 Dengshen Zhang
Dengshen Zhang Yiran Cao
Yiran Cao Daxing Liu
Daxing Liu Jian Zhang1
Jian Zhang1 Yingqiang Guo
Yingqiang Guo- 1Department of Cardiovascular Surgery, Affiliated Hospital of Zunyi Medical University, Zunyi, China
- 2Department of Cardiovascular Surgery, West China Hospital, Sichuan University, Chengdu, China
Mounting evidence suggests that the phenotypic transformation of venous smooth muscle cells (SMCs) from differentiated (contractile) to dedifferentiated (proliferative and migratory) phenotypes causes excessive proliferation and further migration to the intima leading to intimal hyperplasia, which represents one of the key pathophysiological mechanisms of vein graft restenosis. In recent years, numerous miRNAs have been identified as specific phenotypic regulators of vascular SMCs (VSMCs), which play a vital role in intimal hyperplasia in vein grafts. The review sought to provide a comprehensive overview of the etiology of intimal hyperplasia, factors affecting the phenotypic transformation of VSMCs in vein graft, and molecular mechanisms of miRNAs involved in SMCs phenotypic modulation in intimal hyperplasia of vein graft reported in recent years.
Introduction
Coronary artery disease (CAD) is well-established as a significant threat to human health. With the rapid development of society and changes in lifestyle, the incidence of CAD has rapidly escalated in recent years, with the total number of CAD patients in China estimated to be more than 11 million (1). Coronary artery bypass grafting (CABG) surgery is a surgical intervention for CAD, and the grafted vessels often consist of the internal mammary artery, radial artery, and human saphenous vein (HSV). Arterial grafts are often inadequate for recanalization of CABG due to the limitations for harvesting, number and length. Therefore, HSV remains the most widely used for CABG in complex cases with multivessel disease since it is easier to manipulate. Unfortunately, the incidence of vein graft restenosis after CABG is high, with vein graft failure rates of up to 50% within 10 years of surgery, which seriously affects the long-term outcome of CABG surgery (2, 3). An increasing body of evidence suggests that intimal hyperplasia plays a major part in the pathophysiology of vein graft restenosis (4, 5). Indeed, exploring the molecular mechanisms underlying the occurrence of intimal hyperplasia in vein graft is of significant concern in CAD prevention and treatment.
Studies have shown that phenotypic transformation of VSMCs causing excessive proliferation of VSMCs and further migration to the intima is one of the primary pathological mechanisms that contribute to intimal hyperplasia in vein grafts (6–8). Therefore, there is an urgent need to identify novel molecules that can effectively control the phenotypic transformation of VSMCs. To date, miRNAs have been identified as phenotypic regulators of VSMCs with significant specificity, and most of them are endogenous small interfering RNAs with potential clinical applications.
The Phenotypes of Vascular Smooth Muscle Cells (VSMCs) and Intimal Hyperplasia in Vein Grafts
Vein graft restenosis is generally divided into 3 stages: early thrombosis, early to mid-stage intimal hyperplasia and accelerated atherosclerosis, with each of these stages having distinct properties while interweaving and overlapping to promote pathogenesis (4, 5). It is well-established that VSMCs control the vascular microenvironment and secure vascular morphology and function from the intima-media of the vessel wall. VSMCs can switch between differentiated (contractile) and dedifferentiated (synthesis) phenotypes depending on their functions. The dedifferentiated phenotype exhibits a higher proliferative, migratory and synthetic rate of extracellular matrixs (ECMs), thereby being termed the proliferative-migratory or synthetic phenotype. Differentiated VSMCs are marked by the expression of smooth muscle α-actin (SMα-actin), smooth muscle 22α protein (SM22α), myosin heavy chain (SM-MHC), calponin and other molecules, which play an essential role in regulating vascular physiological status such as vasodilation and contraction (9). Dedifferentiated VSMCs often express proteins such as osteopontin (OPN) and proliferating cell nuclear antigen (PCNA) in large amounts, acknowledged as markers of dedifferentiated VSMCs (10, 11). VSMCs within the vein exhibit a quiescent phenotype in their natural state, equipped with contractile and low proliferative and migratory capacities. VSMCs originate from the mesoderm with prodigious plasticity (12, 13). When various injuries and pathological factors stimulate vessels, VSMCs can transform from a differentiated to a dedifferentiated state, accompanied by decreased expression of the above-mentioned differentiation-specific markers and augmented expression of dedifferentiated molecules. Meanwhile, VSMCs become abnormally active in proliferation and migration and secrete many ECMs and cytokines. This process is referred to as the phenotypic switch of VSMCs (14–16). There is ample evidence suggesting that excessive proliferation and migration of dedifferentiated VSMCs and the synthesis of excessive ECMs participate in the pathogenesis of intimal hyperplasia in vein grafts (Figure 1) (6–8).
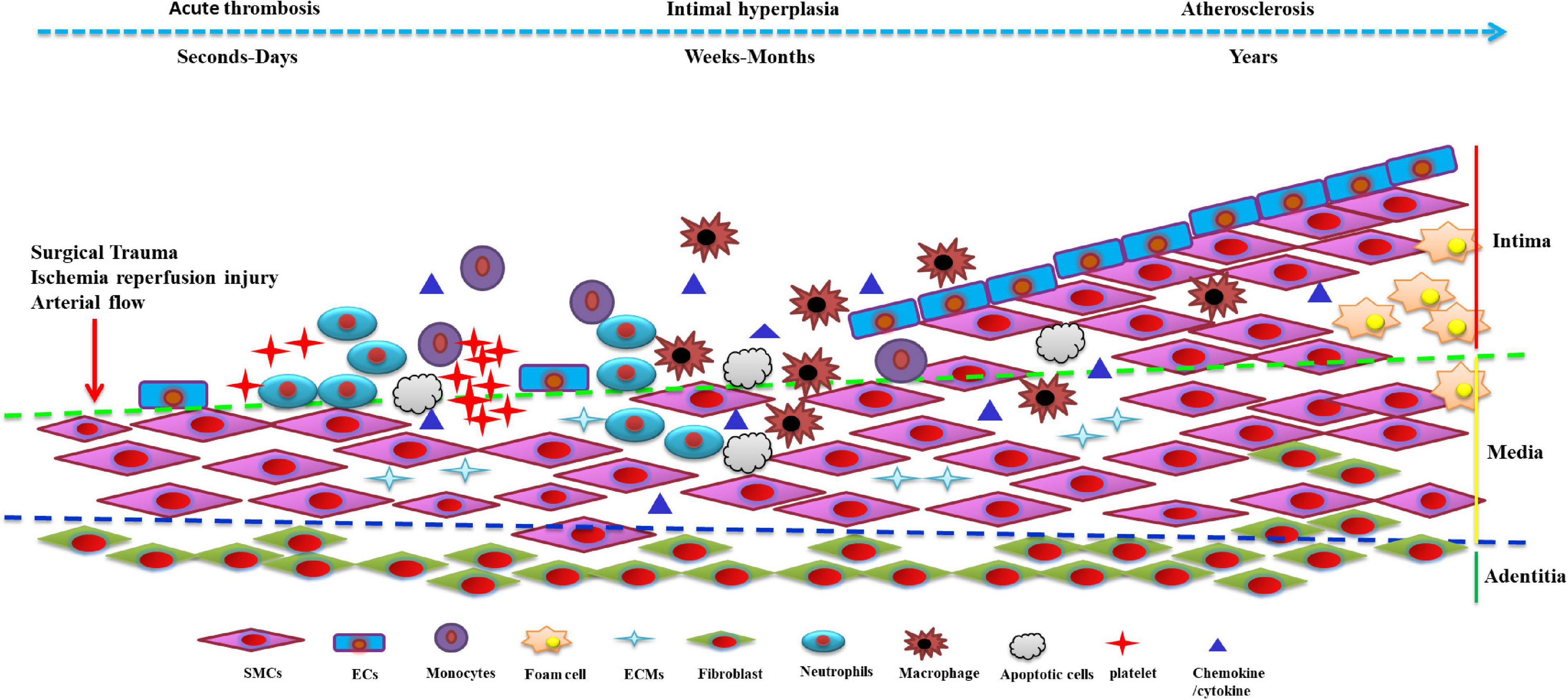
Figure 1. The phenotype of venous smooth muscle cells (SMCs) in the development of vein graft restenosis: Stimulated by various injuries, pathological factors and stress alterations, venous SMCs can transform from a differentiated to a dedifferentiated state, accompanied by a decrease in the expression of the above-mentioned differentiation-specific markers. Venous vascular smooth muscle cells (VSMCs) secrete many extracellular matrix (ECMs) and cytokines, causing excessive proliferation and further migration to the intima leading to intimal hyperplasia resulting in vein graft restenosis. SMCs, smooth muscle cells; ECs, endothelial cells; ECMs, extracellular matrix.
