 Naresh Ganesh1
Naresh Ganesh1 Emiel P. C. van der Vorst2,3,4
Emiel P. C. van der Vorst2,3,4 Jens Spiesshöfer5Shun He6
Jens Spiesshöfer5Shun He6 Mathias Burgmaier1
Mathias Burgmaier1 Hannes Findeisen7Michael Lehrke1Filip K. Swirski8
Hannes Findeisen7Michael Lehrke1Filip K. Swirski8 Nikolaus Marx1
Nikolaus Marx1 Florian Kahles1*
Florian Kahles1*- 1Department of Cardiology, Angiology and Intensive Care Medicine, University Hospital Aachen, Aachen, Germany
- 2Interdisciplinary Center for Clinical Research (IZKF) and Institute for Molecular Cardiovascular Research (IMCAR), University Hospital Aachen, Aachen, Germany
- 3Department of Pathology, Cardiovascular Research Institute Maastricht (CARIM), Maastricht University Medical Centre, Maastricht, Netherlands
- 4Institute for Cardiovascular Prevention (IPEK), Ludwig-Maximilians-University Munich, Munich, Germany
- 5Department of Pneumology and Intensive Care Medicine, University Hospital Aachen, Aachen, Germany
- 6School of Medicine, Southern University of Science and Technology, Shenzhen, China
- 7Department of Cardiology I—Coronary and Peripheral Vascular Disease, Heart Failure, University Hospital Münster, Münster, Germany
- 8Icahn School of Medicine at Mount Sinai, Cardiovascular Research Institute, New York, NY, United States
Despite scientific and clinical advances during the last 50 years cardiovascular disease continues to be the main cause of death worldwide. Especially patients with diabetes display a massive increased cardiovascular risk compared to patients without diabetes. Over the last two decades we have learned that cardiometabolic and cardiovascular diseases are driven by inflammation. Despite the fact that the gastrointestinal tract is one of the largest leukocyte reservoirs of our bodies, the relevance of gut immune cells for cardiovascular disease is largely unknown. First experimental evidence suggests an important relevance of immune cells in the intestinal tract for the development of metabolic and cardiovascular disease in mice. Mice specifically lacking gut immune cells are protected against obesity, diabetes, hypertension and atherosclerosis. Importantly antibody mediated inhibition of leukocyte homing into the gut showed similar protective metabolic and cardiovascular effects. Targeting gut immune cells might open novel therapeutic approaches for the treatment of cardiometabolic and cardiovascular diseases.
Introduction
Scientific discoveries, advances in public health and innovations in medical care (including modern heart failure, lipid-lowering, and antiplatelet therapies) have contributed to a steady decline of mortality from cardiovascular disease in the last 50 years. Despite these improvements, cardiovascular diseases (such as coronary artery disease, myocardial infarction, or heart failure) continue to be the main cause of death worldwide (1, 2). Especially patients with diabetes are more vulnerable compared to those without diabetes and face a very high risk to die from cardiovascular disease (3). Recent evidence showed that sodium glucose linked transporter 2 (SGLT-2) inhibitors (4) and glucagon-like peptide 1 (GLP-1) receptor agonists (5) improved cardiovascular prognosis of patients with diabetes and high cardiovascular risk. Importantly, SGLT-2 inhibitors reduced heart failure endpoints in patients with or without diabetes (6, 7). Thus, SGLT-2 inhibitors emerged as first-line heart failure therapy in current guidelines (8). Despite these recent advances in heart failure and diabetes therapy there is still an urgent unmet clinical need to further reduce cardiovascular morbidity and mortality. Novel therapeutic approaches especially targeting atherosclerosis development and plaque stability must be identified and translated into promising drug targets. Targeting the immune system might be a novel therapeutical approach to protect against cardiovascular disease and its life-threatening complications.
The role of inflammation in cardiovascular disease
Data from the last two decades have shown that leukocytes and inflammatory processes are important players during the development of cardiovascular diseases such as atherosclerosis, myocardial infarction, and heart failure (9–11). Elegant experimental studies have identified inflammatory organ cross talk networks between the heart, the bone marrow, the spleen, and the sympathetic nervous system which could explain how immune cells control and accelerate the development and progression of cardiovascular disease. These observations suggest that after myocardial infarction enhanced sympathetic nervous activity releases noradrenalin in the bone marrow niche, which results in increased hematopoietic stem cell (HSC) activity and emigration to extramedullary sites. Increased production of myeloid immune cells (myelopoiesis) in the bone marrow results in higher numbers of circulating myeloid immune cells that are recruited to atherosclerotic plaques in higher numbers, accelerating plaque growth and inflammation and decreasing plaque stability thus increasing the risk of re-infarction (12–14). This feedback loop and vicious cycle might be one of the explanations why patients after a myocardial infarction are at increased risk to experience re-infarction.
