 Xuhan Liu
Xuhan Liu Weihua Zhang
Weihua Zhang- Department of Cardiovascular Medicine, The First Hospital of Jilin University, Changchun, China
Pulmonary arterial hypertension, or PAH, is a condition that is characterized by pulmonary artery pressures above 20 mmHg (at rest). In the treatment of PAH, the pulmonary vascular system is regulated to ensure a diastolic and contraction balance; nevertheless, this treatment does not prevent or reverse pulmonary vascular remodeling and still causes pulmonary hypertension to progress. According to Warburg, the link between metabolism and proliferation in PAH is similar to that of cancer, with a common aerobic glycolytic phenotype. By activating HIF, aerobic glycolysis is enhanced and cell proliferation is triggered. Aside from glutamine metabolism, the Randle cycle is also present in PAH. Enhanced glutamine metabolism replenishes carbon intermediates used by glycolysis and provides energy to over-proliferating and anti-apoptotic pulmonary vascular cells. By activating the Randle cycle, aerobic oxidation is enhanced, ATP is increased, and myocardial injury is reduced. PAH is predisposed by epigenetic dysregulation of DNA methylation, histone acetylation, and microRNA. This article discusses the abnormal metabolism of PAH and how metabolic therapy can be used to combat remodeling.
Introduction
Idiopathic pulmonary arterial hypertension (IPAH) is a rare type of pulmonary hypertension with a poor clinical prognosis, being classified as group 1 PH (1, 2). The pathologic mechanism of PAH is complex and involves epigenetic factors (3). It is usually dominated by the remodeling of the pulmonary artery or pulmonary vein, or by pulmonary artery combined with pulmonary vein remodeling (4). IPH usually results from pulmonary artery remodeling, whereas pulmonary vein occlusion disease or left heart dysfunction typically results from pulmonary vein remodeling leading to PAH (4). In the vast majority of PAH cases, there is pulmonary vascular remodeling (4, 5). PAH can lead to right ventricular hypertrophy and heart failure, as well as adverse cardiovascular outcomes. In the treatment of pulmonary hypertension, currently available targeted therapeutic agents act on diastolic/contraction balance in the pulmonary arteries. These include SGC stimulants, antagonists of endothelin receptors, and inhibitors of PDE5 (6–8). Despite improving the quality of life in patients with pulmonary hypertension, these drugs did not reverse or delay pulmonary vascular remodeling and still led to the progression of the disease. The cause of pulmonary vascular remodeling can include endothelial cells (EC) dysfunction, cancerous cells proliferating, anti-apoptotic vascular cells, and dysregulated vascular cell metabolism. Researchers initially looked at vascular plexus lesions and the upregulation of HIF-1α in PAH (9) and survivin in IPAH (10). Over the past two decades, the focus of research has shifted from mitochondrial metabolic plasticity to altered intracellular energy metabolism in pulmonary arteries (11, 12). As part of PAH, cellular metabolism is abnormal, including glycolysis, fatty acid metabolism, and glutamine metabolism (13). A metabolic reprogramming mechanism may be involved in PAH pulmonary vascular remodelings. Hence, this review outlines the three abnormal metabolic pathways and identifies possible therapeutic targets for metabolic reprogramming.
Pathophysiology of PAH
Apoptosis and endothelial dysfunction occur in human PAH patients and animal models, attenuating the endothelial-mediated inhibition of smooth muscle cell proliferation. It has also been found that cloning and amplification of anti-apoptotic endothelial cells can jointly cause angioproliferative lesions (14). Remodeling involves endothelial, mesothelial, and epithelial walls (14) as well as complex interactions between epithelial fibroblasts, perivascular inflammatory cells, and the extracellular matrix. Several other factors have also been described previously as causing pulmonary vascular remodeling, such as genetic mutations, signaling pathway abnormalities, and endothelial dysfunction (6, 15–17). In 1926, Dr Otto Warburg proposed similarities between cancer and PAH. Through in vitro and in situ observations (18, 19), researchers confirmed the novel cancer-like hypothesis of PAH despite the initial controversy (18, 19). There is evidence that a wide array of growth factors, cytokines, and chemokines contribute to cancer and PAH development. Of these factors, interleukin 6 (IL-6) and platelet-derived growth factor (PDGF) have been extensively studied (20). Within IPAH plexiform lesions, monoclonal amplification of EC and microsatellite sequence instability have been observed (17, 21).
Patients with PAH and cells cultured in vitro have chromosomal abnormalities (22). Compared to control cells, smooth muscle cells (SMC) and extracellular vesicles (EC) isolated from PAH patients maintain a longer-lasting anti-apoptotic phenotype (22–24). PVCs from PAH patients exhibit metabolic dysregulation both in situ and in vitro (11, 12, 25, 26). In summary, the connection between PAH metabolism and proliferation is similar to that found in cancer (14). Moreover, anti-proliferative or anti-cancer agents may play a role in PAH treatment (13). As a result, the therapeutic potential of numerous anti-neoplastic drugs has been tested in preclinical models, with some of them reaching clinical assays (27). The second-generation tyrosine kinase inhibitor dasatinib improves the symptoms of chronic PAH in multiple animal models (27). However, its administration prior to exposure to PH inducers exacerbates remodeling of the pulmonary vascular system and pulmonary artery pressures. Dasatinib alone had no effect on rats' histology or hemodynamics (27).
Under current experimental conditions, more precise data on pulmonary vascular remodeling are still lacking. As a molecular marker for veins, hepatic ligand protein B4 receptors are currently ineffective at accurately determining pulmonary veins (4). Therefore, it is necessary to develop more effective experimental tools for assessing pulmonary vascular remodeling. Furthermore, many of the vascular cells of PAH in established humans are quiescent and may resist apoptosis. Quiescent cells have different metabolic requirements than aberrantly proliferating cells (4). The field must therefore clarify the cause of metabolic heterogeneity and how it contributes to vascular remodeling in the future (4).
