 Naofumi Amioka1
Naofumi Amioka1 Toru Miyoshi1*Tomoko Yonezawa2Megumi Kondo1
Toru Miyoshi1*Tomoko Yonezawa2Megumi Kondo1 Satoshi Akagi1Masashi Yoshida1Yukihiro Saito1
Satoshi Akagi1Masashi Yoshida1Yukihiro Saito1 Kazufumi Nakamura1Hiroshi Ito1
Kazufumi Nakamura1Hiroshi Ito1- 1Department of Cardiovascular Medicine, Faculty of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, Okayama, Japan
- 2Department of Molecular Biology and Biochemistry, Faculty of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, Okayama, Japan
Background: Abdominal aortic aneurysm (AAA) is a life-threatening disease that lacks effective preventive therapies. This study aimed to evaluate the effect of pemafibrate, a selective peroxisome proliferator-activated receptor alpha (PPARα) agonist, on AAA formation and rupture.
Methods: Experimental AAA was induced by subcutaneous angiotensin II (AngII) infusion in ApoE–/– mice for 4 weeks. Pemafibrate (0.1 mg/kg/day) was administered orally. Dihydroethidium staining was used to evaluate the reactive oxygen species (ROS).
Results: The size of the AngII-induced AAA did not differ between pemafibrate- and vehicle-treated groups. However, a decreased mortality rate due to AAA rupture was observed in pemafibrate-treated mice. Pemafibrate ameliorated AngII-induced ROS and reduced the mRNA expression of interleukin-6 and tumor necrosis factor-α in the aortic wall. Gelatin zymography analysis demonstrated significant inhibition of matrix metalloproteinase-2 activity by pemafibrate. AngII-induced ROS production in human vascular smooth muscle cells was inhibited by pre-treatment with pemafibrate and was accompanied by an increase in catalase activity. Small interfering RNA-mediated knockdown of catalase or PPARα significantly attenuated the anti-oxidative effect of pemafibrate.
Conclusion: Pemafibrate prevented AAA rupture in a murine model, concomitant with reduced ROS, inflammation, and extracellular matrix degradation in the aortic wall. The protective effect against AAA rupture was partly mediated by the anti-oxidative effect of catalase induced by pemafibrate in the smooth muscle cells.
Introduction
Abdominal aortic aneurysm (AAA) is characterized by progressive dilation of the abdominal aorta and associated risk of rupture and sudden death. Multiple factors are associated with the development and fatal rupture of AAA, including infiltration of macrophages that release pro-inflammatory cytokines (1), generation of reactive oxygen species (ROS) (2–5), impairment and apoptosis of vascular smooth muscle cells (VSMCs) (6), and degradation of the extracellular matrix by activated matrix metalloproteinases (MMPs) (7). Unfortunately, effective preventive treatments have not yet been established despite extensive basic and clinical research on AAA. Previous clinical studies have shown the potential of beta-blockers and angiotensin-converting enzyme inhibitors to prevent the development or rupture of AAA; however, their clinical effectiveness for AAA remains controversial (8–11).
Peroxisome proliferator-activated receptors (PPARs) are transcription factors that belong to the nuclear receptor superfamily, which includes the three following subtypes: PPARα, PPARβ/δ, and PPARγ. PPARs bind to PPAR-responsive regulatory elements and regulate energy homeostasis, insulin sensitivity, and lipid metabolism by promoting the expression of various genes (12–15). In addition, the activation of PPARα can regulate the expression of genes involved in inflammation and oxidative stress (16–18). PPARα is expressed in various types of cells in the body, including vascular component cells such as macrophages, vascular smooth muscle cells, and endothelial cells (17, 19–23). Previous studies showed that fibrates, which are PPARα agonists, decrease the production of inflammatory cytokines, infiltration of monocytes, and expression of MMP genes in the aortic wall (24–27). Fibrates have also been reported to attenuate the reduction of anti-oxidative enzymes, including superoxide dismutase and catalase, in the aortic wall impaired by diabetic stress (28). Accumulating evidence showing the counteraction of fibrates on the pathogenesis of AAA suggests the potential of PPARα as a therapeutic target for the development and rupture of AAA (29).
However, the side effects of conventional PPARα agonists, especially off-target effects such as liver damage and elevated serum creatinine levels, are major concerns in clinical practice (30–32). Recently, pemafibrate, a selective PPARα modulator, has been discovered (33, 34). Pemafibrate is more potent in activating PPARα than conventional PPARα agonists, as indicated by its lower effective concentration and higher selectivity toward PPAR subtypes with reduced off-target side effects (35, 36).
In primary studies, angiotensin II (AngII)-infused hypercholesterolemic mice are widely used to develop experimental AAA. AngII infusion has been reported to promote AAA formation in mice by inducing ROS, inflammation, and activating MMPs in the aortic wall (37–40). Thus, AngII infusion in mice is a technically facile animal model that recapitulates multiple facets of AAA in human.
In this study, we investigated the protective effect of pemafibrate on AAA formation and rupture in an experimental murine model, with a focus on AngII-induced ROS production and inflammation.
Materials and Methods
Animals and Treatments
All animal experiments were conducted in accordance with experimental protocols approved by the Institutional Animal Care and Use Committee of Okayama University (OKU-2021372). Male ApoE–/– mice were purchased from Jackson Laboratory (Bar Harbor, ME, United States). Figure 1 shows the experimental animal protocol. For the AAA model, 8-week-old ApoE–/– male mice were stimulated with a continuous infusion of AngII (1,000 ng/min/kg) for 4 weeks. AngII was dissolved in sterile saline and infused using Alzet osmotic pumps (Model 2004, Durect Corp., Cupertino, United States). Osmotic pumps filled with AngII were implanted subcutaneously in the neck under ketamine and xylazine anesthesia. A saline infusion was used as the control. To evaluate the effect of pemafibrate on AAA, treatment with pemafibrate (0.1 mg/kg/day) or vehicle was commenced a week before the administration of AngII or saline. Mice were anesthetized by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (5 mg/kg) before euthanization. To evaluate early changes in the aortic wall after AngII infusion, another set of mice was euthanized at 1 week. Catalase staining, dihydroethidium (DHE) staining, gelatin zymography, and gene expression analysis were performed in mice euthanized at 1 week. In addition, the evaluation of blood pressure, serum lipid profile, the incidence of AAA rupture, the maximum diameter of the abdominal aortas, plaque volume of the thoracic aorta, and histology of the abdominal aorta [elastin van Gieson (EVG) staining] were performed in mice euthanized at 4 weeks. The sample size of each group for the 1-week infusion of saline or AngII was 5. The sample size of each treatment group infused with saline or AngII for 4 weeks was as follows: vehicle-treated saline-infused mice (n = 10), pemafibrate-treated saline-infused mice (n = 10), vehicle-treated AngII-infused mice (n = 25), and pemafibrate-treated AngII-infused mice (n = 25). All mice were euthanized at their respective endpoints under anesthesia.
