 Aleksandra Boikova1†
Aleksandra Boikova1† Megan J. Bywater2,3†
Megan J. Bywater2,3† Gregory A. Quaife-Ryan3Jasmin Straube3Lucy Thompson1Camilla Ascanelli1Trevor D. Littlewood2Gerard I. Evan2
Gregory A. Quaife-Ryan3Jasmin Straube3Lucy Thompson1Camilla Ascanelli1Trevor D. Littlewood2Gerard I. Evan2 James E. Hudson3
James E. Hudson3 Catherine H. Wilson1*
Catherine H. Wilson1*- 1Department of Pharmacology, University of Cambridge, Cambridge, United Kingdom
- 2Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom
- 3QIMR Berghofer Medical Research Institute, Herston, QLD, Australia
Aim: Adult mammalian cardiomyocytes are incapable of significant proliferation, limiting regeneration after myocardial injury. Overexpression of the transcription factor Myc has been shown to drive proliferation in the adult mouse heart, but only when combined with Cyclin T1. As constitutive HRas activity has been shown to stabilise Cyclin T1 in vivo, we aimed to establish whether Myc and HRas could also act cooperatively to induce proliferation in adult mammalian cardiomyocytes in vivo.
Methods and results: Using a genetically modified mouse model, we confirmed that constitutive HRas activity (HRasG12V) increased Cyclin T1 expression. HRasG12V and constitutive Myc expression together co-operate to drive cell-cycle progression of adult mammalian cardiomyocytes. However, stimulation of endogenous cardiac proliferation by the ectopic expression of HRasG12V and Myc also induced cardiomyocyte death, while Myc and Cyclin T1 expression did not.
Conclusion: Co-expression of Cyclin T1 and Myc may be a therapeutically tractable approach for cardiomyocyte neo-genesis post injury, while cell death induced by HRasG12V and Myc expression likely limits this option as a regenerative therapeutic target.
Introduction
The adult heart is one of the least regenerative organs of the human body. Shortly after birth of a mammal, cardiomyocytes exit the cell cycle and are subsequently characterised by a reduced rate of turnover (<1% per year in humans) (1). Consequently, if the adult mammalian heart is damaged (e.g., myocardial infarction), the default response is to replace the lost cardiomyocytes with non-contractile fibrotic scar tissue and cardiomyocyte hypertrophy which frequently lead to heart failure. A number of strategies have been proposed to promote myocardial regeneration post damage, including the direct injection of stem cells or stem cell-derived cardiomyocytes, direct reprogramming of non-myocytes into cardiomyocytes and endogenous cardiomyocyte proliferation (2). Endogenous cardiomyocyte proliferation requires driving the resident quiescent cardiomyocytes into a productive mitogenic cell cycle. Genetic lineage tracing studies in the regenerative neonatal mouse and zebrafish hearts indicate that the majority of newly generated cardiomyocytes are derived from endogenous cardiomyocyte proliferation rather than differentiation from a mesenchymal progenitor (3–5). These studies suggest that forced cardiomyocyte proliferation is a viable strategy for myocardial regeneration and several potential factors capable of reactivating endogenous proliferation of cardiomyocytes in adult hearts have been identified, for example, inhibition of Hippo protein kinase signalling, enforced expression of cell cycle regulators, inactivation of thyroid hormone signalling, and hypoxia, all induce regeneration (6–10). We have recently shown that combined ectopic expression of Myc and Cyclin T1 can lead to extensive cardiomyocyte proliferation in the mouse heart (11), although exploitation of the therapeutic potential requires further exploration.
Myc is a pleiotropic transcription factor that, amongst other activities, regulates cell growth and proliferation in mammalian cells (12). Consequently, Myc expression in normal cells is tightly regulated. Following damage in a regenerative tissue, the release of mitogens stimulates a transient increase in the short-lived Myc protein followed by a transient proliferative response (13). In contrast, Myc transcriptional programmes decrease during postnatal cardiac maturation and fail to reactivate post-infarction (14). Moreover, even when Myc expression is driven ectopically in the adult mammalian heart, it is unable to activate transcription of many target genes and cardiomyocytes remain almost entirely resistant to proliferation (11, 15). We previously established that Myc-driven transcription, and consequently cell proliferation, are critically dependent on the level of Cyclin T1 in cardiomyocytes (11).
Cyclin T1 strongly associates with CDK9 to form the positive transcription elongation factor b (P-TEFb) (16) that phosphorylates paused RNA PolII and the elongation factors DSIF and NELF, leading to productive transcriptional elongation (16, 17). Both components of P-TEFb, Cyclin T1 and CDK9 are tightly regulated by various transcriptional and post-transcriptional mechanisms (18, 19). P-TEFb is also dynamically controlled by an association with an inactivation complex (7SK snRNA, Larp7, MEPCE, and HEXIM) (20, 21). Unlike other cyclin-dependent kinases, CDK9 protein stability is not cell cycle dependent (22), but is primarily determined by binding to Cyclin T1 (23, 24). The level of the Cyclin T1 protein is thus the key factor in controlling the amount of the P-TEFb complex in a cell (23, 24). During postnatal cardiac maturation in mice the level of P-TEFb and phosphorylated RNA PolII steadily decline (11). Transgenic overexpression of Cyclin T1 in the heart leads to increased levels of both CDK9 and phosphorylated RNA PolII and, when expressed throughout development, leads to cardiac hypertrophy (11, 25, 26).
The RAS proteins (K-RAS, N-RAS, and H-RAS in humans) are small GTPases that function as molecular switches, cycling between their “off” GDP-bound and “on” GTP-bound states in response to mitogenic signalling (27). Depending on the cellular context, Ras can activate several downstream pathways that regulate protein synthesis, cell growth, survival, and cell motility (28), in the heart, Ras activation leads to reversible cardiac hypertrophy (29, 30). Hypertrophy, be it transgenically induced by constitutively active RAS or secondary to pressure overload or other hypertrophic stimuli, is accompanied by an increase in total RNA content. This is a result of increased P-TEFb activity and phosphorylation of RNA Pol II that increases total RNA and protein synthesis (25, 31). Using a genetically modified mouse model that combines the elevated expression of HRasG12V and an ectopically switchable Myc allele (MycERT2), we show that expression of constitutively active HRasG12V in the heart leads to increased P-TEFb levels that, when combined with constitutive Myc activity, drives signs of cardiomyocytes proliferation in vivo.
Materials and methods
Mice
All animals were kept under SPF conditions. Mice were maintained on regular diet in a pathogen-free facility on a 12 h light/dark cycle with continuous access to food and water. All mice were euthanised under the schedule 1 method of cervical dislocation. Mouse strain Tg(Myh6-tTA)6Smbf/J was obtained from the Jackson Laboratory, and mouse strain Tg(tetO-HRAS)65Lc/Nci was obtained from the NCI Mouse Repository. Mouse strain R26CAG–c–MycER was produced in house. 1 mg of (Z)-4-hydroxytamoxifen (4-OHT; Sigma, H7904) was i.p injected into adult mice in 10% ethanol and vegetable oil (5 mg/ml), and tissues were collected 4 h post injection. 1 mg of tamoxifen (Sigma, T5648) was i.p injected into adult mice in 10% ethanol and vegetable oil (10 mg/ml) twice over a 24-h period and tissues collected at 24 h post initial i.p. injection. Animals requiring doxycycline treatment were supplied with drinking water containing doxycycline hyclate 100 mg/L (Sigma D9891) in water containing 3% sucrose to increase palatability, this was replenished two times per week in light-protected bottles. Ear biopsies were collected from 2 to 5 weeks old mice and genotyped by PCR with the following oligonucleotide primers: for Rosa26CAG; Universal forward: 5′-CTCTGCTGCCTCCTGGCTTCT-3′ Wild-type reverse: 5′-CGAGGCGGATCACAAGCAATA-3′ and CAG reverse: 5′-TCAATGGGCGGGGGTCGTT-3′. Primers for the TetO-HRas allele were; H003: 5′-TGAAAGTCGAGCTCGGTA-3′ and H004: 5′-CCCGGTGTCTTCTATGGA-3′. Primers for the Myh6-tTA allele were; oIMR8746: 5′-CGCTGTGGG GCATTTTACTTTAG-3′ and oIMR8747: 5′-CATGTCCAGAT CGAAATCGTC-3′.
Immunoblotting
Snap-frozen animal tissues were ground on liquid nitrogen and proteins extracted in buffer containing 1% SDS, 50 mM Tris pH 6.8, and 10% glycerol on ice for 10 min. Lysates were boiled for 10 min, followed by sonication for 15 min at room temperature. Total protein (50 μg) was electrophoresed on an SDS–PAGE gel and transferred onto immobilon-P (Millipore) membranes. These were then blocked in 5% non-fat milk and incubated with primary antibodies overnight at 4°C. Secondary antibodies were applied for 1 h followed by chemiluminescent visualisation (Thermo Scientific, 32106 or Millipore, WBKLS0500). Immunoblots were either developed on Fuji RX X-ray film and scanned on an Epson Perfection V500 Photo flatbed scanner or visualisation was performed on LiCOR Odyssey Fc. Protein quantifications were performed on the LiCOR Odyssey Fc.
Primary antibodies: GAPDH (D16H11) XP® (Cell Signaling Technology, 5174, used at 1:5,000), phospho-Rpb1 CTD (Ser2; E1Z3G; Cell Signaling Technology, 13499, used at 1:2,500), CDK9 (C12F7; Cell Signaling Technology, 2316, used at 1:1,000), Cyclin T1 (D1B6G; Cell Signaling Technology, 81464, used at 1:1,000), p-ERK is phospho-p44/42 MAPK (Thr202/Tyr204, Cell Signaling Technology, 9101, used at 1:500), anti-rabbit IgG HRP (Sigma, A0545, used at 1:10,000). Sample loading was normalised for equal protein content. Expression of GAPDH is included as a confirmation of efficiency of protein isolation and comparable loading between individual tissue samples.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed paraffin-embedded 4 μm sections. Sections were de-paraffinized and rehydrated, followed by antigen retrieval by boiling in 10 mM citrate buffer (pH 6.0) for 10 min. Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide for 30 min. Sections were then treated with rabbit VECTASTAIN Elite ABC horseradish peroxidase kit (Vector Laboratories, PK-6101) following the manufacturer’s protocols. Sections were blocked in normal goat serum for 20 min, followed by 1 h incubation in the primary antibody at room temperature. Sections were then washed three times in PBST and incubated for 1h in secondary antibody, sections were then washed again in PBST and then incubated in ABC complex for 30 min. Sections were developed in DAB (3,3’-diaminobenzidine) for 5 min, counterstained in haematoxylin, dehydrated, and mounted in DPX. Staining was imaged on a Zeiss Axio Imager using the Zeiss ZEN software using the AutoLive setting and interactive white balance.