PAH metabolism
The metabolism of PAHs is accomplished by many cellular and molecular mechanisms with mitochondria at their cores. Metabolic reprogramming is an emerging hallmark of oncogenesis (28). Growing evidence suggests that metabolic dysfunction may be associated with changes in PAH expression and susceptibility (4). Researchers examined metabolite changes in hypoxia-induced PASMCs and PDGF-BB-induced PASMCs (29, 30). A recent report on metabolic alteration was also explored in perioperative period of congenital heart disease associated with pulmonary arterial hypertension (CHD-PAH) patients undergoing repair (31). Furthermore, patients with PAH may be more sensitive to mutagens because of impaired DNA-response mechanisms (32). There was a correlation between PAH and decreased expression of breast cancer 1 protein (BRCA1) and DNA topoisomerase 2-binding protein 1 (TopBP1), both of which are involved in the integrity of the genome (32). Dedicated to overcoming the antiproliferative and lethal effects of DNA damage, PAH-PASMCs have developed a network of highly efficient and complementary mechanisms (33), including sanitation of the oxidized nucleotide pool (the result of this study), DNA damage sensing, and repair (33). Metabolomics studies also provide a detailed understanding of metabolic disturbances in experimental PH challenged by MCT administration or hypoxia (34, 35).
Prior to PAH, cellular cancer-like metabolic abnormalities were thought to include aerobic glycolysis, pentose phosphate pathway activation, mitochondrial dysfunction, altered fatty acid metabolism, and insulin resistance. In PAH, mtHSP90 accumulation contributes to vascular remodeling through its regulation of mitochondrial homeostasis (36). With further research in the area of metabolic dysregulation in PAH, Warburg's principle of dysregulated metabolism can now be included in the metabolic theory of PAH as well as other dysregulated pathways. Earlier studies hypothesized that metabolic dysregulation would occur in the pulmonary vasculature. Even so, increasing evidence suggests that RV metabolism may be present in muscle tissue, which indicates that PAH development may be the result of a combination of paracrine and systemic actions. To develop a comprehensive metabolic theory of PAH, it is necessary to discover the nature of metabolic disorders outside the pulmonary vascular system. PAH has been associated with metabolic dysfunction since its inception, according to many early studies. PAH pathology is characterized not only by hyperproliferative and apoptosis-resistant PVCs, ECs, and SMCs, but also by other types of cells in the vascular system. Cancer-like proliferative increases and resistance to apoptosis are indicative of abnormal mitochondrial metabolism and dynamics (37). A dysregulation of mitochondrial metabolism occurs in the early or late stages of PAH, and its link to reprogramming remains unclear.
Metabolism and inflammation: accumulating evidence suggests a functional role of perivascular inflammation in the initiation and/or progression of pulmonary vascular remodeling (38, 39). There is a correlation between clinical outcomes and high levels of cytokines, chemokines, and inflammatory mediators in PAH patients (38). In PAH lung biopsies, macrophages, mast cells, and T lymphocytes were detected near remodeled pulmonary vasculature, mainly in proximity to macrophages, mast cells, and T lymphocytes (40, 41). Consequentially, several clinical trials have been conducted to investigate inflammation and autoimmunity treatments in PAH patients (42). However, the exact role of inflammation and immunity in PAH-and specifically, its function as a cause, promoter, or downstream bystander—is poorly understood and remains a contentious issue.
RVH is commonly out of adjustment, with two exceptions: ischemia and aerobic glycolysis (43). The Warburg effect is associated with molecular events that adapt to acute cellular stress and prevent apoptosis. In addition to metabolic dysregulation, recent studies have revealed metabolic abnormalities in maladaptive RVH, including aerobic glycolysis, glutamine metabolism, and the Randle cycle (44). Pyruvate kinase muscle isozyme 2 (PKM2), already known for its role in pulmonary artery obliteration, was identified as an integrator, of anaerobic metabolism, oxidative stress, inflammation, and fibrosis in several diseases sharing with RV failure (45). The energy metabolism in PAH needs to be capable of meeting increased metabolic demands, which requires enhanced aerobic glycolysis and reprogramming of multiple molecules. PAH plasticity and adaptation should be combined with the control of cellular stress and protein misfolding, and protein sorting in intracytoplasmic compartments (4), with implications for treating PAH. Below we will describe three types of abnormal metabolism and their relationship with metabolic reprogramming.
Aerobic glycolysis
In PAH, altered intracellular glucose transport may contribute to glucose intolerance. The presence of glycated hemoglobin (46, 47) in IPAH suggests that glucose intracellular influx and insulin resistance are always present when chronic hyperglycemia is present (15, 20, 34, 35). Studies have shown that cancer and PAH share similar features (18, 48–50), including increased cellular glucose uptake and altered glucose metabolism (51). Proliferating cancer cells and pulmonary vascular endothelial cells use a great deal of energy. Under normal oxygen partial pressure, highly proliferating cells change the metabolism from glycolysis to aerobic glycolysis, or the Warburg effect (52). When glucose is available, aerobic glycolysis increases the survival rate of proliferating cells and enhances glucose metabolism. The results of (18F)-fluorodeoxyglucose-PET (FDG-PET) showed that IPAH patients had higher pulmonary glucose uptake and a higher glycolysis ratio than normal subjects (53). Compared to normal cells, endothelial cells had a 3 times higher glycolysis rate, which might improve the proliferation rate of abnormal cells (25). It can therefore be believed that PAH has abnormal aerobic glycolysis, which can result in enhanced glucose utilization, which is used to meet the proliferating cells' energy requirements.
In PAH patients, glucose is mainly metabolized by aerobic glycolysis. In pulmonary vascular cells of PAH animal models and IPAH patients (54), Diebold et al. found hyperpolarized mitochondria, which resulted in diminished oxidative phosphorylation and glucose oxidation. A metabolic pathway that is abnormal in PAH patients, aerobic glycolysis is likely caused by mitochondrial dysfunction (55). In PAH patients and in preclinical PAH models, positron emission tomography has shown increased glucose uptake in the lungs and right ventricle by Warburg metabolism (53). Glucose influx may be caused by increased glucose transporter protein (Glut) expression (56, 57) and expression of a splice variant of terminal pyruvate dehydrogenase kinase (PDK) (57), act together to upregulate the glycolytic response in PAH (56, 57). Glycolysis produces pyruvate. Pyruvate dehydrogenase (PDH) is a key enzyme involved in the complete oxidation of pyruvate to coenzyme A. Inhibits the flow of pyruvate into mitochondria and the oxidation of glucose inside mitochondria. Inhibition of PDK function occurs through phosphorylation of E1-α by PDK (58). Increased expression of PDK inhibits acetyl coenzyme A formation and the Krebs cycle and oxidative phosphorylation in mitochondria in PAH (55). PDK-mediated metabolism impairs cardiac mechanical performance and electrical remodeling, as a result, right heart contractility is reduced, cardiac output is reduced, and RV action potential and QT intervals are prolonged (59). DCA inhibits PDH function by reducing PDH phosphorylation (59), which improves glucose oxidation, RV contractility, and PAH vascular remodeling. Theoretically speaking, DCA corrects pathological inhibition of PDH and glucose oxidation responses only in the lungs and renal vasculature (60), and has little effect on the normal heart and vasculature. Accordingly, mitochondrial hyperpolarization leads to aerobic glycolysis in patients with PAH. Glut expression and PDK overexpression cause an increase in glucose in-flow. DCA targets the lung and RV, affecting energy supply, decreasing RV function, and aggravating ischemia (54, 59). Thus, active control is necessary.
