changsha, China
- 4Department of Anesthesiology, The Second Xiangya Hospital, Central South University, Changsha, China
Background: Desmin is an intermediate filament protein that plays a critical role in the stabilization of the sarcomeres and cell contacts in the cardiac intercalated disk. Mutated DES gene can cause hereditary cardiomyopathy with heterogeneous phenotypes, while the underlying molecular mechanisms requires further investigation.
Methods: We described a Chinese family present with cardiomyopathy and sudden cardiac death (SCD). Whole-exome sequencing (WES) and bioinformatics strategies were employed to explore the genetic entity of this family.
Results: An unknown heterozygote missense variant (c.1300G > A; p. E434K) of DES gene was identified. The mutation cosegregates in this family. The mutation was predicted as pathogenic and was absent in our 200 healthy controls.
Conclusion: We identified a novel DES mutation (p. E434K) in a Chinese family with cardiomyopathy and SCD. Our study not only provided a new case for the study of the relationship between DES mutations and hereditary cardiomyopathy but also broadened the spectrum of DES mutations.
Introduction
Hereditary cardiomyopathy is defined as a primary cardiac disease caused by genetic deficiencies of cardiac muscle (1). Highly heritable but genetically diversely, hereditary cardiomyopathies represent multiple clinical phenotypes, including dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), arrhythmogenic cardiomyopathy (ACM), non-compaction cardiomyopathy (NCM), and other or mixed phenotypes (2). To date, mutations in over 50 genes have been identified as the causative factor for different forms of cardiomyopathies (3). Proteins encoded by these pathogenic genes, such as sarcomere proteins, nuclear envelope proteins, cytoskeletal proteins, desmosomal proteins, and calcium/sodium-handling proteins of sodium channel, voltage-gated, are strongly linked with the occurrence of cardiomyopathy (4). However, there is no clear genotype–phenotype correlations of cardiomyopathy yet, which poses challenges to clinical diagnosis.
Desmin is an important intermediate filament (IF) protein principally expressed in cardiac, skeletal, and smooth muscle tissue (5). Encoded by the gene DES, desmin plays a key role in the mechanical stabilization of the striated muscle sarcomeres and cell contacts within the cardiac intercalated disk (ID) (5). Mutations in DES gene have been reported to be associated with skeletal myopathy and different hereditary cardiomyopathies (6). Nevertheless, the exact molecular mechanism of how DES mutations lead to these diseases, especially in cardiomyopathies, is not fully understood.
In this study, we described a Chinese family with cardiomyopathy and sudden cardiac death (SCD). Employing whole-exome sequencing (WES) technology and bioinformatics strategies, we identified a novel heterozygous mutation in DES gene which may be responsible for the genetic entity of this family.
Case description
Clinical features
A Chinese family with cardiomyopathy and SCD was encountered. The proband (II-5) was a 50-year-old woman from the central south region of China (Hunan Province) (Figure 1A). For more than a decade, she suffered from intermittent chest tightness and palpitation with two syncope episodes experience. According to her medical history, the proband was suspected to be Brugada syndrome in the previous treatment; however, she refused to be further hospitalized for testing and treatment then. During 1 week before her admission, she had increased chest tightness and brief syncope at work, and therefore, she was admitted to our hospital. Physical examination at admission showed high blood pressure (180/130 mmHg). The electrocardiogram (ECG) records showed sinus rhythm, over-load volume of the left ventricle (Figure 1B), but this is not typical for Brugada. The results of ambulatory ECG (AECG) have been shown in Supplementary Figure 1. An echocardiographic assessment revealed the enlargement of left ventricle (LV) and left atrium (LA) (56 and 40 mm, respectively; normal values: <50 and <28 mm, respectively), accompanied by a significant decrease in LV compliance (LVC). Coronary angiography suggested a myocardial bridge and mild stenosis of the left anterior descending artery (Supplementary Figure 2). No myopathies, neurological or bone abnormalities, were found in the proband. Further family history investigation found hypertension in her father (I-1), who died of possible coronary heart disease at age of fifty-two. Her eldest brother (II-1) also suffered from repeated attacks of syncope. Both her eldest brother (II-1) and her third brother (II-3) had SCD. Her sister (II-4) had a history of hypertension. The rest of her family members were healthy and no other malformations were observed in this family (Table 1 and Figure 1A). Given the family history of SCD, the proband received an implantable cardioverter-defibrillator (ICD) and returned for scheduled visits. On the other hand, 200 healthy subjects as described in our previous study were enrolled in this study to exclude polymorphisms (7).
