 Aleksander Turk
Aleksander Turk Tanja Kunej
Tanja Kunej
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 07 July 2022
Sec. Cardio-Oncology
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.931917
This article is part of the Research Topic HF2Cancer: Exploring bidirectional interaction between cardiovascular diseases and cancer View all 18 articles
Cancer and cardiovascular diseases (CVD) account for approximately 27.5 million deaths every year. While they share some common environmental risk factors, their shared genetic risk factors are not yet fully understood. The aim of the present study was to aggregate genetic risk factors associated with the comorbidity of cancer and CVDs. For this purpose, we: (1) created a catalog of genes associated with cancer and CVDs, (2) visualized retrieved data as a gene-disease network, and (3) performed a pathway enrichment analysis. We performed screening of PubMed database for literature reporting genetic risk factors in patients with both cancer and CVD. The gene-disease network was visualized using Cytoscape and the enrichment analysis was conducted using Enrichr software. We manually reviewed the 181 articles fitting the search criteria and included 13 articles in the study. Data visualization revealed a highly interconnected network containing a single subnetwork with 56 nodes and 146 edges. Genes in the network with the highest number of disease interactions were JAK2, TTN, TET2, and ATM. The pathway enrichment analysis revealed that genes included in the study were significantly enriched in DNA damage repair (DDR) pathways, such as homologous recombination. The role of DDR mechanisms in the development of CVDs has been studied in previously published research; however, additional functional studies are required to elucidate their contribution to the pathophysiology to CVDs.
Cancer and cardiovascular diseases (CVD) are among the leading causes of death worldwide. An estimated 17.9 million people die every year as a result of CVDs and approximately 9.6 million deaths per year are caused by cancer (1, 2). While they are two distinct types of illness, they display a significant level of comorbidity (3). A portion of this comorbidity has been attributed to the cardiotoxic effects of chemotherapeutic cancer treatments (4). Cancer and CVD also share contributing risk factors, such as hypertension, obesity, smoking, diabetes mellitus and lifestyle choices, as well as pathophysiological mechanisms, such as oxidative stress, neuro-hormonal activation and inflammation (5). The identification of these common risk factors and mechanisms has led to increased focus on the various possible connections between the two diseases in the field of cardio-oncology. However, much of this research has been focused on the effects of medication and common risk factors. As a result, potential underlying genetic contributors to this comorbidity are poorly understood.
Among the known genetic factors that contribute to the development of both cancer and CVDs is clonal hematopoiesis of intermediate potential (CHIP). CHIP refers to the presence of a sub-population of clonally expanded hematopoietic stem cells within an individual. This may occur as a result of age-related genetic drift or due to genetic variants. While the presence of CHIP is not inherently malignant, it does confer a higher risk for the development of blood malignancies, such as leukemia and CVDs. CHIP has also been associated with worsened heart failure outcomes (6). It has also been shown that patients with congenital heart disease (CHD) carry more damaging gene variants in cancer risk genes, suggesting shared biological pathways (7). Associations between cancer and CVDs on a genetic level have therefore already been identified. Further exploration into their shared genetic background could thus yield valuable results and contribute to our understanding of both cancer and CVDs.
The aim of the present study was to aggregate genetic risk factors associated with the comorbidity of cancer and CVDs. Our goal is to visualize gene-disease interactions in genes with variants associated with cancer and CVD comorbidity. Additionally, the goal is to perform a protein-protein interaction (PPI) analysis for genes included in the study and to perform pathway enrichment analysis to identify biological pathways in which disease-associated genes are involved. This would contribute to the field of cardio-oncology by compiling genetic risk factors and their roles for multiple disease types, which could be used for future development of risk assessment, more accurate prognoses and personalized treatment choices.
The PubMed database was accessed in order to collect relevant articles. Four database searches were conducted, containing common keywords pertinent to the subject matter:
1. Cardiac, mutations, risk, prevalence, predispose*, cancer
2. Heart, cancer, mutation, risk, cardiooncology
3. Cardiovascular, propensity, mutation, risk factor, cancer
4. Heart, cancer, gene*, risk, cardiooncology
The articles within the search results were then manually reviewed and screened for studies that fit two criteria:
1. Both cancer and CVD phenotypes were observed in patients
2. The study is concerned with shared genetic risk factors for cancer and CVDs
Studies that fit both screening criteria were then included in the study. Cancer type, CVD type, chemotherapeutic methods, as well as names of genes with disease-associated variants, were retrieved from studies. STRING software was then used to identify protein-protein interactions (PPIs) between proteins encoded by genes included in the present study (8). Only PPIs categorized as “retrieved from curated databases” or “experimentally validated” were included. The PPIs, genes, and their associated cancer and cardiovascular conditions were then visualized as a network using Cytoscape software (9). Secondary literature review was then conducted for genes with the highest number of disease-associated interactions in the network.
A pathway enrichment analysis using Enrichr was performed on genes included in this study, drawing from the BioPlanet 2019, KEGG 2021 and WikiPathway 2021 databases (10). In order to include the most reliable enrichments, pathway enrichment analysis results were then sorted by lowest adjusted P-value. The 10 lowest P-value results of each database were included in the study. A workflow diagram of the study is presented in Figure 1.