Associated with PAH metabolism disorders caused by a variety of changes in the structure and content of glucose derivatives. Patients with COPD had altered structures of glycoprotein glycans (61). O-GlcNAc level and function are altered in IPAH as a result of altered glucose uptake (62). The hypoxia-induced enhancement of glycosaminoglycan (GAG) synthesis in human lung fibroblasts may contribute to the regulation of GAG synthesis/deposition in PAH. UDP-GlcNAc regulates the synthesis of HA, a large GAG (63). Plasma HA levels are elevated in patients with IPAH (64) and may be related to vascular remodeling in PAH and inflammatory response processes (65). The HSPGs perlecan and agrin are elevated in PAH patients (66). The perlecan content of the EC-PASMC junction is higher (67), and it may be involved in EC barrier function in PAHs, EC-PASMC interactions, and cell proliferation inhibition (67). There may be an association between changes in the structure and content of various glucose derivatives in PAH patients and inflammation and vascular remodeling, as well as vascular plexiform lesions. Despite the late start to the study of the above substances in PAH, more research is needed to determine their specific roles and therapeutic targets. According to Warburg in 1926, aerobic glycolysis, a metabolic change in the pulmonary vascular system, is regulated by the redox state of the activating transcription factors. In addition to HIF-1α, episodic silencing of superoxide dismutase 2 (SOD2) and mitochondrial fission/fusion are also associated with PAH. The oxidative metabolism is inhibited and sustained glycolysis is favored (68).
HIF
Oxygen-sensitive prolyl hydroxylase structural domain-containing enzymes (PHDs) regulate HIF subunit expression (69), which are hydroxylated under aerobic conditions. Von Hipple-Lindau protein (VHL) ubiquitinates targeted HIF subunits (69).
HIF-1α activation may result from abnormalities in mitochondrial metabolism in PAH (70). The abnormalities reduce hydrogen peroxide production and eliminate inhibition of HIF-1α activation (71). DNA methyltransferase in the lung activates epigenetic silencing of superoxide dismutase (SOD2) (71), interfering with gene transcription and reducing hydrogen peroxide levels in the blood, activating HIF-1α and initiating glycolysis. Under normoxia, inhibition of SOD2 expression with siRNA activated HIF-1α in normal PASMC (71). By applying 5-azacytidine for demethylation, SOD2 expression was restored and PASMC proliferation was inhibited (72). A dysregulation of DNA methyltransferase was observed in PAH assays, which may be tissue specific.
In PAH, HIF-1α is closely related to mitochondria. HIF-1α activation with cobalt or desferrioxamine results in mitochondrial fission in human and rodent PAH smooth muscle cells (73), suggesting its association with mitochondrial plasticity. In addition, it regulates mitochondrial dynamics, which results in a decrease in mitochondria and reduced NO utilization in IPAH cells (9). The HIF promotes further lung endothelial damage by mobilizing hematopoietic precursors in PAH. In the mitochondrial respiratory chain, HIF-1α promotes the expression of cytochrome oxidase subunit 4.2 (74). Furthermore, it upregulates PDK transcription (75) and inhibits PDH function to produce a glycolytic state (76). The source of HIF-1α stability is unknown, but it has been demonstrated to be inherited (70). Studies have recently confirmed the significance of HIF-2 in PAH animals. HIF-2 is upregulated and BMPR2 and Cav1 are downregulated in PHD-2 deficient EC under normoxia (77). HIF-2 facilitates the upregulation of the chemokine CXCL12, which stimulates PASMC proliferation (77). HIF-2 deletion also reverses PAH (77). Deletion of HIF-2 is associated with reduced expression of arginase (Arg1), and Arg1 deletion also reverses PAH (72). Transcriptional factor POU5F1 (or OCT4) targets HIF-2 (78), and hypoxia and inflammatory factors (such as IL-1b and IL-6) can upregulate miR-130/301 in EC and PASMC, respectively. Rodent models and patients' lungs and plasma have confirmed this (78). An analysis of bioinformatics data revealed that members of the miR-130/301 family regulate other miRNAs and cellular phenotypes in PAH. In vitro expression of miR-130a in human EC and PASMC has demonstrated this effect. MiR-130/301 inhibits peroxisome proliferator-activated receptor-γ (PPARγ). PPARγ is a target of BMP signaling (79), and inhibition causes abnormal proliferation of vascular endothelium. It may also regulate STAT3-miR-204-SRC pathways (80) and apelin-miR-424/503-FGF2 pathways, maintaining EC homeostasis and inhibiting vascular endothelial proliferation (81). In the glycolytic state, HIF-1 and HIF-2 act together through different mechanisms in the pulmonary vascular system. As a result of HIF-2, miRNAs are important players in glycolysis, and several miRNA inhibitors or mimics have shown therapeutic efficacy in animal models. Despite their lack of tissue specificity, inability to penetrate and degrade cells, and potential toxicity, much preliminary work is still needed before they can be used for deep tissue regeneration (Figure 1).