Primary antibodies: p-ERK is phospho-p44/42 MAPK (Thr202/Tyr204, Cell Signalling Technology, 9101, used at 1:500), and anti-Cyclin T1 (AbCam, ab238940, use at 1:500).
Immunofluorescence
Sections were pre-processed followed by antigen retrieval in the same way as for immunohistochemistry. Sections were blocked in 2.5% goat serum, and 1% BSA in PBST for 20 mins at room temperature. Primary antibody, made up of blocking buffer, was added for 1 h at room temperature, followed by three 5 min PBST washes, and secondary antibodies were added for 1 h at room temperature. Nuclei were visualised using Hoechst (Sigma, 861405) and sections mounted in ProLong™ Gold Antifade Mountant (Thermo Fisher, P36930). Antibodies: anti-phospho-Histone H3 (Ser10; Merck Millipore, 06-570, use at 1:500), cardiac troponin T (13-11; Thermo Fisher, MA5-12969, use at 1:100) and (CT3, Santa Cruz Biotechnology, sc-20025, use at 1:50), anti-Aurora B antibody (Abcam, ab2254, use at 1:200), anti-Mklp1 (Abcam, ab174304, use at 1:400), anti-CD206 (R&D Systems, AF2535, use at 1:100), anti-PCM1 (Sigma-Aldrich, HPA023370, use at 1:100), anti-Ki67 (SolA15; Thermo Fisher, use at 1:100) and (Atlas Antibodies, HPA023374, use at 1:100), Alexa Fluor 555 goat anti-rabbit IgG (H+L; Life Technologies, A21428), Alexa Fluor 555 goat anti-mouse IgG (H+L; Life Technologies, A21422), Alexa Fluor 555 goat anti-rat IgG (H+L; Life Technologies, A21434), Alexa Fluor 488 goat anti-rabbit IgG (H+L; Life Technologies, A11008), Alexa Fluor 488 goat anti-rat IgG (H+L; Life Technologies, A11006), Alexa Fluor 350 goat anti-mouse (H+L; Life Technologies A11045), wheat germ agglutinin, Alexa Fluor™ 488 conjugate (Thermo Fisher, W11261). TUNEL staining we performed following the manufactures instructions using the ApopTag® Fluorescein In Situ Apoptosis Detection Kit (S7110 Sigma-Aldrich). Staining was imaged on a Zeiss Axio Imager using the Zeiss ZEN software using the AutoLive setting or Leica Stellaris 5 confocal microscope. Quantification was performed by counting the number of positive cardiomyocytes for at least five images per organ/mouse, the mean of five raw counts was calculated and is represented by each data point per graph. Cardiomyocyte cell areas were quantified by encircling individual cardiomyocytes stained with cardiac troponin and wheat germ agglutinin.
Quantitative RT-PCR
Total RNA was isolated using TRIzol Reagent (Thermo Fisher, 15596-018) following manufacturer’s instructions. cDNA was synthesised from 1 μg of RNA using the High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Thermo Fisher, 4374966) following manufacturer’s instructions. qRT-PCR reactions were performed on an Applied Biosystems QuantStudio 5 Real-Time PCR System using Fast SYBR Green Master Mix (Thermo Fisher, 4385612), following manufacturer’s instructions. Primers: Ccnd1; forward 5′- GCGTACCCTGACACCAATCTC-3′, reverse 5′-CTCCTCTTCGCACTTCTGCTC-3. Cdk4; forward 5′-ATGGCTGCCACTCGATATGAA-3′, reverse 5′-TC CTCCATTAGGAACTCTCACAC-3′. Cdk1; forward 5′- AGAA GGTACTTACGGTGTGGT-3′, reverse 5′-GAGAGATTTCCC GAATTGCAGT-3′. Ccnb1; forward 5′-AAGGTGCCTGT GTGTGAACC-3′, reverse 5′-GTCAGCCCCATCATCTGCG-3′. Cad; forward 5′- CTGCCCGGATTGATTGATGTC-3′, reverse 5′-GGTATTAGGCATAGCACAAACCA-3′. Pinx1; forward 5′-AGCAAGGAGCCACAGAACATA-3′, reverse 5′-GGTGAGCAATCCAGTTGTCTT-3′. Bzw2; forward 5′-AGC GACTGTCTCAGGAATGC-3′, reverse 5′-CTGTTTCCGGAA GGTCGTT-3′. Polr3d; forward 5′-AAAAGCGTGAACGGGAC AGG-3′, reverse 5′-AATGGGACTGGATCACTTCCG-3′. St6galnac4; forward 5′-TGGTCTACGGGGATGGTCA-3′, reverse 5′-CTGCTCATGCAAACGGTACAT-3′. Actin; forward 5′-GACGATATCGCTGCGCTGGT-3′, reverse 5′-CCACGATG GAGGGGAATA-3′. Gapdh; forward 5′-AGGTCGGTGTGA ACGGATTTG-3′, reverse 5′-TGTAGACCATGTAGTTGAGG TCA-3′. Fosl1; forward 5′-ATGTACCGAGACTACGGGGAA-3′, reverse 5′-CTGCTGCTGTCGATGCTTG-3′. Egr3; forward 5′-CCGGTGACCATGAGCAGTTT-3′, reverse 5′-TAATGGG CTACCGAGTCGCT-3′.
Bulk RNA sequencing
Total RNA was purified using PureLink RNA Mini Kit (Thermo Scientific). On-column DNA digestion was performed using PureLink DNase (Thermo Scientific). 1 μg of purified RNA was treated with Ribozero rRNA removal kit (Illumina). RNA quality and removal of rRNA were checked with the Agilent 2100 Bioanalyser (Agilent Technologies). Libraries for RNA-seq were then prepared with the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina (NEB) following the manufacturer’s instructions starting from the RNA fragmentation step.
Bulk RNA sequencing data analysis
Sequence reads were adapter trimmed using Cutadapt (32) (version 1.11) and aligned using BWA aln for short reads (version 2.5.2a) to the GRCm38 (mm10) assembly with the gene, transcript, and exon features of the Ensembl (release 70) gene model. Expression was estimated using RSEM (33) (version 1.2.30). Transcripts with zero read counts across all samples were removed prior to analysis. Normalisation of RSEM expected read counts was performed by dividing by million reads mapped to generate counts per million (CPM), followed by the trimmed mean of M-values (TMM) method from the edgeR package (34). Differential expression analysis was performed using edgeR.
RNA sequencing comparison to myocardial infarction reversion network
RNA-seq files from E-MTAB-7595, E-MTAB-7636, and GSE95755 were combined into a single count matrix. Before remapping, poor quality sequences (<30 phred score) and adapter sequences were trimmed with Trimmomatic (35). Reads were mapped with STAR (36) to the reference mouse genome sequence GCRm38.p4/mm10 using default settings. The combined count matrix was generated using HTSeq-count (37) on union mode. Differential expression analysis was performed with EdgeR(v3.20.8) (34) using the glmLRT function. Differential expression comparisons were considered significant if FDR p < 0.05. Gene ontological analysis was performed using DAVID and Heatmaps were assembled using GENE-E (Broad Institute).
Statistical analysis
Bioinformatic and statistical analyses were performed using R with Bioconductor packages and comEpiTools packages (38, 39). Gene lists from RNA and ChIP sequencing were analysed in Enrichr or GSEA/MSigDB1 (40). Statistical analyses for IHC and q-RT-PCR were performed using GraphPad Prism v9.0d (GraphPad Software, Inc., San Diego, CA, USA) as indicated with p ≤ 0.05 considered to be statistically significant. Venn diagrams were drawn in the Lucidchart.2 Heatmaps were drawn in R and Morpheus.3
Results
Cardiac HRasG12V expression leads to hypertrophy and amplified P-TEFb expression
We employed the cardiac-specific, doxycycline-controlled transgenic line TetO-HRas [Tg(tetO-HRAS)65Lc/Nci]; Myh6tTA [Tg(Myh6-tTA)6Smbf/Jm] (30) to overexpress HRasG12V specifically in cardiomyocytes. Mice were generated and removed from doxycycline treatment at 4 weeks of age and the subsequent expression of constitutively active HRasG12V led to an increase in hypertrophic cardiomyocytes (∼10%, Supplementary Figure 1A) interspersed with CD206 positive macrophages and cardiomyocytes of normal volume (Figure 1A), as previously described (30). Hypertrophic cardiomyocytes displayed elevated pERK immunohistochemical staining (Figure 1A) and qRT-PCR analysis confirmed increased expression of the ERK-AP1 target genes Egr3 and Fos1L (Figure 1B), confirming enhanced HRas signalling. Despite the heterogeneity of the model, whole hearts expressing HRasG12V displayed a moderate increase in Cyclin T1 and Phosphorylated RNA Pol II (Figures 1A,C and Supplementary Figure 1B).