Etiology of Intimal Hyperplasia in Vein Graft and Phenotypes of Vascular Smooth Muscle Cells (VSMCs)
Intimal hyperplasia in vein graft results from the interplay of multiple factors. The widely acknowledged causes of intimal hyperplasia in vein grafts are injury from surgery, ischemia-reperfusion injury, and stress alterations in venous arterialization, which are associated with dysfunction and phenotypic switch of VSMCs.
Vascular Injury and Phenotypic Switch of Vascular Smooth Muscle Cells (VSMCs) due to Surgical Operations
In patients requiring CABG, procedures such as manipulating, harvesting, examining and dilating the HSV can cause vein injury. The endothelium impedes platelet-derived growth factor production (17), suppresses inflammation (18), prevents platelet aggregation (19, 20), and inhibits VSMCs activity (21). Hence, the integrity of the endothelial monolayer is the premise for preserving its function. When the endothelium is damaged, the subendothelial matrix components become exposed to blood, which induces platelet aggregation and adhesion of inflammatory cells, resulting in the activation of coagulation pathways and acute thrombus formation (22). Moreover, the release of high amounts of tissue factors causes altered behavior of VSMCs, which provides the basis for later restenosis (4). Damage to the endothelium also jeopardizes nitric oxide (NO) secretion. Endothelial-derived NO inhibits the synthesis of tissue factors and the expression of metalloproteinases (MMPs) (23, 24). On the one hand, tissue factor activates VSMCs, driving intimal hyperplasia in the grafted vein (4, 25). On the other hand, MMPs give rise to ECMs degradation and remodeling, facilitating the migration of VSMCs across the ECMs layer to the intimal, exacerbating intimal hyperplasia of vein graft (26). Additionally, vascular injury promotes the synthesis and expression of platelet-derived growth factor-BB (PDGF-BB), transforming growth factor-β (TGF-β), fibroblast growth factor (bFGF), insulin growth factor (IGF-1), angiotensin (Ang-II), tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β), driving the activation and altered behaviors of VSMCs, enhancing the proliferative and migratory capacity, eventually leading to intimal hyperplasia in vein graft (25–28).
Ischemia-Reperfusion Injury and Phenotypic Switch of Vascular Smooth Muscle Cells (VSMCs)
During HSV harvesting and transplantation into the coronary artery, the vein graft vessels are in an ischemic-hypoxic state. After surgery, superoxides produced by ischemia-reperfusion injury represent one of the mechanisms by which IH occurs in vein grafts. Superoxides reduce NO synthesis, promote MMPs expression, and accelerate the phenotypic switch of VSMCs (29, 30). During the harvesting of the HSV, the traditional operation requires stripping the perivascular tissue and fat of the venous epithelium, which disconnects and damages the vasa vasorum. Accordingly, this procedure is an essential element leading to an ischemic and hypoxic state in vein grafts (31). In addition, we suggest that the ischemic-hypoxic phase of vessels should include angiogenesis of vasa vasorum in grafted veins. A favorable long-term patency rate could be reached by applying the “no-touch” technique for HSV harvesting via preoperative ultrasound localization, which preserves as much as possible vasa vasorum around the HSV and reduces ischemic-hypoxic injury (32). Moreover, studies have demonstrated that hypoxic injury can drive remodeling and phenotypic switch of VSMCs, triggering their early abnormal proliferation (33). Taken together, the above findings suggest that the occurrence of intimal hyperplasia in grafted veins may be due to the phenotypic switch of VSMCs caused by ischemic-hypoxic injury.
Stress Alterations in Venous Arterialization and Phenotypic Switch of Vascular Smooth Muscle Cells (VSMCs)
It is well-established that blood flow yields contrasting stress stimuli on the vessel wall (34). The mechanical milieu maintains vascular functional homeostasis and serves as a predisposing contributor to many cardiovascular diseases. The vessel wall bears complex stress, mainly subject to shearing stress along the direction of blood flow and circumferential stress from blood pulsation (35). VSMCs play a crucial role in resisting challenges in the vascular mechanical microenvironment and regulating vascular function (36). The shear stress and circumferential stress of venous flow in the physiological state are 1.5–6 dyn/cm2 and 1%, respectively, while the arteries are about 20–70 dyn/cm2 and 10–15% (8, 37–40). It has been shown that vein grafts are subjected to chronic shear and circumferential stresses in the arterial circulation after implantation (37–39). The neointima forms approximately 2 weeks after surgery, indicating complete repair of venous ECs, but intimal hyperplasia still occurs, suggesting the lesion is still present. Besides, stress alterations persist, justifying its role in vein graft restenosis. Morphological, genetic and metabolic differences between vein and artery lead to distinct responses to stress (40). The HSV is infrequently stenosed unless grafted in an arterial setting. When used as a graft vessel, the internal thoracic artery exhibits patency at 10 years in 90% of cases, whereas most veins fail to stay patent for the same period (41). These studies substantiate that hemodynamic alterations participate in vein graft intimal hyperplasia. When a vein graft is connected to an artery, it sustains stress from the arterial flow. ECs in the vein graft undergo high-flow shear washout and dislodgement, and this stress subsequently stimulates VSMCs in the grafted vein are directly exposed to a circumferential arterial stretch, which can fuel the phenotypic transformation of VSMCs (36–39), translating into massive proliferation and migration of VSMCs, and leading to intimal hyperplasia of grafted vein (6–8). Intimal hyperplasia formation accelerates atherosclerosis as a prelude to vein graft occlusion.
In summary, the three factors described above do not exist alone and often interact to promote intimal hyperplasia of vein grafts. During CABG, the direct injury during vein harvesting, subsequent ischemia-reperfusion injury, and sustained stress alterations due to vein arterialization propel function remodeling and phenotypic switch of VSMCs into a dedifferentiated state with excessive replication, migration to the intima, and synthesis of large amounts of ECMs, conducive to the development of intimal hyperplasia in vein grafts (25, 32, 33, 39). Accordingly, unveiling the molecular pathogenesis of phenotypic transformation of VSMCs in grafted veins and identifying prevention and treatment strategies are important to minimize venous graft stenosis after CABG.
Molecular Mechanism of Intimal Hyperplasia in Vein Graft and Phenotypic Switch of Vascular Smooth Muscle Cells (VSMCs)
Tissue Factor and Growth Factor and Phenotypic Switch of Vascular Smooth Muscle Cells (VSMCs)
Many growth factors and cytokines such as PDGF-BB, TGF-β, bFGF, and IGF-1 can promote VSMCs dedifferentiation (42–45). PDGF-BB is a pro-cytokine that stimulates cell division and is the most potent inducer of phenotypic transformation of VSMCs investigated (46, 47). PDGF-BB earned its name for the secretion of platelet α granules. Current evidence suggests that PDGF-BB can be synthesized and secreted by VSMCs, inflammatory cells, fibroblasts and ECs in response to tissue injury or stimuli (48, 49). Studies have shown that PDGF-BB levels are firmly correlated with the degree of intimal hyperplasia in grafted veins (50, 51). PDGF-BB synthesis is increased in vein grafts and remains stable in arterial grafts (48). Interestingly, it has been shown that blocking PDGF-BB inhibits intimal hyperplasia in HSV (52), and cultures have shown that PDGF-BB induces replication and migration of VSMCs of HSV but rarely affects VSMCs in internal thoracic arteries (53, 54). Dong et al. found that PDGF-BB involving ERK1/2 signal pathway activation could upregulate vascular cell adhesion molecule-1(VCAM-1) level, which led to the phenotypic transformation of VSMCs and exacerbated intimal hyperplasia in balloon injury of rat carotid arteries (55). Xiang et al. found that PDGF-BB could regulate the expression of the inflammatory and adhesion factor VCAM-1 via the JAK2/STAT3 signal pathway, which affected intimal hyperplasia in grafted veins (25). For this reason, PDGF-BB has become the most widely used method for inducing phenotypic switch of VSMCs and harming vascular cells.
Metalloproteinases, Extracellular Matrix and Phenotypic Switch of Vascular Smooth Muscle Cells (VSMCs)
It has been shown that MMPs affect the synthesis and degradation of ECMs such as collagen fibrils and glycoproteins under physiological conditions. During vascular remodeling, ECMs can be degraded by MMPs and remodeled to facilitate VSMCs migration through the ECMs layer to the intima, causing vascular intimal hyperplasia (56, 57). MMPs are widely expressed during intimal hyperplasia of grafted veins, and MMP-2/9 could promote VSMCs migration to the HSV, which can be inhibited by MMP-2/9 knockdown (58–60). Clinical studies have shown that MMP-2 expression in vein grafts is associated with CABG prognosis (61). Moreover, tissue inhibitors of metalloproteinases (TIMPs) alleviate intimal hyperplasia in vein graft by reducing MMPs expression and thus reducing VSMCs migration (62). The aforementioned studies provide important insights into the functions of MMPs and ECMs in the phenotypic transformation of VSMCs.