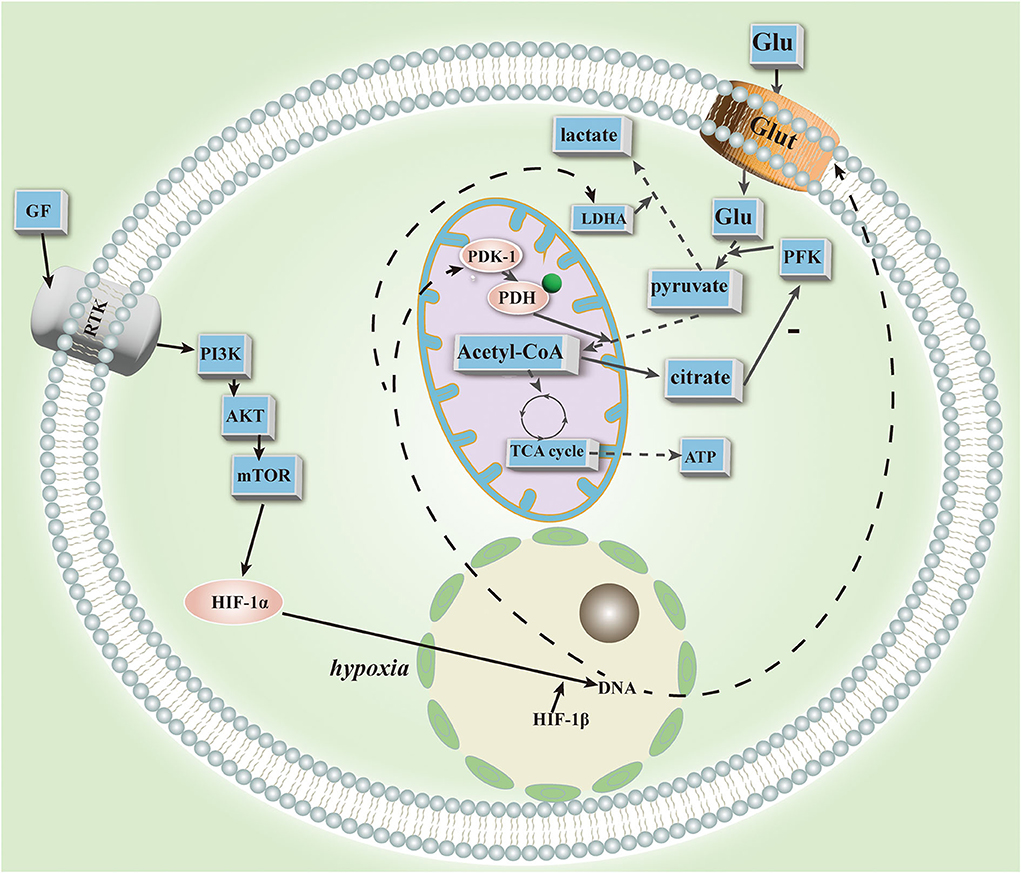
Figure 1. This figure depicts the major steps and key substrates in aerobic glycolysis.
Glutamine metabolism
Research examining mitochondrial dysfunction and aerobic glycolysis partially explains the aberrant energy metabolism in PAH. YAP (Yes-associated protein 1) was recently found to be correlated with glutamine metabolism and vascular proliferation. Yes-associated protein 1 (YAP), when containing a PDZ-binding gene sequence (TAZ), stimulates pulmonary vascular smooth muscle and endothelial proliferation (82). There was increased expression of GLS1 in small pulmonary arteries of rats treated with wild lily base (MCT)-induced PAH and decreased glutamine in isolated EC in this model (82). An additional study found increased glutamine catabolism in patients with PAH and in rats implanted with MCT-induced RV (83). Krebs cycle increases glutamine catabolism while also reshaping glutamate metabolism, possibly contributing to the loss of compensatory cardiac remodeling response. Both monkey HIV-associated PAH and human HIV-mediated PAH were associated with elevated GLS1 expression in the lungs of rhesus monkeys (84). In addition, glutamine catabolism results in proline hydroxylation and α-ketoglutarate activation of mammalian target of rapamycin (mTOR) proteins to promote collagen stability and translation and enhance fibrosis (85), leading to vascular hyperproliferation. Contrary to this, pharmacological inhibition of GLS1 activity affects the Krebs cycle and reduces MCT-induced arterial remodeling in rat PAH (83). GLS1 was inhibited and PAH improved in clinical trials with anticancer drugs (NCT02071862) (86), MK-801 (an NMDAR) (81), and vetiprofen (a YAP) (82). A treatment option that could be considered is reprogramming glutamine metabolism.
There is a significant association between mutations in the bone morphogenetic protein receptor type II gene (BMPR2) and hereditary PAH (37). It has been suggested that aberrant bone morphogenetic protein signaling and epistasis promote aerobic glycolysis for cell proliferation (37). Egnatchik et al. found that PAH patients with aberrant BMPR2 gene mutations had lower trans-lung glutamine concentrations.
BMPR2 gene mutations may affect glutamine metabolism based on the large reduction in glutamine. Yelamanchi et al. suggested that EC with BMPR2 mutations fails to tolerate glutamine-limited survival conditions (87). They suggested that glutamine dependence is caused by mitochondrial damage. Mutations of BMPR2 drive downstream and lead to the formation of ecdysone (87). When Ecdysone is removed from the body, glutamine metabolism returns to normal. It stabilizes HIF-1 and inhibits Sirtuin-3.
Cancer-like glycolysis results when PAH metabolism is dysregulated, causing a shift from oxidative phosphorylation (OXPHOS) to aerobic glycolysis (Warburg effect), and glutamine metabolism, and increased fatty acid oxidation in mitochondria. Based on earlier studies, pulmonary vasculature could provide sufficient glucose for aerobic glycolysis, which would supply energy for cell proliferation. But as glycolysis is enhanced in the Krebs cycle, there is a decrease in the supply of carbon substrates for nucleotides and proteins, and a concomitant decrease in the carbon intermediates not involved in ATP formation, which is not consistent with increased vascular cell proliferation. Thus, it has been hypothesized that glutamine catabolism is essential for backfilling the Krebs cycle with carbon intermediates. Catabolizing glutamine provides carbon intermediates for the Krebs cycle, which provides lipids and amino acids for nucleotide biosynthesis (88). The upregulation and uptake of glutamine by GLS1 drive PAH. PAH is then utilized for glutamate to produce *-ketoglutarate for the Krebs cycle. On the other hand, YAP and TAZ are essential for glutaminase upregulation and hydrolysis. The activation of YAP/TAZ leads to metabolic reprogramming of PAECs and PASMCs in vitro, enhancing glycolysis and glutamine catabolism. This process plays an important role in maintaining a proliferating cell phenotype (78).
