 Li-Li Zhang
Li-Li Zhang Rui-Jie Tang2†
Rui-Jie Tang2† Yue-Jin Yang
Yue-Jin Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 08 August 2022
Sec. Cardiovascular Metabolism
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.964034
This article is part of the Research Topic Investigating Novel Mechanisms and Treatments of Metabolic Disorders Associated with Cardiovascular Disease View all 13 articles
Cardiovascular diseases (CVDs) have been attracting the attention of academic society for decades. Numerous researchers contributed to figuring out the core mechanisms underlying CVDs. Among those, pathological decompensated cellular loss posed by cell death in different kinds, namely necrosis, apoptosis and necroptosis, was widely regarded to accelerate the pathological development of most heart diseases and deteriorate cardiac function. Recently, apart from programmed cell death revealed previously, ferroptosis, a brand-new cellular death identified by its ferrous-iron-dependent manner, has been demonstrated to govern the occurrence and development of different cardiovascular disorders in many types of research as well. Therefore, clarifying the regulatory function of ferroptosis is conducive to finding out strategies for cardio-protection in different conditions and improving the prognosis of CVDs. Here, molecular mechanisms concerned are summarized systematically and categorized to depict the regulatory network of ferroptosis and point out potential therapeutic targets for diverse cardiovascular disorders.
Cardiovascular diseases (CVDs) mean an array of diseases, including atherosclerosis, hypertension, acute coronary syndrome, heart failure (HF), cardiomyopathy, arrhythmia, etc., as the major cause of morbidity and mortality in the global population, imposing heavy economic pressure on families and community. Therefore, to prevent the occurrence and development of CVDs, its pathological mechanisms have become the focus of medical research in recent years.
Iron is an essential microelement for multiple life processes, and the imbalance of iron homeostasis can cause many adverse consequences. Iron overload is associated with iron deposition in various tissues including the heart, in 1981, Sullivan et al. formally proposed the hypothesis of iron-derived heart disease (1). Since then, iron overload was shown to play a pivotal role in the pathogenesis of CVDs and enhance the risk of cardiovascular morbidity (2). Iron overload can induce a novel iron-dependent form of cell death that is neither apoptosis nor necrosis, called ferroptosis (3). Cell death results in the onset and deterioration of acute or chronic disorders, often accompanied by dysregulation of inflammation, cellular dysfunction, and tissue damage. Acute cardiomyocytes (CM) death can cause myocardial infarction (MI) and ischemia-reperfusion (I/R) injury, while chronic progressive CMs death can cause compensatory hypertrophy, eventually leading to HF or cardiomyopathy (4). Ferroptosis is distinguished from other forms of death. And its main feature is the oxidative modification of phospholipid membranes by iron-dependent mechanisms, coupled with a substantial buildup of lipid peroxides. The pathological role of ferroptosis in atherosclerosis, I/R injury, cardiomyopathy, HF and other CVDs has been widely reported, and targeted intervention of ferroptosis effectively prevented the occurrence and development of these diseases (5–9). This review focuses on the underlying pathological mechanism of ferroptosis in CVDs, regarding oxidative stress, inflammation, metabolic disorders and mitochondrial damage, and provides targeted evidence for treatment.
Dixon et al. (3) showed that the selectively RAS-lethal small molecule erastin not only modulated the mitochondrial voltage-dependent anion channel (VDAC), but also inhibited the function of SLC7A11, an important subunit of the cystine/glutamate reverse transporter system Xc– (xCT), reducing intracellular glutathione (GSH) synthesis, further increasing the accumulation of iron-dependent lipid peroxide and inducing the death of RAS mutant fibrosarcoma cell lines. They formally named this new form of regulated cell death triggered by erastin as ferroptosis: an iron-dependent non-apoptotic form of cell death caused by the imbalance of intracellular synthesis and degradation of ROS and lipid peroxides (3). Different from typical cell death form, ferroptosis cannot be inhibited by inhibitors of apoptosis and pyroptosis, but can be inhibited by iron chelators (e.g., deferoxamine), antioxidants (e.g., tea polyphenol) (10, 11). Moreover, ferroptosis has unique morphological features and biochemical markers. Morphologically, there was no cell shrinkage, chromatin condensation, formation of apoptotic bodies, or disintegration of the cytoskeleton, but significant shrinkage of mitochondria and reduction of mitochondrial cristae were observed (3). Regarding distinguishing biomarkers, they are associated with the accumulation of intracellular iron ions and ROS accompanied by the decrease in GSH metabolism, as exemplified in Table 1.

Table 1. Major regulatory indicators and potential markers of ferroptosis.
Iron overload is a prerequisite for ferroptosis, and various iron chelators can inhibit ferroptosis. Transferrin receptor 1 (TfR1) can promote the transfer of extracellular iron into cells, and the silencing of its encoding gene prevented ferroptosis caused by erastin. Iron supplementation accelerated erastin-induced ferroptosis (12). Treatment of dopaminergic cells with ferric ammonium citrate to mimic the iron overload showed that ferroptosis occurred before apoptosis and could be rescued by ferroptosis inhibitors (13). These results further demonstrated the necessity of iron overload in the process of ferroptosis.
The accumulation of lipid peroxide is another crucial element in the execution of ferroptosis. Cellular or organelle membranes are especially vulnerable to attack by ROS due to their high polyunsaturated fatty acids, called lipid peroxidation. An important source of ROS in mammalian cells is mitochondria. Among mitochondrial metabolic activities, tricarboxylic acid (TCA) cycle starts with acetyl-CoA from glucose or fatty acid metabolism, and transfers electrons to the electron transport chain (ETC), completing oxidative phosphorylation (OXPHOS). Once the electrons are leaked from ETC complexes I and III, superoxide will be produced and converted to H2O2 by superoxide dismutase (SOD) (14). Subsequently, Fe2+ released via divalent metal transporter 1 (DMT1) can react with H2O2 to yield hydroxyl (⋅OH) or alkoxy (RO⋅) radicals, known as “Fenton reaction,” resulting in the production of reactive poisonous aldehydes, in the form of 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA) (15–17). In addition, ROS can induce autophagy to accelerate the degradation of ferritin and enhance the expression of TfR1 leading to ferroptosis (18). In turn, ferroptosis can also significantly increase ROS production, disrupt mitochondrial membrane potential, and promote mitochondrial fission (19). Accordingly, mitochondrial damage and energy metabolism disorders exacerbate the vicious cycle between lipid peroxidation and ferroptosis.
As confirmed by Dixon et al., xCT-mediated cystine absorption from extracellular space accompanied by the export of intracellular glutamate could regulate ferroptosis (3). In the cytoplasm, cystine turns into cysteine, a main raw material of GSH synthesis. GSH produced above serves as an electron donor to reduce toxic lipid peroxides into non-toxic alcohols under the enzymolysis of glutathione peroxidase 4 (GPX4) (20). Hence, the xCT-GSH-GPX4 axis conducts the dominant defensive mechanism against cellular ferroptosis. Upregulation of p53 significantly reduced the expression of SLC7A11, which inhibited xCT mediated-cystine uptake and resulted in ferroptosis (21). RSL3, erastin and sulfasalazine are direct targets of GPX4 and make cancer cells more sensitive to ferroptosis (22). It can be inferred that any methods causing xCT inhibition, GSH or GPX4 depletion might promote ferroptosis. In addition, ferroptosis suppressor protein 1 (FSP1), formerly known as apoptosis-inducing factor mitochondria-associated 2, protects cells from GPX4 deletion-induced ferroptosis (23). Treatment of lung cancer cells with iFSP1, an inhibitor of FSP1, potentiated ferroptosis (24). Therefore, antagonism of FSP1 can lead to the occurrence of ferroptosis.
Moreover, ferroptosis as a type of necrotic cell death is mostly accompanied by inflammatory manifestations. Ferroptosis-related necroinflammation was observed in mice with acute kidney injury (25). Ferroptotic cells can release damage-associated molecular patterns (DAMPs) to activate immune cells, which further amplify ferroptosis by releasing inflammatory mediators such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α. In addition, during ferroptosis, polyunsaturated fatty acids, especially arachidonic acids, are prone to peroxidation. The lipoxygenase (LOX) and cyclooxygenase (COX)-generated metabolites of arachidonic acid (hydroxyeicosatetraenoic acid, leukotriene, and prostaglandins) are also involved in inflammatory response (26). The above shows that ferroptosis is closely related to inflammation and immunity, and the persistence of inflammation may form a self-amplifying circuit of ferroptosis.
In sum, oxidative stress, inflammation, metabolic disorders, and mitochondrial damage are the main cellular and molecular pathological mechanisms in ferroptosis.
Oxidative stress is involved in assorted CVDs like MI, I/R injury, and HF. Excessive production of ROS impairs cellular lipids, resulting in ferroptosis. In the cardiovascular system, the antioxidant system controls intracellular ROS and interacts with biological components to maintain redox equilibrium. This system contains counteroxidant enzymes such as SOD, catalase, GPX, and thioredoxin (Trx), and non-enzymatic antioxidants such as tocopherol and coenzyme Q10 (CoQ10) are shown in Figure 1. This system eliminates ROS by chelating metal ions, enhances the production of endogenous antioxidants, and defends against ferroptosis in the cardiac tissues (27–30).
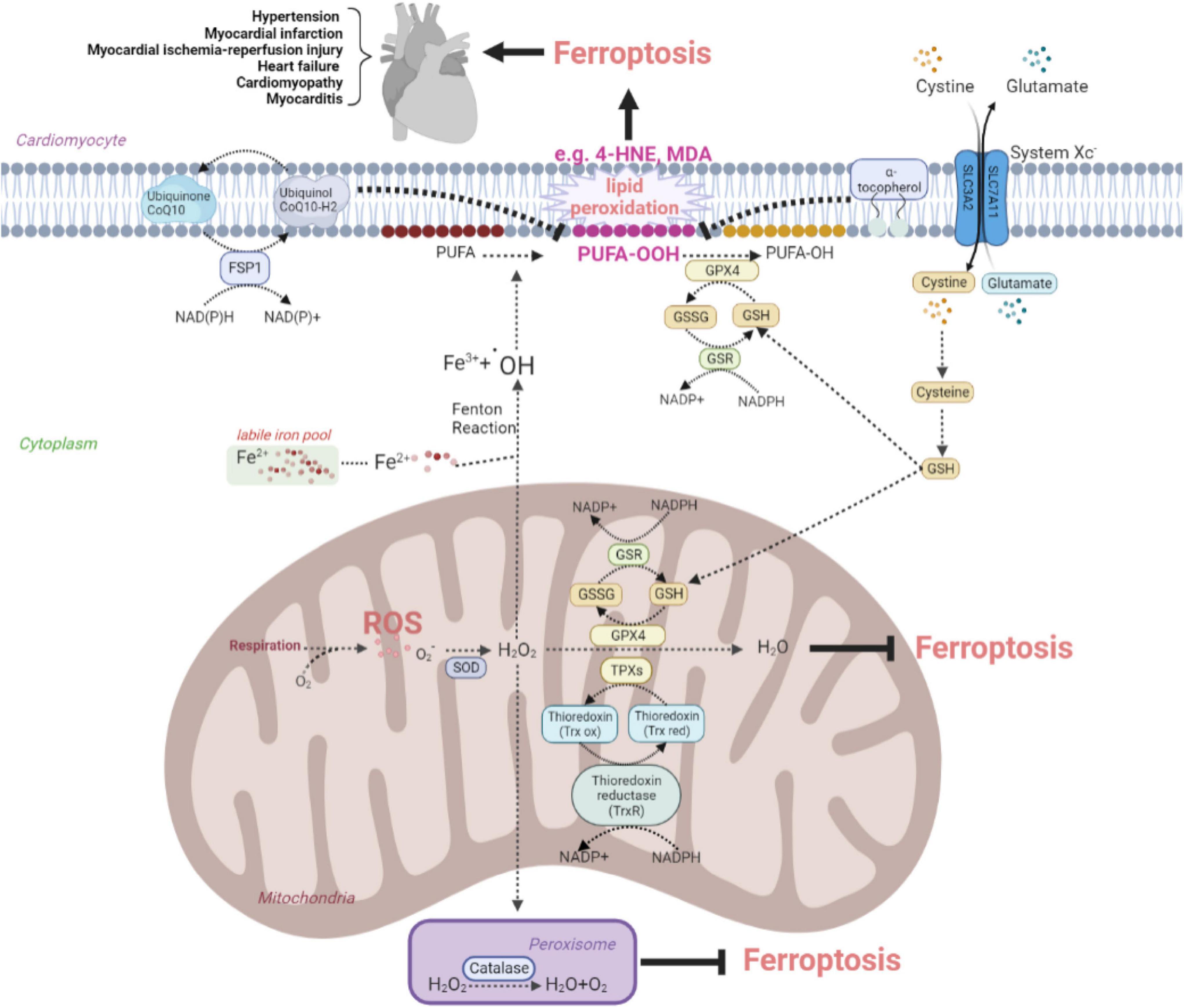
Figure 1. The anti-oxidative system inhibits ferroptosis to protect against cardiovascular disease by eliminating oxidative stress. The figure shows the protective effects of the SOD family, catalase, Trx recycles, system Xc–-GSH-GPX4 pathway, FSP1-CoQ10-NAD(P)H pathway, and tocopherol on the elimination of ROS and reducing the conversion to lipid peroxide, such as toxic MDA and 4-HNE. ROS, reactive oxygen species; SOD, superoxide dismutase; FSP, ferroptosis suppressor protein; CoQ, coenzyme Q; GSH, glutathione; GSSG, oxidized glutathione; GSR, glutathione reductase; GPX, glutathione peroxidase; 4-HNE, 4-hydroxynonenal; MDA, malondialdehyde; TPX, thioredoxin peroxidase; Trx red, reduced thioredoxin; Trx ox, oxidized thioredoxin; PUFA, polyunsaturated fatty acid.