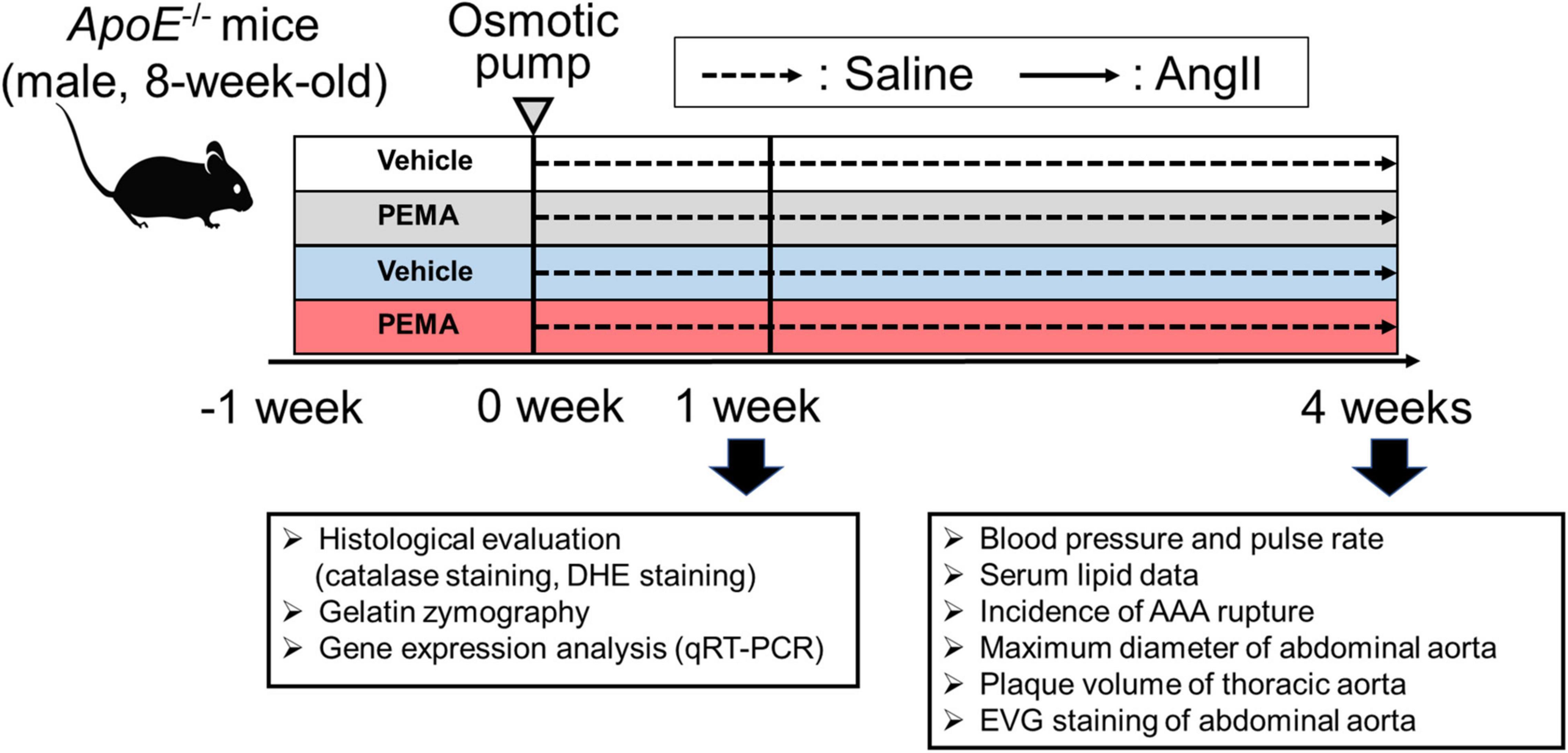
Figure 1. Animal experiment protocol. To develop an experimental abdominal aortic aneurysm (AAA) model, 8-week-old ApoE-/- male mice were infused with angiotensin II (AngII) (1,000 ng/min/kg) or saline for 4 weeks. To assess the effect of pemafibrate, treatment with vehicle or pemafibrate (PEMA) (0.1 mg/kg/day) was initiated 1 week before administration of AngII or saline, resulting in four treatment groups: vehicle-treated saline-infused (n = 10), pemafibrate-treated saline-infused (n = 10), vehicle-treated AngII-infused (n = 25), and pemafibrate-treated AngII-infused (n = 25). Five mice in each group were euthanized to evaluate the early changes after AngII infusion. Blood pressure and heart rate were measured using the tail-cuff method before and 4 weeks after AngII infusion. All mice were euthanized at their respective endpoints under anesthesia. DHE, dihydroethidium; EVG, Elastica van Gieson; qRT-PCR, quantitative reverse transcription polymerase chain reaction.
Measurement of Blood Pressure
Blood pressure was measured using the tail-cuff method (MK-2000; Muromachi, Tokyo, Japan) at the start of the experiment (baseline) and 4 weeks after AngII infusion.
Analysis of Serum Lipid Profile
Mice were fasted overnight at the endpoint of this study (4 weeks after the beginning of AngII or saline infusion). Blood was collected from the right ventricle. Serum samples were separated by centrifugation of blood samples at 3,000 rpm for 20 min. The serum was stored at -80°C. The general serum lipid profile was analyzed using high-performance liquid chromatography (Skylight Biotech Inc., Akita, Japan).
Evaluation of the Incidence of Aortic Rupture, Abdominal Aortic Diameter, and Extent of Atherosclerosis
To assess the incidence of aortic rupture, each mouse was monitored intensively every day to ensure immediate dissection of dead mice. After termination, the abdominal aortic diameter was measured at the suprarenal lesion of the aorta using ex vivo imaging. In addition, thoracic aortas were used for Oil Red O staining (Sigma-Aldrich, St. Louis, MO, United States) to compare the percentage of plaque areas among the treatment groups. The plaque area was quantified using the ImageJ software (National Institutes of Health, Bethesda, MD, United States).
Histological Assessment
Suprarenal abdominal aortic segments were fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 5-μm-thick sections. Three sets of serial sections obtained at 500 μm intervals were stained with EVG using a standard protocol to evaluate the abdominal aorta longitudinally. To detect catalase in the aortic wall, immunostaining was performed using the following method. First, deparaffinized tissue sections were incubated with 0.3% H2O2 in Tris buffer for 15 min at room temperature (RT), 0.025% Triton in Tris buffer for three times, and streptavidin/biotin blocking kit (SP-2002, Vector Labs, Burlingame, CA, United States) for 15 min, respectively, at RT. Subsequently, the slides were incubated with blocking solution (X0909, DAKO, Santa Clara, CA, United States) for 15 min at RT and then incubated with a rabbit anti-mouse catalase primary antibody (1 μg/mL, ab16731, Abcam, Cambridge, United States) overnight at 4°C. Next, the sections were incubated for 30 min at room temperature with a biotinylated swine anti-rabbit IgG secondary antibody (1:500, E0353, DAKO). Finally, a biotinylated protein detection kit (SA-5704, Vector Labs) and diaminobenzidine (DAB) substrate (SK-4105, Vector Labs) were used. EVG- and catalase-stained samples were photographed using an Axioskop 2 Plus light microscope (Zeiss, Oberkochen, Germany). The percentage of catalase-positive areas in the tunica media in five different fields and 7–8 different samples were analyzed using ImageJ software.