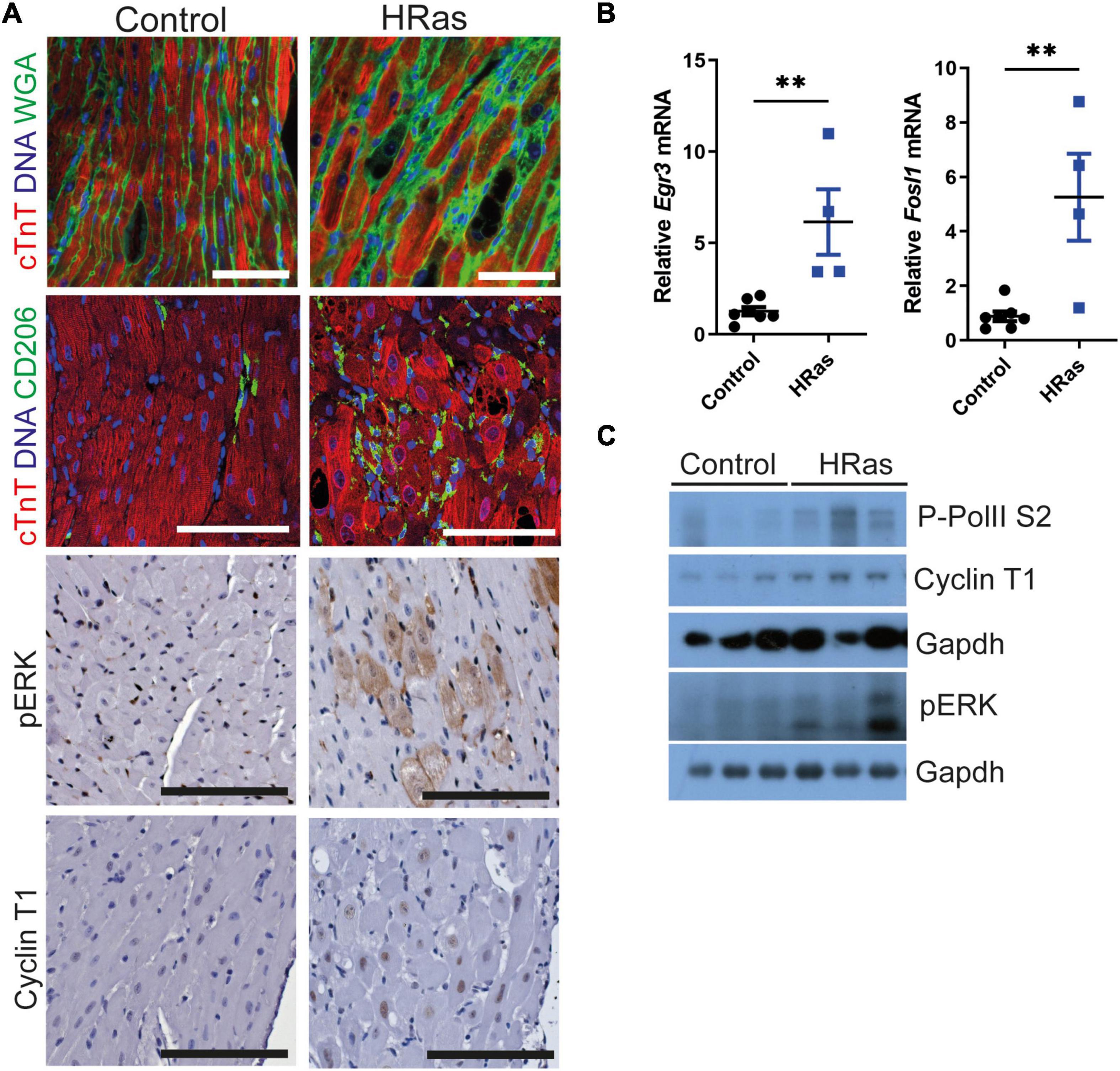
Figure 1. Ectopic cardiomyocyte HRasG12V results in increased Cyclin T1 expression. (A) Top- Immunofluorescent staining of cardiac troponin (red) and wheat germ agglutinin or CD206 (green). middle- immunohistochemical analysis of pERK, bottom- immunohistochemical analysis of Cyclin T1 in hearts from wild type (Control) and TetO-HRas; Myh6-tTA; R26+/+(HRas) mouse hearts 4 weeks post withdrawal of doxycycline. Bars are 100 μm. Representative images based on analysis of 5 images per mouse. (B) Quantitative RT-PCR analysis of control (R26+/+; TetO-HRas, R26+/+; Myh6-tTA or R26+/+, n = 7) and TetO-HRas; Myh6-tTA; R26+/+(R26+/+;HRas, n = 4) mouse hearts 4 weeks post withdrawal of doxycycline. Expression is normalised to Actin and Gapdh and relative to the respective control. Replicate samples are derived from independent mice. Error bars show s.e.m. Two Way ANOVA with multiple comparisons test. **p < 0.01. (C) Immunoblot analysis of the C-terminal domain of phosphorylated RNA Polymerase II [p-Rpb1(S2)], Cyclin T1 and pERK protein expression in hearts isolated from control (R26+/+; TetO-HRas, R26+/+; Myh6-tTA or R26+/+) and TetO-HRas; Myh6-tTA; R26+/+(HRas) mice 4 weeks post withdrawal of doxycycline. Expression of Gapdh is included as a loading control, for completeness, multiple Gapdh blots represent different gels (top – Cyclin T1 and P-RNA PolII S2, bottom – pERK). Replicate samples are derived from independent mice.
HRasG12V enables Myc-driven transcription in cardiomyocytes
To test the hypothesis that persistent HRasG12V expression renders heart tissue Myc responsive, TetO-HRas; Myh6tTA mice were crossed to the R26CMER/+ (R26CAG–c–MycERT2) allele to generate TetO-HRas; Myh6-tTA; R26CMER/+ mice. In these mice, the MycERT2 fusion protein is constitutively expressed from a CAG-enhanced Rosa26 promoter. MycERT2 is inert in the absence of a ligand but rapidly activated following administration of the ERT2 ligand 4OHT (4-hydroxytamoxifen), a primary metabolite of tamoxifen. In these mice, HRasG12V and MycERT2 proteins can be independently activated by the withdrawal of doxycycline or administration of 4OHT (or tamoxifen), respectively (Figure 2A). Doxycycline was withdrawn from TetO-HRas; Myh6-tTA; R26CMER/+ animals at weaning to induce expression of HRasG12V and, once mice reached 8 weeks of age, MycERT2 was activated by injection of 4OHT for 4 h. We confirmed the elevated expression of Cyclin T1, CDK9, and phosphorylated RNA Pol II in the heart tissue of these animals compared to controls (Figure 2B and Supplementary Figure 1C).
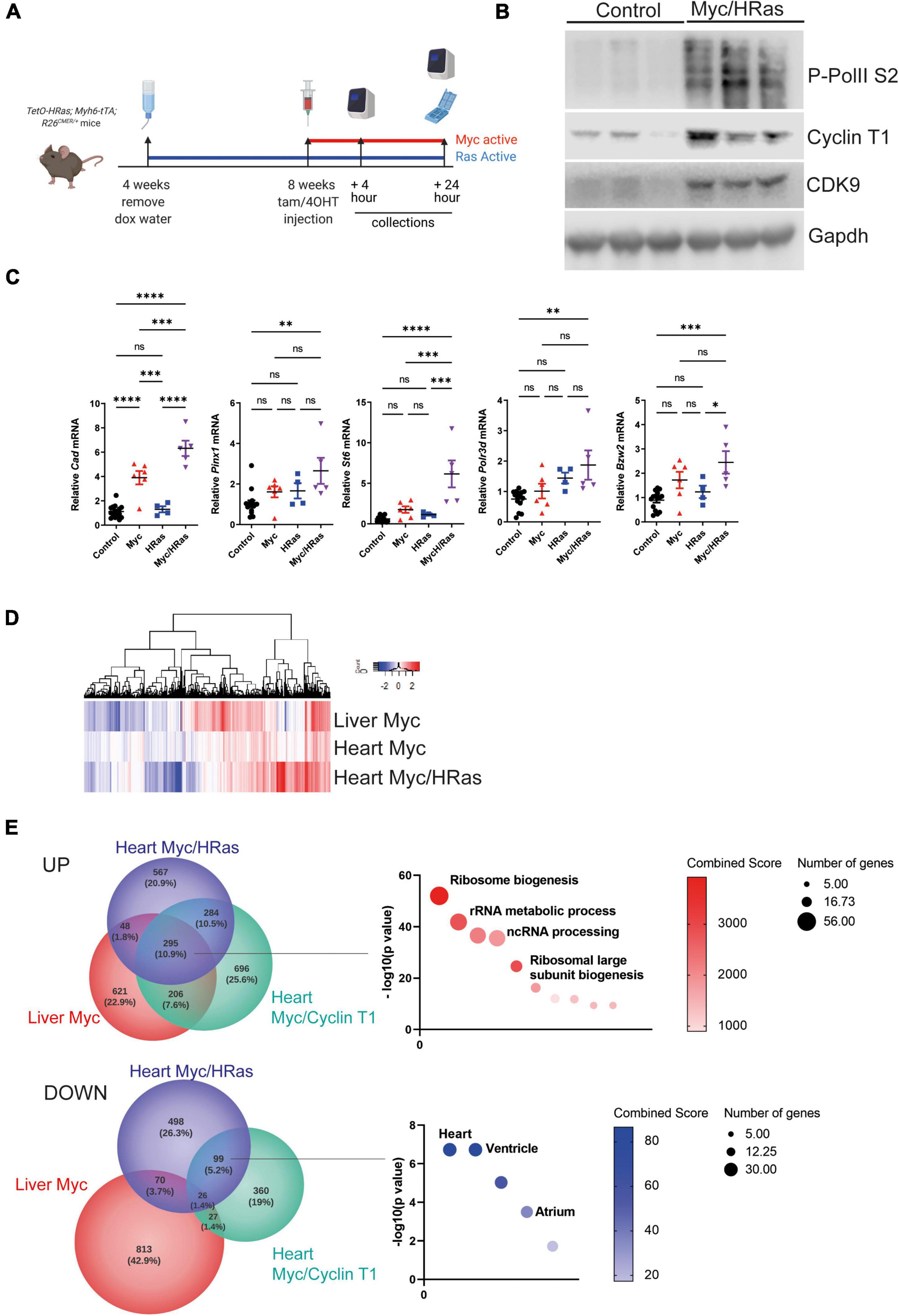
Figure 2. Cardiomyocyte HRasG12V expression enables Myc-driven transcription. (A) Scheme of experimental work. (B) Immunoblot analysis of the C-terminal domain of phosphorylated RNA Polymerase II [p-Rpb1(S2)], Cyclin T1 and CDK9 protein expression in hearts isolated from control (R26+/+; TetO-HRas, R26+/+; Myh6-tTA or R26+/+) and TetO-HRas; Myh6-tTA; R26 CMER/+(HRas/Myc) mice 4 weeks post withdrawal of doxycycline and at 24 h post administration of 4-OHT. Expression of Gapdh is included as a loading control. Replicate samples are derived from independent mice. (C) Quantitative RT-PCR analysis of Cad, Bzw2, Pinx1, Polr3d, St6, and Cdc25a in hearts from control (R26+/+; TetO-HRas, R26+/+; Myh6-tTA or R26+/+, n ≥ 14), TetO-HRas; Myh6-tTA; R26+/+(HRas, n = 4), R26CMER/+ (Myc, n = 6) and TetO-HRas; Myh6-tTA; R26 CMER/+ (MycHRas, n = 5) mice 4 weeks post withdrawal of doxycycline and 4 h post-administration of 4-OHT. Expression is normalised to Actin and Gapdh and relative to the respective control. Error bars show s.e.m. Two Way ANOVA with multiple comparisons test; control vs R26CMER/+: p*** = 0.001 (Cad), control vs TetO-HRas; Myh6-tTA; R26 CMER/+: p** = 0.01 (Bzw2, Pinx1, Polr3d, Cdc25a) p*** = 0.001 (St6). Replicate samples are derived from independent mice. (D) Heat map showing the union of DEGs called in adult liver and adult heart. Shown are mRNA expression fold changes (Log2) upon MycERT2 activation relative to wild-type, as determined by RNA sequencing of R26CMER/+ (n ≥ 3) mice in comparison to wild-type (R26+/+, n ≥ 3) and TetO-HRas; Myh6-tTA; R26 CMER/+ mice (Myc/HRas, n = 3) in comparison to TetO-HRas; Myh6-tTA; R26+/+ mice (n = 3) at 4 h post administration of 4-OHT. (E) Venn diagrams of overlap in the number of genes showing an increase (top) and decrease (bottom) in expression in response to supraphysiological Myc expression. The most significant GO Biological Process and ASCHS4 gene lists that overlap are shown. Comparisons included the liver of R26CMER/+ (Myc liver, n = 3) compared to wild type. The adult mouse heart was isolated 4 weeks post systemic infection with an adeno associated virus encoding Ccnt1 (AAV9-Ccnt1) compared to control and heart from TetO-HRas; Myh6-tTA; R26 CMER/+ (n = 3) 4 weeks after HRasG12D expression and 4 h post Myc activation compared to control, as determined by RNA sequencing (FDR < 0.05 and abs(log2FC) > 0.5).