Immune cells play a crucial role in atherosclerosis formation and subsequent plaque growth. Following intimal lipid accumulation and endothelial dysfunction monocytes migrate into the vessel wall [reviewed in Swirski and Nahrendorf (12)]. Newly-infiltrated monocytes differentiate into macrophages which recognize and ingest lipids that have accumulated in the intima as a consequence of hypercholesterolemia. After lipid ingestion macrophages become lipid- rich “foam cells,” thus activating various inflammatory pathways leading to activation of other immune (including T and B lymphocytes, neutrophils, and dendritic cells) and non-immune cells [including endothelial cells, platelets, smooth muscle cells; reviewed in Swirski and Nahrendorf (12)].
In line with these findings circulating leukocyte numbers and biomarkers of inflammation such as high-sensitivity C-reactive protein and interleukin-6 are associated with an increased risk of cardiovascular events in humans (15–17). In order to prove the inflammatory hypothesis of atherothrombosis several large cardiovascular outcome trials validating drugs which target inflammatory pathways have been conducted. In the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) canakinumab, a therapeutic monoclonal antibody targeting interleukin-1β, showed a 15% risk reduction of non-fatal myocardial infarction (MI), non-fatal stroke, or cardiovascular mortality in patients with previous myocardial infarction and hs-CRP above 2 mg/L (18). In this trial canakinumab was associated with a higher incidence of fatal infection compared to placebo. While this study for the first time demonstrated that an anti-inflammatory drug is able to improve cardiovascular outcomes, the Food and Drug Administration (FDA) rejected clinical approval of canakinumab for cardiovascular disease most likely due to safety concerns and only modest outcome effects. Colchicine, an NACHT, LRR, and PYD domains-containing protein 3 (NLRP3)-inflammasome inhibitor, was investigated in patients with a recent myocardial infarction [Colchicine Cardiovascular Outcomes Trial—COLCOT (19)] and in patients with chronic coronary disease [In the Low-Dose Colchicine 2—LoDoCo2 trial (20)]. In both studies low-dose colchicine significantly reduced cardiovascular events. However, the incidence of death from non-cardiovascular causes was numerically higher in the colchicine group than in the placebo group which requires further investigations. While these studies proved the inflammatory hypothesis of cardiovascular disease, interleukin-1β (canakinumab) or the NLRP3-inflammasome (colchicine) might not be the optimal drug candidates for this purpose. Thus, alternative therapeutical targets modulating inflammation need to be identified to improve prognosis in patients with cardiovascular disease.
Gut immune cells and cardiovascular disease
The gut microbiome and intestinal barrier dysfunction (“leaky gut”) have emerged as potential contributors to the development of cardiovascular disease and are currently intensively studied (21–23). Despite the fact that the gastrointestinal tract (GI tract) is one of the largest leukocyte reservoirs of our bodies containing very high numbers of T lymphocytes, plasma cells, eosinophils, and macrophages (24–26), the relevance of gut immune cells for cardiovascular disease is largely unknown. Integrin β7 (Itgb7) is expressed on circulating leukocytes and mediates immune cell migration selectively into the gut without affecting relevant chemotaxis into other tissues (27–31). Thus, Itgb7−/− (hereafter β7−/− mice) display a useful tool to study selective gut immune cell deficiency, since these mice show strongly reduced leukocyte numbers (especially intraepithelial lymphocytes: αβ and γδ T cells, B cells, and myeloid cells) in the gut. A recently published study investigated whether gut immune cells play a role in atherosclerosis development in mice (32). β7 deficient mice on Ldlr−/− background fed a high cholesterol diet (HCD) showed no difference in weight gain, but had lower levels of plasma total cholesterol, a reduction in circulating Ly-6Chigh and Ly-6Clow monocytes and smaller aortic lesions with an ~50% reduction in plaque size and volume (32). To elucidate the question whether gut immune cells can be therapeutically targeted for cardiovascular disease the authors injected anti-integrin β7 antibodies (which block immune cell homing to the gut) into Ldlr−/− mice and found that these mice had attenuated atherosclerosis (32). These findings show that gut immune cells accelerate lesion growth while antibody mediated integrin β7 neutralization protects against atherosclerosis formation in mice.
