 Kun Zhao
Kun Zhao Yukang Mao1†
Yukang Mao1† Kai Wang
Kai Wang Jing Zhang
Jing Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 26 August 2022
Sec. Cardiovascular Genetics and Systems Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.952949
This article is part of the Research Topic Epigenetic Regulation in Cardiovascular Diseases, Volume II View all 6 articles
Pathological myocardial remodeling was still one of the leading causes of death worldwide with an unmet therapeutic need. A growing number of researchers have addressed the role of epigenome changes in cardiovascular diseases, paving the way for the clinical application of novel cardiovascular-related epigenetic targets in the future. In this review, we summarized the emerged advances of epigenetic regulation, including DNA methylation, Histone posttranslational modification, Adenosine disodium triphosphate (ATP)-dependent chromatin remodeling, Non-coding RNA, and RNA modification, in pathological myocardial remodeling. Also, we provided an overview of the mechanisms that potentially involve the participation of these epigenetic regulation.
Pathological myocardial remodeling, characterized as cardiomyocyte hypertrophy, apoptosis, and excessive fibrosis, is an adaptive growth process that occurs in response to the capacity/pressure load caused by certain diseases, environmental factors, or lifestyles (such as long-term hypertension, atherosclerosis, myocardial infarction, metabolic abnormalities, smoking, drinking, etc.), which ultimately leads to a decrease in heart output and an increased risk of heart failure (1). Thus, it is of great interest to develop novel therapeutic approaches targeting the mechanisms underlying the progression of chronic heart failure-induced pathological myocardial remodeling.
The term “Epigenetic” is a combination of the Greek prefix “epi” (meaning “up,” or “around”), and “genetics.” Thus, epigenetics can be defined as the heritable regulation of gene expression by modifying chromosomal components without altering the genome nucleotide sequence (2). Epigenetic regulation can be divided into five different molecular levels: (1) DNA methylation; (2) Histone posttranslational modification; (3) Adenosine disodium triphosphate (ATP)-dependent chromatin remodeling; (4) Non-coding RNA; (5) RNA modification (3). It maintains cellular characteristics and controls cell differentiation by regulating chromatin structure and gene expression at various molecular levels, which is essential for normal development and disease progression. A growing body of research has shown that epigenome changes may be the markers of various cancers, and several molecules have been identified as the potential targets for some novel therapeutics (4). Notably, recent genetic and biochemical analyses suggested that epigenetic changes play an important role in the occurrence and progression of myocardial remodeling. In this review, we will summarize recent advances, and provide an overview of epigenetic studies targeting pathological myocardial remodeling in human and animal models (Figure 1).
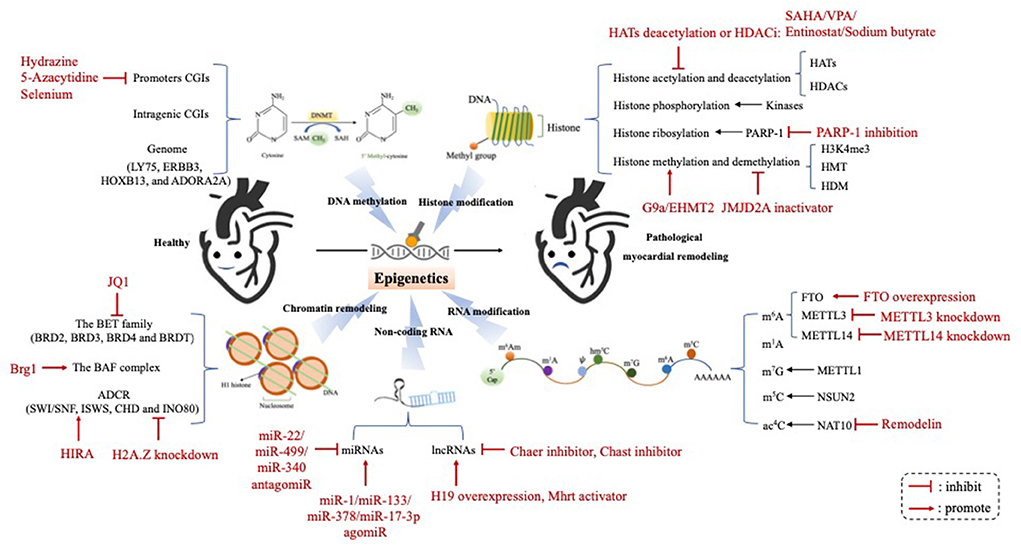
Figure 1. The epigenetic mechanism underlying the occurrence and progression of pathological myocardial remodeling. Epigenetic regulation underlying the development of pathological myocardial remodeling can be divided into four different molecular levels: (1) DNA methylation; (2) Histone modification; (3) Chromatin remodeling; (4) Non-coding RNA; (5) RNA modification. HATs, histone acetyltransferases; HDACs, histone deacetylases; PARP-1, poly (ADP-ribose) polymerase-1; H3K4me3, histone 3 lysine 4 triamcinolone; HMT, histone methyl transferase; HDM, histone trimethyldemethylase; FTO, Fat mass and obesity-associated protein; METTL3, methyltransferase-like 3; METTL14, methyltransferase-like 14; m6A, N6-methyladenosine; m1A, N1-methyladenosine; m7G, N7-methylguanosine; m5C, 5-methylcytosine; ac4C, N4-acetylcytidine; miRNAs, microRNAs; lncRNAs, long ncRNAs; Chast, cardiac hypertrophy-associated transcript; Chaer, cardiac-hypertrophy-associated epigenetic regulator.
DNA methylation in eukaryotic organisms occurs primarily at the 5th carbon atom (5 mC) of the cytosine ring, followed by guanine dinucleotides, where cytosine and guanine are separated by phosphate (CpG). DNA methylation plays a vital role in gene regulation, especially in transcriptional inhibition depending on methylation location. Increased methylation in CpG-enriched regions (CpG islands (CGI) in the gene promoter region) is commonly associated with gene silencing, whereas methylated CpG found in the genome is associated with gene activation (5).
The altered DNA methylation landscape were initially known as critical epidemiological markers underpinning various human diseases, especially in the process of carcinogenesis (6). During the past decade, extensive genome-wide studies have provided robust evidence that DNA methylation is closely involved in the pathogenesis of myocardial remodeling associated with different etiologies, including dilated cardiomyopathy (DCM), ischemic cardiomyopathy (ICM), chronic pressure overload and valve disease (7–11). Movassagh et al. demonstrated for the first time that a huge amount of CGI and promoters are hypomethylated at the end stages of heart failure (7). The previous study determined that the differential DNA methylation in failing and non-failing human myocardium did not occur uniformly throughout the genome using methylated DNA immunoprecipitation (MeDIP) (8).
Recent study found that the overall CpG methylation was increased in DCM hearts compared with the control hearts (12). In 2013, Haas et al. detected different patterns of DNA methylation in left ventricular tissues obtained from DCM patients and reproduced epigenetic regulation patterns of several genes with previously unknown functions in DCM, including lymphocyte antigen 75 (LY75), tyrosine kinase-type cell surface receptor HER3 (ERBB3), Homeobox B13 (HOXB13), and adenosine receptor A2A (ADORA2A) (13). Then the functional correlation of these identified genes in the adaptive or maladaptive pathways in heart failure was further demonstrated in zebrafish (13), suggesting that it is reasonable to investigate their potential as diagnostic and therapeutic targets specifically in DCM but also in heart failure.
Genome-widely DNA methylation analysis in combination with RNA sequencing revealed a robust gene expression pattern that is consistent with suppression of oxidative metabolism, induced anaerobic glycolysis, and deteriorated myocardial remodeling in the ischemic hearts of patients with end-stage heart failure in comparison with non-ischemic control hearts, with the Krüppel-like factor 15 (KLF15) and polycomb methyltransferase enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) being identified as candidate regulators of ICM-associated epigenetic reprogramming (9), indicating that these genes may be relevant druggable targets in myocardial remodeling subsequent to heart failure.
Histones are part of the chromatin complex that forms chromosomes. The histone octamer contains 2 copies of the H2A, H2B, H3, and H4 proteins, forming the core of the nucleosome, which is wrapped 1.67 times by approximately 147 base pairs (14). Unlike DNA methylation, the role of histone modifications in epigenetic regulations appear to be more subtle. The function of histone modification depends on not only the position, but also the type and amount of histone modification. Histone posttranslational modifications include acetylation, methylation, phosphorylation, ribosylation, etc. at different amino acid residues, which may alter the chromatin structure and/or influence the downstream genes expression via recruiting different kinds of regulatory molecules (e.g., histone modifiers, chromatin regulators, and transcription factors) (15, 16). Although it is difficult to decode specific posttranslational modifications at the level of individual histone and mononucleosome, a growing body of evidence has demonstrated that histone modifications may interact to exert transcriptional regulatory effects (17).
Histone acetylation and deacetylation acetylation/deacetylation are the most common histone modifications, which are catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively (18). Findings from previous studies performed on mammalian cells have determined a strong association of histone acetylation and deacetylation with numerous human diseases, including cancer (19), neurological disorders (20) and cardiovascular disease (CVD) (21). Histone acetylation of HATs may disrupt the interactions within and between nucleosomes, thereby “relaxing” chromatin structures and activating transcription. In contrast, HDACs deacetylate histones, which increased histone-DNA interactions, leading to chromatin concentration and gene inhibition (22). It has been demonstrated that specific HATs and HDACs are critical modulators of cardiac hypertrophic response and contribute to the development of pathological myocardial remodeling (21).
In 2007, Montgomery et al. showed that the loss of HDAC1 led to the death of mouse embryos on Day 9.5, but mice lacking HDAC2 survived to the perinatal period and eventually died of heart defects, such as right ventricular cavity occlusion, myocardial cell apoptosis, and bradycardia (23). Besides, cardiac-specific deletion of HDAC1 or HDAC2 resulted in neonatal death, with arrhythmia, or DCM (23).
Also, Trivedi et al. found that the transgenic mice overexpressed with HDAC2 developed exaggerated myocardial hypertrophy (24). In contrast to HDAC2, cardiomyocyte-specific overexpression of HDAC3 in mice increased cardiomyocyte proliferation, rather than promoting cardiac hypertrophy (25). In 2008, Montgomery et al. showed that cardiac-specific HDAC3-deficient mice survived up to 3–4 months after birth, presenting with severe myocardial hypertrophy (26). Moreover, some researchers have found that HDAC4, HDAC5, HDAC7, and HDAC9 can attenuate cardiac hypertrophy through binding to transcriptional factor myocyte enhancer factor 2 (MEF2) and suppressing its activity, supporting the role of these endogenous factors in inhibiting pathological cardiac growth and preserving cardiac function (27–29). On this basis, Helmstadter et al. further demonstrated that calcium/calmodulin-dependent protein kinase II (CaMKII) and protein kinase A (PKA)-dependent phosphorylation co-regulated the nuclear localization of HDAC4 in adult cardiomyocytes, which has significant transcriptional implications in the development of heart failure (30).
In addition, CREB binding protein (CBP)/p300 may drive unrestricted cardiac growth and impaired cardiac geometry by acetylating and increasing the transcriptional activity of MEF2 (31). Cardiac-specific overexpression of p300 in mice increases GATA4 acetylation, resulting in significant centrifugal dilation and contraction dysfunction of the heart (32). In contrast, Oral curcumin (a p300-specific HAT inhibitor) administration was reported to prevent the deterioration of cardiac function in salt-sensitive Dahl rats and surgically induced myocardial infarction (MI) rats (33). Therefore, the deacetylation or inhibition of HATs may be beneficial in ameliorating pathological myocardial remodeling in patients with heart failure.
The pioneering studies demonstrated that the homologs of Silent information regulator 2 (Sir2) (i.e., Sirtuins 1–Sirtuins 7), a family of histone deacetylases located in different cellular compartments whose function depends on nicotinamide adenine dinucleotide (NAD+), play a non-negligible physiopathological role in regulating energy metabolism, senescence, and other biological processes under various pathological cellular processes via acting on different targets in a non-redundant manner (34, 35). The knockdown of Sirtuins may result in epigenetic silencing and defected DNA repair process, leading to shorter lifespan of yeast Saccharomyces cerevisiae and different cardiovascular diseases in mammals (36, 37).
Among the family of Sirtuins, Sirtuins 1 (SIRT1) is the most well-studied one since it is the closest homolog of Sir2 (38). SIRT1 was found to be involved in regulating DNA repair, gene transcription, oxidative stress, and longevity (39, 40). The enhanced activity of SIRT1 was depicted to exert a promising cardioprotective effects against arteriosclerotic cardiovascular disease (ASCVD) by deacetylating the DNA repair machinery components (38, 39).