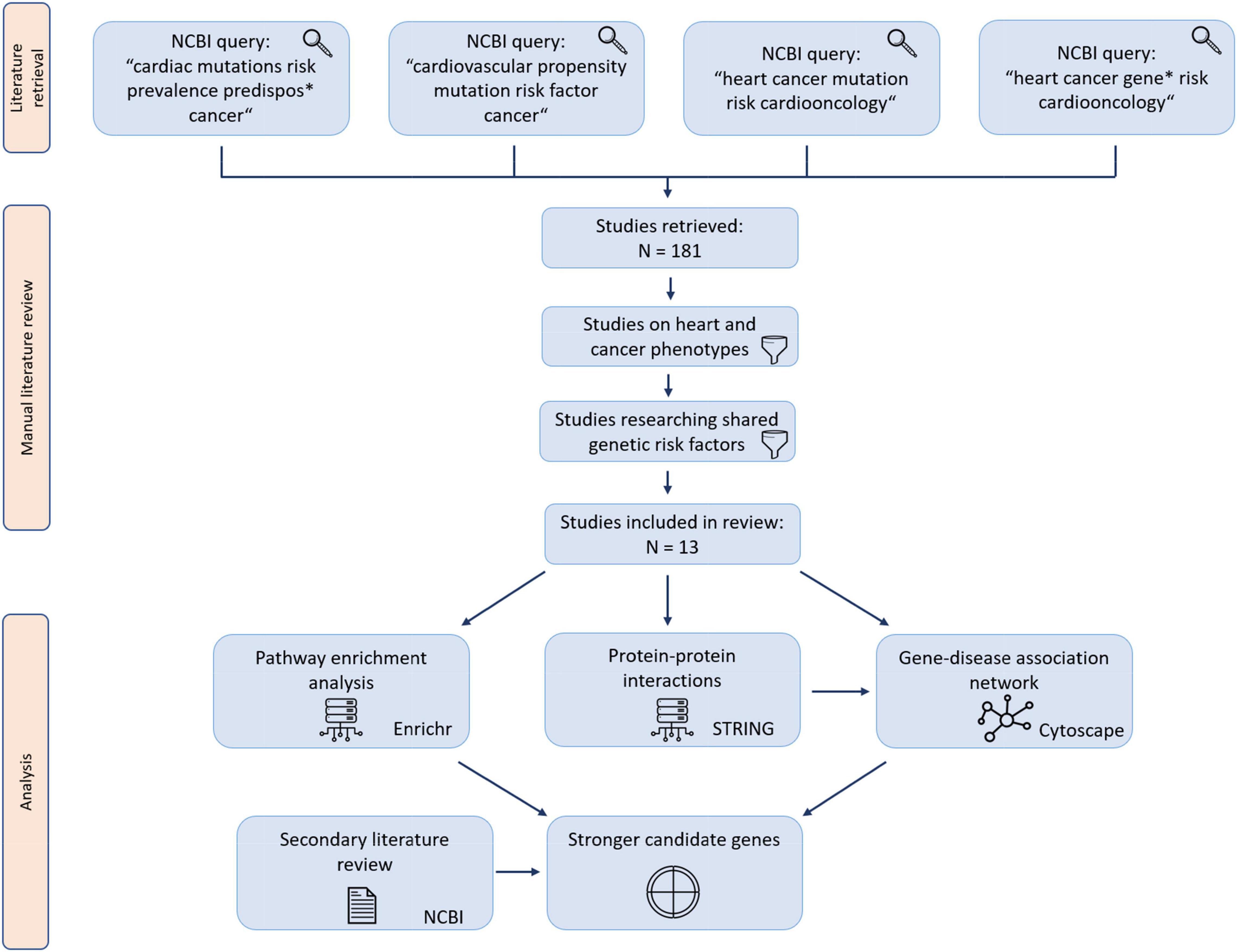
Figure 1. Study workflow diagram. Retrieved studies were manually reviewed for studies matching the screening criteria.
In this study, we conducted manual literature review of genes with variants associated with cancer and CVD comorbidity and then visualized this data as a gene-disease network. Additionally, we expanded upon the network with PPIs obtained from the STRING database. An analysis revealed the obtained gene list was significantly enriched in pathways responsible for DDR. Finally, we conducted secondary literature review of genes identified with the study methodology.
The PubMed database was accessed with four queries containing common keywords for the field of cardio-oncology; the four queries combined yielded 181 articles as search results. After screening and manual review, 13 articles were included in the final data set (11–23). Out of 13 articles included in the study, 10 included chemotherapy as a study parameter. Data extracted from manually reviewed articles is available in Supplementary Table 1. A list of PPIs identified with STRING is available in Supplementary Table 2. Data visualization revealed a large interconnected network of associations between genes, cancer types and CVDs as well as PPIs (Figure 2). The network was composed of 29 loci (28 genes and 1 intergenic region), 13 cancer types and 14 CVDs; thus containing 56 nodes and 146 edges.
