 Miaosen Liu
Miaosen Liu Jialan Lv2
Jialan Lv2 Liding Zhao
Liding Zhao Xiaogang Guo
Xiaogang Guo- 1Clinical Medicine, Zhejiang University School of Medicine, Hangzhou, China
- 2Department of Cardiology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
The ATP consumption in heart is very intensive to support muscle contraction and relaxation. Mitochondrion is the power plant of the cell. Mitochondrial dysfunction has long been believed as the primary mechanism responsible for the inability of energy generation and utilization in heart failure. In addition, emerging evidence has demonstrated that mitochondrial dysfunction also contributes to calcium dysregulation, oxidative stress, proteotoxic insults and cardiomyocyte death. These elements interact with each other to form a vicious circle in failing heart. The role of mitochondrial dysfunction in the pathogenesis of heart failure has attracted increasing attention. The complex signaling of mitochondrial quality control provides multiple targets for maintaining mitochondrial function. Design of therapeutic strategies targeting mitochondrial dysfunction holds promise for the prevention and treatment of heart failure.
Introduction
Mitochondrion is cellular organelle surrounded by two membranes, which is the source of energy production within the cells. Beyond its critical role in supplying energy, mitochondrion is also closely related to reactive oxygen species (ROS) production, Ca2+ homeostasis and related signal transduction (1, 2). Mitochondrion is highly plastic, and the number, shape and function remain relatively stable under normal conditions. Mitochondrial homeostasis is crucial for cell fate (3–5).
Mitochondrion is able to quickly adapt to changing conditions. Mitochondrion adjusts its morphology and function to meet the needs of cell, which is conducive to respond to internal and external stimuli and maintain the physiological function (3). Conversely, mitochondrial dysfunction may in turn cause cell fate transition, leading to the damage of structure, function and metabolism, and even cell death (6, 7), which takes part in the genesis and development of a variety of diseases (8–11). Accumulating studies demonstrated that mitochondrial dysfunction contributes to the pathogenesis of common cardiovascular diseases (CVD) (12–16), culminating in end-stage heart failure (17, 18). In this review, we mainly discuss mitochondrial dysfunction-mediated energy metabolism disturbance, Ca2+ dysregulation, oxidative stress, proteostasis imbalance, and mitophagy deficiency in the development of heart failure.
Mitochondrial quality control
Mitochondrial quality control mainly includes the ubiquitin-proteasome system (UPS) which recognizes and degrades misfolded and damaged proteins to maintain mitochondrial protein homeostasis (19), mitochondrial fusion and fission to ensure the number, morphology and proper intracellular distribution of mitochondria (20), and mitophagy to selectively remove damaged and redundant mitochondria (21). Mitochondrial quality control is of vital importance to maintain mitochondrial homeostasis. Mitochondrial metabolic disorders, morphological and functional changes are closely related to the occurrence, development and treatment of various diseases such as aging, cancer, metabolic disorders, neurodegenerative diseases and CVD (22–24).
Disturbance of mitochondrial energy metabolism in heart failure
Heart failure is a clinical syndrome resulted from a wide range of causes, including ischemic, hypertensive, inflammatory, and toxic heart diseases. The heart is a very high energy consumption organ and must continually generate ATP to support muscle contraction and relaxation. The energy substrates in mitochondria of failing heart are switched. There is about 70% of ATP coming from the beta-oxidation using fatty acids as the primary fuel in mitochondria of healthy adult heart, and the contribution of fatty acids beta-oxidation to overall ATP production can even reach to almost 100% of the total energy requirement of the heart (25). Glucose, lactate, pyruvate, ketone body and amino acid are also substrates for myocardial energy provision (26). During the pathological progression of heart failure, the energy metabolism mode of cardiomyocytes gradually changes, with an increased proportion from glucose and a decreased proportion from fatty acids beta-oxidation (27). This metabolic remodeling of substrate utilization shifts cardiac metabolism to a fetal energy metabolism (28, 29). Compared with fatty acids, the oxygen consumption of energy production is lessened when glucose is used as the substrate (30). To some extent, this switch helps to maintain cardiac function in the process of chronic heart disease.
The oxidative phosphorylation in mitochondria is impaired and energy starvation is observed in the human biopsies of failing heart (31). Heart failure is mainly divided into ischemic and non-ischemic heart failure. Ischemic heart failure is related to coronary artery diseases especially myocardial infarction and accounts for about 50% of heart failure (32). The energy substrates adaptively shift toward glucose, which increases the stoichiometric ratio of ATP production to oxygen consumption and improves cardiac function in ischemic heart (33, 34). Thus replacing fatty acids by glucose is considered to be able to increase oxygen efficiency and benefit energy provision in failing heart. The “adaptive” mechanism during the progression of heart failure is not well understood. Amorim et al. reported that insulin-induced phosphorylation of Akt is normal and the expression of glucose transporter type 4 is unchanged, while the expression of the genes regulating fatty acid oxidation, e.g., long-chain-acyl-coenzyme A dehydrogenase, carnitine palmitoyltransferase 1 (CPT I) and peroxisome proliferator-activated receptor-α, is reduced in the infarcted hearts (35). Oxidative phosphorylation is badly damaged due to severe hypoxia in heart with acute myocardial infarction. Anaerobic glycolysis is inefficient to produce adequate ATP to meet the energy requirement of the heart and also results in the accumulation of lactate (36, 37). ATP depletion and acidosis lead to impaired myocardial contractility and injured membrane pumps and ion channels. The alterations of membrane pumps and ion channels cause mitochondrial swelling, Ca2+ accumulation and the opening of mitochondrial permeability transition pore (mPTP), which is prominent in cardiomyocyte of myocardial ischemic injury and has been demonstrated to play a critical role in various forms of cell death in myocardial ischemia/reperfusion injury (38, 39).
Non-ischemic heart failure is mainly related to hypertrophic cardiomyopathy and dilated cardiomyopathy (DCM) (34). Mitochondrial energy substrates switch is prevalent in non-ischemic heart failure, which is driven by energy metabolic reprogramming leading to altered enzymes and substrate flux (29). Accumulating evidence has indicated that enzymes involved in fatty acids beta-oxidation pathway decrease significantly in the myocardium from heart failure patients. Martin et al. reported that total CPT and CPT II activities decrease in failing heart, and carnitine deficiency is related to ventricle dysfunction (40). Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency is the most common defect of mitochondrial long-chain fatty acid β-oxidation. Mitochondrial energy metabolism is impaired in VLCAD–/– mice, and hypertrophic cardiomyopathy is a typical manifestation of VLCAD deficiency in human (41, 42). The changes in energy metabolic gene expression and substrates progressively lead to energy starvation. On the other hand, the pathological remodeling increases energy consumption in diseased heart. The impairment of mitochondrial energy metabolism leads to energy deficiency, which is in contradiction with the increased energy demand in hypertrophic heart caused by pathological remodeling.
