 Anjali Rao1,2,3Andrew Stewart3,4
Anjali Rao1,2,3Andrew Stewart3,4 Mahmoud Eljalby3,4
Mahmoud Eljalby3,4 Praveen Ramakrishnan5
Praveen Ramakrishnan5 Larry D. Anderson Jr.5,6
Larry D. Anderson Jr.5,6 Farrukh T. Awan5Alvin Chandra1,2,3
Farrukh T. Awan5Alvin Chandra1,2,3 Srilakshmi Vallabhaneni1,2,3Kathleen Zhang1,2,3
Srilakshmi Vallabhaneni1,2,3Kathleen Zhang1,2,3 Vlad G. Zaha1,2,3*
Vlad G. Zaha1,2,3*- 1Division of Cardiology, Department of Internal Medicine, UT Southwestern Medical Center, Dallas, TX, United States
- 2Cardio-Oncology Program, Harold C. Simmons Comprehensive Cancer Center, UT Southwestern Medical Center, Dallas, TX, United States
- 3Parkland Health and Hospital System, Dallas, TX, United States
- 4Department of Internal Medicine, UT Southwestern Medical Center, Dallas, TX, United States
- 5Division of Hematology and Oncology, Department of Internal Medicine, UT Southwestern Medical Center, Dallas, TX, United States
- 6Myeloma, Waldenstrom's, and Amyloidosis Program, Harold C. Simmons Comprehensive Cancer Center, UT Southwestern Medical Center, Dallas, TX, United States
Chimeric antigen receptor T-cell (CAR T) therapy is a revolutionary personalized therapy that has significantly impacted the treatment of patients with hematologic malignancies refractory to other therapies. Cytokine release syndrome (CRS) is a major side effect of CAR T therapy that can occur in 70–90% of patients, with roughly 40% of patients at grade 2 or higher. CRS can cause an intense inflammatory state leading to cardiovascular complications, including troponin elevation, arrhythmias, hemodynamic instability, and depressed left ventricular systolic function. There are currently no standardized guidelines for the management of cardiovascular complications due to CAR T therapy, but systematic practice patterns are emerging. In this review, we contextualize the history and indications of CAR T cell therapy, side effects related to this treatment, strategies to optimize the cardiovascular health prior to CAR T and the management of cardiovascular complications related to CRS. We analyze the existing data and discuss potential future approaches.
Introduction
The power of the immune system in treating neoplastic diseases has long been recognized in the medical community. However, starting from adoptive cell transfer, the precursor of CAR T, various cardiovascular toxic side effects have also been identified. Herein we review the available data, and propose a strategy for prevention, surveillance and management of cardiovascular toxicity in patients receiving immune cellular therapies.
Adoptive cell transfer
Adoptively acquired immunity is the process through which active immune tissues are transferred from a donor to a recipient (1–3). Initial studies performed in the 1950's demonstrated in mouse that immune tissue (i.e., spleen or lymph nodes) but not antigens or peripheral cells from a primary transplant tolerant host induced sustained resistance to rejection in a secondary host (1). In a landmark paper published in 1957, E. Donnall Thomas and colleagues demonstrated a sustained response after bone marrow infusion in several patients with bone marrow deficiency following radiation and chemotherapy (3). This led to the first allogeneic bone marrow transplantations in the early 1960s, using bone marrow from twin siblings. With the subsequent development of autologous stem cell transplantation, adoptive cellular therapies have become a mainstay in the treatment of hematologic malignancies (4).
Modern development of cellular therapies
Following the historic success of bone marrow transplantation, the next phase of adoptive cell transfer came in the 1980s with the emergence of tumor-infiltrating lymphocytes (TIL) (5–8). In this therapy, B- and T-cells isolated from the tumor biopsy are expanded in a laboratory and subsequently infused back into the original host after a dose of chemotherapy (5, 6). TIL were combined with interleukin-2, a key cytokine in the proliferation and differentiation of effector T cells, to enhance their antitumor effects (5, 6).
With the advent of gene-transfer techniques, the potential of peripheral blood T cells was further harnessed through genetic modifications that increase their specificity and augment their function (9, 10). These “first-generation” genetically modified T cells were engineered to express a chimeric antigen receptor (CAR)—composed of an extracellular single-chain variable fragment (scFv) that serves as the targeting moiety, a transmembrane spacer, and intracellular signaling/activation domain(s)—to target surface-exposed tumor-associated antigens (10–12). Over time, CARs evolved to more complex “second-” and “third-generation” CARs that have augmented T cell persistence and proliferation (13–16).
Chimeric antigen receptor T-cell therapy mechanism and indications
The development of CAR T cell therapy triggered a paradigm shift in cancer immunotherapy, demonstrating remarkable success particularly in CD-19 expressing malignancies, as the first genetically engineered personalized therapy option. This therapeutic option has become a viable and commercially available treatment option for several hematologic malignancies (Table 1). Promising results emerged from the initial CAR T trials of tisagenlecleucel (tisa-cel) and axicabtagene ciloleucel (axi-cel) in 2017 (17). Tisa-cel was the first anti-CD-19 CAR T product approved by the Food and Drug Administration (FDA), for patients up to 25 years of age with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL) in 2017 (17). Axi-cel, an anti-CD-19 targeting CAR T-cell, approval followed soon after in 2017 for patients with relapsed or refractory diffuse large B-cell lymphoma (18). Axi-cel was subsequently also approved for the management of patients with relapsed or refractory follicular lymphoma after 2 prior lines of therapy (19). Since then, the FDA has approved 6 total CAR T therapies for the treatment of hematologic malignancies, including lisocabtagene maraleucel (liso-cel) for relapsed or refractory diffuse large B-cell lymphoma, brexucabtagene autoleucel (brexu-cel) for relapsed or refractory mantle cell lymphoma and relapsed or refractory ALL, and idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cita-cel) for relapsed and refractory multiple myeloma (18, 20–23) (Table 1). Responses for all these agents average around 60 to 80% with complete remissions achieved in approximately 40 to 60% of the patients (17–19, 21, 22). These results are especially striking given the failure of conventional chemotherapy, including high-dose chemotherapy and stem cell transplantation in this population.