Patients with diabetes and obesity show more severe atherosclerosis and higher cardiovascular risk than those without diabetes. Based on the promising findings on targeting intestinal leukocytes to treat atherosclerosis it is important to investigate the functional relevance of gut immune cells in cardiometabolic diseases. Several studies in obese mice and men demonstrated major changes to the intestinal immune cell landscape in the obese state compared with the lean state. During metabolic disease the gut contains increased numbers of γδ T cells, macrophages, dendritic cells, NK cells, CD8+ T cells (αβ TCR), and Th1 T cells and a reduction in Treg T cells, anti-inflammatory IgA+ antibody-secreting cells (ASCs) and eosinophils. These changes in immune compartments are associated with an inflammatory environment that is linked with intestinal barrier dysfunction, intestinal dysbiosis and a loss in bacterial diversity during diet-induced obesity (33–40). Interestingly, mice lacking gut immune cells (β7−/−, Integrin alpha E knockout—Itgae−/− or C-C chemokine receptor type 9 knockout—Ccr9−/− mice) fed a diet high in fat, sodium and sugar showed improved glucose tolerance, less weight gain, and reduced white adipose tissue inflammation (reduced numbers of Ly-6Chigh monocytes, neutrophils, and macrophages) (32, 35).
In contrast to wild-type control mice, β7−/− mice were also protected from hypertension (a key feature of the metabolic syndrome), indicating that gut immune cells aggravate adverse cardiometabolic consequences of high-fat diet (32). Mechanistically β7−/− mice on chow diet showed improved glucose tolerance by increased insulin secretion, expended more energy, produced more heat and had lower levels of fasting triglycerides without differences in hepatic secretion of triglycerides or fat absorption (32). These results suggest metabolic rate to be increased in the absence of gut immune cells. While diet-induced obesity is linked with intestinal barrier dysfunction and a loss in bacterial diversity (33–40), the beneficial metabolic effects in β7−/− mice were resistant to antibiotic treatment and not associated with gut permeability abnormalities (32). These findings support a direct effect of gut immune cells on metabolic and cardiovascular disease independent of gut barrier integrity or microbiome changes (Figure 1).
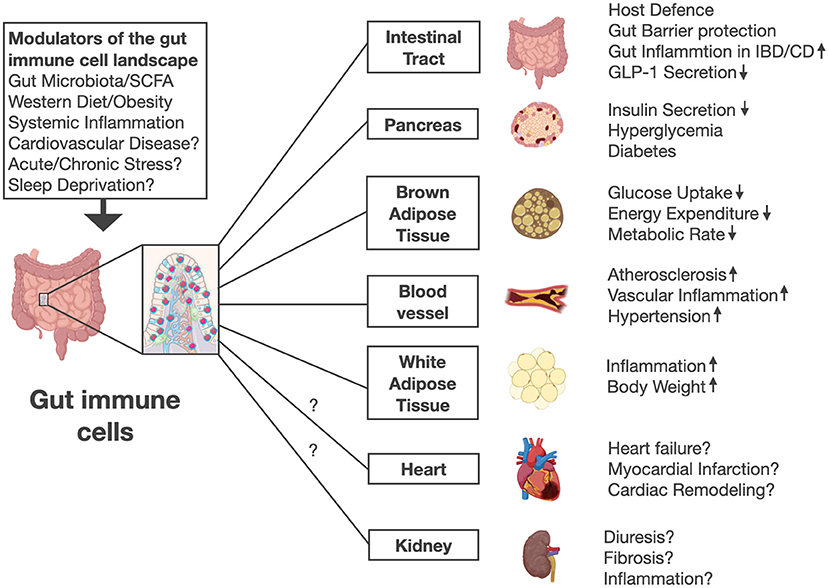
Figure 1. Gut immune cells are modulated by gut microbiota, short chain fatty acids (SCFA), diet and systemic inflammation in mice. Future work is needed to investigate whether other stimuli and lifestyle factors including stress or sleep disorders affect gut immune cell numbers and activation. Intestinal immune cells mediate pleiotropic effects in various organs beyond their role in host defense and gut barrier protection. Mice lacking gut immune cells show increased insulin secretion, improved glucose control, higher energy expenditure, lower body weight, less high fat diet-induced adipose tissue inflammation, and increased circulating GLP-1 levels. Thus, gut immune cell deficient mice were protected against diabetes, obesity, hypertension and atherosclerosis. Future work needs to address whether gut immune cells might affect other diseases including heart failure, myocardial infarction, and kidney function. The illustration was modified from Biorender (https://Biorender.com).