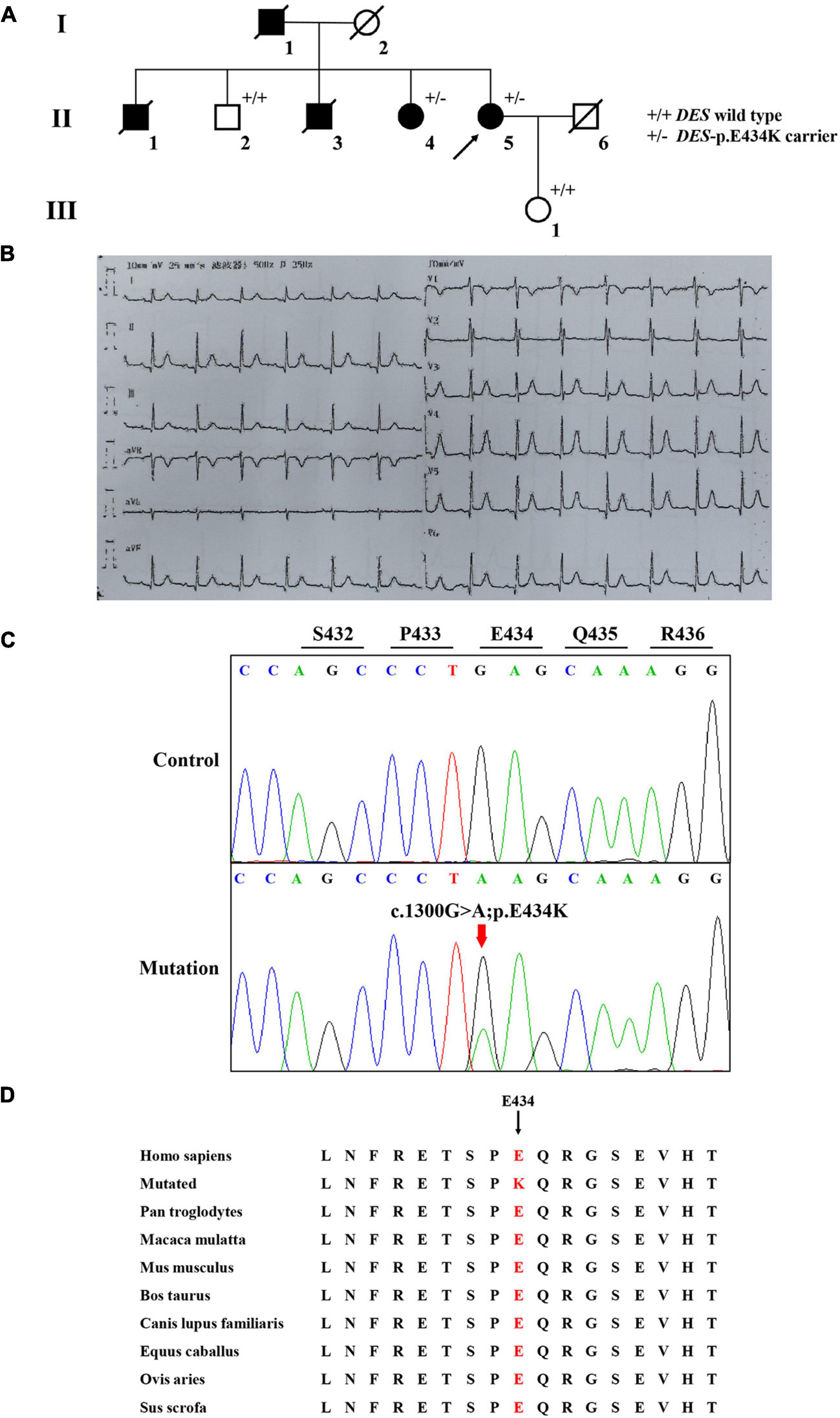
Figure 1. The clinical and sequencing data of this family. (A) The genealogy of this family. Squares indicate male family members and circles indicate female members. The black symbols represent the clinically affected members. The white symbols represent unaffected and healthy members. The arrow shows the proband. Crossed-out symbols stand for deceased relatives. ± represents heterozygous DES-p. E434K variant. + / + represents a wild type. (B) ECG of the proband. (C) Sanger DNA sequencing chromatogram detected a heterozygous mutation (c.1300G > A; p. E434K) of DES gene in the proband. (D) Alignment of multiple DES protein sequences across species. Letters in red show the E434 site is evolutionarily conserved.
