
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 17 June 2022
Sec. Heart Failure and Transplantation
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.910100
This article is part of the Research TopicEdema in Heart Failure with Reduced Ejection FractionView all 7 articles
 Roxana Mihaela Chiorescu1,2†
Roxana Mihaela Chiorescu1,2† Roxana-Daiana Lazar3*†
Roxana-Daiana Lazar3*† Sándor-Botond Buksa2
Sándor-Botond Buksa2 Mihaela Mocan1,2†
Mihaela Mocan1,2† Dan Blendea1,3†
Dan Blendea1,3†From a pathogenetic point of view, heart failure (HF) is characterized by the activation of several neurohumoral pathways with a role in maintaining the cardiac output and the adequate perfusion pressure in target organs and tissues. Decreased cardiac output in HF with reduced ejection fraction causes activation of the sympathetic nervous system, the renin angiotensin aldosterone system, arginine-vasopressin system, natriuretic peptides, and endothelin, all of which cause water and salt retention in the body. As a result, patients will present clinically as the main symptoms: dyspnea and peripheral edema caused by fluid redistribution to the lungs and/or by fluid overload. By studying these pathophysiological mechanisms, biomarkers with a prognostic and therapeutic role in the management of edema were identified in patients with HF with low ejection fraction. This review aims to summarize the current data from the specialty literature of such biomarkers with a role in the pathogenesis of edema in HF with low ejection fraction. These biomarkers may be the basis for risk stratification and the development of new therapeutic means in the treatment of edema in these patients.
Cardiovascular diseases continue to represent a major health problem, being responsible for approximately one-third of global mortality and 45% of all deaths across European countries (1, 2). HFrEF plays a significant role in cardiovascular burden, and despite being extensively studied, its complex pathophysiology remains incompletely understood. Whatever the cause, symptomatic HFrEF is characterized by excessive accumulation of interstitial fluid due to sodium retention, with various implications in the quality of life, prognosis, and therapeutic management of these patients. Moreover, edema was reported to mark Stage C of HF (3, 4). Given the pathophysiology of edema, four theories have been suggested as contributing to extracellular fluid expansion, namely: a reduction in glomerular filtration rate (GFR), a stimulation of the renin-angiotensin-aldosterone pathway, activation of renal sympathetic nerve fibers and inhibition of natriuretic component (5).
Comprehending these mechanisms allows both the understanding of the HF clinic and a logical therapeutic approach, because the treatment aims to correct the excessive compensatory mechanisms, which themselves became the cause of perpetuation and aggravation of the patient's suffering.
Sodium and water renal retention is the first response mechanism that leads to edema formation and a redistribution of body fluids in HF. Additionally, adjustments in the venous abdominal reservoir and central cardiopulmonary vascular beds were also reported to contribute to clinical congestion, impacting right heart hemodynamics that can be used to track alterations in volume status (6). Throughout the years, various biomarkers have been proposed to guide the management of HF. However, there is less data on how biomarkers are linked to edema formation, and in which extend can be used for prognostic, clinical or therapeutical purposes.
HFrEF is characterized by a decrease in heart rate because of various factors that act upon its main determinants (contractility, overload, post-pregnancy, frequency). The decrease in cardiac output in HF is perceived by the body similarly to the decrease in volume. A cascade of mechanisms gets activated and determines the renal retention of water and salt. These mechanisms consist mainly of the redistribution of the cardiac output, which causes changes in the renal hemodynamics and neuroendocrine stimulation. These two main mechanisms (redistribution of the cardiac output and neuroendocrine stimulation) are fundamental for the retention of water and salt in the body and cause edema (7). Also, in HF there is a redistribution of the cardiac output to the essential organs to the detriment of the others. Thus, the flow of the coronary circulation is maintained, the cerebral flow decreases a little and there is a significant drop in the cutaneous and renal circulation. The decrease in cardiac output and its redistribution causes a reduction in renal flow and as a result a decrease in glomerular filtration is seen. Hence, intrarenal blood redistribution occurs and juxtaglomerular nephrons with high concentration capacity will be irrigated preferentially. Normal tubular resorption will result in the formation of a reduced urinary output and in the retention of water and salt (8, 9).
The decrease in renal flow causes an increase in renin release and angiotensin formation, stimulating the secretion of adrenal aldosterone. As a result, the sodium-potassium tubular exchange increases with sodium retention. Decreased renal flow increases the secretion of antidiuretic hormone with water retention. The neuroendocrine mechanism and renal hemodynamic changes cause hydro-saline retention. As a result of the hydro-saline retention when under an increased venous and lymphatic capillary pressure, edema occurs, which in turn lead to the increase in the interstitial pressure with the compression of the small vessels and the increase in the rigidity of the arterial walls (10).
Triggering and coordinating the compensatory mechanisms at the central and peripheral level with the realization of an initially useful adaptive reaction, but which, after being ended, becomes pathological because of the activation of complex neuroendocrine mechanisms. Some of these are vasoconstrictors and others are vasodilators. Neuroendocrine activity causes not only hemodynamic adaptation, and hydro-saline retention, but also cardiac and vascular remodeling. The main vasoconstrictor mechanisms are: the sympathetic stimulation, RAAS, arginine-vasopressin system, endothelins, and the main vasodilatory mechanisms are: the natriuretic system, endothelial vasodilator factor, prostaglandins. The vasodilator systems have an antiproliferative and apoptosis-reducing action, while the vasoconstrictor ones trigger cardiac remodeling processes that involve the proliferation of muscle and nerve tissue and trigger apoptosis (11). Main pathophysiological mechanisms involved in the production of edema in HFrEF are synthetized in Figure 1.
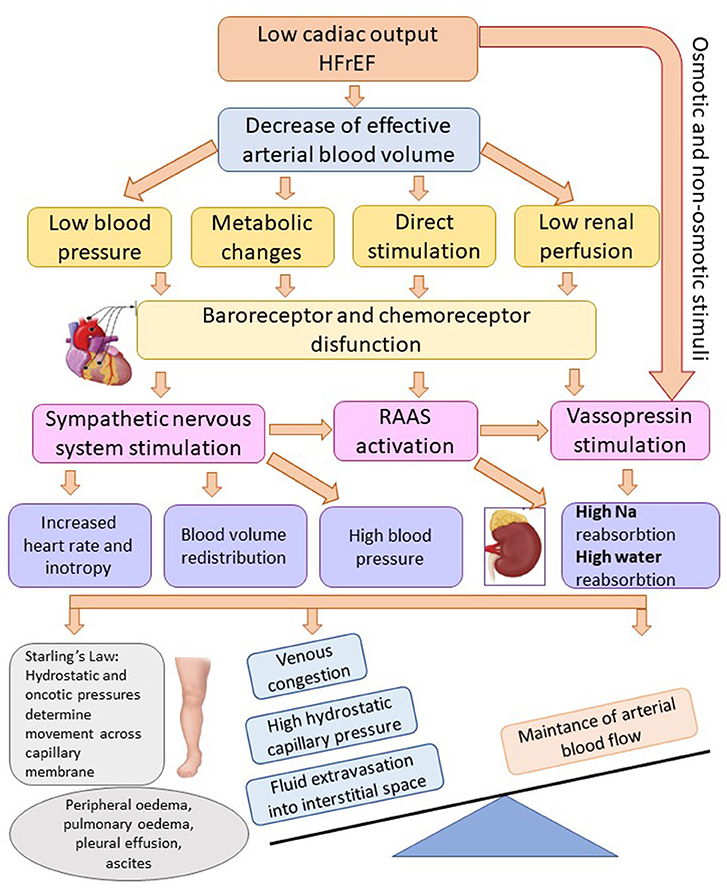
Figure 1. Mechanisms of fluid overload and edema in heart failure with reduced ejection fraction. Adapted after Adams et al. (8) (HFrEF, heart failure with reduced ejection fraction; Na, sodium; RAAS, renin-angiotensin-aldosterone system).
• The sympathetic stimulation is triggered by the stimulation of vascular baroreceptors stimulated by tissue hypoxia. The sympathetic stimulation causes an increase in the release of norepinephrine from the postganglionic terminals and adrenaline from the adrenal medulla. The sympathetic stimulation determines tachycardia and increased contractility at the central level while at the peripheral level it causes vasoconstriction with redistribution of the cardiac output to the periphery, redistribution of the renal plasma flow to the juxtamedullary nephrons, and at the endocrine level the stimulation of renin secretion and other mediators (7).
• The RAAS is activated by: the decrease in the prefusion pressure of the afferent glomerular arteriole, decrease in the sodium concentration at the level of the dense macula, and by the sympathetic activation. Circulating renin converts liver angiotensinogen to angiotensin I (AT I), which is converted by the enzyme converting angiotensin to the active mediator - angiotensin II (AT II). AT II has a potent vasoconstricting actin, increases the release of catecholamines from the nerve endings and the adrenal medulla, and stimulates the release of aldosterone. Thus, the RAAS participates in the redistribution of the cardiac output, salt retention and it maintains the sympathetic stimulation (12).
