
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 05 August 2022
Sec. Heart Failure and Transplantation
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.957006
This article is part of the Research Topic The Heart Lung Disease: A need for novel definitions and understanding of pathological overlaps in the COVID-19 era and beyond View all 5 articles
 Langjiao Liu1Haijiao Jing1Xiaoming Wu1Mengqi Xiang1Valerie A. Novakovic2Shuye Wang1*†
Langjiao Liu1Haijiao Jing1Xiaoming Wu1Mengqi Xiang1Valerie A. Novakovic2Shuye Wang1*† Jialan Shi1,2,3*†
Jialan Shi1,2,3*†The pandemic respiratory illness SARS-CoV-2 has increasingly been shown to be a systemic disease that can also have profound impacts on the cardiovascular system. Although associated cardiopulmonary sequelae can persist after infection, the link between viral infection and these complications remains unclear. There is now a recognized link between endothelial cell dysfunction and thrombosis. Its role in stimulating platelet activation and thrombotic inflammation has been widely reported. However, the procoagulant role of microparticles (MPs) in COVID-19 seems to have been neglected. As membrane vesicles released after cell injury or apoptosis, MPs exert procoagulant activity mainly by exposing phosphatidylserine (PS) on their lipid membranes. It can provide a catalytic surface for the assembly of the prothrombinase complex. Therefore, inhibiting PS externalization is a potential therapeutic strategy. In this paper, we describe the pathophysiological mechanism by which SARS-CoV-2 induces lung and heart complications through injury of endothelial cells, emphasizing the procoagulant effect of MPs and PS, and demonstrate the importance of early antithrombotic therapy. In addition, we will detail the mechanisms underlying hypoxia, another serious pulmonary complication related to SARS-CoV-2-induced endothelial cells injury and discuss the use of oxygen therapy. In the case of SARS-CoV-2 infection, virus invades endothelial cells through direct infection, hypoxia, imbalance of the RAAS, and cytokine storm. These factors cause endothelial cells to release MPs, form MPs storm, and eventually lead to thrombosis. This, in turn, accelerates hypoxia and cytokine storms, forming a positive feedback loop. Given the important role of thrombosis in the disease, early antithrombotic therapy is an important tool for COVID-19. It may maintain normal blood circulation, accelerating the clearance of viruses, waning the formation of MPs storm, and avoiding disease progression.
SARS-CoV-2 was initially thought to be a disease that primarily affected the lungs, but as data accumulated, it emerged that it could also affect other organs and cause multiple organ dysfunction. The most prominent extrapulmonary complication is cardiovascular disease (1). And its occurrence is often closely associated with lung disease (2). While in some patients, the virus may directly infect the target organ through the angiotensin-converting-enzyme 2 (ACE2) receptor, resulting in extrapulmonary complications, in most patients, SARS-CoV-2 first invades the lungs and then progresses to more severe multi-organ failure. Because many patients show only respiratory symptoms (fever, cough, etc.) in the initial stage of infection (3). Thrombosis plays an integral role in pulmonary and cardiovascular complications (4). Ackermann et al. reported that histologic analysis of COVID-19 patients showed widespread thrombosis with microangiopathy, and that the prevalence of microthrombi in COVID-19 patients was nine times higher than in flu patients (5). The same was true for cardiovascular disease. In the laboratory, myocardial injury is associated with coagulation indexes such as elevated D-dimer (6). An experimental study also indicated that myocardial fibrin microthrombosis was common in patients with COVID-19 (7).
Dysfunction of endothelial cells is considered to be the main factor for thrombosis (8). Under normal conditions, endothelial cells play an anticoagulant role through anti-platelet adhesion, synthesis of antithrombin and other coagulation factors, promotion of fibrinolysis and barrier protection. Viral infection causes endothelial cell damage, resulting in disorders of the coagulation and fibrinolytic systems, ultimately leading to thrombosis (9). However, we found that MPs were rarely mentioned in COVID-19 studies. MPs are membrane vesicles released when endothelial cells are activated, damaged or undergoing apoptosis (10). MPs participate in inflammation and coagulation through cellular components such as proteins, lipids, nucleic acids and PS (11–13). The PS exposed on the surface of MPs provides binding sites for endogenous and exogenous FXase complexes and the prothrombin complex, provides a platform for the coagulation cascade, thereby promoting clot formation. Our team has shown in previous studies that when endothelial cells are damaged, PS exposure results leads to abnormalities in the blood coagulation system (14, 15). Therefore, understanding the physiology of endothelial cells injury and the pro-coagulant activity of PS externalization could provide a new prospect for the treatment of COVID-19.
Before there were no specific drugs, antithrombotic therapy for COVID-19 reached a consensus (16). Many studies have also shown that thrombotic-related sequelae (such as pulmonary fibrosis, chest pain, etc.) can occur in COVID-19 patients during convalescence (17, 18). The normal function of blood circulation is to provide oxygen and nutrients to the body and to remove metabolites. After thrombosis, blood vessel blockage hampers the clearance of metabolites and viruses in a timely and efficient manner, aggravating the progression of the disease. Therefore, early antithrombotic therapy is especially important. Unobstructed blood circulation speeds recovery by facilitating the removal of viruses and cytokines, preventing COVID-19 patients from developing serious complications and therefore reducing mortality. Rentsch's study has shown that early initiation of prophylactic anticoagulation reduced 30-day mortality with no increased risk of serious bleeding (19), supporting our opinion.
Lungs are the primary target of SARS-CoV-2 invasion. Most people with mild symptoms have fever, cough and difficulty breathing, but a small number of patients, 5–10%, will develop severe respiratory failure and acute respiratory distress syndrome (ARDS) (20–22). According to relevant literature, the mortality rate of ARDS in COVID-19, at 26–61.5%, is higher than that of ARDS secondary to other diseases (23). Many studies suggest that pulmonary embolism is also very common in COVID-19 patients. Poissy et al. showed that pulmonary embolism (PE) incidence was 20.6% in COVID-19 patients in 1 month in 2020, which was significantly higher than the incidence of PE for other causes of admission during the same period (24). More frightening, many data confirm that some patients still have long-term pulmonary sequelae after discharge from the hospital, including some patients diagnosed with only mild acute phase disease (25). A cohort study by Mo et al. noted that impaired diffusion ability was the most common abnormal lung function among discharged COVID-19 survivors (47.2%) (26). Frija-masson et al. reported an isolated reduction in lung diffusion capacity in 13 of 50 patients (26%) (27). In addition, Cueto-Robledo et al. found severe pulmonary hypertension in patients discharged from hospital (28). The pathogenesis of long COVID-19 is still unknown. Persistent viral shedding (29), inflammatory environment (17), and coagulation issues may be involved in sequelae-related pathologic processes (30).