The glutamate receptors (GluR) are responsible for the function of glutamate, a non-essential amino acid (89). NMDAR and mGluR5 have been found to be located in the pulmonary vasculature and RVs, suggesting they may play a role in the pathogenesis of PAH. The ionotropic glutamate receptor (iGluR) is a glutamate-activated ion channel. iGluR-associated membrane channels, including Na+, K+, and Ca2+ channels, exhibit certain physiological and pharmacological characteristics. By causing Ca2+ ion inward flow and activating protein kinase C and its AKT/MAPK pathway, the NMDA receptor (NMDAR), a type of pulmonary vascular iGluR, causes PAH remodeling. Proliferation and anti-apoptosis are the results. By inhibiting NMDAR, the progression of PAH could be stopped and vascular remodeling reversed. The metabolic glutamate receptors (mGluRs) are another type of glutamate receptor, classified into four groups. The mGluRs are attached to intramembrane G proteins and act on second messenger synthesis or ion channels by activating GTP-binding proteins. Group I mGluR stimulates inositol triphosphate (IP3) metabolism and Ca2+ transfer (90). This signaling pathway activates IP3 and stimulates the intracellular release of Ca2+. mGluR5 is involved in this pathway. Adenylate cyclase (AC) is associated with Group II and III mGluR. Vasodilation is promoted by prostacyclin (PGI2). AC activation increases the level of cAMP, which activates protein kinase A (PKA), which restores the activity of the phosphorylation pathway, and reduces the metabolic shift toward glycolysis. Accordingly, it appears that GluR plays an important role in the pathogenesis of PAH and may therefore affect its prognosis by regulating GluR.
There are targeted therapies for tumors that inhibit glutamine metabolism, such as the GLS inhibitors BPTES and CB-829 (in phase I clinical trials: NCT02071862 and NCT02771626), that suppress tumors in preclinical animal models (91). Studies in preclinical and clinical settings have shown that glutamate dehydrogenase inhibitors, transaminase inhibitors, and glutamine analogs reduce the production of α-ketoglutarate and nucleotides in the Krebs cycle (92). 2-Aminobicyclo(2,2,1)-heptane-2-carboxylic acid (BCH), an inhibitor of the glutamine transporter protein SLC7A, is also a great therapeutic option. By inhibiting the mTOR signaling pathway, BCH prevents glutamine-induced fibrosis from forming (93). As a result, it blocks activation of the mTOR pathway by *-ketoglutarate and inhibits proline hydroxylation by PAH (85). 000In conclusion, glutamine metabolism replenishes carbon intermediates used in aerobic glycolysis and promotes PAH progression. Clinical trials and in vitro studies have investigated glutamine metabolism's antitumor effects. We should examine in more depth in the future the reprogramming of glutamine metabolism, the inhibition of glutaminolytic enzymes, and the glutamate receptors as therapeutic targets.
Randle cycle
Throughout the adult heart, fatty acid oxidation (FAO) supplies ATP. The Randle cycle (94) occurs when fat in the blood inhibits cellular glucose metabolism. FAO produces citrate, which inhibits phosphofructokinase, increasing glucose-6-phosphate levels, inhibiting hexokinase, and reducing pyruvate production. In addition, FAO production of acetyl coenzyme A inhibits PDH. Stephen L. et al. measured direct myocyte metabolism and discovered that FAO and GO inhibition led to a metabolic shift to glycolysis in vivo (68). Two FAO inhibitors, trimetazidine, and ranolazine, did not prolong the QTc interval in a rat model of PAH with ligated pulmonary arteries and increased GO and raised ATP levels, suggesting that FAO is detrimental (95). FAO inhibition activates the Randle cycle (95). Hou posited that FAS inhibition protects mice from PAH caused by hypoxia through activation of P13/AKT signaling through C73 (96). The amount of glycogen in the RV increases when GO is inhibited, and it decreases when FAO is inhibited (95). Guarnieri suggested that trimetazidine has a positive effect on the MCT-induced PAH model (96). The absence of malonyl coenzyme A decarboxylase (MCD) inhibits FAO oxidation and promotes glycolysis, preventing the “glycolytic shift” that occurs during hypoxia. Trimetazidine is metabolized similarly to PAH in rats treated with trimetazidine (26). Inhibiting FAO metabolism, increasing GO metabolism, and activating the Randle cycle will therefore benefit PAH treatment. The use of trimetazidine and ranolazine for the treatment of PAH patients, as well as the combination of FAO inhibitors with PDK inhibitors, requires further study.
Conclusion and prospect
Previously, PAH studies focused on the NO-TAX2-ET1 pathway, but in recent years, metabolic dysregulation and reprogramming have gradually gained attention. Spermine synthase inhibition inhibited platelet-derived growth factor-BB-mediated PASMC proliferation in vitro and reduced monocrotaline-induced pulmonary hypertension in rats in vivo. A therapy for PAH that inhibits spermine synthesis would promote pulmonary vascular remodeling (97). Glut overexpression resulted in increased glucose in-flow and PDK overexpression resulted in mitochondrial hyperpolarization related to aerobic glycolysis. A large amount of carbon intermediates is consumed during aerobic glycolysis, which does not produce enough energy. Energy and carbon intermediates are replenished by glutamine metabolism, which are supplied to proliferating cells and anti-apoptotic cells. In cardiac myocytes, FAO is responsible for the majority of energy, and inhibiting FAO will enhance GO and prevent PAH. The major source of energy in cardiac myocytes is FAO, and inhibiting FAO will increase GO and prevent hypoxia that leads to PAH. Consequently, therapeutic agents targeting the three metabolic pathways and metabolic reprogramming hold great promise in PAH. The use of certain drugs for the treatment of diabetes and metabolic disorders may also be appropriate for PAH. It is still necessary to study their safety and potential side effects further.