Three isoforms of SOD system in mammals are SOD1, SOD2, and SOD3, as the first line of defense against oxygen-derived free radicals in the cardiovascular system. Under the exposure to oxidative stress, they can be rapidly activated to catalyze superoxides into oxygen and H2O2. Among three members of SOD family, SOD1, an isoform located in the cytoplasm containing Cu2+ and Zn2+, outweighs SOD2 and SOD3 in the expression of abundance. A cohort study showed that SOD1 allelic mutations in the population were linked with an elevated risk of death from cardiovascular complications (sudden death, fatal MI, or stroke) (31). SOD1 deletion promoted the occurrence of ferroptosis (32). Sustained delivery of SOD1 could improve myocardial I/R injury (33). SOD2 is a manganese-containing isoenzyme found in mitochondria, also associated with ferroptosis (34). Deficiency of SOD2 increases ROS in CMs, leading to subsequent overproduction of 4-HNE inside mitochondria and impairment of mitochondrial bioenergy production, which is one of the causes of lethal dilated cardiomyopathy (35). The structure of SOD3 is similar to SOD1, and upregulation of SOD3 is conducive to rid the vascular system of oxidative damage, alleviating the severity of hypertension and coronary arteriosclerosis (36). Therefore, the SOD system may protect from the occurrence of ferroptosis in CVDs by reducing superoxides.
Catalase located in the peroxisome varies in different developmental stages of organisms, which is manipulated by peroxisome proliferator-activated receptors (PPARs) and decomposes H2O2 into H2O and oxygen (37). Upregulation of catalase has been reported to increase ferroptosis resistance in xCT-deficient mouse blast fibroblasts. Besides, catalase activation is cardioprotective to H2O2-induced stress in myocardial I/R injury by alleviating ferroptosis (38, 39).
Glutathione peroxidase 4 family contain eight members, namely GPX1–GPX8. The similar denominator among diverse GPXs gifts their abilities to reduce H2O2 and hydroperoxides to H2O. GPX4 is unique in the GPXs family which can reduce phospholipid hydroperoxides of membranes using redox equivalents from GSH. A proteomic analysis showed that GPX4 downregulation using specific siRNA or chemical inhibitor RSL3 caused CMs ferroptosis during MI (40). Activation of nuclear factor erythroid 2-related factor 2/xCT/GPX4 signaling pathway attenuates CMs ferroptosis in doxorubicin-induced cardiomyopathy and myocardial I/R injury by inhibiting oxidative stress (41, 42). Besides, the ferroptosis inhibitor ferrostatin-1 markedly prevented pathological myocardial remodeling and fibrosis in angiotensin II-induced hypertensive cardiomyopathy by attenuating the upregulation of ferrous ion levels and lipid peroxidation in mouse microvascular endothelial cells (ECs) through modulating the xCT/GPX4 signaling (43). The Chinese herbal medicine, Tongxinluo, prevented atherosclerosis, and the xanthohumol reduced myocardial I/R injury as well, both through modulating GPX4 expression to decrease ROS (44, 45). Thus, chemicals targeting GPX4 signaling pathway could act as potential drugs to various CVDs by suppressing ferroptosis.
Thioredoxin reductase (TrxR), Trx and NADPH together constitute the Trx system. TrxR is an NADPH-dependent dimeric selenozyme required for reducing and recycling the oxidized Trx1. TrxR was verified to reduce cell sensitivity to erastin (46). Cardiac-specific TrxR knockout (KO) aged mice were more prone to dilated cardiomyopathy due to increased ROS and dysregulated mitochondrial energy metabolism (47). Additionally, Trx-1 as a small molecule protein participates in redox reactions. Overexpression of Trx-1 could inhibit ferroptosis by increasing GPX4 and GSH (48), which indicated the GSH-Trx cross-talk ensured cell survival. In Trx1 KO mice, increased oxidation of the mammalian target of the rapamycin (mTOR) signaling leads to impaired phosphorylation of substrates and mitochondrial respiratory dysfunction, manifesting as systolic dysfunction in HF (49). The Trx system delivers electrons for thioredoxin peroxidase (TPX), whose function is similar to the GPX to remove ROS by reducing H2O2 and hydroperoxides at a rapid reaction rate (50). Thus, it is speculated that the Trx system alleviates ferroptosis to exert cardioprotective effects by modulating the reduction/oxidative balance.
Vitamin E has eight isomers that inhibit lipid peroxidation by donating phenolic hydrogen to peroxyl radicals, producing inactive tocopherol radicals. Alpha-tocopherol was the most prevalent and physiologically type of vitamin E (51). In vitro, alpha-tocopherol was demonstrated to rescue GPX4-deficient cells from ferroptosis, indicating that GPX4 and vitamin E synergistically maintain lipid redox equilibrium (52). Alpha-tocopherol may decrease myeloperoxidase expression, reducing oxidative and inflammatory reactions induced by I/R damage in mice, ultimately, preserving heart function (53). However, Keith et al. conducted a clinical trial showed that vitamin E supplementation significantly elevated alpha-tocopherol plasma concentrations in the treated group but failed to affect any other markers of oxidative stress and prognosis or function in class III or IV advanced HF (54). It is possible that alpha-tocopherol supplementation alone is not sufficient to combat severe oxidative stress and may achieve the desired therapeutic effect in combination with other natural antioxidants.
CoQ10 is named because of the mammalian CoQ containing 10 isoprene units. CoQ10 mainly exists in two forms, namely ubiquinone (oxidized form) and ubiquinol (reduced form). Ubiquinone can be transformed into ubiquinol by losing two electrons, and ubiquinol plays the main anti-oxidant role. FSP1 participates in the regulation of CoQ10 antioxidant system to alleviate ferroptosis, in a GSH- and GPX4-independent manner. The N-terminal end of FSP1 containing a typical myristoylation motif can bind to the phospholipid bilayer of cell membrane. The reduced form, ubiquinol, converts to the oxidative form, ubiquinone, after chemically reacting with lipid peroxide radicals released from the metabolism of membrane lipid, and eventually shut down the ferroptosis. Furthermore, under the catalysis of FSP1, ubiquinone can be reduced to ubiquinol in the cell membrane using NAD(P)H (23). Recently, an in vivo experiment has proven that FSP1 upregulation alleviates myocardial injury induced by septicemia through inhibition of ferroptosis (54). In short, the FSP1-CoQ10-NAD(P)H axis underlies another indispensable mechanism for myocardium protection under CVDs.
Damaged or dead CMs can generate or release host-derived danger signaling molecules DAMPs, which have been identified as endogenous dangerous signals for the innate immune system (55). The high mobility group box 1 (HMGB1) protein is a type of DAMP that can be released by ferroptotic cells. Ferroptosis inducers, erastin, sorafenib and RSL3, all trigger the release of HMGB1, which recruits pro-inflammatory macrophages and microglia via activating receptors for advanced glycation end products (RAGE)-nuclear factor-kappa B (NF-κB) pathway. Upregulation of HMGB1 expression has been shown to contribute to chronic HF and ischemic heart disease (56–59). Besides, inflammasomes are multi-protein complexes involving intracytoplasmic pattern recognition receptors. Inflammasomes were activated by DAMPs, followed by activating pro-inflammatory proteases and producing the corresponding mature cytokines (60, 61). Englestrin, a selective sodium-glucose cotransporter 2 inhibitor, significantly improved cardiac function in adriamycin cardiomyopathy by attenuating CMs ferroptosis through inhibition of the NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasomes related pathways (62). DAMPs are also processed by antigen-presenting cells like dendritic cells (DCs) through Toll-like receptors (TLRs) and presented to T cells to initiate an immune response. Besides, DAMPs released from ferroptotic ECs facilitates neutrophil adhesion to coronary vessels via activating the TLR4/TIR-domain-containing adapter-inducing interferon-β (TRIF)/interferon I (IFN I) pathway, resulting in neutrophil recruitment to the damaged myocardium (63). These discoveries shed light on the contribution of the immune cells activated by DAMPs from ferroptotic cells leading to CVDs, as summarized in Figure 2.
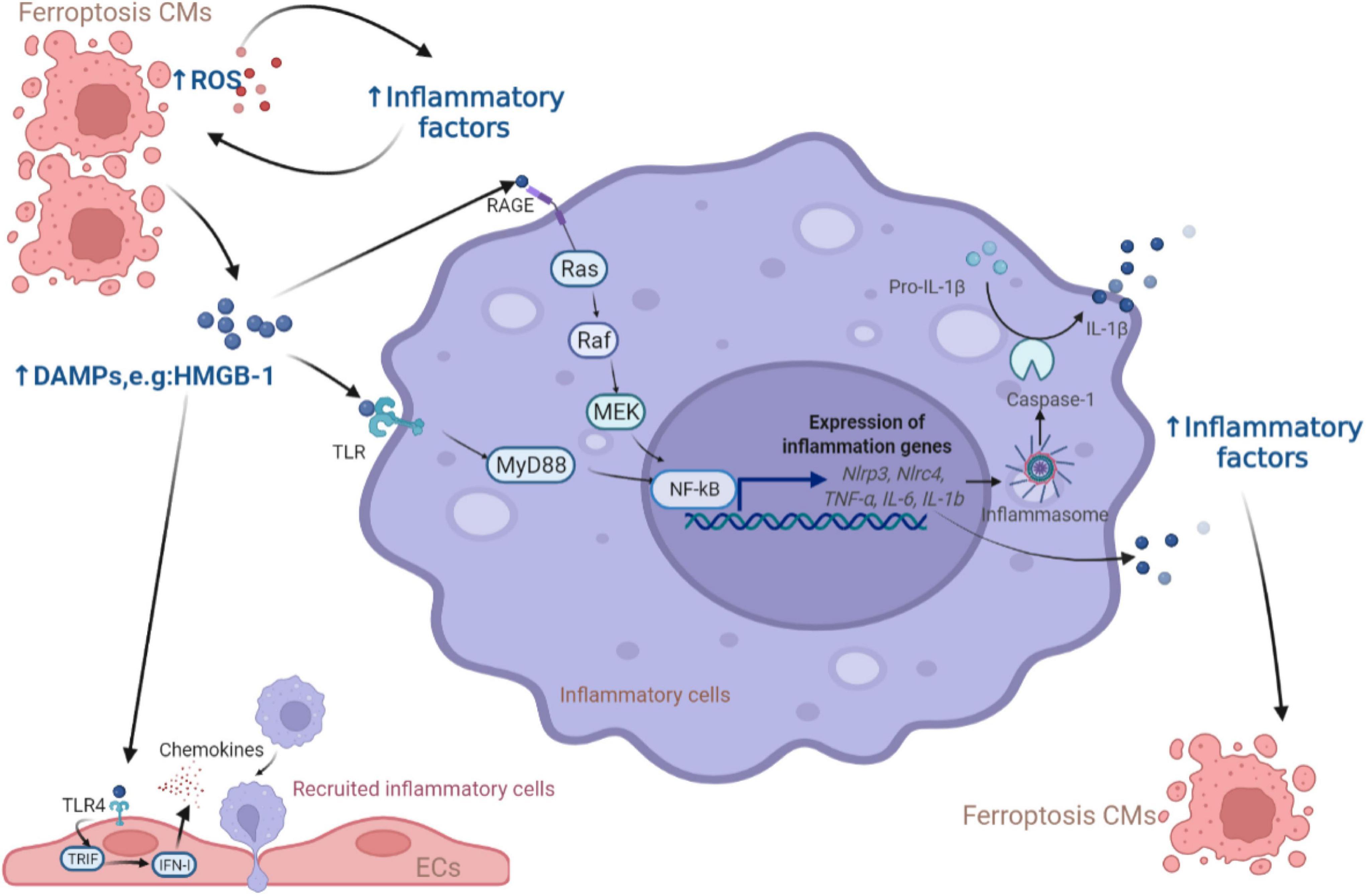
Figure 2. The role of DAMPs released from ferroptotic CMs in inflammation. The release of DAMPs from ferroptotic cells activates the downstream NF-κB inflammatory pathway through the TLR and RAGE receptors of inflammatory cells, leading to the production of inflammatory factors (e.g., NF-κB, IL-6, IFN-γ, TNF-α, and IL-1β) which were also produced due to the increase of ROS to induce more CMs ferroptosis. In addition, ECs respond to DAMPs via TLR4 receptors and release chemokines to promote more inflammatory cells entering the myocardial microenvironment. CM, cardiomyocyte; ROS, reactive oxygen species; DAMP, damage-associated molecular pattern; HMGB, high mobility group box; RAGE, receptor for advanced glycation end products; TLR, Toll-like receptors; MEK, mitogen-activated protein kinase kinase; NF-κB, nuclear factor-kappa B; NLRP, NOD-like receptor thermal protein domain associated protein; NLRC, NLR family CARD domain containing; MyD88, myeloid differentiation factor 88; IL, interleukin; TRIF, TIR-domain-containing adapter-inducing interferon-β; IFN, interferon; EC, endothelial cell.