• The arginine-vasopressin system is stimulated by osmotic receptors, baroreceptors or directly by AT II. The activation of this system contributes to vasoconstriction and water retention (13, 14).
• The endothelin system is stimulated by tissue hypoxia. It causes vasoconstriction, increases inotropism and has a proliferative effect on myocytes and fibroblasts (15).
• Prostaglandins are activated because of increased vasoconstrictor activity. Vasoconstrictor systems are responsible for blood flow redistribution. Thus, at the renal level, the increase in the Ang II concentrations and the renal ischemia determines the increase in the prostaglandins with local vasodilating effect (16, 17).
• Natriuretic peptides (NPs) have vasodilating and antiproliferative action. They are synthesized in the cells of the cardiovascular system:
- Natriuretic peptide A (ANP) from the atrial and ventricular myocardium, the natural stimulus is atrial dilation.
- Natriuretic B peptide (BNP) released by the ventricular myocardium in response to the increase in the filling pressure;
- Endothelial cell peptide C under stimulation of parietal shear force caused by the blood flow.
Natriuretic peptides increase the excretion of sodium and water, have vasodilating action, inhibit the secretion of norepinephrine, renin and endothelin (18).
The diagnosis, prognosis, management and even risk stratification of the patients with HFrEF was synthetized both in the newly published guidelines of AHA/ACC/HFSA in 2022 (19) and in ESC guidelines in 2021 (20).
The new classifications of HF based on left ventricular ejection fraction (LVEF) as recommended by the 2022 AHA/ACC/HFSA Guidelines are as follows:
• HFrEF corresponding to LVEF ≤ 40%.
• HF with improved EF (HFimpEF) corresponding to previous LVEF ≤ 40% and a follow-up measurement of LVEF >40%.
• HFmrEF corresponding to LVEF 41%−49% and evidence of spontaneous or provokable increased LV filling pressures (19).
Heart failure with preserved ejection fraction (HFpEF) corresponds to LVEF ≥50% and presents echocardiographic evidence of spontaneous or provokable increased LV filling pressures.
Under the new guidance, the risk for HF was stratified in stages. The first stage identifies the risk factors for HF early (stage A), and those at risk of HF. In addition, the guidelines will help provide treatment before structural changes or signs of decreased heart function occur (stage B). In other words, stage A is defined as “at risk for HF,” stage B as “pre-HF,” stage C as “symptomatic HF,” and stage D as “advanced HF” (19). In the ESC Guidelines, the recommended biomarkers for the positive diagnosis of HFrEF are NPs (BNP and NT-proBNP). In oncologic patients both Troponins and NPs were proved to be useful for the early detection of cardiac disfunction and follow-up during the chemotherapy (particularly anthracyclines or trastuzumab). Both guidelines clearly confirm the clinical utility of serum NPs (BNP or and NT-proBNP) for establishing disease and prognosis in chronic HF, regardless of etiology (20).
Clear recommendations of biomarkers use were introduced in the new guidance of 2022 AHA/ACC/HFSA. An entire section is dedicated to NPs and their use in emergency for the etiology of the dyspnoea and exclusion of HF, for risk stratification in chronic HF, to establish the prognosis at admission and at discharged after an episode of acute HF, and at last to stratify the patients with risk for developing HF. However, there are insufficient evidence to inform specific guideline recommendations related to NPs-guided therapy or serial measurement of BNP or NT-proBNP to reduce hospitalizations or mortality (19).
Persistence of pulmonary congestion after hospitalization is a negative prognostic factor in patients with decompensated acute HF. Quantification of pulmonary congestion can be done by quantifying the radiological score of congestion, using a systematic approach in six lung areas. Clinical studies have shown that this parameter better identifies the risk of readmission of these patients compared to BNP, physical assessment (NYHA class) and echocardiographic parameters (21). Patients with HFpEF develop exertional pulmonary congestion. Determining the degree of this can be done with the help of exercise pulmonary ultrasound by quantifying the B lines in patients with elevated BNP and echocardiographic parameters of diastolic dysfunction (22).
Unfortunately, the role of other biomarkers of myocardial fibrosis such as ST2 or galectin-3 was not well established, and they were not introduced in clinical practice.
As for the management of HF, 2022 AHA/ACC/HFSA Guidelines included sodium-glucose cotransporter-2 inhibitors (SGLT2i) in the guidelines reflecting the availability of more proven treatment options for both HFpEF and HFrEF. The treatment of hypertension and atrial fibrillation has been updated, along with updates recommending avoidance of routine use of nitrates or phosphodiesterase-5 inhibitors. Moreover, the therapeutic options were classified in low-value, intermediate value and high-value treatments. Tafamidis for cardiac amyloidosis was the only low-value therapy identified. ARNi, ACEi, ARB, beta blocker, MRA, implantable cardioverter-defibrillator, and cardiac resynchronization therapy were classified as high value, while SGLT2i inhibitors and cardiac transplantation were classified as intermediate value therapies (19).
Patients with advanced HF should be addressed to a specialty team in order to increase life expectancy, comorbidities like iron deficiency, anemia, high blood pressure, sleep disorders, type 2 diabetes, atrial fibrillation, coronary artery disease, and cancer should receive special attention (19). Benefits of physical activity and increase in quality of life was observed after systematic rehabilitation programs in patients with HFrEF, but at the moment the biomarkers are not used for the surveillance after rehabilitation (20, 23).
Overall, the new definitions, classifications, and risk stratification in stages of HF are the first steps for us to achieve clarity and uniformity of care to timely diagnose and target appropriate guideline therapies.
The onset of neurohormonal mechanisms in HF has an adaptive role and succeeds in the first phase of compensated heart failure - stage B the control of the symptoms. If the stimulation of these neurohormonal mechanisms continues excessively, their ability to compensate for the pathophysiological changes in heart failure will be overcome and will lead to disease progression. Thus, prompt therapeutic intervention, at the right moment, on these neurohormonal mechanisms will prevent the progression of the disease (20).
During physiological conditions, cardiac activity is modulated by the sympathetic and parasympathetic nervous system via chronotropic, inotropic, dromotropic and lusitropic properties. Adding to this the balance in the peripheral resistance, we get cardiac output (24). Stress conditions are associated with an increased release of catecholamines [CA; adrenalin (AD) and noradrenaline (NA)] from brainstem catecholaminergic neurons, spinal cord efferent nerves and adrenal medulla. The functional consequence of the sympathetic overdrive, seen in chronic HF, is represented by a chronic elevation in both circulating levels of plasma CA and their urinary excretion (24, 25). Thus, the myocardium has to adapt to current cardiovascular demands (both energetic and metabolic) expressed by tachycardia, vasoconstriction and sodium and water retention, in order to maintain heart's function of cardiac pump. Nevertheless, these mechanisms have deleterious effects on cardiac structure and performance, leading not only to symptomatic HF, but also to HF decompensations. In addition, higher levels of plasma circulating NA seen from Stage B in HF, were strongly linked to a worsening of systolic dysfunction and a progression of interstitial fluid accumulation, marking the onset of edema formation at biochemical/molecular level (26–30).
Results of experimental studies have shown that the suppression of central sympathetic activity by renal denervation (ablation of renal nerves, also used as an alternative for the treatment of resistant hypertension in humans), had therapeutic effects on HF, reducing tissue edema (9, 27).
Furthermore, some studies revealed that renal denervation improved natriuretic and diuretic response, especially in conditions predisposing to acute volume expansion. Nevertheless, as the results from denervated HF patients were significantly different from the denervated control specimen (rats), the authors suggested that besides this procedure, other factors/biomarkers that contribute to the improvement in sodium retention during HF should be explored (8, 28).
Catestatin (CTS) and its precursor, chromogranin A (CgA), are other biomarkers implicated in sympathetic activation. Several studies focused on the establishment of their physiological role in patients with HF. Being produced by nerve fibers and neuroendocrine tissues, they exert a downregulation of CA release, providing cardioprotective effects against excessive sympathochromaffin overactivation. CTS, in addition to its prevalent vasoreactivity, expresses cardiosupressive effects (negative inotropic and lusitropic actions) via β2- and α-adrenoceptors found on the myocardial cells surface (31). In other words, CTS's modulatory actions have a direct impact on cardiac cells intrinsic health.
Specifically, CgA demonstrated to be a powerful predictive marker of mortality and morbidity in HF subjects (28). It inhibits cardiac remodeling, reduce arterial blood pressure and decrease endothelial and vascular inflammation (24, 28, 29). The last two mentioned effects may suggest its involvement in body fluid homeostasis, thus an increased circulating plasma level of CgA observed in HF patients, would translate as a compensatory mechanism meant to limit acute/chronic fluid overload. However, further studies to evaluate its relationship with edema formation, are needed.