Table 1. Summary of current FDA-approved CAR T generic names, trade names, and indications.
Chimeric antigen receptor T-cell therapy induction and administration
The administration of CAR T requires the identification of optimal patients who would generally be considered healthy and fit to undergo this procedure. While there is no established consensus on the optimal patient profile that would be considered suitable, various guidelines suggest utilizing established fitness and morbidity scores to determine eligibility (24–26). After harvesting the peripheral blood product through a routine apheresis procedure, the cells typically require processing and manufacturing which can take up to 4–6 weeks. During this interval, patients frequently require “bridging therapy” to ensure that they do not have rapid and symptomatic disease progression. Following successful manufacturing and receipt of the product, patients undergo lymphodepleting chemotherapy typically with fludarabine and cyclophosphamide over 3 days for up to a week prior to reinfusion of the cells. Patients are subsequently monitored closely for the development of cytokine release syndrome (CRS) and neurotoxicity which can manifest for approximately the first month after reinfusion of cells (24, 25). Because of the risks noted with CRS, patients must enter a risk evaluation and mitigation strategy (REMS) program and stay within 2 h of the CAR T center for the first month and must not drive for 2 months following CAR T.
Immune cell-related adverse events
Robust systemic release of a high level of cytokines following overwhelming T cell activation as well as specific interactions between the CAR and its target antigen expressed by non-malignant cells are two mechanisms thought to mediate CAR T toxicities (27). One of the most common CAR T cell-related adverse events is CRS. CRS is a multisystem inflammatory response mediated by a surge of cytokines triggered by an infusion of CAR T cells. Among other toxic phenomena, CRS, in particular, affects 37–93% of patients with lymphoma (28), and 77–93% of patients with leukemia (28–31). Clinical manifestations can range from fevers and constitutional symptoms to hypoxia, hypotension, end-organ damage, and even sepsis-like syndrome or death in severe cases (29). CRS is thought to result from widespread simultaneous activation of T-cells and release of cytokines and chemokines (30, 32). CRS has been associated with elevation of interleukin (IL)-6, IL-8, IL-10, IL-15, GM-CSF, interferon (IFN)-g, MCP-1, MIP-1b, ferritin, CRP, and in severe cases soluble IL-2 receptor (28, 33). Management includes supportive care and antipyretics in mild cases, administration of IL-6-receptor antagonists like tocilizumab in moderate CRS or those not responding to supportive care, and corticosteroids like dexamethasone in more severe cases of CRS (34, 35). CRS can occasionally mimic macrophage activation syndrome (MAS) or hemophagocytic lymphohistiocytosis (HLH) in severe cases, which is often treated with anakinra, an IL-1 receptor antagonist, if the above measures are not effective (36–39). Serum inflammatory markers (acute phase reactants) including c-reactive protein (CRP) and ferritin may be followed clinically to help aid in prediction of impending CRS or to monitor response to therapy, though cytokine levels are not often readily available in real time (39).
CRS may contribute to the development of immune cell-associated neurotoxicity syndrome (ICANS), which can manifest along a spectrum from mild delirium with confusion to cerebral edema, seizures, and even death (34, 40). Cardiovascular manifestations of CRS Although the underlying mechanism of ICANS is incompletely understood compared to CRS, studies have also shown a correlation with elevated levels of inflammatory cytokines like IL-6, IFN-γ and TNFα (33, 41, 42). These signals are postulated to cause endothelial damage and activation with disruption of the blood brain barrier and capillary leak. It requires careful monitoring, frequent assessments, and promptly initiated therapy. ICANS has also been associated with sinus bradycardia that is often self-limited without need for intervention but should be monitored closely (43). Other constitutional, hematologic, renal, gastrointestinal, and dermatologic toxicities have also been observed (28, 41, 44–46).
Cardiovascular complications of cellular imunotherapies
While there has been a consistent trend of improvement in the survival following both autologous and allogeneic hematopoietic cell transplantation bone marrow transplant therapies decade over decade (47, 48), cardiovascular toxicities (49) continue to be frequent complications, along with infections and graft vs. host disease. This has resulted in evolving practice guidelines targeting preventive evaluations pretransplant, monitoring peri-transplant, and surveillance in long term survivors (50, 51). With regard to CAR T therapy, the current information about cardiovascular side effects related to CAR T therapies is limited to a few retrospective studies (Table 2), but concepts established for other adoptive cell transfers likely apply. In particular, with the growing prevalence of cardiovascular disease combined with the increase in available CAR T cell therapies for the treatment of hematologic malignancies, attempting to understand the mechanisms of these complications is essential as this may help guide interventions.