Cardiovascular complications in COVID-19 occur at a high rate and tend to have a poor prognosis. Observational studies have shown that about 5.3% of patients with COVID-19 developed new-diagnosed secondary acute myocardial infarction (AMI) (31). In addition, related reports have shown that 33.3–39.3% were diagnosed with non-obstructive coronary artery disease (32, 33). This suggests that COVID-19 itself may be associated with endothelial dysfunction and hypercoagulability (34). Arrhythmias are a common manifestation of COVID-19 cardiovascular complications. In a study of 138 patients in China, new atrial fibrillation, heart block and ventricular arrhythmias are common in COVID-19, with an incidence of 44.4% in 36 intensive care unit (ICU) patients (35). Arrhythmia is caused by multiple factors. In addition to drugs (such as hydroxychloroquine) and inflammation (36), it can also be secondary to myocardial injury. According to the survey, malignant arrhythmias are more frequent in patients with elevated TnT levels during hospitalization (37). An increased risk of clinical acute myocardial ischemia was also associated with COVID-19. In a large, controlled trial, the incidence of acute myocardial ischemia after COVID-19 diagnosis was five times higher than in the control group (38). Heart failure, myocarditis, and cardiogenic shock have also been reported (39–41). There are also many reports about the sequelae associated with cardiovascular thrombosis. Tschöpe et al. and Fan et al. reported on several cases of coronary angiography of patients discharged from hospital with COVID-19, including left anterior descending artery obstruction in one patient and left anterior descending artery obstruction (in 25% of participants) (42, 43). Ayoubkhani and others also reported a composite outcome (major adverse cardiovascular event), including heart failure, myocardial infarction, stroke and arrhythmia (44).
Pulmonary and cardiovascular complications in COVID-19 are often correlated. Heart and lung are inseparable organs in that they relate to the two major circulation pathways of the human body: systemic circulation and pulmonary circulation. As lung disease progresses, it can cause heart stress and vice versa. Mancini et al. found that circulatory damage caused by changes in long COVID related cardiac function also included reduced pulmonary perfusion (suggesting that it might be related to dyspnea), which in turn resulted in reduced cardiac output and further burdened the heart (45). Several other studies have identified right ventricular dysfunction and diastolic dysfunction as being a result of fibrous lung injury, pulmonary hypertension, and clot burden in critically ill patients (46–48). Guo et al. found that ARDS occurred more frequently in COVID-19 patients with myocardial injury [30 [57.7%] vs. 16 [11.9%]] (49). This evidence confirms the heart-lung connection. As previously mentioned, thrombosis is a major factor in cardiopulmonary complications in COVID-19 patients with endothelial cells injury being a common mechanism bridging the two systems. Von Willebrand Factor (vWF), a major marker of endothelial cell dysfunction, was found to be significantly elevated in laboratory examination of COVID-19 patients, and its value was positively correlated with the severity of the disease (50, 51). The virus invades the lung through the respiratory tract and mainly infects alveolar type II epithelial cells, causing immune cells to release inflammatory mediators and form cytokine storms, resulting in local hypoxia (52). At the same time, some of the virus escapes the phagocytosis of immune cells and further invades the adjacent pulmonary endothelial cells (Figure 1A). Under the direct invasion of virus, hypoxia, imbalance of Renin-angiotensin-aldosterone system (RAAS) and cytokine storm, the integrity and permeability of endothelial cells are damaged (Figure 1B). Damaged endothelial barrier combined with cytokine storm can promote the formation of acute endodermatitis in the lung and extend into systemic circulation, resulting in intravascular blockage and thrombosis that may lead to myocardial injury, ischemic heart disease and other diseases (Figure 1C). The blockage of the blood vessels in turn increases dead-space ventilation, further exacerbating hypoxia, creating a vicious cycle (53).
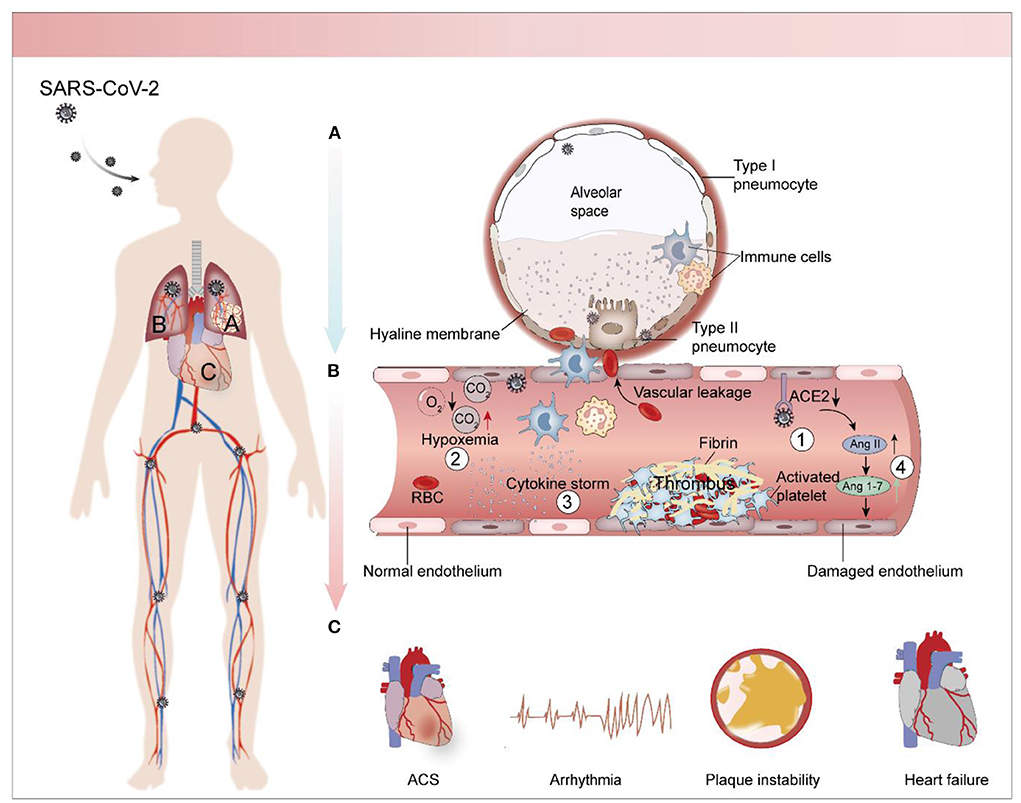
Figure 1. Pathological mechanism of cardiopulmonary complications caused by SARS-CoV-2. (A) Pathophysiology of SARS-CoV-2-infected lungs. The virus infects alveolar type II epithelial cells and pulmonary capillary endothelial cells through the respiratory tract and stimulates immune cells to release inflammatory factors, causing cytokine storm. As viral replication enters into the bloodstream, it stimulates endothelial cell shrinkage and narrows the pulmonary capillary lumen. This causes a local lack of oxygen to the lungs. Endothelial cells damage worsens as the disease progresses, stimulating clotting cascades in the pulmonary capillaries, which leads to further increases in capillary pressure and resulting in pulmonary hypertension. This causes the release of plasma, along with inflammatory cells and coagulation factors, into the alveolar cavities. Evaporation concentrates these substances and forms a peptone-like translucent film in the alveolar cavity. (B) Endothelial cells injury and thrombosis. Endothelial cells injury caused by SARS-CoV-2 is multifactorial. Some viruses infect endothelial cells through ACE2 receptor, and indirect mechanisms including hypoxia, RAAS imbalance and cytokine storm also aggravate endothelial cells injury. The injured endothelial cells stimulate the coagulation cascade and promote the formation of thrombosis. (C) Cardiac complications associated with COVID-19. Including arrhythmias, heart failure, coronary syndrome, which are often closely associated with thrombosis.
The main pathophysiological mechanisms of SARS-CoV-2 injury to endothelial cells include direct mechanisms (direct viral invasion) and indirect mechanisms (hypoxia, RASS imbalance, cytokine storm).
