 Wen Ai
Wen Ai Haihua Yu
Haihua Yu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med. , 17 October 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.910288
This article is part of the Research Topic Case Reports in Atherosclerosis and Vascular Medicine: 2022 View all 10 articles
Introduction: Degos disease, also known as malignant atrophic papulosis (MAP), is a rare systemic obstructive vascular disease with unknown pathophysiology, which can affect multiple systems, especially gastrointestinal tract and central nervous system. Intestinal perforations with MAP is associated with high mortality rate and ambiguous treatment outcomes.
Case presentation: Here we report a missed-opportunity case of Degos disease characterized by generalized skin eruption and multiple intestinal perforations. Definite diagnosis of Degos disease was finally concluded after two exploratory laparotomy operations and skin biopsies. Due to the delayed diagnosis and treatment, the patient died after being discharged automatically in spite of application of aspirin and low-dose subcutaneous heparin. In view of such circumstances, we searched the Pubmed using “Degos [Title] OR Malignant Atrophic Papulosis [Title]” AND “perforation [Title] OR perforations [Title]” and make a detailed analysis of the result.
Conclusions: Degos disease is a rare systemic obstructive vascular disease with unknown pathologic mechanism and unavailable treatment methods. Diagnosis is usually based on the presence of pathognomonic skin lesions and tissue biopsy. Gastrointestinal involvement can cause serious and lethal conditions with high mortality. Currently, how to achieve a satisfying prognosis of MAP with intestinal perforations becomes the most urgent problem in front of medical staff.
Degos disease, also known as malignant atrophic papulosis (MAP), is a rare systemic obstructive vascular disease with unknown pathophysiology, which can affect multiple systems, mainly involving gastrointestinal tract and central nervous system, leading to high mortality (1). Here we report a missed-opportunity case of Degos disease characterized by generalized skin eruption and multiple intestinal perforations and make a literature review of other analogous cases.
A 48-year-old man was admitted to our hospital's emergency department with a 2-month history of recurrent abdominal pain and a 2-week history of aggravation. He underwent an exploratory laparotomy for acute diffuse peritonitis 50 days ago at the local hospital. Intraoperative findings revealed that numerous 0.2–0.4 cm yellow-white tubercles were interspersed on the surface of congestive small intestine, colon, appendix, greater omentum and mesenterium, especially the distal ileum (Figure 1a), without apparent perforation. Rapid pathology of appendix and omentum showed acute nonspecific inflammation and intestinal tuberculosis was suspected initially. Therefore large amount of saline and metronidazole was used for irrigation and abdominal drainage was placed. The patient discharged from hospital with intermittent slight abdominal pain on the 15th day after operation.
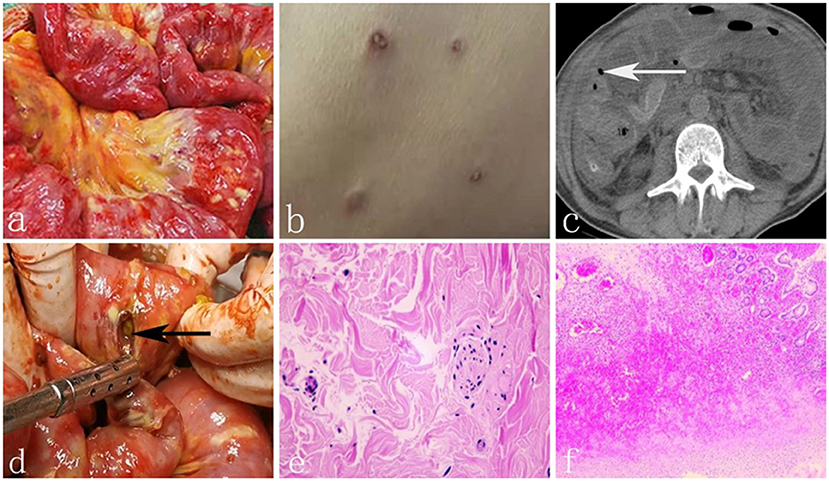
Figure 1. (a) Numerous yellow-white tubercles were interspersed on the surface of small intestine. (b) Generalized skin emption with porcelain - white centers surrounded by erythematous borders. (c) The white arrow shows free air around the intestine. (d) The black arrow shows one of the perforations of the small intestine. (e) The biopsy of skin eruption showed atrophy of epidermis, hyperplasia and collagen of dermal fibrous tissue. (f) Histopathologic result of the resected intestine showed acute and chronic inflammatory cell infiltration.