Interestingly, SIRT1 expression which decreases with aging correlated with the onset and development of heart failure and even ST-segment elevation myocardial infarction (STEMI) (41–43). Heart failure-induced miRNA-138-5p expression may downregulate the activity of SIRT1, thereby enhancing the acetylation of p53, which triggers cardiomyocyte apoptosis (44). Also, pharmacological activation of SIRT1 could improve cardiac contractile dysfunction by inhibiting the acetylation of SERCA2a at K492 (45). Besides, the in-vitro experiments found that sodium-dependent glucose transporters 2 (SGLT2) inhibitors may play a cardioprotective role under hyperglycemia conditions through activating SIRT1/PGC-1α/FGF21 signaling pathway (46).
Sirtuins 3 (SIRT3), a deacetylase which may regulate mitochondrial function, also plays a cardioprotective effects against energy dyshomeostasis and cardiac dysfunction (47). Like SIRT1, the declined activity of SIRT3 associated with aging is associated with endothelial dysfunction, hypertension, and heart failure (48). Emerging evidence demonstrated that the stimulated SIRT3 activity could ameliorate cell death, fibrosis, and the inflammatory response of cardiomyocytes under the hypertrophic stimulus, thus improving health and lifespan (49–51). Besides, silencing SIRT3 may sensitize overload-induced heart failure by worsening cardiomyocyte glucose utilization (52), whereas the impaired SIRT3-mediated endothelium cell metabolism could even result in cardiac diastolic dysfunction (53).
In 1964, histone methylation was first described by Murray (54). The first evidence linking histone methylation to transcription was presented in 1999 by reporting that histone H3 arginine-specific methyltransferase CARM1 interacted with the steroid hormone receptor co-activator GRIP-1 during transcription initiation (55).
The activation or inhibition of histone methylation to genes depends on which residue occurs on histone. For example, histone 3 lysine 4 triamcinolone (H3K4me3) is associated with active promoter activity, whereas trimethylation of H3K9me3 is usually related to transcription inhibition (15, 56).
During heart failure, H3K4me3 increased in the promoter and transcription region of myosin heavy chain (β-MHC), while H3K4me3 decreased at the α-MHC promoter (57), suggesting that “classic” transcription reprogramming that occurred during myocardial remodeling involved not only the reactivation of the fetal heart genes, but also the changes of methylation patterns at the same gene site that are reactivated under pathological stress.
It has been shown that histone methyl transferase (HMT), G9a/EHMT2, was considered essential for homeostasis of cardiomyocytes in the adult heart (58). The study found that conditional abnormalities of G9a silencing in 2-month-old mouse myocardium caused various cardiac dysfunctions in the first 4 weeks of knockout induction. G9a, which inhibited the key cardiac regulatory genes through H3K9me2, was upregulated during the initial stages of heart failure. In addition, the chemical inactivation of G9a in mice undergoing aortic narrowing surgery (TAC) improved cardiac function and inhibited the development of remodeling (58). Thus, G9a acted as an anti-hypertrophy regulator in healthy hearts, and a hypertrophy activator in stress-induced hearts.
Zhang et al. demonstrated that histone trimethyldemethylase (HDM), JMJD2A, promoted myocardial remodeling in TAC-induced mice (59). After TAC surgery, JMJD2A bound to the promoter regions of Four and a Half LIM Domains Protein 1 (FHL1), and reduced H3K9me3 levels in the heart, which may account for the ameliorative effects of JMJD2A inactivation on myocardial remodeling.
Finally, mis-regulation of histone methylation cofactors also affected myocardial remodeling. Specific deletion of the PAX transcriptional activation domain interaction protein (PTIP), a cofactor of H3K4 methylation, in adult mice resulted in a reduction in H3K4me3 (60).
Protein kinases deliver extracellular signals from the cell surface to the nucleus. Kinases could not only phosphorylate cellular proteins, and transcription factors, but also regulate major cellular processes such as transcription, mitosis, DNA damage, and apoptosis via mediating chromatin dynamics. Although histones can be phosphorylated by stress signals, most studies still focused on the role of histone acetylation and methylation in controlling transcription of cardiac genes. Thus, histone phosphorylation was poorly studied and understood. Since many kinases are key mediators of myocardial remodeling, however, whether they acted as physiological histone kinases to regulate transcription remains debatable (61).
Histone ribosylation is mediated by poly (ADP-ribose) polymerase-1 (PARP-1), which transfers the ADP-ribose group from nicotinamide adenine dinucleotide (NAD +) to the receptor substrate (62). Substrates for PARP-1 include PARP-1 itself and the tails of histones H1, H2A, H2B, H3, and H4 (63). Histone ribosylation could induce relaxation of chromatin structure, which helps recruit repair enzymes to damaged DNA sites. In addition to playing a role in DNA repair, PARP also regulates cell death, cell cycle progression, and genomic stability (64). In myocardium, PARP-1 was reported to promote myocardial remodeling, and increased PARP-1 activity led to heart failure in mice and humans (65). In contrast, PARP-KO mice were resistant to angiotensin II-induced myocardial remodeling (65, 66). The mechanism of PARP-1 inducing pathological hypertrophy was achieved by interacting with HDAC and the chromatin remodeler BRG-1 (67). In addition to its effects on fetal cardiac genes, PARP also controlled cardiac hyperplasia via regulating the P38 MAP kinase, ERK1/2, PI3 kinase-Akt-GSK3β, and JNK signaling pathways (68, 69).
At present, several genome-wide studies of myocardial histone posttranslational modifications have been performed in animal models or humans. In 2009, Kaneda et al. published the first genome-wide study of myocardial histone methylation in humans and rodents, showing the significant differences in the distribution patterns of H3K4me3 and H3K9me3 between healthy and failing hearts (70). In 2011, Movassagh et al. also demonstrated that H3K36me3 showed different methylation patterns and enrichment between normal human myocardium and cardiomyopathy hearts (7). In 2013, Papait et al. analyzed seven histone epigenetic markers in the heart tissues of control and TAC mice by ChIP sequencing, which found that histone acetylation levels (e.g., H3K9ac and H3K27ac) were reduced around down-regulated gene loci in TAC mice (71). Besides, the more active the transcription of differentially expressed genes in TAC mice, the fewer histone inhibitory markers (e.g., H3K27me3, H3K9me2, and H3K9me3) they observed.
In 2018, Gilsbach et al. used nuclear staining and fluorescence-assisted sorting to isolate human myocardial cells from normal fetuses, infants, and adults' normal and failing hearts for genome-wide epigenetics (72). The results showed that the whole-transcriptomic regulation of normally methylated cells during normal development was determined mainly by the histones commonly observed on cis-acting elements at the distal end of the genome. Another interesting finding of this study was that the expression of genes associated with heart failure was regulated by activity-enhanced histone markers, but not by DNA methylation.
The dynamic epigenome modifications, including histone posttranslational modification and DNA methylation, may trigger chromatin remodeling, which is considered an important process for controlling cell fate and determining organ development (73). Chromatin regulators, such as histone modifiers, cofactors, and transcription regulators, could recruit other chromatin regulators and/or transcription factors to regulate genes expression. By collecting all the necessary regulators, the distal cis-acting elements (such as enhancers) could form chromatin rings that interact with the proximal promoter region, thereby initiating and enhancing the transcriptional activity of the genes with underlying transcription complexes (73).
Studies regulating the function of bromine domain (BET) proteins, acetylate lysine binding proteins and epigenetic readers have shown that chromatin structures were critical for regulating the dynamics of gene expression in the failed heart (74, 75). The BCD domain (BET) family includes BRD2, BRD3, BRD4, and BRDT. Spiltoir et al. found that hypertrophic stimulation promoted recruitment of BRD4 to the transcription start site (TSS) of the gene encoding atrial natriuretic factors (ANF) (74).
In addition to the histones and coactivators mentioned above, the ATP-dependent chromatin remodeling complex (ADCR) is another regulator that may play a key role in chromatin remodeling (76). ADCR contains a subunit of adenosine triphosphate (ATPase) belonging to the SNF2 protein superfamily, which uses ATP hydrolysis as an energy source to alter or disrupt Histone-DNA interactions. In mammals, four ADCR families (SWI/SNF, ISWS, CHD and INO80) have been identified (77). Among them, the role of SWI/SNF in cardiac development and remodeling has been extensively studied over the past decade (78–82). These studies found that the BAF complex (the mammalian analog of the SWI/SNF complex) was generally abundant in the embryonic heart but down-regulated in the adult myocardium. Myosin heavy chain (MHC) is an important molecule that ensures the normal operation of the heart. There are two main subtypes, namely α-MHC and β-MHC, which are expressed specifically in the mammalian heart and located on the same chromosome (83). α-MHC, which has higher ATPase activity than β-MHC, is mainly expressed in adult cardiomyocytes, while β-MHC is expressed in embryonic cardiomyocytes.
How does the BAF complex regulate the expression of these two homogenous types? Based on the study of the mouse model of myocardial-specific ablation of BRG1 (a nucleosome remodeling factor) at different stages of development, it was found that BRG1 was an important ATPase subunit of the BAF complex. BRG1 could interact with HDAC and PARP-1, thereby inhibiting the α-MHC of the adult hearts, and activating the β-MHC of the embryonic hearts. Since the fact that the BAF complex was down-regulated in adult cardiomyocytes, the inhibition of α-MHC was eliminated as well. Thus, MHC (α-MHC) expression was upregulated in the adult hearts (67). In hypertrophic and failing hearts, subunits of the BAF complex and their binding partners, as well as the fetal MHC, were increased (67).
In addition, the effects of BRG1 on recruiting histone methyltransferase G9a/DNA methyltransferase (DMNT3) complex to the promoter regions of Myh6 gene encoding α-MHC was reported to lead to the deposition of H3K9me3, and CpG methylation in the Myh6 promoter regions, which may subsequently activate the inhibitory mechanism of the Myh6 gene in the adult failing hearts (84). Besides, cardiac myocyte-specific ablation of BRG1 attenuated myocardial remodeling in TAC-induced mice. Thus, BRG1 may act as a stress-activated chromatin remodeler that controls fetal reprogramming of cardiac genes (67).
Previous report demonstrated that SWI/SNF chromatin remodeling complex SWR1 could exchange H2A with H2A.Z at specific genomic loci to maintain genomic integrity and facilitate transcriptional initiation (85). In spit to the upregulated expression of the histone variant H2A.Z in the hearts of TAC mice, shRNA knockdown of H2A.Z was also reported to attenuate myocardial remodeling and down-regulate expression of growth-related genes, such as cyclin-dependent kinase 7 and ribosomal S6 in cultured rat cardiomyocytes (86). In addition, cardiomyocyte-specific conditional knockout of the histone chaperone HIRA in mice induced pathological myocardial remodeling, as well as the impaired intramuscular integrity (87). The above studies showed that ATP-dependent chromatin remodeling may play a crucial role in the pathogenesis of myocardial remodeling under different pathological stimuli.
Protein-coding genes account for a tiny percentage of (3%) of the human genome (88). Recent advances in transcriptomics and bioinformatics have revealed that, remaining 97% untranslated RNAs, defined as non-coding RNAs (ncRNAs), plays a critical role at transcriptional and translational level in both physiological and pathological conditions. Apart from some traditional and well-established ncRNAs, including transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), the other unidentified ncRNAs can be classified into two major categories based on their sizes, namely, short ncRNAs, which comprises fewer than 200 nucleotides and mainly includes microRNAs (miRNAs), small interfering RNA (siRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), small cytoplasmic RNAs (scRNAs), extracellular RNA (exRNAs), piwi-interacting RNAs (piRNAs), and long ncRNAs (lncRNAs) consisting of more than 200 nucleotides (89).