The entry of SARS-CoV-2 into cells is mediated by the recognition of the host cell ACE2 receptor, which requires the mediation of transmembrane protease receptor serinase 2 (TMPRSS2) (54). ACE2 receptors are expressed in multiple organs, such as the lung, heart, kidney, intestine, etc., primarily on endothelial cells (55, 56). Varga's team found viral particles in the renal endothelial cells of COVID-19 patients and noted that direct viral infection through the endothelium may lead to extensive endothelial dysfunction and apoptosis (57). Similarly, Ackermann et al. examining autopsies of lungs from patients who died of COVID-19 demonstrated that viral infection increases endothelial ACE2 expression (5). However, it remains controversial whether SARS-CoV-2 directly infects endothelial cells through ACE2 receptor. Some studies have argued against this view, suggesting that endothelial ACE2 has a low basal expression level and may escape SARS-CoV-2-mediated cell entry (58). Yet there's also support for the presence of ACE2 receptors on endothelial cells. In one study, Ahmetaj-Shala et al. reported detectable levels of ACE2 transcripts in human endothelial cells by reanalyzing the human tissue transcriptome database (59). A representative study by Lei et al. showed that the S protein of SARS-CoV-2 could damage vascular endothelial cells by down-regulating ACE2 (60). Qin et al. demonstrated SARS-CoV-2 in capillary endothelial cells using RNA-seq analysis in a mouse model of COVID-19 (61). A single-cell RNA-seq analysis of non-human primates also showed increased expression of SARS-CoV-2 receptor ACE2 in alveolar epithelial barriers, cardiomyocytes, and vascular endothelial cells (62). Therefore, the role of direct viral infection in endothelial cell injury needs to be further studied.
In the early stage of COVID-19, a small number of SARS-CoV-2 viruses infect nasal cilia cells and secretory cells through the air, activating the innate immune response. Further some active viruses escape the clearance of immune cells and enter the alveolar cavity through the trachea. The virus then infects the type II epithelial cells of the alveoli and the adjacent endothelial cells of the air-blood barrier. This process causes alveolar space damage, recruiting a variety of immune cells which release inflammatory cytokines and cause a cytokine storm (63). Alveolar capillary endothelial cell injury and cytokine storm activate the coagulation cascade to form pulmonary microthrombosis, resulting in an imbalance of pulmonary ventilation and blood flow ratio, leading to local pulmonary hypoxia (64). When viral replication reaches the bloodstream, it stimulates endothelial cell shrinkage, narrowing the pulmonary capillary lumen. This increases pressure in the capillary network, eventually resulting in pulmonary hypertension, which forces plasma into the alveolar cavities along with inflammatory cells and blood coagulation factors. As some of the water in the alveolar cavity evaporates, these substances form a peptone-like translucent film in the alveolar cavity, severely impairing the function of the gas and blood barrier (65). This causes blood with low oxygen saturation to move from the capillary network of the alveolar wall into systemic circulation and causing serious hypoxia.
RAAS is a pressure-boosting regulation network produced by the kidneys, causing vascular smooth muscle contraction, water and sodium retention and resulting in pressure-boosting effects. Under normal circumstances, ACE2 promotes the degradation of Ang II and maintains equilibrium (66). However, due to ACE2 depletion in COVID-19 patients, AngII levels are elevated, causing vasoconstriction and enhanced vascular permeability, thereby damaging endothelial cells (67).
Cytokine storm can not only promote the formation of hypoxia, but also directly cause endothelial injury (68). Wu et al. confirmed that IL-1β and TNF-α (TH17 and TH1 cells express) promote TH17 response. They increase endothelial permeability and vascular leakage (69). High levels of TNF-α and IL-1β may also down-regulate the expression of Kluber-like factor 2 (KLF2) in human endothelial cells, leading to monocyte adhesion and endodermatitis (70). In addition, activated neutrophils have been shown to release neutrophil extracellular traps (Nets) in COVID-19 patients. In an experimental study of COVID-19, Nets production was positively associated with disease severity and mortality. Originally produced to kill pathogens, Nets can also activate endothelial cells when produced in excess (71). Meanwhile, Laforge et al. pointed out that in COVID-19, neutrophils produce excessive ROS, which intensifies the host's immunopathological response, as well as tissue damage, leading to more severe disease (72).
After the activation or injury of endothelial cells, the most serious consequence is the formation of thrombi. First, endothelial cells are damaged and shed, exposing collagen, activating platelets and promoting their adhesion and aggregation through vWF released by endothelial cells, forming platelet thrombosis. At the same time, MPs released from damaged endothelial cells participate in the clotting response by exposing PS on their surfaces, promoting fibrinogen production and further consolidating platelet thrombosis. In addition to procoagulant factors, damaged endothelial cells can secrete fibrinogen activator inhibitor 1 (PAI-1), which inhibits fibrinolysis (73). Thrombin can also induce activation of endothelial cells through protease activated receptor 1 (PAR1) (74), which in turn aggravates the injury of endothelial cells.
PS is a glycerolipid that makes up part of the cell membrane. Under normal conditions, PS is mainly distributed in the inner layer of the plasma membrane. This asymmetric distribution is maintained through the action of three enzymes: ATP-dependent flippase and floppase, and ATP-independent but Ca2+ dependent scramblase. When endothelial cells are damaged, the two ATP-dependent enzymes are unable to function due to energy depletion, but intracellular Ca2+ concentration increases. The activated scramblase flips PS to the outer membrane where it initiates coagulation (75). The specific mechanism is as follows: on the one hand, PS provides a scaffold for the activity of factors VIII and V, which binding membranes through their C2 domains, and factors II, VII, IX, and X, which bind using their Gla domains. These factors combine to form the intrinsic (FXIa-FVIIIa-Ca2+-PL) factor X enzyme complex and the prothrombin complex (FXa-FVa-Ca2+-PL) (76–80). On the other hand, damaged endothelial cells can upregulate the expression of tissue factor, which is activated in the presence of membrane PS and then forms the TF-VIIa-Ca2+-PL complex that participates in exogenous coagulation (81). The coagulation cascade leads to the conversion of prothrombin to thrombin which then catalyzes the formation of a fibrin network (Figure 2A). In this way, fibrin and platelet thrombosis form a stronger thrombus. With endothelial cell damage in severe COVID-19 patients, a large number of PS+ MPs are released, forming an MPs storm and ultimately causing serious thrombotic events (Figure 2B).