We have previously determined that Myc-driven transcription is limited by the low level of endogenous P-TEFb within the adult heart. To establish if an HRasG12V-driven increase in P-TEFb levels could enhance Myc-driven transcription, we quantified the expression of a panel of Myc target genes post MycERT2 activation. Cad, a Myc target gene previously shown to be transcribed following MycERT2 activation in all tissues, including the heart, showed increased expression in the heart tissue of both Myc activated (R26CMER/) and HRasG12V/Myc activated (TetO-HRas; Myh6-tTA; R26CMER/+) tissues, confirming the functionality of the MycERT2 fusion in both conditions (Figure 2C). We then determined the expression of a panel of Myc target genes that we have previously observed as unchanged in the adult heart in response to MycERT2 activation alone. The co-expression of HRasG12V sensitised the heart tissue to Myc-dependent expression of these targets over controls (Figure 2C). We also compared changes in global gene expression in heart of HRasG12V (TetO-HRas; Myh6-tTA; R26+/+) mice and Myc/HRasG12V (TetO-HRas; Myh6-tTA; R26 CMER/+) mice 4 h after MycERT2 activation. We have previously observed marked induction of a Myc-driven transcriptional programme in the liver of MycERT2 expressing (R26 CMER/+) mice in the absence of co-expressed HRasG12V (11) (Figure 2D). In contrast to the weak transcriptional response elicited by MycERT2 activation alone, in the presence of HRasG12V, MycERT2 resulted in a marked transcriptional response (Figure 2D). MycERT2 and HRasG12V expressing hearts displayed 1198 up-regulated DEGs and 693 down regulated DEGs (Supplementary Table 1) in comparison to HRasG12V expression alone. Myc target gene identities overlapped with genes induced by MycERT2 in the adult liver, and in the adult heart in the presence of elevated Cyclin T1 expression (AAV9-driven cardiomyocyte-specific) (11) (Figure 2E). These genes are Myc targets, involved in direct Myc-regulated processes such as ribosomal biogenesis and RNA metabolic processes (Figure 2E, Supplementary Figure 1D, and Supplementary Table 2). In contrast, there was little overlap between downregulated genes observed following MycERT2 and HRasG12V activation in the heart and genes downregulated by MycERT2 alone in the liver, suggesting a level of tissue specificity for Myc-dependent transcriptional repression. Consistent with this, genes downregulated in response to HRasG12V and MycERT2 overlapped with those downregulated in the MycERT2 and Cyclin T1 expressing heart – these genes were characteristic of a homeostatic heart transcriptional programme, such as HCN4, MYOT (Figure 2E and Supplementary Table 3).
To determine if the transcriptional changes observed in response to combined activation of MycERT2 and HRasG12V in the adult heart were similar to that observed in a regenerating neonatal heart, we analysed previous data to compare the transcriptional profiles of cardiomyocytes isolated from surgically infarcted hearts at different stages of development (14). Our unbiased comparison revealed that the P1 “neonatal regeneration gene expression signature” correlated most closely with the expression changes within the TetO-HRas; Myh6-tTA; R26CMER/+ hearts (Figure 3A). Upregulated genes that overlapped with those also elevated in the “neonatal regeneration gene expression signature” enriched for GO terms including cell cycle control (Figure 3B). Interestingly, downregulated genes that overlapped with those also reduced in the “neonatal regeneration gene expression signature” contained genes involved in the negative regulation of mTOR signalling and we have previously observed a broad reprogramming of metabolism with a transition from neonatal to adult cardiac maturation (41). Furthermore, when regulated metabolic genes from our analysis are overlaid on the KEGG metabolic pathways TetO-HRas; Myh6-tTA; R26CMER/+ hearts show broad reversion and reprogramming to neonatal metabolic pathways (Supplementary Figure 2A). Consistent with this reversion, gene set enrichment analysis (GSEA) indicated MycERT2 and HRasG12V expressing hearts display enrichment of genes involved in glycolysis, characteristic of neonatal cardiomyocyte metabolism, and a reduction in genes involved with oxidative phosphorylation and fatty acid metabolism characteristic of adult cardiomyocyte metabolism (Figure 3C).
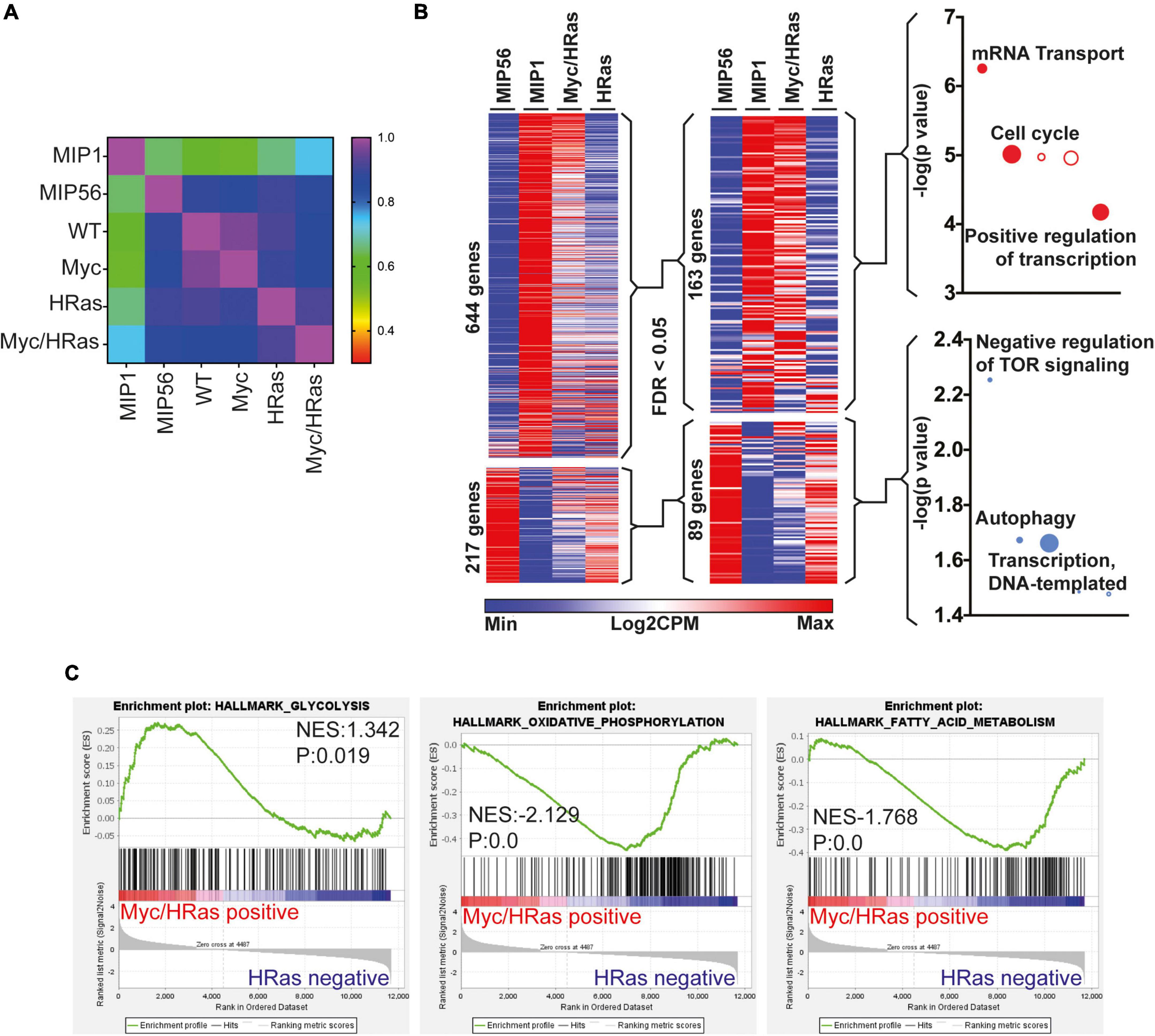
Figure 3. Cardiac MycERT2 and HRasG12V expression result in signalling characteristic of neonatal cardiomyocytes. (A) Pearson correlative analysis of regenerative network genes (described in 14) expressed in day 1 myocardial infarcted heart myocytes (MIP1), adult day 56 myocardial infarcted heart myocytes (MIP56), adult R26+/+ (WT) hearts, adult R26 CMER/+ (Myc) hearts, adult TetO-HRas; Myh6-tTA; R26+/+ (HRas) and adult TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas) RNA-seq datasets. (B) Heat map depicts gene expression of the regeneration network genes (described in 14) from RNA-seq of TetO-HRas; Myh6-tTA; R26+/+ (HRas, n = 3) and TetO-HRas; Myh6-tTA; R26 CMER/+ P60 adult hearts (Myc/HRas, n = 3). MIP1.Myo and MIP56.Myo denotes neonatal (P1) or adult (P56) cardiomyocytes isolated from myocardial infarction-operated hearts (n = 4). Regeneration network genes were filtered for genes differentially expressed (DEGs) between TetO-HRas; Myh6-tTA; R26+/+ (HRas) and TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas) hearts (FDR < 0.05). (C) Gene set enrichment analysis of glycolysis, oxidative phosphorylation, and fatty acid metabolism gene lists in comparison to differential gene expression observed between TetO-HRas; Myh6-tTA; R26 CMER/+ mice (Myc/HRas, n = 3) and TetO-HRas; Myh6-tTA; R26+/+ mice (n = 3) at 4 h post administration of 4-OHT.