Adrenomedullin (ADM) represents a hormone secreted mainly by the gastrointestinal cells that exerts several physiological functions, among which the most relevant are vasodilation, anti-inflammation, tissue repair and organ protection (30, 32). ADM is normally found in healthy individuals' plasma in low concentrations (33). Its effects are believed to be related to the modulation of sympathetic nervous system during stress conditions. Moreover, ADM release was shown to be stimulated by fluid overload, in order to combat further vascular leakage, thus limiting congestion and maintaining the endothelial barrier function (34). Taking these into account, several studies revealed that higher levels of plasma ADM are markedly increased in HF, corelating with the aggravation of signs and symptoms of remaining congestion, and in consequence, with disease severity (30, 34, 35). In addition, a rise in the use of loop diuretics at discharge and greater risk of readmission due to decompensated HF, were predicted by increased levels of ADM. These results proved that ADM could be a useful marker to identify HF subjects at risk of rehospitalization due to suboptimal decongestion (34). Another study proposed that the stabilizing endothelial barrier effect, could be achieved by administrating 15 ng/kg/min of ADM, thus reducing pulmonary congestion without significant hemodynamic effects (36). Unfortunately, since ADM plasma levels are also very elevated in severe infections (especially in septic status), its use in HF patients must be carefully interpreted. From another point of view, ADM could also be used as a target therapy in HF. The ACCOST-HH Trial hypothesized that Adrecizumab (ADZ), a non-neutralizing anti-ADM antibody, binds to ADM, prolonging its half-life, reinforcing the fact that it is not an inhibiting antibody (37, 38). In theory, ADZ allows shifting of ADM from the interstitial space into the blood vessels (33, 38). Consequently, the bioavailability of ADM will increase and its beneficial effects will be enhanced. However, since an increased plasma level of biologically active ADM (bio-ADM), correlates with a high mortality risk, whilst its plasma level normalization, correlates with a decrease in mortality (39), it becomes a controversial subject of whether supplemental administration of ADM would really be beneficial among HF patients.
Renin-angiotensin-aldosterone system (RAAS) enhances and contributes to the built of the congestive syndrome seen in HF via its chronic activation. The pathophysiologic impact of the RAAS is mainly guided by AT II and aldosterone. The adverse cardiovascular events are similar to those observed in sustained sympathetic activation and excessive CA release, amongst which, vasoconstriction, and sodium and water retention, are included (8). Moreover, the congestive syndrome seen in HF associated with renal dysfunction, is usually difficult to evaluate and tends to a worsen HF. Nevertheless, signs and symptoms of excessive volume overload may be present even in patients with HF and preserved EF due to renal impairment (14), proving a close interrelation between the kidneys, through RAAS activation, and sympathetic nervous system (SNS) (26). This is also proved by, low circulating levels of AT I and II that were seen after radiofrequency renal denervation (9). The implementation of RAAS blockers in the last decades, has led to a significant reduction in the rates of hospitalization and mortality among HF patients (40).
With regard to the impact of renin in the congestive HF, Sullivan et al. (41) concluded that before the onset of Stage C HF, which is associated with fluid retention, experimental animals expressed higher levels of plasma renin activity. It is the first structure released from the juxtaglomerular apparatus in the RAAS cascade. Studies have shown that the progression of HF is held back due to a decrease in sodium and water reabsorption, amongst patients with normalized plasma renin activity. Ergo, despite the fact that the first two generations of direct renin inhibitors (DRI) failed to show valid clinical benefits, Aliskiren proved to delay progression of HF from stage B to stage C, as a result of significant reduction in the development of edema (41). Also, adding DRI to the standard HF therapy was shown to carry great potential benefits in HFrEF patients with elevated plasma renin activity (26).
Aldosterone acts on sodium handling in the renal distal tubules and it is considered to be a key-hormone in the progression of HF, through its effects on the vessels, heart and brain (26, 42). Even though its production is stimulated by AT II, other hormones and mediators have been shown to act on its production and release. In that matter, angiotensin converting enzyme inhibitors (ACEi) proved to produce a transient drop in plasma levels of aldosterone (8). Regarding its potential benefits in combating edemas, Girerd et al. (40) reported that in patients hospitalized for severe diuretic resistance and concerning hypokalemia, intravenous mineralocorticoids antagonists (MRAs), such as canrenoate, attained decongestion and could be used as a rescue therapy. Furthermore, aldosterone blockade could be also achieved using aldosterone synthase inhibitors (ASIs) as an alternative to MRAs. The advantage of this novel approach lies within the fact that ASIs do not induce negative feedback and eventually increased cortisol (which in turn exert various side effects, increasing even cardiovascular risk via prolonged activation), as opposed to MRAs. Their effects were studied in patients with primary aldosteronism, displaying a reversible dose-dependent reduction in aldosterone concentrations, both plasmatic and urinary. Also, clinical tolerance and biological safety were comparable to those of eplerenone (43). However, these blockades are not always sufficient since there were described cases in which a rebound increase in circulating aldosterone was described when using MRA combined with ACE inhibitors (44). This phenomenon, called aldosterone breakthrough, is still to be cleared up.
Angiotensin and aldosterone physiologically increase the apical permeability of the collecting ducts to sodium, modulating the activity of epithelial sodium channels (ENaC) (45). Since these biomarkers show elevated plasma concentrations in HF, it was hypothesized that their chronic overdrive might generate a malfunction of ENaC. Zheng et al. (5) proved in an experimental study, that rats with HF expressed an abundance of protein subunits of ENaC in the cortex and medulla of the kidneys.
AT II is the major effector of RAAS (37). AT II plays an important role in the short-term maintenance of circulatory homeostasis, but its sustained production has maladaptive effects and leads to fibrosis of the heart, kidneys, and other organs, as well as to the worsening of neurohormonal activation by increasing the release of norepinephrine from the sympathetic nerve endings by stimulating the adrenal medulla for aldosterone release. Aldosterone supports short-term circulation by renal reabsorption of sodium in exchange for potassium, but in the long-term it has negative effects through hypertrophy and fibrosis at the myocardial and vascular level, decreasing vascular compliance and increasing the myocardial rigidity (8).
Angiotensin converting enzyme 2 (ACE2) is a counter regulator of RAAS with the role of opposing the effects of AT II by transforming it into AT I (10), which has a protective role on the cardiovascular system (11). The value of ACE2 is increased in patients with HF and systolic dysfunction alongside an unfavorable evolution. Thus, ACE2 is a biomarker with a prognostic role for patients with HFrEF (12). New drugs that target ACE2 were shown to attenuate diastolic disfunction in HF subjects. SARS-CoV-2 virus uses host-receptors of ACE2 to attach and invade human cells and cause multi-organ lesions. Comorbidities such as HF and coronary artery disease express elevated levels of ACE2, thus leading to an increased susceptibility and disease severity in SARS-CoV-2 infection. Also, the binding of the virus to ACE2 receptors determines endothelial dysfunction through hyperinflammation, elevated plasma levels of prothrombin, and as a consequence the risk of arterial and venous thrombosis increases (46–48).
ACEi use in SARS-CoV-2 infected patients has been controverted, since various authors have raised concern about the fact that they will increase infection susceptibility due to raised expression of ACE2. Despite this, ARBs were shown to be superior to ACEi in this category of patients, and treatment continuation has been proposed. However, other authors claimed that ACEi can be in fact benefic against pulmonary lesions induced by the virus and should be introduced in the treatment strategy of COVID-19 patients (49).
Additionally, a meta-regression analyses shows that ARBs/ACEi use, correlates with a significant drop in mortality in COVID-19 patients, after sex, age, cardiovascular disease, high blood pressure and chronic kidney disease adjusting. Moreover, treatment management of COVID-19 patients from current guidelines also recommend continuation with these pharmacological agents in patients with cardiovascular comorbidities (46).
The apelin/APJ system also has a role in counter-regulating RAAS by antagonizing AT II activity (50). Apelin is a peptide encoded by the APLN gene, located on the long arm of the X chromosome. It is originally produced as a pre-pro-apelin consisting of 77 amino acids. Apelin is subsequently cleaved into smaller C-terminal fragments with biological activity (apelin-13,−16,−17,−19,−36) (51). Apelin-13 is the shortest fragment derived from pre-pro-apelin and it is the most potent peptide from all the fragments cleaved by angiotensin-converting enzyme from this 77-amino acid protein. At the cellular level, apelin exerts its effects by activating B-protein kinase pathways (Akt), Cε protein phosphatidyl-inositol-3 kinase (PI3K) and regulated extracellular signaling kinases (Erk), proteins known to play a role in preventing apoptosis and cell proliferation and migration. Apelin has complex functions in the body, being a potent inotropic, vasodilator and diuretic agent. The apelin/APJ system has many physiological actions on the hydric balance. The latter effect is produced by inhibition of the vasopressin secretion at the neuronal level and by actions on renal microcirculation and tubular function. During the evolution of HF, the plasma level of apelin decreases gradually after an initial rise in the early stage of the disease (52, 53). There is evidence that ACE2 is the enzyme which fragments apelin by removing the C-terminus, the main area through which it exerts its effect on the APJ receptor (51, 54). In patients with HF with a low ejection fraction and an unfavorable prognosis, apelin has reduced values, thus being a biomarker with a prognostic role (55, 56).