Inflammatory Factors and Phenotypic Switch of Vascular Smooth Muscle Cells (VSMCs)
The inflammatory cascade is vital for triggering vein graft intimal hyperplasia, which participates in the pathogenesis of late stages of vein graft atherosclerosis (63, 64). Increased permeability and disrupted function of ECs drive leukocyte activation and aggregation, followed by secretion of multiple chemokines and cytokines, which subsequently promote the activation of inflammatory cells. The knock-on effect induces VSMCs to proliferate and migrate (65, 66). Interestingly, in an external jugular vein-abdominal aortic graft rat model, nuclear transcription factor kappa B (NF-κB) remained active at high levels 4 weeks after surgery. After siRNA silenced NF-κB, vein graft intimal hyperplasia was significantly ameliorated (67). Since NF-κB activation is the core of inflammation, it is reasonable to speculate that inhibiting the inflammatory cascade could stabilize the differentiated phenotype of venous VSMCs to exert an anti- intimal hyperplasia effect. NF-κB is a critical transcriptional regulator in the transcription of inflammatory factors such as IL-1, IL-6, TNF-α, the C-C motif chemokine ligand 2 (CCL2/MCP-1), ICAM-1 and VCAM-1 (68, 69). These elements indicate that cytokines, chemokines and adhesion molecules mediate the recruitment and infiltration of inflammatory cells into the grafted vessel wall. CCL2/MCP-1 is an important mediator of vascular inflammation and the most potent chemotactic factor for monocytes. After vein grafting, the CCL2/MCP-1 level increases markedly and persists for several weeks (70). Blocking CCL2/MCP-1 and its receptor can inhibit monocyte adhesion in the vessel wall, attenuating artery intimal hyperplasia and atherosclerosis (71). Knocking down the expression of CCL2/MCP-1 by siRNA can dramatically reduce the proliferation and migration of VSMCs (72). Moreover, it has been shown that VCAM-1 interacts with integrin α4β1 on the leukocyte surface and triggers inflammatory cascade signaling (73). VCAM-1 promotes VSMCs to proliferate and migrate, and restricting VCAM-1 expression was found to attenuate intimal hyperplasia in a rat model of carotid artery injury (74). Besides, VCAM-1 participates in leukocyte exudation, and ICAM-1-siRNA delivered via ultrasound microbubbles can reportedly effectively retard arterial intimal hyperplasia (75). Intriguingly, Huang et al. found that ICAM-1 antibodies could inhibit the replication of VSMCs (76). Taken together, chemokines, adhesion factors and inflammatory cytokines orchestrate the phenotypic transformation of VSMCs and the development of vascular intimal hyperplasia.
Discovery, Characteristics and Functions of miRNAs
In 1993, researchers identified a non-coding RNA that could repress the expression of lin-14 and thus affect nematode development (77). Subsequently, let-7, the second miRNA, was also discovered in nematodes (78). Since then, more and more miRNAs have been identified in human, animal, and plant genomes, and scholars have unveiled that miRNAs are widely present in eukaryotes and participate in the biological effects. miRNAs represent endogenous and non-coding RNAs with independent transcription units, containing about 22 nucleotides that function mainly by recognizing and binding to the 3’ untranslated region (3’UTR) of the messenger RNA of their targets via complementary base pairing. miRNAs are evolutionarily conserved, homologous and tissue-specific in species (79). The base pair between nucleotides 2 to 8 at the 5’-end of miRNAs serves as the most central and conservative region, called the seed sequence. The seed sequence is the core region that binds to the 3’ UTR of the targets (80). The slice and maturation of miRNA undergo three main processes: the generation of primary miRNA (pri-miRNA), the generation of precursor miRNA (pre-miRNA) and its transportation out of the nucleus, and the maturation of miRNAs. It has been established that more than 2000 miRNAs are encoded by the human genome, regulating one-third of human genes (81). In addition, evidence from studies indicates some atypical examples of miRNAs functionality and localization, including interactions with proteins beyond the argonaute family and transcriptional regulation in the nucleus and in mitochondria (82). It further expands the importance of its role in regulatory function of organism. miRNAs engage in many biological processes, including growth, aging, angiogenesis, immune regulation, metabolism, cell proliferation, apoptosis, cell migration, tumorigenesis, metastasis, and drug resistance.
Role of miRNAs in the Phenotypic Switch of Vascular Smooth Muscle Cells (VSMCs) in Intimal Hyperplasia of Vein Graft
miRNAs are widely expressed in the blood vascular system and regulate vascular cell growth and function. Knock-out of dicer (an enzyme essential for miRNAs maturation) showed arrest of embryogenesis due to the inability to generate normal blood vessels, suggesting that miRNAs may be essential for angiogenesis (83). miR-145-5p is the most abundant miRNA in VSMCs, and it promotes the differentiation of mesenchymal stem cells (MSCs) to VSMCs through the Kruppl-like factor 4 (KLF4) pathway (84). Antagonizing miR-1 expression could lead into VSMCs-specific markers and inhibit the differentiation of embryonic stem cell-derived VSMCs via the KLF4 pathway (85). Each miRNA can regulate the expression and function of multiple targets via the same signal pathway. Indeed, it should be borne in mind that a miRNA could regulate many targets. Currently, many miRNAs are involved in the regulation of VSMCs. Many miRNAs with regulatory effects on the function and phenotype of vascular VSMCs have been identified in recent years. miR-1, miR-15b/16-5p, miR-21, miR-22, miR-23b, miR-34a, miR-125b, miR-126-3p, miR-132, miR-133(miR-133a-1/miR-133a-2), miR-143-5p/145-5p, miR-195, miR-214, miR-223, miR-548f-5p, miR-638, miR-663, and miR-1298-5p miRNAs help maintain the differentiated phenotype of VSMCs and curb the proliferation and migration of VSMCs (84–102). Among above-mentioned miRNAs, miR-23b, miR-25, miR-125, miR-143-5p/145-5p, miR-221/222, miR-214, miR-638 and miR-663 can mediate the function and phenotype switch of VSMCs induced by PDGF-BB, and all of them participate in the regulation of intimal hyperplasia in injured vessels.
The etiologies, factors and molecular mechanisms described above do not exist alone and often interact to influence VSMCs functions in vessel disease. Interesting, the study by Zahedi et al. shows that dicer activity controls neointimal hyperplasia by reducing VSMCs proliferation after vascular injury. The levels of miR-147-3p, miR-143-3p, miR-100-5p, miR-99a-5p, and miR-27a-3p were most significantly reduced in dicer knockout mice, upon further study, Zahedi et al. found that these miRNAs may be involved in this process of VSMCs phenotypic regulation by influencing inflammation-induced growth factor signaling (103). It is suggests that general suppression of miRNA synthesis and growth factor signaling play a part role in regulation of VSMCs phenotype. The study by Sun et al. indicates that miR-133a-3p is reduced significancely in carotid artery ligation-induced vascular injury, Salusin-β is upregulated, MMP9 expression and reactive oxygen species (ROS) production is increased, promotes VSMCs migration and neointima formation in response to vascular injury (104). It has been shown that ischemic-hypoxic injury with ROS production and oxidative stress is an important cause of vascular remodeling (105). Previous studies have showed that miR-21, miR-24 miR-31-5p and miR-210 regulates ischemia/hypoxia stress-induced VSMCs functions in vascular disease (105–108). Differences in miRNA expression patterns as a function of hemodynamic forces have been detected, which is named the term “mechano-miRNAs.” miRNAs such as miR-33, miR-126 and miR-143-5p/145-5p are counted amongst those mechano-miRNAs (8, 45, 92). In particular, miR-126 is an endothelial enriched miRNA, Jansen et al. found that the transfer of miR-126-3p mediated by endothelial particles into recipient VSMCs could inhibit the its proliferation, migration and subsequent neointima formation of injuried artery (92). The study by Santovito et al. showed that, under high shear stress, the nuclear import of miR-126-5p is reduced, promotes ECs apoptosis and exacerbates atherosclerosis (109). On the one hand, it indicates that mechano-miRNAs is vital in phenotype transformation of VSMCs. On the other hand, it reveals a non-canonical mechanism by which miRNAs including miR-126-5p may modulate protein function in VSMCs, as already proven in ECs for caspase-3 (109). Moreover, miR-126-5p has been shown to promote the contractile phenotype of VSMCs (110). It is well known that miR-143-5p/145-5p has been confirmed to control VSMCs phenotype in a large number of studies. It is called the regulator of VSMCs. The study by Climent et al. showed that vessel stress triggers miR-143-5p/145-5p transfer from VSMCs to their neighboring ECs to modulate the angiogenesis and vascular homeostasis (111). Hergenreider et al. research indicated that extracellular vesicles secreted by shear-stress-stimulated ECs are enriched in miR-143-5p/145-5p and regulate target gene expression in co-cultured VSMCs (112). These interesting studies suggest that some miRNAs such as miR-143-5p, miR-145-5p act as communication molecules between VSMCs and ECs exposure to shear stress. It also suggests that some miRNAs plays an important role in phenotypic regulation of VSMCs causing by non-physiological shear stress.