Recent studies revealed that mitochondrial pyruvate carrier (MPC), which transports pyruvate into the mitochondria, is reduced in failing human and mouse hearts (43, 44). Cardiac assist device was found to increase MPC expression in the myocardium and promote myocardial recovery in patients with chronic heart failure (45). Mice with cardiac-specific deletion of MPC1 or/and MPC2 resulted in cardiac hypertrophy, dilated cardiomyopathy, and contractile dysfunction (43–46). These results indicate an important role of pyruvate metabolism in myocardial metabolism and function. Pyruvate dehydrogenase (PDH), an enzyme converts pyruvate into acetyl-CoA, was reported to be inactivated in advanced pathological conditions of heart failure (47, 48). However, a recent study found that PDH is activated at an early phase before the down-regulation of fatty acid oxidation and tricarboxylic acid (TCA) cycle, suggesting that PDH activation is one of the earliest events to compensate for metabolic impairment from myocardial damage (49). Increased PDH expression and activity were evident with decreased expression of PDH kinase 4, MPC1 and MPC2, sustaining the capacity for PDH to facilitate glucose metabolism in end-stage systolic heart failure (50).
Dysregulation of Ca2+ homeostasis in heart failure
As a second messenger, Ca2+ plays a central role in myocardial excitation-contraction coupling (51). Using the energy of ATP hydrolysis, Ca2+ pump transports Ca2+ ions from the cytoplasm into the sarcoplasmic reticulum (SR) or out of the cell. Therefore, energy deficiency always couples with the dysregulation of Ca2+ transportation in failing heart, which consequently leads to excitation-contraction uncoupling and cardiac dysfunction (52). Dysregulation of Ca2+ homeostasis is also associated with abnormal leak of Ca2+ from SR through ryanodine receptors (RyR) and results in increased cytosolic Ca2+ at baseline but reduced cytosolic Ca2+ transients during excitation (53, 54). Overwhelming studies revealed that Ca2+ leaked from the SR via RyR2 causes mitochondrial Ca2+ overload, which plays a key role in heart failure (55).
Mitochondrial Ca2+ uptake and efflux also influence cytosolic Ca2+ homeostasis (56). Mitochondrial Ca2+ uptake depends on the mitochondrial Ca2+ uniporter (MCU) (57, 58), while mitochondrial Ca2+ efflux is mainly regulated by the mitochondrial Na+/Ca2+/Li+ exchanger (56). Mitochondria can sense the change of cytosol free Ca2+ and maintain intracellular Ca2+ homeostasis by regulating the opening and closing of MCU and Na+/Ca2+/Li+ exchanger, so as to prevent cytosolic Ca2+ overload and cell damage (56). However, excessive mitochondrial Ca2+ uptake causes mitochondrial Ca2+ overload, which impairs mitochondrial function, leading to decreased ATP generation and increased mitochondrial ROS (mtROS) production, and is a key determinant in heart failure (55). MCU is also a mitochondrial redox sensor and MCU oxidation enhances its channel activity leading to mitochondrial Ca2+ overload, increased mtROS, and even cell death (59). In addition, increased intracellular Ca2+ leads to the activation of calpain, which mediates mitochondrial damage in diseased hearts (60). Recent studies revealed that the activation of calpain results in mitochondrial damage and subsequent cardiac dysfunction by impairing mitophagy and promoting mitochondrial fission and apoptosis (13, 61).
On the other hand, if the level of mitochondrial Ca2+ falls below a critical threshold level, Ca2+-dependent activation of tricarboxylic acid (TCA) cycle enzymes is disrupted, and consequently ATP generation is impaired whereas NADH oxidation is increased. This pathological change was demonstrated in both myocytes isolated from failing heart (62) and in an animal model of heart failure (63). Therefore, mitochondrial Ca2+ homeostasis is critical for normal heart function.
Accumulation of mitochondrial reactive oxygen species in heart failure
Mitochondria are one of the main sources of ROS production within the cell. TCA cycle-dependent electron transfer complexes are distributed in the inner membrane of mitochondria. Most electron transfer process is coupled with the generation of ATP, and only 1–2% of the electrons are transferred to produce superoxide anion, which can be scavenged by superoxide dismutase (64). The mtROS is mainly generated from the complex I and complex III in the electron transport chain (65). The basal level of mtROS can fulfill essential physiological functions by acting as signaling molecule. It plays an important physiological role in mediating gene expression, regulating cell cycle and cell differentiation, and is of great significance for stress response, cell survival, cell proliferation, etc. (1).
However, excessive accumulation of mtROS plays an important role in pathological process of heart failure. Dai et al. demonstrated that mtROS contributes to angiotensin II-induced cardiac hypertrophy and heart failure (66). Chouchani et al. showed that mtROS generation during the early stages of reperfusion promotes myocardial ischemia/reperfusion injury (67). It is recognized that mtROS drives acute cardiovascular events such as electrical instability and chronic proteome remodeling in heart failure (68). Dysfunction of the mitochondrial respiratory chain is accompanied by increased generation of mtROS, leading to oxidative stress and resulting in mitochondrial protein and DNA damage, membrane lipid peroxidation, and the opening of the mPTP. The opening of the mPTP causes the release of cytochrome C and cell apoptosis, consequently promotes the progression of heart failure (69). It is well believed that apoptosis is mainly responsible for the cumulative loss of cardiomyocyte in failing heart, and causally contributes to myocardial dysfunction progression (70). Recent study also demonstrated that mtROS induced NLRP3 inflammasome activation and cardiomyocyte pyroptosis in DCM, uncovering a novel event in the initiation and progression of heart failure (71).
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) is one of the predominant sources of ROS in the heart. Accumulating data indicate that complex crosstalk and interaction exist between NOX and mtROS (72, 73). Under conditions of oxidative stress, NOX-derived ROS can cause lipid peroxidation of mitochondrial membrane and the opening of redox-sensitive mitochondrial ATP-sensitive K+ channel (mitoKATP), leading to mtROS generation from the electron transport chain (73).
Dysregulation of mitochondrial proteostasis in heart failure
Mitochondria have their own genome encoding mitochondrial specific proteins. The mitochondrial proteome is composed of 1,000∼1,500 proteins, encoded by both mitochondrial and nuclear genomes (74, 75). Mitochondrial proteostasis is essential for the function of cell. Normal mitochondrial function requires coordinated gene expression in the nucleus and in mitochondria (19). The UPS controls protein transport across the mitochondrial outer membrane, and primarily degrades outer membrane proteins that are improperly imported or damaged/mislocalized (19). Furthermore, mitochondrial chaperones and proteases govern protein folding and degrade damaged proteins inside mitochondria. Dysregulation of mitochondrial proteostasis leads to proteotoxic insults and eventually cell death (19).
Under cellular stress conditions, both mitochondrial-encoded and nuclear-encoded proteins are misfolded and dysfunctional. The generation of mtROS compromises protein integrity and folding. The proteotoxic stress within mitochondria activates mitochondrial unfolded protein response (UPRmt), which induces the transcription of mitochondrial chaperones (e.g., heat shock protein 60), antioxidants (e.g., thioredoxin 2) and proteases (e.g., caseinolytic mitochondrial matrix peptidase proteolytic subunit, CLPP; YME1 like 1 ATPase, YME1L; OMA1 zinc metallopeptidase, OMA1) (76). UPRmt re-establish mitochondrial proteostasis by facilitating protein folding or repairing misfolded proteins, and degrading unrepairable proteins (75).