Cell Culture
Human aortic VSMCs were obtained from Lonza (Basel, Germany) and cultured in SmGM-2 Bullet Kit medium (Lonza) supplemented with 5% fetal bovine serum (FBS), 0.2% human fibroblast growth factor-B, 0.1% gentamicin/amphotericin B solution, 0.1% human epidermal growth factor, and 0.1% insulin. To analyze the effect of pemafibrate on ROS, cells were first grown in a reduced-serum medium for 24 h and then cultured in the respective basal medium supplemented with 1% FBS. The cells were treated with pemafibrate (0.1–10 μM) or vehicle (dimethyl sulfoxide) for 24 h. To evaluate the anti-oxidative effect of catalase activity induced by pemafibrate, 3-amino-1,2,4-triazole (3AT) (Sigma Aldrich) (50 mM), a catalase inhibitor, was co-administered with pemafibrate.
Small Interfering Ribonucleic Acid Transfection
Gene silencing experiments were performed by transfecting VSMCs with 10 μM small interfering ribonucleic acid (siRNAs) (Ambion, Life Technologies, Darmstadt, Germany) targeting catalase (CAT), PPARα, or the negative control, using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, United States), and Opti-MEM (Gibco, Waltham, MA, United States) for 24 h, before each treatment.
Catalase Activity
The catalase activity of VSMCs was determined using a simple visual assay as described previously (41). Briefly, catalase powder (Sigma Aldrich) dissolved in 100 μL of distilled water was used to prepare catalase standards. Next, VSMCs (1.0 × 107 cells) were suspended in 100 μL of phosphate-buffered saline (PBS). Catalase standard or sample solution (100 μL) was transferred to a Pyrex test tube (13 mm diameter × 100 mm height), and 100 μL each of 1% Triton X-100 (MP Biomedicals, Santa Ana, CA, United States) and 30% hydrogen peroxide (Wako, Osaka, Japan) were added, mixed gently, and incubated at 20°C. After the completion of the reaction, the height of the O2-forming foam, which remained constant for 15 min, was measured in millimeters using a ruler.
Detection of Reactive Oxygen Species
The presence of ROS in the aortic walls of mice and VSMCs was analyzed using DHE staining (Molecular Probes, Eugene, United States). Briefly, VSMCs were plated on glass coverslips placed in 12-well plates. Subsequently, sub-confluent cells were stimulated with AngII (100 nM) for 30 min, washed with PBS, incubated with DHE (5 μM) for 30 min, and analyzed for fluorescence. Red fluorescence intensity (FI) (585 nm) was measured using an OLYMPUS IX71 fluorescence microscope (Tokyo, Japan). The mean FIs of 10–20 nuclei per image, 5 images per coverslip, and 3 coverslips per sample were measured using the ImageJ software. The aortas of mice treated for 1 week were perfused with PBS (pH 7.4) for 5 min at 4°C. Subsequently, the aortic tissue was harvested from the abdominal aorta, embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek United States, Torrance, CA, United States), and snap-frozen. Freshly cut frozen aortic sections (5 μm) were incubated with DHE for 30 min at 37°C to detect ROS. For in vivo experiments, the mean FI of 10–20 nuclei per image and five images per sample were analyzed using the ImageJ software.
Gelatin Zymography
The enzymatic activities of MMP-2 and MMP-9 were analyzed in the aortic walls of mice treated with vehicle or pemafibrate for 1 week. Briefly, 10 μg of total protein isolated from the abdominal aorta was subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis containing 1 mg/mL gelatin added before heating. After electrophoresis, the gel was washed with 2.5% Triton X-100 solution for 30 min and then incubated in a solution containing 50 mM Tris-HCl, 5 mM CaCl2, and 1 μM ZnCl2 for 16 h at 37°C. Following incubation, the gel was stained with 0.05% Coomassie Brilliant Blue R-250 for 30 min at room temperature, washed with buffer, and photographed. Pro-MMP-2, active MMP-2, and pro-MMP-9 were visualized as colorless bands against a blue background. The color density of the band formation area was determined using the ImageJ software.
Quantitative Reverse Transcription-Polymerase Chain Reaction
RNA was extracted from the aortic tissue of mice in the acute model or from VSMCs using the RNeasy Mini Kit (Qiagen, Valencia, United States). Complementary diribonucleic acid (cDNA) was synthesized from 1.0 μg of extracted total RNA using ReverTra Ace (TOYOBO, Osaka, Japan). The synthesized cDNAs were subjected to polymerase chain reaction (PCR) using the TaqMan Gene Expression Master Mix (Applied Biosystems, Foster City, United States) and predesigned gene-specific primer and probe sets (TaqMan Gene Expression Assays; Applied Biosystems). TaqMan gene expression probe-and-primer sets for PPARα, superoxide dismutases (SOD1 and SOD2), nicotinamide adenine dinucleotide phosphate oxidases (NOX2 and NOX4), CAT, heme oxygenase-1 (HO-1), interleukin-6 (IL-6), tumor necrosis factor-α (TNFα), and transforming growth factor-β1 (TGF-β1) were purchased from Applied Biosystems. Real-time PCR was performed in triplicate for each sample using the QuantStudio 1 real-time PCR System (Applied Biosystems) described previously (42). The results were quantified using the relative Ct method and normalized to the internal control, glyceraldehyde 3-phosphate dehydrogenase. See Supplementary Table 1 for PCR primer details.
Statistical Analyses
All analyses were performed using EZR version 1.41 (Saitama Medical Center, Jichi Medical University, Saitama, Japan) (43), a graphical user interface of R (The R Foundation for Statistical Computing, Vienna, Austria), or SigmaPlot version 14.5 (Systat Software Inc., San Jose, CA, United States). For normally distributed continuous variables, results are expressed as the mean ± standard error of the mean. One-way analysis of variance with Bonferroni post-hoc test was used to examine the differences among groups. Non-normally distributed continuous variables were expressed as median (interquartile range) and analyzed using the Kruskal–Wallis tests. Categorical variables are presented as absolute values and frequencies, and categorical variables were compared using the chi-square test. Kaplan–Meier survival curves were used to evaluate survival rates, and significance was assessed using the log-rank test. In some experiments, technical replicates were performed as described in each figure legend. Differences with p < 0.05 were considered statistically significant.