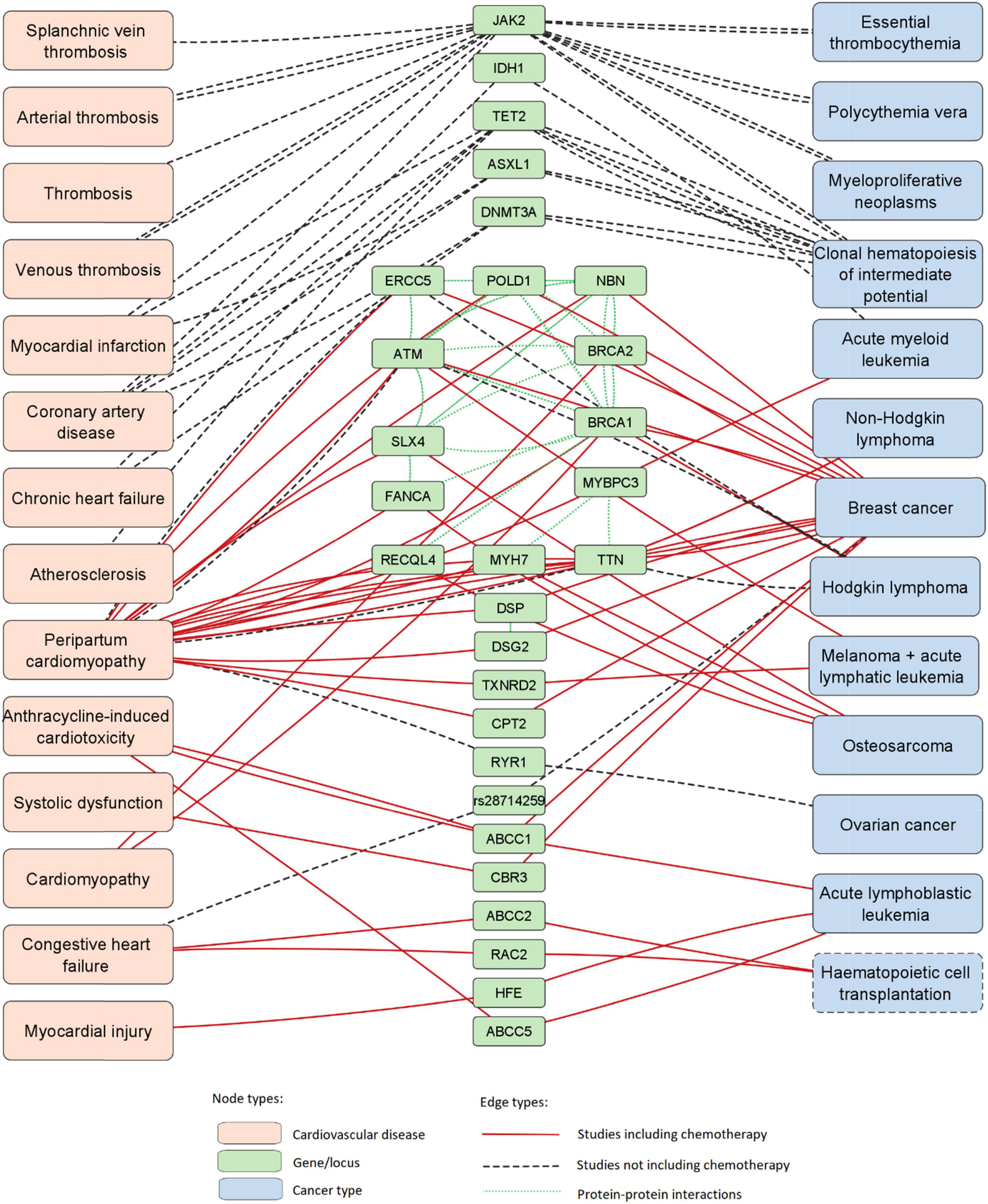
Figure 2. Network of associations between 29 loci (28 genes and 1 SNP), cancer types and CVDs. This network was expanded with data of PPIs. Hematopoietic cell transplantation is marked with a dashed node outline as it is not a disease.
Genes in the network with the most disease-connected edges were JAK2, TTN, TET2, and ATM, with 16, 12, 8, and 6 edges, respectively. Diseases with the highest number of gene-connected edges were peripartum cardiomyopathy, breast cancer, clonal hematopoiesis of intermediate potential (CHIP) and coronary artery disease, with 24, 15, 10, and 5 edges, respectively.
Furthermore, we conducted a pathway enrichment analysis of the 28 disease-associated genes. Of the 30 pathway enrichments, 28 enrichments had an adjusted P-value ≤ 0.05 (Table 1 and Supplementary Table 3). Among them, 3 were associated with CVDs, 4 were associated with cancer, and 16 were associated with DNA damage repair. Other enrichments include nucleotide methylation, meiosis or meiotic recombination, membrane transport proteins and other functions.