Copeptin represents a C-terminal subdivision of the precursor pre-pro-vasopressin, and similar to NT-proBNP, directly reflects arginine-vasopressin (AV) levels (which are difficult to measure and not used in clinical practice). Its secretion is stimulated by hyperosmolarity and exposure of the body to different stages of endogenous stress, as a result of AV system activation (57). The MOLITOR study (58) showed that high copeptin level at admission proved to be the best prognostic marker of rehospitalization and 90-day mortality rate, in conjunction with the worst prognosis at 3 months follow-up of patients with increased copeptin serum levels during the first 3 days of hospitalization. Also, its predictive value was much the same as NT-proBNP's in terms of all-cause-mortality, or even superior (59, 60). When combined with sodium, copeptin provided increased prognostic information for HF mortality and correlated with other markers of congestion, proving its involvement in pathophysiological mechanism of fluid retention (60).
In the distal nephrons, the kinin–kallikrein system (KKS) modulates water and sodium reabsorption by acting on the ENaC gating. Increased kallikrein urinary concentrations following administration of antihypertensive drugs displays KKS's interaction with other biologically active molecules such as prostaglandins and RAAS, as well as its involvement in their mechanism of action (61). Evidence suggests that bradykinin (BK) acutely repress ENaC activity in a reversible and dose-dependent manner (62). Moreover, BK was shown to generate a decrease in ENaC opening probability, thus blocking sodium and water reabsorption. Hence, in experimental studies on mice, augmented ENaC action with subsequent hypertension were observed following genetic ablation of BK receptors. Likewise, increased BK signaling revealed a powerful antihypertensive effect, proving its value in the biological natriuretic response and clinical additional benefits in decongestion (61).
Also, in spite of being a paradox given its name, angiotensin converting enzyme (ACE) seems to be a much more potent catalyst in cleaving kinins, than AT I in AT II (15). This became unquestionable when ACE inhibition was followed by a considerable increase in plasma concentration of BK. On the bright side, among BK's therapeutic effects, vasodilation and reduction of sympathetic overdrive were just “the tip of the iceberg” (8). Therefore, suspicions were raised on whether the clinical improvement seen in congestive HF patients should be attributable to the inhibition of AT II production, or much likely to the augmentation of BK.
Finally, other proteases are noteworthy due to their effect on the activation of ENaC. A recent study revealed that patients with HF and signs of clinical congestion presented significantly elevated urinary concentrations in plasmin, furin and prostasin, confirming their involvement in edema formation (63).
Natriuretic peptides have long been used to assess the level of congestion in HF. Their plasma concentrations are increased as a response to volume and pressure overload in the myocardial walls. The diuretic and natriuretic effects of NPs as an attempt to reduce blood volume, is generated by antagonizing the actions of AT II on vascular walls (reducing peripheral vasculature resistance), aldosterone secretion, and sodium and water reabsorption (9, 64). Also, Bagshaw et al. (10) observed that NPs proved to be useful in assessing interstitial fluid overload, that eventually leads to acute decompensations of HF, associated by clinical exacerbation, hospitalization, and death. Herein, the use of these biomarkers could be translated into earlier therapeutical interventions and better clinical outcomes.
Nevertheless, a prospective design study containing 565 cases of sudden cardiac deaths (SCD) disclosed that even a subclinical volume and pressure overload triggers NT-proBNP and leads to an increased risk of SCD before the onset of clinical signs and symptoms of HF, also reflecting its prognostic value as a marker of myocardial injury and hemodynamic stress (65). Finally, from a statistic point of view, NT-proBNP's diagnostic accuracy for HF shows a specificity of 90%, a negative predictive value of 95%, and a positive predictive value of 85% (66).
Sacubitril/valsartan combination acts as first line blocker of angiotensin-neprilysin receptor (ARNi) approved for HF treatment. On one hand, sacubitril determines a reduction in the preload and ventricular remodeling via its natriuretic, diuretic and vasodilator effects. On the other hand, valsartan expresses its benefic cardiovascular effects via selective blockade of AT I receptor and subsequent AT II inhibition. As a result, sodium and water retention and cardiorenal fibrosis are repressed (67).
Nevertheless, isolated utilization of neprilysin inhibitors enhanced plasma levels of AT I and AT II, proving the need of concomitant use of an angiotensin receptor blocker (ARBs) in order to achieve cardiovascular and renal benefic effects, and also the superiority of the combination in the treatment of HF patients (67, 68).
Its most important roles are natriuresis, diuresis, vasodilatation, inhibition of the renin-angiotensin-aldosterone system and blockage of the sympathetic nervous system (69, 70). As a general review, NPs act on three types of guanylyl cyclase receptors (NPR): NPR-A, NPR-B, NPR-C (69). Many factors can stimulate the secretion of ANP: increased intravascular volume, increased wall tension, chronic hypoxia, increased osmolarity (69). Given its sensitivity, we can use it to differentiate non-cardiac dyspnea from the cardiac one. We can consider, that it is a useful biomarker to diagnose HF (both chronic and acute) especially in outpatients with type 2 diabetes (69, 71), being also useful in predicting mortality, onset of atrial fibrillation and renal regression (69). Evidence suggest that its elevated plasma levels could also indicate a higher risk of major cardiovascular events and mortality, organ dysfunction and sepsis (70). The release of ANP depends on many factors, such as catecholamines, endothelins, arginine vasopressin and thyroid hormones (69).
Corin represents a pro-natriuretic peptide convertase that together with NPs, plays an important role in the compensatory mechanism of maintaining fluid homeostasis in HF. Nonetheless, this compensatory mechanism seems to be limited since reduced plasma concentration of corin coupled with augmented plasma levels of NPs due to enzymatic downregulation (72), were observed in several studies amongst HF patients (26, 73, 74). For instance, levels of uncleaved pro-atrial natriuretic peptide (pro-ANP) were significantly (p = 0.01) elevated in HF patients and pre-clinical models, contributed to acute decompensations due to fluid disbalance (73, 74). Furthermore, Tripathi et al. (73) showed that on a genetic level, NPs transcripts increase in the last two stages of HF, while their plasma concentrations increased only in terminal HF (stage D). In contrast, cardiac corin transcripts begun to decline at early-stage B of HF (when patient do not exert symptoms of congestion), and remained low in evolution to stages C and D. To support this finding, another study exposed that experimental corin transcript restoration, significantly attenuates HF progression, in both HF and dilatative patients (75). The puzzling aspect in this last study is represented by the fact that corin, even in its catalytically inactive state, decreased congestion, thus proving that cardiac protective effects of corin do not necessarily require its protease status. Ultimately, corin evaluation in HF patients could stand as a marker of “disease progression” on a molecular level when other biomarkers such as NPs are not yet altered. This provides new insights in terms of early therapeutical intervention to prevent disease progression and onset of clinical symptoms. In addition, very low corin plasma levels combined with impaired pro-ANP cleavage and subsequently, very high levels of NPs (74), were found to be inversely corelated with the accumulation of lung edema and with the degree of decompensation.
Neprilysin, a metalloprotease enzyme, is responsible for the degradation of the NPs, AT I and bradykinin, thus becoming a potential target for the treatment of congestive HF. Therefore, its inhibition was proposed as an adjuvant in diuretic therapy due to the rise in plasma concentrations of NPs with increased natriuresis, diuresis, and vasorelaxation (43). In this matter, following renal denervation, the beneficial effect was attributed to a significant inhibition of neprilysin in the kidneys due to its autocrine effects that exert cardioprotective actions and also combat congestion (9).
Galectin-3 is a complex biomarker with pleiotropic physiological functions and documented pro-fibrotic activity myofibroblast proliferation that leads to ventricular remodeling and cardiac myocyte stress (76, 77). Studies on experimental animals proved that upregulation of galectin-3 was prone to exacerbate HF (77). Its role in clinical congestion and fluid disbalance may be attributable to its involvement in the inflammatory cascade following cardiac injury. As a matter of fact, genetic knockout of galectin-3 showed a reduction in cardiac inflammation and a slower disease progression thanks to a resistance of the left ventricle at pressure and volume overload (76, 78). Additionally, an extensive metanalysis including 7,057 patients with acute HF, presented elevated serum levels of galectin-3, which correlated with increased risk of all-cause mortality and cardiovascular mortality (79). In terms of prognostic value, it appears that galectin-3 has a superior power in predicting 60-day mortality as compared to the standard biomarker, NT-proBNP (78, 80). Surely, most recent findings agree that this novel biological molecule could be of great use in identifying patients at high risk of forthcoming events.
Mechanical strain on cardiac fibroblasts and cardiomyocytes greatly induces the ST2 gene, according to genomic research. A membrane-bound receptor (ST2L) as well as a soluble version of ST2 are produced by this gene (sST2) (81). In instances of cardiac stress such as high blood pressure and myocardial hypertrophy (82) levels of sST2 are significantly elevated. Furthermore, when associated with other biomarkers of myocardial injury such as high-sensitive Troponin (hsTn) and NT-proBNP, in unstable patients with acute HF, sST2 increased the risk of death with a hazard ratio (HR) of 2.64 (66). The precise mechanism of sST2 in HF remodeling and decompensation, however, is unknown, but it was established that its levels are elevated both in HFpEF and in HFrEF (83). Acute and chronic HF are also responsible for higher levels of sST2 and it has been linked to poor prognosis and unfavorable LV remodeling (84). As for therapeutical implications, Gaggin et al. showed that sST2 levels are specifically identified various strata of risk based on subsequent achievement of beta-blocker dosing, expanding the observation that sST2 values are changed by beta-blocker dosing. Our findings open the possibility of prospectively investigating the use of sST2-guided beta-blocker treatment in HFrEF, since researchers in the field of chronic HF are currently tentatively examining the potential for biomarker-guided HF care (85).