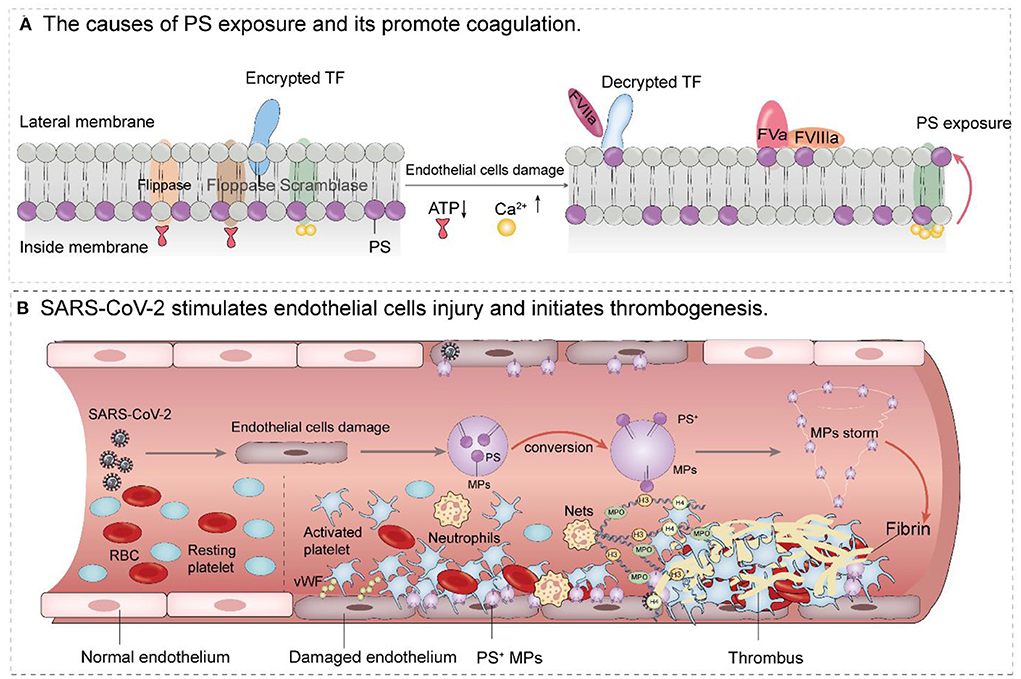
Figure 2. Mechanism of PS+ MPs promoting thrombosis after endothelial cells injury. (A) When endothelial cells are damaged, the two ATP-dependent enzymes are unable to function due to energy depletion. However, as intracellular Ca2+ concentration, the activated scramblase flips PS to the outer membrane where it initiates coagulation PS provides a scaffold for the activity of factors VIII and V, which binding membranes through their C2 domains, and factors II, VII, IX, and X, which bind using their Gla domains. These factors combine to form the intrinsic (FXIa-FVIIIa-Ca2+-PL) factor X enzyme complex and the prothrombin complex (FXa-FVa-Ca2+-PL) Damaged endothelial cells can also upregulate the expression of tissue factor, which is activated in the presence of membrane PS and then forms the TF-VIIa complex that participates in exogenous coagulation. (B) SARS-CoV-2 causes endothelial dysfunction and thrombosis. After endothelial cell injury, stimulated platelets were transformed from the resting state to the activated state, and PS+ microparticles were released from the surface of endothelial cells to form a particle storm. Platelet adhesion, coupled with PS exposure, stimulates the activation of the clotting cascade, which ultimately leads to thrombosis (The involvement of neutrophils and Nets in endothelial cell injury and thrombosis is also shown in B).
In conclusion, whether in patients with pre-existing comorbidities or in previously healthy patients, the heart and lung complications in COVID-19 are essentially caused by thrombosis. We have mentioned that the virus causes the endothelial cells destruction which leads to thrombosis. In turn, thrombosis exacerbates the vicious cycle of hypoxia and endothelial damage which eventually aggravates the progression of the disease. Therefore, in the absence of specific drugs for the elimination of SARS-CoV-2, in addition to conventional treatments such as antiviral drugs, attention should be paid to antithrombotic therapies, oxygenation and preventing endothelial cells damage.
The therapeutic benefits of anticoagulation in COVID-19 are widely accepted. Low molecular weight heparin (LWMN) is currently the most commonly used anticoagulant for COVID-19. Heparin is recommended because aside from its powerful blood-thinning properties, it also has anti-inflammatory and antiviral effects, and can be injected subcutaneously to reduce the adverse reactions of the drug (82) (a study has confirmed that SARS-CoV-2 spike protein S1 subunit contains a receptor binding domain structure that can interact with heparin) (83). Meanwhile, inhibition of platelet activation has also been reported to have beneficial effects. A recent study demonstrated that in-hospital aspirin use was associated with a lower incidence of in-hospital death (84). Antiplatelet drugs include the thrombotic inhibitor aspirin, P2Y12 inhibitor clopidogrel, phosphodiester RAASe inhibitor dipyridamole, among others (85). Aspirin is the drug of choice for the prevention of arterial thrombosis in high-risk patients with cardiovascular risk factors (86). Thrombolytic therapy is more controversial. In one study however, the use of tissue plasminogen activator (tPA) improved oxygenation in patients with severe COVID-19 (87). Therefore, more exploration of the use of thrombolytic therapy in severely ill patients is warranted.
However, the timing of anticoagulant therapy remains controversial. A randomized controlled trial (RCT) showed that therapeutic anticoagulant therapy did not reduce mortality in severe COVID-19 patients (88). Similarly, another study showed that in patients with severe COVID-19, treatment with antiplatelet drugs was less likely to improve the number of days without organ support over 21 days than treatment without antiplatelet drugs (89). In contrast, in another report of 465 patients with moderate COVID-19, therapeutic heparin reduced mortality at 28 days (23 patients with therapeutic heparin (10.1%) vs. 38 patients (16.0%) given prophylactic heparin). And the risk of major bleeding seems low [Two (0.9%) patients received therapeutic heparin and four (1.7%) patients received prophylactic heparin] (90). Another retrospective analysis of 413 patients showed that early anticoagulant therapy with relatively high doses of LMWH improved clinical outcomes and shortened hospital stays for COVID-19 patients (91). Gonzalez-ochoa et al. reported that early use of Sulodexide improved clinical outcomes. Compared with the control group, sulodexide had a lower hospitalization rate (17.7 vs. 29.4% [P = 0.03]) and fewer patients requiring supplemental oxygen therapy (30 vs. 42% [P = 0.05]) (30% vs. 42% [P = 0.05]). After 2 weeks, d-dimer levels >500 ng/dL were reduced (22 vs. 47% [P < 0.01]) and there was no difference in thromboembolic events, major bleeding, and mortality (92). The above results highlight the necessity of early antithrombotic therapy. In fact, although the symptoms of early-stage patients are mild, and D-dimer is normal or only slightly increased, it is still possible that fibrinolytic activity is present and that the body is in the early stages of coagulation activation. Early endothelial cells damage and PS externalization may be too subtle to be detectible at this stage. Our main task is to prevent further externalization of PS to prevent further clotting response. Therefore, early antithrombotic therapy is necessary. It guarantees the alveolar has effective blood perfusion, avoids endothelial to activation, protects the air/blood exchange, ensures unobstructed pulmonary circulation, and maintains saturated blood oxygen levels throughout the systemic circulation, which prevents endothelial cells damage and thrombosis. In patients with advanced stage disease, there is already severe thrombosis in the body, thus cytokines and viruses cannot be cleared in time, further aggravating the difficulty of treatment.
Vaccination is well-known to lessen the severity of acute phase COVID-19, and some reports also indicate that Long-COVID symptoms are less prevalent and milder in vaccinated people. A case-control study involving 1.2 million people who received two doses of vaccine had a lower chance of having symptoms lasting 28 days or longer (odds ratio 0.51 [95% CI 0.32–0.82]; P = 0.0060) (93). However, long-term sequelae are still found in some vaccinated patients (94). The main reason why vaccination is ineffective for some patients is that the pathogenesis of COVID-19 and Long COVID is multifactorial. Persistent viral infection and thrombosis are the main factors. Therefore, we still advocate early antithrombotic therapy in vaccinated patients.