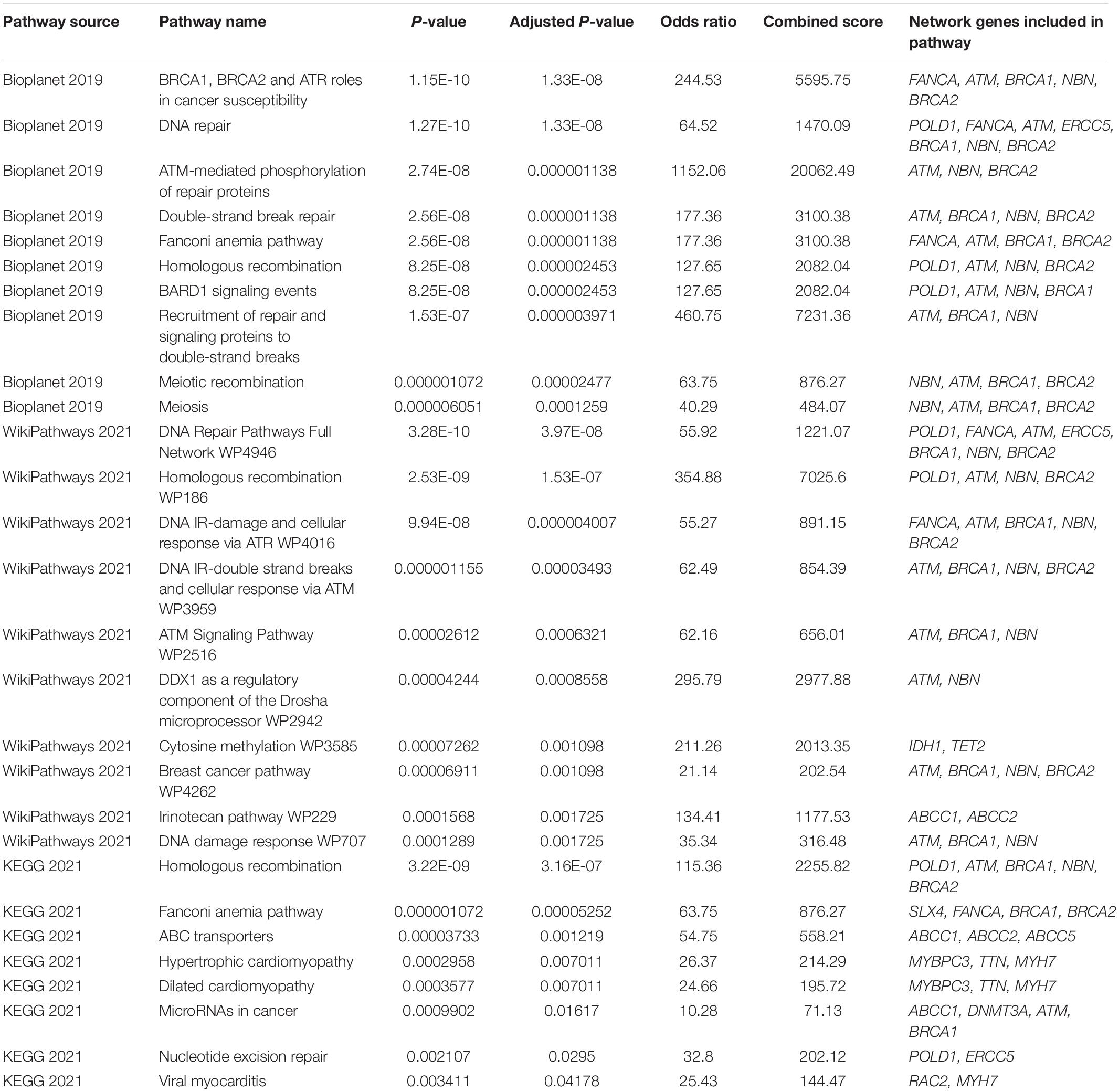
Table 1. Pathway enrichment analysis for 28 genes associated with cancer and CVDs.
In this study we conducted a database search and literature review for genes associated with cancer and CVD comorbidity, including chemotherapy-induced cardiotoxicity. Of the 181 manually reviewed articles, 13 were included in the analysis. We then formed a network of gene-disease associations, which was expanded upon with PPI data. Analysis revealed a large interconnected network containing a single subnetwork with 56 nodes and 146 edges. Enrichment analysis results also showed that genes were enriched in pathways associated with DNA damage repair, cancer and CVDs. Genes in the gene-disease association network with the highest number of edges were JAK2, TTN, TET2, and ATM.
Janus kinase 2 (JAK2) is encoded by the JAK2 gene. It serves as a kinase for cytokine receptors and is part of the JAK-STAT signaling pathway, which allows the cell to respond to various stress factors, including endotoxins, hypoxia, UV radiation and hyperosmolarity (24). The pathway is also important for the regulation of the immune system through the polarization of T helper cells (25). JAK2 plays an important role in hematopoiesis, as JAK2 gene disruptions in animal models are embryonically lethal due to a lack of definitive erythropoiesis (26). JAK2 has been associated with the copresence of CHIP and myocardial infarction (16). A variant of JAK2 has also been associated with an increased risk of developing myeloproliferative neoplasms such as polycythemia vera, essential thrombocythemia and primitive myelofibrosis (18, 27). As such JAK2 has been proposed as a potential target for the treatment or management of these diseases (28).
The TTN gene encodes titin (TTN), a large protein that plays a structural, scaffolding and signaling role in sarcomeres of striated muscle tissue (29). TTN spans from the Z-disc to the M-band, functioning as a molecular spring and giving muscles passive stiffness (30). The protein is also present in the heart’s muscle tissue. While its mechanical and scaffolding properties are critical for sarcomere function, its signaling role is also important. TTN binds to telethonin via its NH2 terminal domain, thus recruiting the muscle LIM protein (MLP) to the Z-line, where it interacts with other signaling molecules (31, 32). TTN could also be involved in hypertrophic signaling through its N2-A domain. This domain interacts with multiple signaling molecules, including muscle-ankyrin-repeat-proteins (MARPs), which are stress-response molecules and signal transducers (31). TTN variants have been associated with several CVDs, including dilated cardiomyopathy, fatal cardiomyopathy and heart failure (29, 33, 34). TTN has also been associated with peripartum cardiomyopathy, Hodgkin lymphoma, non-Hodgkin lymphoma and breast cancer (12). Furthermore, it has been suggested that the mutation load of TTN is indicative of a high tumor mutation burden (35).