Twenty days later, the patient developed severe abdominal pain suddenly with nausea and vomiting accompanied by fever (39°C). He was admitted to the local hospital again and transferred to our intensive care unit (ICU) for deterioration of his condition. Physical examination showed evident abdominal tenderness and rebound tenderness with weakened bowel sounds. Meanwhile, generalized skin eruptions with an atrophic porcelain-white center surrounded by erythematous rim were visible over trunk and extremities, measuring 0.2–1.2 cm (Figure 1b). No chronic diseases, no alcohol use, no family history, no herbal agents, or no suspected drug use were reported. Relevant laboratory tests and radiographic results were as follows: WBC 8.65*109 (3.5–9.5*109), NEUT% 0.938↑ (0.40–0.75), procalcitonin 171.45 (0–0.05 ng/ml), HBV-DNA 9.15E+08↑ (<5.0E+02 IU/ml). Tuberculin test, Widder test, anti SS-A, and SS-B antibodies were negative. ANA was 1:100 weakly positive and Anti Ro-52 antibodies was positive too. Computed tomography showed a small amount of free air bubble, edema and thickening of the small bowel wall and massive seroperitoneum (Figure 1c). An exploratory laparotomy was performed on account of primary diagnosis of Crohn's disease with intestinal perforations. In the operation, we found more than 100 whitish-yellow plaques, dozens of intestinal perforations and purulent materials (Figure 1d). We resected 2 meters of the perforated small bowel and sutured the seromuscular layer in the wafery areas followed by enterostomy and abdominal drainage tube placement. The diagnosis of Crohn's disease was doubtful for the eccentric appearance of the intestine conditions, but there was still no definite diagnosis.
On the 5th day after operation, intestinal contents drained again, which signified recurrence of perforation. At this point, we eventually started to pay attention to the correlation between the perforation and skin lesions. Biopsy of skin eruption was conducted and the pathology showed atrophy of epidermis, hyperplasia and collagen of dermal fibrous tissue, degeneration of elastic fibers, significantly reduction and partial necrosis of skin appendages, fibrinoid necrosis in small vessels of the deep dermis and thrombosis in local lumen (Figure 1e). Histopathology of the resected intestine showed edema and hyperaemia, thrombus organization of both tiny artery and veins lumen with acute and chronic inflammatory cell infiltration (Figure 1f). Finally, definite diagnosis of Degos disease was concluded. The patient was given aspirin (100 mg/day) and low-dose subcutaneous heparin (5,000 U/dy), but his body condition went from bad to worse because of the severe abdominal inafection. He requested automatic discharge and died 3 days later.
Malignant atrophic papulosis (MAP) was first described by Köhlmeier in 1941 (2) and defined by Degos in 1942 (3), which usually occurs in the 20–50 age group with a slight male dominance (4). Previous appellation way of MAP was chaotic until Theodoridis et al. (5) renamed and divided it into two categories in 2014: benign atrophic papulosis (BAP) and malignant atrophic papulosis (MAP). The former is more common and characterized by cutaneous form, with atrophic porcelain-white center in size of 0.2–1 cm, distributed over the trunk and extremities, rarely on the face and scalp. The papules are red in early stage and expand gradually with atrophy in the center and leave white scars after deflorescence. The latter is characterized by involvement of internal organs, especially gastrointestinal tract (50%) and central nerves system (20%) (1). Median survival time of MAP is 2–3 years and five-year survival rate is less than 50% (6). However, these two forms can't be easily distinguished because involvement of inner organs may occur with skin lesions simultaneously or not (7).