Inflammation is tightly associated with oxidative stress, which triggers a range of pro-inflammatory factors such as NF-κB, IL-6, IFN-γ, TNF-α (64–67). Besides, studies have confirmed that the NF-κB pathway and IL-6/JAK2/STAT3 pathway activation could induce ferroptosis (68, 69). IFN-γ could down-regulate the expression of two subunits of xCT, SLC3A2 and SLC7A11, through the ASK1/JNK or JAK1-2/STAT1 pathway, reducing cellular uptake of cystine and thus promoting ferroptosis (70, 71). In cardiovascular-related diseases, the inhibition of these inflammatory pathways could rescue the ferroptotic CMs or ECs. For example, the ferroptosis inhibitor ferrostatin-1 improved sepsis-induced cardiac systolic dysfunction partly through alleviating the TLR4/NF-κB signaling pathway and reducing TNF-α and IL-6 (72). A hormone elabela significantly attenuated ferroptosis in hypertensive mice by inhibiting the cardiac IL-6/STAT3 signaling pathway (43). Nevertheless, the involvement of IFN-γ in the occurrence of ferroptosis has not been investigated in CVDs and needs to be further studied.
Furthermore, membrane-bound arachidonic acid was mainly found in the phospholipids of cell membranes and subject to COX, LOX and cytochrome P450 to yield a series of inflammatory mediators, like prostaglandins and leukotrienes, causing aseptic inflammation (73, 74). Reducing intracellular levels of arachidonoyl in CMs blocked neutrophil replenishment and prevented the onset of ferroptosis. Eliminating oxidative productions from arachidonic acid could also inhibit NF-κB pathway activation (75). Thus, agents against metabolites of arachidonic acid can play an anti-inflammatory or cytoprotective role in CVDs caused by ferroptosis.
During myocardial injury, different types of immune cells interact with each other, and with CMs, fibroblasts, and ECs to elicit inflammatory responses. Among these immune cells, the macrophages and T cells are the two most studied types in the cardiovascular field. Here, we focus on the possible effects of these two types of cells on ferroptosis in CVDs, shown in Figure 3.
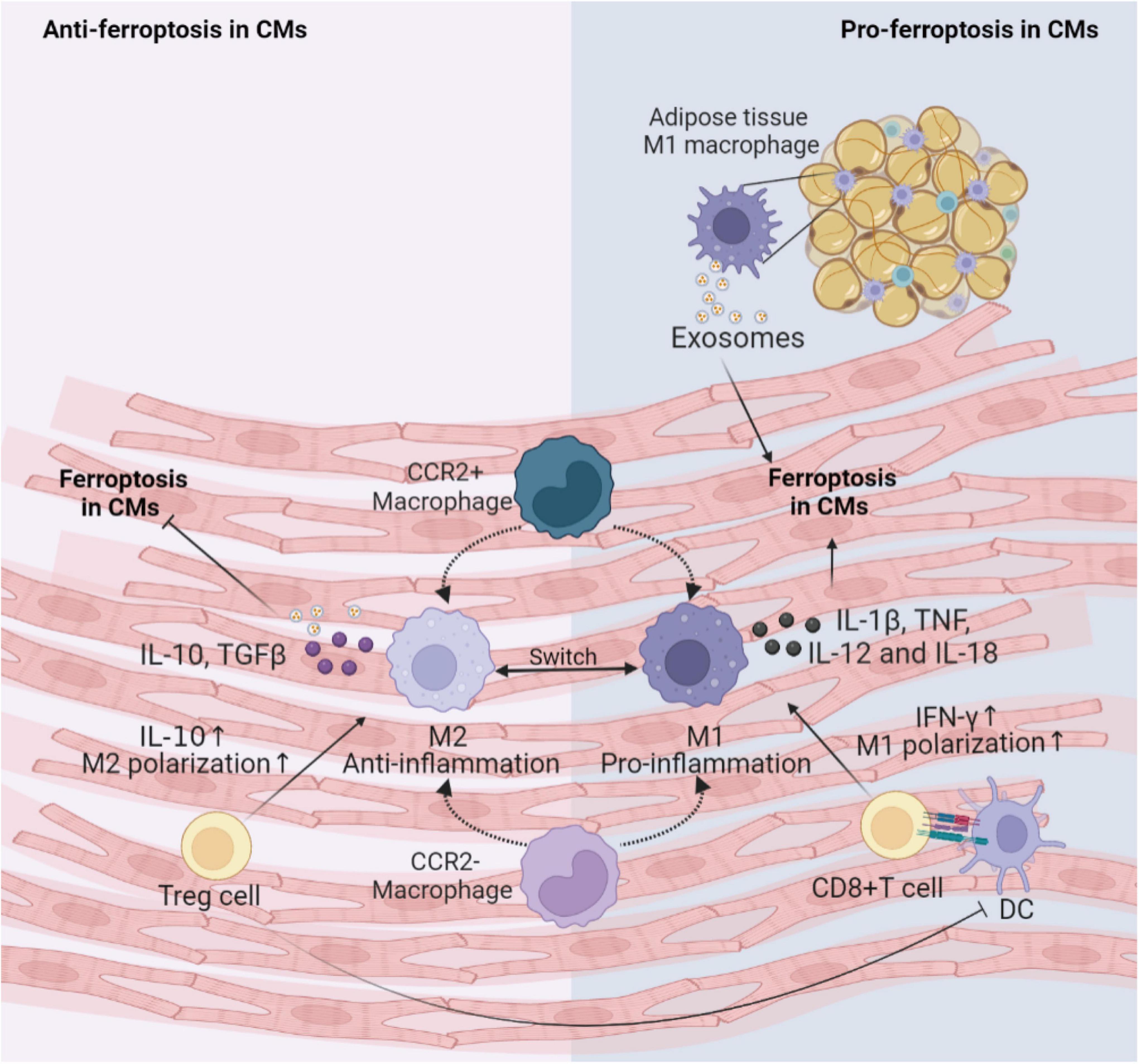
Figure 3. Immune cells involved in the regulation of CMs ferroptosis. This figure summarizes macrophages and T cells activity regulating the ferroptosis following cardiac injury. The cross-talk between these immune cells and CMs depends on various inflammatory factors and exosomes. DC, dendritic cell; CM, cardiomyocyte; IL, interleukin; TNF, tumor necrosis factor; IFN, interferon; TGF, transforming growth factor; CCR, C-C motif chemokine receptor 2.
Generally, the first immune cell type to respond to myocardial injury is the tissue-resident macrophages. Tissue-resident C-C motif chemokine receptor 2 (CCR2)– macrophages originated from the embryonic developmental stage are distinguished from tissue-resident CCR2+ macrophage repopulated by monocytes extravasating into the myocardium. Unlike CCR2+ macrophages, tissue-resident CCR2– macrophages could inhibit monocyte recruitment (76). Apart from preventing fibrosis and stimulating angiogenesis (77), CCR2– macrophages also improve cardiac repair via promoting clearance of apoptotic CMs, up-regulating anti-inflammatory mediators IL-10 and TGF-β, and down-regulating pro-inflammatory mediators IL-1β, TNF-α, IL-6 and IFN-γ after MI (78). Consumption of resident CCR2– macrophages leads to activation of inflammasomes in CMs, abnormal mitochondrial accumulation, and postinfarction ventricular dysfunction (79). It can be inferred that cardiac resident CCR2– macrophages may engulf CMs undergoing ferroptosis and reduce the inflammation to inhibit ferroptosis. Subsequently, the circulating CCR2+ mononuclear macrophages infiltrate into the myocardial injury environment in greater number than the CCR2– macrophages and polarize to M1 pro-inflammatory phenotype and M2 reparative phenotype (80). Iron overload promotes polarization of M1 macrophages (81). M1 macrophages are more resistant than M2 macrophages to ferroptosis due to high intracellular levels of inducible nitric oxide synthase and nitric oxide radicals which inhibit lipid peroxidation (82, 83). M1 macrophages secrete large amounts of pro-inflammatory factors such as IL-1β, TNF, IL-12, and IL-18, increase ROS and have the potential to induce ferroptosis in adventitial hypoxic CMs (84, 85). Besides, exosomes derived from M1 macrophages in obese adipose tissue carry miR-140-5p to induce ferroptosis in CMs by targeting xCT to inhibit GSH synthesis, leading to abnormal left ventricular contraction in obese mice (86). While it has been shown that M2 macrophage-derived exosomes inhibit cell ferroptosis (87). Hence, M1 macrophages may be a key inducer of ferroptosis in the microenvironment of myocardial inflammation. Future studies are needed to prove the effects of macrophages on the onset of ferroptosis in CMs.
Infiltrating T cells activated by dendritic cells (DCs) may also regulate the occurrence of ferroptosis in CVDs. Cytotoxic CD8+ T cells can lead to poor cardiac remodeling after ischemia, acute hypertensive heart injury, acute rejection of allogeneic heart retransplantation and other CVDs, possibly by inducing CMs ferroptosis through releasing IFN-γ and promoting M1 polarization (88). For CD4+ effector T cells, the effects of different subtypes are pleiotropic. T helper (Th)1 cells could activate cardiac fibroblasts to induce fibrosis; while regulatory Treg cells could reduce the number of myofibroblasts through inhibiting inflammation via anti-inflammatory cytokine IL-10 in hypertension and HF (89–91). Additionally, Treg cells beneficially influenced wound healing after MI by regulating the polarization of M2 macrophages and inhibiting proinflammatory cytokines production (92). Paracrine action of Treg cells also promoted fetal and maternal CMs proliferation to improve the prognosis of MI during pregnancy (93). Thus, Treg cells may alleviate cellular ferroptosis in the myocardial microenvironment by interacting with CMs and inflammatory cells.