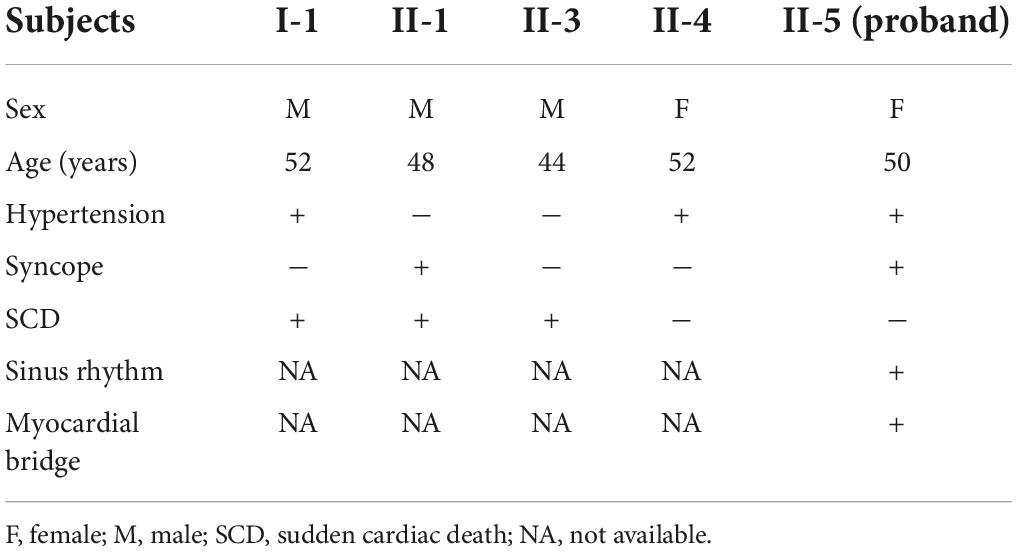
Table 1. Clinical data of patients in this family.
Genetic analysis
Whole-exome sequencing yielded 11.7 Gb of data with 99.5% coverage of the target region and 99.1% of the target covered over 10 ×. Data quality control steps, co-segregation, and bioinformatics analysis were performed following the published literature and our previous published studies (8–11). After preliminary screening, variants were further filtered by cardiomyopathy-related genes list as described in our previous study (12). A set of eight variants in eight genes were detected and were further analyzed. Bioinformatics annotations of the eight variants are shown in Table 2. Sanger sequencing was performed on all available family members and showed that a novel heterozygous mutation (c.1300G > A; p. E434K) of the DES gene (NM_001927.4) may underlie the genetic factor of this family (Figure 1C). Sanger sequencing confirmed that all living affected members in this family, including the proband (II-5) and her sister (II-4), harbored this heterozygous missense mutation in DES. DNA samples of other affected family members (I-1, II-1, and II-3) were unavailable. This mutation was neither identified in the unaffected living family members (II-2 and III-1) (Table 2), nor in our 200 healthy controls. Alignment of desmin amino acid sequences revealed that E434 is conserved in different species (Figure 1D). Modeling of proteins before and after missense mutation was performed by the SWISS-MODEL.1 Results revealed that the missense mutation at E434K may lead to the changing of protein surface charge as marked by red arrow in Figure 2A. In the WT desmin protein, E434 is hydrophobic with S432 and R436. In the mutated desmin protein, the hydrophobic effect with S432 disappeared after E434K mutation, while the hydrophobic effect with R436 remained (Figure 2A). Furthermore, MetaDome software2 indicated that the affected residue, E434, is located in an intolerant region of desmin (Figure 2B).