Intestinal perforations with MAP is an uncommon phenomenon with a rate of 2.1% reported (8). We searched the Pubmed using “Degos [Title] OR Malignant Atrophic Papulosis [Title]” AND “perforation[Title] OR perforations[Title]” and found 11 cases. Nevertheless, the detailed data of 1 case reported by G H Evans (9) couldn't be enquired, so only the 10 cases were exhibited in Table 1 (10–19). As shown in the literature, the median age was 41.5 (range 7–56) with a male 60% proportion and only three of them were alive at follow-up. Table 2 showed four case study series ever published (5, 18, 20, 21). The age at diagnosis ranges from 34 to 37.9 years old and MAP accounts for 65% of the total cases (155:239). Two studies (5, 18) reported 0% mortality and another (20)reported 3% mortality for BAP while the mortality for MAP is 65.3–75%. Given MAP's high mortality rate, Theodoridis et al. (5) put forward a follow-up plan: whole-skin examination and skin biopsy for histological examination for BAP and colonoscopy/gastroscopy/laparoscopy if organ symptomatology suspected, the follow-up frequency was twice yearly for 0–7 years and once yearly for 7–10 years.
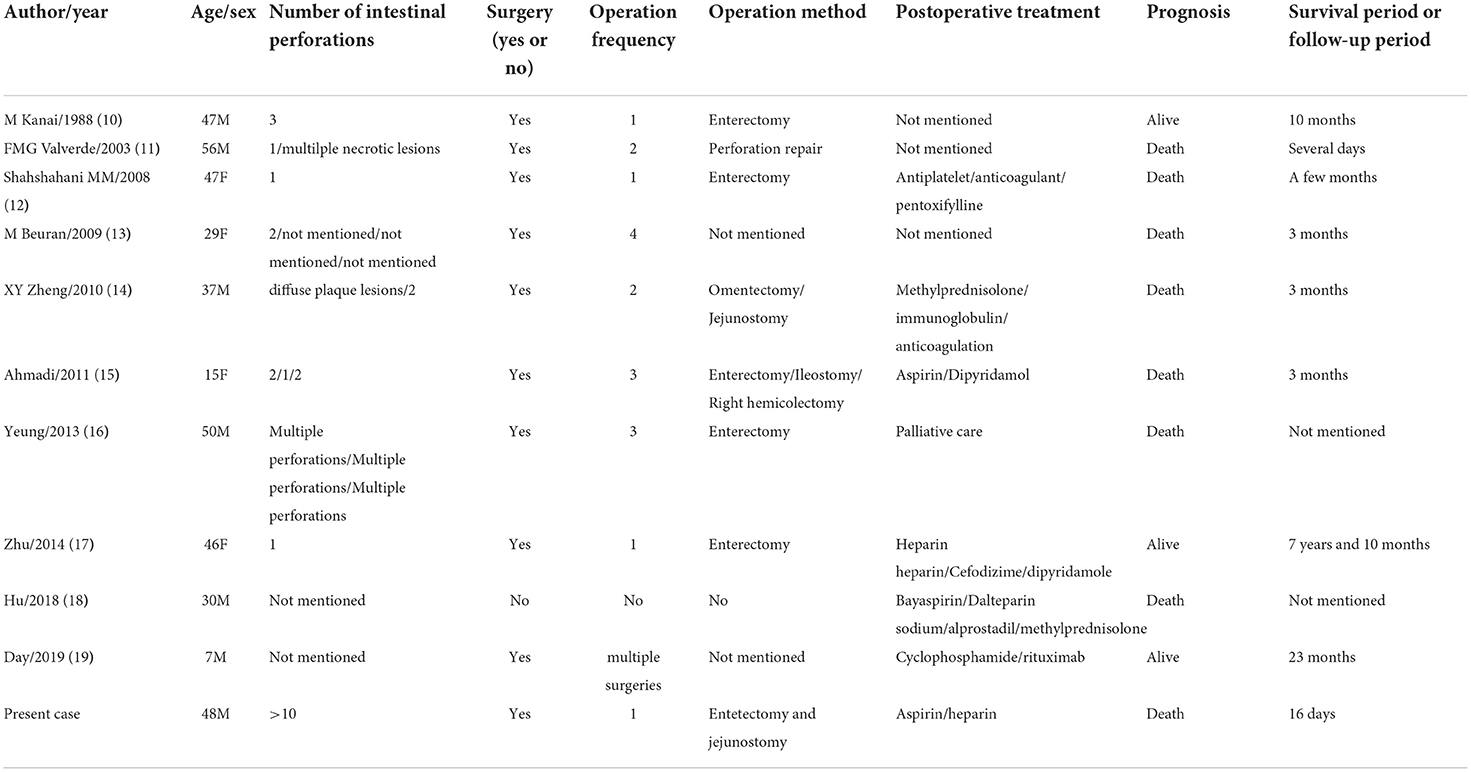
Table 1. Previous case reports of MAP and intestinal perforations.