MicroRNAs (miRNAs) can be defined as short ncRNAs ranging 18–25 nucleotides in length, which are emerging as key regulators involved in the development and progression of CVDs, predominantly due to inhibiting translation or degrading target messenger RNAs (mRNAs) (90). A large amount of miRNAs appear to be differentially expressed between hypertrophic and non-hypertrophic cardiac tissues and regulate different cell types and pathways during myocardial remodeling. Sayed et al. found that miR-1 was downregulated at an early stage in a mouse model of myocardial remodeling induced by transverse aortic constriction (TAC), and that overexpression of miR-1 in cultured cardiomyocytes decreased the levels of growth-related factors including Ras GTPase-activating protein (RasGAP), cyclin-dependent kinase 9 (Cdk9), Ras homolog enriched in brain (Rheb) and fibronectin (91). Similarly, miR-133 expression level has been shown to be significantly augmented in human and murine models of myocardial remodeling. Both in vitro and in vivo functional studies indicated that miR-133 silence induced by adenoviral transfection or antagomiR led to exacerbated myocardial remodeling and impaired cardiac function, while its overexpression showed opposite effects, with Rhoa, Cdc42, and NELFA/Whsc2 being identified as target genes of miR-133 by bioinformatics analysis (92). Also, miR-21 overexpression was reported to trigger cardiac fibroblasts activation and promote cardiomyocytes proliferation against stress overloaded conditions (93). In PE- or AngII-induced cardiomyocyte hypertrophic model, miR-22 is the most remarkably upregulated miRNA, increases the cell size and regulates the fetal gene expression (nppa and myh6), with PTEN acting as its potential downstream target (94). Cai et al. indicated that miR-765 expression was enhanced in human falling hearts, and that overexpression of miR-765 resulted in reduced inhibitor-1 expression, which subsequently suppressed cardiomyocyte contractile function and calcium cycling through regulation of the PP-1 signaling axis (95). miR-499 levels were increased in human and mouse failing hearts after chronic pressure overload, and its upregulation adversely affect the cardiac function and geometry (96). miR-340 is a pro-eccentric hypertrophy miRNA, which plays a pivotal role, at least in part though targeting cardiomyocyte structure protein DMD, in the development of myocardial remodeling and heart failure associated with DCM, and whose expression can be largely attributed to cytokine CT-1 activation (97). Interestingly, some miRNAs have been documented to stimulate cardiomyocyte proliferation, and thus may function as promising therapeutic targets in cardiac regeneration. Xiao et al. and Shi et al. have demonstrated that miR-31 and miR-17 promotes postnatal cardiomyocyte proliferation and cardiac repair after ischemia-reperfusion injury, with RhoBTB1 and TIMP3 being the main target gene, respectively (98, 99).
Recently, a novelty research identified that Angiotensin receptor/Neprilysin inhibitor (ARNI) may improve maladaptive myocardial remodeling in cardiac resynchronization therapy with defibrillator (CRTd) non-responder patients via modulating miRNAs including miR-18, miR-145, and miR-181 (100).
The mechanisms by which lncRNAs regulate gene expression seem to be complex and multifaceted, which may involve chromatin modification, transcription, post-transcriptional processing and affecting miRNA-mediated gene regulation (101). Kumarswamy and co-workers reported that the level of LIPCAR, a mitochondria-derived lncRNA, was markedly upregulated in cardiac tissues from patients with left ventricular remodeling and chronic heart failure, and that high LIPCAR levels were strongly associated with a higher risk of cardiovascular death (102). RNA sequencing analysis revealed a distinct cardiac ncRNAs expression profile in which lncRNA H19 and its encoded miR-675 comprised the most dominantly upregulated ncRNAs since the early phase after TAC-induced myocardial remodeling in mice. Adenovirus-mediated overexpression and a siRNA-mediated silencing of H19 suppressed hypertrophic growth in neonatal cardiomyocytes, whereas knockdown of H19 accentuated myocardial remodeling (103). Moreover, Wang et al. identified a heart-enriched lncRNA, termed as cardiac-hypertrophy-associated epigenetic regulator (Chaer), which plays a crucial role in the development of myocardial remodeling via interacting with catalytic subunit of polycomb repressor complex 2 (PRC2). The interactions between Chaer and PRC2 inhibits histone H3 lysine 27 methylation (H3K27me3) at the promoter regions of genes, thus attenuating myocardial remodeling in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), neonatal rat cardiomyocytes and mouse model after TAC surgery (104).
A new cluster of lncRNA transcripts, namely myosin heavy-chain-associated RNA transcripts (Mhrt), has been found to be derived from a two-exon gene located on Myh7 loci by Han et al. with Mhrt779 being the most abundant species, and characterized by high cardiac-specificity, high abundance in the adult heart and potent cardioprotective effects. To be noted, a diminished Mhrt expression was observed in TAC-induced hypertrophic mouse hearts, which may be partially attributed to Brg1-Hdac-Parp complex. Collectively, these findings provide novel insights into the regulatory roles of Mhrt in myocardial remodeling, implying the property for Mhrt as a promising therapeutic target in pathological cardiac remodeling (105).
Using whole-genome sequencing, Viereck et al. identified another cardiac-specifically expressed lncRNA—cardiac hypertrophy-associated transcript (Chast)—as a candidate molecule that may influence myocardial remodeling. Chast was significantly upregulated in cardiomyocytes isolated from TAC-induced hypertrophic mice and patients with aortic stenosis. Subsequent in vitro and in vivo functional studies support the role of Chast as an endogenous prohypertrophic lncRNA (106).
The above studies emphasized the valuable diagnostic role of ncRNAs in the pathological myocardial remodeling. Meanwhile, understanding these entangled networks of RNA regulatory and functional interactions appeared to have promising implications in the characterization of the molecular events that control the progression of CVDs. Importantly, the manipulation of ncRNAs expression levels by either inhibiting or activating illustrates future novel therapeutic strategies in the pathological myocardial remodeling.
Similar to DNA modification in epigenetics, accumulating evidence has implicated the presence of over one hundred types of RNA modification (107). RNA modification is an universal phenomenon of epigenetic regulation in eukaryotes, which endows RNA molecules with structural and functional diversity (108). One of the most common RNA modifications is methylation, which is indispensable in life and plays critical roles in biological functions. As key molecule linking DNA with protein, mRNA acts as a major carrier of genetic information, and thereby representing the most widely-studied populations in RNA methylation modification. In eukaryotic mRNA, different types of methylation modification have been documented, such as N7-methylguanosine (m7G), N6-methyladenosine (m6A), 5-methylcytosine (m5C), N1-methyladenosine (m1A), N4-acetylcytidine (ac4C), and 2'-O-methylation (Nm), with m6A being the most abundant modification in mRNA (Table 1).
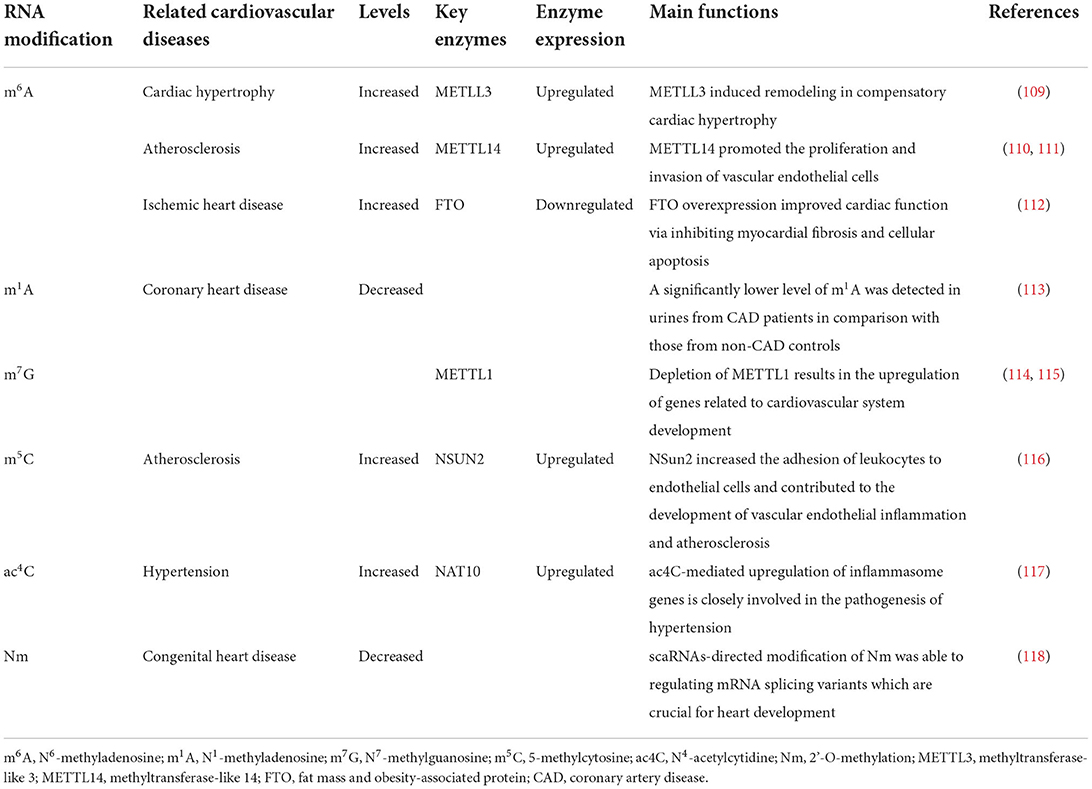
Table 1. The roles of RNA modifications in CVDs.
N6-methyladenosine (m6A) is generated by adding a methyl group at the N6 position of adenosine, which is highly conversed in sequence and ubiquitously found in most eukaryotes RNAs, such as yeast, plants, flies, mammals, and even viral RNAs, and plays an essential role in mRNA post-transcriptional modification and metabolism (119). m6A was originally detected in mammalian mRNAs in 1974 (120), then Jia et al. identified fat mass and obesity-associated protein (FTO) for the first time as m6A demethylase, implicating the dynamic and reversible nature of m6A methylation modification (121).
As a typical representative of epigenetic transcriptomics, m6A has profound effects on a series of RNA metabolism processes, including mRNA transcription, alternative splicing, transport, stability, degradation and miRNA processing (122), which are all confirmed to be closely involved in biological events such as stem cell renewal and differentiation, body growth and development, circadian rhythms, adipogenesis, spermatogenesis and carcinogenesis (123–125). Recently, m6A has been demonstrated to be catalyzed by a methyltransferase complex, which consists of Wilms' tumor 1-associating protein (WTAP), RNA-binding motif protein 15/15B (RBM15/15B), methyltransferase-like 3 (METTL3), and 14 (METTL14), and is demethylated by demethylases ALKB homo-log 5 (ALKBH5) and fat mass and obesity-associated protein (FTO) (126).
METTL3 and METTL14 are known as core components of the methyltransferase complex, with METTL3 functioning as a catalytic subunit, while METTL14 being responsible for substrate recognition, and they play a pivotal role in cardiovascular diseases. Dorn et al. have recorded that m6A methylation is increased in cardiomyocytes in response to hypertrophic stimulation, and enriched on genes involving protein kinases and intracellular signaling pathways. METTL3-mediated m6A methylation leads to compensated myocardial remodeling without interfering with cardiac function, whereas reduced m6A induced by METTL3 knockdown drives eccentric myocardial remodeling and dysfunction (109).
METTL3 was significantly upregulated in mouse model of myocardial infarction (MI) and TGF-β1-treated cardiac fibroblasts (CFs). In vitro and in vivo functional experiments showed that METTL3 overexpression can induce cardiac fibroblast proliferation and cardiac fibrosis, while knockdown of METTL3 exerted anti-fibrotic effects. Furthermore, RNA sequencing (RNA-seq) and m6A sequencing (m6A-seq) analyses revealed that METTL3 silencing caused an overall decrease of expression level and m6A level of fibrosis-related genes, implicating the pro-fibrotic property of METTL3 (127).
METTL3 was also found to be vital in maintaining the proliferative capacity of neonatal cardiomyocytes (128). Besides, METTL3 improved cardiomyocyte proliferation upon MI by upregulating miR-17-3p expression (129).
Interestingly, METTL14 has been reported to promote vascular endothelial cell proliferation and inflammation by increasing the m6A methylation modification of pre-miR-19a and forkhead box O1 (FOXO1), respectively, thus contributing to atherosclerosis. Both genetic ablation and siRNA-mediated knockdown of METTL4 can inhibit the development of atherosclerotic plaques (110, 111).
FTO is highly expressed in fetal heart ventricles and has been demonstrated by numerous studies to be closely involved in the pathogenesis of congenital heart disease [including hypertrophic cardiomyopathy (HCM), atrial septal defect (ASD) and ventricular septal defect (VSD)] (130), arrhythmias (131), coronary artery disease (CAD) (132), and metabolism-related heart disease (including obesity and type 2 diabetes) (133, 134). Mathiyalagan et al. utilized heart samples from human, pig and mouse as well as cultured cardiomyocytes to explore the potential role of m6A modification and FTO in failing hearts, and they concluded that FTO had diminished expression in mammalian failing hearts and hypoxia-induced cardiomyocyte models, which subsequently increases m6A RNA modification and impairs cardiomyocyte contractile function as a result, and that FTO overexpression in mouse models of MI can attenuate cardiac fibrosis and enhance angiogenesis, suggesting that FTO-dependent m6A methylation is of utmost importance in preserving cardiac function during heart failure (112).