UPRmt is of great benefit to the preservation of ATP production, reduction of mtROS accumulation, and inhibition of mitochondria-mediated cell death, and showed a protective effect in chronic and acute cardiac injury (77). Activating transcription factor associated with stress-1 (ATFS-1), an UPRmt-inducing transcription factor, was showed to preserve ATP production by promoting the assembly and function of oxidative phosphorylation components during mitochondrial stress (78). Upregulation of mitochondrial Lon protease (LONP1), a component of UPRmt, protected the myocardium from cardiac stress and limited ischemia/reperfusion injury (79). Recent study found that intensive sympatho-excitation leads to pathological cardiac hypertrophy and fibrosis, which is coupled with decreased UPRmt and increased mitochondrial proteotoxic stress (80). These myocardial morphological alterations is closely associated with heart failure with preserved ejection fraction (HFpEF) (81). Smyrnias et al. demonstrated that UPRmt is activated during chronic pressure overload and pharmacological enhancement of the UPRmt alleviates mitochondrial and contractile dysfunction in the stressed heart (82). Xu et al. also found that choline attenuated the mito-nuclear protein imbalance and activated UPRmt to preserve the ultrastructure and function of mitochondria in hypertrophic heart (83). Further studies confirmed that pharmacological UPRmt activation exerts cardioprotective effect in an ATF5-dependent manner in mouse models of ischemia-reperfusion injury and transverse aortic constriction-induced cardiac hypertrophy (84, 85). All these indicate that UPRmt may play an important protective role in stressed heart and may be potential therapeutic target for heart failure.
Reciprocal regulation between mitochondrial proteases YME1L and OMA1 is critical in UPRmt. They antagonistically regulate mitochondrial proteostasis by ATP-dependent network. When mitochondrial depolarization with preserved ATP levels, YME1L is further activated and OMA1 is degraded through a mechanism involving YME1L. Whereas in the absence of ATP, OMA1 is activated and stabilized by membrane depolarization, and subsequently promotes YME1L degradation (86, 87). It was reported that pressure overload decreases YME1L expression and cardiac-specific overexpression of YME1L improves cardiac function in pressure overload-induced heart failure (88). Acin-Perez demonstrated that OMA1 ablation averts cardiomyocyte death in three different mouse models of heart failure: tachycardiomyopathy, heart failure with preserved left ventricular ejection fraction, and left ventricular myocardial ischemia and hypertrophy (89). All these suggested that the regulation of YME1L and OMA1 is potential target for preventing the progression of heart failure associated with distinct types of etiologies.
However, there is contradictory evidence that UPRmt may be associated with adverse events within the heart. The release of mitochondrial chaperone molecule heat shock protein 60 was observed to promote proinflammatory tumor necrosis factor-α, which correlated with increased myocyte apoptosis in heart failure (90). The mitochondrial matrix protease CLPP plays a central role in the activation of the UPRmt, deletion of CLPP in heart increased the synthesis of oxidative phosphorylation subunits and attenuated the mitochondrial cardiomyopathy (91). Cao et al. showed that parvostatin improved left ventricular function and slowed the progression of heart failure in mice by blocking UPRmt activator c-Jun (92). The contradictory effects of UPRmt on heart may due to its intensive degree. A moderate activation of UPRmt may maintain normal mitochondrial and cardiac function by removing/repairing damaged mitochondrial proteins, while an excessive UPRmt may exacerbate mitochondrial dysfunction and cardiac dysfunction due to a massive cleavage of mitochondrial proteins (77).
Moreover, hyperacetylation of mitochondrial proteins has been found in the myocardium of patients and animal model with heart failure (93). The hyperacetylation of TCA cycle enzymes and electron transfer enzymes results in decreased activity of these enzymes, leading to disruption of mitochondrial bioenergetics and redox homeostasis (93, 94). Reduced fatty acid beta-oxidation is associated with increased short-chain acyl-CoA in the failing heart, indicating an imbalance in the utilization and supply of acetyl-CoA may lead to increased acetylation of mitochondrial proteins (17). Sirtuin 3 (SIRT3), a key deacetylase in mitochondria, has been demonstrated to be downregulated in the failing heart, and SIRT3 knockout mice are susceptible to develop transverse aortic constriction-induced heart failure (95). This result suggests that reduced protein deacetylation may be also responsible for protein hyperacetylation in mitochondria of the failing heart.
Imbalance of mitochondrial dynamics in heart failure
Mitochondrial dynamics including fusion and fission determines the number, morphology and distribution of mitochondria, and modulates mitochondrial functions to respond properly to body demands (20). Imbalance of mitochondrial dynamics disturbs energy and mtROS generation, Ca2+ homeostasis and proteostasis, even induces cell death in the heart (96). Mitofusin 1 (MFN1) and MFN2 are the core components of the mitochondrial fusion machinery and coordinately regulate mitochondrial fusion (97). Mitochondrial fission requires fission protein dynamin-related protein 1 (Drp1) (98).
The balance between fusion and fission ensures the number and morphology of mitochondria in cardiomyocytes. In addition, mitochondria are tightly packed between the sarcomere myofibrils or closely localized to the SR in order to provide crosstalk between sarcomeres and efficient SR-mitochondria during excitation–contraction coupling (99). Downregulation of mitochondrial dynamics regulator MFN2 has been found resulting in mitochondrial fragmentation and contributing to the development of heart failure in rats and in patients with pulmonary arterial hypertension (PAH) (100). Decreased MFN2 expression and excessive mitochondrial fission were also observed in diabetic mice and promoted the development of diabetic cardiomyopathy, indicating mitochondrial dynamics is therapeutic target for intervention in diabetic cardiomyopathy (101).
Activation of fission protein Drp1 and aberrant mitochondrial fission were observed in PAH, and the Drp1 inhibitor Mdivi-1 attenuated mitochondrial fragmentation and improved exercise capacity, right ventricular function, and hemodynamics in experimental PAH (102). Recent study showed that lipid overload induced Drp1 acetylation and eventually resulted in cardiomyocyte death and heart dysfunction (103). Phosphorylated activation of Drp1 and mitochondrial fission contributed to cardiomyocyte pyroptosis in non-ischemic DCM mice, and culminating in end-stage heart failure (71).
Impairment of mitophagy in heart failure
Mitophagy is a selective autophagic process that eliminates dysfunctional mitochondria. It is essential for mitochondrial quality control and cell function (21). Mitophagic deficiency results in the accumulation of dysfunctional/damaged mitochondria, which reduces the capacity of ATP production and increases the generation of mtROS (21). Continuous constitutive autophagy has a crucial role in maintaining cardiac structure and function. The phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)/Parkin pathway is an important pathway in regulating mitophagy (104). Mitophagy is impaired in aged and doxorubicin-treated hearts, and inhibition of Parkin-mediated mitophagy subsequently promotes cardiac dysfunction and overexpression of Parkin attenuates the functional decline in mouse hearts (105). Overexpression of Parkin also protects cardiac myocytes against hypoxia-mediated cell death and Parkin protein deficiency aggravates myocardial damage and reduces survival following myocardial infarction (106). In addition, AMP-activated protein kinase α2 (AMPKα2) prevents the progression of heart failure by promoting mitophagy via PINK1 phosphorylation (107).