Hypoxemia induced by COVID-19 plays an important role in the process of endothelial cells damage, such that the improvement of blood oxygen saturation can also improve the course of the disease. A controlled trial showed that high-flow oxygen significantly reduces the need for mechanical ventilation support in severe COVID-19. Compared with those receiving conventional oxygen therapy, the incidence of endotracheal intubation was reduced (34.3 vs. 51.0 %) and the median 28-day clinical recovery time was shorter (11 vs. 14 days) in patients receiving high-flow oxygen therapy (95).
Our group has previously demonstrated that lactadherin can specifically bind to PS. Because lactadherin contains a C2 domain homologous to those on some clotting factors, it can act both as a probe and a competitive inhibitor to factor V and VIII binding. The structure of lactadherin also includes an RGD domain that has been shown to act as an “eat me” signal that could aid in the clearance of PS-exposing damaged or apoptotic cells. Although there are few reports on the application of lactadherin in COVID-19, the procoagulant effect of PS in COVID-19 cannot be ignored. Therefore, lactadherin is a promising agent to study in terms of inhibiting the effects of PS externalization (79, 80, 96).
In previous influenza studies, statins have been shown to reduce endothelial cell activation, inhibit procoagulant pathways, and enhance antithrombotic effects. A retrospective study has shown that in-hospital statin use is associated with a reduced risk of death among COVID-19 patients. In this article, Cox models were used to find that the 28-day all-cause mortality was 5.2% in the statin group and 9.4% in the non-statin group, with an adjusted hazard ratio of 0.58 (97).
ACE2 and TMPRSS2, as important vectors of SARS-CoV-2, are potential therapeutic targets. Damir Bojadzic et al. demonstrated that several small molecule inhibitors targeting the SARS-CoV-2 spike protein and human ACE2 (hACE2) prevented two different SARS-CoV-2-S-expressing pseudoviruses from entering hACE2-expressing cells (98). In a mouse model, TMPRSS2 inhibitor (nafamostat) protected mice against SARS-CoV-2 infection and subsequent COVID-19 lung disease. These results represent preclinical evidence for the use of such treatments in COVID-19 (99).
Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II-1 receptor blockers (ARBs) are widely used in the treatment of arterial hypertension, heart failure and chronic kidney disease. Zhang et al. have confirmed that, the inpatient use of ACE inhibitor/ARB was associated with a lower all-cause mortality compared with non-users among hospitalized COVID-19 patients with hypertension (100).
Cytokine storm is an important mechanism of COVID-19. Currently, the research drugs for COVID-19 are mainly focused on corticosteroids. A study involving 2,104 patients with COVID-19 showed that patients with invasive mechanical ventilation or oxygen therapy and without invasive mechanical ventilation had a significantly lower 28-day mortality after receiving glucocorticoids (6 mg/d, for up to 10 days) compared to the normal care group (101).
As the COVID-19 pandemic persists and with the continued high incidence of cardiopulmonary complications in COVID-19 patients, we emphasize the key role that endothelial cells injury plays. By understanding its specific physiological and pathological mechanism and the current research into experimental clinical drugs, we believe that the rate of patient survival will be significantly improved. As many patients present with existing cardiovascular disease, care must be taken to perform a comprehensive evaluation paying particular attention to the risk of adverse drug interactions. More importantly, a review of previous pandemics has shown that thrombosis occurs whenever endothelial cell damage is present. The crucial role of PS in activating coagulation has led us to conclude that inhibiting PS externalization with early antithrombotic therapy can reduce mortality.
LL prepared figures and wrote the manuscript. HJ, XW, and MX provided helpful comments and acquired data. JS and SW came up with the project, designed the study, contributed to successive drafts, and reviewed this manuscript. VN gave the revision advice and polished this review. All authors read and approved the final manuscript.
We thank all participants for their contribution in our study and the reviewers for the suggestions provided.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lan Y, Liu W, Zhou Y. Right ventricular damage in COVID-19: association between myocardial injury and COVID-19. Front Cardiovasc Med. (2021) 8:606318. doi: 10.3389/fcvm.2021.606318
2. Puri G, Singh VP, Naura AS. COVID-19 severity: lung-heart interplay. Curr Cardiol Rev. (2021) 17:e230421189016. doi: 10.2174/1573403X16999201210200614
3. Ning Q, Wu D, Wang X, Xi D, Chen T, Chen G, et al. The mechanism underlying extrapulmonary complications of the coronavirus disease 2019 and its therapeutic implication. Signal Transduct Target Ther. (2022) 7:57. doi: 10.1038/s41392-022-00907-1
4. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. (2020) 135:2033–40. doi: 10.1182/blood.2020006000
5. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
6. Italia L, Tomasoni D, Bisegna S, Pancaldi E, Stretti L, Adamo M, et al. COVID-19 and heart failure: from epidemiology during the pandemic to myocardial injury, myocarditis, and heart failure sequelae. Front Cardiovasc Med. (2021) 8:713560. doi: 10.3389/fcvm.2021.713560
7. Bois MC, Boire NA, Layman AJ, Aubry MC, Alexander MP, Roden AC, et al. COVID-19-associated nonocclusive fibrin microthrombi in the heart. Circulation. (2021) 143:230–43. doi: 10.1161/CIRCULATIONAHA.120.050754
8. Jin Y, Ji W, Yang H, Chen S, Zhang W, Duan G. Endothelial activation and dysfunction in COVID-19: from basic mechanisms to potential therapeutic approaches. Signal Transduct Target Ther. (2020) 5:293. doi: 10.1038/s41392-020-00454-7
9. Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. (2020) 41:3038–44. doi: 10.1093/eurheartj/ehaa623
10. Che Mohd Nassir CMN, Hashim S, Wong KK, Abdul Halim S, Idris NS, Jayabalan N, et al. COVID-19 infection and circulating microparticles-reviewing evidence as microthrombogenic risk factor for cerebral small vessel disease. Mol Neurobiol. (2021) 58:4188–215. doi: 10.1007/s12035-021-02457-z
11. Rausch L, Lutz K, Schifferer M, Winheim E, Gruber R, Oesterhaus EF, et al. Binding of phosphatidylserine-positive microparticles by PBMCs classifies disease severity in COVID-19 patients. J Extracell Vesicles. (2021) 10:e12173. doi: 10.1002/jev2.12173