N1-methyladenosine (m1A) was first extracted from rat liver by Dunn (135), and later identified as another naturally occurring RNA methylation modification manner different from m6A. Due to the nature of a positively charged methylated nucleoside (136), m1A markedly alter RNA structure and protein-RNA interactions through binding to translation initiation sites and the first splice site of mRNA under physiological conditions, and, especially, plays a critical role in promoting translation of methylated mRNA (137, 138). m1A is highly conversed in mammalian cells and is reversible by ALKBH1 and ALKBH3 in response to multiple cellular signal (139). A significantly lower level of m6A and m1A was detected in urines of CAD patients in comparison with those of non-CAD controls, indicating that methylated nucleoside may represent a novel and promising species of biomarkers for early diagnosis of CVD (113).
The 7-methylguanosine cap added to the 5' end of mRNA is essential for efficient gene expression and cell viability (140). N7-methylguanosine (m7G) modification is prevalent in all eukaryotic organisms in which it acts as a key regulator involved in different stages of gene expression, including transcription, mRNA splicing, nuclear export, translation and mRNA stability (141). Based on advanced high-throughput sequencing approaches developed for transcriptome-wide mapping of internal m7G modification, m7G DiseaseDB database is constructed and designated to identify the disease phenotypes that are most enriched with m7G variants. Among them, a considerable amount of variants are related to cardiovascular phenotypes, suggesting a strong linkage between m7G modification and CVD (142). Recently, a study have noted that METTL1-mediated m7G modification is necessary for self-renewal and differentiation of mouse embryonic stem cells (mESCs) and human induced pluripotent stem cells (hiPSCs), while depletion of METTL1 results in the upregulation of genes related to cardiovascular system development such as angiogenesis, thus implicating the potential of m7G modification as a therapeutic target in CVD therapy (114, 115).
The presence of 5-methylcytosine (m5C) in DNA and its role as an epigenetic regulator of genome activity has been well-established. In fact, m5C is also widely distributed in eukaryotic RNAs, mainly confined to tRNAs and rRNAs, as well as in mRNAs and other ncRNAs, and is involved in multiple cellular functions such as cell cycle control, cell proliferation and differentiation, and development (143). The important role of m5C post-transcriptional modification in mRNA metabolism focus mainly on the regulation of mRNA export and translation (144–146). As the major mRNA m5C methyltransferase, NSun2 upregulates the expression of ICAM-1 by methylating ICAM-1 mRNA, thereby increasing the adhesion of leukocytes to endothelial cells and contributing to the development of vascular endothelial inflammation and atherosclerosis (116).
In contrast to other RNA modifications, N4-acetylcytidine (ac4C) remains poorly understood. Initially described in the bacterial tRNAmet anticodon several decades ago (147), ac4C was subsequently detected in eukaryotic tRNAs and 18S rRNA (148). More recently, Arango et al. documented for the first time the presence of ac4C modification in mRNAs, and they determined that mRNA acetylation within coding sequences can promote translation and mRNA stability (149). In all cases above, N-acetyltransferase 10 (NAT10) is the only known enzyme responsible for ac4C production (150). There has been robust evidence that ac4C-mediated upregulation of inflammasome genes, such as NLRC4 and NLRP3, is closely involved in the pathogenesis of cardiovascular disorders (151, 152). ac4C and adenine, when applied in combination, can activate the NLRC4 inflammasome, induce the IL-1β production in monocytes and neutrophils, enhance platelets and neutrophil activation as well as elevate blood pressure in mice (117). Moreover, the pro-inflammatory effect of NLRP3/IL-1β axis has been fully elucidated in the development of various CVDs, including atherosclerosis (153), atrial fibrillation (154), myocardial infarction (155) and ischemia-reperfusion injury (156), indicating that ac4C modification may be a promising therapeutic target in the future.
2'-O-methylation (Nm) is one of the most prevalent RNA modifications, which can occur within any ribonucleotide (Am, Cm, Gm, Um) (157, 158). Nm acts as an important epigenetic regulators of gene expression, which has recently been found to stabilize mRNA and inhibit its translation in mammalian cells (159, 160). To date, there has been accumulating evidence that Nm modification is strongly associated with CVD (161). According to the latest research, scaRNAs-directed modification of Nm is able to regulating mRNA splicing variants which are crucial for heart development, and disruption of which would alter regulatory processes leading to defective heart development and CHDs (118).
The exploration and understanding of epigenetic landscapes during cardiometabolic disturbances furnish novel epigenetic targets that may become promising pharmacological interventions to mitigate the development of CVDs (162). Multiple preclinical studies have shown the cardioprotective effects of various epigenetic-based therapies (“epidrugs”), including pharmacological DNA methylation inhibitors, histone deacetylase inhibitors (HDACi), BET inhibitors, and so on (163) (Table 2).
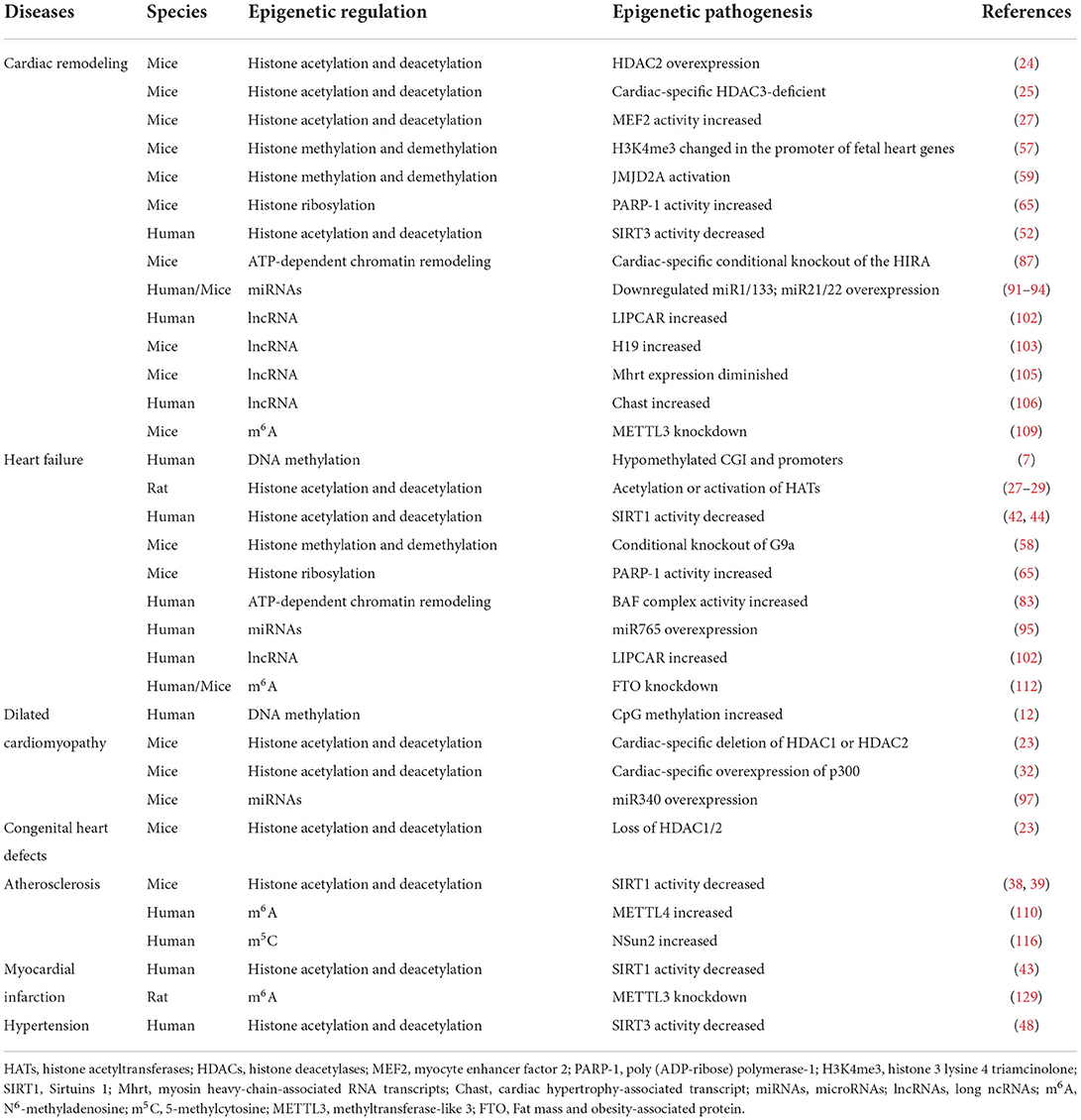
Table 2. The multiple cardiac diseases and their corresponding epigenetic pathogenesis.
The reversible methylation status and expression levels of target genes offers researchers a promising prospect for developing the potential effects of DNA methyltransferase inhibitors or related epidrugs on improving pathological myocardial remodeling. Hydrazine, a drug originally designed to lower blood pressure, was found to maintain Ca (2+) homeostasis and improve cardiac function by enhancing myocardial sarcoplasmic reticulum Ca (2+)-ATPase (SERCA2a) activity and inducing DNA demethylation in the gene promoter in cardiomyocytes (164). The supplementation of selenium which is an essential trace element showed obvious cardioprotective effects toward advanced glycation end products (AGEs)-induced heart failure via inhibiting DNMT2-induced DNA methylation of glutathione peroxidase 1 (GPX1) gene promoter in myocytes (165). Moreover, as an inhibitor of DNA methylation, 5-Azacytidine (5aza) has been shown to be effective in attenuating pressure overload-induced myocardial remodeling (166).
HDACi, which exerted anti-proliferative and anti-inflammatory effects, are the largest class of epidrugs that may be highly correlated with the prevention and treatment of CVDs (167). Indeed, HDACs could modulate acetylation status of histone proteins. The current preclinical studies indicated that HDACi could mitigate cardiac pathological remodeling and improve myocardial function via acetylating or deacetylating target genes (168, 169). The available HDACi, suberoylanilide hydroxamic acid (SAHA), was reported to attenuate ischemia/reperfusion-induced cardiac injure through the induction of cardiomyocyte autophagic flux (170). Also, sodium butyrate has been identified to blunt myocardial I/R injury and preserve cardiac performance by inhibiting the activity of histone deacetylases (171, 172). Aliphatic acids like valproic acid (VPA), an inhibitor of class I HDACs, has been reported to attenuate MI-induced myocardial remodeling by inhibiting the activities of histone deacetylases and subsequently accumulating hyperacetylated histone tails (173). Entinostat (MS-275A), another inhibitor of class I HDACs, has also been reported to play a protective role in ischemia reperfusion injury-induced myocardial remodeling (174). Besides, the class II HDACi could prevent myocardial hypertrophy and cardiac fibrosis (175). However, Trichostatin A (TSA), an inhibitor of both class I and II HDACs, showed some severe side effects including DNA damage, which limited its clinical use (176).
The effects of some compounds (i.e., folates, apicidin, and valproic acid) on regulating chromatin accessibility that changed in cardiometabolic disease states by modulating epigenetic marks on DNA or histones aroused the interest of developing them as CVD-related epidrugs (177, 178). Apabetalone (RVX-208), a direct epidrug, was shown to mitigate the development of CVDs and reduce major adverse cardiovascular event (MACE) risk by binding to the BET family member BRD4 (179, 180). The transcriptional regulation of RVX-208 targeting BRD4 could modulate cholesterol metabolism and vascular inflammation, contributing to its effects on attenuating cardiac hypertrophy and fibrosis (181, 182). Also, in mice or cultured cells, the small-molecule inhibitor JQ1 (a small molecular inhibitor of BET acetylycine reader protein) could inhibit norepinephrine-induced cell hypertrophy. It has also been reported that the use of JQ1 had therapeutic effects on animal models of pathological myocardial remodeling (183).
The rapid development of the nucleotide gene therapies which includes antisense oligonucleotide (ASO) and siRNA may usher in the potential breakthrough of the treatment targeting non-coding RNAs in ameliorating the symptoms and prognosis of patients with CVDs (184, 185). Notably, the easy-synthesized mimic or analogs or inhibitors of non-coding RNA are considered low toxic when transfected in vivo (184), which may pave the way for the use of exosome miRNAs and/or lncRNAs as new tools for diagnosing and treating pathological myocardial remodeling.
Multiple potent and selective small molecule compounds targeting RNA modification have been confirmed and applied mainly in cancer-related fields (186). Recently, a few activators or inhibitors of m6A enzymes including METTL3–METTL14 complex have also been developed (187, 188). Remodelin, a small molecule inhibitor of NAT10, was reported to alleviate cancers or inflammatory diseases (189, 190). However, the effectiveness of these compounds in CVDs remain to be demonstrated in the future. Moreover, epitranscriptome editing, a novel genome editing tool, could modify m6A via editing mutated or dysregulated functional m6A sites, making it worthy of being a promising approach to the clinical treatment of pathological myocardial remodeling subsequent to heart failure (191).