Results
Pemafibrate Reduced Angiotensin II-Induced Abdominal Aortic Rupture
At 4 weeks, no fatal aortic rupture was observed in vehicle- or pemafibrate-treated mice without AngII infusion (Figure 2A). The incidence of fatal aortic rupture in the pemafibrate-treated AngII-infused group was significantly lower than that in the vehicle-treated AngII-infused group [3/25 (12%) vs. 11/25 (44%), p = 0.012]. Immediate dissection of dead mice confirmed that all ruptures had occurred in the suprarenal abdominal aorta. Kaplan–Meier analysis indicated significantly increased survival in the pemafibrate-treated AngII-infused group compared with that in the vehicle-treated AngII-infused group (log-rank p = 0.009) (Figure 2B). The maximum abdominal aortic diameter of surviving mice was significantly increased by AngII infusion; however, this increase was not significantly changed by pemafibrate at 4 weeks (Figures 2C,D). Figure 2E shows representative findings of the thoracic aorta stained with Oil Red O. There was no significant difference in the percentage of plaque area between the vehicle- and pemafibrate-treated AngII-infused mice (Figure 2F). Representative findings of EVG staining of the abdominal aorta at 4 weeks are shown in Figures 2G–N. Degradation of the elastic lamina was observed in the aortic walls of vehicle- and pemafibrate-treated AngII-infused mice.
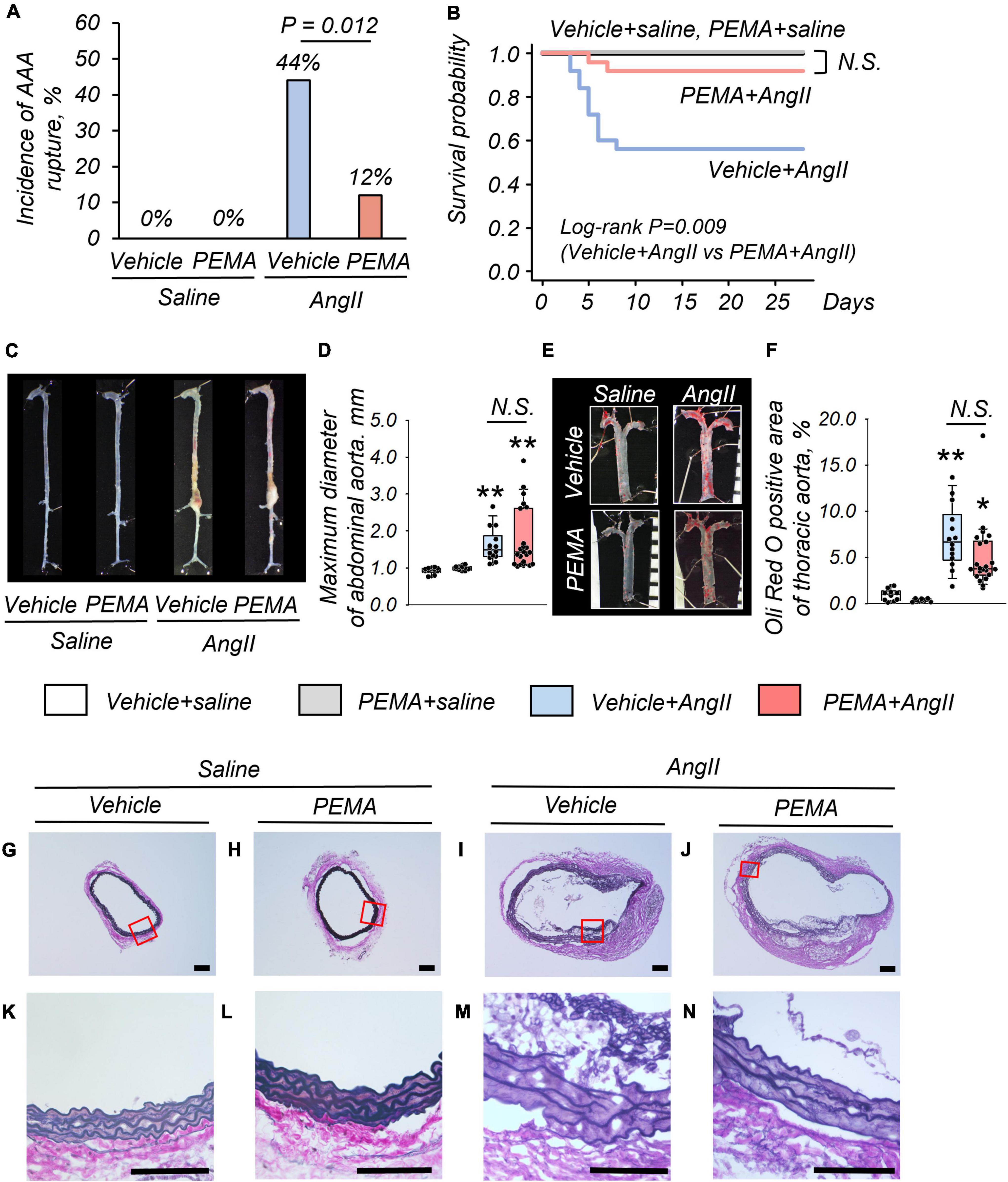
Figure 2. Impact of pemafibrate (PEMA) on abdominal aortic aneurysm (AAA) in a murine model. Incidence of aortic rupture in the vehicle-treated saline-infused (vehicle + saline) (n = 10), PEMA-treated saline-infused (PEMA + saline) (n = 10), vehicle-treated angiotensin II-infused (vehicle + AngII) (n = 25), and PEMA-treated angiotensin II-infused (PEMA + AngII) (n = 25) groups (A). Kaplan–Meier survival curve for AngII- or saline-infused mice in the presence or absence of PEMA (B). Representative findings of aortas (C) and maximum diameter of suprarenal abdominal aortas (D) extracted from surviving mice. Representative findings of Oil Red O staining (E) and Oil Red O-positive area (F) of thoracic aortas extracted from surviving mice [n = 10 (vehicle + saline group), n = 10 (PEMA + saline group), n = 14 (vehicle + AngII group), and n = 22 (PEMA + AngII group)]. Representative results of Elastica van Gibson staining (G–N). Data are expressed as frequencies for categorical variables and analyzed by chi-square test (A) or as median (interquartile range) and analyzed using the Kruskal–Wallis test followed by Bonferroni corrections (D,F). Survival probabilities were compared using the log-rank test (B). *p < 0.01 vs. vehicle + saline group; **p < 0.001 vs. vehicle + saline. N.S., not significant. Scale bar = 100 μm.
Pemafibrate Did Not Influence Serum Lipid Profiles and Blood Pressure
Compared with the baseline of the experiment (Figures 3A,B), an increase in systolic and diastolic blood pressure by AngII infusion was observed at 4 weeks (Figures 3C,D). Pemafibrate did not ameliorate this increase in blood pressure. Analyses of serum triglyceride, total cholesterol, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol levels indicated no significant difference among all groups of mice at 4 weeks (Figures 3E–H).