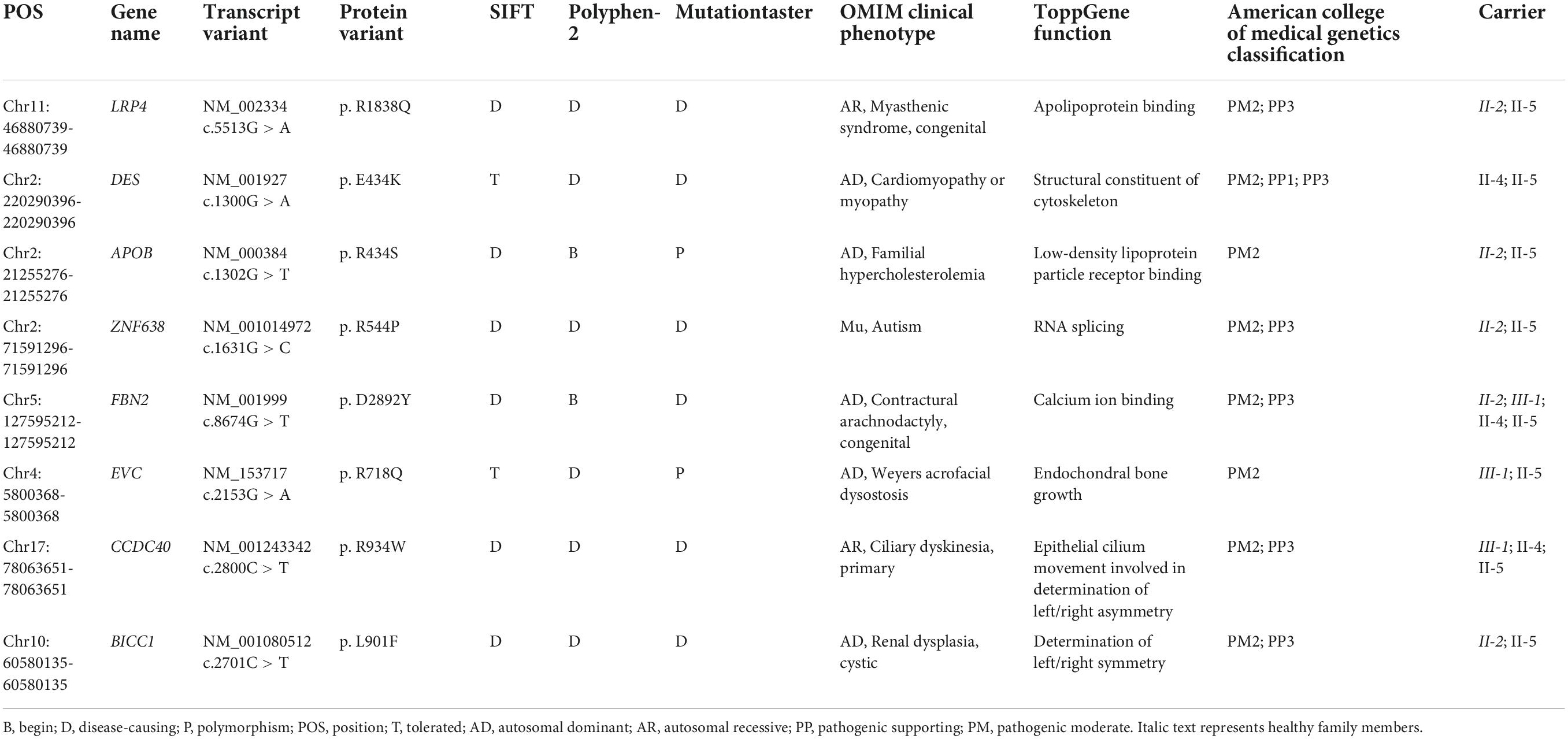
Table 2. Variants identified by WES in this family.