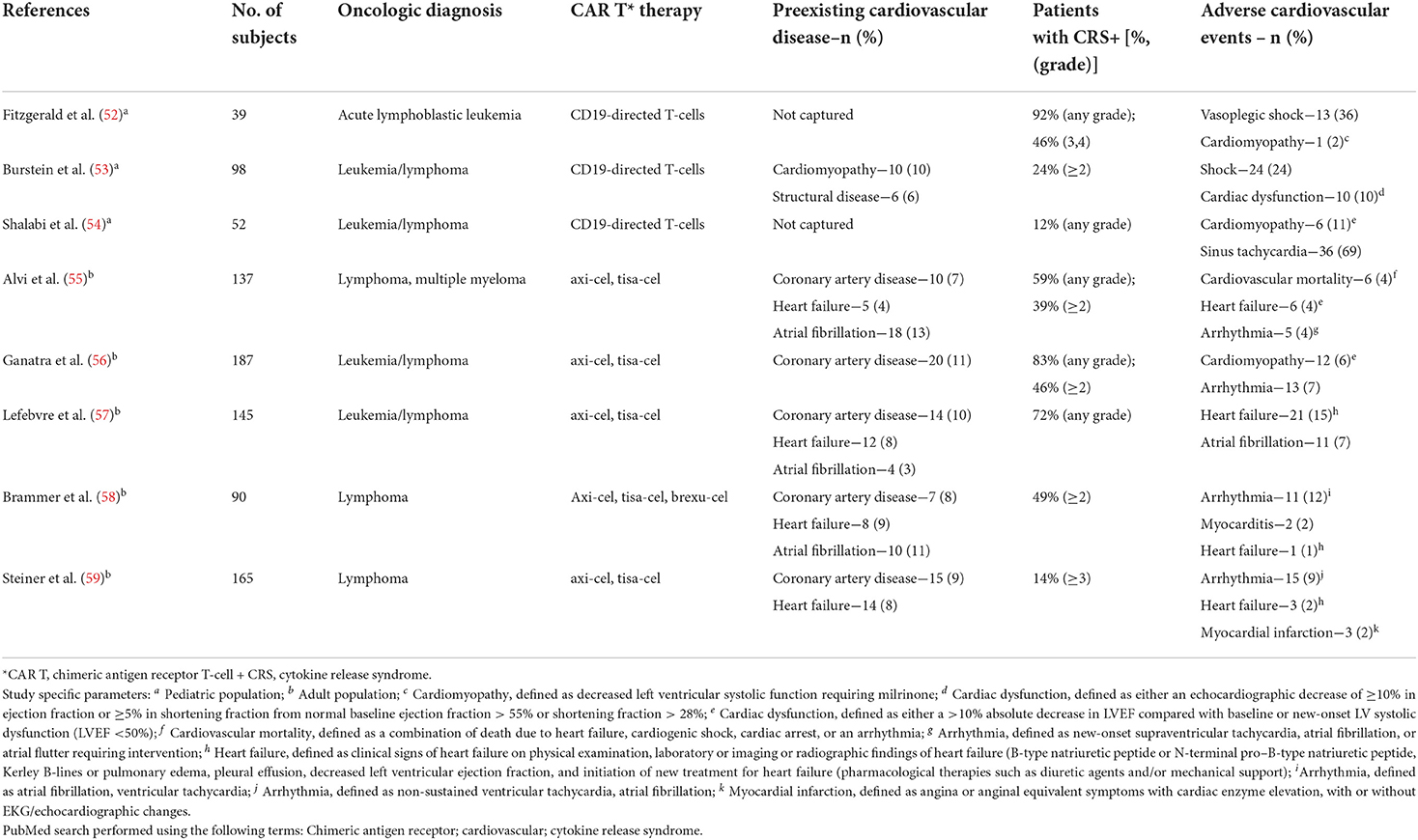
Table 2. Summary of pediatric and adult studies investigated cardiovascular complications and CAR T-cell therapy.
The impact of CAR T cell therapy on the cardiovascular system manifests as hemodynamic compromise, myocardial injury/dysfunction, and/or cardiac arrhythmias (60, 61). There is also the potential for pericardial complications, such as in a case report (62) describing a patient with high-grade lymphoma who developed a pericardial effusion and tamponade with cardiogenic shock after CAR T therapy. Higher-grade CRS appears to be linked to adverse cardiovascular events of all types. This is likely driven by the release of inflammatory cytokines into the bloodstream with CAR T therapy, particularly the secretion of interleukin-6 (IL-6). This cytokine is a mediator of systemic inflammation, leading to hemodynamic compromise and even circulatory collapse in CRS. Of the studies published so far in patients treated with CAR T cell therapy, cardiovascular monitoring was performed in 3 pediatric studies and 5 adult studies (52–59). All studies in adult populations were retrospective, single-center observational cohort studies. Across all studies, cardiovascular complications have been inconsistently monitored. In children, transient and reversible hypotension in the setting of high-grade CRS was more commonly noted. In studies that monitored for cardiovascular complications in adults, the most frequently observed were cardiac arrhythmias and heart failure, albeit with relatively low event rates overall. Interestingly, preexisting cardiovascular disease (including heart failure) has not been shown to be reliably associated with the development of cardiovascular complications after CAR T cell therapy in one cohort study (57). In contrast, in another cohort study (55), troponin elevation was notably associated with cardiovascular adverse events in patients undergoing CAR T cell therapy. The patients with troponin elevation in this study were older and had more traditional cardiovascular risk factors. In both these cohort studies cardiovascular complications occurred with increased frequency at higher grades of CRS (2 or greater). As such, additional studies in larger cohorts are needed to establish risk factors, biomarker elevation patterns, imaging findings, event rates, and outcomes after CAR T cell therapy.