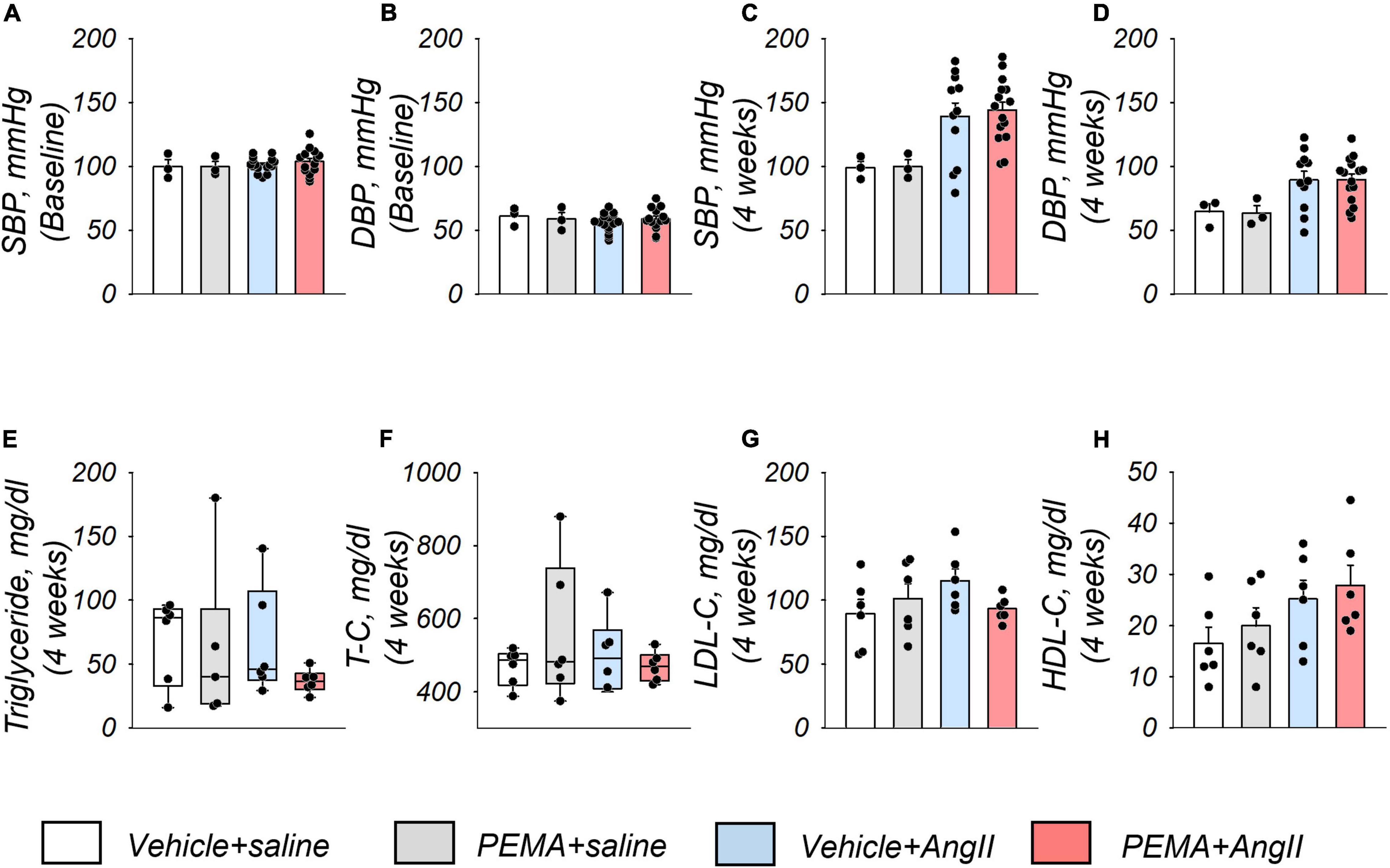
Figure 3. Blood pressure and serum lipid profiles of study mice. Systolic blood pressure (SBP) and diastolic blood pressure (DBP) at the start of the experiment (baseline) (A,B) and 4 weeks after the administration of AngII or saline infusion (C,D) in each group. Sample size: n = 3 (vehicle + saline and PEMA + saline group] and n = 11–16 (vehicle + AngII and PEMA + AngII group) in triplicate. Samples that matched the median body weights of each group were selected. Serum levels of triglycerides, total cholesterol (T-C), low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C) 4 weeks after the administration of AngII or saline infusion in each group (E–H), respectively. Sample size: n = 6 per group. Samples that matched the median body weights of each group were selected. Data are expressed as mean ± standard error of the mean and analyzed by one-way analysis of variance for normally distributed continuous variables (A–D,G,H) or expressed as median (interquartile range) and analyzed using the Kruskal–Wallis test (E,F). p < 0.05.
Pemafibrate Increased the Expression of Catalase and Reduced Angiotensin II-Induced Reactive Oxygen Species in the Aortic Wall
Representative findings of anti-catalase staining of abdominal aortic tissue extracted from mice at 1 week after AngII infusion are shown in Figures 4A,B. Pemafibrate significantly increased the expression of catalase in the tunica media of the aortic wall compared with that in control mice (Figure 4C). Figures 4D–G represents DHE staining findings in the tunica media of abdominal aorta extracted from mice 1 week after AngII infusion. In addition, pemafibrate pre-treatment significantly attenuated the increase in ROS induced by AngII, as indicated by FI (Figure 4H).
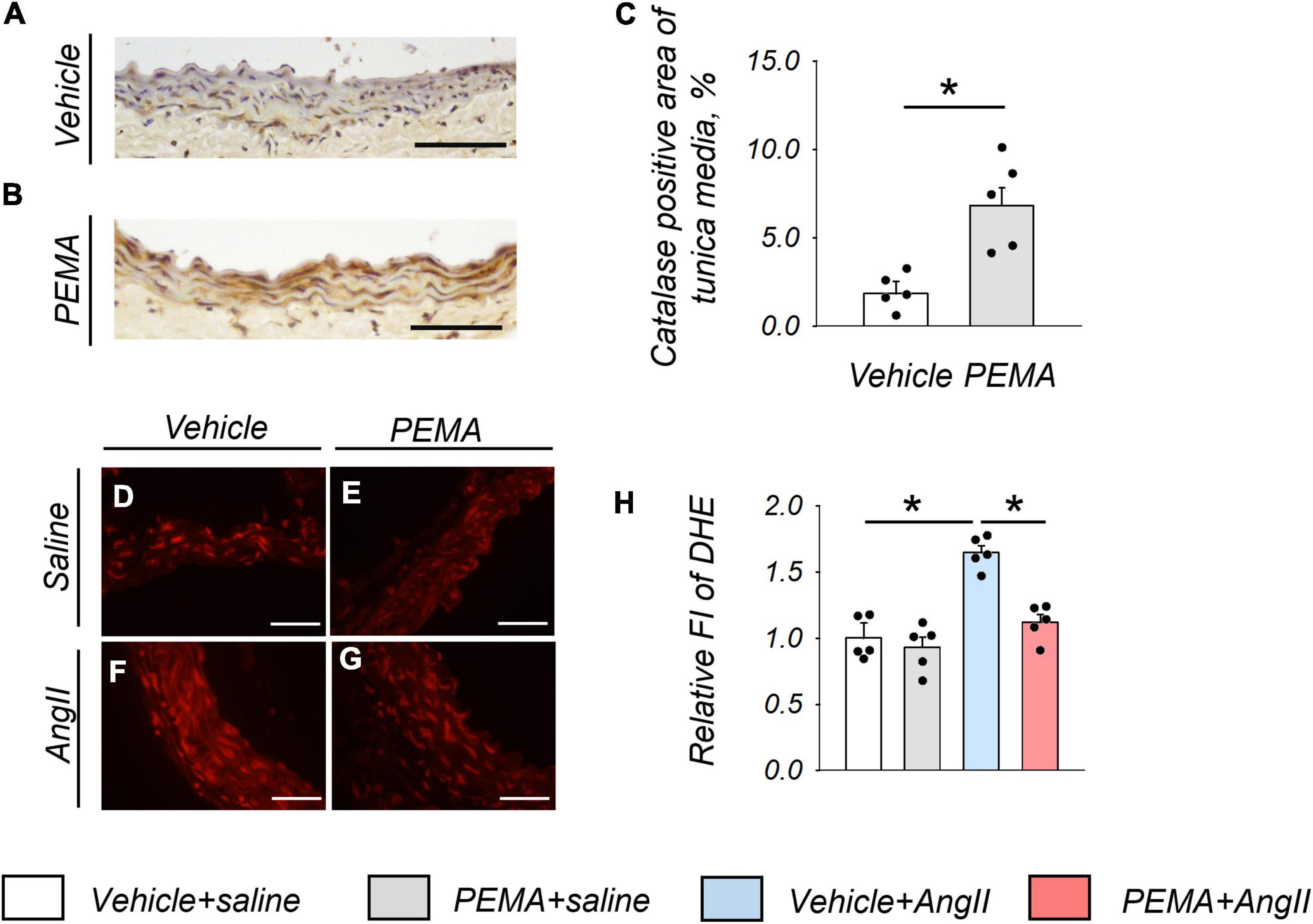
Figure 4. Catalase expression and the reduction in reactive oxygen species (ROS) in the aortic wall at 1 week after angiotensin (AngII) infusion. Representative findings of anti-catalase staining (A,B) and quantitative evaluation of catalase expression (C) in the suprarenal aorta from vehicle-treated saline-infused mice and in pemafibrate (PEMA)-treated saline-infused mice (n = 5 per group, performed in duplicate). Representative results of dihydroethidium (DHE) staining (D–G) and relative fluorescence intensity (FI) of DHE staining (n = 5 per group, performed in duplicate) (H) in the suprarenal aorta extracted from vehicle-treated saline-infused, PEMA-treated saline-infused, vehicle-treated AngII-infused, and PEMA-treated AngII-infused mice. Data are expressed as the mean ± standard error of the mean. All analyses were performed using analysis of variance and Bonferroni corrections. *p < 0.01. Scale bar = 50 μm.