Author contributions
XL: methodology, investigation, formal analysis, and writing-original draft. LZ: writing-review and editing. WZ: project administration and supervision. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Provencher S, Boucherat O, Potus F, Bonnet S. Pulmonary hypertension thresholds: time to lower further? Lancet Respir Med. (2020) 8:834–6. doi: 10.1016/S2213-2600(20)30326-X
2. Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Kardiol Pol. (2015) 73:1127–206. doi: 10.5603/KP.2015.0242
3. Yan Y, He Y-Y, Jiang X, Wang Y, Chen J-W, Zhao J-H, et al. DNA methyltransferase 3B deficiency unveils a new pathological mechanism of pulmonary hypertension. Sci Adv. (2020) 6:eaba2470. doi: 10.1126/sciadv.aba2470
4. Tuder RM, Archer SL, Dorfmüller P, Erzurum SC, Guignabert C, Michelakis E, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. (2013) 62:D4–12. doi: 10.1016/j.jacc.2013.10.025
5. Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines. the diagnosis and treatment of pulmonary hypertension. Rev Esp Cardiol. (2016) 69:177. doi: 10.1016/j.recesp.2016.01.002
6. Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ, Nicolls MR. Pathobiology of pulmonary arterial hypsertension and right ventricular failure. Eur Respir J. (2012) 40:1555–65. doi: 10.1183/09031936.00046612
7. Zhao M, Austin ED, Hemnes AR, Loyd JE, Zhao Z. An evidence-based knowledgebase of pulmonary arterial hypertension to identify genes and pathways relevant to pathogenesis. Mol Biosyst. (2014) 10:732–40. doi: 10.1039/C3MB70496C
8. Lan NSH, Massam BD, Kulkarni SS, Lang CC. Pulmonary arterial hypertension: pathophysiology and treatment. Diseases. (2018) 6:38. doi: 10.3390/diseases6020038
9. Fijalkowska I, Xu W, Comhair SAA, Janocha AJ, Mavrakis LA, Krishnamachary B, et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol. (2010) 176:1130–8. doi: 10.2353/ajpath.2010.090832
10. McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, et al. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. (2005) 115:1479–91. doi: 10.1172/JCI23203
11. Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JGN, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv15 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. (2008) 294:H570–8. doi: 10.1152/ajpheart.01324.2007
12. Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med. (2012) 185:260–6. doi: 10.1164/rccm.201108-1536PP
13. Mprah R, Adzika GK, Gyasi YI, Ndzie Noah ML, Adu-Amankwaah J, Adekunle AO, et al. Glutaminolysis: a driver of vascular and cardiac remodeling in pulmonary arterial hypertension. Front Cardiovasc Med. (2021) 8:667446. doi: 10.3389/fcvm.2021.667446
14. Thompson AAR, Lawrie A. Targeting vascular remodeling to treat pulmonary arterial hypertension. Trends Mol Med. (2017) 23:31–45. doi: 10.1016/j.molmed.2016.11.005
15. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. (2012) 122:4306–13. doi: 10.1172/JCI60658
16. Guignabert C, Dorfmüller P. Pathology and pathobiology of pulmonary hypertension. Semin Respir Crit Care Med. (2017) 38:571–84. doi: 10.1055/s-0037-1606214
17. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. (2019) 53:1801887. doi: 10.1183/13993003.01887-2018
18. Rai PR, Cool CD, King JAC, Stevens T, Burns N, Winn RA, et al. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. (2008) 178:558–64. doi: 10.1164/rccm.200709-1369PP
19. Sakao S, Tatsumi K. Vascular remodeling in pulmonary arterial hypertension: multiple cancer-like pathways and possible treatment modalities. Int J Cardiol. (2011) 147:4–12. doi: 10.1016/j.ijcard.2010.07.003
20. Boucherat O, Vitry G, Trinh I, Paulin R, Provencher S, Bonnet S. The cancer theory of pulmonary arterial hypertension. Pulm Circ. (2017) 7:285–99. doi: 10.1177/2045893217701438
21. Aldred MA, Comhair SA, Varella-Garcia M, Asosingh K, Xu W, Noon GP, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. (2010) 182:1153–60. doi: 10.1164/rccm.201003-0491OC
22. Tu L, De Man FS, Girerd B, Huertas A, Chaumais M-C, Lecerf F, et al. A critical role for p130Cas in the progression of pulmonary hypertension in humans and rodents. Am J Respir Crit Care Med. (2012) 186:666–76. doi: 10.1164/rccm.201202-0309OC
23. Masri FA, Xu W, Comhair SAA, Asosingh K, Koo M, Vasanji A, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L548–54. doi: 10.1152/ajplung.00428.2006
24. Tu L, Dewachter L, Gore B, Fadel E, Dartevelle P, Simonneau G, et al. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am J Respir Cell Mol Biol. (2011) 45:311–22. doi: 10.1165/rcmb.2010-0317OC
25. Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. (2007) 104:1342–7. doi: 10.1073/pnas.0605080104
26. Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. (2010) 2:44ra58. doi: 10.1126/scitranslmed.3001327
27. Awada C, Grobs Y, Wu W-H, Habbout K, Romanet C, Breuils-Bonnet S, et al. R-Crizotinib predisposes to and exacerbates pulmonary arterial hypertension in animal models. Eur Respir J. (2021) 57:2. doi: 10.1183/13993003.03271-2020
28. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
29. He Y-Y, Xie X-M, Zhang H-D, Ye J, Gencer S, van der Vorst EPC, et al. Identification of hypoxia induced metabolism associated genes in pulmonary hypertension. Front Pharmacol. (2021) 12:753727. doi: 10.3389/fphar.2021.753727
30. Yan Y, Jiang R, Yuan P, Wen L, Pang X-B, Jing Z-C, et al. Implication of proliferation gene biomarkers in pulmonary hypertension. Animal Model Exp Med. (2021) 4:369–80. doi: 10.1002/ame2.12191
31. He Y-Y, Yan Y, Chen J-W, Liu S, Hua L, Jiang X, et al. Plasma metabolomics in the perioperative period of defect repair in patients with pulmonary arterial hypertension associated with congenital heart disease. Acta Pharmacol Sin. (2022) 43:1710–20. doi: 10.1038/s41401-021-00804-3
32. Ranchoux B, Meloche J, Paulin R, Boucherat O, Provencher S, Bonnet S. DNA damage and pulmonary hypertension. Int J Mol Sci. (2016) 17:990. doi: 10.3390/ijms17060990
33. Vitry G, Paulin R, Grobs Y, Lampron M-C, Shimauchi T, Lemay S-E, et al. Oxidized DNA precursors cleanup by NUDT1 contributes to vascular remodeling in pulmonary arterial hypertension. Am J Respir Crit Care Med. (2021) 203:614–27. doi: 10.1164/rccm.202003-0627OC