Tet methylcytosine dioxygenase 2 (TET2), encoded by TET2, plays a role in epigenetic DNA modification by converting 5-methylcytosine to 5-hydroxymethylcytosine, thus promoting DNA demethylation (36). Through its regulatory functions TET2 affects hematopoiesis by promoting the self-renewal of stem cells, lineage commitment and monocyte differentiation (37). Li et al. report that 8% of TET2-knockout mice developed lethal myeloid malignancies within their first year of life (38). TET2 variants have also been associated with increased risk of myeloid malignancies. These malignancies include B- and T-cell lymphomas, myeloproliferative neoplasms, chronic myelomonocytic leukemia, acute myeloid leukemia, myelodysplastic syndrome, CHIP and others (16, 19, 36, 38). Sano et al. have shown that TET2 deficiency in mice is associated with greater cardiac dysfunction and heart failure, accelerated by TET2-induced clonal hematopoiesis. This is likely caused by the influence of TET2 deficiency on the IL-1β/NLRP3 inflammatory pathway, as treatment with an NLRP3 inflammation inhibitor protected against the development of heart failure (39).
The ATM gene encodes the ATM serine/threonine kinase (ATM), which regulates multiple processes, including DNA damage response, oxidative stress levels and mitochondrial homeostasis (40). ATM plays a role in the cellular response to double-stranded breaks (DSB) (41). It phosphorylates TP53 – one of the most important tumor suppressor proteins (41, 42). Furthermore, ATM has also been shown to phosphorylate BRCA1, NBN and other tumor suppressor proteins (43, 44). ATM variants have been associated with a predisposition for developing ischemic heart disease (45). Beside CVDs, ATM variants have been associated with conveying an increased risk of developing some cancers, including chronic lymphocytic leukemia, breast cancer, pancreatic cancer and mantle cell lymphoma (12, 46, 47). While ATM has several tumor suppressing functions, ATM-induced signaling can cause tumor progression via the αvβ3 integrin pathway in some cancers (48). ATM-dependent signaling in tumor cells can also increase resistance to radiotherapy and chemoresistance (49). Due to its role in signaling pathways and DNA damage repair, ATM has been proposed as a potential target for the development of future chemotherapies (50).
In the present study, a pathway analysis revealed that the genes included in the analysis were enriched in pathways associated with DDR. Among them were pathways such as Nucleotide excision repair, DNA damage response WP707 and Double-strand break repair. While variants of DDR genes are known to contribute to cancer development, they have also been associated with some CVDs, such as heart failure and atherosclerosis (51). Results of a study of DDR in experimental heart failure in rats conducted by Yndestad et al. suggest that DDR mechanisms could play an important part in counteracting CVD-related genotoxic stress and damage to tissue (52). DDR mechanisms are important for counteracting the effects of oxidative stress on the cell (51). Among other shared risk factors, increased oxidative stress contributes to the development of both cancer and CVDs (53). Variants of DDR genes could thus be related to an increased susceptibility to damage from endogenous oxidative stress. Additional functional research is necessary to determine the role of DDR mechanisms in CVDs.
Several genes included in this study were identified in research concerned with the effects of chemotherapies on CVD development. Chemotherapeutics are a diverse group of molecules that remains a widely used approach in cancer treatment. These molecules often target rapidly replicating cancer cells by inducing DNA damage (54). However, this treatment also affects other cells, leading to various side effects, including cardiotoxicity. A common class of chemotherapeutics are anthracyclines and are known to have dose-dependent cardiotoxic side effects (54). The main mechanism of anthracycline-induced cardiotoxicity (ACT) is thought to affect heart cells by topoisomerase 2β inhibition, leading to cardiomyocyte apoptosis and mitochondrial biogenesis inhibition (55). The mechanisms of cardiotoxicity are not equally understood in all chemotherapeutics, however. The pathophysiology of HER-2-targeting agents, used for the treatment of breast cancer, is currently unclear (56). Further studies into the mechanisms of chemotherapy-induced cardiotoxicity for various treatments could serve to improve future therapeutic developments.
Genes included in this study could serve as potential variant screening targets for chemotherapy-induced cardiotoxicity risk assessment. Aminkeng et al. have identified variants of several genes associated with ACT, which could also serve as potential screening targets (57). Gene panels may be more cost effective than whole genome sequencing (WGS) for this purpose, but lack the option of re-analysis as knowledge of genetic risk factors advances (58). Pharmacogenomic testing is recommended for childhood cancer patients with indication for the anthracyclines daunorubicin and doxorubicin for specific variants of RARG, SLC28A3 and UGT1A6*4. However, testing is not currently recommended for adult patients or children with indications for other anthracycline treatment (57). As the cost of WGS and gene panels fall and risk assessment strategies improve, screening a larger number of patients may become viable.
Accurately predicting cardiotoxic effects in patients and elucidating their pathophysiological mechanisms are of vital importance in cardio-oncology. This requires processing large data quantities, which is a task well-suited for machine learning. In cardio-oncology studies, machine learning has been used to perform retrospective cardiotoxicity risk assessment (59) and to estimate the safety of hERG blockers (60). Computational approaches could also have potential to improve cardiovascular imaging (61). Current research also suggests that these methods could be useful for predicting the cardiotoxic effects of drugs (62, 63). The use of machine learning may, in the future, assist clinicians in identifying probable pathogenic pathways and the treatment option with the highest likelihood of success.
In this study we conducted manual literature review and data synthesis of genetic factors associated with cancer and CVD comorbidity. We visualized a network of gene-disease associations based on data extracted from 13 articles. Additionally, we added information on PPIs of proteins encoded by genes in this study and included the PPIs in the network. The results revealed a large, highly interconnected network of known genetic risk factors shared between cancer and CVDs. Pathway enrichment analysis indicate that the genes included in this study are enriched in various DDR pathways, such as homologous recombination. While prior literature has associated some DDR gene variants with cancer and CVD comorbidity, the mechanism behind this has not yet been sufficiently studied. Additional functional studies are required to elucidate the role of DDR gene variants in the development of both cancer and CVDs and chemotherapy-induced cardiotoxicity. In the future, gene variants screening before cancer treatment could improve outcomes for patients at risk of cardiotoxicity.