As the Figure 4 shows that under aerobic circumstances, glucose is initially transformed into pyruvate through the glycolytic pathway and subsequently undergoes the TCA cycle and mitochondrial OXPHOS processes to complete metabolism (94, 95). At the initial stage of glycolysis, the glucose molecule is phosphorylated by hexokinase to produce glucose 6-phosphate, which is then further converted to fructose 6-phosphate by glucose 6-phosphate isomerase, to subsequently produce fructose 1,6-diphosphate catalyzed by fructokinase 6-phosphate kinase 1, the main rate-limiting enzyme in the glycolytic process. Simultaneously, fructose 6-phosphate is bypassed by glucose-6-phosphate dehydrogenase and enter the pentose phosphate pathway to produce the essential biosynthetic precursor and NADPH, 60% of which required in animals is produced in this manner. NADPH participates in multiple anabolic reactions, responsible for the recycling of GSH, ubiquinone and Trx, etc., which is crucial for ensuring the redox homeostasis in the body (96). Pyruvate produced by glycolysis enters the mitochondria to generate acetyl-CoA, which enters the TCA cycle to generate NADH, FADH2 coupled with OXPHOS. Mitochondrial OXPHOS is accompanied by the transfer of electrons in the ETC to molecular oxygen, leading to the generation of ROS. Therefore, it can be speculated that glucose metabolism in CMs is closely linked to ferroptosis. Echoing the above hypothesis, appropriate blockade of glycolysis or TCA significantly inhibited ferroptosis induced by erastin, cystine depletion or RSL3 treatment, as manifested by stabilizing mitochondrial membrane potential and decreasing lipid peroxides accumulation (97, 98). According to the metabolomic analysis, CMs exposed to the ferroptosis inducer RSL3 showed markedly elevated isocitric acid and ketoglutarate (KG) contents (99). Accumulation of some TCA circulating metabolites, alpha-KG, succinate, fumarate and malate, can all contribute to lipid ROS accumulation and ferroptosis. Additionally, alpha-KG dehydrogenase/succinate/aconitase activity was enhanced during the onset of ferroptosis due to cysteine deprivation (97, 100). In the TCA cycle, glutamine can be catabolized to glutamate by glutaminase, which is further metabolized to alpha-KG for replenishment. The glutamine-fueled intracellular metabolic pathway is required for ferroptosis induced by cysteine deprivation (101). Inhibition of glutamine catabolism reduces cardiac injury triggered by I/R, offering a prospective therapy (102). These results suggest that glucose metabolism products enhance the mitochondrial ROS generation, further amplifying the ferroptosis signal.
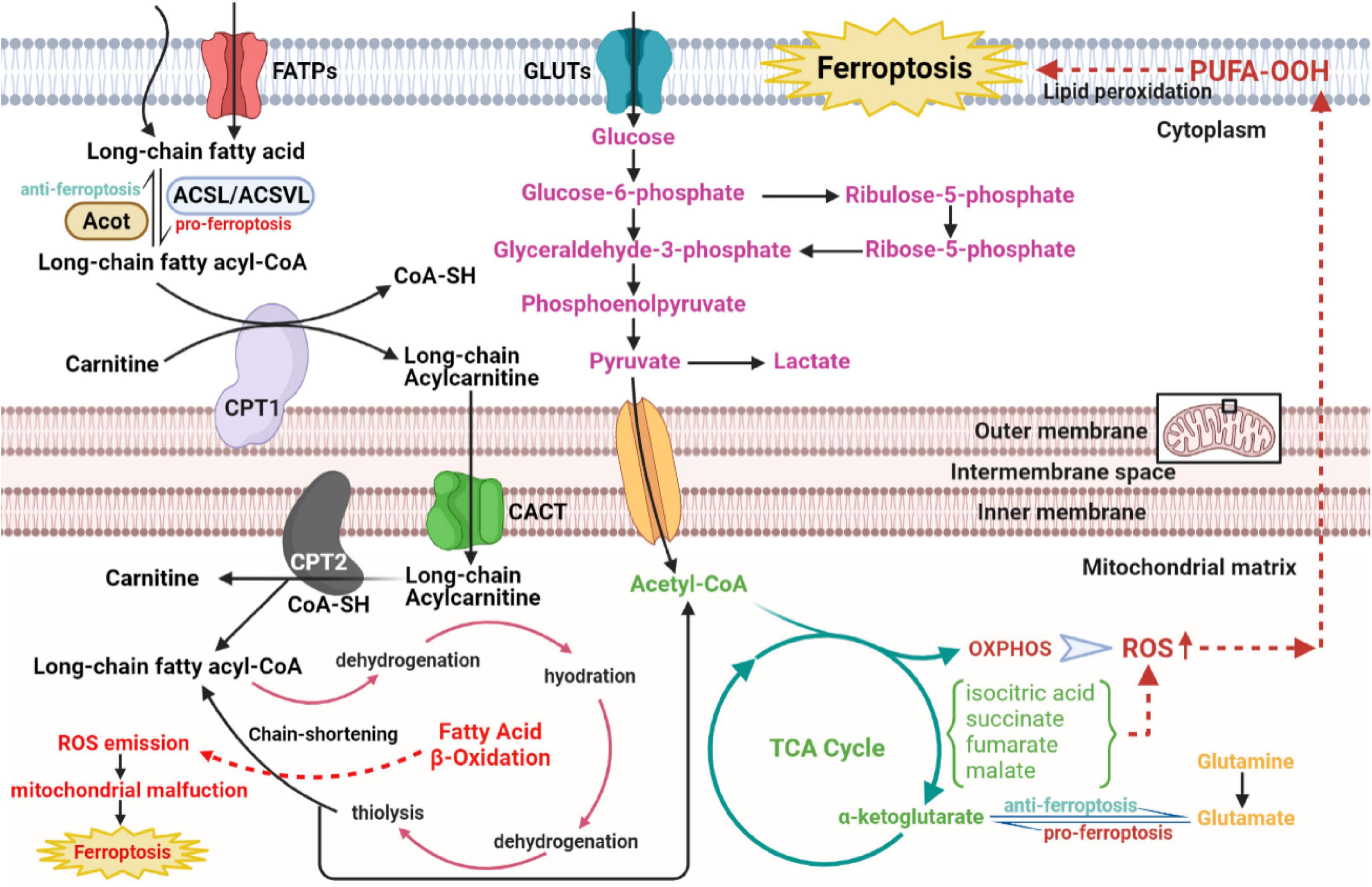
Figure 4. The role of glucose and fatty acid metabolic disorders in the regulatory network of ferroptosis of CVDs. The long-chain fatty acid is activated and transformed into long-chain fatty acyl-CoA with the catalytic activity of ACSL/ACSVL. The long-chain fatty acyl-CoA combines with carnitine and enters fatty acid β-oxidation in the mitochondrial matrix with help of CPT1, CPT2, and CACT. Then acetyl-CoA from the β-oxidation cycle, anaerobic glycolysis, and glutaminolysis gather to strengthen the metabolism of the TCA cycle in mitochondrial matrix, which activates downstream OXPHOS and ROS, eventually resulting in cellular ferroptosis. CVDs, cardiovascular diseases; ACSL, long-chain acyl-CoA synthases; ACSVL, very long-chain acyl-CoA synthases; CoA, coenzyme A; CPT1, carnitine palmitoyltransferase1; CPT2, carnitine palmitoyltransferase2; CACT, carnitine-acylcarnitine translocase; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species.
Given that fatty acids are the core energy source for the myocardium, concerns about lipid metabolism have crucial implications in ferroptosis from CVDs (Figure 4). According to the basic perspectives on fatty acid metabolism, three stages are involved in this process, named activation, translocation and β-oxidation. Some biomolecule evolved in the above three stages, either metabolites or relative enzymes, can spark cellular ferroptosis. As the first reaction of fatty acid catabolism, long-chain acyl-CoA synthetases (ACSLs) and very long-chain acyl-CoA synthetases (ACSVLs) converts different kinds of long-chain fatty acid to corresponding fatty acyl-CoA. Among these enzymes, ACSL4 mostly activates the long-chain fatty acids and subsequently enriches cellular membrane with polyunsaturated ones. Specifically, two long-chain fatty acids, arachidonic acid, and adrenoic acid, can be both converted to arachidonoyl-CoA and adrenoyl-CoA via ACSL4 and coenzyme A. Following the activation of long-chain fatty acids, arachidonoyl-CoA and adrenoyl-CoA are used to synthesize phosphatidylethanolamine and phosphatidylinositol, composing negatively charged cell membrane. Phosphatidylethanolamine can often be oxidized, triggering the ferroptosis of cells, particularly under the treatment with RSL3 (103). Consistent with the above findings, ACSL4, iron and MDA level in reperfusion myocardium gradually rose with the prolongation of myocardial ischemic time (104). Serving as the functionally well-matched adversary to ACSLs, Acyl-CoA thioesterases (ACOTs) accelerate fatty acid synthesis with the substrate fatty acyl-CoA, and the balance between ACSLs and ACOTs governs the dynamic equilibrium of fatty acid metabolism. As reported in cardiovascular basic researches, ACOT1, an isoform of ACOTs working in cytoplasm, hydrolyzes fatty acyl-CoAs, varying from short-chain ones to very long-chain ones, to free fatty acids and conenzyme A in myocardium (105, 106). Under the condition of doxorubicin-induced cardiomyopathy, Acot1 knockdown sensitizes CMs to ferroptosis, whereas ACOT1 overexpression poses defensive effect on cellular ferroptosis. This beneficial effect is conducted through ACOTs’ function of lipid components alteration and acyl-CoA hydrolysis, evidenced by increased anti-oxidative ω-3 polyunsaturated fatty acids and docosahexaenoic acid abundance in the hearts of ACOT1-overexpressed mice (107). Meanwhile, in the stage of fatty acid activation, the adipokine chemerin deriving from adipose tissue inhibits fatty acid oxidation and maintains fatty acid levels to confer ferroptosis resistance. Chemerin inhibition shuts down the anti-oxidative CoQ system, followed by ROS explosion, additionally, its deficiency remarkably attenuates glycerophospholipid species accumulation, including phosphatidic acid, phosphatidylcholine, phosphatidylglycerol and intact sphingolipids, with an immediate increase in the replenishment of their corresponding oxidative and catabolic products (108, 109). Subsequent to the translocation stage, the substrate, fatty acyl-CoA, enters the β-oxidation cycle in mitochondrial matrix. After every four steps of β-oxidation, the carbon chain of acyl-CoA is shortened by two carbons, with acetyl-CoA released. Then, explosive acetyl-CoA production from fatty acid β-oxidation accelerates downstream TCA cycle and excites ROS-induced ferroptosis. Hence, ideal component remodeling and maintenance of free fatty acids metabolic balance are predicted to improve the sensitivity of CMs to ferroptosis.
The balance of iron homeostasis in the body depends on the metabolic regulation of iron absorption, recycling and release. The absorption of dietary iron into the blood is regulated by DMT1 of intestinal epithelial cells. Iron recycling is obtained from senescent red blood cells phagocytosed by reticuloendothelial macrophages. Ferroportin (FP) is the only known mammalian iron exporter capable of releasing iron into the circulation. Hepcidin, a liver-derived hormone, can degrade FP and reduce the release of iron to the circulation. Therefore, in addition to nutritional iron deficiency, the disorders of the process mentioned above such as FP mutation and abnormal hepcidin levels (affected by inflammation, hypoxia, hemochromatosis protein, TfR, and other factors) can lead to the imbalance of iron homeostasis in the body (110).
At the level of individual CMs, the metabolic mechanisms maintaining iron homeostasis are mainly illustrated in Figure 5, associated with CMs ferroptosis. The uptake and transport of iron in the CMs mainly depend on TfR1, which takes in the Fe3+-transferrin (Tf) complex into the cytoplasm via endocytosis, and finally Fe3+ mainly stores in the form of ferritin and hemosiderin. Ferritin controls the distribution and storage of iron in cells, preventing iron-catalyzed Fenton reaction and free radical production. Ferritin consists of 24 subunits, including ferritin light chain (Ftl), and ferritin heavy chain (Fth), the latter with ferrous oxidase activity can oxidize poly(C)-binding protein (PCBP)-carried Fe2+ to Fe3+. In ferritin knock-out mice, ferroptosis induced by decreased xCT expression in CMs predisposed the mice to cardiomyopathy (9). BTB and CNC Homology 1 (BACH1) is a heme-binding transcription factor required for the appropriate regulation of oxidative stress responses and metabolic pathways associated with heme and iron. BACH1 inhibited Fth1 and Ftl1 encoding ferritin, exacerbating MI by promoting ferroptosis (111). Degradation of ferritin, especially Fth1, through nuclear receptor coactivator 4 (NCOA4) –mediated ferritinophagy resulted in the release of ferritin-bound iron to replenish free Fe2+ (112). Inhibition of NCOA4-mediated ferritinophagy contributed to myocardial hypertrophy through mitochondrial iron overload (113). Regarding Fe2+, it can be taken up into the CMs by DMT1. Additionally, Fe2+ can be transformed by Fe3+ deoxygenation under the catalysis of the iron oxide reductase six-transmembrane epithelial antigen of the prostate 3 (STEAP3), followed by releasing from the endosome mediated by DMT1 into the ferroptosis-associated unstable iron pool in the cytoplasm. Furthermore, loss of cardiac iron exporter FP1 rapidly caused an eventual fatal impairment of cardiac function in mice, which is associated with intracellular iron deposition in the myocardium (114). Salvia miltiorrhiza injection decreased cardiac iron deposition and inhibited cardiac oxidation by down-regulating DMT1 and TfR1 expression and up-regulating FP1 protein levels (115). The above evidence suggests that reducing cardiac iron uptake and increasing iron excretion is one of the essential mechanisms for enhancing cardiac function.