Combined HRas and Myc expression drives proliferation of cardiomyocytes in vivo
To determine if this Myc-dependent “regenerative transcriptional response” results in productive proliferation, MycERT2/HRasG12V expressing mice (TetO-HRas; Myh6-tTA; R26CMER/+) or mice expressing either HRasG12V or MycERT2 alone were administered tamoxifen via intraperitoneal injection at 8 weeks of age to activate MycERT2 and hearts collected after 24 h. Transcriptional analysis indicated that activation of MycERT2 in the presence of HRasG12V induced robust expression of multiple cell cycle genes (Cdk4, Ccnd1, Cdk1, Ccnb1) that were not induced by HRasG12V or Myc activation alone (except Cdk4 which is a known Myc target, Figure 4A). Furthermore, markers of cytokinesis were upregulated in Myc and HRasG12V expressing hearts (Figure 4B) and GSEA indicated that these hearts displayed significant enrichment for genes involved in cell cycle-related pathways such as E2F targets and G2/M checkpoints (Supplementary Figure 2B). These transcriptional events also underpinned significant markers of cell-cycle progression indicated by Ki67 (general cell cycle) and p-H3 (mitotic) positive cardiomyocytes specifically marked by PCM1 and cardiac troponin T, respectively (Figures 4C,D). In addition, Aurora B kinase displayed staining representative of cell cycle progression (prophase, metaphase, late anaphase/telophase, and mid body localisation) combined with features of disassembled sarcomeres that become marginalised to the cell periphery (4, 42) (Figure 4E and Supplementary Figure 3). Furthermore, Mklp1 expression was observed in cardiomyocytes at the cleavage furrow (Figure 4F).
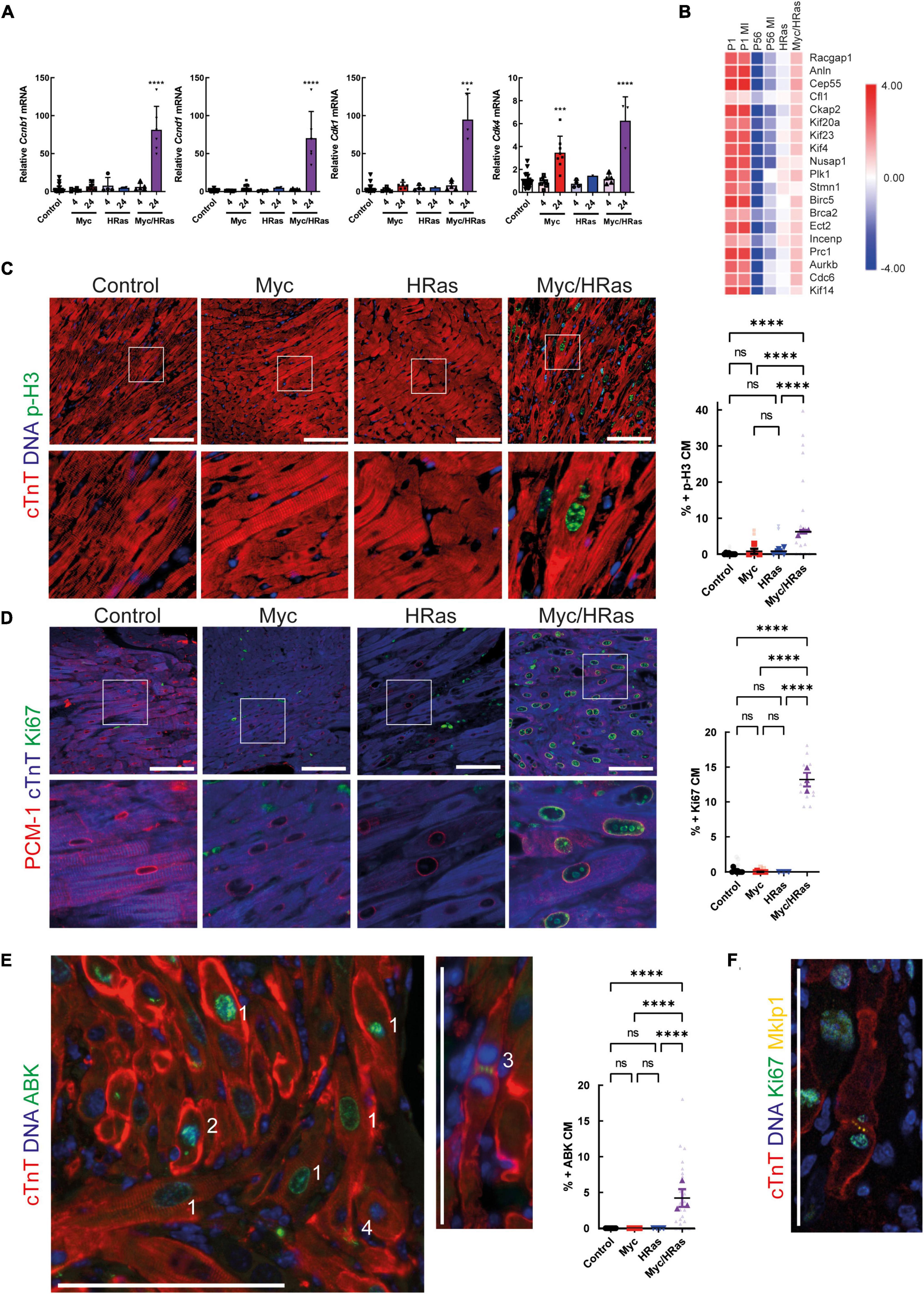
Figure 4. MycERT2 and HRasG12V expression drive cardiomyocyte proliferation. (A) Quantitative RT-PCR analysis of Cyclin D1, Cdk4, Cyclin B1, and Cdk1 in hearts of control (R26+/+; TetO-HRas, R26+/+; Myh6-tTA or R26+/+, n ≥ 24), TetO-HRas; Myh6-tTA; R26+/+(R26+/+;HRas, n ≥ 5), R26CMER/+ (n ≥ 14) and TetO-HRas; Myh6-tTA; R26 CMER/+ (R26 CMER/+;HRas, n = 8) mice 4 weeks post withdrawal of doxycycline and 4 or 24 h post administration of 4-OHT. Expression is normalised to Actin and Gapdh and relative to the respective control. Error bars show s.e.m. One Way ANOVA with multiple comparisons test; control vs 24 h R26CMER/+: p = 0.001*** (Cdk4), control vs 24 h TetO-HRas; Myh6-tTA; R26 CMER/+: p**** = 0.0001 (Cyclin D1, Cdk4, Cyclin B1, Cdk1). Replicate samples are derived from independent mice. (B) Heat map depicts gene expression (LogCPM) of cytokinesis genes in the regeneration network (described in 14) from RNA-seq of adult TetO-HRas; Myh6-tTA; R26+/+ (HRas, n = 3) and adult TetO-HRas; Myh6-tTA; R26 CMER/+ hearts (Myc/HRas, n = 3), neonatal (P1) or adult (P56) cardiomyocytes isolated from sham and myocardial infarction-operated (MI) hearts (n = 4). (C) Left- immunofluorescent staining of heart from control, TetO-HRas; Myh6-tTA; R26+/+ (HRas), R26CMER/+ (Myc) and TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas) mice 4 weeks post-withdrawal of doxycycline and 24 h post administration of tamoxifen. Cardiac troponin (red) and p-H3 positive mitotic nuclei (green). Representative images based on analysis of at least 3 independent mice. Right- quantification of percent p-H3 positive cardiomyocyte in hearts isolated from control (R26+/+; TetO-HRas, R26+/+; Myh6-tTA or R26+/+, black, n = 8), TetO-HRas; Myh6-tTA; R26+/+ (HRas, red, n = 3), R26CMER/+ (Myc, blue, n = 4) and TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas, purple, n = 3) mice 4 weeks post withdrawal of doxycycline and 24 h post administration of tamoxifen. Bold symbols = average per mouse heart, small faint symbols = each individual field quantified. Bars are 100 μm. Representative images based on analysis of 5 images per mouse; error bars show s.e.m. One Way ANOVA with multiple comparisons test: p < 0.0001****. (D) Left- Immunofluorescent staining of PCM-1 (red) and Ki67 (green) in the heart from control, TetO-HRas; Myh6-tTA; R26+/+ (HRas), R26CMER/+ (Myc) and TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas) mice 4 weeks post withdrawal of doxycycline and 24 h post administration of tamoxifen. Right Myc/HRas image shows relocation of PCM1 and cardiac troponin signal in mitotic cardiomyocytes. Right- quantification of percent Ki67 positive cardiomyocytes in hearts isolated from control (R26+/+; TetO-HRas, R26+/+; Myh6-tTA or R26+/+, black, n = 10), TetO-HRas; Myh6-tTA; R26+/+ (HRas, red, n = 3), R26CMER/+ (Myc, blue, n = 7) and TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas, purple, n = 3) mice 4 weeks post withdrawal of doxycycline and 24 h post administration of tamoxifen. Bold symbols = average per mouse heart, small faint symbols = each individual field quantified. Bars are 100 μm. Representative images based on analysis of at least 3 independent mice. One Way ANOVA with multiple comparisons test: p < 0.0001****. (E) Left- Immunofluorescent staining of cardiac troponin (red) and Aurora B positive mitotic nuclei (green) in the heart of TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas) mice 24 h post administration of tamoxifen. Numbers indicate differences in Aurora B localisation throughout the cell cycle. 1- Prophase and Prometaphase, 2- Metaphase, 3- Late anaphase/early telophase, 4 – centrally located mid-body. Right- quantification of percent ABK positive cardiomyocytes in hearts isolated from control (R26+/+; TetO-HRas, R26+/+; Myh6-tTA or R26+/+, black, n = 8), TetO-HRas; Myh6-tTA; R26+/+ (HRas, red, n = 4), R26CMER/+ (Myc, blue, n = 7) and TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas, purple, n = 3) mice 4 weeks post withdrawal of doxycycline and 24 h post administration of tamoxifen. Bold symbols = average per mouse heart, small faint symbols = each individual field quantified. Bars are 100 μm. Representative images based on analysis of at least 3 independent mice. Kruskal–Wallis with Dunn’s multiple comparisons: p < 0.01**, p < 0.001***. (F) Immunofluorescent staining of cardiac troponin (red), Ki67 (green) and Mklp1 (orange) in the heart of TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas) mice 24 h post administration of tamoxifen. Bars = 100 μm.