A recent study exposed that cBIN1, a key mechanism in maintaining normal heart contraction, relaxation, and cardiomyocyte health, expresses various alterations in HF. Moreover, gene therapy with exogenous cBIN1 rescued cardiac lusitropic and inotropic properties, proving not only its great therapeutic potential in failing hearts (86), but also serving as a biomarker of cardiomyocyte remodeling (87).
Furthermore, Food and Drug Administration included Ischemia-Modified Albumin, electrocardiogram and the cardiac Troponin I as ischemia markers for the diagnosis of acute coronary syndromes (ACS), cumulating altogether a 95% sensitivity. Another biomarker of myocardial necrosis, Heart-type Fatty Acid-Binding Protein was proposed as an extra tool for the diagnosis of ACS, expressing only additive diagnostic value (66). However, its prognostic value for myocardial damage was established due to the fact that it proved to be the earliest marker for ACS confirmation (88).
Cathepsins are lysosomal proteases contributing to autophagic degradation of cellular substrates. They are composed of 11 members (cathepsin B, C, F, H, K, L, O, S, V, X, and W). Various findings support the notion that cathepsins could participate in cardiac remodeling by mediating extracellular matrix degradation. Cathepsin S and K were found increased in pressure overload-induced myocardium of patients with HF (89). Cysplatin-C is a potent cysteine protease inhibitor, that plays an essential role in vascular pathophysiology. It is an inhibitor of cathepsin S and K. The activity of cathepsin S is tightly regulated by its endogenous inhibitor, cystatin C, which also has a role in antigen presentation. Cathepsin S is a lysosomal enzyme that belongs to the papain-like protease of cysteine proteases, playing an active role in blood vessels permeability and angiogenesis. In normal situation, kidneys excrete Cysplatin-S. Any kind of kidney disease (glomerulopathy, tubulopathy, vasculopathy, interstitial disease) is first observed by high concentration of Cysplatin-C (90).
There was demonstrated, that Cysplatin-C has a major prognostic value in HFrEF. Cysplatin-C is a strong predictor of events in stable chronic HF, independent of traditional risk factors and BNP. Moreover, serum Cysplatin-C levels are correlated with glomerular filtration rate, so it can be useful in detecting kidney diseases. Recent studies demonstrated a direct correlation between high levels of Cysplatin-C and hospitalization rate and mortality. We can affirm, that Cysplatin-C could be a double predictor for acute/chronic kidney diseases and initial phase of HF (88).
Biomarkers of different physiopathological pathways responsible for volume overload and edema HFrEF are synthetized in Figure 2.
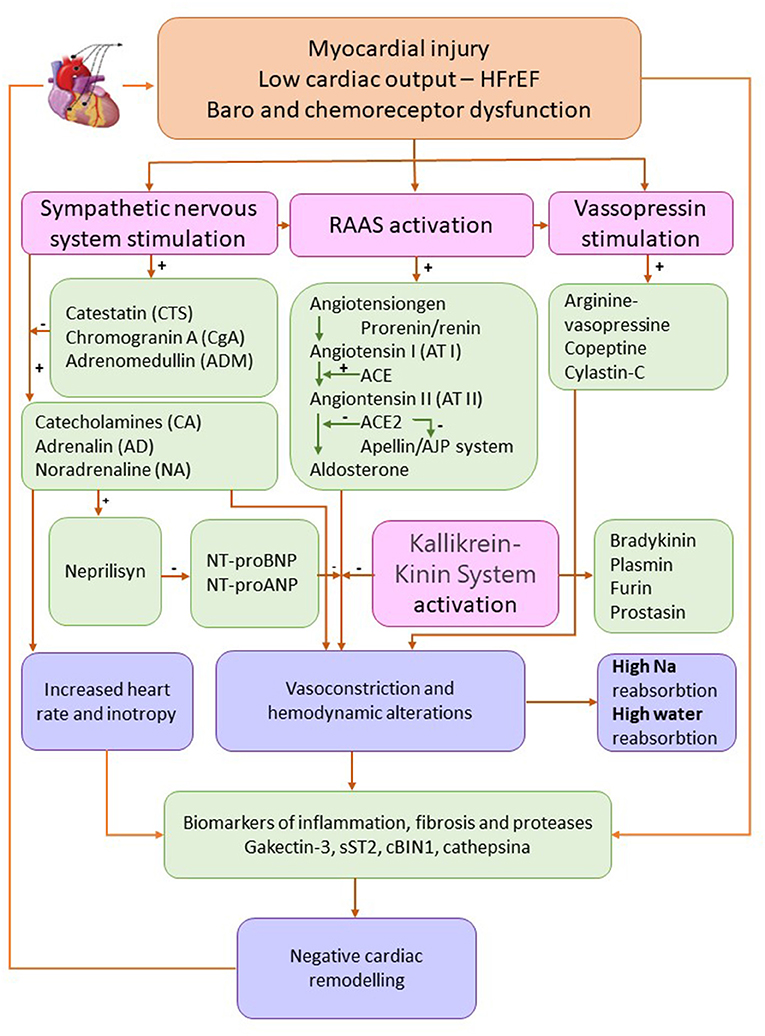
Figure 2. Biomarkers of different physiopathological pathways responsible for volume overload and edema heart failure with reduced ejection fraction. Adapted from Ibrahim et al. (84) and Oikonomou et al. (91) (HFrEF, heart failure with reduced ejection fraction; Na, sodium; RAAS, renin-angiotensin-aldosterone system).
This review summarizes the current data from the specialty literature regarding biomarkers that play a significant role in the pathogenesis of edema in HFrEF. Our work is meant to help clinicians working in cardiovascular medicine, express new targeted approaches in terms of pathophysiology, diagnosis, research clinical trials and new drugs development to reduce the morbidity and mortality in HF patients.
Several biomarkers have been studied in only few, limited scientific papers, underlying the need for continuous and rigorous further research. Hence, the lack of metanalysis focusing on their prognostic value as multimarker strategy in HF patients, encompasses our study limitation.
The biomarkers with clinical implications for edema in HFrEF are synthetized in Table 1.
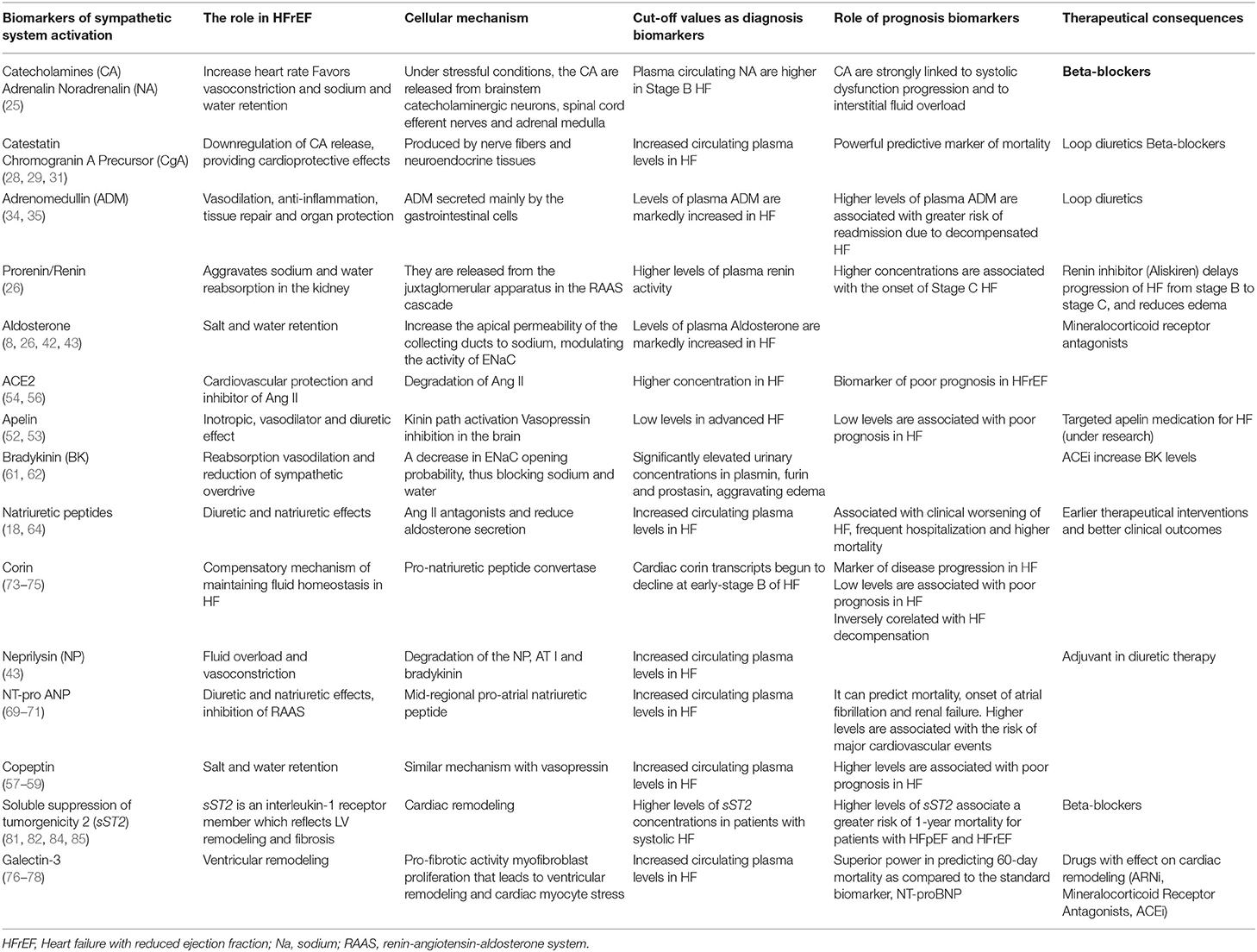
Table 1. Biomarkers with clinical implications for edema in HFrEF.