In brief, miRNAs play a crucial role in the phenotype transformation of VSMCs, and in-depth research on their functions, targets and interaction networks can help understand proliferative diseases of VSMCs such as intimal hyperplasia after vessel injury, intimal hyperplasia in vein graft after CABG surgery, and atherosclerosis from a new perspective, offering therapies for prevention and treatment. The widely acknowledged causes of intimal hyperplasia in vascular disease are injury from surgery, ischemic-hypoxic injury, and stress alterations, which are associated with dysfunction and phenotypic switch of VSMCs. Intimal hyperplasia caused by VSMCs phenotype and function change results from the interplay of multiple factors. At present, the literature reports are very rare that the regulation of miRNAs on inflammatory factors, metalloproteinases, extracellular matrix, tissue factor and growth factor influencing the phenotypic switch of venous VSMCs in vein graft. The further research is still required.
It is widely acknowledged that miRNAs are essential for regulating the proliferation and differentiation of VSMCs during embryogenesis (113). Many cardiovascular diseases are relevant to the abnormal expression and function of miRNAs (114). As shown in Figure 2, miRNAs exhibit regulatory effects on the function and phenotype of venous SMCs (115–121). Furthermore, many studies have shown that miRNA regulation of phenotypic transformation of VSMCs is vital for intimal hyperplasia in vein grafts. miR-21 and miR-145 are the most plentiful miRNAs expressed on the vessels. Studies in rats have shown that miR-21 is significantly upregulated after vessel damage with decreased endogenous c-Ski and increased VSMC proliferation (87). In addition, the Phosphatase and Tensin Homolog (PTEN) gene serves as a target for miR-21, and overexpression of miR-21 inactivates PTEN, minimizing apoptosis and maximizing the proliferation of VSMCs (105). Interestingly, some researchers have documented a significantly elevated miR-21 level in intimal hyperplasia of vein graft in mouse and porcine models, also observed in human decaying grafted veins. Focal knockdown of miR-21 in vein grafts could limit the number of VSMCs in the nascent intima and attenuate intimal hyperplasia (116). In this regard, miR-21 has been documented to regulate the phenotype of VSMCs, involving the occurrence of intimal hyperplasia in grafted veins. Likewise, miR-145 propels the expression of differentiation genes to maintain the differentiated state of VSMCs via the TGF-β signaling pathway (117). miR-145 fosters the expression of SM22α that a specific marker of differentiated phenotype, and inhibits the phenotype transformation of VSMCs and their ability to proliferate and migrate by curbing the expression of the KLF4 transcription factor (95). The overexpression of miR-145 impedes the phenotypic transformation of VSMCs and ameliorates the intimal hyperplasia of vein graft in a rabbit model (118). The above-mentioned studies conclude that miR-21 has a pro-VSMCs dedifferentiation effect, and miR-145 can stimulate cell differentiation and maintain the differentiated phenotype of venous SMCs. The vasculature represents a typical mechanical system comprised of VSMCs, which are the primary cells of the vessel wall and are vital for enduring pressure changes and maintaining vascular functional homeostasis. Exposure of venous SMCs to a 10% 1.25 Hz cyclic stretch yielded waning expression of miR-33 and a waxing proliferation of venous SMCs. This phenomenon was verified in rat models; the downregulation of miR-33 and upregulation of phosphorylated smad2/5 upregulated VSMCs replication and exacerbated intimal hyperplasia in vein grafts. Overexpression of miR-33 was found to remarkably hinder the formation of intimal hyperplasia in vein graft (8). Interestingly, difference from only one miR-33 isoform in rodents which is conserved with human miR-33a, miR-33 exist two isoforms in humans: miR-33a and miR-33b. Therefore, we should pay attention to the different functions of the miRNAs ‘ isoforms in different species and cells. Moreover, Wang et al. found that after Matrigel-127 mixed adenovirus transfection to silence miR-221 in rat models, the proliferation of venous SMCs was reduced by approximately 20% with improved blood flow of vein graft (119). Using the above method to overexpress miR-365, Cao et al. found that it downregulated the expression of G1/S-specific cyclin D1 and amplified the expression of differentiation-type markers in VSMCs, thereby inhibiting intimal hyperplasia in the grafted vein (120). Intriguingly, miR-16-5p was downregulated in rat vein grafts with alterations in phenotypic markers, and miR-16-5p overexpression suppressed intimal hyperplasia. Mechanistically, miR-16-5p could disrupt the expression of zyxin, a mechanotransducer of biological signals, via binding to the 3’UTR of the zyxin- mRNA, which inhibited the proliferation and migration of cultured VSMCs by preventing the switch from a contractile to a synthetic phenotype. Our study implies that miR-16-5p is a potential therapeutic target for combating intimal hyperplasia in vein grafts (121). miRNAs can be used as targets for preventing VSMCs from switching phenotype and preventing intimal hyperplasia after vein grafting.
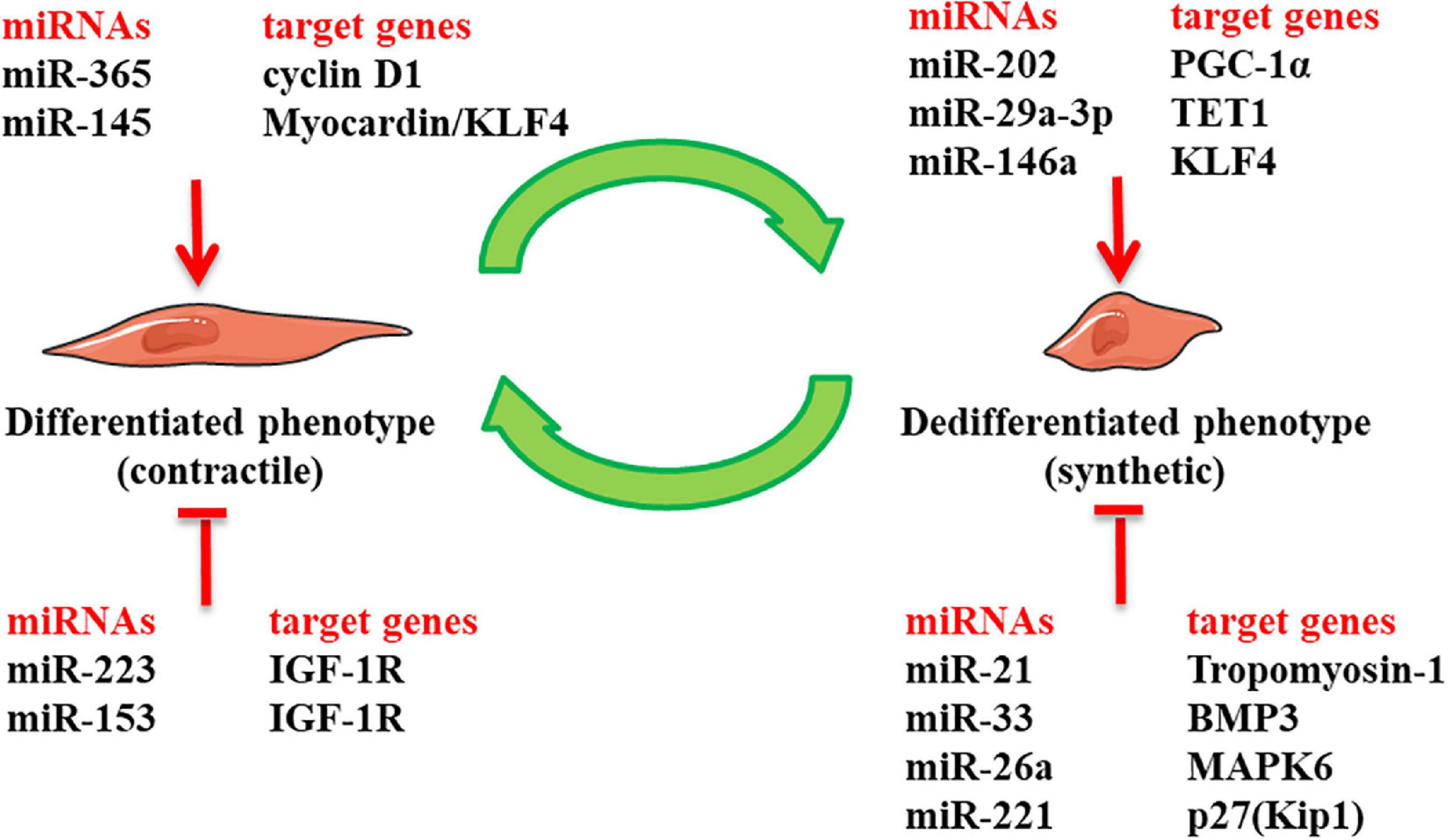
Figure 2. The role of miRNAs in regulating phenotypic switch of venous smooth muscle cells (SMCs) in intimal hyperplasia of vein graft: it presents miRNAs that have been identified bearing regulatory effects on the phenotype of venous vascular smooth muscle cells (VSMCs) in intimal hyperplasia of vein graft. Klf4, Kruppl-like factor 4; IGF-1R, insulin-like growth factor-1 receptor; PCG-1α, peroxisome proliferator-activated-receptor-γ coactivator-1α; TET1, Ten-eleven translocation methylcytosine dioxygenase 1; BMP3, bone morphogenetic protein 3; MAPK6, mitogen-Activated Protein Kinase 6; p27(Kip1), p27KIP1 gene.