Table 2. Demographic and clinical data of four previous studies.
Etiology of Degos disease remains unknown up to now. Viral infection, autoimmune disease, coagulopathy, collagen vascular disorders and genetic defects may be some of the underlying causes (12). In this case, the level of patient's HBV-DNA may be an explanation of MAP since it is 9.15E+08↑ IU/ml, significantly higher than normal. Autosomal dominant trait of MAP is hypothesized in view of reports of clusters of patients among members of the same family and first-degree relatives (22). Diagnosis of MAP depends mainly on presence of pathognomonic skin lesions and tissue biopsy pathology containing wedge necrosis of superficial and deep dermis, with arteriolar wall inflammatory cell infiltration, epidermal atrophy and dermal collagen rigidification. Dermoscopy reveals dendritic vessels, loop and irregular vessels, with central unstructured area but it lacks specificity (23).
At present, there is still no clear guideline for the treatment of MAP. Immuno-suppressor, such as azathioprine and cyclophosphamide are proved to be invalid (24). Antiplatelet drugs (aspirin or clopidogrel) or anticoagulants (warfarin or heparin) may be helpful (25). In 2011 Magro CM reported the pathologic findings of extensive deposits of C5b−9 within the cutaneous vasculature, and proposed that inhibition of C5 might be a therapeutic approach (26), and in 2013 he discovered the use of eculizumab as salvage therapy in critically ill patients with thrombotic micro angiopathy (27). Shapiro LS thought that treprostinil may offer a second effective treatment approach to individuals with MAP or rescue therapy to those in whom eculizumab treatment has failed (28). Unfortunately, large sample data results are still lacking, especially for gastrointestinal tract perforations.
In conclusion, Degos disease is a rare systemic obstructive vascular disease with unknown pathologic mechanism and unavailable treatment methods. Diagnosis is usually based on the presence of the pathognomonic skin lesions and tissue biopsy. Gastrointestinal involvement can cause serious and lethal conditions with high mortality (29).The patient's diagnosis was delayed because we ignored the relation between skin changes and peritonitis. Although current pharmacological treatments have limited value for MAP with intestinal perforations, early diagnosis and treatment play an important role in improving prognosis and survival rate.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the participant/s for the publication of this case report. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
WA wrote the manuscript. ZL and FL searched literatures. HY revised and approved the final manuscript. All authors have read and approved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MAP, malignant atrophic papulosis; BAP, benign atrophic papulosis; ICU, intensive care unit; WBC, white blood cell; NEUT, neutrophil; HBV-DNA, hepatitis B virus-DNA; ANA, antinuclear antibodies.