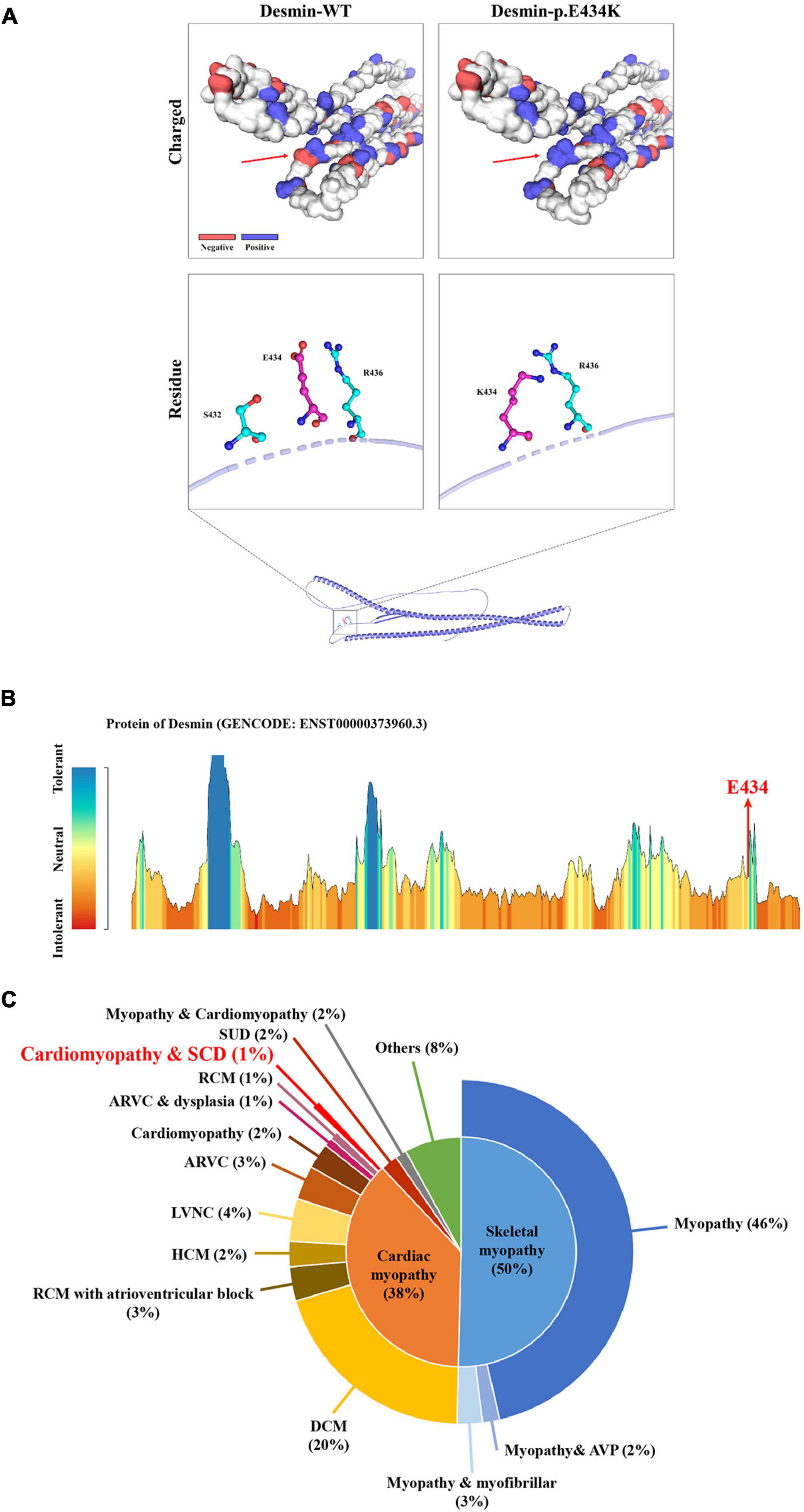
Figure 2. The bioinformatics analysis of mutations. Structure prediction of the mutant protein. The wild-type DES (DES-WT) protein structure and the p. E434K mutant DES (DES-p. E434K) protein structure were predicted by SWISS-MODEL online software. (A) Red arrow shows the charged change of desmin protein. In the DES-WT protein, E434 is hydrophobic with S432 and R436. In the DES-p.E434K protein, the hydrophobic effect with S432 disappeared after E434K mutation, while the hydrophobic effect with R436 remained (B) MetaDome server was used to identify the intolerant regions in DES. As depicted, the affected nucleotide/residue is located in an intolerant region. (C) Overview of all reported phenotypes caused by DES mutations. ARVC, arrhythmogenic right ventricular cardiomyopathy; AVP, autophagic vacuolar pathology; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVNC, left ventricular non-compaction; RCM, restrictive cardiomyopathy; SCD, sudden cardiac death; SUD, sudden unexplained death.