CRS monitoring and grading
Most patients undergoing CAR T can be managed on the regular cell therapy hospital floor with only a minority requiring ICU care, but close monitoring and specialty care is. due to rapid onset of CRS, it is recommended that this therapy is given at a specialized center with CAR T experience and credentialling.
Grading of CRS is now done per the American Society of Transplantation and Cellular Therapy (ASTCT) consensus guidelines (Table 3) (34).

Table 3. American Society of Blood and Marrow Transplantation (ASBMT) consensus grading of cytokine release syndrome (CRS) severity (34).
CRS management
Rates of CRS and median time to onset vary depending on the particular CAR T product and disease burden. For example, in the KarMMa study of ide-cel for relapsed/refractory multiple myeloma (22), CRS was seen in 84% of patients, but most cases were only grade 1 or 2, with only 5% of patients developing grade 3–5. Median time to onset of CRS in the KarMMa study was 1 day (range 1–12 days) with a median duration of 5 days (range 1–63).
Management of CRS required tocilizumab in 52% patients, but only 15% required glucocorticoids (22, 63). On the other hand, in the Zuma-1 study of axi-cel for relapsed/refractory large B-cell lymphomas, CRS was a nearly universal side effect, with 93% of patients experiencing any grade CRS and 11% with grade 3 or higher, and hypotension was seen in 63%, tachycardia in 40%, and hypoxia in 34% (64). The median time to onset of CRS was 2 days (range 1–12) with a median duration of 8 days (65). All patients had resolution of their CRS, except for one patient who died from complications of HLH, and another patient who died of cardiac arrest with ongoing CRS. Tocilizumab was given in 43% and corticosteroids were required in 27% of Zuma-1 patients; however, more recently the FDA has issued a new label change for axi-cel allowing the prophylactic use of 3 days of corticosteroids based on a study showing much less severe CRS and ICANS without impairment of lymphoma response rates (66). The decision regarding inpatient vs. outpatient care and aggressive early therapy vs. minimal therapy for CRS is not only made based on the track record of the particular CAR T product but also based on risk factors such as age, frailty, and tumor burden, as higher tumor burden consistently correlates with increased incidence and severity of CRS (67).
Surveillance for cardiovascular toxicity
At our institution, cardiovascular (CV) surveillance for CAR-T therapy begins with CV risk stratification prior to infusion. Patients with CV comorbidities (especially heart failure, coronary artery disease, arrhythmias) or new/worsening CV symptoms (i.e., chest pain, dyspnea on exertion, lower extremity edema) represent a high CV risk group. Older age and prior cardiotoxic cancer therapy (i.e., anthracyclines, chest radiation) may also raise the risk of CV toxicity after treatment (68). In these high CV risk patients, standard baseline testing should include a 12-lead electrocardiogram, cardiac biomarkers (troponin, NT-proBNP), and transthoracic echocardiography. In some cases, cardiac MRI may clarify features of cardiac structure and/or function that would guide optimization of CV therapy. Cardioprotective therapies such as beta-blockers and renin-angiotensin-aldosterone system blockers, diuretics, and/or antiarrhythmics should be utilized as clinically indicated. In addition, any patient with the above cardiovascular comorbidities, and whose baseline electrocardiogram or transthoracic echocardiogram is abnormal, should be considered for cardio-oncology referral pre-CAR T therapy.
Inpatient monitoring after CAR-T infusion is strongly recommended for patients with increased baseline CV risk. Figure 1 shows our institutional algorithm for surveillance and monitoring in this population. Standard monitoring protocols after CAR-T infusion include daily blood counts and metabolic profiling, physical examination, and screening for CRS (69). Patients at high baseline CV risk should additionally be monitored on telemetry with close monitoring of oral and intravenous fluid input, urine output, and daily body weight measurement. Given the observed association between CRS and CV events after CAR-T (55, 57), all patients with grade 3 or 4 CRS should also be placed on these CV monitoring protocols.
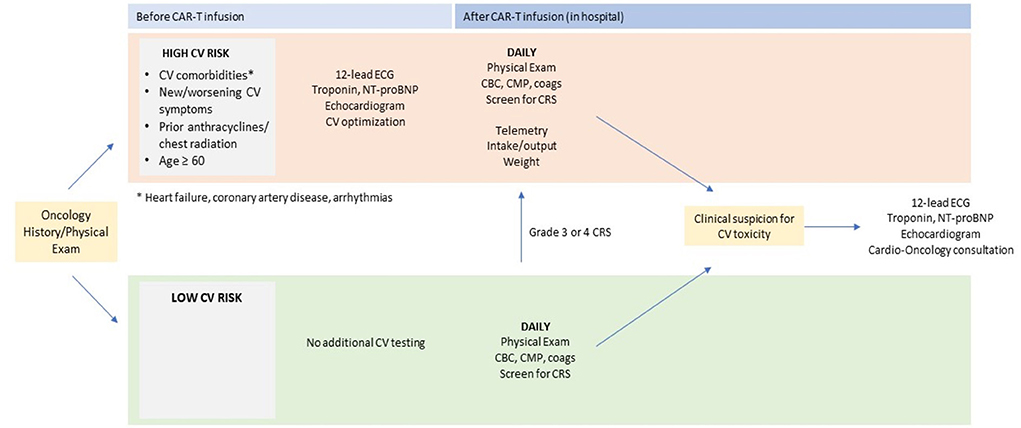
Figure 1. Proposed pre- and post-CAR T cardiac screening. CAR, chimeric antigen receptor; ECG, electrocardiography; NT-proBNP, N-terminal pro–B-type natriuretic peptide. *arrhythmias, coronary artery disease, heart failure.