FUN14 domain-containing protein 1 (FUNDC1) is an outer mitochondrial membrane protein and serves as a receptor to mediate mitophagy (104). It was reported that hypoxic preconditioning induces FUNDC1-dependent mitophagy, regulates mitochondrial homeostasis, and protects the heart from ischemia/reperfusion injury (108). Autophagy protein 5 (Atg5) deficiency decreases mitophagy, leading to increased ROS production and NF-κB activity, thereby contributing to cardiac inflammation and injury (109, 110). Wang et al. found that mitophagy coordinates the UPRmt to attenuate sepsis-mediated myocardial injury, and endogenous UPRmt is a downstream signal of mitophagy to maintain mitochondrial homeostasis in the case of mitophagy inhibition (111). Insufficient mitophagy has been associated with multiple forms of cardiomyopathy, including age-related cardiomyopathy (112), obesity-associated cardiomyopathy (113) and diabetic cardiomyopathy (114). Moreover, pharmacological or genetic inhibition of mitophagy often exacerbates the progression of heart failure in multiple animal models of cardiovascular diseases (104, 115).
Targeting mitochondrial dysfunction as a therapeutic strategy
Classic treatments recommended in patients with heart failure include diuresis to reduce cardiac preload, vasodilation to reduce the pressure load, inhibition of angiotensin II to block the pathological remodeling, Digoxin or inotropes to enhance myocardial contractility, and so on (116). Although recent evidence showed cardiac myosin activator Omecamtiv Mecarbil (117) and the soluble guanylate cyclase stimulator Vericiguat (118) can benefit patients with heart failure, the outcome of heart failure especially HFpEF is still difficult to change fundamentally.
Mitochondrial dysfunction results in energy metabolism disturbance, Ca2+ dysregulation, oxidative stress and proteotoxic insults, and leads to a final outcome-cardiomyocyte death in the heart (Figure 1). The loss of cardiomyocyte contributes to reduced ventricular systolic dysfunction, and is of central importance in the development of heart failure (71, 119, 120). Mitochondrial dysfunction-related cell death may be potential therapeutic targets for heart failure. Several of microRNAs have been shown to inhibit mitochondrion-mediated apoptosis and improve cardiac function by regulating mitochondrial fission and fusion (121–123). Targeting mitochondrial dysfunction-related pyroptosis is also showed to improve cardiac function in DCM mice (71). However, these preliminary findings mainly derived from in vitro and preclinical animal models, and lots of work need to be done to further evaluate their potential of cardioprotection in human.
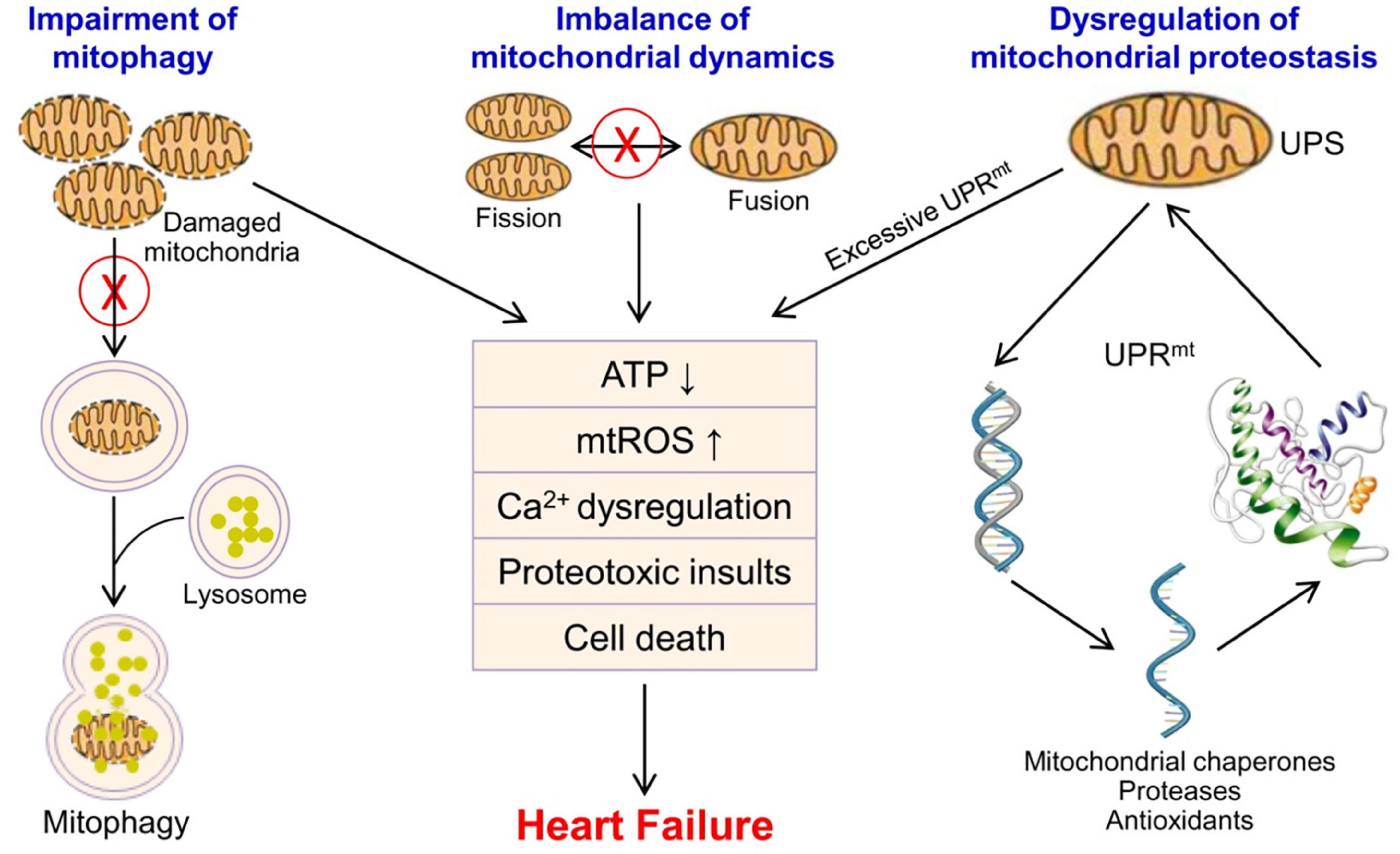
Figure 1. Mitochondrial quality control and mitochondrial dysfunction in the pathophysiology of heart failure. Mitochondrial quality control mainly includes UPS/UPRmt, mitochondrial fusion and fission, and mitophagy to ensure the number, morphology and function of mitochondria. Mitochondrial dysfunction leads to energy metabolism disturbance, Ca2+ dysregulation, oxidative stress, proteotoxic insults, and cardiomyocyte death in the heart, and contributes to the progression of heart failure. mtROS, mitochondrial reactive oxygen species; UPS, ubiquitin-proteasome system; UPRmt, mitochondrial unfolded protein response.
It was reported that the overexpression of catalase targeted to mitochondria but not wild type catalase in peroxisomes ameliorates cardiac hypertrophy and diastolic dysfunction in mice (66). Mitochondria-targeted antioxidant MitoQ was found to decrease heart dysfunction after myocardial ischemia-reperfusion (124), and attenuate hypertension-induced cardiac hypertrophy (125). Recent study also showed that mitoTEMPOL, an mtROS scavenger, attenuates nicotine-induced myocardial remodeling and cardiac dysfunction (126). Coenzyme Q10, an electron carrier in mitochondria, is thought to reduce oxidative stress because of its antioxidant activity. Al Saadi et al. included 11 studies to review the efficacy of coenzyme Q10 in heart failure, and concluded that the included studies provide moderate-quality evidence for the benefit of coenzyme Q10 in reducing all-cause mortality and hospitalization related to heart failure. However, there was low-quality evidence to conclude whether coenzyme Q10 improves either left ventricular ejection or exercise capacity (127). Large-scale and high quality clinical trials are needed to further evaluate the efficacy of coenzyme Q10. Due to the essential role of mtROS in cell function and survival, a precise control between ROS production and detoxification is a decisive issue need to be solved.