Discussion
In this case report, we reported a Chinese family with cardiomyopathy and SCD. The proband suffered from hypertension and recurrent syncope. Employing WES combined with cardiomyopathy-related gene-filtering, an unknown heterozygous mutation (c.1300G > A; p. E434K) of DES gene was detected and might be the pathogenic genetic factor in this family. Of note, the case history investigation of proband’s sister (II-4) revealed that she had hypertension with occasional chest tightness while without a history of syncope. Since recent studies have pointed out that mutations in DES gene may cause cardiac myopathies with a broad spectrum of pathological phenotypes within the same family (13). The same is suspected in this family of the present study. Unfortunately, the proband’s sister (II-4) refused to cooperate with further tests because of financial reasons.
Desmin is an IF protein that is prominently expressed in cardiac, skeletal, and smooth muscle tissue (5). As a kind of typical IF proteins, desmin consists of a head domain, a central homologous rod domain, and a tail domain (14). The structure of desmin is related to the assembly mechanism of IF. Different disease-causing DES mutations interfere at different stages within this assembly process (15, 16). In our study, the missense mutation (p. E434K) locates in the tail domain that alters the acidic glutamic acid at position 434 to a basic lysine acid. This missense mutation at E434 site may lead to the charged change in desmin protein. The hydrophobic effect of amino acid residues near the mutant site may also be affected (Figure 2A). Although the exact molecular function of the desmin tail domain is not well understood, it is suggested that the tail domain participates in width control of unit length filaments and mediates Ca2+- or Mg2+-dependent cross-linking (17, 18). Moreover, changes in hydrophobicity of amino acid residues may also affect the formation of a hydrophobic seam, which further leads to filament formation defects (6). Thus, we suggested that the missense mutation (p. E434K) detected in DES gene may be disease-causing, in line with the previous studies.
According to published literature, mutations in DES gene can cause myopathies or cardiomyopathies with heterogeneous phenotypes (19). Some patients with DES mutations may also present a combined skeletal and cardiac myopathy. For cases with cardiomyopathies, the spectrum of cardiac phenotypes associated with DES mutations ranges from DCM, HCM, RCM, arrhythmogenic right ventricular cardiomyopathy (ARVC), and left ventricular cardiomyopathy (LVNC). Most of the DES mutations were detected in DCM. The associated clinical phenotypes of some mutations overlap significantly. For example, DES-p. K144* has been reported in both DCM and HCM. Moreover, DES-p. P419S and DES-p. R454W have been reported in different cardiomyopathies, including DCM, HCM, ARVC, and RCM (6). These DES mutations may be associated with an incomplete penetrance and diverse expressivity. Recent studies have pointed out that mutations in DES gene may cause cardiac myopathies with a broad spectrum of pathological phenotypes within the same family (20–24). Some complex or mixed phenotypes had been also reported in rare cases with atypical or unknown cardiomyopathy (6). Nevertheless, it is still not clear why phenotypes caused by DES mutations are diverse. A brief review of these reported phenotypes according to the Human Gene Mutation Database (HGMD)3 is shown in Figure 2C, which may help the diagnosis of diseases associated with DES mutations. It is worth noting that less than 1% of cases reported cardiomyopathy as coexists with SCD, which suggested that the family reported in our study is relatively rare. Of note, the mutation (c.1300G > A; p. E434K) of DES gene identified in the present study has not been published, therefore, is considered novel.
Conclusion
We used WES to explore the genetic entity in a Chinese family with cardiomyopathy and SCD. A novel heterozygous missense mutation (c.1300G > A; p. E434K) of DES gene was detected. Our study not only provided a new case for the study of the relationship between DES mutations and hereditary cardiomyopathy but also broadened the spectrum of DES mutations.
Data availability statement
The original contributions are included in the Supplementary material and the following database: GSE-Human repository, accession number: HRA002969 (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA002969).