Combined HRasG12V and MycERT2 expression leads to apoptosis of cardiomyocytes
It has been previously noted that HRasG12V hypertrophic hearts display increased levels of cell death, mononuclear cell infiltration, and damage that ultimately lead to heart failure (30). We observed an increase in the presence of mononuclear cell infiltration such as CD206 positive macrophages (Figure 1A) and the apoptosis marker cleaved caspase 3 (Figure 5A). We also observed increased cleaved caspase 3 when elevated HRasG12V expression was combined with MycERT2 activation (Figure 5A) and an apoptotic gene expression signature was present (Supplementary Figure 1D). Therefore, longer-term analysis and cardiomyocyte dissociation and counting of these mice hearts were not possible. Although elevated levels of Myc expression are associated with apoptosis in some tissues (43) we did not observe apoptosis in hearts expressing activated MycERT2 in the absence of HRasG12V (Figure 5A). Using a different system, we next determined if the cell death observed with HRasG12V and Myc together was recapitulated by co-activation of MycERT2 and Cyclin T1. We infected, R26CMER/+, and R26LSL–CMER/+;Myh6Cre mice with cardiac-specific (cTnT promotor-dependent) overexpression of Cyclin T1 using AAV9-cTnT-Ccnt1 or a control β-galactosidase virus (AAV9-cTnT-LacZ). In contrast, to control mice, MycERT2 activation in the presence of Cyclin T1 increased cardiomyocyte proliferation as previously described (11), increased heart weight to tibia size ratio, increased number of Ki67 and p-H3 positive cardiomyocyte nuclei (Supplementary Figure 4) (11). However, we found no evidence of apoptosis (cleaved caspase 3) in cardiomyocytes in hearts overexpressing Myc and Cyclin T1 (Figure 5B). To confirm negativity we also performed Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) for cell death which also indicated that elevated Myc and Cyclin T1 activity does not induce cardiomyocyte cell death within 48 h (Figure 5B). While the method for overexpressing Cyclin T1 differed, we compared the upregulated gene expression profile of Myc/HRas expressing hearts and Myc/Cyclin T1 expressing hearts and found the GSEA hallmark signatures vastly overlapped. Myc/Ras expressing hearts showed additional gene set enrichment for apoptosis (including the pro-apoptotic proteins Bak and Bok), epithelial mesenchymal transition and inflammatory response (Figure 5C and Supplementary Table 4). The only GSEA pathway that was enriched in Myc/Cyclin T1 expressing hearts was Wnt/Beta Catenin. These data indicate that cell death is specific to HRasG12V activation, not simply an inevitable consequence of enforced endogenous cardiomyocyte proliferation.
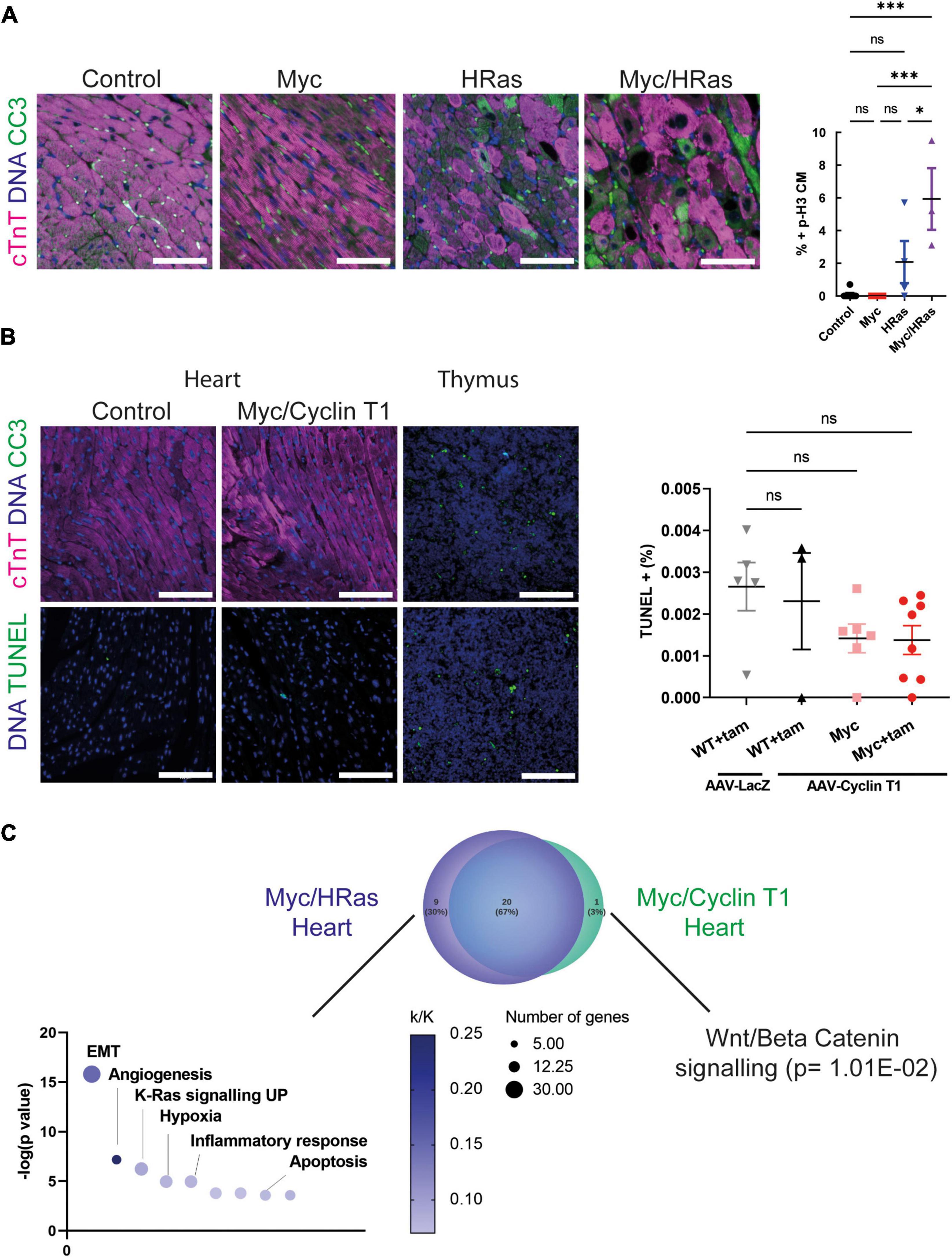
Figure 5. Cardiac HRasG12V and MycERT2 expression results in cardiomyocyte apoptosis. (A) Left- immunofluorescent staining of cleaved caspase 3 (green) and cardiac troponin (pink) in heart from control, TetO-HRas; Myh6-tTA; R26+/+ (HRas), R26CMER/+ (Myc) and TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas) mice 4 weeks post withdrawal of doxycycline and 24 h post administration of tamoxifen. Representative images based on analysis of at least 3 independent mice. Right- quantification of CC3 positive cardiomyocytes in hearts isolated from control (R26+/+; TetO-HRas, R26+/+; Myh6-tTA or R26+/+, black, n = 8), TetO-HRas; Myh6-tTA; R26+/+ (HRas, red, n = 4), R26CMER/+ (Myc, blue, n = 4) and TetO-HRas; Myh6-tTA; R26 CMER/+ (Myc/HRas, purple, n = 3) mice 4 weeks post withdrawal of doxycycline and 24 h post administration of tamoxifen. Bars are 100 μm Representative images based on analysis of 5 images per mouse; error bars show s.e.m. One-Way ANOVA with multiple comparisons test; Myc/HRas vs control: p = 0.0003, Myc/HRas vs Myc: P = 0.0006, Myc/HRas vs HRas; Myh6-tTA; R26+/+: p = 0.0295. (B) Left- immunofluorescent staining of heart from Control and Myh6-Cre; R26LSL– CMER/+ (Myc/Cyclin T1) mice 4 weeks post systemic infection with an AAV-Cyclin T1 adeno-associated virus and 48 post administration of tamoxifen (tam) at adulthood. Top: Control is R26+/+, cleaved caspase 3 (green) and cardiac troponin (pink), Bottom: Control is Myh6-Cre; R26LSL– CMER/+ without tam treatment. TUNEL (green). Representative images based on analysis of 5 independent mice. Right- quantification of TUNEL positive cells in hearts isolated from R26+/+ (WT) mice infected with AAV-LacZ and or AAV-Cyclin T1 and Myh6-Cre; R26LSL– CMER/+ (Myc) mice 4 weeks post systemic infection with an adeno-associated virus and 48 post administration of tamoxifen (tam). Bars are 100 μm. Representative images based on analysis of 5 images per mouse; error bars show s.e.m. Thymus is shown as a positive control. One-Way ANOVA with multiple comparisons test. ns = not significant. (C) Venn diagram of the overlap of the GSEA hallmark signatures obtained from the upregulated gene expression profile of Myc/HRas expressing hearts or Myc/Cyclin T1 expressing hearts. Adult mouse hearts were either isolated 4 weeks post systemic infection with an adeno associated virus encoding Ccnt1 (AAV9-Ccnt1) compared to control and heart from TetO-HRas; Myh6-tTA; R26 CMER/+ (n = 3) 4 weeks after HRasG12D expression and 4 h post Myc activation compared to control, as determined by RNA sequencing (FDR < 0.05 and abs(log2FC) > 0.5). The significant non-overlapping GSEA hallmark signatures are shown below.
Discussion
Myc is a transcription factor that serves as a pivotal instructor of tissue regeneration following injury in a regenerative organ (13), however, in the heart both Myc transcription and Myc-driven transcription are attenuated. When Myc is acutely overexpressed in the heart it competently binds to DNA but only drives a limited transcriptional programme, and cell-cycle progression is not observed within 48 h (11). Protracted Myc expression in cardiomyocytes eventually leads to DNA synthesis, and myocyte hypertrophy but not to cardiomyocyte cytokinesis (15). We have previously shown that the ability of Myc to drive all the transcriptional programmes necessary for cell division in cardiomyocytes depends on the level of P-TEFb (11). We and others have shown that the level of P-TEFb is dependent on the level of Cyclin T1 and elevated Cyclin T1 leads to a corresponding increase in CDK9 and phosphorylated RNA PolII (11, 25, 26). Since HRasG12V upregulates P-TEFb in cardiomyocytes (31) resulting in cardiac muscle hypertrophy, we hypothesised that Myc and Ras would co-operate to drive cardiomyocyte proliferation.