GLP-1: Mechanistic link connecting gut immune cells and cardiovascular disease?
In response to food intake the gut incretin hormone GLP-1 (glucagon-like peptide-1) is secreted by intestinal L cells leading to insulin secretion and glucose control (41). Pharmacological activation of the GLP-1 receptor is currently used for the treatment of patients with type 2 diabetes (42). Beyond their glucoregulatory function GLP-1 receptor agonists exert pleiotropic protective cardiovascular effects in different organ systems (43) and improved cardiovascular outcomes in diabetic patients at high cardiovascular risk (44–49). Besides food intake GLP-1 secretion is induced by acute inflammatory stimuli including LPS, IL-6, and IL1b. This was found to be mediated by IL-6 signaling in L-cells (50, 51). Consistently mice and patients with acute [sepsis (51) or myocardial infarction (52)] or chronic inflammatory cardiovascular diseases [coronary artery disease (53) or heart failure (54)] show increased circulating GLP-1 levels independent of food intake. Elevated GLP-1 levels were independently associated with mortality in patients with sepsis or myocardial infarction (55, 56). Mechanistic experimental studies suggest upregulation of GLP-1 secretion to be an endogenous protective counter-regulatory response circuit (52). Interestingly gut immune cell deficient mice (β7−/−) which are protected from atherosclerosis, hypertension, obesity and diabetes have higher plasma GLP-1 levels (32, 35). Since gut immune cells (in particular intraepithelial αβ and γδ T lymphocytes) express high levels of the GLP-1 receptor (32, 57) it was elucidated whether these cells directly regulate GLP-1 secretion from intestinal L cells to modulate cardiometabolic disease. For this purpose the authors of a previous study generated mixed bone marrow chimeras (bmGlp1r−/−β7−/−) with selective GLP-1 receptor deficiency in β7+ gut immune cells and unaffected GLP-1 receptor expression on all other cell types. Mice with selective gut immune cell GLP-1 receptor deficiency showed higher plasma levels of GLP-1, were more glucose tolerant, presented with less hypercholesterolemia and developed smaller atherosclerotic plaques with fewer aortic leukocytes (32). Further mechanistic experiments identified a negative feedback mechanism in which gut immune cell GLP-1 receptor activation was found to directly inhibit L cell derived GLP-1 secretion through a still unknown gut immune cell derived mediator (32). These findings suggest that GLP-1 receptor deficiency in gut immune cells limits development of cardiovascular and cardiometabolic disease through upregulation of systemic GLP-1 availability (Figure 2). In other words GLP-1 might be one of the underlying mechanistic links connecting gut immune cells and cardiovascular disease.
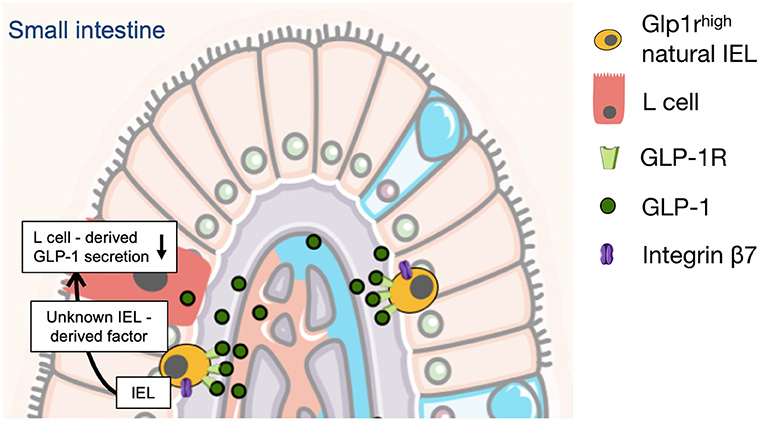
Figure 2. L-cell derived GLP-1 secretion is controlled and limited by gut intraepithelial (IEL) lymphocytes in mice. This is mediated through a negative feedback mechanism in which gut IEL GLP-1 receptor activation directly inhibits L cell derived GLP-1 secretion through a still unknown IEL-derived mediator (32). The illustration was modified from Servier Medical Art (https://smart.servier.com/).