Ethics statement
The studies involving human participants were reviewed and approved by the Review Board of the Xiangya Hospital of the Central South University in China. The patients/participants provided their written informed consent to participate in this case study. Written informed consent was obtained from the individual(s) for the publication of this care report and any potentially identifiable images or data included in this article.
Author contributions
RY enrolled the family members. YS performed DNA isolation and sanger sequencing. Y-XL and L-LF performed genetic analysis. Y-XL wrote the manuscript. L-LF and YD supported the project. All authors reviewed the manuscript.
Funding
This study was supported by National Natural Science Foundation of China (81800220), National Natural Science Foundation of Hunan province (2019JJ50890), Research Project of Hunan Provincial Health Commission (202103012102), the Fundamental Research Funds for the Central Universities of Central South University (2021zzts0079), and Hunan Provincial Innovation Foundation for Postgraduate (CX20210177).
Acknowledgments
We thank all subjects for participating in this study. We also thank Shuai Guo from the University of Texas, MD Anderson Cancer Center for the help with language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.971501/full#supplementary-material
Footnotes
- ^ https://swissmodel.expasy.org/
- ^ https://stuart.radboudumc.nl/metadome/dashboard
- ^ http://www.hgmd.cf.ac.uk/docs/login.html
References
1. Kimura A. Molecular etiology and pathogenesis of hereditary cardiomyopathy. Circ J. (2008) 72:A38–48. doi: 10.1253/circj.cj-08-0050
2. Yuan SM. Cardiomyopathy in the pediatric patients. Pediatr Neonatol. (2018) 59:120–8. doi: 10.1016/j.pedneo.2017.05.003
3. Mook OR, Haagmans MA, Soucy JF, van de Meerakker JB, Baas F, Jakobs ME, et al. Targeted sequence capture and GS-FLX titanium sequencing of 23 hypertrophic and dilated cardiomyopathy genes: implementation into diagnostics. J Med Genet. (2013) 50:614–26. doi: 10.1136/jmedgenet-2012-101231
4. Zhao Y, Feng Y, Zhang YM, Ding XX, Song YZ, Zhang AM, et al. Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int J Mol Med. (2015) 36:1479–86. doi: 10.3892/ijmm.2015.2361
5. Hnia K, Ramspacher C, Vermot J, Laporte J. Desmin in muscle and associated diseases: beyond the structural function. Cell Tissue Res. (2015) 360:591–608. doi: 10.1007/s00441-014-2016-4
6. Brodehl A, Gaertner-Rommel A, Milting H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys Rev. (2018) 10:983–1006. doi: 10.1007/s12551-018-0429-0
7. Xiang R, Fan LL, Huang H, Cao BB, Li XP, Peng DQ, et al. A novel mutation of GATA4 (K319E) is responsible for familial atrial septal defect and pulmonary valve stenosis. Gene. (2014) 534:320–3.
8. Wang T, Liu YX, Luo FM, Dong Y, Li YL, Fan LL. A novel homozygous variant of TMEM231 in a case with hypoplasia of the cerebellar vermis and polydactyly. Front Pediatr. (2021) 9:774575. doi: 10.3389/fped.2021.774575
9. Huang H, Chen Y, Jin J, Du R, Tang K, Fan L, et al. CSRP3, p.Arg122*, is responsible for hypertrophic cardiomyopathy in a Chinese family. J Gene Med. (2022) 24:e3390. doi: 10.1002/jgm.3390
10. Tavasoli AR, Memar E, Ashrafi MR, Hosseini S, Haghighi R, Ghabeli H, et al. Primary and secondary microcephaly, global developmental delay, and seizure in two siblings caused by a novel missense variant in the ZNF335 gene. J Mol Neurosci. (2022) 2022:1955. doi: 10.1007/s12031-021-01955-y
11. Fan LL, Liu JS, Huang H, Du R, Xiang R. Whole exome sequencing identified a novel mutation (p.Ala1884Pro) of beta-spectrin in a Chinese family with hereditary spherocytosis. J Gene Med. (2019) 21:e3073. doi: 10.1002/jgm.3073