However, despite a variety of experimental evidence demonstrated above, the degree to which these preclinical observations or findings could be translated into humans is still relatively unknown, which were looking forward to being tested in more standardized clinical trials.
The repurposed drugs with a possible indirect epigenetic interference, including metformin, statins, sodium glucose transporter inhibitors 2 (SGLT2i), and omega 3 polyunsaturated fatty acids (PUFAs), have been identified by a number of preclinical and clinical experiments as having the cardioprotective effects in cardiac cells (163, 192). Specifically, it was reported that metformin could ameliorate cardiac fibrosis (193). Metformin have epigenetic-oriented effects by inhibiting the activity and expression of multiple histone methyltransferases, HATs, and DNMTs, thus affecting the transcription of target genes implicated in some vital signaling pathways (194). Statins may also participate in the prevention and treatment of CVDs through histone modifications (195). There are still few randomized trials have been carried out to evaluate whether the potential epigenetic-oriented effects of these non-canonical repurposed drugs, which were not originally developed as epidrugs, were involved in their cardioprotective effects against different clinical phenotypes of heart failure (163).
The detrimental effects of epigenetically altered gene expression profiles elicited by undesirable environmental conditions are increasingly posing an overwhelming threat to human health status, leading to higher prevalence of CVD worldwide. In this mini-review, we summarize the current knowledge about mechanistic basis for epigenetic regulation, which includes DNA methylation, histone modifications, ATP-dependent chromatin structural remodeling, non-coding RNAs and RNA modification, and further discuss the potential role of epigenetic regulation in pathological myocardial remodeling.
Despite advancements in this new realm, however, there still exist some restrictions that hamper the therapeutic exploitation of epigenetic regulators in clinical applications.
• First to be noted, most systemic epigenetic regulators possess limited cardiac-specificity, and instead, they alter gene expression in a relatively routine and comprehensive manner, which may result in an array of adverse effects.
• Second, due to the complexity of cell type composition in cardiac tissue and cell type-specific epigenetic characteristics in disease progression, studies in which changes in epigenetic profile of each cellular component in hypertrophic heart are still lacking. Pursuing cell-specific epigenetic information that changes in the cardiometabolic processes may advance individualized risk assessment and individualized pharmacotherapies for patients.
• Third, the establishment and refinement of appropriate targeted therapeutic strategies with high effectiveness and safety and low incidence of off-target mutations based on epigenetic mechanisms remain a great challenge to date.
• Forth, more research is needed to acquire a comprehensive understanding of how epigenetic regulation perform on cardiovascular disease so as to open up new avenues for pathological myocardial remodeling therapy.
• Finally, precisely defined patient cohorts in whom one pathomechanism is the sole or dominant cause of disease development and progression as well as defined selection criteria and suitable clinical outcome parameters needs to be highly considered in future clinical applications.
In the near future, with the tremendous changes in disease spectrum, rapid emergence of advanced sequencing technologies and increasing demand for novel pathological myocardial remodeling therapy, these gaps could be partly overcome using innovative bioinformatic tools designed to disentangle the epigenetic landscape of myocardial remodeling in man, which may provide improved precision medicine and personalized therapeutic approaches for patients at high risk of myocardial remodeling. In parallel, the continuous research and development on the molecular mechanisms underlying the cardioprotective effects of epidrugs would definitely broaden their application prospects for beneficial to patients with pathological myocardial remodeling or cardiometabolic disturbances.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. (2018) 15:387–407. doi: 10.1038/s41569-018-0007-y
2. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. (2004) 429:457–63. doi: 10.1038/nature02625
3. Liang M. Epigenetic mechanisms and hypertension. Hypertension. (2018) 72:1244–54. doi: 10.1161/HYPERTENSIONAHA.118.11171
4. Liu CF, Tang WHW. Epigenetics in cardiac hypertrophy and heart failure. JACC Basic Transl Sci. (2019) 4:976–93. doi: 10.1016/j.jacbts.2019.05.011
5. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. (2012) 13:484–92. doi: 10.1038/nrg3230
6. Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet. (2021) 37:1012–27. doi: 10.1016/j.tig.2021.05.002
7. Movassagh M, Choy MK, Goddard M, Bennett MR, Down TA, Foo RS. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PloS ONE. (2010) 5:e8564. doi: 10.1371/journal.pone.0008564
8. Movassagh M, Choy MK, Knowles DA, Cordeddu L, Haider S, Down T, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. (2011) 124:2411–22. doi: 10.1161/CIRCULATIONAHA.111.040071
9. Pepin ME, Ha CM, Crossman DK, Litovsky SH, Varambally S, Barchue JP, et al. Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab Investig. (2019) 99:371–86. doi: 10.1038/s41374-018-0104-x
10. Mayer SC, Gilsbach R, Preissl S, Monroy Ordonez EB, Schnick T, Beetz N, et al. Adrenergic repression of the epigenetic reader MeCP2 facilitates cardiac adaptation in chronic heart failure. Circ Res. (2015) 117:622–33. doi: 10.1161/CIRCRESAHA.115.306721
11. Pan S, Lai H, Shen Y, Breeze C, Beck S, Hong T, et al. DNA methylome analysis reveals distinct epigenetic patterns of ascending aortic dissection and bicuspid aortic valve. Cardiovasc Res. (2017) 113:692–704. doi: 10.1093/cvr/cvx050
12. Cheedipudi SM, Matkovich SJ, Coarfa C, Hu X, Robertson MJ, Sweet M, et al. Genomic reorganization of lamin-associated domains in cardiac myocytes is associated with differential gene expression and DNA methylation in human dilated cardiomyopathy. Circ Res. (2019) 124:1198–213. doi: 10.1161/CIRCRESAHA.118.314177
13. Haas J, Frese KS, Park YJ, Keller A, Vogel B, Lindroth AM, et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med. (2013) 5:413–29. doi: 10.1002/emmm.201201553
14. Zhang Y, Sun Z, Jia J, Du T, Zhang N, Tang Y, et al. Overview of histone modification. Adv Exp Med Biol. (2021) 1283:1–16. doi: 10.1007/978-981-15-8104-5_1
15. Zhao Y, Garcia BA. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb Perspect Biol. (2015) 7:a025064. doi: 10.1101/cshperspect.a025064
16. Papait R, Serio S, Condorelli G. Role of the epigenome in heart failure. Physiol Rev. (2020) 100:1753–77. doi: 10.1152/physrev.00037.2019
17. Héberlé É, Bardet AF. Sensitivity of transcription factors to DNA methylation. Essays Biochem. (2019) 63:727–41. doi: 10.1042/EBC20190033
18. Chelladurai P, Boucherat O, Stenmark K, Kracht M, Seeger W, Bauer UM, et al. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol. (2021) 178:54–71. doi: 10.1111/bph.14932
19. Boggs AE, Vitolo MI, Whipple RA, Charpentier MS, Goloubeva OG, Ioffe OB, et al. α-Tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration. Cancer Res. (2015) 75:203–15. doi: 10.1158/0008-5472.CAN-13-3563
20. Toker L, Tran GT, Sundaresan J, Tysnes OB, Alves G, Haugarvoll K, et al. Genome-wide histone acetylation analysis reveals altered transcriptional regulation in the Parkinson's disease brain. Mol Neurodegener. (2021) 16:31. doi: 10.1186/s13024-021-00450-7
21. Qin J, Guo N, Tong J, Wang Z. Function of histone methylation and acetylation modifiers in cardiac hypertrophy. J Mol Cell Cardiol. (2021) 159:120–129. doi: 10.1016/j.yjmcc.2021.06.011
22. Gray SG, Teh BT. Histone acetylation/deacetylation and cancer: an “open” and “shut” case? Curr Mol Med. (2001) 1:401–29. doi: 10.2174/1566524013363537
23. Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. (2007) 21:1790–802. doi: 10.1101/gad.1563807
24. Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. (2007) 13:324–31. doi: 10.1038/nm1552
25. Trivedi CM, Lu MM, Wang Q, Epstein JA. Transgenic overexpression of Hdac3 in the heart produces increased postnatal cardiac myocyte proliferation but does not induce hypertrophy. J Biol Chem. (2008) 283:26484–9. doi: 10.1074/jbc.M803686200
26. Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, et al. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. (2008) 118:3588–97. doi: 10.1172/JCI35847
27. Backs J, Worst BC, Lehmann LH, Patrick DM, Jebessa Z, Kreusser MM, et al. Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4. J Cell Biol. (2011) 195:403–15. doi: 10.1083/jcb.201105063
28. He T, Huang J, Chen L, Han G, Stanmore D, Krebs-Haupenthal J, et al. Cyclic AMP represses pathological MEF2 activation by myocyte-specific hypo-phosphorylation of HDAC5. J Mol Cell Cardiol. (2020) 145:88–98. doi: 10.1016/j.yjmcc.2020.05.018
29. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. (2002) 110:479–88. doi: 10.1016/S0092-8674(02)00861-9
30. Helmstadter KG, Ljubojevic-Holzer S, Wood BM, Taheri KD, Sedej S, Erickson JR, et al. CaMKII and PKA-dependent phosphorylation co-regulate nuclear localization of HDAC4 in adult cardiomyocytes. Basic Res Cardiol. (2021) 116:11. doi: 10.1007/s00395-021-00850-2
31. Slepak TI, Webster KA, Zang J, Prentice H, O'Dowd A, Hicks MN, et al. Control of cardiac-specific transcription by p300 through myocyte enhancer factor-2D. J Biol Chem. (2001) 276:7575–85. doi: 10.1074/jbc.M004625200
32. Yanazume T, Hasegawa K, Morimoto T, Kawamura T, Wada H, Matsumori A, et al. Cardiac p300 is involved in myocyte growth with decompensated heart failure. Mol Cell Biol. (2003) 23:3593–606. doi: 10.1128/MCB.23.10.3593-3606.2003
33. Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. (2008) 118:868–78. doi: 10.1172/JCI33160
34. Liberale L, Kraler S, Camici GG, Lüscher TF. Ageing and longevity genes in cardiovascular diseases. Basic Clin Pharmacol Toxicol. (2020) 127:120–31. doi: 10.1111/bcpt.13426
35. Singh CK, Chhabra G, Ndiaye MA, Garcia-Peterson LM, Mack NJ, Ahmad N. The role of sirtuins in antioxidant and redox signaling. Antioxid Redox Signal. (2018) 28:643–61. doi: 10.1089/ars.2017.7290
36. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. (1999) 13:2570–80. doi: 10.1101/gad.13.19.2570
37. Pan X, Pi C, Ruan X, Zheng H, Zhang D, Liu X. Mammalian sirtuins and their relevance in vascular calcification. Front Pharmacol. (2022) 13:907835. doi: 10.3389/fphar.2022.907835
38. Ministrini S, Puspitasari YM, Beer G, Liberale L, Montecucco F, Camici GG. Sirtuin 1 in endothelial dysfunction and cardiovascular aging. Front Physiol. (2021) 12:733696. doi: 10.3389/fphys.2021.733696
39. Chen C, Zhou M, Ge Y, Wang X. SIRT1 and aging related signaling pathways. Mech Ageing Dev. (2020) 187:111215. doi: 10.1016/j.mad.2020.111215