The utility of routine cardiac biomarker testing for detection of CV toxicity after CAR-T is uncertain. Where there is clinical suspicion for a CV event after CAR-T infusion based on symptoms or monitoring, initial evaluation should include cardiac biomarkers (troponin, NT-proBNP), 12-lead electrocardiogram (ECG), and transthoracic echocardiography (TTE). Cardio-oncology consultation should be obtained, if available, to direct further diagnostic evaluation and management.
Data are limited regarding the optimal surveillance and testing protocol for patients undergoing CAR T cell therapy. Current standards of practice have been previously published by Hayden et al. (26), Ghosh et al. (60), and Totzeck et al. (70), with similar approaches to our institution and each other with regard to screening and surveillance while on CAR T therapy. Ghosh et al. propose that all patients undergo baseline cardiac magnetic resonance imaging (CMR) with follow-up CMR in patients with abnormal biomarkers, ECG, and/or TTE. We generally agree with these publications on the initial evaluation of patients after a cardiovascular event with CAR-T infusion, including cardiac blood biomarkers (troponin, NT-proBNP), ECG, and TTE, with judicious use of CMR in appropriate cases. By contrast, there is some variability in the post-CAR T surveillance and monitoring approaches proposed by the other consensus approaches. For example, Ghosh et al. recommend for all patients to follow-up with cardio-oncology 3 months after CAR T cell therapy, whereas the other two consensus recommendations propose a 7-day follow-up visit. We propose a patient-specific approach depending on the type of cardiovascular event that patient experienced. The utility of monitoring for late effects (i.e., at 3 months post CAR-T and beyond) and the potential for long-term CV consequences of CAR-T itself stand out as areas for future study.
Future directions
Current targets of CAR T are malignant immune cells, but new targets continue to develop. There has been an expanding focus on targeting solid tumors, and overall, nearly 600 clinical trials are underway (71–73). Multiple new endeavors are focusing on solid tumor surface antigens such as carcinoembryonic antigen (CEA), ganglioside GD2 subtype, mesothelin, interleukin-13 receptor α (IL-13Rα), human epidermal growth factor receptor 2 (HER2), fibroblast activation protein (FAP), and L1 cell adhesion molecule (L1CAM) (16, 74–79).
Multiple trials are currently ongoing evaluating various CAR T products in different disease entities including allogeneic products utilizing various T-cell and NK-cell engineering and manufacturing procedures. Moreover, the well-documented side effects of CAR T–most notably, CRS–have spurred the recent discussion surrounding CAR NK-cell therapy, a potential avenue to mitigatehe systemic immune effects (73). CAR T has been shown to effectively target and remove activated cardiac fibroblasts in mice, suggesting potential applications to address myocardial scar and fibrosis (80, 81). At the same time, early signals have raised concerns about the unique dangers of systemic immune effects in patients with preceding cardiovascular diseases or cardiovascular risks, with limited information about cardiotoxicity available from the initial CAR T trials. Clinical practice guidelines are emerging to address immune cell-related adverse events (82). Next steps also include validated risk prediction tools for cardiovascular complications after CAR-T, elucidate mechanisms of these immune-mediated complications, development of preventative therapies by integrating timelines of cardiac blood biomarkers and immunophenotyping in this population.
Conclusions
The rapid development of immunocellular personalized therapeutic modalities is creating unprecedented opportunities for treatment of cancers. To optimize the cardiovascular outcomes in patients treated with CAR T several lessons learned from other anticancer therapies and from early CAR T studies may be beneficial. While early studies have established the specific indications for these therapies, cardiovascular risk profiles will need to be defined further during their real-life application. The awareness of interactions between the cardiovascular risks, underlying cardiovascular problems and the cytokine release syndrome is prompting the definition of systematic assessments before and during CAR T therapy. Yet unknown potential latent effects, such as vascular inflammation seen after other immunotherapeutic interventions (i.e., immune checkpoint inhibitor therapies) will need to be taken in consideration for long-term cardiovascular surveillance. Inclusion of cardiovascular endpoints in trials, as well as broad collaborative, prospective clinical registries have the potential to provide new information about these risks. And not the least, the further investigation of such observations in targeted research studies has the potential to refine this technology and expand its safe applicability.
Author contributions
AR and VZ organized the outline and the components of the manuscript, as well. AR, AS, and ME performed an extensive literature search of cardiovascular disease and chimeric antigen receptor T cell therapy. PR, LA, and FA wrote individual sections on CAR T therapy and provided feedback on the manuscript. AC, SV, and KZ created the figure and wrote the section on surveillance and monitoring. All authors contributed to the writing efforts and the editing of this manuscript.
Funding
VZ received support from the Cancer Prevention Research Institute of Texas (RP180404).