Patient chemotherapy information was extracted from articles during manual literature review. Some articles analyzed in the present study did not include chemotherapeutics as a study parameter, as it was not the intended focus of the studies. While treatment was not a study parameter, patients may have undergone chemotherapeutic treatment, which could contribute to CVD development. Therefore, it is possible that chemotherapies contributed to disease development in these studies as well. Moreover, in this study we focused on changes at the DNA level. In the future, taking into account other omics levels, such as transcriptomics, proteomics and epigenomics may yield a more holistic view of the study field. It is also possible that some relevant research articles may not have been included based on the present study’s queries. Searches for specific associations between CVD and cancer types may yield more publications as a database search output. We highlighted a few genes with the highest number of edges in the gene-disease association network. It should be noted, however, that an increased number of connections may be due to research interest and it is possible that other genes in the network could also be of great significance.
Variants of genes involved in DDR pathways appear to contribute to cancer and CVD comorbidity in cancer patients undergoing chemotherapy. Further functional studies would contribute to the understanding of the underlying disease mechanisms and allow for the development of precision medicine approaches to disease treatment.
The present study could serve as a basis for the development of a cardio-oncology database. Such a database could prove invaluable for the understanding of the molecular relationship between diseases and identifying potential novel molecular targets for treatment. Additionally, it could serve to identify biomarkers, which could be used for risk assessment and optimal treatment choices.
While cardio-oncology is a rapidly expanding field, many genetic risk factors shared by cancers and CVDs are currently not yet known. Thus, the network formed in this study is incomplete and will require follow-up studies as more research in the field is conducted.
The original contributions presented in this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the patients/participants or patients/participants legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
AT conducted literature review, screening and analysis, wrote the manuscript, and created graphics. AT and TK designed study methodology and conducted manuscript review. Both authors contributed to the article and approved the submitted version.
This research was funded by the Slovenian Research Agency, research program P4-0220.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.931917/full#supplementary-material
1. World Health Organization. Cardiovascular Diseases. (n.d.). Available online at: https://www.who.int/health-topics/cardiovascular-diseases (accessed March 7, 2022).
2. World Health Organization. Cancer. (n.d.). Available online at: https://www.who.int/health-topics/cancer (accessed March 7, 2022).
3. Finke D, Heckmann MB, Frey N, Lehmann LH. Cancer—a major cardiac comorbidity with implications on cardiovascular metabolism. Front Physiol. (2021) 12:729713. doi: 10.3389/fphys.2021.729713
4. Stoltzfus KC, Zhang Y, Sturgeon K, Sinoway LI, Trifiletti DM, Chinchilli VM, et al. Fatal heart disease among cancer patients. Nat Commun. (2020) 11:2011. doi: 10.1038/s41467-020-15639-5
5. Boer RA, Meijers WC, Meer P, Veldhuisen DJ. Cancer and heart disease: associations and relations. Eur J Heart Fail. (2019) 21:1515–25. doi: 10.1002/ejhf.1539
6. Libby P, Sidlow R, Lin AE, Gupta D, Jones LW, Moslehi J, et al. Clonal hematopoiesis. J Am Coll Cardiol. (2019) 74:567–77. doi: 10.1016/j.jacc.2019.06.007
7. Morton SU, Shimamura A, Newburger PE, Opotowsky AR, Quiat D, Pereira AC, et al. Association of damaging variants in genes with increased cancer risk among patients with congenital heart disease. JAMA Cardiol. (2021) 6:457. doi: 10.1001/jamacardio.2020.4947
8. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. (2021) 49:D605–12. doi: 10.1093/nar/gkaa1074
9. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. (2003) 13:2498–504. doi: 10.1101/gr.1239303
10. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. (2016) 44:W90–7. doi: 10.1093/nar/gkw377
11. Kattih B, Shirvani A, Klement P, Garrido AM, Gabdoulline R, Liebich A, et al. IDH1/2 mutations in acute myeloid leukemia patients and risk of coronary artery disease and cardiac dysfunction—a retrospective propensity score analysis. Leukemia. (2021) 35:1301–16. doi: 10.1038/s41375-020-01043-x
12. Pfeffer TJ, Schlothauer S, Pietzsch S, Schaufelberger M, Auber B, Ricke-Hoch M, et al. Increased cancer prevalence in peripartum cardiomyopathy. JACC CardioOncology. (2019) 1:196–205. doi: 10.1016/j.jaccao.2019.09.008
13. Schneider BP, Shen F, Gardner L, Radovich M, Li L, Miller KD, et al. Genome-wide association study for anthracycline-induced congestive heart failure. Clin Cancer Res. (2017) 23:43–51. doi: 10.1158/1078-0432.CCR-16-0908
14. Semsei AF, Erdelyi DJ, Ungvari I, Csagoly E, Hegyi MZ, Kiszel PS, et al. ABCC1 polymorphisms in anthracycline-induced cardiotoxicity in childhood acute lymphoblastic leukaemia. Cell Biol Int. (2012) 36:79–86. doi: 10.1042/CBI20110264