40. Hwang JW, Yao H, Caito S, Sundar IK, Rahman I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic Biol Med. (2013) 61:95–110. doi: 10.1016/j.freeradbiomed.2013.03.015
41. Meng X, Tan J, Li M, Song S, Miao Y, Zhang Q. Sirt1: role under the condition of ischemia/hypoxia. Cell Mol Neurobiol. (2017) 37:17–28. doi: 10.1007/s10571-016-0355-2
42. Lu TM, Tsai JY, Chen YC, Huang CY, Hsu HL, Weng CF, et al. Downregulation of Sirt1 as aging change in advanced heart failure. J Biomed Sci. (2014) 21:57. doi: 10.1186/1423-0127-21-57
43. D'Onofrio N, Sardu C, Paolisso P, Minicucci F, Gragnano F, Ferraraccio F, et al. MicroRNA-33 and SIRT1 influence the coronary thrombus burden in hyperglycemic STEMI patients. J Cell Physiol. (2020) 235:1438–52. doi: 10.1002/jcp.29064
44. Sun S, Wang C, Weng J. MicroRNA-138-5p drives the progression of heart failure via inhibiting sirtuin 1 signaling. Mol Med Rep. (2021) 23:276. doi: 10.3892/mmr.2021.11915
45. Gorski PA, Jang SP, Jeong D, Lee A, Lee P, Oh JG, et al. Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca(2+)-ATPase in heart failure. Circ Res. (2019) 124:e63–80. doi: 10.1161/CIRCRESAHA.118.313865
46. Packer M. Cardioprotective effects of sirtuin-1 and its downstream effectors: potential role in mediating the heart failure benefits of SGLT2 (sodium-glucose cotransporter 2) inhibitors. Circ Heart Fail. (2020) 13:e007197. doi: 10.1161/CIRCHEARTFAILURE.120.007197
47. Cao M, Zhao Q, Sun X, Qian H, Lyu S, Chen R, et al. Sirtuin 3: emerging therapeutic target for cardiovascular diseases. Free Radic Biol Med. (2022) 180:63–74. doi: 10.1016/j.freeradbiomed.2022.01.005
48. He X, Zeng H, Chen JX. Emerging role of SIRT3 in endothelial metabolism, angiogenesis, and cardiovascular disease. J Cell Physiol. (2019) 234:2252–65. doi: 10.1002/jcp.27200
49. Palomer X, Román-Azcona MS, Pizarro-Delgado J, Planavila A, Villarroya F, Valenzuela-Alcaraz B, et al. SIRT3-mediated inhibition of FOS through histone H3 deacetylation prevents cardiac fibrosis and inflammation. Signal Transduct Target Ther. (2020) 5:14. doi: 10.1038/s41392-020-0114-1
50. Silaghi CN, Farca? M, Crăciun AM. Sirtuin 3 (SIRT3) pathways in age-related cardiovascular and neurodegenerative diseases. Biomedicines. (2021) 9:1574. doi: 10.3390/biomedicines9111574
51. Chen J, Chen S, Zhang B, Liu J. SIRT3 as a potential therapeutic target for heart failure. Pharmacol Res. (2021) 165:105432. doi: 10.1016/j.phrs.2021.105432
52. Zeng H, He X, Chen JX. Endothelial sirtuin 3 dictates glucose transport to cardiomyocyte and sensitizes pressure overload-induced heart failure. J Am Heart Assoc. (2020) 9:e015895. doi: 10.1161/JAHA.120.015895
53. Zeng H, Chen JX. Sirtuin 3, endothelial metabolic reprogramming, and heart failure with preserved ejection fraction. J Cardiovasc Pharmacol. (2019) 74:315–23. doi: 10.1097/FJC.0000000000000719
54. Murray K. The occurrence of epsilon-n-methyl lysine in histones. Biochemistry. (1964) 3:10–5. doi: 10.1021/bi00889a003
55. Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, et al. Regulation of transcription by a protein methyltransferase. Science. (1999) 284:2174–7. doi: 10.1126/science.284.5423.2174
56. Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. (2014) 15:93–106. doi: 10.1038/nrg3607
57. Pandya K, Kohro T, Mimura I, Kobayashi M, Wada Y, Kodama T, et al. Distribution of histone3 lysine 4 trimethylation at T3-responsive loci in the heart during reversible changes in gene expression. Gene Expr. (2012) 15:183–98. doi: 10.3727/105221612X13372578119698
58. Papait R, Serio S, Pagiatakis C, Rusconi F, Carullo P, Mazzola M, et al. Histone methyltransferase G9a is required for cardiomyocyte homeostasis and hypertrophy. Circulation. (2017) 136:1233–46. doi: 10.1161/CIRCULATIONAHA.117.028561
59. Zhang QJ, Chen HZ, Wang L, Liu DP, Hill JA, Liu ZP. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest. (2011) 121:2447–56. doi: 10.1172/JCI46277
60. Stein AB, Jones TA, Herron TJ, Patel SR, Day SM, Noujaim SF, et al. Loss of H3K4 methylation destabilizes gene expression patterns and physiological functions in adult murine cardiomyocytes. J Clin Invest. (2011) 121:2641–50. doi: 10.1172/JCI44641
61. Mahmoud SA, Poizat C. Epigenetics and chromatin remodeling in adult cardiomyopathy. J Pathol. (2013) 231:147–57. doi: 10.1002/path.4234
62. Wang Y, Luo W, Wang Y. PARP-1 and its associated nucleases in DNA damage response. DNA Repair. (2019) 81:102651. doi: 10.1016/j.dnarep.2019.102651
63. Pandey N, Black BE. Rapid detection and signaling of DNA damage by PARP-1. Trends Biochem Sci. (2021) 46:744–757. doi: 10.1016/j.tibs.2021.01.014
64. Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. (2006) 125:315–26. doi: 10.1016/j.cell.2006.02.041
65. Pillai JB, Gupta M, Rajamohan SB, Lang R, Raman J, Gupta MP. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. (2006) 291:H1545–53. doi: 10.1152/ajpheart.01124.2005
66. Bartha E, Solti I, Kereskai L, Lantos J, Plozer E, Magyar K, et al. PARP inhibition delays transition of hypertensive cardiopathy to heart failure in spontaneously hypertensive rats. Cardiovasc Res. (2009) 83:501–10. doi: 10.1093/cvr/cvp144
67. Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, et al. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. (2010) 466:62–7. doi: 10.1038/nature09130
68. Palfi A, Toth A, Kulcsar G, Hanto K, Deres P, Bartha E, et al. The role of Akt and mitogen-activated protein kinase systems in the protective effect of poly(ADP-ribose) polymerase inhibition in Langendorff perfused and in isoproterenol-damaged rat hearts. J Pharmacol Exp Ther. (2005) 315:273–82. doi: 10.1124/jpet.105.088336
69. Palfi A, Toth A, Hanto K, Deres P, Szabados E, Szereday Z, et al. Sumegi B, Toth K, Halmosi R. PARP inhibition prevents postinfarction myocardial remodeling and heart failure via the protein kinase C/glycogen synthase kinase-3beta pathway. J Mol Cell Cardiol. (2006) 41:149–59. doi: 10.1016/j.yjmcc.2006.03.427
70. Kaneda R, Takada S, Yamashita Y, Choi YL, Nonaka-Sarukawa M, Soda M, et al. Genome-wide histone methylation profile for heart failure. Genes Cells. (2009) 14:69–77. doi: 10.1111/j.1365-2443.2008.01252.x
71. Papait R, Cattaneo P, Kunderfranco P, Greco C, Carullo P, Guffanti A, et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci USA. (2013) 110:20164–9. doi: 10.1073/pnas.1315155110
72. Gilsbach R, Schwaderer M, Preissl S, Gruning BA, Kranzhofer D, Schneider P, et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun. (2018) 9:391. doi: 10.1038/s41467-017-02762-z
73. Nodelman IM, Bowman GD. Biophysics of chromatin remodeling. Annu Rev Biophys. (2021) 50:73–93. doi: 10.1146/annurev-biophys-082520-080201
74. Spiltoir JI, Stratton MS, Cavasin MA, Demos-Davies K, Reid BG, Qi J, et al. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J Mol Cell Cardiol. (2013) 63:175–9. doi: 10.1016/j.yjmcc.2013.07.017
75. Borck PC, Guo LW, Plutzky J. BET Epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res. (2020) 126:1190–208. doi: 10.1161/CIRCRESAHA.120.315929
76. Crosswhite PL. ATP-dependent chromatin remodeling complexes in embryonic vascular development and hypertension. Am J Physiol Heart Circ Physiol. (2019) 317:H575–80. doi: 10.1152/ajpheart.00147.2019
77. Centore RC, Sandoval GJ, Soares LMM, Kadoch C, Chan HM. Mammalian SWI/SNF chromatin remodeling complexes: emerging mechanisms and therapeutic strategies. Trends Genet. (2020) 36:936–50. doi: 10.1016/j.tig.2020.07.011
78. Banerjee R, Bultman SJ, Holley D, Hillhouse C, Bain JR, Newgard CB, et al. Non-targeted metabolomics of Brg1/Brm double-mutant cardiomyocytes reveals a novel role for SWI/SNF complexes in metabolic homeostasis. Metabolomics. (2015) 11:1287–301. doi: 10.1007/s11306-015-0786-7
79. Bevilacqua A, Willis MS, Bultman SJ. SWI/SNF chromatin-remodeling complexes in cardiovascular development and disease. Cardiovasc Pathol. (2014) 23:85–91. doi: 10.1016/j.carpath.2013.09.003
80. Bultman SJ, Holley DW, G de Ridder G, Pizzo SV, Sidorova TN, Murray KT, et al. BRG1 and BRM SWI/SNF ATPases redundantly maintain cardiomyocyte homeostasis by regulating cardiomyocyte mitophagy and mitochondrial dynamics in vivo. Cardiovasc Pathol. (2016) 25:258–69. doi: 10.1016/j.carpath.2016.02.004
81. Vieira JM, Howard S, Villa Del Campo C, Bollini S, Dubé KN, Masters M, et al. BRG1-SWI/SNF-dependent regulation of the Wt1 transcriptional landscape mediates epicardial activity during heart development and disease. Nat Commun. (2017) 8:16034. doi: 10.1038/ncomms16034
82. Lei I, Tian S, Chen V, Zhao Y, Wang Z. SWI/SNF component BAF250a coordinates OCT4 and WNT signaling pathway to control cardiac lineage differentiation. Front Cell Dev Biol. (2019) 7:358. doi: 10.3389/fcell.2019.00358
83. England J, Loughna S. Heavy and light roles: myosin in the morphogenesis of the heart. Cell Mol Life Sci. (2013) 70:1221–39. doi: 10.1007/s00018-012-1131-1
84. Han P, Li W, Yang J, Shang C, Lin CH, Cheng W, et al. Epigenetic response to environmental stress: assembly of BRG1-G9a/GLP-DNMT3 repressive chromatin complex on Myh6 promoter in pathologically stressed hearts. Biochim Biophys Acta. (2016) 1863(Pt. B):1772–81. doi: 10.1016/j.bbamcr.2016.03.002
85. Billon P, Cote J. Precise deposition of histone H2A.Z in chromatin for genome expression and maintenance. Biochim Biophys Acta. (2013) 1819:290–302. doi: 10.1016/j.bbagrm.2011.10.004
86. Chen IY, Lypowy J, Pain J, Sayed D, Grinberg S, Alcendor RR, et al. Histone H2A.z is essential for cardiac myocyte hypertrophy but opposed by silent information regulator 2alpha. J Biol Chem. (2006) 281:19369–77. doi: 10.1074/jbc.M601443200
87. Valenzuela N, Fan Q, Fa'ak F, Soibam B, Nagandla H, Liu Y, et al. Cardiomyocyte-specific conditional knockout of the histone chaperone HIRA in mice results in hypertrophy, sarcolemmal damage and focal replacement fibrosis. Dis Model Mech. (2016) 9:335–45. doi: 10.1242/dmm.022889
88. Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. (2013) 9:e1003569. doi: 10.1371/journal.pgen.1003569
89. Panni S, Lovering RC, Porras P, Orchard S. Non-coding RNA regulatory networks. Biochim Biophys Acta Gene Regul Mech. (2020) 1863:194417. doi: 10.1016/j.bbagrm.2019.194417
90. Zhu L, Li N, Sun L, Zheng D, Shao G. Non-coding RNAs: the key detectors and regulators in cardiovascular disease. Genomics. (2021) 113(Pt. 2):1233–246. doi: 10.1016/j.ygeno.2020.10.024
91. Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. (2007) 100:416–24. doi: 10.1161/01.RES.0000257913.42552.23
92. Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nature medicine. (2007) 13:613–8. doi: 10.1038/nm1582
93. Surina S, Fontanella RA, Scisciola L, Marfella R, Paolisso G, Barbieri M. miR-21 in human cardiomyopathies. Front Cardiovasc Med. (2021) 8:767064. doi: 10.3389/fcvm.2021.767064
94. Xu XD, Song XW, Li Q, Wang GK, Jing Q, Qin YW. Attenuation of microRNA-22 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. J Cell Physiol. (2012) 227:1391–8. doi: 10.1002/jcp.22852
95. Cai WF, Liu GS, Lam CK, Florea S, Qian J, Zhao W, et al. Up-regulation of micro-RNA765 in human failing hearts is associated with post-transcriptional regulation of protein phosphatase inhibitor-1 and depressed contractility. Eur J Heart Fail. (2015) 17:782–93. doi: 10.1002/ejhf.323