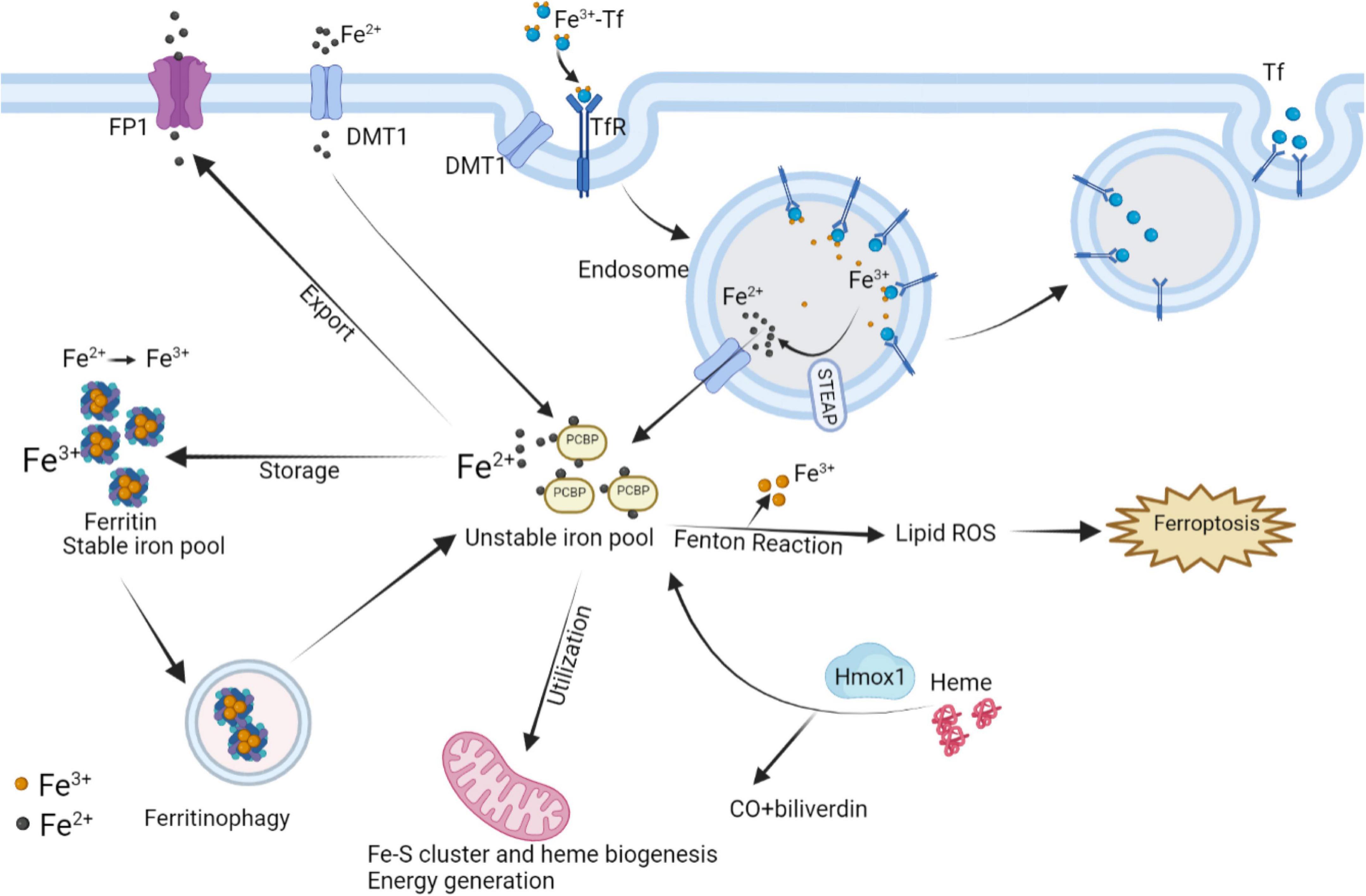
Figure 5. Interaction of intracellular iron metabolism and ferroptosis. Fe3+ and Tf bind to form Fe3+-Tf complex and enter the cell by means of TfR1 via endocytosis. In endosomes, Fe3+ is liberated from Tf and catalyzed by STEAP to Fe2+, while Tf is released extracellularly by exocytosis. The process Fe2+ released from endosomes or absorbed into the cells depends on DMT1. In mitochondria, the utilization of Fe2+ is mainly the synthesis of Fe–S clusters and heme, which can be degraded by Hmox1 to regenerate Fe2+. Excess Fe2+ can store in ferritin by converting into Fe3+, while ferritinophagy can re-release free iron. Intracellular Fe2+ can also be exported through FP1. The above processes participate in the regulation of intracellular Fe2+ content to control Fenton reaction, thereby regulating the occurrence of ferroptosis. FP1, ferroportin1; DMT1, divalent metal transporter 1; Tf, transferrin; TfR1, transferrin receptor 1; Hmox1, Heme oxygenase 1; STEAP3, six-transmembrane epithelial antigen of the prostate 3.
Heme oxygenase 1 (Hmox1) is a rate-limiting enzyme in the metabolism of the iron porphyrin compound heme, which is broken down into carbon monoxide, biliverdin, and Fe2+. The myocardium is rich in heme for the synthesis of myoglobin, cytochrome, etc., therefore, Hmox1 plays an important role in the regulation of myocardial iron metabolism. Hmox1 mediates ferroptosis induced by the release of Fe2+ from heme and is responsible for adriamycin-induced cardiotoxicity or cardiac injury with sickle cell disease in mice (7, 116). While Hmox1 also regulates intracellular oxidation levels by stabilizing hypoxia-inducible factor 1α to prevent ischemia-mediated injury (117). Consequently, excessive up-regulation of Hmox1 is toxic, while moderate up-regulation of Hmox1 may have a positive cytoprotective effect on the myocardium.
Apart from serving as the primary organelle for intracellular energy generation, mitochondria also play the central role in utilization, catabolism, and anabolisms for iron. Cellular iron homeostasis and mitochondrial homeostasis are interdependent. Mitochondria must import and incorporate iron ions to form iron–sulfur clusters, heme, and certain mitochondrial proteins that underpin cellular respiration (Figure 5) (118–120). While the deficiency in mitochondrial iron-sulfur cluster biosynthesis also triggered intracellular uptake and redistribution of iron (121). The absence of certain RNA-binding proteins in mitochondria can disrupt cellular iron homeostasis and lead to apoptosis (122). Therefore, the following sections elaborated on the association between mitochondria and ferroptosis in CVDs.
Mitochondrial morphology is an essential judge of cell death. They are generally short rod-shaped or spherical, 1–2 μm long, and consist of double membranes, an outer membrane and an inner membrane with a cristae structure. Ferroptosis is correlated with significant morphological alterations in the mitochondria. After the onset of the ferroptotic stimulus, fragmentation, condensed membrane density, lower volume, reduced or absent cristae and rupture of the outer membrane shortly occurs in the mitochondria (3, 123, 124). The tendency for reduction in mitochondrial membrane potential leads to mitochondrial dysfunction, as evidenced by increased ROS production, mitochondrial membrane hyperpolarization and mitochondrial swelling, ultimately leading to hypertrophy of CMs (113, 125–127). Both Fe3+ and Fe2+ overload can potentially cause mitochondrial disorders as described above, but Fe2+ has been proven to have more destructive effects than Fe3+ (128). Tadokoro et al. (129) revealed that excessive accumulation of free Fe2+ in mitochondria of CMs in mice with doxorubicin cardiomyopathy or I/R-induced HF, and that chelating Fe2+ in mitochondria prevented ferroptosis in CMs. Hence, mitochondrial damage due to Fe2+ overload was the main mechanism of cardiac injury (129). On the inner membrane of the mitochondria, the respiratory chain carries out electron transfer via four protein complexes (I, II, III, and IV) to produce ATP, which is the most basic energy metabolism in the organism. Chronic iron overload could additionally inflict damage on mitochondrial DNA and impair the synthesis of respiratory chain subunit complexes I and IV. These results explained chronic progressive cardiac hypertrophy and cardiac insufficiency (130). Treatment with the ferroptosis inhibitor liproxstatin-1 reduced myocardial infarcted size in mice by controlling the entry and exit of mitochondrial ROS via diminishing VDAC1, a pore-like protein in the outer mitochondrial membrane (131). Thus, ferroptosis accounts for impaired cardiac function by causing deterioration of mitochondrial function and interfering with mitochondrial dynamics.
Mitochondrial metabolism in the pathological state actively facilitates the generation of lipid ROS, and remarkably increases the level of loosely bound unstable iron ions, which is a prerequisite for ferroptosis (132). Mitochondria contains an abundance of GSH, accounting for about 10–15% of total intracellular GSH. Two carrier proteins, mitochondrial dicarboxylate and 2-oxoglutarate, have been identified as GSH transporters (133). Inhibition of these two carrier proteins resulted in the depletion of GSH and ultimately ferroptosis in CMs. In detail, when GSH was depleted rapidly, hyperpolarization of the mitochondrial membrane potential caused the release of cytochrome C and the activation of caspase, thus explaining the role of these two carriers in regulating ferroptosis (97, 134). In mitochondria, there are also organic compounds mitigating ferroptosis. Dihydrolipoic acid acts as an essential cofactor for several mitochondrial enzyme complexes, allowing the direct reduction of pro-ferroptotic peroxidized phospholipids to hydroxyphospholipids and scavenging oxygen radicals (135). It is speculated that more effective pharmacological interventions to protect mitochondria could be new therapeutic targets, improving clinical outcomes in the treatment of CVDs.
Mitophagy is a process that selectively scavenges damaged mitochondria and maintains normal mitochondrial activity. Specifically, the serine/threonine kinase PTEN-inducible kinase 1 (PINK1) and the E3 ubiquitin-protein ligase Parkin synergistically sense the functional state of mitochondria and label damaged mitochondria. The damaged mitochondria are encapsulated into autophagosomes and fused with lysosomes, thus completing mitochondrial degradation. Mitophagy was detected to protect against the development of HF, myocardial I/R injury, and obese cardiomyopathy through the Pink1/Parkin pathway (136–138). Mitophagy receptor deficiency can lead to cardiac remodeling and dysfunction through ACSL4-mediated activation of ferroptosis (134). However, overactivation of mitophagy may also induced HF by activating TLR4/NADPH oxidase 4 pathway (8), and mitophagy was also reported to trigger necroptosis and ferroptosis in melanoma cells (139). Therefore, mitophagy has diverse features under different conditions and the regulation of ferroptosis in CMs mediated by mitophagy may need to be further explored.
Several studies have come to the opposite conclusion that mitochondria are dispensable for the execution of ferroptosis induced by GPX4 inhibition, and mitochondria-deficient cell lines show no difference in the susceptibility of ferroptosis from control (3). Possible explanations are that firstly, once GPX4 was eliminated, small amounts of •O2–, •OH, and H2O2 will be quickly magnified by Fenton reaction, causing full-scale ferroptosis whatever the activity of mitochondria, and secondly, there may be some differences in metabolism between cell lines, with varying degree of dependence on mitochondria.