1. Hohwy T, Jensen MG, Tøttrup A, Steiniche T, Fogh K, A. fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leinden mutation and lupus anticoagulant. Acta Derm Venereol. (2006) 86:245–7. doi: 10.2340/00015555-0068
2. Kohlmeier W. Multiple hautnekrosen bei thrombangiitis obliterans. Arch Dermatol Syph. (1941) 181:783–92. doi: 10.1007/BF01828450
3. Degos R, Delort J, Tricot R. Dermatite papule-squameuse atrophiante. Bull Soc Fr Dermatol Syphiligr. (1942) 49:148–50.
4. Snow JL, Muller SA. Degos syndrome: malignant atrophic papulosis. Semin Dermatol. (1995) 14:99–105. doi: 10.1016/S1085-5629(05)80004-5
5. Theodoridis A, Konstantinidou A, Makrantonaki E, Zouboulis CC. Malignant and benign forms of atrophic papulosis (Köhlmeier-Degos disease): systemic involvement determines the prognosis. Br J Dermatol. (2014) 170:110–5. doi: 10.1111/bjd.12642
6. Tummidi S, Nagendran P, Gedela S, Ramani JR, Shankaralingappa A. Degos disease: a case report and review of the literature. J Med Case Rep. (2020) 14:204. doi: 10.1186/s13256-020-02514-6
7. Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease) - a review. Orphanet J Rare Dis. (2013) 8:10. doi: 10.1186/1750-1172-8-10
8. Vardeh H, Margo C, Rosen S. Gastrointestinal pathology associated with dermatomyositis: presentation of three cases and a general review. AJ Surg Pathol Rev Rep. (2010) 21:293–300.
9. Evans GH, Ribeiro BF. Degos' disease. A rare cause of multiple intestinal perforation. J R Coll Surg Edinb. (1987) 32:371–2.
10. Kanai M, Kondoh S, Kuriki H, Mukaiyama H, Mori K, Tanno T, et al. report of an atypical case of Degos' disease with multiple perforations of the stomach and small intestine. Nihon Geka Gakkai Zasshi. (1988) 89:1127–31.
11. González Valverde FM, Menarguez Pina F, Ruiz JA, Gómez Ramos MJ, Mauri Barbera F, Luri Prieto P, et al. Presentation of Degos syndrome as acute small-bowel perforation. Arch Surg. (2003) 138:57–8. doi: 10.1001/archsurg.138.1.57
12. Shahshahani MM, Hashemi P, Nemati R, Nikoo A, Mazoochy H, Rashidi A, et al. case of Degos disease with pleuropericardial fibrosis, jejunal perforation, hemiparesis, and widespread cutaneous lesions. Int J Dermatol. (2008) 47:493–5. doi: 10.1111/j.1365-4632.2008.03394.x
13. Beuran M, Chiotoroiu AL, Morteanu S, Chilie A, Avram M, Roşu O, et al. <jabb>Boala köhlmeier-Degos (Papuloza Atrofiantă Malignă): cauză de perforatii intestinale multiple iterative–prezentare de caz [Köhlmeier-Degos disease (Malignant Atrophic Papulosis): a cause of recurrent multiple intestinal perforations]. Chirurgia. (2009) 104:765−72.
14. Zheng XY, Huang DY, Xin Y, Wang XF. Malignant atrophic papulosis with severe gastrointestinal perforation and omental necrosis: a case report. J Int Med Res. (2010) 38:1164–9. doi: 10.1177/147323001003800346
15. Ahmadi M, Rafi SA, Faham Z, Azhough R, Rooy SB, Rahmani O, et al. A fatal case of Degos' disease which presented with recurrent intestinal perforation. World J Gastrointest Surg. (2011) 3:156–8. doi: 10.4240/wjgs.v3.i10.156
16. Yeung JT, Ma JK, Yung AW. Degos' syndrome complicated by bowel perforation: focus on radiological findings. Hong Kong Med J. (2013) 19:174–7.