In conclusion, miRNAs are pronounced regulators in the biological processes of differentiation, proliferation and migration of VSMCs, formulate a phenotype switch of VSMCs and influence the occurrence of intimal hyperplasia in grafted veins, which broaden the therapeutic horizons for vein graft restenosis in clinical practice.
Prospect
Restenosis induced by intimal hyperplasia of the graft veins after CABG is the main factor affecting surgery outcomes in CAD patients. In the process of surgical procedures, there is sufficient time for genetic manipulation, so that controlling molecular alterations in the initial stage of injury, making it possible to prevent intimal hyperplasia of the graft veins. Moreover, focal gene interventions during the procedure shun the side effects of systemic administration, which is ideal for prophylaxis of vein graft degeneration. VSMCs are one of the most important cells of the vessel wall, and the phenotypic transformation of VSMCs plays an important part in the mechanism of vein graft intimal hyperplasia-induced atherosclerosis. miRNAs are well-studied non-coding RNAs with independent transcription units and stable expression, involved in many biological processes. miRNAs have been discovered to regulate the switch of VSMC phenotypes. By intervening and maintaining the differentiated phenotype of VSMCs, miRNAs are poised to provide a novel strategy of local gene therapy for the prophylaxis and treatment of vein graft failure and other cardiovascular diseases. miRNAs represents a promising technique for silencing genes that cause above-mentioned intimal hyperplasia in vein graft after CABG surgery as well as other cardiovascular diseases caused by phenotypic switch of VSMCs, such as aterial intimal hyperplasia after vessel injury, pulmonary hypertension, hypertension and atherosclerosis.
miRNAs represents a promising technique for silencing genes that cause above-mentioned cardiovascular diseases. The successful delivery of miRNAs to the vascular wall faces multiple challenges (122). Firstly, these challenges include cell specificity, targeted delivery of miRNAs, anatomical tissue barriers. Secondly, there is no report of studies to translate findings of miRNAs in proliferative cardiovascular diseases from rodent models into human studies. This may be due to face risks of toxicity and side effects due to a wide range of target genes and cells. In addition, the selection of the target genes has become a difficult problem. There are considerable number of miRNAs that regulate the proliferative of VSMCs in cardiovascular diseases. Which miRNA is the most effective molecule to regulate the phenotype of VSMCs or whether multiple genes are required to work together in the process of phenotypic switch of VSMCs, and what is the detailed and specific mechanism and so on, a further research is still required.
The role of miRNAs in cardiovascular diseases is a rising research filed. Elucidating the regulatory mechanism of miRNA in the organism can help understand proliferative cardiovascular diseases of VSMCs such as intimal hyperplasia after vessel injury, intimal hyperplasia in vein graft after CABG surgery, pulmonary hypertension, hypertension and atherosclerosis from a new perspective, offering miRNA therapy.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (82160060 and 81970345), the doctoral startup project of Affiliated Hospital of Zunyi Medical University [(2021) No.4], and Guizhou Provincial Science and Technology Projects ZK(2022)YB669.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhao D, Liu J, Wang M, Zhang X, Zhou M. Epidemiology of cardiovascular disease in China: current features and implications. Nat Rev Cardiol. (2019) 16:203–12.
2. Raza S, Blackstone EH, Houghtaling PL, Rajeswaran J, Riaz H, Bakaeen FG, et al. Influence of diabetes on long-term coronary artery bypass graft patency. J Am Coll Cardiol. (2017) 70:515–24.
3. Weintraub WS, Grau-Sepulveda MV, Weiss JM, O’Brien SM, Peterson ED, Kolm P, et al. Comparative effectiveness of revascularization strategies. N Engl J Med. (2012) 366:1467–76.
4. de Vries MR, Simons KH, Jukema JW, Braun J, Quax PH. Vein graft failure: from pathophysiology to clinical outcomes. Nat Rev Cardiol. (2016) 13:451–70.
5. Ward AO, Caputo M, Angelini GD, George SJ, Zakkar M. Activation and inflammation of the venous endothelium in vein graft disease. Atherosclerosis. (2017) 265:266–74.
6. Wadey K, Lopes J, Bendeck M, George S. Role of smooth muscle cells in coronary artery bypass grafting failure. Cardiovasc Res. (2018) 114:601–10.
7. Quist WC, LoGerfo FW. Prevention of smooth muscle cell phenotypic modulation in vein grafts: a histomorphometric study. J Vasc Surg. (1992) 16:225–31.
8. Huang K, Bao H, Yan ZQ, Wang L, Zhang P, Yao QP, et al. MicroRNA-33 protects against neointimal hyperplasia induced by arterial mechanical stretch in the grafted vein. Cardiovasc Res. (2017) 113:488–97.
9. Shanahan CM, Weissberg PL, Metcalfe JC. Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ Res. (1993) 73:193–204.
10. Jiang H, Lun Y, Wu X, Xia Q, Zhang X, Xin S, et al. Association between the hypomethylation of osteopontin and integrin β3 promoters and vascular smooth muscle cell phenotype switching in great saphenous varicose veins. Int J Mol Sci. (2014) 15:18747–61. doi: 10.3390/ijms151018747
11. Hu WY, Fukuda N, Satoh C, Jian T, Kubo A, Nakayama M, et al. Phenotypic modulation by fibronectin enhances the angiotensin II-generating system in cultured vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2000) 20:1500–5. doi: 10.1161/01.atv.20.6.1500
12. Frismantiene A, Philippova M, Erne P, Resink TJ. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. (2018) 52:48–64. doi: 10.1016/j.cellsig.2018.08.019
13. Liu M, Gomez D. Smooth muscle cell phenotypic diversity. Arterioscler Thromb Vasc Biol. (2019) 39:1715–23.
14. Yoshiyama S, Chen Z, Okagaki T, Kohama K, Nasu-Kawaharada R, Izumi T, et al. Nicotine exposure alters human vascular smooth muscle cell phenotype from a contractile to a synthetic type. Atherosclerosis. (2014) 237:464–70. doi: 10.1016/j.atherosclerosis.2014.10.019
15. Davis-Dusenbery BN, Wu C, Hata A. Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arterioscler Thromb Vasc Biol. (2011) 31:2370–7. doi: 10.1161/ATVBAHA.111.226670
16. Spin JM, Maegdefessel L, Tsao PS. Vascular smooth muscle cell phenotypic plasticity: focus on chromatin remodelling. Cardiovasc Res. (2012) 95:147–55. doi: 10.1093/cvr/cvs098
17. Qi YX, Jiang J, Jiang XH, Wang XD, Ji SY, Han Y, et al. PDGF-BB and TGF-β1 on cross-talk between endothelial and smooth muscle cells in vascular remodeling induced by low shear stress. Proc Natl Acad Sci U S A. (2011) 108:1908–13. doi: 10.1073/pnas.1019219108
18. Aslan M, Freeman BA. Oxidases and oxygenases in regulation of vascular nitric oxide signaling and inflammatory responses. Immunol Res. (2002) 26:107–18.
19. Xu X, Liu Y, Li K, Wang P, Xu L, Yang Z, et al. Intensive atorvastatin improves endothelial function and decreases ADP-induced platelet aggregation in patients with STEMI undergoing primary PCI: a single-center randomized controlled trial. Int J Cardiol. (2016) 222:467–72. doi: 10.1016/j.ijcard.2016.07.223
20. Schiattarella GG, Carrizzo A, Ilardi F, Damato A, Ambrosio M, Madonna M, et al. Rac1 modulates endothelial function and platelet aggregation in diabetes mellitus. J Am Heart Assoc. (2018) 7:e007322. doi: 10.1161/JAHA.117.007322
21. Ribback S, Pavlovic D, Herbst D, Nedeljkov-Jancic R, Wendt M, Nedeljkov V, et al. Effects of amitriptyline, fluoxetine, tranylcypromine and venlafaxine on rat vascular smooth muscle in vitro–the role of the endothelium. J Physiol Pharmacol. (2012) 63:119–25.
22. Kumar RA, Moake JL, Nolasco L, Bergeron AL, Sun C, Dong JF, et al. Enhanced platelet adhesion and aggregation by endothelial cell-derived unusually large multimers of von Willebrand factor. Biorheology. (2006) 43:681–91.
23. Murad F. What are the molecular mechanisms for the antiproliferative effects of nitric oxide and cGMP in vascular smooth muscle? Circulation. (1997) 95:1101–3.