Recent study showed that the administration of sodium-glucose co-transporter 2 (SGLT2) inhibitor Empagliflozin significantly reduced cardiovascular death and hospitalization for heart failure in patients regardless of the presence or absence of diabetes (128). It is speculated that SGLT2 inhibitor might improve mitochondrial energetics in the heart by offering β-hydroxybutyrate as an attractive substrate for oxidation and protect against heart failure (129). Empagliflozin was also proved to reduce cytosolic Na+ and increase mitochondrial Ca2+ in cardiomyocytes independent of SGLT inhibition, probably by inhibition of Na+/H+ exchanger (130). Further evaluation is needed to elucidate whether this effect contributes to the beneficial effect of Empagliflozin on heart failure.
CGP-37157, a selective inhibitor of the mitochondrial Na+/Ca2+ exchanger, was shown to maintain mitochondrial Ca2+ and ameliorate pathological myocardial remodeling, left ventricular dysfunction and arrhythmias (63). By targeting mitochondrial electron transfer chain, a novel strategy normalizing NAD+/NADH redox balance was showed to improve myocardial energetics and cardiac function in failing mouse heart (131). NAD+ precursors may have the potential and worth to evaluate their effect on heart failure (132). Moreover, Perhexiline has been reported to improve cardiac energetics and symptom status in heart failure patients (133, 134). It is speculated that the modification of myocardial energy substrate by inhibiting CPT is the primary mechanism underlying the benefit of Perhexiline. However, this hypothesis is not supported by all available evidence. Pleiotropic or unanticipated mechanisms may be relevant to the action of Perhexiline in heart failure (135, 136).
Accumulating evidence identified that MPC abundance mediated pathological cardiac hypertrophy and overexpression of MPC protected against cardiac hypertrophy and dysfunction (44, 45), highlighting the potential of MPC in preventing cardiac remodeling and heart failure.
Conclusion
Mitochondrial dysfunction is a well-known hallmark of heart diseases, and strongly linked to the pathological development of cardiac dysfunction (17). The complicated signaling of mitochondrial quality control and related cell death offers diverse targets for inhibiting mitochondrial dysfunction. Design of therapeutic strategies targeting the signal molecules in mitochondrial dysfunction holds promise for the prevention and treatment of heart failure (Table 1).

Table 1. Therapeutic strategies targeting mitochondria for treatment of heart failure.
Author contributions
ML prepared the table and wrote the manuscript. JL and ZP drew the figure and revised the manuscript. DW and LZ contributed to the original idea of the review. XG supervised the work and provided research funding support. All authors have read and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (82170331 and U21A20337) and the Key Research and Development Plan of Zhejiang Province (2020C03017).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. (2015) 163:560–9. doi: 10.1016/j.cell.2015.10.001
2. Bravo-Sagua R, Parra V, Lopez-Crisosto C, Diaz P, Quest AF, Lavandero S. Calcium transport and signaling in mitochondria. Compr Physiol. (2017) 7:623–34. doi: 10.1002/cphy.c160013
3. Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell Dev Biol. (2020) 8:467. doi: 10.3389/fcell.2020.00467
4. Breda CNS, Davanzo GG, Basso PJ, Saraiva Camara NO, Moraes-Vieira PMM. Mitochondria as central hub of the immune system. Redox Biol. (2019) 26:101255. doi: 10.1016/j.redox.2019.101255
5. Bahat A, Gross A. Mitochondrial plasticity in cell fate regulation. J Biol Chem. (2019) 294:13852–63. doi: 10.1074/jbc.REV118.000828
6. Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. (2018) 69:62–72. doi: 10.1016/j.ceca.2017.05.003
7. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. (2011) 333:1109–12. doi: 10.1126/science.1201940
8. Miguel V, Tituana J, Herrero JI, Herrero L, Serra D, Cuevas P, et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest. (2021) 131:e140695. doi: 10.1172/JCI140695
9. Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med. (2013) 62:90–101. doi: 10.1016/j.freeradbiomed.2012.11.014
10. Dusabimana T, Kim SR, Kim HJ, Park SW, Kim H. Nobiletin ameliorates hepatic ischemia and reperfusion injury through the activation of SIRT-1/FOXO3a-mediated autophagy and mitochondrial biogenesis. Exp Mol Med. (2019) 51:1–16. doi: 10.1038/s12276-019-0245-z
11. Wang Z, White A, Wang X, Ko J, Choudhary G, Lange T, et al. mitochondrial fission mediated cigarette smoke-induced pulmonary endothelial injury. Am J Respir Cell Mol Biol. (2020) 63:637–51. doi: 10.1165/rcmb.2020-0008OC
12. Tyrrell DJ, Blin MG, Song J, Wood SC, Zhang M, Beard DA, et al. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ Res. (2020) 126:298–314. doi: 10.1161/CIRCRESAHA.119.315644
13. Guan L, Che Z, Meng X, Yu Y, Li M, Yu Z, et al. MCU Up-regulation contributes to myocardial ischemia-reperfusion Injury through calpain/OPA-1-mediated mitochondrial fusion/mitophagy Inhibition. J Cell Mol Med. (2019) 23:7830–43. doi: 10.1111/jcmm.14662
14. Chiang S, Braidy N, Maleki S, Lal S, Richardson DR, Huang ML. Mechanisms of impaired mitochondrial homeostasis and NAD(+) metabolism in a model of mitochondrial heart disease exhibiting redox active iron accumulation. Redox Biol. (2021) 46:102038. doi: 10.1016/j.redox.2021.102038
15. Wohlgemuth SE, Calvani R, Marzetti E. The interplay between autophagy and mitochondrial dysfunction in oxidative stress-induced cardiac aging and pathology. J Mol Cell Cardiol. (2014) 71:62–70. doi: 10.1016/j.yjmcc.2014.03.007
16. Zhu H, Tan Y, Du W, Li Y, Toan S, Mui D, et al. Phosphoglycerate mutase 5 exacerbates cardiac ischemia-reperfusion injury through disrupting mitochondrial quality control. Redox Biol. (2021) 38:101777. doi: 10.1016/j.redox.2020.101777
17. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. (2018) 128:3716–26. doi: 10.1172/JCI120849
18. Kumar AA, Kelly DP, Chirinos JA. Mitochondrial Dysfunction in Heart Failure With Preserved Ejection Fraction. Circulation. (2019) 139:1435–50. doi: 10.1161/CIRCULATIONAHA.118.036259
19. Song J, Herrmann JM, Becker T. Quality control of the mitochondrial proteome. Nat Rev Mol Cell Biol. (2021) 22:54–70. doi: 10.1038/s41580-020-00300-2
20. Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. (2015) 4:6–13. doi: 10.1016/j.redox.2014.11.006
21. Doblado L, Lueck C, Rey C, Samhan-Arias AK, Prieto I, Stacchiotti A, et al. Mitophagy in Human Diseases. Int J Mol Sci. (2021) 22:3903. doi: 10.3390/ijms22083903
22. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. (2012) 148:1145–59. doi: 10.1016/j.cell.2012.02.035
23. Sorrentino V, Menzies KJ, Auwerx J. Repairing mitochondrial dysfunction in disease. Annu Rev Pharmacol Toxicol. (2018) 58:353–89. doi: 10.1146/annurev-pharmtox-010716-104908
24. Chiu HY, Tay EXY, Ong DST, Taneja R. Mitochondrial dysfunction at the center of cancer therapy. Antioxid Redox Signal. (2020) 32:309–30. doi: 10.1089/ars.2019.7898
25. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
26. Glatz JFC, Nabben M, Young ME, Schulze PC, Taegtmeyer H, Luiken J. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165579. doi: 10.1016/j.bbadis.2019.165579
27. Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. (1994) 267:H742–50. doi: 10.1152/ajpheart.1994.267.2.H742
28. Chen L, Song J, Hu S. Metabolic remodeling of substrate utilization during heart failure progression. Heart Fail Rev. (2019) 24:143–54. doi: 10.1007/s10741-018-9713-0
29. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. (2014) 7:1022–31. doi: 10.1161/CIRCHEARTFAILURE.114.001469
30. Abozguia K, Clarke K, Lee L, Frenneaux M. Modification of myocardial substrate use as a therapy for heart failure. Nat Clin Pract Cardiovasc Med. (2006) 3:490–8. doi: 10.1038/ncpcardio0583
31. Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Mol Cell Biochem. (1998) 180:171–7.