12. Garnier Y, Claude L, Hermand P, Sachou E, Claes A, Desplan K, et al. Plasma microparticles of intubated COVID-19 patients cause endothelial cell death, neutrophil adhesion and netosis, in a phosphatidylserine-dependent manner. Br J Haematol. (2022) 196:1159–69. doi: 10.1111/bjh.18019
13. Chatterjee V, Yang X, Ma Y, Wu MH, Yuan SY. Extracellular vesicles: new players in regulating vascular barrier function. Am J Physiol Heart Circ Physiol. (2020) 319:H1181–96. doi: 10.1152/ajpheart.00579.2020
14. Cao M, Li T, He Z, Wang L, Yang X, Kou Y, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood. (2017) 129:1855–64. doi: 10.1182/blood-2016-09-739334
15. He Z, Si Y, Jiang T, Ma R, Zhang Y, Cao M, et al. Phosphotidylserine exposure and neutrophil extracellular traps enhance procoagulant activity in patients with inflammatory bowel disease. Thromb Haemost. (2016) 115:738–51. doi: 10.1160/TH15-09-0710
16. McFadyen JD, Stevens H, Peter K. The emerging threat of (micro)thrombosis in COVID-19 and its therapeutic implications. Circ Res. (2020) 127:571–87. doi: 10.1161/CIRCRESAHA.120.317447
17. Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med. (2021) 27:601–15. doi: 10.1038/s41591-021-01283-z
18. Dhawan RT, Gopalan D, Howard L, Vicente A, Park M, Manalan K, et al. Beyond the clot: perfusion imaging of the pulmonary vasculature after COVID-19. Lancet Respir Med. (2021) 9:107–16. doi: 10.1016/S2213-2600(20)30407-0
19. Rentsch CT, Beckman JA, Tomlinson L, Gellad WF, Alcorn C, Kidwai-Khan F, et al. Early initiation of prophylactic anticoagulation for prevention of coronavirus disease 2019 mortality in patients admitted to hospital in the United States: cohort study. BMJ. (2021) 372:n311. doi: 10.1136/bmj.n311
20. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
21. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
22. Li X, Zeng W, Li X, Chen H, Shi L, Li X, et al. CT imaging changes of corona virus disease 2019(COVID-19): a multi-center study in Southwest China. J Transl Med. (2020) 18:154. doi: 10.1186/s12967-020-02324-w
23. Hashemian SM, Shafigh N, Afzal G, Jamaati H, Tabarsi P, Marjani M, et al. Plasmapheresis reduces cytokine and immune cell levels in COVID-19 patients with acute respiratory distress syndrome (ARDS). Pulmonology. (2021) 27:486–92. doi: 10.1016/j.pulmoe.2020.10.017
24. Poissy J, Goutay J, Caplan M, Parmentier E, Duburcq T, Lassalle F, et al. Pulmonary embolism in patients with COVID-19: awareness of an increased prevalence. Circulation. (2020) 142:184–6. doi: 10.1161/CIRCULATIONAHA
25. Lund LC, Hallas J, Nielsen H, Koch A, Mogensen SH, Brun NC, et al. Post-acute effects of SARS-CoV-2 infection in individuals not requiring hospital admission: a Danish population-based cohort study. Lancet Infect Dis. (2021) 21:1373–82. doi: 10.1016/S1473-3099(21)00211-5
26. Mo X, Jian W, Su Z, Chen M, Peng H, Peng P, et al. Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur Respir J. (2020) 55:2001217. doi: 10.1183/13993003.01217-2020
27. Frija-Masson J, Debray MP, Gilbert M, Lescure FX, Travert F, Borie R, et al. Functional characteristics of patients with SARS-CoV-2 pneumonia at 30 days post-infection. Eur Respir J. (2020) 56:2001754. doi: 10.1183/13993003.01754-2020
28. Cueto-Robledo G, Porres-Aguilar M, Puebla-Aldama D, Barragán-Martínez MDP, Jurado-Hernández MY, García-César M, et al. Severe pulmonary hypertension: an important sequel after severe post-acute COVID-19 pneumonia. Curr Probl Cardiol. (2022) 47:101004. doi: 10.1016/j.cpcardiol.2021.101004
29. Tarhini H, Recoing A, Bridier-Nahmias A, Rahi M, Lambert C, Martres P, et al. Long-term severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infectiousness among three immunocompromised patients: from prolonged viral shedding to SARS-CoV-2 superinfection. J Infect Dis. (2021) 223:1522–7. doi: 10.1093/infdis/jiab075
30. Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19 (pasc): an overview of biological factors that may contribute to persistent symptoms. Front Microbiol. (2021) 12:698169. doi: 10.3389/fmicb.2021.698169
31. Li Y, Liu T, Tse G, Wu M, Jiang J, Liu M, et al. Electrocardiograhic characteristics in patients with coronavirus infection: a single-center observational study. Ann Noninvasive Electrocardiol. (2020) 25:e12805. doi: 10.1111/anec.12805 Epub 2020 Sep 20
32. Stefanini GG, Montorfano M, Trabattoni D, Andreini D, Ferrante G, Ancona M, et al. ST-elevation myocardial infarction in patients with COVID-19: clinical and angiographic outcomes. Circulation. (2020) 141:2113–6. doi: 10.1161/CIRCULATIONAHA.120.047525
33. Bangalore S, Sharma A, Slotwiner A, Yatskar L, Harari R, Shah B, et al. ST-segment elevation in patients with COVID-19-a case series. N Engl J Med. (2020) 382:2478–80. doi: 10.1056/NEJMc2009020
34. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
35. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
36. Mazzanti A, Briani M, Kukavica D, Bulian F, Marelli S, Trancuccio A, et al. Association of hydroxychloroquine with qtc interval in patients with COVID-19. Circulation. (2020) 142:513–5. doi: 10.1161/CIRCULATIONAHA.120.048476
37. Khalid M, Awan S, Jatoi NN, Jatoi HN, Yasmin F, Ochani RK, et al. Cardiac manifestations of the coronavirus disease-19: a review of pathogenesis, clinical manifestations, diagnosis, and treatment. Pan Afr Med J. (2021) 39:173. doi: 10.11604/pamj.2021.39.173.27802
38. Modin D, Claggett B, Sindet-Pedersen C, Lassen MCH, Skaarup KG, Jensen JUS, et al. Acute COVID-19 and the incidence of ischemic stroke and acute myocardial infarction. Circulation. (2020) 142:2080–2. doi: 10.1161/CIRCULATIONAHA.120.050809
39. Adeghate EA, Eid N, Singh J. Mechanisms of COVID-19-induced heart failure: a short review. Heart Fail Rev. (2021) 26:363–9. doi: 10.1007/s10741-020-10037-x
40. Siripanthong B, Nazarian S, Muser D, Deo R, Santangeli P, Khanji MY, et al. Recognizing COVID-19-related myocarditis: the possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm. (2020) 17:1463–71. doi: 10.1016/j.hrthm.2020.05.001
41. Chau VQ, Giustino G, Mahmood K, Oliveros E, Neibart E, Oloomi M, et al. Cardiogenic shock and hyperinflammatory syndrome in young males with COVID-19. Circ Heart Fail. (2020) 13:e007485. doi: 10.1161/CIRCHEARTFAILURE.120.007485
42. Tschöpe C, Sherif M, Anker MS, Geisel D, Kuehne T, Kelle S. COVID-19-convalescence phase unmasks a silent myocardial infarction due to coronary plaque rupture. ESC Heart Fail. (2021) 8:971–3. doi: 10.1002/ehf2.13186