Conclusion and future perspectives
Cardiometabolic diseases are characterized and driven by systemic inflammation. Recent data indicate that in cardiovascular and metabolic diseases inflammation is not limited to the vessel wall, bone marrow, spleen or adipose tissue. First experimental evidence suggests an important relevance of immune cells in the intestinal tract for the development of metabolic and cardiovascular disease in mice. Mice specifically lacking gut immune cells are protected against obesity, diabetes, hypertension and atherosclerosis. Importantly anti-Integrin-β7 antibody mediated inhibition of leukocyte homing into the gut showed similar protective metabolic and cardiovascular effects as genetic models. Based on these observations targeting gut immune cells and in particular Integrin-β7 might open novel therapeutic approaches for the treatment of cardiometabolic and cardiovascular disease. Therapeutic targeting of gut immune cell trafficking is well-established and safe in patients suffering from inflammatory bowel diseases (IBDs), including Crohn's disease (CD), and ulcerative colitis (UC). Blocking intestinal immune cell homing by Vedolizumab, an anti-α4β7 antibody, has become an important pillar of IBD therapy (58–61). Therefore, it is tempting to speculate that antibody-mediated blockage of intestinal leukocyte homing might be a promising drug target for patients with cardiovascular disease. Especially the recent identification of a previously unrecognized crosstalk between gut immune cells and the incretin system (increased GLP-1 secretion in the absence of gut immune cells) might open new therapeutic avenues for vulnerable high-risk patients with diabetes and cardiovascular disease. However, several key questions remain: Can we translate the experimental observations on gut immune cells and cardiovascular and cardiometabolic effects to patients? Is intestinal immune cell trafficking and activation relevant for human cardiovascular disease? How exactly do gut leukocytes accelerate atherosclerosis formation, hypertension, and diabetes? Is GLP-1 the only mechanistic link between gut immune cells and cardiovascular actions or do other incretin hormones such as GIP (glucose-dependent insulinotropic peptide) or GLP-2 or any other as of yet unknown factors play a role? Does gut immune cell trafficking or activation affect plaque inflammation by increasing circulating monocytes and neutrophils (due to upregulated hematopoiesis)? Do gut immune cells control local macrophage proliferation in atherosclerotic lesions? Beyond their role in atherogenesis what is the effect of blocking intestinal leukocyte homing on other manifestations of cardiovascular disease like cardiac remodeling after myocardial infarction or heart failure? What is the effect of cardiovascular events (i.e., myocardial infarction, stroke, or decompensated heart failure) on intestinal immune cell homing and activation? Are gut immune cell numbers and or activation status associated with cardiovascular risk in patients?
Answering these questions may be essential to better understand how intestinal leukocytes control development of cardiometabolic and cardiovascular disease and to evaluate whether the above mentioned promising experimental observations could the translated into clinical practice. Targeting gut immune cells might be a future therapeutical approach to prevent and suppress the devastating consequences of cardiovascular disease.
Author contributions
FK and NG drafted the manuscript, which was critically revised by all authors. All authors provided intellectual input and gave final approval.
Funding
This study was supported by grants from the European Research Area Network on Cardiovascular Diseases (ERA-CVD and BMBF, Grant No. JTC-2019, MyPenPath-01KL2004) and from The European Foundation for the Study of Diabetes (EFSD)/Novo Nordisk Foundation (NNF2OSA0066111) to FK and grants from the Deutsche Forschungsgemeinschaft (SFB TRR 219 M-03) and Interreg V-A grant EURlipids to ML. NM was supported by grants from the Deutsche Forschungsgemeinschaft (German Research Foundation; TRR 219; Project-ID 322900939 [M03, M05]) as well as the CORONA Stiftung, Germany. EV was supported by a grant from the Interdisciplinary Center for Clinical Research within the faculty of Medicine at the RWTH Aachen University and NWO-ZonMw Veni (91619053).