32. Liang P, Ye F, Hou CC, Pi L, Chen F. Mesenchymal stem cell therapy for patients with ischemic heart failure- past, present, and future. Curr Stem Cell Res Ther. (2021) 16:608–21. doi: 10.2174/1574888X15666200309144906
33. Morrow DA, Givertz MM. Modulation of myocardial energetics: emerging evidence for a therapeutic target in cardiovascular disease. Circulation. (2005) 112:3218–21. doi: 10.1161/CIRCULATIONAHA.105.581819
34. Gupte AA, Hamilton DJ. Mitochondrial function in non-ischemic heart failure. Adv Exp Med Biol. (2017) 982:113–26. doi: 10.1007/978-3-319-55330-6_6
35. Amorim PA, Nguyen TD, Shingu Y, Schwarzer M, Mohr FW, Schrepper A, et al. Myocardial infarction in rats causes partial impairment in insulin response associated with reduced fatty acid oxidation and mitochondrial gene expression. J Thorac Cardiovasc Surg. (2010) 140:1160–7. doi: 10.1016/j.jtcvs.2010.08.003
36. Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol. (2014) 171:2080–90. doi: 10.1111/bph.12475
37. Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, Lin YH, Hausenloy DJ. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. (2020) 57:102884. doi: 10.1016/j.ebiom.2020.102884
38. Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. (2001) 276:2571–5. doi: 10.1074/jbc.M006825200
39. Liu LF, Qian ZH, Qin Q, Shi M, Zhang H, Tao XM, et al. Effect of melatonin on oncosis of myocardial cells in the myocardial ischemia/reperfusion injury rat and the role of the mitochondrial permeability transition pore. Genet Mol Res. (2015) 14:7481–9. doi: 10.4238/2015.July.3.24
40. Martin MA, Gomez MA, Guillen F, Bornstein B, Campos Y, Rubio JC, et al. Myocardial carnitine and carnitine palmitoyltransferase deficiencies in patients with severe heart failure. Biochim Biophys Acta. (2000) 1502:330–6. doi: 10.1016/s0925-4439(00)00061-2
41. Tucci S, Flogel U, Hermann S, Sturm M, Schafers M, Spiekerkoetter U. Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD(-/-)) mice. Biochim Biophys Acta. (2014) 1842:677–85. doi: 10.1016/j.bbadis.2014.02.001
42. Seminotti B, Leipnitz G, Karunanidhi A, Kochersperger C, Roginskaya VY, Basu S, et al. Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum Mol Genet. (2019) 28:928–41. doi: 10.1093/hmg/ddy403
43. Mccommis KS, Kovacs A, Weinheimer CJ, Shew TM, Koves TR, Ilkayeva OR, et al. Nutritional modulation of heart failure in mitochondrial pyruvate carrier-deficient mice. Nat Metab. (2020) 2:1232–47. doi: 10.1038/s42255-020-00296-1
44. Fernandez-Caggiano M, Kamynina A, Francois AA, Prysyazhna O, Eykyn TR, Krasemann S, et al. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat Metab. (2020) 2:1223–31. doi: 10.1038/s42255-020-00276-5
45. Cluntun AA, Badolia R, Lettlova S, Parnell KM, Shankar TS, Diakos NA, et al. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab. (2021) 33:629–48.e10. doi: 10.1016/j.cmet.2020.12.003
46. Zhang Y, Taufalele PV, Cochran JD, Robillard-Frayne I, Marx JM, Soto J, et al. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat Metab. (2020) 2:1248–64. doi: 10.1038/s42255-020-00288-1
47. Mori J, Basu R, Mclean BA, Das SK, Zhang L, Patel VB, et al. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ Heart Fail. (2012) 5:493–503. doi: 10.1161/CIRCHEARTFAILURE.112.966705
48. Atherton HJ, Dodd MS, Heather LC, Schroeder MA, Griffin JL, Radda GK, et al. Role of pyruvate dehydrogenase inhibition in the development of hypertrophy in the hyperthyroid rat heart: a combined magnetic resonance imaging and hyperpolarized magnetic resonance spectroscopy study. Circulation. (2011) 123:2552–61. doi: 10.1161/CIRCULATIONAHA.110.011387
49. Nakai G, Shimura D, Uesugi K, Kajimura I, Jiao Q, Kusakari Y, et al. Pyruvate dehydrogenase activation precedes the down-regulation of fatty acid oxidation in monocrotaline-induced myocardial toxicity in mice. Heart Vessels. (2019) 34:545–55. doi: 10.1007/s00380-018-1293-3
50. Sheeran FL, Angerosa J, Liaw NY, Cheung MM, Pepe S. Adaptations in protein expression and regulated activity of pyruvate dehydrogenase multienzyme complex in human systolic heart failure. Oxid Med Cell Longev. (2019) 2019:4532592. doi: 10.1155/2019/4532592
51. Eisner DA, Caldwell JL, Kistamas K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. (2017) 121:181–95. doi: 10.1161/CIRCRESAHA.117.310230
52. Luo M, Anderson ME. Mechanisms of altered Ca(2)(+) handling in heart failure. Circ Res. (2013) 113:690–708. doi: 10.1161/CIRCRESAHA.113.301651
53. Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci USA. (2006) 103:511–8. doi: 10.1073/pnas.0510113103
54. Lehnart SE, Wehrens XH, Marks AR. Defective ryanodine receptor interdomain interactions may contribute to intracellular Ca2+ leak: a novel therapeutic target in heart failure. Circulation. (2005) 111:3342–6. doi: 10.1161/CIRCULATIONAHA.105.551861
55. Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci USA. (2015) 112:11389–94. doi: 10.1073/pnas.1513047112
56. Pathak T, Trebak M. Mitochondrial Ca(2+) signaling. Pharmacol Ther. (2018) 192:112–23. doi: 10.1016/j.pharmthera.2018.07.001
57. Perocchi F, Gohil VM, Girgis HS, Bao XR, Mccombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. (2010) 467:291–6. doi: 10.1038/nature09358
58. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. (2011) 476:341–5. doi: 10.1038/nature10234
59. Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, et al. Mitochondrial Ca(2+) uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell. (2017) 65:1014–28.e7. doi: 10.1016/j.molcel.2017.01.032
60. Zhang M, Wang G, Peng T. Calpain-mediated mitochondrial damage: an emerging mechanism contributing to cardiac disease. Cells. (2021) 10:2024. doi: 10.3390/cells10082024
61. Chen Q, Thompson J, Hu Y, Dean J, Lesnefsky EJ. Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion. Am J Physiol Cell Physiol. (2019) 317:C910–21. doi: 10.1152/ajpcell.00190.2019
62. Liu T, O’rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. (2008) 103:279–88. doi: 10.1161/CIRCRESAHA.108.175919
63. Liu T, Takimoto E, Dimaano VL, Demazumder D, Kettlewell S, Smith G, et al. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res. (2014) 115:44–54. doi: 10.1161/CIRCRESAHA.115.303062
64. Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med. (2019) 44:3–15. doi: 10.3892/ijmm.2019.4188