15. Vulsteke C, Pfeil AM, Maggen C, Schwenkglenks M, Pettengell R, Szucs TD, et al. Clinical and genetic risk factors for epirubicin-induced cardiac toxicity in early breast cancer patients. Breast Cancer Res Treat. (2015) 152:67–76. doi: 10.1007/s10549-015-3437-9
16. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
17. Pearson EJ, Nair A, Daoud Y, Blum JL. The incidence of cardiomyopathy in BRCA1 and BRCA2 mutation carriers after anthracycline-based adjuvant chemotherapy. Breast Cancer Res Treat. (2017) 162:59–67. doi: 10.1007/s10549-016-4101-8
18. Vannucchi A, Guglielmelli P. JAK2 mutation-related disease and thrombosis. Semin Thromb Hemost. (2013) 39:496–506. doi: 10.1055/s-0033-1343890
19. Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. (2019) 4:25. doi: 10.1001/jamacardio.2018.3965
20. Armenian SH, Ding Y, Mills G, Sun C, Venkataraman K, Wong FL, et al. Genetic susceptibility to anthracycline-related congestive heart failure in survivors of haematopoietic cell transplantation. Br J Haematol. (2013) 163:205–13. doi: 10.1111/bjh.12516
21. Hertz DL, Caram MV, Kidwell KM, Thibert JN, Gersch C, Seewald NJ, et al. Evidence for association of SNPs in ABCB1 and CBR3, but not RAC2, NCF4, SLC28A3 or TOP2B, with chronic cardiotoxicity in a cohort of breast cancer patients treated with anthracyclines. Pharmacogenomics. (2016) 17:231–40. doi: 10.2217/pgs.15.162
22. Krajinovic M, Elbared J, Drouin S, Bertout L, Rezgui A, Ansari M, et al. Polymorphisms of ABCC5 and NOS3 genes influence doxorubicin cardiotoxicity in survivors of childhood acute lymphoblastic leukemia. Pharmacogenomics J. (2016) 16:530–5. doi: 10.1038/tpj.2015.63
23. Lipshultz SE, Lipsitz SR, Kutok JL, Miller TL, Colan SD, Neuberg DS, et al. Impact of hemochromatosis gene mutations on cardiac status in doxorubicin-treated survivors of childhood high-risk leukemia. Cancer. (2013) 119:3555–62. doi: 10.1002/cncr.28256
24. Dudley AC, Thomas D, Best J, Jenkins A. The STATs in cell stress-type responses. Cell Commun Signal. (2004) 2:8. doi: 10.1186/1478-811X-2-8
25. Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal. (2017) 15:23. doi: 10.1186/s12964-017-0177-y
26. Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essentialdevelopmental checkpoint in definitivehematopoiesis. Cell. (1998) 93:397–409. doi: 10.1016/S0092-8674(00)81168-X
27. Mahjoub S, Baccouche H, Sahnoun M, Kaabi H, Manai Z, Slama H, et al. The JAK2 mutation in myeloproliferative disorders: a predictive factor of thrombosis. Tunis Med. (2015) 93:474–7.
28. Reddy MM, Deshpande A, Sattler M. Targeting JAK2 in the therapy of myeloproliferative neoplasms. Expert Opin Ther Targets. (2012) 16:313–24. doi: 10.1517/14728222.2012.662956
29. Azad A, Poloni G, Sontayananon N, Jiang H, Gehmlich K. The giant titin: how to evaluate its role in cardiomyopathies. J Muscle Res Cell Motil. (2019) 40:159–67. doi: 10.1007/s10974-019-09518-w
30. Granzier HL, Labeit S. The giant protein titin. Circ Res. (2004) 94:284–95. doi: 10.1161/01.RES.0000117769.88862.F8
31. Linke WA, Krüger M. The giant protein titin as an integrator of myocyte signaling pathways. Physiology. (2010) 25:186–98. doi: 10.1152/physiol.00005.2010
32. Knöll R, Hoshijima M, Chien KR. Muscle LIM protein in heart failure. Exp Clin Cardiol. (2002) 7:104–5.
33. Carmignac V, Salih MAM, Quijano-Roy S, Marchand S, Al Rayess MM, Mukhtar MM, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol. (2007) 61:340–51. doi: 10.1002/ana.21089
34. Li S, Zhang C, Liu N, Bai H, Hou C, Song L, et al. Titin-truncating variants are associated with heart failure events in patients with left ventricular non-compaction cardiomyopathy. Clin Cardiol. (2019) 42:530–5. doi: 10.1002/clc.23172
35. Oh J-H, Jang SJ, Kim J, Sohn I, Lee J-Y, Cho EJ, et al. Spontaneous mutations in the single TTN gene represent high tumor mutation burden. NPJ Genomic Med. (2020) 5:33. doi: 10.1038/s41525-019-0107-6
36. Feng Y, Li X, Cassady K, Zou Z, Zhang X. TET2 function in hematopoietic malignancies, immune regulation, and DNA repair. Front Oncol. (2019) 9:210. doi: 10.3389/fonc.2019.00210
37. Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The ten-eleven translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia. (2014) 28:485–96. doi: 10.1038/leu.2013.337
38. Li Z, Cai X, Cai C-L, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. (2011) 118:4509–18. doi: 10.1182/blood-2010-12-325241
39. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J Am Coll Cardiol. (2018) 71:875–86. doi: 10.1016/j.jacc.2017.12.037
40. Shiloh Y, Ziv Y. The ATM protein: the importance of being active. J Cell Biol. (2012) 198:273–5. doi: 10.1083/jcb.201207063
41. Canman CE, Lim D-S, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM Kinase by Ionizing Radiation and Phosphorylation of p53. Science. (1998) 281:1677–9. doi: 10.1126/science.281.5383.1677
42. Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, et al. ATM associates with and phosphorylates p53: mapping the region of interaction. Nat Genet. (1998) 20:398–400. doi: 10.1038/3882
43. Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of BRCA1 in the DNA damage response to double-strand breaks. Science. (1999) 286:1162–6. doi: 10.1126/science.286.5442.1162
44. Kim S-T, Lim D-S, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. (1999) 274:37538–43. doi: 10.1074/jbc.274.53.37538
45. Thrasher P, Singh M, Singh K. Ataxia-telangiectasia mutated kinase: role in myocardial remodeling. J Rare Dis Res Treat. (2017) 2:32–7.
46. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. (2016) 15:1781–91. doi: 10.1158/1535-7163.MCT-15-0945
47. Goldgar DE, Healey S, Dowty JG, Da Silva L, Chen X, Spurdle AB, et al. Rare variants in the ATMgene and risk of breast cancer. Breast Cancer Res. (2011) 13:R73. doi: 10.1186/bcr2919
48. Stucci S, Tucci M, Passarelli A, Silvestris F. Avβ3 integrin: pathogenetic role in osteotropic tumors. Crit Rev Oncol Hematol. (2015) 96:183–93. doi: 10.1016/j.critrevonc.2015.05.018
49. Stucci LS, Internò V, Tucci M, Perrone M, Mannavola F, Palmirotta R, et al. The ATM gene in breast cancer: its relevance in clinical practice. Genes (Basel). (2021) 12:727. doi: 10.3390/genes12050727
50. Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. (2015) 149:124–38. doi: 10.1016/j.pharmthera.2014.12.001
51. Shukla PC, Singh KK, Yanagawa B, Teoh H, Verma S. DNA damage repair and cardiovascular diseases. Can J Cardiol. (2010) 26:13A–6. doi: 10.1016/S0828-282X(10)71055-2
52. Yndestad A, Neurauter CG, Øie E, Forstrøm RJ, Vinge LE, Eide L, et al. Up-regulation of myocardial DNA base excision repair activities in experimental heart failure. Mutat Res Mol Mech Mutagen. (2009) 666:32–8. doi: 10.1016/j.mrfmmm.2009.03.008
53. Koene RJ, Prizment AE, Blaes A, Konety SH. Shared risk factors in cardiovascular disease and cancer. Circulation. (2016) 133:1104–14. doi: 10.1161/CIRCULATIONAHA.115.020406
54. Woods D, Turchi JJ. Chemotherapy induced DNA damage response. Cancer Biol Ther. (2013) 14:379–89. doi: 10.4161/cbt.23761
55. Henriksen PA. Anthracycline cardiotoxicity: an update on mechanisms, monitoring and prevention. Heart. (2018) 104:971–7. doi: 10.1136/heartjnl-2017-312103
56. Ruddy KJ, Patel SR, Higgins AS, Armenian SH, Herrmann J. Cardiovascular health during and after cancer therapy. Cancers (Basel). (2020) 12:3737. doi: 10.3390/cancers12123737
57. Aminkeng F, Ross CJD, Rassekh SR, Hwang S, Rieder MJ, Bhavsar AP, et al. Recommendations for genetic testing to reduce the incidence of anthracycline-induced cardiotoxicity. Br J Clin Pharmacol. (2016) 82:683–95. doi: 10.1111/bcp.13008
58. Cirino AL, Lakdawala NK, McDonough B, Conner L, Adler D, Weinfeld M, et al. A comparison of whole genome sequencing to multigene panel testing in hypertrophic cardiomyopathy patients. Circ Cardiovasc Genet. (2017) 10:e001768. doi: 10.1161/CIRCGENETICS.117.001768
59. Zhou Y, Hou Y, Hussain M, Brown S, Budd T, Tang WHW, et al. Machine learning–based risk assessment for cancer therapy–related cardiac dysfunction in 4300 longitudinal oncology patients. J Am Heart Assoc. (2020) 9:e019628. doi: 10.1161/JAHA.120.019628
60. Yang P-C, DeMarco KR, Aghasafari P, Jeng M-T, Dawson JRD, Bekker S, et al. A computational pipeline to predict cardiotoxicity: from the atom to the rhythm. Circ Res. (2020) 126:947–64. doi: 10.1161/CIRCRESAHA.119.316404
61. Madan N, Lucas J, Akhter N, Collier P, Cheng F, Guha A, et al. Artificial intelligence and imaging: opportunities in cardio-oncology. Am Hear J Plus Cardiol Res Pract. (2022) 15:100126. doi: 10.1016/j.ahjo.2022.100126
62. Cai C, Guo P, Zhou Y, Zhou J, Wang Q, Zhang F, et al. Deep learning-based prediction of drug-induced cardiotoxicity. J Chem Inf Model. (2019) 59:1073–84. doi: 10.1021/acs.jcim.8b00769
Keywords: cancer, cardiovascular disease, risk factor, chemotherapy, comorbidity, genetic variant, interaction network, cardio-oncology
Citation: Turk A and Kunej T (2022) Shared Genetic Risk Factors Between Cancer and Cardiovascular Diseases. Front. Cardiovasc. Med. 9:931917. doi: 10.3389/fcvm.2022.931917
Received: 29 April 2022; Accepted: 21 June 2022;
Published: 07 July 2022.
Edited by:
Canan G. Nebigil, INSERM U1260 Nanomedicine Régénératrice (RNM), FranceReviewed by:
Daniela Cardinale, European Institute of Oncology (IEO), ItalyCopyright © 2022 Turk and Kunej. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tanja Kunej, dGFuamEua3VuZWpAYmYudW5pLWxqLnNp
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.