Water retention and edema in HFrEF is a consequence of the redistribution of cardiac output and neurohormonal activation involving mainly the sympathetic system, the renin-angiotensin aldosterone system as well as the systems of quinines, endothelin, neuropeptides, and arginine-vasopressin. The in-depth study of the activation of these systems in HFrEF allows the identification of new markers with a prognostic and therapeutic value. The quantification of these biomarkers plays an important role in the timely initiation of an appropriate therapy against water retention and edema and thus it helps to prevent rehospitalizations and impaired cardiac function since these are considered strong determinants of mortality in those patients. Among the new biomarkers studied with a negative prognostic role, we mention the increase of the serum values of: catestatin, chromogranin A, adrenomedullin, ACE2, copeptin, MR-proANP, galectin-3, sST2 as well as the decrease in apelin. Furthermore, the determination of these system-activation-specific biomarkers with a pathophysiological role in water and sodium retention, allows both the adequate modulation of the medication doses recommended by the guidelines and the development of new therapeutic means.
RC, R-DL, and DB research the literature. RC, R-DL, MM, and DB studied and analyzed the articles. RC, R-DL, and MM wrote the paper. All authors contributed to the article and approved the submitted version.
This work was granted by project PDI-PFE-CDI 2021, entitled Increasing the Performance of Scientific Research, Supporting Excellence in Medical Research and Innovation, PROGRES, no. 40PFE/30.12.2021 and by a grant of the Romanian Ministry of Education 492 and Research, CCCDI - UEFISCDI, project number PN III-P2-2.1-PED-2019-1057, within PNCDI III.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Timmis A, Townsend N, Gale C, Grobbee R, Maniadakis N, Flather M, et al. European Society of Cardiology: cardiovascular disease statistics 2017. Eur Heart J. (2018) 39:508–77. doi: 10.1093/eurheartj/ehx628
2. Townsend N, Kazakiewicz D, Lucy Wright F, Timmis A, Huculeci R, Torbica A, et al. Epidemiology of cardiovascular disease in Europe. Nat Rev Cardiol. (2022) 19:133–43. doi: 10.1038/s41569-021-00607-3
3. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American college of cardiology foundation/American Heart Association task force on practice guidelines. Circulation. (2013) 128:1810–52. doi: 10.1161/CIR.0b013e31829e8807
4. Bozkurt B, Coats AJS, Tsutsui H, Abdelhamid CM, Adamopoulos S, Albert N, et al. Universal definition and classification of heart failure: a report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. Eur J Heart Fail. (2021) 23:352–80. doi: 10.1002/ejhf.2115
5. Zheng H, Liu X, Rao US, Patel KP. Increased renal ENaC subunits and sodium retention in rats with chronic heart failure. Am J Physiol Ren Physiol. (2011) 300:641–9. doi: 10.1152/ajprenal.00254.2010
6. Miller WL. Fluid volume overload and congestion in heart failure. Circ Hear Fail. (2016) 9:e002922. doi: 10.1161/CIRCHEARTFAILURE.115.002922
7. Borovac JA, D'Amario D, Bozic J, Glavas D. Sympathetic nervous system activation and heart failure: Current state of evidence and the pathophysiology in the light of novel biomarkers. World J Cardiol. (2020) 12:373–408. doi: 10.4330/wjc.v12.i8.373
8. Adams KF. Pathophysiologic role of the renin-angiotensin-aldosterone and sympathetic nervous systems in heart failure. Am J Health Syst Pharm. (2004) 61(Suppl 2):S4–13. doi: 10.1093/ajhp/61.suppl_2.S4
9. Sharp Iii TE, Lefer DJ. Renal denervation to treat heart failure. Annu Rev Physiol. (2021) 83:39–58. doi: 10.1146/annurev-physiol-031620-093431
10. Bagshaw SM, Cruz DN. Fluid overload as a biomarker of heart failure and acute kidney injury. Contrib Nephrol. (2010) 164:54–68. doi: 10.1159/000313721
11. Hartupee J, Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol. (2016) 14:30–8. doi: 10.1038/nrcardio.2016.163
12. Ames MK, Atkins CE, Pitt B. The renin-angiotensin-aldosterone system and its suppression. J Vet Intern Med. (2019) 33:363–82. doi: 10.1111/jvim.15454
13. Simões e Silva AC, Teixeira MM. ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol Res. (2016) 107:154–62. dx. doi: 10.1016/j.phrs.2016.03.018
14. Muneer K, Nair A. Angiotensin-converting enzyme inhibitors and receptor blockers in heart failure and chronic kidney disease - demystifying controversies. Indian Heart J. (2017) 69:371–4. doi: 10.1016/j.ihj.2016.08.007
15. Scicli AG, Carretero OA. Renal kallikrein-kinin system. Kidney Int. (1986) 29:120–30. doi: 10.1038/ki.1986.14
16. Docherty KF, Vaduganathan M, Solomon SD, McMurray JJV. Sacubitril/Valsartan. JACC Hear Fail. (2020) 8:800–10. doi: 10.1016/j.jchf.2020.06.020
17. Packer M, Anker SD, Butler J, Filippatos G, Ferreira JP, Pocock SJ, et al. Empagliflozin in patients with heart failure, reduced ejection fraction, and volume overload: EMPEROR-reduced trial. J Am Coll Cardiol. (2021) 77:1381–92. doi: 10.1016/j.jacc.2021.01.033
18. Lieberthal W, Bernard DB. Clinical relevance of the natriuretic peptides in edematous states. Curr Opin Nephrol Hypertens. (1993) 2:653–61. doi: 10.1097/00041552-199307000-00016
19. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure. J Am Coll Cardiol. (2022) 79:e263–421. doi: 10.1016/j.jacc.2021.12.012
20. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2021) 42:3599–726.