Conflict of interest
Author FA has provided consultancy to: Genentech, Astrazeneca, Abbvie, Janssen, Pharmacyclics, Gilead sciences, Kite pharma, Celgene, Karyopharm, MEI Pharma, Verastem, Incyte, Beigene, Johnson and Johnson, Dava Oncology, BMS, Merck, Cardinal Health, ADCT therapeutics, and Epizyme.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ALL, acute lymphocytic leukemia; CAD, coronary artery disease; CAR T-cell, chimeric antigen receptor T-cell; CAR NK-cell, chimeric antigen receptor natural killer cell; CEA, carcinoembryonic antigen; CHF, congestive heart failure; CMR, cardiac magnetic resonance; CRS, cytokine release syndrome; CV, cardiovascular; CVD, cardiovascular disease; ECG, electrocardiogram; FAP, fibroblast activation protein; FDA, Food and Drug Administration; GD2, disialoganglioside 2; HER2, human epidermal growth factor receptor 2; HLH, hemophagocytic lymphohystiocytosis; ICANS, immune cell-associated neurotoxicity syndrome; IFN-γ, interferon-gamma; IL, interleukin; L1CAM, L1 cell adhesion molecule; MCP-1, monocyte chemoattractant protein-1; MI, myocardial infarction; MIP-1β, macrophage inflammatory protein-1 beta; REMS, risk evaluation and mitigation strategy; TIL, tumor-infiltrating lymphocytes; TNFα, tumor necrosis factor alpha; TTE, transthoracic echocardiogram.
References
1. Billingham RE, Brent L, Medawar PB. Quantitative studies on tissue transplantation immunity. II. The origin, strength and duration of actively and adoptively acquired immunity. Proc R Soc Lond B Biol Sci. (1954) 143:58–80. doi: 10.1098/rspb.1954.0054
2. Lorenz E, Uphoff D, Reid TR, Shelton E. Modification of irradiation injury in mice and guinea pigs by bone marrow injections. J Natl Cancer Inst. (1951) 12:197–201.
3. Thomas ED, Lochte HL Jr, Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med. (1957) 257:491–6. doi: 10.1056/NEJM195709122571102
4. Thomas ED. A history of haemopoietic cell transplantation. Br J Haematol. (1999) 105:330–9. doi: 10.1111/j.1365-2141.1999.01337.x
5. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. (1985) 313:1485–92. doi: 10.1056/NEJM198512053132327
6. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. (1986) 233:1318–21. doi: 10.1126/science.3489291
7. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
8. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. (2020) 17:807–21. doi: 10.1038/s41423-020-0488-6
9. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183
10. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. (2011) 118:4817–28. doi: 10.1182/blood-2011-04-348540
11. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
12. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
13. Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. (2013) 21:2122–9. doi: 10.1038/mt.2013.154
14. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
15. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. (2015) 21:524–9. doi: 10.1038/nm.3833
16. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
17. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
18. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
19. Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. (2022) 23:91–103. doi: 10.1016/S1470-2045(21)00591-X
20. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
21. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
22. Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
23. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. (2021) 398:314–24. doi: 10.1016/S0140-6736(21)00933-8
24. Yakoub-Agha I, Chabannon C, Bader P, Basak GW, Bonig H, Ciceri F, et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: best practice recommendations of the european society for blood and marrow transplantation (EBMT) and the joint accreditation committee of ISCT and EBMT (JACIE). Haematologica. (2020) 105:297–316. doi: 10.3324/haematol.2019.229781
25. Hayden PJ, Sirait T, Koster L, Snowden JA, Yakoub-Agha I. An international survey on the management of patients receiving CAR T-cell therapy for haematological malignancies on behalf of the chronic malignancies working party of EBMT. Curr Res Transl Med. (2019) 67:79–88. doi: 10.1016/j.retram.2019.05.002
26. Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, et al. Management of adults and children receiving CAR T-cell therapy: 2021 best practice recommendations of the european society for blood and marrow transplantation (EBMT) and the joint accreditation committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol. (2022) 33:259–75. doi: 10.1016/j.annonc.2021.12.003
27. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
28. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. (2014) 20:119–22. doi: 10.1097/PPO.0000000000000035
29. Frey N, Porter D. Cytokine release syndrome with chimeric antigen receptor T cell therapy. Biol Blood Marrow Transplant. (2019) 25:e123–e7. doi: 10.1016/j.bbmt.2018.12.756
30. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. (2016) 3:16011. doi: 10.1038/mto.2016.11
31. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