96. Matkovich SJ, Hu Y, Eschenbacher WH, Dorn LE, Dorn GW II. Direct and indirect involvement of microRNA-499 in clinical and experimental cardiomyopathy. Circ Res. (2012) 111:521–31. doi: 10.1161/CIRCRESAHA.112.265736
97. Zhou J, Gao J, Zhang X, Liu Y, Gu S, Zhang X, et al. microRNA-340-5p functions downstream of cardiotrophin-1 to regulate cardiac eccentric hypertrophy and heart failure via target gene dystrophin. Int Heart J. (2015) 56:454–8. doi: 10.1536/ihj.14-386
98. Xiao J, Liu H, Cretoiu D, Toader DO, Suciu N, Shi J, et al. miR-31a-5p promotes postnatal cardiomyocyte proliferation by targeting RhoBTB1. Exp Mol Med. (2017) 49:e386. doi: 10.1038/emm.2017.150
99. Shi J, Bei Y, Kong X, Liu X, Lei Z, Xu T, et al. miR-17-3p Contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. Theranostics. (2017) 7:664–676. doi: 10.7150/thno.15162
100. Sardu C, Massetti M, Scisciola L, Trotta MC, Santamaria M, Volpicelli M, et al. Angiotensin receptor/Neprilysin inhibitor effects in CRTd non-responders: from epigenetic to clinical beside. Pharmacol Res. (2022) 182:106303. doi: 10.1016/j.phrs.2022.106303
101. Wang L, Wang J, Li G, Xiao J. Non-coding RNAs in physiological cardiac hypertrophy. Adv Exp Med Biol. (2020) 1229:149–61. doi: 10.1007/978-981-15-1671-9_8
102. Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A, et al. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res. (2014) 114:1569–75. doi: 10.1161/CIRCRESAHA.114.303915
103. Liu L, An X, Li Z, Song Y, Li L, Zuo S, et al. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc Res. (2016) 111:56–65. doi: 10.1093/cvr/cvw078
104. Wang Z, Zhang XJ, Ji YX, Zhang P, Deng KQ, Gong J, et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. (2016) 22:1131–39. doi: 10.1038/nm.4179
105. Han P, Li W, Lin CH, Yang J, Shang C, Nuernberg ST, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. (2014) 514:102–6. doi: 10.1038/nature13596
106. Viereck J, Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M, et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med. (2016) 8:326ra22. doi: 10.1126/scitranslmed.aaf1475
107. Jonkhout N, Tran J, Smith MA, Schonrock N, Mattick JS, Novoa EM. The RNA modification landscape in human disease. Rna. (2017) 23:1754–1769. doi: 10.1261/rna.063503.117
108. Schaefer MR. The regulation of RNA modification systems: the next frontier in epitranscriptomics? Genes. (2021) 12:345. doi: 10.3390/genes12030345
109. Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, et al. The N(6)-Methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation. (2019) 139:533–45. doi: 10.1161/CIRCULATIONAHA.118.036146
110. Zhang BY, Han L, Tang YF, Zhang GX, Fan XL, Zhang JJ, et al. METTL14 regulates M6A methylation-modified primary miR-19a to promote cardiovascular endothelial cell proliferation and invasion. Eur Rev Med Pharmacol Sci. (2020) 24:7015–23. doi: 10.26355/eurrev_202006_21694
111. Jian D, Wang Y, Jian L, Tang H, Rao L, Chen K, et al. METTL14 aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics. (2020) 10:8939–56. doi: 10.7150/thno.45178
112. Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N, et al. FTO-Dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation. (2019) 139:518–32. doi: 10.1161/CIRCULATIONAHA.118.033794
113. Li Y, Yu H, Zhao W, Xu X, Zhou J, Xu M, et al. Analysis of urinary methylated nucleosides of patients with coronary artery disease by high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom. (2014) 28:2054–8. doi: 10.1002/rcm.6986
114. Lin S, Liu Q, Lelyveld VS, Choe J, Szostak JW, Gregory RI. Mettl1/Wdr4-Mediated m(7)G tRNA Methylome Is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol Cell. (2018) 71:244–55.e5. doi: 10.1016/j.molcel.2018.06.001
115. Deng Y, Zhou Z, Ji W, Lin S, Wang M. METTL1-mediated m(7)G methylation maintains pluripotency in human stem cells and limits mesoderm differentiation and vascular development. Stem Cell Res Ther. (2020) 11:306. doi: 10.1186/s13287-020-01814-4
116. Luo Y, Feng J, Xu Q, Wang W, Wang X. NSun2 Deficiency protects endothelium from inflammation via mRNA methylation of ICAM-1. Circ Res. (2016) 118:944–56. doi: 10.1161/CIRCRESAHA.115.307674
117. Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med. (2017) 23:174–84. doi: 10.1038/nm.4267
118. Nagasawa C, Ogren A, Kibiryeva N, Marshall J, O'Brien JE, Kenmochi N, et al. The Role of scaRNAs in adjusting alternative mRNA splicing in heart development. J Cardiovasc Dev Dis. (2018) 5:26. doi: 10.3390/jcdd5020026
119. Akhtar J, Lugoboni M, Junion G. m(6)A RNA modification in transcription regulation. Transcription. (2021) 12:266–76. doi: 10.1080/21541264.2022.2057177
120. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. (1974) 71:3971–5. doi: 10.1073/pnas.71.10.3971
121. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. (2011) 7:885–7. doi: 10.1038/nchembio.687
122. Oerum S, Meynier V, Catala M, Tisné C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. (2021) 49:7239–255. doi: 10.1093/nar/gkab378
123. Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. (2013) 155:793–806. doi: 10.1016/j.cell.2013.10.026
124. Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. (2013) 155:1409–21. doi: 10.1016/j.cell.2013.10.047
125. Zhao K, Yang CX, Li P, Sun W, Kong XQ. Epigenetic role of N6-methyladenosine (m6A) RNA methylation in the cardiovascular system. J Zhejiang Univ Sci B. (2020) 21:509–23. doi: 10.1631/jzus.B1900680
126. Chen YS, Ouyang XP, Yu XH, Novák P, Zhou L, He PP, et al. N6-Adenosine methylation (m(6)A) RNA modification: an emerging role in cardiovascular diseases. J Cardiovasc Transl Res. (2021) 14:857–72. doi: 10.1007/s12265-021-10108-w
127. Li T, Zhuang Y, Yang W, Xie Y, Shang W, Su S, et al. Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J. (2021) 35:e21162. doi: 10.1096/fj.201903169R
128. Zhao K, Yang C, Zhang J, Sun W, Zhou B, Kong X, et al. METTL3 improves cardiomyocyte proliferation upon myocardial infarction via upregulating miR-17-3p in a DGCR8-dependent manner. Cell Death Discov. (2021) 2021/10/13;7:291. doi: 10.1038/s41420-021-00688-6
129. Yang C, Zhao K, Zhang J, Wu X, Sun W, Kong X, et al. Comprehensive analysis of the transcriptome-wide m6A methylome of heart via MeRIP after birth: day 0 vs. Day 7. Front Cardiovasc Med. (2021) 8:633631. doi: 10.3389/fcvm.2021.633631
130. Boissel S, Reish O, Proulx K, Kawagoe-Takaki H, Sedgwick B, Yeo GS, et al. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet. (2009) 85:106–11. doi: 10.1016/j.ajhg.2009.06.002
131. Carnevali L, Graiani G, Rossi S, Al Banchaabouchi M, Macchi E, Quaini F, et al. Signs of cardiac autonomic imbalance and proarrhythmic remodeling in FTO deficient mice. PLoS ONE. (2014) 9:e95499. doi: 10.1371/journal.pone.0095499
132. Gustavsson J, Mehlig K, Leander K, Lissner L, Björck L, Rosengren A, et al. FTO genotype, physical activity, and coronary heart disease risk in Swedish men and women. Circ Cardiovasc Genet. (2014) 7:171–7. doi: 10.1161/CIRCGENETICS.111.000007
133. Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. (2010) 42:1086–92. doi: 10.1038/ng.713
134. Shen F, Huang W, Huang JT, Xiong J, Yang Y, Wu K, et al. Decreased N(6)-methyladenosine in peripheral blood RNA from diabetic patients is associated with FTO expression rather than ALKBH5. J Clin Endocrinol Metab. (2015) 100:E148–54. doi: 10.1210/jc.2014-1893
135. Dunn DB. The occurrence of 1-methyladenine in ribonucleic acid. Biochim Biophys Acta. (1961) 46:198–200. doi: 10.1016/0006-3002(61)90668-0
136. Agris PF. The importance of being modified: roles of modified nucleosides and Mg2+ in RNA structure and function. Prog Nucleic Acid Res Mol Biol. (1996) 53:79–129. doi: 10.1016/S0079-6603(08)60143-9
137. Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. (2016) 530:441–6. doi: 10.1038/nature16998
138. Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol. (2016) 12:311–6. doi: 10.1038/nchembio.2040
139. Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, et al. ALKBH1-Mediated tRNA demethylation regulates translation. Cell. (2016) 167:816–28.e16. doi: 10.1016/j.cell.2016.09.038
140. Cowling VH. Regulation of mRNA cap methylation. Biochem J. (2009) 425:295–302. doi: 10.1042/BJ20091352
141. Enroth C, Poulsen LD, Iversen S, Kirpekar F, Albrechtsen A, Vinther J. Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res. (2019) 47:e126. doi: 10.1093/nar/gkz736
142. Song B, Tang Y, Chen K, Wei Z, Rong R, Lu Z, et al. m7GHub: deciphering the location, regulation and pathogenesis of internal mRNA N7-methylguanosine (m7G) sites in human. Bioinformatics. (2020) 36:3528–36. doi: 10.1093/bioinformatics/btaa178
143. Dubin DT, Taylor RH. The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res. (1975) 2:1653–68. doi: 10.1093/nar/2.10.1653
144. Bourgeois G, Ney M, Gaspar I, Aigueperse C, Schaefer M, Kellner S, et al. Eukaryotic rRNA modification by yeast 5-methylcytosine-methyltransferases and human proliferation-associated antigen p120. PLoS ONE. (2015) 10:e0133321. doi: 10.1371/journal.pone.0133321
145. Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. (2012) 40:5023–33. doi: 10.1093/nar/gks144
146. Yang X, Yang Y, Sun BF, Chen YS, Xu JW, Lai WY, et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. (2017) 27:606–25. doi: 10.1038/cr.2017.55
147. Oashi Z, Murao K, Yahagi T, Von Minden DL, McCloskey JA, Nishimura S. Characterization of C + located in the first position of the anticodon of Escherichia coli tRNA Met as N4-acetylcytidine. Biochim Biophys Acta. (1972) 262:209–13. doi: 10.1016/0005-2787(72)90234-1
148. Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. (2018) 46:D303–7. doi: 10.1093/nar/gkx1030
149. Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell. (2018) 175:1872–86.e24. doi: 10.1016/j.cell.2018.10.030
150. Jin G, Xu M, Zou M, Duan S. The processing, gene regulation, biological functions, and clinical relevance of N4-acetylcytidine on RNA: a systematic review. Mol Ther Nucleic Acids. (2020) 20:13–24. doi: 10.1016/j.omtn.2020.01.037
151. Darisipudi MN, Thomasova D, Mulay SR, Brech D, Noessner E, Liapis H, et al. Uromodulin triggers IL-1β-dependent innate immunity via the NLRP3 inflammasome. J Am Soc Nephrol. (2012) 23:1783–9. doi: 10.1681/ASN.2012040338
152. Duan J, Zhang Q, Hu X, Lu D, Yu W, Bai H. N(4)-acetylcytidine is required for sustained NLRP3 inflammasome activation via HMGB1 pathway in microglia. Cell Signal. (2019) 58:44–52. doi: 10.1016/j.cellsig.2019.03.007
153. Paramel Varghese G, Folkersen L, Strawbridge RJ, Halvorsen B, Yndestad A, Ranheim T, et al. NLRP3 Inflammasome expression and activation in human atherosclerosis. J Am Heart Assoc. (2016) 5:e003031. doi: 10.1161/JAHA.115.003031
154. Yao C, Veleva T, Scott L Jr, Cao S, Li L, et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation. (2018) 138:2227–42. doi: 10.1161/CIRCULATIONAHA.118.035202
155. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. (2018) 15:203–14. doi: 10.1038/nrcardio.2017.161
156. Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. (2013) 99:164–74. doi: 10.1093/cvr/cvt091
157. Helm M, Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet. (2017) 18:275–91. doi: 10.1038/nrg.2016.169