21. Kobayashi M, Watanabe M, Coiro S, Bercker M, Paku Y, Iwasaki Y, et al. Mid-term prognostic impact of residual pulmonary congestion assessed by radiographic scoring in patients admitted for worsening heart failure. Int J Cardiol. (2019) 289:91–8. doi: 10.1016/j.ijcard.2019.01.091
22. Coiro S, Simonovic D, Deljanin-Ilic M, Duarte K, Carluccio E, Cattadori G, et al. Prognostic value of dynamic changes in pulmonary congestion during exercise stress echocardiography in heart failure with preserved ejection fraction. Circ Hear Fail. (2020) 13:e006769. doi: 10.1161/CIRCHEARTFAILURE.119.006769
23. Farcaş AD, Năstasă LE, Anton FP, Stoia MA, Goidescu M, Mocan Hognogi DL, et al. Quality of life – an important parameter of cardiac rehabilitation in heart failure patients. Balneo Res J. (2018) 9:288–90. doi: 10.12680/balneo.2018.198
24. Bencivenga L, Palaia ME, Sepe I, Gambino G, Komici K, Cannavo A, et al. Why do we not assess sympathetic nervous system activity in heart failure management: might GRK2 serve as a new biomarker? Cells. (2021) 10:457. doi: 10.3390/cells10020457
25. Mahata SK, Zheng H, Mahata S, Liu X, Patel KP. Effect of heart failure on catecholamine granule morphology and storage in chromaffin cells. J Endocrinol. (2016) 230:309. doi: 10.1530/JOE-16-0146
26. Sullivan RD, Mehta RM, Tripathi R, Reed GL. Gladysheva IP. Renin activity in heart failure with reduced systolic function-new insights. Int J Mol Sci. (2019) 20:3182. doi: 10.3390/ijms20133182
27. Sano M. Sodium glucose cotransporter (SGLT)-2 inhibitors alleviate the renal stress responsible for sympathetic activation. Ther Adv Cardiovasc Dis. (2020) 14:1753944720939383. doi: 10.1177/1753944720939383
28. Di Comite G, Morganti A. Chromogranin A: a novel factor acting at the cross road between the neuroendocrine and the cardiovascular systems. J Hypertens. (2011) 29:409–14. doi: 10.1097/HJH.0b013e328341a429
29. Bozic J, Kumric M, Ticinovic Kurir T, Urlic H, Martinovic D, Vilovic M, et al. Catestatin as a biomarker of cardiovascular diseases: a clinical perspective. Biomedicines. (2021) 9:1757. doi: 10.3390/biomedicines9121757
30. Polyakova EA, Mikhaylov EN, Sonin DL, Cheburkin YV, Galagudza MM. Neurohumoral, cardiac and inflammatory markers in the evaluation of heart failure severity and progression. J Geriatr Cardiol. (2021) 18:47–66. doi: 10.11909/j.issn.1671-5411.2021.01.007
31. Angelone T, Quintieri AM, Brar BK, Limchaiyawat PT, Tota B, Mahata SK, et al. The antihypertensive chromogranin a peptide catestatin acts as a novel endocrine/paracrine modulator of cardiac inotropism and lusitropism. Endocrinology. (2008) 149:4780. doi: 10.1210/en.2008-0318
32. Bustos B, Martínez-Herrero S. Martínez A. Adrenomedullin: not just another gastrointestinal peptide. Biomolecules. (2022) 12:156. doi: 10.3390/biom12020156
33. Voors AA, Kremer D, Geven C, Ter Maaten JM, Struck J, Bergmann A, et al. Adrenomedullin in heart failure: pathophysiology and therapeutic application. Eur J Heart Fail. (2019) 21:163–71. doi: 10.1002/ejhf.1366
34. Pandhi P, Ter Maaten JM, Emmens JE, Struck J, Bergmann A, Cleland JG, et al. Clinical value of pre-discharge bio-adrenomedullin as a marker of residual congestion and high risk of heart failure hospital readmission methods and results. Eur J Heart Fail. (2020) 22:683–91. doi: 10.1002/ejhf.1693
35. Nishikimi T, Saito Y, Kitamura K, Ishimitsu T, Eto T, Kangawa K, et al. Increased plasma levels of adrenomedullin in patients with heart failure. J Am Coll Cardiol. (1995) 26:1424–31. X doi: 10.1016/0735-1097(95)00338-X
36. Kita T, Kitamura K. Translational studies of adrenomedullin and related peptides regarding cardiovascular diseases. Hypertens Res. (2022) 45:389–400. doi: 10.1038/s41440-021-00806-y
37. Blet A, Deniau B, Geven C, Sadoune M, Caillard A, Kounde P-R, et al. Adrecizumab, a non-neutralizing anti-adrenomedullin antibody, improves haemodynamics and attenuates myocardial oxidative stress in septic rats. Intensive Care Med Exp. (2019) 7:1–13. doi: 10.1186/s40635-019-0255-0
38. Skurk C, Rottbauer W, Kessler M, Akin I, Kluge S, Burdelski C, et al. The ACCOST-HH trial. Eur Heart J. (2020) 41:4296–8. doi: 10.1093/eurheartj/ehaa757
39. Tolppanen H, Rivas-Lasarte M, Lassus J, Sans-Roselló J, Hartmann O, Lindholm M, et al. Adrenomedullin: a marker of impaired hemodynamics, organ dysfunction, and poor prognosis in cardiogenic shock. Ann Intensive Care. (2017) 7:6. doi: 10.1186/s13613-016-0229-2
40. Girerd N, Aubry M, Lantelme P, Huttin O, Rossignol P. Intravenous mineralocorticoid receptor antagonist use in acutely decompensated heart failure with diuretic resistance. Int Heart J. (2021) 62:193–6. doi: 10.1536/ihj.20-442
41. Sullivan RD, Mehta RM, Tripathi R, Gladysheva IP, Reed GL. Normalizing plasma renin activity in experimental dilated cardiomyopathy: effects on edema, cachexia, and survival. Int J Mol Sci Artic. (2019) 20:3886. doi: 10.3390/ijms20163886
42. Weber KT. Aldosterone in congestive heart failure. N Engl J Med. (2001) 345:1689–97. doi: 10.1056/NEJMra000050
43. George J, Struthers AD, Lang CC. Modulation of the renin-angiotensin-aldosterone system in heart failure. Curr Atheroscler Rep. (2014) 16:403. doi: 10.1007/s11883-014-0403-7
44. Ghionzoli N, Gentile F, Anna M, Franco M. Del, Castiglione V, Aimo A, et al. Current and emerging drug targets in heart failure treatment. Heart Fail Rev. (2021) 1:3. doi: 10.1007/s10741-021-10137-2
45. Basu R, Poglitsch M, Yogasundaram H, Thomas J, Rowe BH, Oudit GY. Roles of angiotensin peptides and recombinant human ACE2 in heart failure. J Am Coll Cardiol. (2017) 69:805–19. doi: 10.1016/j.jacc.2016.11.064
46. Lee T, Cau A, Cheng MP, Levin A, Lee TC, Vinh DC, et al. Angiotensin receptor blockers and angiotensin-converting enzyme inhibitors in COVID-19: meta-analysis/meta-regression adjusted for confounding factors. CJC Open. (2021) 3:965–75. doi: 10.1016/j.cjco.2021.03.001
47. Beyerstedt S, Casaro EB, Rangel ÉB. COVID-19: angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur J Clin Microbiol Infect Dis. (2021) 40:905–19. doi: 10.1007/s10096-020-04138-6
48. Pathangey G, Fadadu PP, Hospodar AR, Abbas AE. Angiotensin-converting enzyme 2 and COVID-19: Patients, comorbidities, and therapies. Am J Physiol Lung Cell Mol Physiol. (2021)320: L301–30. doi: 10.1152/ajplung.00259.2020
49. Vidal-Petiot E, Gault N. Renin-angiotensin system blockers and COVID-19. BMC Med. (2021) 19:19–21. doi: 10.1186/s12916-021-02012-6
50. Japp AG, Newby DE. The apelin-APJ system in heart failure. Pathophysiologic relevance and therapeutic potential. Biochem Pharmacol. (2008) 75:1882–92. doi: 10.1016/j.bcp.2007.12.015
51. Yu XH, Tang Z Bin, Liu LJ, Qian H, Tang SL, Zhang DW, et al. Apelin and its receptor APJ in cardiovascular diseases. Clin Chim Acta. (2014) 428:1–8. doi: 10.1016/j.cca.2013.09.001
52. Maguire JJ, Kleinz MJ, Pitkin SL, Davenport AP. [Pyr1]apelin-13 identified as the predominant apelin isoform in the human heart: vasoactive mechanisms and inotropic action in disease. Hypertension. (2009) 54:598–604. doi: 10.1161/HYPERTENSIONAHA.109.134619
53. Goidescu CM, Vida-Simiti LA. The apelin-APJ system in the evolution of heart failure. Clujul Med. (2015) 88:3–8. doi: 10.15386/cjmed-380
54. Sato T, Suzuki T, Watanabe H, Kadowaki A, Fukamizu A, Liu PP, et al. Apelin is a positive regulator of ace2 in failing hearts. J Clin Invest. (2013) 123:5203–11. doi: 10.1172/JCI69608
55. Chandrasekaran B, Kalra PR, Donovan J, Hooper J, Clague JR, McDonagh TA. Myocardial apelin production is reduced in humans with left ventricular systolic dysfunction. J Card Fail. (2010) 16:556–61 doi: 10.1016/j.cardfail.2010.02.004
56. Goidescu CM, Chiorescu RM, Diana MHL, Mocan M, Stoia MA, Anton FP, et al. ACE2 and apelin-13: biomarkers with a prognostic value in congestive heart failure. Dis Markers. (2021) 22:5569410. doi: 10.1155/2021/5569410
57. Dobša L, Cullen Edozien K. Copeptin and its potential role in diagnosis and prognosis of various diseases. Biochem Med. (2013) 23:172–90. doi: 10.11613/BM.2013.021