We employed a previously described mouse model of HRasG12V overexpression in which oncogenic HRasG12V is under the control of a tetracycline response element (TetO-HRas) harbouring a cardiomyocyte-specific reverse tetracycline transactivator (Myh6-tTA). Withdrawal of doxycycline at 4 weeks of age leads to cardiomyocyte-restricted HRasG12V overexpression and pathogenic myocardial hypertrophy. Cessation of HRasG12V activity after induction of hypertrophy leads to hypertrophy resolution (30). We confirmed that long-term overexpression of HRasG12V in cardiomyocytes led to increased HRas signalling and cardiomyocyte hypertrophy as previously reported (29, 30, 44–46). Four weeks after HRasG12V expression higher levels of CDK9, Cyclin T1, and phosphorylated RNA Pol II in HRasG12V were observed compared to control animals, confirming that long-term HRasG12V expression upregulates the transcriptional elongation machinery. The exact mechanism of increasing P-TEFb activity by Ras is not known, however, recently the downstream Ras effector AP-1 (heterodimeric transcription factors comprising members of the Fos and Jun families) has been shown to be critical for both Zebrafish and Xenopus tropicalis heart regeneration (47, 48). Fosl1 plays an essential role in cardiomyocyte proliferation by interacting with JunB and binding the Ccnt1 promoter, increasing the expression of Cyclin T1 (48). Here we show that HRasG12V overexpression in cardiomyocytes leads to increased Fosl1 and an increase in Cyclin T1 protein levels. We have previously demonstrated that Myc co-operates with elevated Cyclin T1 expression to drive cardiomyocyte proliferation in vivo (10), therefore, we hypothesised that Myc and HRas would also co-operate in cardiomyocyte proliferation. In the presence, but not the absence, of HRasG12V, Myc activation competently elicited transcriptional programmes involved in cell growth, biogenesis, and metabolism that led to a cardiomyocyte proliferative response. Overlap of the genes altered in Myc and HRas activated hearts with those altered during regeneration of P1 neonatal cardiomyocytes indicated a strong correlation, suggesting that common regenerative transcriptional pathways are activated.
Quantification of cardiomyocyte division is notoriously challenging as cell-cycle re-entry does not necessarily lead to cell division (49–51), and the cell death observed in the HRas model prohibited long-term experimentation and cardiomyocyte number estimation by dissociation techniques. Ki67 is expressed during all active phases of the cell cycle while p-H3 expression begins in G2 and is present throughout mitosis. These markers relay no information regarding cytokinesis and hence they are technically markers of cell cycling and not necessarily proliferation. Here, we observe both Ki67 and p-H3 expression in cardiomyocytes (Figure 4D) demonstrating that combined Myc and HRas overexpression drive cardiomyocytes into the cell cycle. Aurora B kinase is expressed during G2, anaphase, metaphase, telophase, and cytokinesis, where it is detected within the mid-body and can be used as a marker of cardiomyocyte division. However, it is present in an asymmetrical location within cardiomyocytes undergoing bi-nucleation (49) and caution is needed when interpreting staining patterns (50). Here we observed Aurora B kinase localisation in cardiomyocytes in prophase, metaphase, anaphase/telophase and symmetrical mid-bodies. Mitotic kinesin-like protein 1 (Mklp1) is an additional marker of cardiomyocyte cytokinesis where it accumulates at the cleavage furrow (52, 53). Here we observed Mklp1 staining at the cleavage furrow of cardiomyocytes in Myc/HRas overexpressing hearts. While these data provide some evidence that Myc and HRas together drive cardiomyocytes’ entry into the cell cycle with positivity for markers of cytokinesis, productive cell division cannot be formally confirmed without direct cardiomyocyte counting.
In healthy adult mammalian heart apoptosis is rare with only 0.01–0.001% TUNEL-positive cardiomyocytes observed. This rises to 2–12% apoptotic cardiomyocytes in MI ischaemia and reperfusion injury (54–56). Apoptotic death is also a common feature of cardiomyocytes driven into the cycle by overexpression of some cell cycle regulators such as E2F1, CDK1, and CCNB (8, 57). Paradoxically oncogenes, particularly Myc and Ras, promote both pro-proliferative and pro-apoptotic signals, and the cell fate outcome depends very much on the cell type and context (58). In cardiomyocytes, PI3K and ERK activation downstream of HRas has strong anti-apoptotic effects, promoting cardiomyocyte survival in pressure-overload hypertrophy and heart failure (59). However, hypertrophic cardiomyopathy resulting from HRas mutation associated with chronic constitutively activated Ras/Raf/MEK/ERK pathway and pathological hypertrophy leads to apoptosis (60), similar to the phenotype seen in TetO-HRas; Myh6-tTA mice used in this study. We show that MycER expression alone did not lead to cell death in agreement with similar models (15). However, prolonged/persistent ectopic HRasG12V signalling leads to apoptotic cell death, which was exacerbated by transient Myc signalling. Transcriptional profiling indicated Myc/HRas hearts expressed increased levels of the pro-apototic factors Bak, Bok and enhanced inflammatory response, which may be promoting apoptosis. These data indicate endogenous cardiac regeneration is unlikely to be effective with the combination of Myc and HRas. More importantly, the data suggest patients with elevation in Ras signalling in hypertrophic hearts may not benefit or may be harmed, from driving endogenous cardiomyocyte regeneration via activation of cell cycle genes such as Myc. Here, we activated HRas for 4 weeks, further studies are required to establish whether a shorter period of HRas expression results in similar transcriptional changes and proliferation without hypertrophy and apoptosis.
More encouragingly, elevated Cyclin T1 expression, absent of HRas activation, co-operated with activated Myc to drive cardiomyocyte proliferation in the absence of apoptosis, suggesting that co-expression of Cyclin T1 and Myc may be a therapeutically tractable approach for heart regeneration after injury. Myc/Cyclin T1 hearts expressed an increased Wnt/Beta catenin signature, which has recently been shown to be cardioprotective in an adult mammalian setting (61), deciphering the key protective and destructive pathways will be key to translating these findings. As with all genes able to drive proliferation in cardiomyocytes the expression must be tightly regulated and localised. Promising modes of delivery are being developed including cell-specific viral expression vectors and transient modified mRNA technologies.
Study limitations
HRasG12V is restricted to the cardiomyocytes in the TetO-HRas; Myh6-tTA; R26CMER/+ mice, but Myc is expressed across the whole animal, so care must be taken to draw conclusions from the bulk transcriptional data because the interaction between Myc activated non-myocytes and cardiomyocytes are possible. The different systems used to overexpress HRas (TetO-HRas; Myh6-tTA; R26CMER/+ mice) and Cyclin T1 (AAV9-cTnT-Ccnt1) mean the apoptosis and proliferation efficiencies between these groups cannot be directly compared.
Data availability statement
All datasets generated and used in this study, have been deposited in ArrayExpress (www.ebi.ac.uk/arrayexpress) under accession codes: E-MTAB-7595, E-MTAB-8462, and E-MTAB-7636. Further information and requests for resources and reagents should be directed to, and will be fulfilled, by CW, Y2h3MzlAY2FtLmFjLnVr.
Ethics statement
All experimental procedures received ethical approval and were conducted in accordance with the Home Office UK guidelines, under project licences 70/7586 and 80/2396 (GE) and PP2054013 (CW) that were evaluated and approved by the Animal Welfare and Ethical Review Body at the University of Cambridge.
Author contributions
MB and CW conceptualised the study. AB, MB, GQ-R, CA, and LT performed the experimental work. GQ-R and JS performed the sequencing analysis. AB, MB, and CW wrote the manuscript. GE and TL edited the manuscript. GE, JH, and CW obtained the funding. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Cancer Research UK (Programme Grant CRUK-19013 to GE). MB was funded by an EMBO Long-term fellowship and an Australian National Health and Medical Research Council Early Career Fellowship. This work was also supported by funding from the Wellcome Trust Institutional Strategic Support Fund (204845/Z/16/Z to CW).
Acknowledgments
We thank the support staff in the Cambridge University Biomedical Services at the Gurdon Institute, CRUK Cambridge Institute and the Anne McLaren Building. We also like to thank the NGS library facility at the Wellcome – MRC Cambridge Stem Cell Institute and Steven Lane for laboratory support. Images were created with BioRender.com.
Conflict of interest
Authors MB and CW were named inventors on a patent (PCT/GB2020/050350) relating to data in this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.948281/full#supplementary-material
Footnotes
- ^ http://www.gsea-msigdb.org/gsea/index.jsp
- ^ www.lucidchart.com
- ^ https://software.broadinstitute.org/morpheus
References
1. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé- F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science. (2009) 324:98–103.
3. Jopling C, Sleep E, Raya M, Martí M, Raya A, Belmonte JCI. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. (2010) 464:606–9.
4. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. (2011) 331:1078–80.
5. Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, et al. Primary contribution to zebrafish heart regeneration by gata4+ cardiomyocytes. Nature. (2010) 464:601–5. doi: 10.1038/nature08804
6. Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, et al. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature. (2017) 550:260–4.
7. Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, et al. Hypoxia induces heart regeneration in adult mice. Nature. (2017) 541:222–7.
8. Mohamed T, Ang Y-S, Radzinsky E, Zhou P, Huang Y, Elfenbein A, et al. Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell. (2018) 173:104–16.
9. Hirose K, Payumo AY, Cutie S, Hoang A, Zhang H, Guyot R, et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science. (2019) 364:184–8.