24. Huang J, Lin SC, Nadershahi A, Watts SW, Sarkar R. Role of redox signaling and poly (adenosine diphosphate-ribose) polymerase activation in vascular smooth muscle cell growth inhibition by nitric oxide and peroxynitrite. J Vasc Surg. (2008) 47:599–607. doi: 10.1016/j.jvs.2007.11.006
25. Xiang S, Liu J, Dong N, Shi J, Xiao Y, Wang Y, et al. Suppressor of cytokine signaling 3 is a negative regulator for neointimal hyperplasia of vein graft stenosis. J Vasc Res. (2014) 51:132–43. doi: 10.1159/000355193
26. Mountain DJ, Freeman BM, Kirkpatrick SS, Beddies JW, Arnold JD, Freeman MB, et al. Androgens regulate MMPs and the cellular processes of intimal hyperplasia. J Surg Res. (2013) 184:619–27. doi: 10.1016/j.jss.2013.05.070
27. Wolff RA, Ryomoto M, Stark VE, Malinowski R, Tomas JJ, Stinauer MA, et al. Antisense to transforming growth factor-beta1 messenger RNA reduces vein graft intimal hyperplasia and monocyte chemotactic protein 1. J Vasc Surg. (2005) 41:498–508. doi: 10.1016/j.jvs.2004.12.037
28. Jia G, Aggarwal A, Yohannes A, Gangahar DM, Agrawal DK. Cross-talk between angiotensin II and IGF-1-induced connexin 43 expression in human saphenous vein smooth muscle cells. J Cell Mol Med. (2011) 15:1695–702. doi: 10.1111/j.1582-4934.2010.01161.x
29. Jeremy JY, Dashwood MR, Mehta D, Izzat MB, Shukla N, Angelini GD. Nitric oxide, prostacyclin and cyclic nucleotide formation in externally stented porcine vein grafts. Atherosclerosis. (1998) 141:297–305. doi: 10.1016/s0021-9150(98)00183-x
30. Hattori K, Yamanouchi D, Banno H, Kobayashi M, Yamamoto K, Kajikuri J, et al. Celiprolol reduces the intimal thickening of autogenous vein grafts via an enhancement of nitric oxide function through an inhibition of superoxide production. J Vasc Surg. (2007) 46:116–23. doi: 10.1016/j.jvs.2007.03.044
31. Ramos De Souza D, Dashwood MR, Samano N. Saphenous vein graft harvesting and patency: no-touch harvesting is the answer. J Thorac Cardiovasc Surg. (2017) 154:1300–1.
32. Verma S, Lovren F, Pan Y, Yanagawa B, Deb S, Karkhanis R, et al. Pedicled no-touch saphenous vein graft harvest limits vascular smooth muscle cell activation: the PATENT saphenous vein graft study. Eur J Cardiothorac Surg. (2014) 45:717–25. doi: 10.1093/ejcts/ezt560
33. Tan X, Feng L, Huang X, Yang Y, Yang C, Gao Y. Histone deacetylase inhibitors promote eNOS expression in vascular smooth muscle cells and suppress hypoxia-induced cell growth. J Cell Mol Med. (2017) 21:2022–35. doi: 10.1111/jcmm.13122
34. Owens CD. Adaptive changes in autogenous vein grafts for arterial reconstruction: clinical implications. J Vasc Surg. (2010) 51:736–46. doi: 10.1016/j.jvs.2009.07.102
35. Mavromatis K, Fukai T, Tate M, Chesler N, Ku DN, Galis ZS. Early effects of arterial hemodynamic conditions on human saphenous veins perfused ex vivo. Arterioscler Thromb Vasc Biol. (2000) 20:1889–95.
36. Momin A, Sharabiani MT, Wendler O, Angelini GD, Desai J. Arterial blood pressure and vascular function in human saphenous vein. Perfusion. (2015) 30:233–8.
37. DeSart KM, Butler K, O’Malley KA, Jiang Z, Berceli SA. Time and flow-dependent changes in the p27(kip1) gene network drive maladaptive vascular remodeling. J Vasc Surg. (2015) 62:1296–302.e2. doi: 10.1016/j.jvs.2014.05.015
38. Meirson T, Orion E, Di Mario C, Webb C, Patel N, Channon KM, et al. Flow patterns in externally stented saphenous vein grafts and development of intimal hyperplasia. J Thorac Cardiovasc Surg. (2015) 150:871–8. doi: 10.1016/j.jtcvs.2015.04.061
39. Berard X, Déglise S, Alonso F, Saucy F, Meda P, Bordenave L, et al. Role of hemodynamic forces in the ex vivo arterialization of human saphenous veins. J Vasc Surg. (2013) 57:1371–82.
40. Wolf K, Hu H, Isaji T, Dardik A. Molecular identity of arteries, veins, and lymphatics. J Vasc Surg. (2019) 69:253–62.
41. Harskamp RE, Alexander JH, Schulte PJ, Brophy CM, Mack MJ, Peterson ED, et al. Vein graft preservation solutions, patency, and outcomes after coronary artery bypass graft surgery: follow-up from the PREVENT IV randomized clinical trial. JAMA Surg. (2014) 149:798–805. doi: 10.1001/jamasurg.2014.87
42. Chadjichristos CE, Morel S, Derouette JP, Sutter E, Roth I, Brisset AC, et al. Targeting connexin 43 prevents platelet-derived growth factor-BB-induced phenotypic change in porcine coronary artery smooth muscle cells. Circ Res. (2008) 102:653–60. doi: 10.1161/CIRCRESAHA.107.170472
43. Latif N, Sarathchandra P, Chester AH, Yacoub MH. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur Heart J. (2015) 36:1335–45.
44. DeLong SA, Moon JJ, West JL. Covalently immobilized gradients of bFGF on hydrogel scaffolds for directed cell migration. Biomaterials. (2005) 26:3227–34. doi: 10.1016/j.biomaterials.2004.09.021
45. Li Q, Fu J, Xia Y, Qi W, Ishikado A, Park K, et al. Homozygous receptors for insulin and not IGF-1 accelerate intimal hyperplasia in insulin resistance and diabetes. Nat Commun. (2019) 10:4427.
46. Duchek P, Somogyi K, Jékely G, Beccari S, Rørth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. (2001) 107:17–26.
47. Li L, Blumenthal DK, Terry CM, He Y, Carlson ML, Cheung AK. PDGF-induced proliferation in human arterial and venous smooth muscle cells: molecular basis for differential effects of PDGF isoforms. J Cell Biochem. (2011) 112:289–98. doi: 10.1002/jcb.22924
48. Sterpetti AV, Lepidi S, Borrelli V, Di Marzo L, Sapienza P, Cucina A, et al. Growth factors and experimental arterial grafts. J Vasc Surg. (2016) 64:1444–9.
49. Wang C, Liu Y, He D. Diverse effects of platelet-derived growth factor-BB on cell signaling pathways. Cytokine. (2019) 113:13–20.
50. Sterpetti AV, Cucina A, Lepidi S, Randone B, Stipa F, Aromatario C, et al. Progression and regression of myointimal hyperplasia in experimental vein grafts depends on platelet-derived growth factor and basic fibroblastic growth factor production. J Vasc Surg. (1996) 23:568–75.
51. Hoch JR, Stark VK, Turnipseed WD. The temporal relationship between the development of vein graft intimal hyperplasia and growth factor gene expression. J Vasc Surg. (1995) 22:51–8.
52. George SJ, Williams A, Newby AC. An essential role for platelet-derived growth factor in neointima formation in human saphenous vein in vitro. Atherosclerosis. (1996) 120:227–40. doi: 10.1016/0021-9150(95)05717-x
53. Yang Z, Oemar BS, Carrel T, Kipfer B, Julmy F, Lüscher TF. Different proliferative properties of smooth muscle cells of human arterial and venous bypass vessels: role of PDGF receptors, mitogen-activated protein kinase, and cyclin-dependent kinase inhibitors. Circulation. (1998) 97:181–7. doi: 10.1161/01.cir.97.2.181
54. Turner NA, Ho S, Warburton P, O’Regan DJ, Porter KE. Smooth muscle cells cultured from human saphenous vein exhibit increased proliferation, invasion, and mitogen-activated protein kinase activation in vitro compared with paired internal mammary artery cells. J Vasc Surg. (2007) 45:1022–8. doi: 10.1016/j.jvs.2007.01.061
55. Dong LH, Wen JK, Miao SB, Jia Z, Hu HJ, Sun RH, et al. Baicalin inhibits PDGF-BB-stimulated vascular smooth muscle cell proliferation through suppressing PDGFRβ-ERK signaling and increase in p27 accumulation and prevents injury-induced neointimal hyperplasia. Cell Res. (2010) 20:1252–62. doi: 10.1038/cr.2010.111
56. Uglow EB, Slater S, Sala-Newby GB, Aguilera-Garcia CM, Angelini GD, Newby AC, et al. Dismantling of cadherin-mediated cell-cell contacts modulates smooth muscle cell proliferation. Circ Res. (2003) 92:1314–21. doi: 10.1161/01.RES.0000079027.44309.53
57. Anstadt MP, Franga DL, Portik-Dobos V, Pennathur A, Bannan M, Mawulawde K, et al. Native matrix metalloproteinase characteristics may influence early stenosis of venous versus arterial coronary artery bypass grafting conduits. Chest. (2004) 125:1853–8. doi: 10.1378/chest.125.5.1853
58. Thomas AC, Newby AC. Effect of matrix metalloproteinase-9 knockout on vein graft remodelling in mice. J Vasc Res. (2010) 47:299–308.