158. Vandivier LE, Gregory BD. Reading the epitranscriptome: new techniques and perspectives. Enzymes. (2017) 41:269–98. doi: 10.1016/bs.enz.2017.03.004
159. Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m(6)A(m) in the 5' cap controls mRNA stability. Nature. (2017) 541:371–5. doi: 10.1038/nature21022
160. Elliott BA, Ho HT, Ranganathan SV, Vangaveti S, Ilkayeva O, Abou Assi H, et al. Modification of messenger RNA by 2'-O-methylation regulates gene expression in vivo. Nat Commun. (2019) 10:3401. doi: 10.1038/s41467-019-11375-7
161. Li M, Yang Y, Wang Z, Zong T, Fu X, Aung LHH, et al. Piwi-interacting RNAs (piRNAs) as potential biomarkers and therapeutic targets for cardiovascular diseases. Angiogenesis. (2021) 24:19–34. doi: 10.1007/s10456-020-09750-w
162. Costantino S, Mohammed SA, Ambrosini S, Paneni F. Epigenetic processing in cardiometabolic disease. Atherosclerosis. (2019) 281:150–8. doi: 10.1016/j.atherosclerosis.2018.09.029
163. Napoli C, Bontempo P, Palmieri V, Coscioni E, Maiello C, Donatelli F, et al. Epigenetic therapies for heart failure: current insights and future potential. Vasc Health Risk Manag. (2021) 17:247–54. doi: 10.2147/VHRM.S287082
164. Kao YH, Cheng CC, Chen YC, Chung CC, Lee TI, Chen SA, et al. Hydralazine-induced promoter demethylation enhances sarcoplasmic reticulum Ca2+ -ATPase and calcium homeostasis in cardiac myocytes. Lab Invest. (2011) 91:1291–7. doi: 10.1038/labinvest.2011.92
165. Zhu H, Wang X, Meng X, Kong Y, Li Y, Yang C, et al. Selenium supplementation improved cardiac functions by suppressing DNMT2-mediated GPX1 promoter DNA methylation in AGE-induced heart failure. Oxid Med Cell Longev. (2022) 2022:5402997. doi: 10.1155/2022/5402997
166. Russell-Hallinan A, Neary R, Watson CJ, Baugh JA. Repurposing from oncology to cardiology: low-dose 5-azacytidine attenuates pathological cardiac remodeling in response to pressure overload injury. J Cardiovasc Pharmacol Ther. (2021) 26:375–85. doi: 10.1177/1074248420979235
167. Lyu X, Hu M, Peng J, Zhang X, Sanders YY. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther Adv Chronic Dis. (2019) 10:2040622319862697. doi: 10.1177/2040622319862697
168. Ooi JY, Tuano NK, Rafehi H, Gao XM, Ziemann M, Du XJ, et al. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics. (2015) 10:418–30. doi: 10.1080/15592294.2015.1024406
169. Zhang L, Qin X, Zhao Y, Fast L, Zhuang S, Liu P, et al. Inhibition of histone deacetylases preserves myocardial performance and prevents cardiac remodeling through stimulation of endogenous angiomyogenesis. J Pharmacol Exp Ther. (2012) 341:285–93. doi: 10.1124/jpet.111.189910
170. Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. (2014) 129:1139–51. doi: 10.1161/CIRCULATIONAHA.113.002416
171. Hu X, Zhang K, Xu C, Chen Z, Jiang H. Anti-inflammatory effect of sodium butyrate preconditioning during myocardial ischemia/reperfusion. Exp Ther Med. (2014) 8:229–32. doi: 10.3892/etm.2014.1726
172. Chen Y, Du J, Zhao YT, Zhang L, Lv G, Zhuang S, et al. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol. (2015) 14:99. doi: 10.1186/s12933-015-0262-8
173. Lee TM, Lin MS, Chang NC. Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am J Physiol Heart Circ Physiol. (2007) 293:H968–77. doi: 10.1152/ajpheart.00891.2006
174. Aune SE, Herr DJ, Mani SK, Menick DR. Selective inhibition of class I but not class IIb histone deacetylases exerts cardiac protection from ischemia reperfusion. J Mol Cell Cardiol. (2014) 72:138–45. doi: 10.1016/j.yjmcc.2014.03.005
175. Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, et al. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. (2008) 22:3549–60. doi: 10.1096/fj.08-108548
176. Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. (2011) 17:330–9. doi: 10.1038/nm.2305
177. Title LM, Ur E, Giddens K, McQueen MJ, Nassar BA. Folic acid improves endothelial dysfunction in type 2 diabetes–an effect independent of homocysteine-lowering. Vasc Med. (2006) 11:101–9. doi: 10.1191/1358863x06vm664oa
178. Plutzky J. The PPAR-RXR transcriptional complex in the vasculature: energy in the balance. Circ Res. (2011) 108:1002–16. doi: 10.1161/CIRCRESAHA.110.226860
179. Wasiak S, Gilham D, Tsujikawa LM, Halliday C, Calosing C, Jahagirdar R, et al. Downregulation of the complement cascade in vitro, in mice and in patients with cardiovascular disease by the bet protein inhibitor apabetalone (RVX-208). J Cardiovasc Transl Res. (2017) 10:337–47. doi: 10.1007/s12265-017-9755-z
180. Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci USA. (2013) 110:19754–9. doi: 10.1073/pnas.1310658110
181. Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell. (2013) 154:569–82. doi: 10.1016/j.cell.2013.07.013
182. Song S, Liu L, Yu Y, Zhang R, Li Y, Cao W, et al. Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-β-induced endothelial-mesenchymal transition and cardiac fibrosis. J Mol Cell Cardiol. (2019) 127:83–96. doi: 10.1016/j.yjmcc.2018.12.002
183. Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med. (2017) 9:eaah5084. doi: 10.1126/scitranslmed.aah5084
184. Bernardo BC, Ooi JY, Lin RC, McMullen JR. miRNA therapeutics: a new class of drugs with potential therapeutic applications in the heart. Future Med Chem. (2015) 7:1771–92. doi: 10.4155/fmc.15.107
185. Schmidt MF. miRNA targeting drugs: the next blockbusters? Methods Mol Biol. (2017) 1517:3–22. doi: 10.1007/978-1-4939-6563-2_1
186. Han X, Wang M, Zhao YL, Yang Y, Yang YG. RNA methylations in human cancers. Semin Cancer Biol. (2021) 75:97–115. doi: 10.1016/j.semcancer.2020.11.007
187. Cully M. Chemical inhibitors make their RNA epigenetic mark. Nat Rev Drug Discov. (2019) 18:892–4. doi: 10.1038/d41573-019-00179-5
188. Selberg S, Blokhina D, Aatonen M, Koivisto P, Siltanen A, Mervaala E, et al. Discovery of small molecules that activate RNA methylation through cooperative binding to the METTL3-14-WTAP complex active site. Cell Rep. (2019) 26:3762–71.e5. doi: 10.1016/j.celrep.2019.02.100
189. Dalhat MH, Mohammed MRS, Ahmad A, Khan MI, Choudhry H. Remodelin, a N-acetyltransferase 10 (NAT10) inhibitor, alters mitochondrial lipid metabolism in cancer cells. J Cell Biochem. (2021) 122:1936–45. doi: 10.1002/jcb.30155
190. Wu Y, Cao Y, Liu H, Yao M, Ma N, Zhang B. Remodelin, an inhibitor of NAT10, could suppress hypoxia-induced or constitutional expression of HIFs in cells. Mol Cell Biochem. (2020) 472:19–31. doi: 10.1007/s11010-020-03776-w
191. Xia Z, Tang M, Ma J, Zhang H, Gimple RC, Prager BC, et al. Epitranscriptomic editing of the RNA N6-methyladenosine modification by dCasRx conjugated methyltransferase and demethylase. Nucleic Acids Res. (2021) 49:7361–74. doi: 10.1093/nar/gkab517
192. Nishitani S, Fukuhara A, Shin J, Okuno Y, Otsuki M, Shimomura I. Metabolomic and microarray analyses of adipose tissue of dapagliflozin-treated mice, and effects of 3-hydroxybutyrate on induction of adiponectin in adipocytes. Sci Rep. (2018) 8:8805. doi: 10.1038/s41598-018-27181-y
193. Zhao Q, Song W, Huang J, Wang D, Xu C. Metformin decreased myocardial fibrosis and apoptosis in hyperhomocysteinemia -induced cardiac hypertrophy. Curr Res Transl Med. (2021) 69:103270. doi: 10.1016/j.retram.2020.103270
194. Bridgeman SC, Ellison GC, Melton PE, Newsholme P, Mamotte CDS. Epigenetic effects of metformin: from molecular mechanisms to clinical implications. Diabetes Obes Metab. (2018) 20:1553–62. doi: 10.1111/dom.13262
195. Storino Farina M, Rojano Rada J, Molina Garrido A, Martínez X, Pulgar A, Paniagua R, et al. Statins and atherosclerosis: the role of epigenetics. Medwave. (2015) 15:e6324. doi: 10.5867/medwave.2015.10.6324
5mC, 5th carbon atom; CGI, CpG islands; MeDIP, Methylated DNA immunoprecipitation; DCM, Dilated cardiomyopathy; LY75, Lymphocyte antigen 75; ERBB3, Tyrosine kinase-type cell surface receptor HER3; HOXB13, Homeobox B13; ADORA2, Aadenosine receptor A2A; HATs, Histone acetyltransferases; HDACs, Histone deacetylation enzymes; MEF2, Myocyte enhancer factor 2; CaMKII, Calcium/calmodulin-dependent protein kinase II; PKA, Protein kinase A; CBPCREB, Binding protein; MI, Myocardial infarction; H3K4me3, Histone 3 lysine 4 triamcinolone; β-MHC, Myosin heavy chain; HMT, Histone methyl transferase; TAC, Aortic narrowing surgery; HDM, Histone trimethyldemethylase; FHL1, Four and a Half LIM Domains Protein 1; PTIP, PAX transcriptional activation domain interaction protein; PARP-1, Poly (ADP-ribose) polymerase-1; NAD +, Nicotinamide adenine dinucleotide; BET, Bromine domain/BCD domain; TSS, Transcription start site; ANF, Atrial natriuretic factors; ADCR, ATP-dependent chromatin remodeling complex; ATPase, Adenosine triphosphate; MHC, Myosin heavy chain; DMNT3, DNA methyltransferase; ncRNAs, Non-coding RNAs; tRNAs, Transfer RNAs; rRNAs, Ribosomal RNAs; miRNAs, MicroRNAs; siRNAs, Small interfering RNA; snRNAs, Small nuclear RNAs; snoRNAs, Small nucleolar RNAs; scRNAs, Small cytoplasmic RNAs; exRNAs, Extracellular RNAs; piRNAs, Piwi-interacting RNAs; lncRNAs, Long ncRNAs; CVD, Cardiovascular disease; mRNAs, Messenger RNAs; TAC, Transverse aortic constriction; RasGAP, Ras GTPase-activating protein; Cdk9, Cyclin-dependent kinase 9; Rheb, Ras homolog enriched in brain; Chaer, Cardiac-hypertrophy-associated epigenetic regulator; PRC2, Polycomb repressor complex 2; H3K27me3, Histone H3 lysine 27 methylation; hiPSC-CMs, Pluripotent stem cell-derived cardiomyocytes; Mhrt, Myosin heavy-chain-associated RNA transcripts; Chast, Cardiac hypertrophy-associated transcript; m7G, N7-methylguanosine; m6A, N6-methyladenosine; m5C, 5-methylcytosine; m1A, N1-methyladenosine; ac4C, N4-acetylcytidine; Nm, 2'-O-methylation; FTO, Fat mass and obesity-associated protein; WTAP, Wilms' tumor 1-associating protein; RBM15/15B, RNA-binding motif protein 15/15B; METTL3, Methyltransferase-like 3; METTL14, Methyltransferase-like 14; ALKBH5, ALKB homo-log 5; CFs, Cardiac fibroblasts; RNA-seq, RNA sequencing; m6A-seq, m6A sequencing; FOXO1, Forkhead box O1; HCM, Hypertrophic cardiomyopathy; ASD, Atrial septal defect; VSD, Ventricular septal defect; CAD, Coronary artery disease; m1A, N1-methyladenosine; m7G, N7-methylguanosine; mESCs, Mouse embryonic stem cells; hiPSCs, Human induced pluripotent stem cells; m5C, 5-methylcytosine; ac4C, N4-acetylcytidine; NAT10, N-acetyltransferase 10; Nm, 2'-O-methylation.
Keywords: epigenetic regulation, pathological myocardial remodeling, DNA methylation, histone posttranslational modification, adenosine disodium triphosphate (ATP)-dependent chromatin remodeling, RNA modification
Citation: Zhao K, Mao Y, Li Y, Yang C, Wang K and Zhang J (2022) The roles and mechanisms of epigenetic regulation in pathological myocardial remodeling. Front. Cardiovasc. Med. 9:952949. doi: 10.3389/fcvm.2022.952949
Received: 25 May 2022; Accepted: 10 August 2022;
Published: 26 August 2022.
Edited by:
Suowen Xu, University of Science and Technology of China, ChinaReviewed by:
Giuseppe Paolisso, University of Campania Luigi Vanvitelli, ItalyCopyright © 2022 Zhao, Mao, Li, Yang, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Zhang, empfbmptdUAxNjMuY29t; Kai Wang, bmptdXdrQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.