17. Zhu M, Jiang Z, Jiang R, Chong X, Sun J. Degos disease with an intestinal perforation: a case report. Int J Dermatol. (2014) 53:631–4. doi: 10.1111/j.1365-4632.2012.05602.x
18. Hu P, Mao Z, Liu C, Hu X, Kang H, Zhou F. Malignant atrophic papulosis with motor aphasia and intestinal perforation: a case report and review of published works. J Dermatol. (2018) 45:723–6. doi: 10.1111/1346-8138.14280
19. Day W, Gabriel C, Kelly RE Jr., Magro CM, Williams JV, Werner A, et al. Juvenile dermatomyositis resembling late-stage Degos disease with gastrointestinal perforations successfully treated with combination of cyclophosphamide and rituximab: case-based review. Rheumatol Int. (2020) 40:1883–90. doi: 10.1007/s00296-019-04495-2
20. Burg G, Vieluf D, Stolz W, Landthaler M, Braun-Falco O. Maligne atrophische Papulose (Morbus Köhlmeier-Degos) [Malignant atrophic papulosis (Köhlmeier-Degos disease)]. Hautarzt. (1989) 40:480–5.
21. Assier H, Chosidow O, Piette JC, Boffa MC, Youinou P, Thomas L, et al. Absence of antiphospholipid and anti-endothelial cell antibodies in malignant atrophic papulosis: a study of 15 cases. J Am Acad Dermatol. (1995) 33:831–3. doi: 10.1016/0190-9622(95)91843-4
22. Katz SK, Mudd LJ, Roenigk HH Jr. Malignant atrophic papulosis (Degos' disease) involving three generations of a family. J Am Acad Dermatol. (1997) 37(3 Pt1):480–4. doi: 10.1016/S0190-9622(18)30753-9
23. Anker JP, Kaminska-Winciorek G, Lallas A, Nicoletti S, Januszewski K, Mazzei ME, et al. The dermoscopic variability of Degos disease at different stages of progression. Dermatol Pract Concept. (2014) 4:59–61. doi: 10.5826/dpc.0403a11
24. Gmuca S, Boos MD, Treece A, Narula S, Billinghurst L, Bhatti T, et al. Degos disease mimicking primary vasculitis of the CNS. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e206. doi: 10.1212/NXI.0000000000000206
25. Gutiérrez-Pascual M, Hernández-Martín A, Colmenero I, García-Peñas JJ, López-Pino MA, Torrelo A. Malignant atrophic papulosis: a case report with severe visual and neurological impairment. Pediatr Dermatol. (2011) 28:302–5. doi: 10.1111/j.1525-1470.2010.01287.x
26. Magro CM, Poe JC, Kim C, Nuovo G, Crow MK, Crow YJ. Degos disease: a C5b-9/interferon-α-mediated endotheliopathy syndrome. Am J Clin Pathol. (2011) 135:599–610. doi: 10.1309/AJCP66QIMFARLZKI
27. Magro CM, Wang X, Garrett-Bakelman F, Shapiro LS, DeSancho MT. The effects of Eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. (2013) 8:185. doi: 10.1186/1750-1172-8-185
28. Shapiro LS, Toledo-Garcia AE, Farrell JF. Effective treatment of malignant atrophic papulosis (Köhlmeier-Degos disease) with treprostinil–early experience. Orphanet J Rare Dis. (2013) 8:52. doi: 10.1186/1750-1172-8-52
Keywords: Degos disease, malignant atrophic papulosis, intestinal perforations, skin eruption, case report
Citation: Ai W, Liang Z, Li F and Yu H (2022) Degos disease with multiple intestinal perforations: A missed-opportunity case report and literature review. Front. Cardiovasc. Med. 9:910288. doi: 10.3389/fcvm.2022.910288
Received: 24 April 2022; Accepted: 30 September 2022;
Published: 17 October 2022.
Edited by:
Masanori Aikawa, Brigham and Women's Hospital and Harvard Medical School, United StatesReviewed by:
Sijan Basnet, Reading Hospital, United StatesCopyright © 2022 Ai, Liang, Li and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haihua Yu, eWhoc2QyMDAzMDhAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.