34. Zhao J-H, He Y-Y, Guo S-S, Yan Y, Wang Z, Ye J, et al. Circulating plasma metabolomic profiles differentiate rodent models of pulmonary hypertension and idiopathic pulmonary arterial hypertension patients. Am J Hypertens. (2019) 32:1109–17. doi: 10.1093/ajh/hpz121
35. Zheng H-K, Zhao J-H, Yan Y, Lian T-Y, Ye J, Wang X-J, et al. Metabolic reprogramming of the urea cycle pathway in experimental pulmonary arterial hypertension rats induced by monocrotaline. Respir Res. (2018) 19:94. doi: 10.1186/s12931-018-0800-5
36. Boucherat O, Peterlini T, Bourgeois A, Nadeau V, Breuils-Bonnet S, Boilet-Molez S, et al. Mitochondrial HSP90 accumulation promotes vascular remodeling in pulmonary arterial hypertension. Am J Respir Crit Care Med. (2018) 198:90–103. doi: 10.1164/rccm.201708-1751OC
37. Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. (2018) 360:j5492. doi: 10.1136/bmj.j5492
38. Hu Y, Chi L, Kuebler WM, Goldenberg NM. Perivascular inflammation in pulmonary arterial hypertension. Cells. (2020) 9:2338. doi: 10.3390/cells9112338
39. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. (2014) 115:165–75. doi: 10.1161/CIRCRESAHA.113.301141
40. Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 186:261–72. doi: 10.1164/rccm.201201-0164OC
41. Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 186:897–908. doi: 10.1164/rccm.201202-0335OC
42. Goldenberg NM, Rabinovitch M, Steinberg BE. Inflammatory basis of pulmonary arterial hypertension: implications for perioperative and critical care medicine. Anesthesiology. (2019) 131:898–907. doi: 10.1097/ALN.0000000000002740
43. Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, et al. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. (2005) 45:1849–55. doi: 10.1016/j.jacc.2005.02.065
44. Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. (2014) 115:176–88. doi: 10.1161/CIRCRESAHA.113.301129
45. Shimauchi T, Boucherat O, Yokokawa T, Grobs Y, Wu W, Orcholski M, et al. PARP1-PKM2 axis mediates right ventricular failure associated with pulmonary arterial hypertension. JACC Basic Transl Sci. (2022) 7:384–403. doi: 10.1016/j.jacbts.2022.01.005
46. Belly MJ, Tiede H, Morty RE, Schulz R, Voswinckel R, Tanislav C, et al. HbA1c in pulmonary arterial hypertension: a marker of prognostic relevance? J Heart Lung Transplant. (2012) 31:1109–14. doi: 10.1016/j.healun.2012.08.014
47. Pugh ME, Robbins IM, Rice TW, West J, Newman JH, Hemnes AR. Unrecognized glucose intolerance is common in pulmonary arterial hypertension. J Heart Lung Transplant. (2011) 30:904–11. doi: 10.1016/j.healun.2011.02.016
48. Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res. (2014) 115:148–64. doi: 10.1161/CIRCRESAHA.115.301130
49. Cool CD, Kuebler WM, Bogaard HJ, Spiekerkoetter E, Nicolls MR, Voelkel NF. The hallmarks of severe pulmonary arterial hypertension: the cancer hypothesis-ten years later. Am J Physiol Lung Cell Mol Physiol. (2020) 318:L1115–L30. doi: 10.1152/ajplung.00476.2019
50. Guignabert C, Tu L, Le Hiress M, Ricard N, Sattler C, Seferian A, et al. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev. (2013) 22:543–51. doi: 10.1183/09059180.00007513
51. Pedersen PL. Warburg me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers' most common phenotypes, the “Warburg Effect”, ie, elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. (2007) 39:211–22. doi: 10.1007/s10863-007-9094-x
52. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
53. Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, et al. Lung 18F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 185:670–9. doi: 10.1164/rccm.201108-1562OC
54. Diebold I, Hennigs JK, Miyagawa K, Li CG, Nickel NP, Kaschwich M, et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. (2015) 21:596–608. doi: 10.1016/j.cmet.2015.03.010
55. Ryan JJ, Archer SL. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part I: metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation. (2015) 131:1691–702. doi: 10.1161/CIRCULATIONAHA.114.006979
56. Michelakis ED, McMurtry MS, Wu X-C, Dyck JRB, Moudgil R, Hopkins TA, et al. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. (2002) 105:244–50. doi: 10.1161/hc0202.101974
57. Zhang H, Wang D, Li M, Plecitá-Hlavatá L, D'Alessandro A, Tauber J, et al. Metabolic and proliferative state of vascular adventitial fibroblasts in pulmonary hypertension is regulated through a MicroRNA-124/PTBP1 (polypyrimidine tract binding protein 1)/pyruvate kinase muscle axis. Circulation. (2017) 136:2468–85. doi: 10.1161/CIRCULATIONAHA.117.028069
58. Patel KP, O'Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. (2012) 106:385–94. doi: 10.1016/j.ymgme.2012.03.017
59. Piao L, Fang Y-H, Cadete VJJ, Wietholt C, Urboniene D, Toth PT, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med. (2010) 88:47–60. doi: 10.1007/s00109-009-0524-6
60. Guignabert C, Tu L, Izikki M, Dewachter L, Zadigue P, Humbert M, et al. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22alpha-targeted overexpression of the serotonin transporter. FASEB J. (2009) 23:4135–47. doi: 10.1096/fj.09-131664
61. Mészáros B, Járvás G, Farkas A, Szigeti M, Kovács Z, Kun R, et al. Comparative analysis of the human serum N-glycome in lung cancer, COPD and their comorbidity using capillary electrophoresis. J Chromatogr B Analyt Technol Biomed Life Sci. (2020) 1137:121913. doi: 10.1016/j.jchromb.2019.121913
62. Ferrer CM, Sodi VL, Reginato MJ. O-GlcNAcylation in cancer biology: linking metabolism and signaling. J Mol Biol. (2016) 428:3282–94. doi: 10.1016/j.jmb.2016.05.028
63. Vigetti D, Deleonibus S, Moretto P, Karousou E, Viola M, Bartolini B, et al. Role of UDP-N-acetylglucosamine (GlcNAc) and O-GlcNAcylation of hyaluronan synthase 2 in the control of chondroitin sulfate and hyaluronan synthesis. J Biol Chem. (2012) 287:35544–55. doi: 10.1074/jbc.M112.402347