Pemafibrate Enhanced the Expression of Catalase and Suppressed the Expression of Genes Associated With Inflammation in the Aortic Tissue
The administration of pemafibrate increased CAT mRNA abundance in the aortic wall of mice infused with saline for 1 week. This increase was further enhanced in mice infused with AngII (Figure 5A). The mRNA expression levels of HO-1, NOX-2, and NOX-4 in the vehicle-treated AngII-infused group were also significantly increased compared with those in the vehicle-treated group; however, these increases were not affected by pemafibrate treatment (Figures 5B,E,F). There were no statistically significant differences in the mRNA expression level of SOD-1 and SOD-2 between any of the groups (Figures 5C,D). Pemafibrate significantly suppressed the enhanced mRNA expression of IL-6, TNF-α, and TGF-β1 induced by AngII infusion (Figures 5G–I). MMP-2 activity was significantly increased by AngII infusion, and this increase was attenuated by pemafibrate treatment (Figures 5J–L). The activity of pro-MMP-9 was not affected by AngII infusion or pemafibrate treatment (Figures 5J,M).
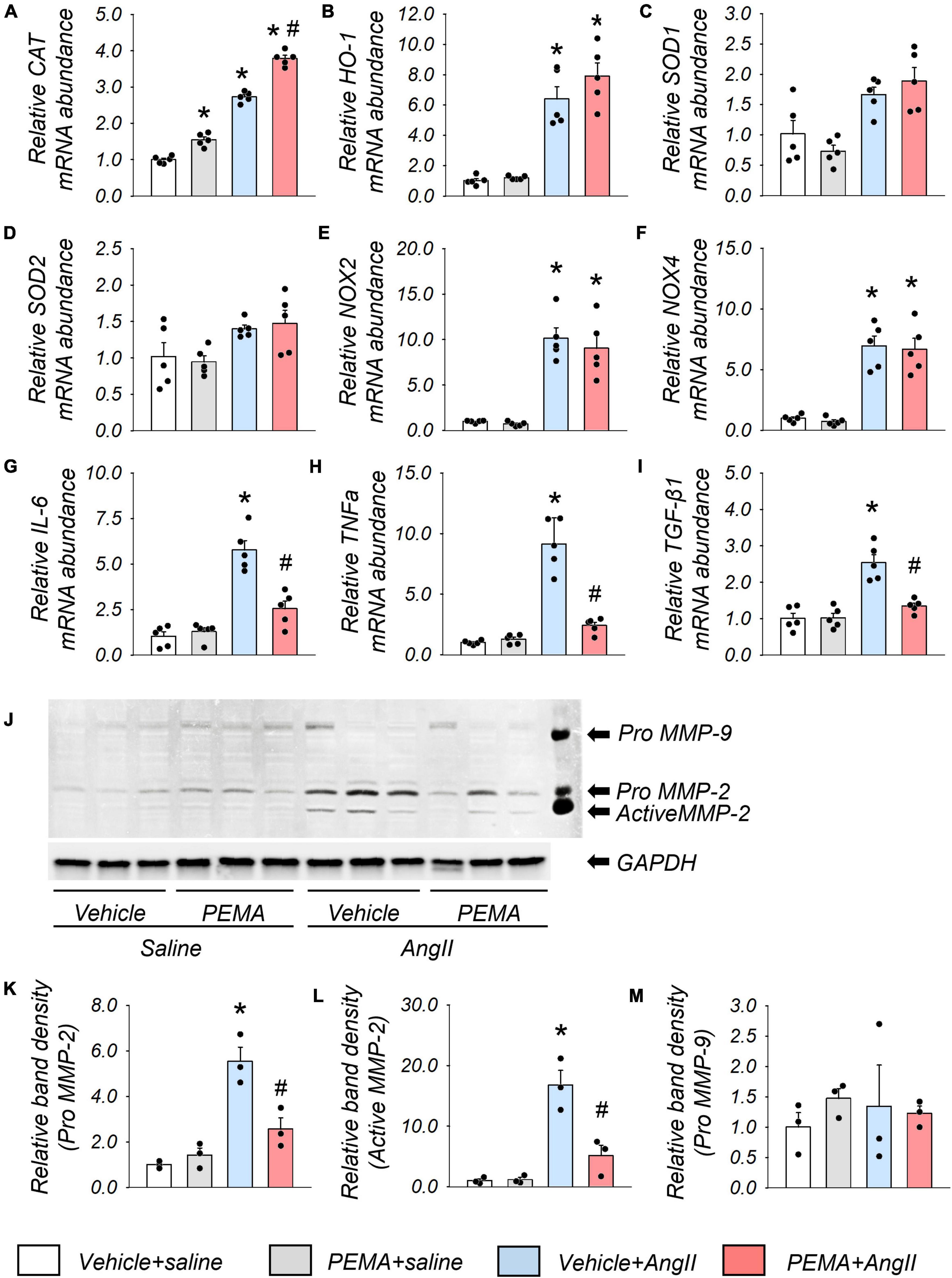
Figure 5. Gene expression and matrix metalloproteinase (MMP) activity in the aortic tissues extracted from the acute model of ApoE–/– mice. (A–I) mRNA expression levels of catalase (CAT), heme oxygenase-1 (HO-1), superoxide dismutase 1 (SOD-1), SOD-2, nicotinamide adenine dinucleotide phosphate oxidases (NOX2 and NOX4), Interleukin-6 (IL-6), tumor necrosis factor-α (TNFα), and transforming growth factor-β1 (TGF-β1) in the suprarenal aortic tissues extracted from vehicle-treated saline-infused (vehicle + saline), pemafibrate-treated saline-infused (PEMA + saline), vehicle-treated angiotensin II-infused (vehicle + AngII), and PEMA-treated angiotensin II-infused (PEMA + AngII) mice (n = 5 per group, performed in duplicates). All gene expression analyses were conducted using quantitative reverse transcription-polymerase chain reaction, and each gene expression level was normalized using glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (J) MMP-2, and MMP-9 activities in the abdominal aorta of mice infused with saline or AngII for 4 weeks were analyzed using gelatin zymography. Quantitative analysis of pro-MMP-2 (K), active MMP-2 (L), and pro-MMP-9 (M) in the abdominal aorta (n = 3 per group, performed in duplicates). For gelatin zymography, the samples matched to the median body weight of each group were selected. Data are expressed as mean ± standard error of the mean. All analyses were performed using analysis of variance and Bonferroni corrections. *p < 0.05 vs. the vehicle-treated saline-infused group; #p < 0.05 vs. the vehicle-treated AngII-infused group.