Rapid advances in current evidence-based medicine for the treatment of CVDs firmly support the effectiveness of several drugs, such as angiotensin-converting enzyme inhibitors, beta-blockers and aldosterone receptor antagonists. Nevertheless, certain adverse side effect of these drugs has caused the administration restricted in specific patients. Recently, targeting iron homeostasis and ferroptosis has become a new direction for clinical treatment of CVDs. Two randomized clinical trials (FAIR-HF and Confirm-HF) have provided clear evidence for the benefit of iron supplementation in patients with chronic HF and iron deficiency. Although oral iron supplementation has no apparent effect, intravenous ferric carboxymaltose can improve hospitalization and mortality from cardiovascular events in iron-deficient HFrEF (LVEF ≤ 45%) patients (140). In terms of the secondary prevention of CVDs, a meta-analysis failed to identify participants more likely to derive clinical benefits from iron nutritional supplements in reducing the risk of cardiovascular outcomes (141). The benefit of iron supplementation in patients of acute CVDs without iron deficiency is unclear, and this treatment may exacerbate (I/R) injury. Conversely, administration of deferoxamine which chelates redox-active iron to inhibit ferroptosis has been shown to improve blood oxidative stress markers as an adjunctive therapy for patients with ST-segment elevation MI (142). Intravenous infusion of deferoxamine mesylate at the initiation of thrombolysis was also effective in ischemic stroke patients, with no apparent adverse effects (143). Therefore, pharmacological treatment to maintain iron homeostasis and inhibit ferroptosis may serve as a new therapeutic strategy for CVDs.
Additionally, although indicators of ferroptosis have predictive capacity on the prognosis of amyotrophic lateral sclerosis (144), ferroptosis biomarkers for predicting early diagnosis and prognosis of CVDs have not yet been reported clinically. Classic CVD-related ferroptosis biomarkers validated in animal models include GPX4, ROS, 4-HNE, COX-2, and iron levels, but there are still some limitations of detection (35, 45, 86). First, the activity of ferroptosis metabolites is time-sensitive and should be detected in time after the onset of CVDs. Second, oxidative stress also occurs during necrosis and apoptosis, and simple ROS detection cannot explain the occurrence of CM ferroptosis. Third, the great challenge for COX-2 as a biomarker of ferroptosis is that upregulation of COX-2 has been observed under a variety of inflammatory conditions, except in the case of myocardial ferroptosis. Finally, iron levels may not represent the level of ferroptosis and need to be measured in combination with other indicators. Other potential ferroptosis biomarkers associated with CVDs can be referred to Table 1. A recent bioinformatics analysis also determined elevated key genes associated with MI, ferroptosis, and hypoxia, such as Atf3, Socs3, Hspa1b, Cxcl2, and Myd88, which could serve as new biomarkers for MI (145). Thus, indicators of pathophysiology based on ferroptosis may have predictive potential for CVDs in the future, providing better stratification for patient personalized care and resource allocation.
In summary, ferroptosis-related pathological mechanisms such as oxidative stress, inflammation, metabolic disorders and mitochondrial damage are involved in the development of CVDs. As research proceeds, inhibition of ferroptosis may be an effective strategy for the treatment of CVDs.
L-LZ and R-JT determined the topic, wrote the initial manuscript, searched the related literatures, and created the figures of the manuscript. Y-JY supervised the planning and execution of the research activity. All authors have approved to the final version of the manuscript, responsible for the accuracy, and authenticity of the article.
This study was supported by the National Key Research and Development Program (No. 2017YFC1700503) and the National Natural Science Foundation of China (Nos. 81573957, 81874461, and 82070307).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. (1981) 1:1293–4. doi: 10.1016/s0140-6736(81)92463-6
2. Oudit GY, Trivieri MG, Khaper N, Husain T, Wilson GJ, Liu P, et al. Taurine supplementation reduces oxidative stress and improves cardiovascular function in an iron-overload murine model. Circulation. (2004) 109:1877–85. doi: 10.1161/01.Cir.0000124229.40424.80
3. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
4. Liu B, Zhao C, Li H, Chen X, Ding Y, Xu S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem Biophys Res Commun. (2018) 497:233–40. doi: 10.1016/j.bbrc.2018.02.061
5. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med. (2020) 160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026
6. Wang XD, Kang S. Ferroptosis in myocardial infarction: Not a marker but a maker. Open Biol. (2021) 11:200367. doi: 10.1098/rsob.200367
7. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U.S.A. (2019) 116:2672–80. doi: 10.1073/pnas.1821022116
8. Chen X, Xu S, Zhao C, Liu B. Role of Tlr4/Nadph oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun. (2019) 516:37–43. doi: 10.1016/j.bbrc.2019.06.015
9. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of cardiac ferritin h facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. (2020) 127:486–501. doi: 10.1161/circresaha.120.316509
10. Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res. (2019) 14:532–41. doi: 10.4103/1673-5374.245480
11. Mu M, Wang Y, Zhao S, Li X, Fan R, Mei L, et al. Engineering a Ph/glutathione-responsive tea polyphenol nanodevice as an apoptosis/ferroptosis-inducing agent. ACS Appl Bio Mater. (2020) 3:4128–38. doi: 10.1021/acsabm.0c00225
12. Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. (2018) 22:3826–36. doi: 10.26355/eurrev_201806_15267
13. Zhang P, Chen L, Zhao Q, Du X, Bi M, Li Y, et al. Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Radic Biol Med. (2020) 152:227–34. doi: 10.1016/j.freeradbiomed.2020.03.015
14. Ishihara G, Kawamoto K, Komori N, Ishibashi T. Molecular hydrogen suppresses superoxide generation in the mitochondrial complex I and reduced mitochondrial membrane potential. Biochem Biophys Res Commun. (2020) 522:965–70. doi: 10.1016/j.bbrc.2019.11.135
15. Winterbourn CC. Toxicity of iron and hydrogen peroxide: The fenton reaction. Toxicol Lett. (1995) 82-83:969–74. doi: 10.1016/0378-4274(95)03532-x
16. Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W, et al. Fenton-reaction-acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano. (2018) 12:11355–65. doi: 10.1021/acsnano.8b06201
17. Liu T, Liu W, Zhang M, Yu W, Gao F, Li C, et al. Ferrous-supply-regeneration nanoengineering for cancer-cell-specific ferroptosis in combination with imaging-guided photodynamic therapy. ACS Nano. (2018) 12:12181–92. doi: 10.1021/acsnano.8b05860
18. Park E, Chung SW. Ros-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. (2019) 10:822. doi: 10.1038/s41419-019-2064-5
19. Liu M, Fan Y, Li D, Han B, Meng Y, Chen F, et al. Ferroptosis inducer erastin sensitizes nsclc cells to celastrol through activation of the ros-mitochondrial fission-mitophagy axis. Mol Oncol. (2021) 15:2084–105. doi: 10.1002/1878-0261.12936
20. Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. (1982) 710:197–211. doi: 10.1016/0005-2760(82)90150-3
21. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a P53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
22. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to Gpx4 inhibition. Nature. (2017) 551:247–50. doi: 10.1038/nature24297
23. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The coq oxidoreductase Fsp1 acts parallel to Gpx4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
24. Jo A, Bae JH, Yoon YJ, Chung TH, Lee EW, Kim YH, et al. Plasma-activated medium induces ferroptosis by depleting Fsp1 in human lung cancer cells. Cell Death Dis. (2022) 13:212. doi: 10.1038/s41419-022-04660-9
25. Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Niño MD, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced aki. J Am Soc Nephrol. (2017) 28:218–29. doi: 10.1681/asn.2015121376
26. Kiesel L, Przylipiak A, Rabe T, Przylipiak M, Runnebaum B. Arachidonic acid and its lipoxygenase metabolites stimulate prolactin release in superfused pituitary cells. Hum Reprod. (1987) 2:281–5. doi: 10.1093/oxfordjournals.humrep.a136535
27. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. Ros and ros-mediated cellular signaling. Oxid Med Cell Longev. (2016) 2016:4350965. doi: 10.1155/2016/4350965
28. Lillo-Moya J, Rojas-Solé C, Muñoz-Salamanca D, Panieri E, Saso L, Rodrigo R. Targeting ferroptosis against ischemia/reperfusion cardiac injury. Antioxidants. (2021) 10:667. doi: 10.3390/antiox10050667
29. Gasparetto C, Malinverno A, Culacciati D, Gritti D, Prosperini PG, Specchia G, et al. Antioxidant vitamins reduce oxidative stress and ventricular remodeling in patients with acute myocardial infarction. Int J Immunopathol Pharmacol. (2005) 18:487–96. doi: 10.1177/039463200501800308
30. Venardos KM, Perkins A, Headrick J, Kaye DM. Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: A review. Curr Med Chem. (2007) 14:1539–49. doi: 10.2174/092986707780831078
31. Neves AL, Mohammedi K, Emery N, Roussel R, Fumeron F, Marre M, et al. Allelic variations in superoxide dismutase-1 (Sod1) gene and renal and cardiovascular morbidity and mortality in type 2 diabetic subjects. Mol Genet Metab. (2012) 106:359–65. doi: 10.1016/j.ymgme.2012.04.023
32. Wang T, Tomas D, Perera ND, Cuic B, Luikinga S, Viden A, et al. Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis. Cell Death Differ. (2021) 29:1187–98. doi: 10.1038/s41418-021-00910-z
33. Seshadri G, Sy JC, Brown M, Dikalov S, Yang SC, Murthy N, et al. The delivery of superoxide dismutase encapsulated in polyketal microparticles to rat myocardium and protection from myocardial ischemia-reperfusion injury. Biomaterials. (2010) 31:1372–9. doi: 10.1016/j.biomaterials.2009.10.045
34. Chang WT, Bow YD, Fu PJ, Li CY, Wu CY, Chang YH, et al. A marine terpenoid, heteronemin, induces both the apoptosis and ferroptosis of hepatocellular carcinoma cells and involves the ros and mapk pathways. Oxid Med Cell Longev. (2021) 2021:7689045. doi: 10.1155/2021/7689045
35. Sharma S, Bhattarai S, Ara H, Sun G, St Clair DK, Bhuiyan MS, et al. Sod2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox Biol. (2020) 37:101740. doi: 10.1016/j.redox.2020.101740
36. Decharatchakul N, Settasatian C, Settasatian N, Komanasin N, Kukongviriyapan U, Intharapetch P, et al. Association of combined genetic variations in Sod3, Gpx3, Pon1, and Gstt1 with hypertension and severity of coronary artery disease. Heart Vessels. (2020) 35:918–29. doi: 10.1007/s00380-020-01564-6
37. Chen T, Jin X, Crawford BH, Cheng H, Saafir TB, Wagner MB, et al. Cardioprotection from oxidative stress in the newborn heart by activation of Pparγ is mediated by catalase. Free Radic Biol Med. (2012) 53:208–15. doi: 10.1016/j.freeradbiomed.2012.05.014
38. Hwang JS, Kim E, Lee HG, Lee WJ, Won JP, Hur J, et al. Peroxisome proliferator-activated receptor δ rescues xct-deficient cells from ferroptosis by targeting peroxisomes. Biomed Pharmacother. (2021) 143:112223. doi: 10.1016/j.biopha.2021.112223
39. Liu X, Qi K, Gong Y, Long X, Zhu S, Lu F, et al. Ferulic acid alleviates myocardial ischemia reperfusion injury via upregulating Ampkα2 expression-mediated ferroptosis depression. J Cardiovasc Pharmacol. (2021) 79:489–500. doi: 10.1097/fjc.0000000000001199
40. Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW, et al. Quantitative proteomic analyses reveal that Gpx4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. (2019) 10:835. doi: 10.1038/s41419-019-2061-8
41. Wang Y, Yan S, Liu X, Deng F, Wang P, Yang L, et al. Prmt4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/Gpx4 pathway. Cell Death Differ. (2022). [Onlie ahead of print].