32. Dai H, Wang Y, Lu X, Han W. Chimeric antigen receptors modified T-cells for cancer therapy. J Natl Cancer Inst. (2016) 108:djv439. doi: 10.1093/jnci/djv439
33. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. (2017) 130:2295–306. doi: 10.1182/blood-2017-06-793141
34. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
35. Thompson JA, Schneider BJ, Brahmer J, Andrews S, Armand P, Bhatia S, et al. Management of Immunotherapy-Related Toxicities, Version 1.2019. J Natl Compr Canc Netw. (2019) 17:255–89. doi: 10.6004/jnccn.2019.0013
36. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
37. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
38. Reagan PM, Neelapu SS. How I manage: pathophysiology and management of toxicity of chimeric antigen receptor T-cell therapies. J Clin Oncol. (2021) 39:456–66. doi: 10.1200/JCO.20.01616
39. Greenbaum U, Strati P, Saliba RM, Torres J, Rondon G, Nieto Y, et al. CRP and ferritin in addition to the EASIX score predict CAR-T-related toxicity. Blood Adv. (2021) 5:2799–806. doi: 10.1182/bloodadvances.2021004575
40. Sheth VS, Gauthier J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. (2021) 56:552–66. doi: 10.1038/s41409-020-01134-4
41. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
42. Hunter BD, Jacobson CA. CAR T-cell associated neurotoxicity: mechanisms, clinicopathologic correlates, and future directions. J Natl Cancer Inst. (2019) 111:646–54. doi: 10.1093/jnci/djz017
43. Catala E, Iacoboni G, Vidal-Jordana A, Oristrell G, Carpio C, Vilaseca A, et al. Neurotoxicity-associated sinus bradycardia after chimeric antigen receptor T-cell therapy. Hematol Oncol. (2022) 40:482–7. doi: 10.1002/hon.2976
44. Fischer JW, Bhattarai N. CAR-T cell therapy: mechanism, management, and mitigation of inflammatory toxicities. Front Immunol. (2021) 12:693016. doi: 10.3389/fimmu.2021.693016
45. Penack O, Koenecke C. Complications after CD19+ CAR T-Cell therapy. Cancers (Basel). (2020) 12:12113445. doi: 10.3390/cancers12113445
46. Rubin DB, Vaitkevicius H. Neurological complications of cancer immunotherapy (CAR T cells). J Neurol Sci. (2021) 424:117405. doi: 10.1016/j.jns.2021.117405
47. Bhatia S, Dai C, Landier W, Hageman L, Wu J, Schlichting E, et al. Trends in late mortality and life expectancy after allogeneic blood or marrow transplantation over 4 decades: a blood or marrow transplant survivor study report. JAMA Oncol. (2021) 7:1626–34. doi: 10.1001/jamaoncol.2021.3676
48. Bhatia S, Dai C, Landier W, Hageman L, Wu J, Schlichting E, et al. Trends in late mortality and life expectancy after autologous blood or marrow transplantation over three decades: a BMTSS report. J Clin Oncol. (2022) 2022:JCO2102372. doi: 10.1200/JCO.21.02372
49. Herrmann J, Lenihan D, Armenian S, Barac A, Blaes A, Cardinale D, et al. Defining cardiovascular toxicities of cancer therapies: an International Cardio-Oncology Society (IC-OS) consensus statement. Eur Heart J. (2022) 43:280–99. doi: 10.1093/eurheartj/ehab674
50. Saad A, de Lima M, Anand S, Bhatt VR, Bookout R, Chen G, et al. Hematopoietic cell transplantation, version 2.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2020) 18:599–634. doi: 10.6004/jnccn.2020.0021
51. Tevaarwerk A, Denlinger CS, Sanft T, Ansbaugh SM, Armenian S, Baker KS, et al. Survivorship, version 1.2021. J Natl Compr Canc Netw. (2021) 19:676–85. doi: 10.6004/jnccn.2021.0028
52. Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, et al. Cytokine release syndrome after chimeric antigen receptor T Cell therapy for acute lymphoblastic leukemia. Crit Care Med. (2017) 45:e124–e31. doi: 10.1097/CCM.0000000000002053
53. Burstein DS, Maude S, Grupp S, Griffis H, Rossano J, Lin K. Cardiac profile of chimeric antigen receptor T cell therapy in children: a single-institution experience. Biol Blood Marrow Transplant. (2018) 24:1590–5. doi: 10.1016/j.bbmt.2018.05.014
54. Shalabi H, Sachdev V, Kulshreshtha A, Cohen JW, Yates B, Rosing DR, et al. Impact of cytokine release syndrome on cardiac function following CD19 CAR-T cell therapy in children and young adults with hematological malignancies. J Immunother Cancer. (2020) 8:1159. doi: 10.1136/jitc-2020-001159
55. Alvi RM, Frigault MJ, Fradley MG, Jain MD, Mahmood SS, Awadalla M, et al. Cardiovascular events among adults treated with chimeric antigen receptor T-cells (CAR-T). J Am Coll Cardiol. (2019) 74:3099–108. doi: 10.1016/j.jacc.2019.10.038
56. Ganatra S, Redd R, Hayek SS, Parikh R, Azam T, Yanik GA, et al. Chimeric antigen receptor T-cell therapy-associated cardiomyopathy in patients with refractory or relapsed non-hodgkin lymphoma. Circulation. (2020) 142:1687–90. doi: 10.1161/CIRCULATIONAHA.120.048100
57. Lefebvre B, Kang Y, Smith AM, Frey NV, Carver JR, Scherrer-Crosbie M. Cardiovascular effects of CAR T cell therapy: a retrospective study. JACC CardioOncol. (2020) 2:193–203. doi: 10.1016/j.jaccao.2020.04.012
58. Brammer JE, Braunstein Z, Katapadi A, Porter K, Biersmith M, Guha A, et al. Early toxicity and clinical outcomes after chimeric antigen receptor T-cell (CAR-T) therapy for lymphoma. J Immunother Cancer. (2021) 9:2303. doi: 10.1136/jitc-2020-002303