43. Fan BE, Umapathi T, Chua K, Chia YW, Wong SW, Tan GWL, et al. Delayed catastrophic thrombotic events in young and asymptomatic post COVID-19 patients. J Thromb Thrombolysis. (2021) 51:971–7. doi: 10.1007/s11239-020-02332-z
44. Ayoubkhani D, Khunti K, Nafilyan V, Maddox T, Humberstone B, Diamond I, et al. Post- COVID syndrome in individuals admitted to hospital with COVID-19: retrospective cohort study. BMJ. (2021) 372:n693. doi: 10.1136/bmj.n693
45. Mancini DM, Brunjes DL, Lala A, Trivieri MG, ContreRAAS JP, Natelson BH. Use of cardiopulmonary stress testing for patients with unexplained dyspnea post-coronavirus disease. JACC Heart Fail. (2021) 9:927–37. doi: 10.1016/j.jchf.2021.10.002
46. Tudoran C, Tudoran M, Pop GN, Giurgi-Oncu C, Cut TG, Lazureanu VE, et al. Associations between the severity of the post-acute COVID-19 syndrome and echocardiographic abnormalities in previously healthy outpatients following infection with SARS-CoV-2. Biology. (2021) 10:469. doi: 10.3390/biology10060469
47. Bende F, Tudoran C, Sporea I, Fofiu R, Bâldea V, Cotrău R, et al. A multidisciplinary approach to evaluate the presence of hepatic and cardiac abnormalities in patients with post-acute COVID-19 syndrome-a pilot study. J Clin Med. (2021) 10:2507. doi: 10.3390/jcm10112507
48. Sonnweber T, Sahanic S, Pizzini A, Luger A, Schwabl C, Sonnweber B, et al. Cardiopulmonary recovery after COVID-19: an observational prospective multicentre trial. Eur Respir J. (2021) 57:2003481. doi: 10.1183/13993003.03481-2020
49. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:811–8. doi: 10.1001/jamacardio.2020.1017
50. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. (2020) 46:1089–98. doi: 10.1007/s00134-020-06062-x
51. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. (2020) 7:e575–82. doi: 10.1016/S2352-3026(20)30216-7
52. Carcaterra M, Caruso C. Alveolar epithelial cell type II as main target of SARS-CoV-2 virus and COVID-19 development via NF-Kb pathway deregulation: a physio-pathological theory. Med Hypotheses. (2021) 146:110412. doi: 10.1016/j.mehy.2020.110412
53. Sandoval Y, Januzzi JL Jr, Jaffe AS. Cardiac troponin for assessment of myocardial injury in COVID-19: JACC review topic of the week. J Am Coll Cardiol. (2020) 76:1244–58. doi: 10.1016/j.jacc.2020.06.068
54. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a Review. JAMA. (2020) 324:782–93. doi: 10.1001/jama.2020.12839
55. Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. (2020) 17:259–60. doi: 10.1038/s41569-020-0360-5
56. Bermejo-Martin JF, Almansa R, Torres A, González-Rivera M, Kelvin DJ. COVID-19 as a cardiovascular disease: the potential role of chronic endothelial dysfunction. Cardiovasc Res. (2020) 116:e132–3. doi: 10.1093/cvr/cvaa140
57. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
58. McCracken IR, Saginc G, He L, Huseynov A, Daniels A, Fletcher S, et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation. (2021) 143:865–8. doi: 10.1161/CIRCULATIONAHA.120.052824
59. Ahmetaj-Shala B, Vaja R, Atanur SS, George PM, Kirkby NS, Mitchell JA. Cardiorenal tissues express SARS-CoV-2 entry genes and basigin (BSG/CD147) Increases with age in endothelial cells. JACC Basic Transl Sci. (2020) 5:1111–23. doi: 10.1016/j.jacbts.2020.09.010
60. Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, et al. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE2. Circ Res. (2021) 128:1323–6. doi: 10.1161/CIRCRESAHA
61. Qin Z, Liu F, Blair R, Wang C, Yang H, Mudd J, et al. Endothelial cell infection and dysfunction, immune activation in severe COVID-19. Theranostics. (2021) 11:8076–91. doi: 10.7150/thno.61810
62. Ma S, Sun S, Li J, Fan Y, Qu J, Sun L, et al. Single-cell transcriptomic atlas of primate cardiopulmonary aging. Cell Res. (2021) 31:415–32. doi: 10.1038/s41422-020-00412-6
63. Gao YM, Xu G, Wang B, Liu BC. Cytokine storm syndrome in coronavirus disease 2019: a narrative review. J Intern Med. (2021) 289:147–61. doi: 10.1111/joim.13144
64. Leisman DE, Deutschman CS, Legrand M. Facing COVID-19 in the ICU: vascular dysfunction, thrombosis, and dysregulated inflammation. Intensive Care Med. (2020) 46:1105–8. doi: 10.1007/s00134-020-06059-6
65. Congiu T, Demontis R, Cau F, PiRAAS M, Fanni D, Gerosa C, et al. Scanning electron microscopy of lung disease due to COVID-19 - a case report and a review of the literature. Eur Rev Med Pharmacol Sci. (2021) 25:7997–8003. doi: 10.26355/eurrev_202112_27650
66. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. (2020) 126:1456–74. doi: 10.1161/CIRCRESAHA.120.317015
67. Brojakowska A, Narula J, Shimony R, Bander J. Clinical implications of SARS-CoV-2 interaction with renin angiotensin system: JACC review topic of the week. J Am Coll Cardiol. (2020) 75:3085–95. doi: 10.1016/j.jacc.2020.04.028
68. Evans PC, Rainger GE, Mason JC, Guzik TJ, Osto E, Stamataki Z, et al. Endothelial dysfunction in COVID-19: a position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc Res. (2020) 116:2177–84. doi: 10.1093/cvr/cvaa230
69. Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor fedratinib. J Microbiol Immunol Infect. (2020) 53:368–70. doi: 10.1016/j.jmii.2020.03.005
70. Xu S, Liu Y, Ding Y, Luo S, Zheng X, Wu X, et al. The zinc finger transcription factor, KLF2, protects against COVID-19 associated endothelial dysfunction. Signal Transduct Target Ther. (2021) 6:266. doi: 10.1038/s41392-021-00690-5
71. Fernández-Pérez MP, Águila S, Reguilón-Gallego L, de Los Reyes-García AM, Miñano A, Bravo-Pérez C, et al. Neutrophil extracellular traps and von Willebrand factor are allies that negatively influence COVID-19 outcomes. Clin Transl Med. (2021) 11:e268. doi: 10.1002/ctm2.268
72. Laforge M, Elbim C, Frère C, Hémadi M, Massaad C, Nuss P, et al. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat Rev Immunol. (2020) 20:515–6. doi: 10.1038/s41577-020-0407-1
73. Huertas A, Montani D, Savale L, Pichon J, Tu L, Parent F, et al. Endothelial cell dysfunction: a major player in SARS-CoV-2 infection (COVID-19)? Eur Respir J. (2020) 56:2001634. doi: 10.1183/13993003.01634-2020
74. Sriram K, Insel PA. Inflammation and thrombosis in COVID-19 pathophysiology: proteinase-activated and purinergic receptors as drivers and candidate therapeutic targets. Physiol Rev. (2021) 101:545–67. doi: 10.1152/physrev.00035.2020
75. Bevers EM, Williamson PL. Getting to the Outer Leaflet: physiology of phosphatidylserine exposure at the plasma membrane. Physiol Rev. (2016) 96:605–45. doi: 10.1152/physrev.00020.2015
76. Stace CL, Ktistakis NT. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta. (2006) 1761:913–26. doi: 10.1016/j.bbalip.2006.03.006