58. Düngen HD, Tscholl V, Obradovic D, Radenovic S, Matic D, Musial Bright L, et al. Prognostic performance of serial in-hospital measurements of copeptin and multiple novel biomarkers among patients with worsening heart failure: results from the MOLITOR study. ESC Heart Fail. (2018) 5:288–296. doi: 10.1002/ehf2.12231
59. Zhong Y, Wang R, Yan L, Lin M, Liu X, You T. Copeptin in heart failure: Review and meta-analysis. Clin Chim Acta. (2017) 475:36–43. doi: 10.1016/j.cca.2017.10.001
60. Ebong I, Mazimba S, Breathett K. Cardiac biomarkers in advanced heart failure: how can they impact our pre-transplant or pre-LVAD decision-making. Curr Heart Fail Rep. (2019) 16:274–84. doi: 10.1007/s11897-019-00447-w
61. Mamenko M, Zaika O, Pochynyuk O. Direct regulation of ENaC by bradykinin in the distal nephron. Implications for renal sodium handling. Curr Opin Nephrol Hypertens. (2014) 23:122–9. doi: 10.1097/01.mnh.0000441053.81339.61
62. Zaika O, Mamenko M. O'neil RG, Pochynyuk O. Bradykinin acutely inhibits activity of the epithelial Na channel in mammalian aldosterone-sensitive distal nephron. Am J Physiol Ren Physiol. (2011) 300:1105–15. doi: 10.1152/ajprenal.00606.2010
63. Zheng H, Liu X, Sharma NM Li Y, Pliquett RU, Patel KP. Urinary proteolytic activation of renal epithelial Na + channels in chronic heart failure. Hypertension. (2016) 67:197–205. doi: 10.1161/HYPERTENSIONAHA.115.05838
64. Koratala A, Kazory A. Natriuretic peptides as biomarkers for congestive states: the cardiorenal divergence. Dis Markers. (2017) 2017:1454986. doi: 10.1155/2017/1454986
65. Everett BM, Moorthy M V, Tikkanen JT, Cook NR, Albert CM. Markers of myocardial stress, myocardial injury, and subclinical inflammation and the risk of sudden death. Circulation. (2020) 142:1148–58. doi: 10.1161/CIRCULATIONAHA.120.046947
66. Jacob R, Khan M. Cardiac biomarkers: what is and what can be. Indian J Cardiovasc Dis women WINCARS. (2018) 3:240–4. doi: 10.1055/s-0039-1679104
67. Dargad R, Prajapati M, Dargad R, Parekh J. Sacubitril/valsartan: a novel angiotensin receptor-neprilysin inhibitor. Indian Heart J. (2018) 70:S102–10. doi: 10.1016/j.ihj.2018.01.002
68. Yandrapalli S, Khan MH, Rochlani Y, Aronow WS. Sacubitril/valsartan in cardiovascular disease: evidence to date and place in therapy. Ther Adv Cardiovasc Dis. (2018) 12:217–31. doi: 10.1177/1753944718784536
69. Idzikowska K, Zielińska M. Midregional pro-atrial natriuretic peptide, an important member of the natriuretic peptide family: potential role in diagnosis and prognosis of cardiovascular disease. J Int Med Res. (2018) 46:3017–29. doi: 10.1177/0300060518786907
70. Yagmur E, Sckaer JH, Koek GH, Weiskirchen R, Trautwein C, Koch A, et al. Elevated MR-proANP plasma concentrations are associated with sepsis and predict mortality in critically ill patients. J Transl Med. (2019) 17:415. doi: 10.1186/s12967-019-02165-2
71. Jensen J, Schou M, Kistorp C, Faber J, Hansen TW, Jensen MT, et al. MR-proANP and incident cardiovascular disease in patients with type 2 diabetes with and without heart failure with preserved ejection fraction. Cardiovasc Diabetol. (2020) 19:180. doi: 10.1186/s12933-020-01155-9
72. Zaidi SS, Ward RD, Ramanathan K, Yu X, Gladysheva IP, Reed GL. Possible enzymatic downregulation of the natriuretic peptide system in patients with reduced systolic function and heart failure: a pilot study. Biomed Res Int. (2018) 2018:7279036. doi: 10.1155/2018/7279036
73. Tripathi R, Wang D, Sullivan R, Fan T-HM, Gladysheva IP, Reed GL. Depressed corin levels indicate early systolic dysfunction before increases of atrial natriuretic peptide/B-type natriuretic peptide and heart failure development. Hypertension. (2016) 67:362–7. doi: 10.1161/HYPERTENSIONAHA.115.06300
74. Ibebuogu UN, Gladysheva IP, Houng AK, Reed GL. Original articles decompensated heart failure is associated with reduced corin levels and decreased cleavage of pro-atrial natriuretic peptide. Circ Heart Fail. (2011) 4:114–20. doi: 10.1161/CIRCHEARTFAILURE.109.895581
75. Tripathi R, Sullivan RD, Fan T-HM, Houng AK, Mehta RM, Reed GL, et al. Cardiac-specific overexpression of catalytically inactive corin reduces edema, contractile dysfunction, and death in mice with dilated cardiomyopathy. Int J Mol Sci. (2019) 21:203. doi: 10.3390/ijms21010203
76. Suthahar N, Meijers WC, Silljé HHW, Ho JE, Liu FT, de Boer RA. Galectin-3 activation and inhibition in heart failure and cardiovascular disease: an update. Theranostics. (2018) 8:593–609. doi: 10.7150/thno.22196
77. Gehlken C, Suthahar N, Meijers WC, de Boer RA. Galectin-3 in heart failure: an update of the last 3 years. Heart Fail Clin. (2018) 14:75–92. doi: 10.1016/j.hfc.2017.08.009
78. Lubrano V, Balzan S. Role of oxidative stress-related biomarkers in heart failure: galectin 3, α1-antitrypsin and LOX-1: new therapeutic perspective? Mol Cell Biochem. (2019) 464:143–52. doi: 10.1007/s11010-019-03656-y
79. Chen H, Chen C, Fang J, Wang R, Nie W. Circulating galectin-3 on admission and prognosis in acute heart failure patients: a meta-analysis. Heart Fail Rev. (2019) 25:331–41. doi: 10.1007/s10741-019-09858-2
80. Gaggin HK, Januzzi JL. Biomarkers and diagnostics in heart failure. Biochim Biophys Acta Mol Basis Dis. (2013) 1832:2442–50. doi: 10.1016/j.bbadis.2012.12.014
81. Shah RV, Chen-Tournoux AA, Picard MH, Van Kimmenade RRJ, Januzzi JL. Serum levels of the interleukin-1 receptor family member ST2, cardiac structure and function, and long-term mortality in patients with acute dyspnea. Circ Heart Fail. (2009) 2:311–9. doi: 10.1161/CIRCHEARTFAILURE.108.833707
82. Farcaş AD, Mocan M, Anton FP, Diana MHL, Chiorescu RM, Stoia MA, et al. Short-term prognosis value of sST2 for an unfavorable outcome in hypertensive patients. Dis Markers. (2020) 2020:8143737. doi: 10.1155/2020/8143737
83. Mocan M, Hognogi LDM, Anton FP, Chiorescu RM, Goidescu CM, Stoia MA, et al. Biomarkers of inflammation in left ventricular diastolic dysfunction. Dis Markers. (2019) 2019:7583690. doi: 10.1155/2019/7583690
84. Ibrahim NE, Januzzi JL. Established and emerging roles of biomarkers in heart failure. Circ Res. (2018) 123:614–629. doi: 10.1161/CIRCRESAHA.118.312706
85. Gaggin HK, Motiwala S, Bhardwaj A, Parks KA, Januzzi JL. Soluble concentrations of the interleukin receptor family member ST2 and β-Blocker therapy in chronic heart failure. Circ Hear Fail. (2013) 6:1209–13. doi: 10.1161/CIRCHEARTFAILURE.113.000457
86. Li J, Richmond B, Hong T. Cardiac T-Tubule cBIN1-microdomain, a diagnostic marker and therapeutic target of heart failure. Int J Mol Sci. (2021) 22:2299. doi: 10.3390/ijms22052299
87. Shaw RM, Nikolova AP, Hitzeman TC, Baum R, Caldaruse AM, Agvanian S, et al. Association of a novel diagnostic biomarker, the plasma cardiac bridging integrator 1 score, with heart failure with preserved ejection fraction and cardiovascular hospitalization. JAMA Cardiol. (2018) 3:1206–10. doi: 10.1001/jamacardio.2018.3539
88. Nigam PK. Biochemical markers of myocardial injury. Indian J Clin Biochem. (2007) 22:10–7. doi: 10.1007/BF02912874
89. Yadati T, Houben T, Bitorina A, Shiri-Sverdlov R. The ins and outs of cathepsins: physiological function and role in disease management. Cells. (2020) 9:1679. doi: 10.3390/cells9071679
90. Hoes MF, Tromp J, Ouwerkerk W, Bomer N, Oberdorf-Maass SU, Samani NJ, et al. The role of cathepsin D in the pathophysiology of heart failure and its potentially beneficial properties: a translational approach. Eur J Heart Fail. (2020) 22:2102–11. doi: 10.1002/ejhf.1674
Keywords: heart failure, volume overload, physiopathology, edema, biomarkers
Citation: Chiorescu RM, Lazar R-D, Buksa S-B, Mocan M and Blendea D (2022) Biomarkers of Volume Overload and Edema in Heart Failure With Reduced Ejection Fraction. Front. Cardiovasc. Med. 9:910100. doi: 10.3389/fcvm.2022.910100
Received: 31 March 2022; Accepted: 23 May 2022;
Published: 17 June 2022.
Edited by:
Ryan D. Sullivan, University of Arizona, United StatesReviewed by:
Weizhong Zhu, Nantong University, ChinaCopyright © 2022 Chiorescu, Lazar, Buksa, Mocan and Blendea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roxana-Daiana Lazar, cm94YW5hZGxhemFyQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.