65. Zhang L, Wang X, Cueto R, Effi C, Zhang Y, Tan H, et al. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. (2019) 26:101284. doi: 10.1016/j.redox.2019.101284
66. Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. (2011) 108:837–46. doi: 10.1161/CIRCRESAHA.110.232306
67. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. (2014) 515:431–5. doi: 10.1038/nature13909
68. Dey S, Demazumder D, Sidor A, Foster DB, O’rourke B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ Res. (2018) 123:356–71. doi: 10.1161/CIRCRESAHA.118.312708
69. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301:H2181–90. doi: 10.1152/ajpheart.00554.2011
70. Moe GW, Marin-Garcia J. Role of cell death in the progression of heart failure. Heart Fail Rev. (2016) 21:157–67. doi: 10.1007/s10741-016-9532-0
71. Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. (2020) 34:101523. doi: 10.1016/j.redox.2020.101523
72. Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. (2010) 1797:897–906. doi: 10.1016/j.bbabio.2010.01.032
73. Zhang Y, Murugesan P, Huang K, Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol. (2020) 17:170–94. doi: 10.1038/s41569-019-0260-8
74. Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. (2008) 134:112–23. doi: 10.1016/j.cell.2008.06.016
75. Jovaisaite V, Mouchiroud L, Auwerx J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol. (2014) 217:137–43. doi: 10.1242/jeb.090738
76. Zhu L, Zhou Q, He L, Chen L. Mitochondrial unfolded protein response: an emerging pathway in human diseases. Free Radic Biol Med. (2021) 163:125–34. doi: 10.1016/j.freeradbiomed.2020.12.013
77. Svagusa T, Martinic M, Kovacevic L, Sepac A, Milicic D, Bulum J, et al. Mitochondrial unfolded protein response, mitophagy and other mitochondrial quality control mechanisms in heart disease and aged heart. Croat Med J. (2020) 61:126–38. doi: 10.3325/cmj.2020.61.126
78. Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell. (2015) 58:123–33. doi: 10.1016/j.molcel.2015.02.008
79. Venkatesh S, Li M, Saito T, Tong M, Rashed E, Mareedu S, et al. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J Mol Cell Cardiol. (2019) 128:38–50. doi: 10.1016/j.yjmcc.2018.12.017
80. Nandi SS, Katsurada K, Mahata SK, Patel KP. Neurogenic hypertension mediated mitochondrial abnormality leads to cardiomyopathy: contribution of UPR(mt) and norepinephrine-miR- 18a-5p-HIF-1alpha axis. Front Physiol. (2021) 12:718982. doi: 10.3389/fphys.2021.718982
81. Severino P, D’amato A, Prosperi S, Fanisio F, Birtolo LI, Costi B, et al. Myocardial tissue characterization in heart failure with preserved ejection fraction: from histopathology and cardiac magnetic resonance findings to therapeutic targets. Int J Mol Sci. (2021) 22:7650. doi: 10.3390/ijms22147650
82. Smyrnias I, Gray SP, Okonko DO, Sawyer G, Zoccarato A, Catibog N, et al. Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J Am Coll Cardiol. (2019) 73:1795–806. doi: 10.1016/j.jacc.2018.12.087
83. Xu M, Xue RQ, Lu Y, Yong SY, Wu Q, Cui YL, et al. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc Res. (2019) 115:530–45. doi: 10.1093/cvr/cvy217
84. Wang YT, Lim Y, Mccall MN, Huang KT, Haynes CM, Nehrke K, et al. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am J Physiol Heart Circ Physiol. (2019) 317:H472–8. doi: 10.1152/ajpheart.00244.2019
85. Zhang B, Tan Y, Zhang Z, Feng P, Ding W, Wang Q, et al. Novel PGC-1alpha/ATF5 axis partly activates UPR(mt) and mediates cardioprotective role of tetrahydrocurcumin in pathological cardiac hypertrophy. Oxid Med Cell Longev. (2020) 2020:9187065. doi: 10.1155/2020/9187065
86. Rainbolt TK, Saunders JM, Wiseman RL. YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep. (2015) 16:97–106. doi: 10.15252/embr.201438976
87. Rainbolt TK, Lebeau J, Puchades C, Wiseman RL. Reciprocal degradation of YME1L and OMA1 adapts mitochondrial proteolytic activity during stress. Cell Rep. (2016) 14:2041–9. doi: 10.1016/j.celrep.2016.02.011
88. Guo Y, Wang Z, Qin X, Xu J, Hou Z, Yang H, et al. Enhancing fatty acid utilization ameliorates mitochondrial fragmentation and cardiac dysfunction via rebalancing optic atrophy 1 processing in the failing heart. Cardiovasc Res. (2018) 114:979–91. doi: 10.1093/cvr/cvy052
89. Acin-Perez R, Lechuga-Vieco AV, Del Mar Munoz M, Nieto-Arellano R, Torroja C, Sanchez-Cabo F. Ablation of the stress protease OMA1 protects against heart failure in mice. Sci Transl Med. (2018) 10:eaan4935. doi: 10.1126/scitranslmed.aan4935
90. Lin L, Kim SC, Wang Y, Gupta S, Davis B, Simon SI, et al. HSP60 in heart failure: abnormal distribution and role in cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. (2007) 293:H2238–47. doi: 10.1152/ajpheart.00740.2007
91. Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, et al. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep. (2016) 17:953–64. doi: 10.15252/embr.201642077
92. Cao S, Zeng Z, Wang X, Bin J, Xu D, Liao Y. Pravastatin slows the progression of heart failure by inhibiting the c-Jun N-terminal kinase-mediated intrinsic apoptotic signaling pathway. Mol Med Rep. (2013) 8:1163–8. doi: 10.3892/mmr.2013.1622
93. Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. (2016) 2:e84897. doi: 10.1172/jci.insight.84897
94. Zhang X, Ji R, Liao X, Castillero E, Kennel PJ, Brunjes DL, et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation. (2018) 137:2052–67. doi: 10.1161/CIRCULATIONAHA.117.030486
95. Chen T, Liu J, Li N, Wang S, Liu H, Li J, et al. Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PLoS One. (2015) 10:e0118909. doi: 10.1371/journal.pone.0118909
96. Marin-Garcia J, Akhmedov AT. Mitochondrial dynamics and cell death in heart failure. Heart Fail Rev. (2016) 21:123–36. doi: 10.1007/s10741-016-9530-2
97. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. (2003) 160:189–200. doi: 10.1083/jcb.200211046
98. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. (2007) 282:11521–9. doi: 10.1074/jbc.M607279200
99. Dorn GW II, Maack C. SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol. (2013) 55:42–9. doi: 10.1016/j.yjmcc.2012.07.015
100. Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E, Luo N, et al. PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med. (2013) 187:865–78. doi: 10.1164/rccm.201209-1687OC
101. Hu L, Ding M, Tang D, Gao E, Li C, Wang K, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics. (2019) 9:3687–706. doi: 10.7150/thno.33684
102. Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. (2012) 110:1484–97. doi: 10.1161/CIRCRESAHA.111.263848
103. Hu Q, Zhang H, Gutierrez Cortes N, Wu D, Wang P, Zhang J, et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res. (2020) 126:456–70. doi: 10.1161/CIRCRESAHA.119.315252
104. Shires SE, Gustafsson AB. Mitophagy and heart failure. J Mol Med. (2015) 93:253–62. doi: 10.1007/s00109-015-1254-6
105. Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. (2013) 4:2308. doi: 10.1038/ncomms3308
106. Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. (2013) 288:915–26. doi: 10.1074/jbc.M112.411363
107. Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, et al. AMPKalpha2 protects against the development of heart failure by enhancing mitophagy via pink1 phosphorylation. Circ Res. (2018) 122:712–29. doi: 10.1161/CIRCRESAHA.117.312317
108. Zhang W, Siraj S, Zhang R, Chen Q. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy. (2017) 13:1080–1. doi: 10.1080/15548627.2017.1300224
109. Zhao W, Li Y, Jia L, Pan L, Li H, Du J. Atg5 deficiency-mediated mitophagy aggravates cardiac inflammation and injury in response to angiotensin II. Free Radic Biol Med. (2014) 69:108–15. doi: 10.1016/j.freeradbiomed.2014.01.002
110. Kanamori H, Takemura G, Goto K, Maruyama R, Ono K, Nagao K, et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am J Physiol Heart Circ Physiol. (2011) 300:H2261–71. doi: 10.1152/ajpheart.01056.2010
111. Wang Y, Jasper H, Toan S, Muid D, Chang X, Zhou H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. (2021) 45:102049. doi: 10.1016/j.redox.2021.102049
112. Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. (2010) 6:600–6. doi: 10.4161/auto.6.5.11947
113. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Alternative mitophagy protects the heart against obesity-associated cardiomyopathy. Circ Res. (2021) 129:1105–21. doi: 10.1161/CIRCRESAHA.121.319377
114. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. (2019) 124:1360–71. doi: 10.1161/CIRCRESAHA.118.314607
115. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. (2007) 13:619–24. doi: 10.1038/nm1574
116. Mcdonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2021) 42:3599–726. doi: 10.1093/eurheartj/ehab368
117. Teerlink JR, Diaz R, Felker GM, Mcmurray JJV, Metra M, Solomon SD, et al. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N Engl J Med. (2021) 384:105–16. doi: 10.1056/NEJMoa2025797
118. Armstrong PW, Pieske B, Anstrom KJ, Ezekowitz J, Hernandez AF, Butler J, et al. Vericiguat in patients with heart failure and reduced ejection fraction. N Engl J Med. (2020) 382:1883–93. doi: 10.1056/NEJMoa1915928
119. Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. (2017) 390:400–14. doi: 10.1016/S0140-6736(16)31713-5
120. Narula J, Haider N, Virmani R, Disalvo TG, Kolodgie FD, Hajjar RJ, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. (1996) 335:1182–9. doi: 10.1056/NEJM199610173351603
121. Wang K, Long B, Jiao JQ, Wang JX, Liu JP, Li Q, et al. miR-484 regulates mitochondrial network through targeting Fis1. Nat Commun. (2012) 3:781. doi: 10.1038/ncomms1770
122. Wang K, Liu CY, Zhang XJ, Feng C, Zhou LY, Zhao Y, et al. miR-361-regulated prohibitin inhibits mitochondrial fission and apoptosis and protects heart from ischemia injury. Cell Death Differ. (2015) 22:1058–68. doi: 10.1038/cdd.2014.200
123. Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. (2011) 17:71–8. doi: 10.1038/nm.2282
124. Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. (2005) 19:1088–95. doi: 10.1096/fj.05-3718com
125. Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cocheme HM, et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. (2009) 54:322–8. doi: 10.1161/HYPERTENSIONAHA.109.130351
126. Ramalingam A, Budin SB, Mohd Fauzi N, Ritchie RH, Zainalabidin S. Targeting mitochondrial reactive oxygen species-mediated oxidative stress attenuates nicotine-induced cardiac remodeling and dysfunction. Sci Rep. (2021) 11:13845. doi: 10.1038/s41598-021-93234-4
127. Al Saadi T, Assef Y, Farwati M, Turkmani K, Al-Mouakeh A, Shebli B, et al. Coenzyme Q10 for heart failure. Cochrane Database Syst Rev. (2021) 2:CD008684. doi: 10.1002/14651858.CD008684.pub3
128. Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. (2020) 383:1413–24. doi: 10.1056/NEJMoa2022190
129. Martens P, Mathieu C, Verbrugge FH. Promise of SGLT2 inhibitors in heart failure: diabetes and beyond. Curr Treat Options Cardiovasc Med. (2017) 19:23. doi: 10.1007/s11936-017-0522-x
130. Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, et al. Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia. (2017) 60:568–73. doi: 10.1007/s00125-016-4134-x
131. Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. (2016) 134:883–94. doi: 10.1161/CIRCULATIONAHA.116.022495
132. Martin AS, Abraham DM, Hershberger KA, Bhatt DP, Mao L, Cui H, et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight. (2017) 2:e93885. doi: 10.1172/jci.insight.93885
133. Abozguia K, Elliott P, Mckenna W, Phan TT, Nallur-Shivu G, Ahmed I, et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. (2010) 122:1562–9. doi: 10.1161/CIRCULATIONAHA.109.934059
134. Beadle RM, Williams LK, Kuehl M, Bowater S, Abozguia K, Leyva F, et al. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail. (2015) 3:202–11. doi: 10.1016/j.jchf.2014.09.009
135. George CH, Mitchell AN, Preece R, Bannister ML, Yousef Z. Pleiotropic mechanisms of action of perhexiline in heart failure. Expert Opin Ther Pat. (2016) 26:1049–59. doi: 10.1080/13543776.2016.1211111
Keywords: heart failure, mitochondria, calcium, reactive oxygen species, mitophagy, fusion and fission
Citation: Liu M, Lv J, Pan Z, Wang D, Zhao L and Guo X (2022) Mitochondrial dysfunction in heart failure and its therapeutic implications. Front. Cardiovasc. Med. 9:945142. doi: 10.3389/fcvm.2022.945142
Received: 16 May 2022; Accepted: 04 August 2022;
Published: 24 August 2022.
Edited by:
Salvatore Pepe, Royal Children’s Hospital, AustraliaReviewed by:
Qun Chen, Virginia Commonwealth University, United StatesShyam Nandi, University of Nebraska Medical Center, United States
Copyright © 2022 Liu, Lv, Pan, Wang, Zhao and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaogang Guo, Z3hnMjIyMjJAemp1LmVkdS5jbg==