59. Steiner RE, Banchs J, Koutroumpakis E, Becnel M, Gutierrez C, Strati P, et al. Cardiovascular events in patients treated with chimeric antigen receptor t-cell therapy for aggressive B-cell lymphoma. Haematologica. (2021) 107:1555–66. doi: 10.3324/haematol.2021.280009
60. Ghosh AK, Chen DH, Guha A, Mackenzie S, Walker JM, Roddie C. CAR T cell therapy-related cardiovascular outcomes and management: systemic disease or direct cardiotoxicity? JACC CardioOncol. (2020) 2:97–109. doi: 10.1016/j.jaccao.2020.02.011
61. Jamal FA, Khaled SK. The cardiovascular complications of chimeric antigen receptor T cell therapy. Curr Hematol Malig Rep. (2020) 15:130–2. doi: 10.1007/s11899-020-00567-4
62. Moriyama S, Fukata M, Yokoyama T, Ueno S, Nunomura T, Mori Y, et al. Case report: cardiac tamponade in association with cytokine release syndrome following CAR-T cell therapy. Front Cardiovasc Med. (2022) 9:848091. doi: 10.3389/fcvm.2022.848091
63. Anderson LD Jr. Idecabtagene vicleucel (ide-cel) CAR T-cell therapy for relapsed and refractory multiple myeloma. Future Oncol. (2022) 18:277–89. doi: 10.2217/fon-2021-1090
64. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. (2019) 20:31–42. doi: 10.1016/S1470-2045(18)30864-7
65. Riedell PA, Bishop MR. Safety and efficacy of axicabtagene ciloleucel in refractory large B-cell lymphomas. Ther Adv Hematol. (2020) 11:2040620720902899. doi: 10.1177/2040620720902899
66. Oluwole OO, Bouabdallah K, Munoz J, De Guibert S, Vose JM, Bartlett NL, et al. Prophylactic corticosteroid use in patients receiving axicabtagene ciloleucel for large B-cell lymphoma. Br J Haematol. (2021) 194:690–700. doi: 10.1111/bjh.17527
67. Yan Z, Zhang H, Cao J, Zhang C, Liu H, Huang H, et al. Characteristics and risk factors of cytokine release syndrome in chimeric antigen receptor T cell treatment. Front Immunol. (2021) 12:611366. doi: 10.3389/fimmu.2021.611366
68. Armenian SH, Lacchetti C, Barac A, Carver J, Constine LS, Denduluri N, et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. (2017) 35:893–911. doi: 10.1200/JCO.2016.70.5400
69. Yanez L, Sanchez-Escamilla M, Perales MA. CAR T cell toxicity: current management and future directions. Hemasphere. (2019) 3:e186. doi: 10.1097/HS9.0000000000000186
70. Totzeck M, Michel L, Lin Y, Herrmann J, Rassaf T. Cardiotoxicity from chimeric antigen receptor-T cell therapy for advanced malignancies. Eur Heart J. (2022) 43:1928–40. doi: 10.1093/eurheartj/ehac106
71. Albinger N, Hartmann J, Ullrich E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. (2021) 28:513–27. doi: 10.1038/s41434-021-00246-w
72. Marofi F, Al-Awad AS, Sulaiman Rahman H, Markov A, Abdelbasset WK, Ivanovna Enina Y, et al. CAR-NK cell: a new paradigm in tumor immunotherapy. Front Oncol. (2021) 11:673276. doi: 10.3389/fonc.2021.673276
73. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
74. Cha SE, Kujawski M, P JY, Brown C, Shively JE. Tumor regression and immunity in combination therapy with anti-CEA chimeric antigen receptor T cells and anti-CEA-IL2 immunocytokine. Oncoimmunology. (2021) 10:1899469. doi: 10.1080/2162402X.2021.1899469
75. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. (2010) 363:1324–34. doi: 10.1056/NEJMoa0911123
76. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
77. Le Q, Castro S, Tang T, Loeb AM, Hylkema T, McKay CN, et al. Therapeutic targeting of mesothelin with chimeric antigen receptor T cells in acute myeloid leukemia. Clin Cancer Res. (2021) 27:5718–30. doi: 10.1158/1078-0432.CCR-21-1546
78. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
79. Hong H, Brown CE, Ostberg JR, Priceman SJ, Chang WC, Weng L, et al. L1 cell adhesion molecule-specific chimeric antigen receptor-redirected human T cells exhibit specific and efficient antitumor activity against human ovarian cancer in mice. PLoS ONE. (2016) 11:e0146885. doi: 10.1371/journal.pone.0146885
80. Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, et al. Targeting cardiac fibrosis with engineered T cells. Nature. (2019) 573:430–3. doi: 10.1038/s41586-019-1546-z
81. Rurik JG, Tombacz I, Yadegari A, Mendez Fernandez PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science. (2022) 375:91–6. doi: 10.1126/science.abm0594
Keywords: chimeric antigen receptor (CAR T), cardio-oncology, immunotherapy, cytokine release syndrome (CRS), cellular therapy, cardiovascular disease
Citation: Rao A, Stewart A, Eljalby M, Ramakrishnan P, Anderson LD Jr, Awan FT, Chandra A, Vallabhaneni S, Zhang K and Zaha VG (2022) Cardiovascular disease and chimeric antigen receptor cellular therapy. Front. Cardiovasc. Med. 9:932347. doi: 10.3389/fcvm.2022.932347
Received: 29 April 2022; Accepted: 16 August 2022;
Published: 23 September 2022.
Edited by:
Feng Cao, People's Liberation Army General Hospital, ChinaReviewed by:
Maria Teresa Palano, MultiMedica (IRCCS), ItalyDaniel Chen, University College London, United Kingdom
Copyright © 2022 Rao, Stewart, Eljalby, Ramakrishnan, Anderson, Awan, Chandra, Vallabhaneni, Zhang and Zaha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vlad G. Zaha, dmxhZC56YWhhQHV0c291dGh3ZXN0ZXJuLmVkdQ==