Conflict of interest
Author FK served as a consultant to Bayer and Novo Nordisk and served as a speaker for Novo Nordisk. Author NM has received support for clinical trial leadership from Boehringer Ingelheim, Novo Nordisk, served as a consultant to Bayer, Boehringer Ingelheim, Merck, Novo Nordisk, AstraZeneca, BMS, received grant support from Boehringer Ingelheim, Merck, Novo Nordisk, and served as a speaker for Bayer, Boehringer Ingelheim, Merck, Novo Nordisk, Lilly, BMS, and AstraZeneca. Author ML received grants and personal fees from Boehringer Ingelheim, MSD, Novo Nordisk and personal fees from Amgen, Sanofi, Astra Zeneca, Bayer, Lilly, Daiichi Sankyo, Novarits, Amarin.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. (2012) 366:54–63. doi: 10.1056/NEJMra1112570
2. GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. (2015) 385:117–71. doi: 10.1016/S0140-6736(14)61682-2
3. Rawshani A, Rawshani A, Franzen S, Eliasson B, Svensson AM, Miftaraj M, et al. Mortality and cardiovascular disease in type 1 and type 2 diabetes. N Engl J Med. (2017) 376:1407–18. doi: 10.1056/NEJMoa1608664
4. Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. (2019) 393:31–9. doi: 10.1016/S0140-6736(18)32590-X
5. Sattar NM, Lee MY, Kristensen SL, Branch RH, Del Prato S, Khurmi NS, et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. (2021) 9:653–62. doi: 10.1016/S2213-8587(21)00203-5
6. Zannad F, Ferreira JP, Pocock SJ, Anker SD, Butler J, Filippatos G, et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet. (2020) 396:819–29. doi: 10.1016/S0140-6736(20)31824-9
7. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Bohm MHP, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. (2021) 385:1451–61. doi: 10.1056/NEJMoa2107038
8. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2021) 42:3599–726. doi: 10.1093/eurheartj/ehab368
9. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
10. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. (2005) 352:1685–95. doi: 10.1056/NEJMra043430
11. Libby P, Ridker PM, Hansson GK, Leducq A. Transatlantic network on, inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. (2009) 54:2129–38. doi: 10.1016/j.jacc.2009.09.009
12. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. (2013) 339:161–6. doi: 10.1126/science.1230719
13. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. (2012) 487:325–9. doi: 10.1038/nature11260
14. Schloss MJ, Hulsmans M, Rohde D, Lee IH, Severe N, Foy BH, et al. B lymphocyte-derived acetylcholine limits steady-state and emergency hematopoiesis. Nat Immunol. (2022) 23:605–18. doi: 10.1038/s41590-022-01165-7
15. Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. (2000) 342:836–43. doi: 10.1056/NEJM200003233421202
16. Sabatine MS, Morrow DA, Cannon CP, Murphy SA, Demopoulos LA, DiBattiste PM, et al. Relationship between baseline white blood cell count and degree of coronary artery disease and mortality in patients with acute coronary syndromes: a TACTICS-TIMI 18 (Treat angina with aggrastat and determine cost of therapy with an invasive or conservative strategy- thrombolysis in myocardial infarction 18 trial) substudy. J Am Coll Cardiol. (2002) 40:1761–8. doi: 10.1016/S0735-1097(02)02484-1
17. Ernst E, Hammerschmidt DE, Bagge U, Matrai A, Dormandy JA. Leukocytes and the risk of ischemic diseases. JAMA. (1987) 257:2318–24. doi: 10.1001/jama.257.17.2318
18. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
19. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381:2497–505. doi: 10.1056/NEJMoa1912388
20. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. (2020) 383:1838–47. doi: 10.1056/NEJMoa2021372
21. Lewis CV, Taylor WR. Intestinal barrier dysfunction as a therapeutic target for cardiovascular disease. Am J Physiol Heart Circ Physiol. (2020) 319:H1227–33. doi: 10.1152/ajpheart.00612.2020
22. Yoo JY, Sniffen S, McGill Percy KC, Pallaval VB, Chidipi B. Gut dysbiosis and immune system in atherosclerotic cardiovascular disease (ACVD). Microorganisms. (2022) 10:108. doi: 10.3390/microorganisms10010108