59. Berceli SA, Jiang Z, Klingman NV, Pfahnl CL, Abouhamze ZS, Frase CD, et al. Differential expression and activity of matrix metalloproteinases during flow-modulated vein graft remodeling. J Vasc Surg. (2004) 39:1084–90. doi: 10.1016/j.jvs.2003.12.031
60. Turner NA, Hall KT, Ball SG, Porter KE. Selective gene silencing of either MMP-2 or MMP-9 inhibits invasion of human saphenous vein smooth muscle cells. Atherosclerosis. (2007) 193:36–43.
61. Perek B, Malinska A, Misterski M, Ostalska-Nowicka D, Zabel M, Perek A, et al. Preexisting high expression of matrix metalloproteinase-2 in tunica media of saphenous vein conduits is associated with unfavorable long-term outcomes after coronary artery bypass grafting. Biomed Res Int. (2013) 2013:730721. doi: 10.1155/2013/730721
62. George SJ, Wan S, Hu J, MacDonald R, Johnson JL, Baker AH. Sustained reduction of vein graft neointima formation by ex vivo TIMP-3 gene therapy. Circulation. (2011) 124(Suppl. 11):S135–42. doi: 10.1161/CIRCULATIONAHA.110.012732
63. Yu B, Kiechl S, Qi D, Wang X, Song Y, Weger S, et al. A cytokine-like protein dickkopf-related protein 3 is atheroprotective. Circulation. (2017) 136:1022–36. doi: 10.1161/CIRCULATIONAHA.117.027690
64. Sharony R, Pintucci G, Saunders PC, Grossi EA, Baumann FG, Galloway AC, et al. Matrix metalloproteinase expression in vein grafts: role of inflammatory mediators and extracellular signal-regulated kinases-1 and -2. Am J Physiol Heart Circ Physiol. (2006) 290:H1651–9. doi: 10.1152/ajpheart.00530.2005
65. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. (2015) 116:1231–44.
66. Ajami NE, Gupta S, Maurya MR, Nguyen P, Li JY, Shyy JY, et al. Systems biology analysis of longitudinal functional response of endothelial cells to shear stress. Proc Natl Acad Sci U S A. (2017) 114:10990–5.
67. Meng XB, Bi XL, Zhao HL, Feng JB, Zhang JP, Song GM, et al. Small interfering RNA targeting nuclear factor kappa B to prevent vein graft stenosis in rat models. Transplant Proc. (2013) 45:2553–8. doi: 10.1016/j.transproceed.2013.03.045
68. Varejckova M, Gallardo-Vara E, Vicen M, Vitverova B, Fikrova P, Dolezelova E, et al. Soluble endoglin modulates the pro-inflammatory mediators NF-κB and IL-6 in cultured human endothelial cells. Life Sci. (2017) 175:52–60. doi: 10.1016/j.lfs.2017.03.014
69. Sanders WG, Morisseau C, Hammock BD, Cheung AK, Terry CM. Soluble epoxide hydrolase expression in a porcine model of arteriovenous graft stenosis and anti-inflammatory effects of a soluble epoxide hydrolase inhibitor. Am J Physiol Cell Physiol. (2012) 303:C278–90. doi: 10.1152/ajpcell.00386.2011
70. Stark VK, Hoch JR, Warner TF, Hullett DA. Monocyte chemotactic protein-1 expression is associated with the development of vein graft intimal hyperplasia. Arterioscler Thromb Vasc Biol. (1997) 17:1614–21. doi: 10.1161/01.atv.17.8.1614
71. Mori E, Komori K, Yamaoka T, Tanii M, Kataoka C, Takeshita A, et al. Essential role of monocyte chemoattractant protein-1 in development of restenotic changes (neointimal hyperplasia and constrictive remodeling) after balloon angioplasty in hypercholesterolemic rabbits. Circulation. (2002) 105:2905–10. doi: 10.1161/01.cir.0000018603.67989.71
72. Schepers A, Eefting D, Bonta PI, Grimbergen JM, de Vries MR, van Weel V, et al. Anti-MCP-1 gene therapy inhibits vascular smooth muscle cells proliferation and attenuates vein graft thickening both in vitro and in vivo. Arterioscler Thromb Vasc Biol. (2006) 26:2063–9. doi: 10.1161/01.ATV.0000235694.69719.e2
73. Preiss DJ, Sattar N. Vascular cell adhesion molecule-1: a viable therapeutic target for atherosclerosis? Int J Clin Pract. (2007) 61:697–701. doi: 10.1111/j.1742-1241.2007.01330.x
74. Qu Y, Shi X, Zhang H, Sun W, Han S, Yu C, et al. VCAM-1 siRNA reduces neointimal formation after surgical mechanical injury of the rat carotid artery. J Vasc Surg. (2009) 50:1452–8. doi: 10.1016/j.jvs.2009.08.050
75. Suzuki J, Ogawa M, Takayama K, Taniyama Y, Morishita R, Hirata Y, et al. Ultrasound-microbubble-mediated intercellular adhesion molecule-1 small interfering ribonucleic acid transfection attenuates neointimal formation after arterial injury in mice. J Am Coll Cardiol. (2010) 55:904–13. doi: 10.1016/j.jacc.2009.09.054
76. Huang X, Xu MQ, Zhang W, Ma S, Guo W, Wang Y, et al. ICAM-1-Targeted liposomes loaded with liver X receptor agonists suppress PDGF-induced proliferation of vascular smooth muscle cells. Nanoscale Res Lett. (2017) 12:322. doi: 10.1186/s11671-017-2097-6
77. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-y
78. Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. (2000) 403:901–6.
79. Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. (2000) 408:86–9.
80. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. (2005) 120:15–20. doi: 10.1016/j.cell.2004.12.035
81. Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. (2011) 39:D152–7.
82. Santovito D, Weber C. Non-canonical features of microRNAs: paradigms emerging from cardiovascular disease. Nat Rev Cardiol. (2022): [Online ahead of print].
83. Yang WJ, Yang DD, Na S, Sandusky GE, Zhang Q, Zhao G. Dicer is required for embryonic angiogenesis during mouse development. J Biol Chem. (2005) 280:9330–5.
84. Yeh YT, Wei J, Thorossian S, Nguyen K, Hoffman C, Del Álamo JC, et al. MiR-145 mediates cell morphology-regulated mesenchymal stem cell differentiation to smooth muscle cells. Biomaterials. (2019) 204:59–69. doi: 10.1016/j.biomaterials.2019.03.003
85. Xie C, Huang H, Sun X, Guo Y, Hamblin M, Ritchie RP, et al. MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem Cells Dev. (2011) 20:205–10. doi: 10.1089/scd.2010.0283
86. Xu F, Ahmed AS, Kang X, Hu G, Liu F, Zhang W, et al. MicroRNA-15b/16 attenuates vascular neointima formation by promoting the contractile phenotype of vascular smooth muscle through targeting YAP. Arterioscler Thromb Vasc Biol. (2015) 35:2145–52. doi: 10.1161/ATVBAHA.115.305748
87. Li J, Zhao L, He X, Yang T, Yang K. MiR-21 inhibits c-Ski signaling to promote the proliferation of rat vascular smooth muscle cells. Cell Signal. (2014) 26:724–9. doi: 10.1016/j.cellsig.2013.12.013
88. Yang F, Chen Q, He S, Yang M, Maguire EM, An W, et al. miR-22 is a novel mediator of vascular smooth muscle cell phenotypic modulation and neointima formation. Circulation. (2018) 137:1824–41. doi: 10.1161/CIRCULATIONAHA.117.027799
89. Iaconetti C, De Rosa S, Polimeni A, Sorrentino S, Gareri C, Carino A, et al. Down-regulation of miR-23b induces phenotypic switching of vascular smooth muscle cells in vitro and in vivo. Cardiovasc Res. (2015) 107:522–33. doi: 10.1093/cvr/cvv141
90. Chen Q, Yang F, Guo M, Wen G, Zhang C, Luong LA, et al. miRNA-34a reduces neointima formation through inhibiting smooth muscle cell proliferation and migration. J Mol Cell Cardiol. (2015) 89(Pt A):75–86. doi: 10.1016/j.yjmcc.2015.10.017
91. Chen Z, Wang M, Huang K, He Q, Li H, Chang G. MicroRNA-125b affects vascular smooth muscle cell function by targeting serum response factor. Cell Physiol Biochem. (2018) 46:1566–80. doi: 10.1159/000489203
92. Jansen F, Stumpf T, Proebsting S, Franklin BS, Wenzel D, Pfeifer P, et al. Intercellular transfer of miR-126-3p by endothelial microparticles reduces vascular smooth muscle cell proliferation and limits neointima formation by inhibiting LRP6. J Mol Cell Cardiol. (2017) 104:43–52. doi: 10.1016/j.yjmcc.2016.12.005
93. Chen WJ, Chen YH, Hsu YJ, Lin KH, Yeh YH. MicroRNA-132 targeting PTEN contributes to cilostazol-promoted vascular smooth muscle cell differentiation. Atherosclerosis. (2018) 274:1–7. doi: 10.1016/j.atherosclerosis.2018.04.030
94. Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. (2011) 109:880–93. doi: 10.1161/CIRCRESAHA.111.240150
95. Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. (2009) 460:705–10.