Pemafibrate Decreased Reactive Oxygen Species by Increasing the Expression and Activity of Catalase in Vascular Smooth Muscle Cells
Treatment of VSMCs with pemafibrate (10 μM) significantly enhanced CAT mRNA expression by 1.6-fold compared with that of the untreated control. Furthermore, this increase in expression was dose-dependent (Figure 6A). However, pemafibrate treatment did not affect the mRNA expression of any of the other genes analyzed in this study (Figures 6B–F). Pemafibrate treatment did not directly influence PPARα mRNA expression, but transfection with siRNA-PPARα significantly suppressed PPARα mRNA expression (Figure 6G). Accordingly, the pemafibrate treatment-mediated increase in catalase expression was significantly attenuated by transfection with siRNA-CAT and siRNA-PPARα in VSMCs (Figure 6H). Catalase activity was enhanced in pemafibrate-treated VSMCs compared with that in the untreated control. This increase in catalase activity was suppressed by adding 3AT, a catalase inhibitor, and was attenuated by siRNA-PPARα (Figure 6I). DHE staining of VSMCs treated with a combination of AngII, pemafibrate, and siRNA of CAT or PPARα is shown in Figures 1J–N. Stimulation with AngII significantly increased ROS by 1.5-fold, which was attenuated by pemafibrate treatment. This effect of pemafibrate treatment was reversed to a moderate extent by transfection with siRNA-CAT or siRNA-PPARα in VSMCs (Figure 6O).
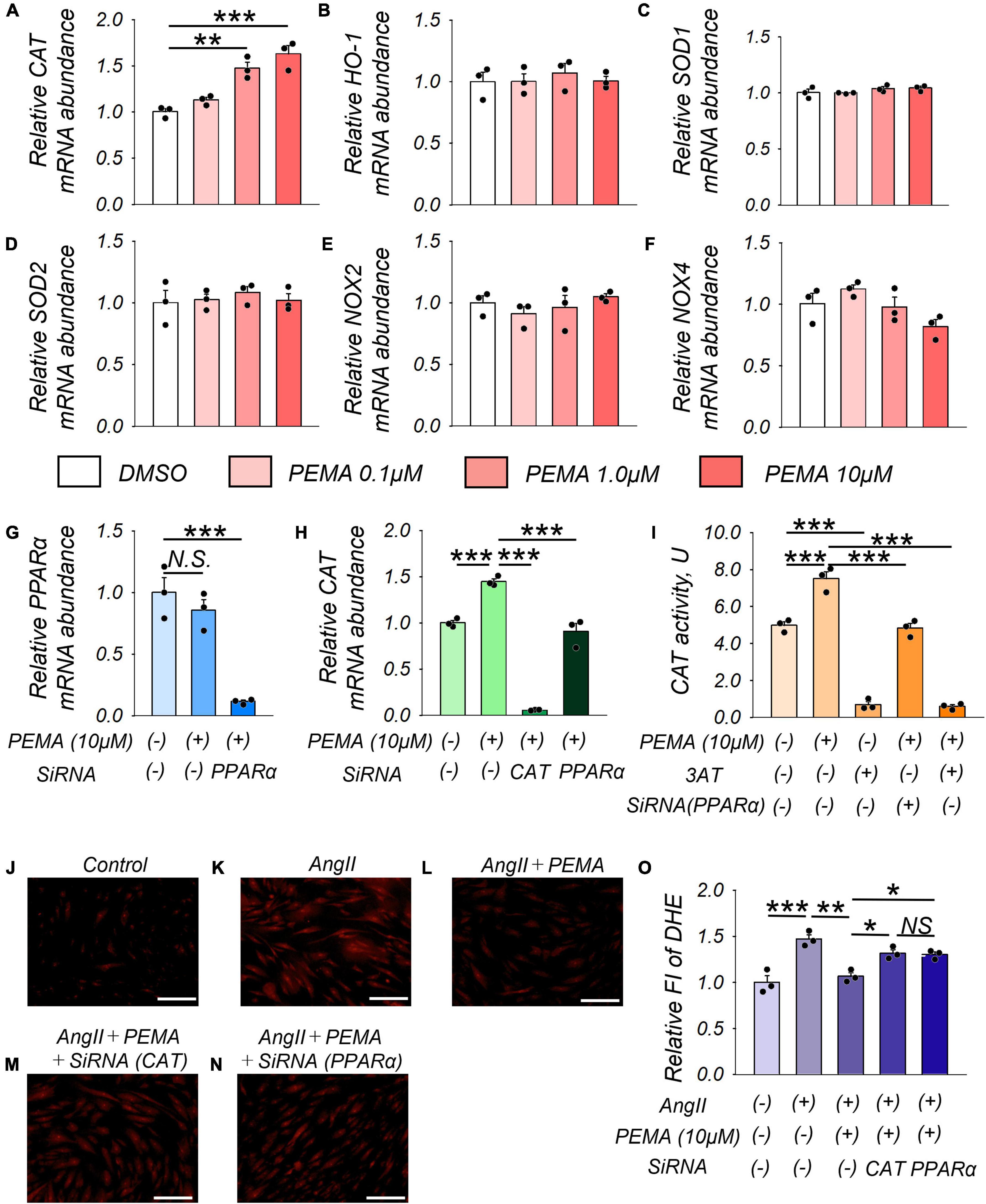
Figure 6. Pemafibrate (PEMA)-mediated gene expression changes and the reduction in ROS in vascular smooth muscle cells (VSMCs). (A–F) mRNA expression levels of catalase (CAT), heme oxygenase-1 (HO-1), superoxide dismutase 1 (SOD-1), SOD-2, nicotinamide adenine dinucleotide phosphate oxidases (NOX2 and NOX4) in VSMCs treated with dimethyl sulfoxide (DMSO) and PEMA (0.1, 1.0, and 10 μM) for 24 h. (G) mRNA expression levels of peroxisome proliferator-activated receptor alpha (PPARα) in VSMCs treated with 10 μM PEMA with or without transfection using SiRNA of PPARα for 24 h before the PEMA administration. (H) mRNA expression levels of PPARα in VSMCs treated with 10 μM PEMA with or without transfection using SiRNA of CAT or PPARα. All gene expression analyses were conducted using quantitative reverse transcription-polymerase chain reaction, and each gene expression level was normalized using glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (I) CAT activity in VSMCs (1.0 × 107 cells) treated with 10 μM PEMA in the presence or absence of 3-amino-1,2,4-triazole (3AT) treatment and/or transfection using SiRNA of PPARα. Representative results of DHE staining (J–N) and relative DHE fluoro-intensity (FI) (O) of VSMCs stimulated with AngII (100 nM) with or without PEMA, and/or transfection using SiRNA of CAT or PPARα. All analyses were performed using analysis of variance and Bonferroni corrections; n = 3 per group, performed in duplicates. Data are expressed as mean ± standard error of the mean. *p < 0.05; **p < 0.01; ***p < 0.001. N.S., not significant.