42. Xu S, Wu B, Zhong B, Lin L, Ding Y, Jin X, et al. Naringenin alleviates myocardial ischemia/reperfusion injury by regulating the nuclear factor-erythroid factor 2-related factor 2 (Nrf2) /System Xc-/ glutathione peroxidase 4 (Gpx4) axis to inhibit ferroptosis. Bioengineered. (2021) 12:10924–34. doi: 10.1080/21655979.2021.1995994
43. Zhang Z, Tang J, Song J, Xie M, Liu Y, Dong Z, et al. Elabela Alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the Il-6/Stat3/Gpx4 signaling. Free Radic Biol Med. (2022) 181:130–42. doi: 10.1016/j.freeradbiomed.2022.01.020
44. Wang Y, Kuang X, Yin Y, Han N, Chang L, Wang H, et al. Tongxinluo prevents chronic obstructive pulmonary disease complicated with atherosclerosis by inhibiting ferroptosis and protecting against pulmonary microvascular barrier dysfunction. Biomed Pharmacother. (2022) 145:112367. doi: 10.1016/j.biopha.2021.112367
45. Lin JH, Yang KT, Lee WS, Ting PC, Luo YP, Lin DJ, et al. Xanthohumol protects the rat myocardium against ischemia/reperfusion injury-induced ferroptosis. Oxid Med Cell Longev. (2022) 2022:9523491. doi: 10.1155/2022/9523491
46. Yang Y, Sun S, Xu W, Zhang Y, Yang R, Ma K, et al. Piperlongumine Inhibits thioredoxin reductase 1 by targeting selenocysteine residues and sensitizes cancer cells to erastin. Antioxidants. (2022) 11:710. doi: 10.3390/antiox11040710
47. Kiermayer C, Northrup E, Schrewe A, Walch A, de Angelis MH, Schoensiegel F, et al. Heart-specific knockout of the mitochondrial thioredoxin reductase (Txnrd2) induces metabolic and contractile dysfunction in the aging myocardium. J Am Heart Assoc. (2015) 4:e002153. doi: 10.1161/jaha.115.002153
48. Bai L, Yan F, Deng R, Gu R, Zhang X, Bai J. Thioredoxin-1 rescues Mpp(+)/Mptp-induced ferroptosis by increasing glutathione peroxidase 4. Mol Neurobiol. (2021) 58:3187–97. doi: 10.1007/s12035-021-02320-1
49. Oka SI, Chin A, Park JY, Ikeda S, Mizushima W, Ralda G, et al. Thioredoxin-1 maintains mitochondrial function via mechanistic target of rapamycin signalling in the heart. Cardiovasc Res. (2020) 116:1742–55. doi: 10.1093/cvr/cvz251
50. Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. (2014) 66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036
52. Hu Q, Zhang Y, Lou H, Ou Z, Liu J, Duan W, et al. Gpx4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. (2021) 12:706. doi: 10.1038/s41419-021-04008-9
53. Wallert M, Ziegler M, Wang X, Maluenda A, Xu X, Yap ML, et al. A -tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. (2019) 26:101292. doi: 10.1016/j.redox.2019.101292
54. Keith ME, Jeejeebhoy KN, Langer A, Kurian R, Barr A, O’Kelly B, et al. A controlled clinical trial of vitamin E supplementation in patients with congestive heart failure. Am J Clin Nutr. (2001) 73:219–24. doi: 10.1093/ajcn/73.2.219
55. Gong T, Liu L, Jiang W, Zhou R. Damp-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
56. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of hmgb1 in ferroptosis. Biochem Biophys Res Commun. (2019) 510:278–83. doi: 10.1016/j.bbrc.2019.01.090
57. Fan H, Tang HB, Chen Z, Wang HQ, Zhang L, Jiang Y, et al. Inhibiting Hmgb1-rage axis prevents pro-inflammatory macrophages/microglia polarization and affords neuroprotection after spinal cord injury. J Neuroinflammation. (2020) 17:295. doi: 10.1186/s12974-020-01973-4
58. Foglio E, Pellegrini L, Germani A, Russo MA, Limana F. Hmgb1-mediated apoptosis and autophagy in ischemic heart diseases. Vasc Biol. (2019) 1:H89–96. doi: 10.1530/vb-19-0013
59. Xiao N, Zhang J, Chen C, Wan Y, Wang N, Yang J. Mir-129-5p improves cardiac function in rats with chronic heart failure through targeting Hmgb1. Mamm Genome. (2019) 30:276–88. doi: 10.1007/s00335-019-09817-0
60. Son S, Yoon SH, Chae BJ, Hwang I, Shim DW, Choe YH, et al. Neutrophils facilitate prolonged inflammasome response in the damp-rich inflammatory milieu. Front Immunol. (2021) 12:746032. doi: 10.3389/fimmu.2021.746032
61. Biasizzo M, Kopitar-Jerala N. Interplay between Nlrp3 inflammasome and autophagy. Front Immunol. (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
62. Quagliariello V, De Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A, et al. The Sglt-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. (2021) 20:150. doi: 10.1186/s12933-021-01346-y
63. Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and Tlr4/trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. (2019) 129:2293–304. doi: 10.1172/jci126428
64. Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. (2009) 81:457–64. doi: 10.1093/cvr/cvn335
65. Siwik DA, Colucci WS. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev. (2004) 9:43–51. doi: 10.1023/b:Hrev.0000011393.40674.13
66. Peng Y, Yang Q, Gao S, Liu Z, Kong W, Bian X, et al. Il-6 protects cardiomyocytes from oxidative stress at the early stage of lps-induced sepsis. Biochem Biophys Res Commun. (2022) 603:144–52. doi: 10.1016/j.bbrc.2022.03.013
67. Emran T, Chowdhury NI, Sarker M, Bepari AK, Hossain M, Rahman GMS, et al. L-carnitine protects cardiac damage by reducing oxidative stress and inflammatory response via inhibition of tumor necrosis factor-alpha and interleukin-1beta against isoproterenol-induced myocardial infarction. Biomed Pharmacother. (2021) 143:112139. doi: 10.1016/j.biopha.2021.112139
68. Li S, He Y, Chen K, Sun J, Zhang L, He Y, et al. Rsl3 drives ferroptosis through Nf-K b pathway activation and Gpx4 depletion in glioblastoma. Oxid Med Cell Longev. (2021) 2021:2915019. doi: 10.1155/2021/2915019
69. Zhao Y, Wang C, Yang T, Wang H, Zhao S, Sun N, et al. Chlorogenic acid alleviates chronic stress-induced duodenal ferroptosis via the inhibition of the Il-6/Jak2/Stat3 signaling pathway in rats. J Agric Food Chem (2022) 70:4353–61. doi: 10.1021/acs.jafc.2c01196
70. Wei TT, Zhang MY, Zheng XH, Xie TH, Wang W, Zou J, et al. Interferon-Γ induces retinal pigment epithelial cell ferroptosis by a Jak1-2/Stat1/Slc7a11 signaling pathway in age-related macular degeneration. FEBS J. (2021) 289:1968–83. doi: 10.1111/febs.16272
71. Zhang H, Jiao W, Cui H, Sun Q, Fan H. Combined exposure of alumina nanoparticles and chronic stress exacerbates hippocampal neuronal ferroptosis via activating Ifn-Γ/Ask1/Jnk signaling pathway in rats. J Hazard Mater. (2021) 411:125179. doi: 10.1016/j.jhazmat.2021.125179
72. Xiao Z, Kong B, Fang J, Qin T, Dai C, Shuai W, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered. (2021) 12:9367–76. doi: 10.1080/21655979.2021.2001913
73. Nelson JR, Raskin S. The eicosapentaenoic acid:Arachidonic acid ratio and its clinical utility in cardiovascular disease. Postgrad Med. (2019) 131:268–77. doi: 10.1080/00325481.2019.1607414
74. Innes JK, Calder PC. Omega-6 fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids. (2018) 132:41–8. doi: 10.1016/j.plefa.2018.03.004
75. Li C, Deng X, Xie X, Liu Y, Friedmann Angeli JP, Lai L. Activation of glutathione peroxidase 4 as a novel anti-inflammatory strategy. Front Pharmacol. (2018) 9:1120. doi: 10.3389/fphar.2018.01120
76. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, et al. Tissue resident Ccr2- and Ccr2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. (2019) 124:263–78. doi: 10.1161/circresaha.118.314028
77. Revelo XS, Parthiban P, Chen C, Barrow F, Fredrickson G, Wang H, et al. Cardiac resident macrophages prevent fibrosis and stimulate angiogenesis. Circ Res. (2021) 129:1086–101. doi: 10.1161/circresaha.121.319737
78. Jia D, Chen S, Bai P, Luo C, Liu J, Sun A, et al. Cardiac resident macrophage-derived legumain improves cardiac repair by promoting clearance and degradation of apoptotic cardiomyocytes after myocardial infarction. Circulation. (2022) 145:1542–56. doi: 10.1161/circulationaha.121.057549
79. Nicolás-Ávila JA, Lechuga-Vieco AV, Esteban-Martínez L, Sánchez-Díaz M, Díaz-García E, Santiago DJ, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell. (2020) 183:94.e–109.e. doi: 10.1016/j.cell.2020.08.031
80. Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, Godwin J, et al. The macrophage in cardiac homeostasis and disease: Jacc macrophage in Cvd series (Part 4). J Am Coll Cardiol. (2018) 72:2213–30. doi: 10.1016/j.jacc.2018.08.2149
81. Zhou Y, Que KT, Zhang Z, Yi ZJ, Zhao PX, You Y, et al. Iron overloaded polarizes macrophage to proinflammation phenotype through Ros/Acetyl-P53 pathway. Cancer Med. (2018) 7:4012–22. doi: 10.1002/cam4.1670
82. Cui Y, Zhang Z, Zhou X, Zhao Z, Zhao R, Xu X, et al. Microglia and macrophage exhibit attenuated inflammatory response and ferroptosis resistance after Rsl3 stimulation via increasing Nrf2 expression. J Neuroinflammation. (2021) 18:249. doi: 10.1186/s12974-021-02231-x
83. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. (2020) 16:278–90. doi: 10.1038/s41589-019-0462-8
84. Wang Y, Qiu Z, Yuan J, Li C, Zhao R, Liu W, et al. Hypoxia-reoxygenation induces macrophage polarization and causes the release of exosomal mir-29a to mediate cardiomyocyte pyroptosis. Vitro Cell Dev Biol Anim. (2021) 57:30–41. doi: 10.1007/s11626-020-00524-8
85. Deng Z, Shi F, Zhou Z, Sun F, Sun MH, Sun Q, et al. M1 macrophage mediated increased reactive oxygen species (Ros) influence wound healing via the mapk signaling in vitro and in vivo. Toxicol Appl Pharmacol. (2019) 366:83–95. doi: 10.1016/j.taap.2019.01.022
86. Zhao X, Si L, Bian J, Pan C, Guo W, Qin P, et al. Adipose tissue macrophage-derived exosomes induce ferroptosis via glutathione synthesis inhibition by targeting Slc7a11 in obesity-induced cardiac injury. Free Radic Biol Med. (2022) 182:232–45. doi: 10.1016/j.freeradbiomed.2022.02.033
87. Xu LC, Cao J, Li WJ, Yang ZM, Zhao R, Zhang JR, et al. [Ferroptosis in laryngeal squamous cell carcinoma and its regulation by M2 macrophage-derived exosomes]. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. (2022) 57:324–32. doi: 10.3760/cma.j.cn115330-20210621-00361
88. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. Cd8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
89. Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. (2017) 214:3311–29. doi: 10.1084/jem.20161791
90. Lu M, Qin X, Yao J, Yang Y, Zhao M, Sun L. Th17/Treg Imbalance modulates rat myocardial fibrosis and heart failure by regulating lox expression. Acta Physiol. (2020) 230:e13537. doi: 10.1111/apha.13537
91. Kanellakis P, Dinh TN, Agrotis A, Bobik A. Cd4+Cd25+Foxp3+ regulatory t cells suppress cardiac fibrosis in the hypertensive heart. J Hypertens. (2011) 29:1820–8. doi: 10.1097/HJH.0b013e328349c62d
92. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+ Cd4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. (2014) 115:55–67. doi: 10.1161/circresaha.115.303895
93. Zacchigna S, Martinelli V, Moimas S, Colliva A, Anzini M, Nordio A, et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat Commun. (2018) 9:2432. doi: 10.1038/s41467-018-04908-z
94. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the tca cycle via circulating lactate. Nature. (2017) 551:115–8. doi: 10.1038/nature24057