77. Vance JE, Steenbergen R. Metabolism and functions of phosphatidylserine. Prog Lipid Res. (2005) 44:207–34. doi: 10.1016/j.plipres.2005.05.001
78. Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. (2003) 42:423–38. doi: 10.1016/s0163-7827(03)00025-0
79. Shi J, Gilbert GE. Lactadherin inhibits enzyme complexes of blood coagulation by competing for phospholipid-binding sites. Blood. (2003) 101:2628–36. doi: 10.1182/blood-2002-07-1951
80. Xie R, Gao C, Li W, Zhu J, Novakovic V, Wang J, et al. Phagocytosis by macrophages and endothelial cells inhibits procoagulant and fibrinolytic activity of acute promyelocytic leukemia cells. Blood. (2012) 119:2325–34. doi: 10.1182/blood-2011-06-362186
81. Langer F, Ruf W. Synergies of phosphatidylserine and protein disulfide isomeRAASe in tissue factor activation. Thromb Haemost. (2014) 111:590–7. doi: 10.1160/TH13-09-0802
82. Shi C, Tingting W, Li JP, Sullivan MA, Wang C, Wang H, et al. Comprehensive landscape of heparin therapy for COVID-19. Carbohydr Polym. (2021) 254:117232. doi: 10.1016/j.carbpol.2020.117232
83. Mycroft-West CJ, Su D, Pagani I, Rudd TR, Elli S, Gandhi NS, et al. Heparin inhibits cellular invasion by SARS-CoV-2: structural dependence of the interaction of the spike S1 receptor-binding domain with heparin. Thromb Haemost. (2020) 120:1700–15. doi: 10.1055/s-0040-1721319
84. Meizlish ML, Goshua G, Liu Y, Fine R, Amin K, Chang E, et al. Intermediate-dose anticoagulation, aspirin, and in-hospital mortality in COVID-19: a propensity score-matched analysis. Am J Hematol. (2021) 96:471–9. doi: 10.1002/ajh.26102
85. Mackman N, Bergmeier W, Stouffer GA, Weitz JI. Therapeutic strategies for thrombosis: new targets and approaches. Nat Rev Drug Discov. (2020) 19:333–52. doi: 10.1038/s41573-020-0061-0
86. Gerotziafas GT, Catalano M, Colgan MP, Pecsvarady Z, Wautrecht JC, Fazeli B, et al. Guidance for the Management of patients with vascular disease or cardiovascular risk factors and COVID-19: position paper from VAS-European Independent Foundation in Angiology/Vascular Medicine. Thromb Haemost. (2020) 120:1597–628. doi: 10.1055/s-0040-1715798
87. Wang J, Hajizadeh N, Moore EE, McIntyre RC, Moore PK, Veress LA, et al. Tissue plasminogen activator (tPA) treatment for COVID-19 associated acute respiratory distress syndrome (ARDS): a case series. J Thromb Haemost. (2020) 18:1752–5. doi: 10.1111/jth.14828
88. REMAP-CAP Investigators, ACTIV-4a Investigators, ATTACC Investigators, Goligher EC, Bradbury CA, McVerry BJ, et al. Therapeutic anticoagulation with heparin in critically ill patients with COVID-19. N Engl J Med. (2021) 385:777–89. doi: 10.1056/NEJMoa2103417
89. REMAP-CAP Writing Committee for the REMAP-CAP Investigators, Bradbury CA, Lawler PR, Stanworth SJ, McVerry BJ, McQuilten Z. Effect of antiplatelet therapy on survival and organ support-free days in critically ill patients with COVID-19: a randomized clinical trial. JAMA. (2022) 327:1247–59. doi: 10.1001/jama.2022.2910
90. Sholzberg M, Tang GH, Rahhal H, AlHamzah M, Kreuziger LB, Áinle FN, et al. Effectiveness of therapeutic heparin versus prophylactic heparin on death, mechanical ventilation, or intensive care unit admission in moderately ill patients with COVID-19 admitted to hospital: RAPID randomised clinical trial. BMJ. (2021) 375:n2400. doi: 10.1136/bmj.n2400
91. Arslan Y, Yilmaz G, Dogan D, Hasirci M, Cetindogan H, Ocal N, et al. The effectiveness of early anticoagulant treatment in COVID-19 patients. Phlebology. (2021) 36:384–91. doi: 10.1177/0268355520975595
92. Gonzalez-Ochoa AJ, Raffetto JD, Hernández AG, Zavala N, Gutiérrez O, Vargas A, et al. Sulodexide in the treatment of patients with early stages of COVID-19: a randomized controlled trial. Thromb Haemost. (2021) 121:944–54. doi: 10.1055/a-1414-5216
93. Antonelli M, Penfold RS, Merino J, Sudre CH, Molteni E, Berry S, et al. Risk factors and disease profile of post-vaccination SARS-CoV-2 infection in UK users of the COVID Symptom Study app: a prospective, community-based, nested, case-control study. Lancet Infect Dis. (2022) 22:43–55. doi: 10.1016/S1473-3099(21)00460-6
94. Ledford H. Do vaccines protect against long COVID? What the data say. Nature. (2021) 599:546–8. doi: 10.1038/d41586-021-03495-2
95. Ospina-Tascón GA, Calderón-Tapia LE, García AF, Zarama V, Gómez-Álvarez F, Álvarez-Saa T, et al. Effect of high-flow oxygen therapy vs conventional oxygen therapy on invasive mechanical ventilation and clinical recovery in patients with severe COVID-19: a randomized clinical trial. JAMA. (2021) 326:2161–71. doi: 10.1001/jama.2021.20714
96. Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Membrane phosphatidylserine regulates surface charge and protein localization. Science. (2008) 319:210–3. doi: 10.1126/science.1152066
97. Zhang XJ, Qin JJ, Cheng X, Shen L, Zhao YC, Yuan Y, et al. In-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19. Cell Metab. (2020) 32:176–87.e4. doi: 10.1016/j.cmet.2020.06.015
98. Bojadzic D, Alcazar O, Chen J, Chuang ST, Condor Capcha JM, Shehadeh LA, et al. Small-molecule inhibitors of the coronavirus spike: ACE2 protein-protein interaction as blockers of viral attachment and entry for SARS-CoV-2. ACS Infect Dis. (2021) 7:1519–34. doi: 10.1021/acsinfecdis.1c00070
99. Li K, Meyerholz DK, Bartlett JA, McCray PB Jr. The TMPRSS2 inhibitor nafamostat reduces SARS-CoV-2 pulmonary infection in mouse models of COVID-19. mBio. (2021) 12:e0097021. doi: 10.1128/mBio.00970-21
100. Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, et al. Association of inpatient use of angiotensin-converting enzyme inhibitors and angiotensin ii receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res. (2020) 126:1671–81. doi: 10.1161/CIRCRESAHA.120.317134
Keywords: COVID-19, cardiopulmonary, phosphatidylserine, microparticles, endothelial cells, antithrombotic, sequelae
Citation: Liu L, Jing H, Wu X, Xiang M, Novakovic VA, Wang S and Shi J (2022) The cross-talk of lung and heart complications in COVID-19: Endothelial cells dysfunction, thrombosis, and treatment. Front. Cardiovasc. Med. 9:957006. doi: 10.3389/fcvm.2022.957006
Received: 30 May 2022; Accepted: 11 July 2022;
Published: 05 August 2022.
Edited by:
Alaa Omar, Mount Sinai St. Luke's Hospital, United StatesReviewed by:
Diana Maria Ronderos, Bronx Lebanon Hospital, United StatesCopyright © 2022 Liu, Jing, Wu, Xiang, Novakovic, Wang and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jialan Shi, amlhbGFuX3NoaUBkZmNpLmhhcnZhcmQuZWR1; c2hpNzM2NjFAZ21haWwuY29t; Shuye Wang, d2Fuc2h1eWUzMzY2QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.