96. Wang YS, Wang HY, Liao YC, Tsai PC, Chen KC, Cheng HY, et al. MicroRNA-195 regulates vascular smooth muscle cell phenotype and prevents neointimal formation. Cardiovasc Res. (2012) 95:517–26. doi: 10.1093/cvr/cvs223
97. Afzal TA, Luong LA, Chen D, Zhang C, Yang F, Chen Q, et al. NCK associated protein 1 modulated by miRNA-214 determines vascular smooth muscle cell migration, proliferation, and neointima hyperplasia. J Am Heart Assoc. (2016) 5:e004629. doi: 10.1161/JAHA.116.004629
98. Shan Z, Qin S, Li W, Wu W, Yang J, Chu M, et al. An endocrine genetic signal between blood cells and vascular smooth muscle cells: role of MicroRNA-223 in smooth muscle function and atherogenesis. J Am Coll Cardiol. (2015) 65:2526–37. doi: 10.1016/j.jacc.2015.03.570
99. Sun Y, Yang Z, Zheng B, Zhang XH, Zhang ML, Zhao XS, et al. A novel regulatory mechanism of smooth Muscle α-Actin expression by NRG-1/circACTA2/miR-548f-5p axis. Circ Res. (2017) 121:628–35.
100. Li P, Liu Y, Yi B, Wang G, You X, Zhao X, et al. MicroRNA-638 is highly expressed in human vascular smooth muscle cells and inhibits PDGF-BB-induced cell proliferation and migration through targeting orphan nuclear receptor NOR1. Cardiovasc Res. (2013) 99:185–93. doi: 10.1093/cvr/cvt082
101. Li P, Zhu N, Yi B, Wang N, Chen M, You X, et al. MicroRNA-663 regulates human vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Circ Res. (2013) 113:1117–27. doi: 10.1161/CIRCRESAHA.113.301306
102. Hu W, Wang M, Yin H, Yao C, He Q, Yin L, et al. MicroRNA-1298 is regulated by DNA methylation and affects vascular smooth muscle cell function by targeting connexin 43. Cardiovasc Res. (2015) 107:534–45. doi: 10.1093/cvr/cvv160
103. Zahedi F, Nazari-Jahantigh M, Zhou Z, Subramanian P, Wei Y, Grommes J, et al. Dicer generates a regulatory microRNA network in smooth muscle cells that limits neointima formation during vascular repair. Cell Mol Life Sci. (2017) 74:359–72. doi: 10.1007/s00018-016-2349-0
104. Sun HJ, Zhao MX, Ren XS, Liu TY, Chen Q, Li YH, et al. Salusin-β promotes vascular smooth muscle cell migration and intimal hyperplasia after vascular injury via ROS/NFκB/MMP-9 pathway. Antioxid Redox Signal. (2016) 24:1045–57.
105. Lin Y, Liu X, Cheng Y, Yang J, Huo Y, Zhang C. Involvement of MicroRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. J Biol Chem. (2009) 284:7903–13. doi: 10.1074/jbc.M806920200
106. Zhang J, Cai W, Fan Z, Yang C, Wang W, Xiong M, et al. MicroRNA-24 inhibits the oxidative stress induced by vascular injury by activating the Nrf2/Ho-1 signaling pathway. Atherosclerosis. (2019) 290:9–18.
107. Zhou B, Wu LL, Zheng F, Wu N, Chen AD, Zhou H, et al. miR-31-5p promotes oxidative stress and vascular smooth muscle cell migration in spontaneously hypertensive rats via inhibiting FNDC5 expression. Biomedicines. (2021) 9:1009. doi: 10.3390/biomedicines9081009
108. Jin Y, Pang T, Nelin LD, Wang W, Wang Y, Yan J, et al. MKP-1 is a target of miR-210 and mediate the negative regulation of miR-210 inhibitor on hypoxic hPASMC proliferation. Cell Biol Int. (2015) 39:113–20. doi: 10.1002/cbin.10339
109. Santovito D, Egea V, Bidzhekov K, Natarelli L, Mourão A, Blanchet X, et al. Noncanonical inhibition of caspase-3 by a nuclear microRNA confers endothelial protection by autophagy in atherosclerosis. Sci Transl Med. (2020) 12:eaaz2294. doi: 10.1126/scitranslmed.aaz2294
110. Shi X, Ma W, Pan Y, Li Y, Wang H, Pan S, et al. MiR-126-5p promotes contractile switching of aortic smooth muscle cells by targeting VEPH1 and alleviates Ang II-induced abdominal aortic aneurysm in mice. Lab Invest. (2020) 100:1564–74. doi: 10.1038/s41374-020-0454-z
111. Climent M, Quintavalle M, Miragoli M, Chen J, Condorelli G, Elia LTGF. β triggers miR-143/145 transfer from smooth muscle cells to endothelial cells, thereby modulating vessel stabilization. Circ Res. (2015) 116:1753–64. doi: 10.1161/CIRCRESAHA.116.305178
112. Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJ, Zeiher AM, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. (2012) 14:249–56.
113. Albinsson S, Suarez Y, Skoura A, Offermanns S, Miano JM, Sessa WC, et al. MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler Thromb Vasc Biol. (2010) 30:1118–26.
115. Horita HN, Simpson PA, Ostriker A, Furgeson S, Van Putten V, Weiser-Evans MC, et al. Serum response factor regulates expression of phosphatase and tensin homolog through a microRNA network in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2011) 31:2909–19. doi: 10.1161/ATVBAHA.111.233585
116. McDonald RA, White KM, Wu J, Cooley BC, Robertson KE, Halliday CA, et al. miRNA-21 is dysregulated in response to vein grafting in multiple models and genetic ablation in mice attenuates neointima formation. Eur Heart J. (2013) 34:1636–43. doi: 10.1093/eurheartj/eht105
117. Zhao N, Koenig SN, Trask AJ, Lin CH, Hans CP, Garg V, et al. MicroRNA miR145 regulates TGFBR2 expression and matrix synthesis in vascular smooth muscle cells. Circ Res. (2015) 116:23–34. doi: 10.1161/CIRCRESAHA.115.303970
118. Ohnaka M, Marui A, Yamahara K, Minakata K, Yamazaki K, Kumagai M. Effect of microRNA-145 to prevent vein graft disease in rabbits by regulation of smooth muscle cell phenotype. J Thorac Cardiovasc Surg. (2014) 148:676–682.e2. doi: 10.1016/j.jtcvs.2013.11.054
119. Wang XW, He XJ, Lee KC, Huang C, Hu JB, Zhou R, et al. MicroRNA-221 sponge therapy attenuates neointimal hyperplasia and improves blood flows in vein grafts. Int J Cardiol. (2016) 208:79–86. doi: 10.1016/j.ijcard.2016.01.006
120. Cao BJ, Zhu L, Wang XW, Zou RJ, Lu ZQ. MicroRNA-365 promotes the contractile phenotype of venous smooth muscle cells and inhibits neointimal formation in rat vein grafts. IUBMB Life. (2019) 71:908–16. doi: 10.1002/iub.2022
121. Zhang D, Shi J, Liang G, Liu D, Zhang J, Pan S, et al. miR-16-5p is a novel mediator of venous smooth muscle phenotypic switching. J Cardiovasc Transl Res. (2022): [Online ahead of print], doi: 10.1007/s12265-022-10208-1
Keywords: miRNAs, VSMCs, phenotypic transformation, intimal hyperplasia, vein graft
Citation: Zhang D, Cao Y, Liu D, Zhang J and Guo Y (2022) The Etiology and Molecular Mechanism Underlying Smooth Muscle Phenotype Switching in Intimal Hyperplasia of Vein Graft and the Regulatory Role of microRNAs. Front. Cardiovasc. Med. 9:935054. doi: 10.3389/fcvm.2022.935054
Received: 03 May 2022; Accepted: 20 June 2022;
Published: 28 July 2022.
Edited by:
Chengxing Shen, Shanghai Jiao Tong University, ChinaReviewed by:
Donato Santovito, National Research Council (CNR), ItalyLingfang Zeng, King’s College London, United Kingdom
Copyright © 2022 Zhang, Cao, Liu, Zhang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingqiang Guo, ZHJndW95cUBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work