12. Zhang SB, Liu YX, Fan LL, Huang H, Li JJ, Jin JY, et al. A novel heterozygous variant p.(Trp538Arg) of SYNM is identified by whole-exome sequencing in a Chinese family with dilated cardiomyopathy. Ann Hum Genet. (2019) 83:95–9. doi: 10.1111/ahg.12287
13. Bergman JE, Veenstra-Knol HE, van Essen AJ, van Ravenswaaij CM, den Dunnen WF, van den Wijngaard A, et al. Two related dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene. Eur J Med Genet. (2007) 50:355–66. doi: 10.1016/j.ejmg.2007.06.003
14. Carlsson L, Thornell LE. Desmin-related myopathies in mice and man. Acta Physiol Scand. (2001) 171:341–8. doi: 10.1046/j.1365-201x.2001.00837.x
15. Brodehl A, Hedde PN, Dieding M, Fatima A, Walhorn V, Gayda S, et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J Biol Chem. (2012) 287:16047–57. doi: 10.1074/jbc.M111.313841
16. Bar H, Mucke N, Kostareva A, Sjoberg G, Aebi U, Herrmann H. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc Natl Acad Sci U.S.A. (2005) 102:15099–104. doi: 10.1073/pnas.0504568102
17. Lin YC, Broedersz CP, Rowat AC, Wedig T, Herrmann H, Mackintosh FC, et al. Divalent cations crosslink vimentin intermediate filament tail domains to regulate network mechanics. J Mol Biol. (2010) 399:637–44. doi: 10.1016/j.jmb.2010.04.054
18. Herrmann H, Haner M, Brettel M, Muller SA, Goldie KN, Fedtke B, et al. Structure and assembly properties of the intermediate filament protein vimentin: the role of its head, rod and tail domains. J Mol Biol. (1996) 264:933–53. doi: 10.1006/jmbi.1996.0688
19. Brodehl A, Pour HS, Stanasiuk C, Ratnavadivel S, Hendig D, Gaertner A, et al. Restrictive cardiomyopathy is caused by a novel homozygous desmin (DES) mutation p.Y122H leading to a severe filament assembly defect. Genes (Basel). (2019) 10:918. doi: 10.3390/genes10110918
20. Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. (2010) 19:4595–607. doi: 10.1093/hmg/ddq387
21. Brodehl A, Dieding M, Biere N, Unger A, Klauke B, Walhorn V, et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J Mol Cell Cardiol. (2016) 91:207–14. doi: 10.1016/j.yjmcc.2015.12.015
22. Marakhonov AV, Brodehl A, Myasnikov RP, Sparber PA, Kiseleva AV, Kulikova OV, et al. Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum Mutat. (2019) 40:734–41. doi: 10.1002/humu.23747
23. Bermudez-Jimenez FJ, Carriel V, Brodehl A, Alaminos M, Campos A, Schirmer I, et al. Novel desmin mutation p.Glu401Asp impairs filament formation, disrupts cell membrane integrity, and causes severe arrhythmogenic left ventricular cardiomyopathy/dysplasia. Circulation. (2018) 137:1595–610. doi: 10.1161/CIRCULATIONAHA.117.028719
Keywords: hereditary cardiomyopathy, SCD, DES, mutation, whole-exome sequencing
Citation: Liu Y-X, Yu R, Sheng Y, Fan L-L and Deng Y (2022) Case report: Whole-exome sequencing identifies a novel DES mutation (p. E434K) in a Chinese family with cardiomyopathy and sudden cardiac death. Front. Cardiovasc. Med. 9:971501. doi: 10.3389/fcvm.2022.971501
Received: 17 June 2022; Accepted: 05 September 2022;
Published: 04 October 2022.
Edited by:
Neil Morgan, University of Birmingham, United KingdomReviewed by:
Andreas Brodehl, Heart and Diabetes Center North Rhine-Westphalia, GermanyRaj Sewduth, VIB KU Leuven Center for Cancer Biology, Belgium
Copyright © 2022 Liu, Yu, Sheng, Fan and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang-Liang Fan, c3dmYW5saWFuZ2xpYW5nQGNzdS5lZHUuY24=; Yao Deng, NjE5MDY1Mzc0QHFxLmNvbQ==
†These authors have contributed equally to this work