23. Yamashita T, Kasahara K, Emoto T, Matsumoto T, Mizoguchi T, Kitano N, et al. Intestinal immunity and gut microbiota as therapeutic targets for preventing atherosclerotic cardiovascular diseases. Circ J. (2015) 79:1882–90. doi: 10.1253/circj.CJ-15-0526
24. Mowat AM, Viney JL. The anatomical basis of intestinal immunity. Immunol Rev. (1997) 156:145–66. doi: 10.1111/j.1600-065X.1997.tb00966.x
25. Lee SH, Starkey PM, Gordon S. Quantitative analysis of total macrophage content in adult mouse tissues. Immunochemical studies with monoclonal antibody F4/80. J Exp Med. (1985) 161:475–89. doi: 10.1084/jem.161.3.475
26. Cerf-Bensussan N, Guy-Grand D. Intestinal intraepithelial lymphocytes. Gastroenterol Clin North Am. (1991) 20:549–76. doi: 10.1016/S0889-8553(21)00570-7
27. Parker CM, Cepek KL, Russell GJ, Shaw SK, Posnett DN, Schwarting R, et al. A family of beta 7 integrins on human mucosal lymphocytes. Proc Natl Acad Sci USA. (1992) 89:1924–8. doi: 10.1073/pnas.89.5.1924
28. Holzmann B, Weissman IL. Peyer's patch-specific lymphocyte homing receptors consist of a VLA-4-like alpha chain associated with either of two integrin beta chains, one of which is novel. EMBO J. (1989) 8:1735–41. doi: 10.1002/j.1460-2075.1989.tb03566.x
29. Holzmann B, McIntyre BW, Weissman IL. Identification of a murine Peyer's patch–specific lymphocyte homing receptor as an integrin molecule with an alpha chain homologous to human VLA-4 alpha. Cell. (1989) 56:37–46. doi: 10.1016/0092-8674(89)90981-1
30. Gorfu G, Rivera-Nieves J, Ley K. Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr Mol Med. (2009) 9:836–50. doi: 10.2174/156652409789105525
31. Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat Rev Immunol. (2011) 11:445–56. doi: 10.1038/nri3007
32. He S, Kahles F, Rattik S, Nairz M, McAlpine CS, Anzai A, et al. Gut intraepithelial T cells calibrate metabolism and accelerate cardiovascular disease. Nature. (2019) 566:115–9. doi: 10.1038/s41586-018-0849-9
33. Monteiro-Sepulveda M, Touch S, Mendes-Sa C, Andre S, Poitou C, Allatif O, et al. Jejunal T cell inflammation in human obesity correlates with decreased enterocyte insulin signaling. Cell Metab. (2015) 22:113–24. doi: 10.1016/j.cmet.2015.05.020
34. Magalhaes I, Pingris K, Poitou C, Bessoles S, Venteclef N, Kiaf B, et al. Mucosal-associated invariant T cell alterations in obese and type 2 diabetic patients. J Clin Invest. (2015) 125:1752–62. doi: 10.1172/JCI78941
35. Luck H, Tsai S, Chung J, Clemente-Casares X, Ghazarian M, Revelo XS, et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab. (2015) 21:527–42. doi: 10.1016/j.cmet.2015.03.001
36. Johnson AM, Costanzo A, Gareau MG, Armando AM, Quehenberger O, Jameson JM, et al. High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS ONE. (2015) 10:e0122195. doi: 10.1371/journal.pone.0122195
37. Garidou L, Pomie C, Klopp P, Waget A, Charpentier J, Aloulou M, et al. The gut microbiota regulates intestinal CD4 T cells expressing RORgammat and controls metabolic disease. Cell Metab. (2015) 22:100–12. doi: 10.1016/j.cmet.2015.06.001
38. Winer DA, Winer S, Dranse HJ, Lam TK. Immunologic impact of the intestine in metabolic disease. J Clin Invest. (2017) 127:33–42. doi: 10.1172/JCI88879
39. Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S, et al. Gut-associated IgA(+) immune cells regulate obesity-related insulin resistance. Nat Commun. (2019) 10:3650. doi: 10.1038/s41467-019-11370-y
40. Khan S, Luck H, Winer S, Winer DA. Emerging concepts in intestinal immune control of obesity-related metabolic disease. Nat Commun. (2021) 12:2598. doi: 10.1038/s41467-021-22727-7
41. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
42. Lehrke M, Marx N. New antidiabetic therapies: innovative strategies for an old problem. Curr Opin Lipidol. (2012) 23:569–75. doi: 10.1097/MOL.0b013e328359b19f
43. Drucker DJ. The cardiovascular biology of glucagon-like peptide-1. Cell Metab. (2016) 24:15–30. doi: 10.1016/j.cmet.2016.06.009
44. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. (2016) 375:1834–44. doi: 10.1056/NEJMoa1607141
45. Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2016) 375:311–22. doi: 10.1056/NEJMoa1603827
46. Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. (2019) 394:121–30. doi: 10.1016/S0140-6736(19)31149-3