10. Monroe TO, Hill MC, Morikawa Y, Rodney GG, Martin JF, Monroe TO, et al. Article YAP partially reprograms chromatin accessibility to directly induce adult cardiogenesis In Vivo YAP partially reprograms chromatin accessibility to directly induce adult cardiogenesis In Vivo. Dev Cell. (2019) 48:765–79. doi: 10.1016/j.devcel.2019.01.017
11. Bywater MJ, Burkhart DL, Sabò A, Straube J, Pendino V, Hudson JE, et al. Reactivation of Myc transcription in the mouse heart unlocks its proliferative capacity. Nat Commun. (2020) 11:1827. doi: 10.1038/s41467-020-15552-x
12. Amati B, Littlewood TD, Evan GI, Land H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. (1993) 12:5083–7. doi: 10.1002/j.1460-2075.1993.tb06202.x
14. Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, et al. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation. (2017) 136:1123–39. doi: 10.1161/CIRCULATIONAHA.117.028252
15. Xiao G, Mao S, Baumgarten G, Serrano J, Jordan MC, Roos KP, et al. Inducible activation of c-Myc in adult myocardium in vivo provokes cardiac myocyte hypertrophy and reactivation of DNA synthesis. Circ Res. (2001) 89:1122–9. doi: 10.1161/hh2401.100742
16. Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. (2015) 16:167–76. doi: 10.1038/nrm3953
17. Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. (2006) 23:297–305. doi: 10.1016/j.molcel.2006.06.014
18. Marshall RM, Salerno D, Garriga J, Graña X, Marshall M, Salerno D, et al. Cyclin T1 expression is regulated by multiple signaling pathways and mechanisms during activation of human peripheral blood lymphocytes. J Immunol. (2005) 175:6402–11. doi: 10.4049/jimmunol.175.10.6402
19. Paparidis NF, Durvale MC, Canduri F. The emerging picture of CDK9/P-TEFb: More than 20 years of advances since PITALRE. Mol Biosyst. (2017) 13:246–76. doi: 10.1039/c6mb00387g
20. Kim YK, Mbonye U, Hokello J, Karn JT-. Cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J Mol Biol. (2011) 410:896–916. doi: 10.1016/j.jmb.2011.03.054
21. Quaresma AJC, Bugai A, Barboric M. Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res. (2016) 44:7527–39. doi: 10.1093/nar/gkw585
22. Garriga J, Bhattacharya S, Calbo J, Marshall M, Truongcao M, Haines DS, et al. CDK9 is constitutively expressed throughout the cell cycle, and its steady-state expression is independent of SKP2. Mol Cell Biol. (2003) 23:5165–73. doi: 10.1128/MCB.23.15.5165-5173.2003
23. O’Keeffe B, Fong Y, Chen D, Zhou S, Zhou Q. Requirement for a kinase-specific chaperone pathway in the production of a Cdk9/Cyclin T1 heterodimer responsible for P-TEFb-mediated tat stimulation of HIV-1 transcription. J Biol Chem. (2000) 275:279–87. doi: 10.1074/jbc.275.1.279
24. Chiu Y, Cao H, Jacque J, Stevenson M, Rana TM. Inhibition of human immunodeficiency virus type 1 replication by RNA interference directed against human transcription elongation factor P-TEFb (CDK9/CyclinT1). J Virol. (2004) 78:2517–29. doi: 10.1128/jvi.78.5.2517-2529.2004
25. Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, et al. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med. (2002) 8:1310–7. doi: 10.1038/nm778
26. Sano M, Wang SC, Scaglia F, Xie M, Sakai S, Tanaka T, et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. (2004) 23:3559–69. doi: 10.1038/sj.emboj.7600351
27. Karnoub AE, Weinberg RA. Ras oncogenes: Split personalities. Nat Rev Mol Cell Biol. (2008) 9:517–31.
28. Sugden PH. Ras, Akt, and mechanotransduction in the cardiac myocyte. Circ Res. (2003) 93:1179–92. doi: 10.1161/01.RES.0000106132.04301.F5
29. Hunter JJ, Tanaka N, Rockman HA, Ross J, Chien KR. Ventricular expression of a MLC-2v-ras fusion gene induces cardiac hypertrophy and selective diastolic dysfunction in transgenic mice*. J Biol Chem. (1995) 270:23173–8. doi: 10.1074/jbc.270.39.23173
30. Wei B-R, Martin PL, Spehalski SBHE, Kumar M, Hoenerhoff MJ, Rozenberg J, et al. Capacity for resolution of ras–MAPK-initiated early pathogenic myocardial hypertrophy modeled in mice. Comp Med. (2011) 61:109–18.
31. Abdellatif M, Packer SE, Michael LH, Zhang D, Charng MJ, Schneider MD. A ras-dependent pathway regulates RNA polymerase II Phosphorylation in cardiac myocytes: Implications for cardiac hypertrophy. Mol Cell Biol. (1998) 18:6729–36. doi: 10.1128/MCB.18.11.6729
32. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. (2011) 17:10. doi: 10.14806/ej.17.1.200
33. Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. (2011) 12:323. doi: 10.1186/1471-2105-12-323
34. Robinson MD, McCarthy DJ, Smyth GK. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. (2009) 26:139–40.
35. Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20.
36. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. Ultrafast universal RNA-seq aligner. Bioinformatics. (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
37. Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. (2015) 31:166–9. doi: 10.1093/bioinformatics/btu638
38. Gentleman R, Carey V, Bates D, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. (2004) 5:R80. doi: 10.1186/gb-2004-5-10-r80
39. Kishore K, de Pretis S, Lister R, Morelli MJ, Bianchi V, Amati B, et al. methylPipe and compEpiTools: A suite of R packages for the integrative analysis of epigenomics data. BMC Bioinformatics. (2015) 16:313. doi: 10.1186/s12859-015-0742-6
40. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. (2013) 14:128. doi: 10.1186/1471-2105-14-128
41. Batho CAP, Mills RJ, Hudson JE. Metabolic regulation of human pluripotent stem cell-derived cardiomyocyte maturation. Curr Cardiol Rep. (2020) 22:73. doi: 10.1007/s11886-020-01303-3
42. Pianca N, Sacchi F, Umansky KB, Chirivì M, Iommarini L, Da Pra S, et al. Glucocorticoid receptor antagonization propels endogenous cardiomyocyte proliferation and cardiac regeneration. Nat Cardiovasc Res. (2022) 1:617–33. doi: 10.1038/s44161-022-00090-0
43. Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, et al. Distinct thresholds govern myc’s biological output in vivo. Cancer Cell. (2008) 14:447–57. doi: 10.1016/j.ccr.2008.10.018
44. Zheng M, Dilly K, Dos Santos Cruz J, Li M, Gu Y, Ursitti JA, et al. Sarcoplasmic reticulum calcium defect in Ras-induced hypertrophic cardiomyopathy heart. Am J Physiol Hear Circ Physiol. (2004) 286:424–33. doi: 10.1152/ajpheart.00110.2003
45. Ruan H, Mitchell S, Vainoriene M, Lou Q, Xie LH, Ren S, et al. Giα1-Mediated cardiac electrophysiological remodeling and arrhythmia in hypertrophic cardiomyopathy. Circulation. (2007) 116:596–605. doi: 10.1161/CIRCULATIONAHA.106.682773
46. Mitchell S, Ota A, Foster W, Zhang B, Fang Z, Patel S, et al. Distinct gene expression profiles in adult mouse heart following targeted MAP kinase activation. Physiol Genomics. (2006) 25:50–9. doi: 10.1152/physiolgenomics.00224.2005
47. Beisaw A, Kuenne C, Guenther S, Dallmann J, Wu CC, Bentsen M, et al. AP-1 contributes to chromatin accessibility to promote sarcomere disassembly and cardiomyocyte protrusion during zebrafish heart regeneration. Circ Res. (2020) 126:1760–78. doi: 10.1161/CIRCRESAHA.119.316167
48. Wu H-Y, Zhou Y-M, Liao Z-Q, Zhong J-W, Liu Y-B, Zhao H, et al. Fosl1 is vital to heart regeneration upon apex resection in adult Xenopus tropicalis. npj Regen Med. (2021) 6:1–16. doi: 10.1038/s41536-021-00146-y
49. Heallen TR, Kadow ZA, Kim JH, Wang J, Martin JF. Stimulating cardiogenesis as a treatment for heart failure. Circ Res. (2019) 124:1647–57.
50. Engel FB, Schebesta M, Keating MT. Anillin localization defect in cardiomyocyte binucleation. J Mol Cell Cardiol. (2006) 41:601–12. doi: 10.1016/j.yjmcc.2006.06.012
51. Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. (2005) 19:1175–87. doi: 10.1101/gad.1306705
52. Capalbo L, Bassi ZI, Geymonat M, Todesca S, Copoiu L, Enright AJ, et al. The midbody interactome reveals unexpected roles for PP1 phosphatases in cytokinesis. Nat Commun. (2019) 10:1–17. doi: 10.1038/s41467-019-12507-9
53. Nishiyama C, Saito Y, Sakaguchi A, Kaneko M, Kiyonari H, Xu Y, et al. Prolonged myocardial regenerative capacity in neonatal opossum. Circulation. (2022) 146:125–39. doi: 10.1161/CIRCULATIONAHA.121.055269
55. Van Empel VPM, Bertrand ATA, Hofstra L, Crijns HJ, Doevendans PA, De Windt LJ. Myocyte apoptosis in heart failure. Cardiovasc Res. (2005) 67:21–9.
56. Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M, et al. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. (2011) 2:1–11.
57. Agah R, Kirshenbaum LA, Abdellatif M, Truong LD, Chakraborty S, Michael LH, et al. Adenoviral delivery of E2F-1 directs cell cycle reentry and p53- independent apoptosis in postmitotic adult myocardium in vivo. J Clin Invest. (1997) 100:2722–8. doi: 10.1172/JCI119817
58. Cox AD, Der CJ. The dark side of Ras: Regulation of apoptosis. Oncogene. (2003) 22:8999–9006. doi: 10.1038/sj.onc.1207111
59. Gallo S, Vitacolonna A, Bonzano A, Comoglio P, Crepaldi TERK. A key player in the pathophysiology of cardiac hypertrophy. Int J Mol Sci. (2019) 20:1–21.
60. Sala V, Gallo S, Leo C, Gatti S, Gelb BD, Crepaldi T. Signaling to cardiac hypertrophy: Insights from human and mouse RASopathies. Mol Med. (2012) 18:938–47.
Keywords: Myc (c-Myc), HRas gene, cardiomyocyte, proliferation, cell-cycle
Citation: Boikova A, Bywater MJ, Quaife-Ryan GA, Straube J, Thompson L, Ascanelli C, Littlewood TD, Evan GI, Hudson JE and Wilson CH (2022) HRas and Myc synergistically induce cell cycle progression and apoptosis of murine cardiomyocytes. Front. Cardiovasc. Med. 9:948281. doi: 10.3389/fcvm.2022.948281
Received: 19 May 2022; Accepted: 27 September 2022;
Published: 20 October 2022.
Edited by:
Ajit Magadum, Temple University, United StatesReviewed by:
Felix B. Engel, University Hospital Erlangen, GermanyMiao Cui, University of Texas Southwestern Medical Center, United States
Gabriele D’Uva, University of Bologna, Italy
Copyright © 2022 Boikova, Bywater, Quaife-Ryan, Straube, Thompson, Ascanelli, Littlewood, Evan, Hudson and Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine H. Wilson, Y2h3MzlAY2FtLmFjLnVr
†These authors have contributed equally to this work