95. Yao X, Li W, Fang D, Xiao C, Wu X, Li M, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells. Adv Sci. (2021) 8:e2100997. doi: 10.1002/advs.202100997
96. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent nadph production. Nature. (2014) 510:298–302. doi: 10.1038/nature13236
97. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354.e–63.e. doi: 10.1016/j.molcel.2018.10.042
98. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-Stress-mediated ampk activation inhibits ferroptosis. Nat Cell Biol. (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
99. Rodríguez-Graciani KM, Chapa-Dubocq XR, Ayala-Arroyo EJ, Chaves-Negrón I, Jang S, Chorna N, et al. Effects of ferroptosis on the metabolome in cardiac cells: The role of glutaminolysis. Antioxidants. (2022) 11:278. doi: 10.3390/antiox11020278
100. Shin D, Lee J, You JH, Kim D, Roh JL. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. (2020) 30:101418. doi: 10.1016/j.redox.2019.101418
101. Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, et al. Mir-137 regulates ferroptosis by targeting glutamine transporter Slc1a5 in melanoma. Cell Death Differ. (2018) 25:1457–72. doi: 10.1038/s41418-017-0053-8
102. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
103. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. Acsl4 Dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
104. Tang LJ, Luo XJ, Tu H, Chen H, Xiong XM, Li NS, et al. Ferroptosis Occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol. (2021) 394:401–10. doi: 10.1007/s00210-020-01932-z
105. Franklin MP, Sathyanarayan A, Mashek DG. Acyl-Coa thioesterase 1 (Acot1) regulates Pparα to couple fatty acid flux with oxidative capacity during fasting. Diabetes. (2017) 66:2112–23. doi: 10.2337/db16-1519
106. Cavalli M, Diamanti K, Dang Y, Xing P, Pan G, Chen X, et al. The thioesterase Acot1 as a regulator of lipid metabolism in type 2 diabetes detected in a multi-omics study of human liver. Omics. (2021) 25:652–9. doi: 10.1089/omi.2021.0093
107. Liu Y, Zeng L, Yang Y, Chen C, Wang D, Wang H. Acyl-Coa thioesterase 1 prevents cardiomyocytes from doxorubicin-induced ferroptosis via shaping the lipid composition. Cell Death Dis. (2020) 11:756. doi: 10.1038/s41419-020-02948-2
108. Tan SK, Mahmud I, Fontanesi F, Puchowicz M, Neumann CKA, Griswold AJ, et al. Obesity-dependent adipokine chemerin suppresses fatty acid oxidation to confer ferroptosis resistance. Cancer Discov. (2021) 11:2072–93. doi: 10.1158/2159-8290.Cd-20-1453
109. Thayyullathil F, Cheratta AR, Alakkal A, Subburayan K, Pallichankandy S, Hannun YA, et al. Acid sphingomyelinase-dependent autophagic degradation of Gpx4 is critical for the execution of ferroptosis. Cell Death Dis. (2021) 12:26. doi: 10.1038/s41419-020-03297-w
110. Lakhal-Littleton S. Mechanisms of cardiac iron homeostasis and their importance to heart function. Free Radic Biol Med. (2019) 133:234–7. doi: 10.1016/j.freeradbiomed.2018.08.010
111. Nishizawa H, Matsumoto M, Shindo T, Saigusa D, Kato H, Suzuki K, et al. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor Bach1. J Biol Chem. (2020) 295:69–82. doi: 10.1074/jbc.RA119.009548
112. Qin X, Zhang J, Wang B, Xu G, Yang X, Zou Z, et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy. (2021) 17:4266–85. doi: 10.1080/15548627.2021.1911016
113. Tang M, Huang Z, Luo X, Liu M, Wang L, Qi Z, et al. Ferritinophagy activation and sideroflexin1-dependent mitochondria iron overload is involved in apelin-13-induced cardiomyocytes hypertrophy. Free Radic Biol Med. (2019) 134:445–57. doi: 10.1016/j.freeradbiomed.2019.01.052
114. Lakhal-Littleton S, Wolna M, Carr CA, Miller JJ, Christian HC, Ball V, et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc Natl Acad Sci U.S.A. (2015) 112:3164–9. doi: 10.1073/pnas.1422373112
115. Zhang Y, Xue Y, Zheng B, Han X, Ma D, Ma Z, et al. Salvia miltiorrhiza (Sm) injection ameliorates iron overload-associated cardiac dysfunction by regulating the expression of Dmt1, Tfr1, and Fp1 in rats. Evid Based Complement Alternat Med. (2021) 2021:6864723. doi: 10.1155/2021/6864723
116. Menon AV, Liu J, Tsai HP, Zeng L, Yang S, Asnani A, et al. Excess Heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood. (2022) 139:936–41. doi: 10.1182/blood.2020008455
117. Dunn LL, Kong SMY, Tumanov S, Chen W, Cantley J, Ayer A, et al. Hmox1 (heme oxygenase-1) protects against ischemia-mediated injury via stabilization of Hif-1α (hypoxia-inducible factor-1α). Arterioscler Thromb Vasc Biol. (2021) 41:317–30. doi: 10.1161/atvbaha.120.315393
118. Alfadhel M, Nashabat M, Abu Ali Q, Hundallah K. Mitochondrial iron-sulfur cluster biogenesis from molecular understanding to clinical disease. Neurosciences. (2017) 22:4–13. doi: 10.17712/nsj.2017.1.20160542
119. Misslinger M, Lechner BE, Bacher K, Haas H. Iron-sensing is governed by mitochondrial, not by cytosolic iron-sulfur cluster biogenesis in aspergillus fumigatus. Metallomics. (2018) 10:1687–700. doi: 10.1039/c8mt00263k
120. Sandoval-Acuña C, Torrealba N, Tomkova V, Jadhav SB, Blazkova K, Merta L, et al. Targeting Mitochondrial iron metabolism suppresses tumor growth and metastasis by inducing mitochondrial dysfunction and mitophagy. Cancer Res. (2021) 81:2289–303. doi: 10.1158/0008-5472.Can-20-1628
121. Dietz JV, Fox JL, Khalimonchuk O. Down the iron path: Mitochondrial iron homeostasis and beyond. Cells (2021) 10:2198. doi: 10.3390/cells10092198
122. Su Y, Yang Y, Huang Y. Loss of Ppr3, Ppr4, Ppr6, or Ppr10 Perturbs iron homeostasis and leads to apoptotic cell death in schizosaccharomyces pombe. FEBS J. (2017) 284:324–37. doi: 10.1111/febs.13978
123. Sedlackova L, Korolchuk VI. Mitochondrial quality control as a key determinant of cell survival. Biochim Biophys Acta Mol Cell Res. (2019) 1866:575–87. doi: 10.1016/j.bbamcr.2018.12.012
124. DeHart DN, Fang D, Heslop K, Li L, Lemasters JJ, Maldonado EN. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem Pharmacol. (2018) 148:155–62. doi: 10.1016/j.bcp.2017.12.022
125. Kumfu S, Chattipakorn S, Fucharoen S, Chattipakorn N. Mitochondrial calcium uniporter blocker prevents cardiac mitochondrial dysfunction induced by iron overload in thalassemic mice. Biometals. (2012) 25:1167–75. doi: 10.1007/s10534-012-9579-x
126. Kim M, Kim J, Cheon CI, Cho DH, Park JH, Kim KI, et al. Increased expression of the F(1)F(O) Atp synthase in response to iron in heart mitochondria. BMB Rep. (2008) 41:153–7. doi: 10.5483/bmbrep.2008.41.2.153
127. Sripetchwandee J, KenKnight SB, Sanit J, Chattipakorn S, Chattipakorn N. Blockade of mitochondrial calcium uniporter prevents cardiac mitochondrial dysfunction caused by iron overload. Acta Physiol. (2014) 210:330–41. doi: 10.1111/apha.12162
128. Chen MP, Cabantchik ZI, Chan S, Chan GC, Cheung YF. Iron overload and apoptosis of Hl-1 cardiomyocytes: Effects of calcium channel blockade. PLoS One. (2014) 9:e112915. doi: 10.1371/journal.pone.0112915
129. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. (2020) 5:e132747. doi: 10.1172/jci.insight.132747
130. Gao X, Qian M, Campian JL, Marshall J, Zhou Z, Roberts AM, et al. Mitochondrial dysfunction may explain the cardiomyopathy of chronic iron overload. Free Radic Biol Med. (2010) 49:401–7. doi: 10.1016/j.freeradbiomed.2010.04.033
131. Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing Vdac1 levels and restoring Gpx4 levels. Biochem Biophys Res Commun. (2019) 520:606–11. doi: 10.1016/j.bbrc.2019.10.006
132. Ibrahim WH, Habib HM, Kamal H, St Clair DK, Chow CK. Mitochondrial superoxide mediates labile iron level: Evidence from Mn-Sod-transgenic mice and heterozygous knockout mice and isolated rat liver mitochondria. Free Radic Biol Med. (2013) 65:143–9. doi: 10.1016/j.freeradbiomed.2013.06.026
133. Chen Z, Lash LH. Evidence for mitochondrial uptake of glutathione by dicarboxylate and 2-oxoglutarate carriers. J Pharmacol Exp Ther. (1998) 285:608–18.
134. Pei Z, Liu Y, Liu S, Jin W, Luo Y, Sun M, et al. Fundc1 insufficiency sensitizes high fat diet intake-induced cardiac remodeling and contractile anomaly through Acsl4-mediated ferroptosis. Metabolism. (2021) 122:154840. doi: 10.1016/j.metabol.2021.154840
135. Jang S, Chapa-Dubocq XR, Tyurina YY, St Croix CM, Kapralov AA, Tyurin VA, et al. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. (2021) 45:102021. doi: 10.1016/j.redox.2021.102021
136. Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, et al. Ampkα2 protects against the development of heart failure by enhancing mitophagy via Pink1 phosphorylation. Circ Res. (2018) 122:712–29. doi: 10.1161/circresaha.117.312317
137. Yao L, Chen H, Wu Q, Xie K. Hydrogen-rich saline alleviates inflammation and apoptosis in myocardial I/R injury Via pink-mediated autophagy. Int J Mol Med. (2019) 44:1048–62. doi: 10.3892/ijmm.2019.4264
138. Wang SH, Zhu XL, Wang F, Chen SX, Chen ZT, Qiu Q, et al. Lncrna H19 governs mitophagy and restores mitochondrial respiration in the heart through Pink1/parkin signaling during obesity. Cell Death Dis. (2021) 12:557. doi: 10.1038/s41419-021-03821-6
139. Basit F, van Oppen LM, Schöckel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ros increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. (2017) 8:e2716. doi: 10.1038/cddis.2017.133
140. Jankowska EA, Drozd M, Ponikowski P. Iron deficiency treatment in patients with heart failure. Handb Exp Pharmacol. (2017) 243:561–76. doi: 10.1007/164_2017_30
141. Khan SU, Khan MU, Riaz H, Valavoor S, Zhao D, Vaughan L, et al. Effects of nutritional supplements and dietary interventions on cardiovascular outcomes: An umbrella review and evidence map. Ann Intern Med. (2019) 171:190–8. doi: 10.7326/m19-0341
142. Chan W, Taylor AJ, Ellims AH, Lefkovits L, Wong C, Kingwell BA, et al. Effect of iron chelation on myocardial infarct size and oxidative stress in st-elevation-myocardial infarction. Circ Cardiovasc Interv. (2012) 5:270–8. doi: 10.1161/circinterventions.111.966226
143. Millán M, DeGregorio-Rocasolano N, Pérez de la Ossa N, Reverté S, Costa J, Giner P, et al. Targeting pro-oxidant iron with deferoxamine as a treatment for ischemic stroke: Safety and optimal dose selection in a randomized clinical trial. Antioxidants. (2021) 10:1270. doi: 10.3390/antiox10081270
144. Devos D, Moreau C, Kyheng M, Garçon G, Rolland AS, Blasco H, et al. A ferroptosis-based panel of prognostic biomarkers for amyotrophic lateral sclerosis. Sci Rep. (2019) 9:2918. doi: 10.1038/s41598-019-39739-5
Keywords: ferroptosis, cardiovascular disease, mechanism, pathology, metabolism
Citation: Zhang L-L, Tang R-J and Yang Y-J (2022) The underlying pathological mechanism of ferroptosis in the development of cardiovascular disease. Front. Cardiovasc. Med. 9:964034. doi: 10.3389/fcvm.2022.964034
Received: 08 June 2022; Accepted: 25 July 2022;
Published: 08 August 2022.
Edited by:
Jian Shi, University of Leeds, United KingdomReviewed by:
Francesca Vinchi, Lindsley F. Kimball Research Institute, United StatesCopyright © 2022 Zhang, Tang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue-Jin Yang, eWFuZ3lqZndAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.