47. Hernandez AF, Green JB, Janmohamed S, D'Agostino RB, Granger CB, Jones NP, et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. (2018) 392:1519–29. doi: 10.1016/S0140-6736(18)32261-X
48. Gerstein HC, Sattar N, Rosenstock J, Ramasundarahettige C, Pratley R, Lopes RD, et al. Cardiovascular and renal outcomes with efpeglenatide in type 2 diabetes. N Engl J Med. (2021) 385:896–907. doi: 10.1056/NEJMoa2108269
49. Husain M, Birkenfeld AL, Donsmark M, Dungan K, Eliaschewitz FG, Franco DR, et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. (2019) 381:841–51. doi: 10.1056/NEJMoa1901118
50. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med. (2011) 17:1481–9. doi: 10.1038/nm.2513
51. Kahles F, Meyer C, Mollmann J, Diebold S, Findeisen HM, Lebherz C, et al. GLP-1 secretion is increased by inflammatory stimuli in an IL-6-dependent manner, leading to hyperinsulinemia and blood glucose lowering. Diabetes. (2014) 63:3221–9. doi: 10.2337/db14-0100
52. Diebold S, Moellmann J, Kahles F, Haj-Yehia E, Liehn EA, Nickel A, et al. Myocardial infarction is sufficient to increase GLP-1 secretion, leading to improved left ventricular contractility and mitochondrial respiratory capacity. Diabetes Obes Metab. (2018) 20:2911–8. doi: 10.1111/dom.13472
53. Piotrowski K, Becker M, Zugwurst J, Biller-Friedmann I, Spoettl G, Greif M, et al. Circulating concentrations of GLP-1 are associated with coronary atherosclerosis in humans. Cardiovasc Diabetol. (2013) 12:117. doi: 10.1186/1475-2840-12-117
54. Hattori A, Kawamura I, Yamada Y, Kanamori H, Aoyama T, Ushikoshi H, et al. Elevated plasma GLP-1 levels and enhanced expression of cardiac GLP-1 receptors as markers of left ventricular systolic dysfunction: a cross-sectional study. BMJ Open. (2013) 3:e003201. doi: 10.1136/bmjopen-2013-003201
55. Lebherz C, Schlieper G, Mollmann J, Kahles F, Schwarz M, Brunsing J, et al. GLP-1 levels predict mortality in patients with critical illness as well as end-stage renal disease. Am J Med. (2017) 130:833–41.e3. doi: 10.1016/j.amjmed.2017.03.010
56. Kahles F, Ruckbeil MV, Mertens RW, Foldenauer AC, Arrivas MC, Moellmann J, et al. Glucagon-like peptide 1 levels predict cardiovascular risk in patients with acute myocardial infarction. Eur Heart J. (2019) 41:882–9. doi: 10.1093/eurheartj/ehz728
57. Yusta B, Baggio LL, Koehler J, Holland D, Cao X, Pinnell LJ, et al. GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R. Diabetes. (2015) 64:2537–49. doi: 10.2337/db14-1577
58. Vermeire S, Sandborn WJ, Danese S, Hebuterne X, Salzberg BA, Klopocka M, et al. Anti-MAdCAM antibody (PF-00547659) for ulcerative colitis (TURANDOT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:135–44. doi: 10.1016/S0140-6736(17)30930-3
59. Picardo S, Panaccione R. Anti-MADCAM therapy for ulcerative colitis. Expert Opin Biol Ther. (2020) 20:437–42. doi: 10.1080/14712598.2020.1691520
60. Peyrin-Biroulet L, Hart A, Bossuyt P, Long M, Allez M, Juillerat P, et al. Etrolizumab as induction and maintenance therapy for ulcerative colitis in patients previously treated with tumour necrosis factor inhibitors (HICKORY): a phase 3, randomised, controlled trial. Lancet Gastroenterol Hepatol. (2021) 7:P128–40. doi: 10.1016/S2468-1253(21)00298-3
Keywords: gut immune cells, intraepithelial lymphocytes, integrin β7, cardiovascular disease, myocardial infarction, atherosclerosis, heart failure, GLP-1
Citation: Ganesh N, van der Vorst EPC, Spiesshöfer J, He S, Burgmaier M, Findeisen H, Lehrke M, Swirski FK, Marx N and Kahles F (2022) Gut immune cells—A novel therapeutical target for cardiovascular disease? Front. Cardiovasc. Med. 9:943214. doi: 10.3389/fcvm.2022.943214
Received: 13 May 2022; Accepted: 01 August 2022;
Published: 15 August 2022.
Edited by:
Mahdi Garelnabi, University of Massachusetts Lowell, United StatesReviewed by:
Christoph E. Hagemeyer, Monash University, AustraliaParham Mardi, Alborz University of Medical Sciences, Iran
Copyright © 2022 Ganesh, van der Vorst, Spiesshöfer, He, Burgmaier, Findeisen, Lehrke, Swirski, Marx and Kahles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florian Kahles, ZmthaGxlc0B1a2FhY2hlbi5kZQ==