64. Aytekin M, Comhair SAA., de la Motte C, Bandyopadhyay SK, Farver CF, Hascall VC, et al. High levels of hyaluronan in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. (2008) 295:L789–L99. doi: 10.1152/ajplung.90306.2008
65. Lauer ME, Aytekin M, Comhair SA, Loftis J, Tian L, Farver CF, et al. Modification of hyaluronan by heavy chains of inter-α-inhibitor in idiopathic pulmonary arterial hypertension. J Biol Chem. (2014) 289:6791–8. doi: 10.1074/jbc.M113.512491
66. Abdul-Salam VB, Wharton J, Cupitt J, Berryman M, Edwards RJ, Wilkins MR. Proteomic analysis of lung tissues from patients with pulmonary arterial hypertension. Circulation. (2010) 122:2058–67. doi: 10.1161/CIRCULATIONAHA.110.972745
67. Chang Y-T, Tseng C-N, Tannenberg P, Eriksson L, Yuan K, de Jesus Perez VA, et al. Perlecan heparan sulfate deficiency impairs pulmonary vascular development and attenuates hypoxic pulmonary hypertension. Cardiovasc Res. (2015) 107:20–31. doi: 10.1093/cvr/cvv143
68. Archer SL, Fang Y-H, Ryan JJ, Piao L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ. (2013) 3:144–52. doi: 10.4103/2045-8932.109960
69. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. (1999) 399:271–5. doi: 10.1038/20459
70. Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. (2010) 121:2661–71. doi: 10.1161/CIRCULATIONAHA.109.916098
71. Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. (1996) 271:32253–9. doi: 10.1074/jbc.271.50.32253
72. Cowburn AS, Crosby A, Macias D, Branco C, Colaço RDDR, Southwood M, et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci USA. (2016) 113:8801–6. doi: 10.1073/pnas.1602978113
73. Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang Y-H, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. (2012) 110:1484–97. doi: 10.1161/CIRCRESAHA.111.263848
74. Shimoda LA, Semenza GL, HIF. and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med. (2011) 183:152–6. doi: 10.1164/rccm.201009-1393PP
75. Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. (2005) 105:659–69. doi: 10.1182/blood-2004-07-2958
76. Patel KP, O'Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. (2012) 105:34–43. doi: 10.1016/j.ymgme.2011.09.032
77. Gordeuk VR, Sergueeva AI, Miasnikova GY, Okhotin D, Voloshin Y, Choyke PL, et al. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. (2004) 103:3924–32. doi: 10.1182/blood-2003-07-2535
78. Bertero T, Lu Y, Annis S, Hale A, Bhat B, Saggar R, et al. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J Clin Invest. (2022) 132:e161077. doi: 10.1172/JCI161077
79. Alastalo T-P, Li M., Perez VdJ, Pham D, Sawada H, Wang JK, et al. Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest. (2011) 121:3735–46. doi: 10.1172/JCI43382
80. Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med. (2011) 208:535–48. doi: 10.1084/jem.20101812
81. Dumas SJ, Bru-Mercier G, Courboulin A, Quatredeniers M, Rücker-Martin C, Antigny F, et al. NMDA-type glutamate receptor activation promotes vascular remodeling and pulmonary arterial hypertension. Circulation. (2018) 137:2371–89. doi: 10.1161/CIRCULATIONAHA.117.029930
82. Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR Yu Q, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest. (2016) 126:3313–35. doi: 10.1172/JCI86387
83. Piao L, Fang Y-H, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med. (2013) 91:1185–97. doi: 10.1007/s00109-013-1064-7
84. Durante W. The emerging role of l-glutamine in cardiovascular health and disease. Nutrients. (2019) 11:2092. doi: 10.3390/nu11092092
85. Ge J, Cui H, Xie N, Banerjee S, Guo S, Dubey S, et al. Glutaminolysis promotes collagen translation and stability via α-ketoglutarate-mediated mTOR activation and proline hydroxylation. Am J Respir Cell Mol Biol. (2018) 58:378–90. doi: 10.1165/rcmb.2017-0238OC
86. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. (2014) 13:890–901. doi: 10.1158/1535-7163.MCT-13-0870
87. Yelamanchi SD, Jayaram S, Thomas JK, Gundimeda S, Khan AA, Singhal A, et al. A pathway map of glutamate metabolism. J Cell Commun Signal. (2016) 10:69–75. doi: 10.1007/s12079-015-0315-5
88. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. (2012) 15:110–21. doi: 10.1016/j.cmet.2011.12.009
89. Nedergaard M, Takano T, Hansen AJ. Beyond the role of glutamate as a neurotransmitter. Nat Rev Neurosci. (2002) 3:748–55. doi: 10.1038/nrn916
90. Litim N, Morissette M, Di Paolo T. Metabotropic glutamate receptors as therapeutic targets in Parkinson's disease: an update from the last 5 years of research. Neuropharmacology. (2017) 115:166–79. doi: 10.1016/j.neuropharm.2016.03.036
91. Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene. (2016) 35:3619–25. doi: 10.1038/onc.2015.447
92. Ahluwalia GS, Grem JL, Hao Z, Cooney DA. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther. (1990) 46:243–71. doi: 10.1016/0163-7258(90)90094-I
93. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. (2009) 136:521–34. doi: 10.1016/j.cell.2008.11.044
94. Randle PJ, Priestman DA, Mistry SC, Halsall A. Glucose fatty acid interactions and the regulation of glucose disposal. J Cell Biochem. (1994) 55(Suppl.):1–11. doi: 10.1002/jcb.240550002
95. Fang Y-H, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle's cycle. J Mol Med. (2012) 90:31–43. doi: 10.1007/s00109-011-0804-9
96. Hou C, Chen J, Zhao Y, Niu Y, Lin S, Chen S, et al. The emerging role of fatty acid synthase in hypoxia-induced pulmonary hypertensive mouse energy metabolism. Oxid Med Cell Longev. (2021) 2021:9990794. doi: 10.1155/2021/9990794
Keywords: PAH, metabolism, mitochondria, randle cycle, glutamine, FAO
Citation: Liu X, Zhang L and Zhang W (2022) Metabolic reprogramming: A novel metabolic model for pulmonary hypertension. Front. Cardiovasc. Med. 9:957524. doi: 10.3389/fcvm.2022.957524
Received: 31 May 2022; Accepted: 26 July 2022;
Published: 26 August 2022.
Edited by:
Olga Tura-Ceide, Hospital Clinic of Barcelona, SpainReviewed by:
Sebastien Bonnet, Laval University, CanadaZhi-Cheng Jing, Peking Union Medical College Hospital (CAMS), China
Copyright © 2022 Liu, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weihua Zhang, d2VpaHVhQGpsdS5lZHUuY24=