Discussion
To the best of our knowledge, this is the first study to demonstrate the protective effect of pemafibrate on the prevention of fatal aortic rupture in an experimental AAA model. The primary finding of this study was that pemafibrate did not ameliorate the size of AngII-induced AAAs but significantly prevented fatal aortic rupture, which may be mediated by its anti-oxidative and anti-inflammatory effects. This protective effect against aortic rupture is partly attributable to the enhanced expression and activity of catalase in VSMCs.
Catalase, a strong antioxidant enzyme, mitigates oxidative stress by converting cellular hydrogen peroxide to water and oxygen. Overexpression of the catalase gene is associated with decreased inflammatory markers, VSMC apoptosis, and MMP activity in the aorta, leading to the prevention of experimental AAA formation in mice (44). PPARα agonists increase the expression and activity of catalase in diverse tissues, including the heart, liver, and kidneys (16, 45–47). In the present study, we demonstrated that pemafibrate treatment significantly decreased ROS levels in AngII-stimulated aortic tissue of mice and VSMCs, both of which were associated with increased catalase gene expression. The ability of pemafibrate to increase catalase activity and gene expression, leading to decreased ROS, was attenuated by knocking down PPARα in VSMCs. PPARα-associated enhanced expression of the catalase gene by pemafibrate is further supported by the presence of PPAR-α-specific binding sites in the promoter region of catalase (48). Pemafibrate-induced reduction in ROS could have contributed to the reduced incidence of aortic rupture in experimental AAA. However, ROS include several components such as superoxide anions and hydrogen peroxide. As DHE staining is mainly used to detect superoxide, further studies are needed to elucidate the impact of other ROS on aortic rupture in experimental AAA.
Inflammation plays a pivotal role in developing cardiovascular diseases, including AAA (49, 50). A previous study showed that pro-inflammatory cytokines, including IL-6, IL-β1, TNF-α, monocyte chemoattractant protein (MCP)-1, and MCP-2, may contribute to pathological changes within the established, pre-ruptured AAA (51). During AAA development, monocytes are recruited into the aortic wall by chemotactic cytokines, including IL-6 and MCP-1 (52). Macrophages produce MMPs, cytokines, and chemokines in mouse and human AAA lesions (1). MMPs secreted by inflammatory cells induce ECM degradation, resulting in a decrease in aortic wall integrity. Our study demonstrated that pemafibrate significantly suppressed the enhanced gene expression of the pro-inflammatory cytokines IL-6, TNF-α, and TGF-β1 in the aortic wall 1 week after AngII infusion. Several studies have reported the preventive effects of PPARα on inflammation and atherosclerosis (53–56). One study showed that PPARα activation could decrease the production of IL-6 (54) and the cytokine-induced expression of adhesion molecules, such as vascular cell adhesion molecule-1, both in vivo and ex vivo (55). However, few studies have demonstrated the anti-inflammatory effects of pemafibrate on the aortic wall. Administration of pemafibrate decreased the gene expression of vascular cell adhesion molecule-1 and IL-6 in atherosclerotic lesions of ApoE2-knock-in mice (53). Previous studies have suggested that pemafibrate may affect the polarization or migration of macrophages through PPARα activation-mediated regulation of gene expression.
Furthermore, pemafibrate attenuated AngII-induced enhancement of MMP-2 activity in the aortic wall of mice. Maintenance of the structural integrity of the aortic wall, together with inhibition of extracellular matrix degradation, is primarily responsible for preventing fatal aortic rupture in AAA (57). ROS are key modulators of MMP activity (37). Accordingly, suppression of ROS in VSMCs protects against AAA formation in mice (58). In this study, MMP-2 activity after 1 week of treatment was significantly reduced in pemafibrate-treated mice compared with that in vehicle-treated mice. Therefore, it is speculated that pemafibrate might prevent extracellular matrix degradation at the early stages in the murine AAA model, thereby ameliorating fatal abdominal aortic rupture.
Pemafibrate is widely used to treat hypertriglyceridemia owing to its high clinical efficacy and minor disadvantages compared with those of conventional PPARα agonists (59). Furthermore, PROMINENT, a large ongoing randomized controlled trial, is investigating the preventive effect of pemafibrate on cardiovascular events in patients with type 2 diabetes mellitus (60). Considering that pemafibrate improved survival in AngII-infused mice by preventing fatal rupture of AAA, it may provide a promising novel treatment strategy for AAA in clinical practice.
This study had several limitations. First, the effects of pemafibrate on different experimental AAA models were not evaluated. It is well known that the AngII-induced AAA model is associated with a higher rate of aortic rupture than other experimental models of AAA induced by CaCl2, CaPO4, or elastase (61). Although it is possible that pemafibrate can attenuate the dilatation and rupture of the abdominal aorta in other AAA models, further studies are required to confirm the reduction in the rupture rate in animal models. Second, this study examined the protection, not regression, of AAA caused by pemafibrate. In clinical practice, treatment is initiated after the establishment of AAA. Prospective clinical studies are required to determine the beneficial effects of pemafibrate on AAA. Third, we did not directly compare the effects of conventional PPARα agonists and pemafibrate on the development and aortic rupture of AAA. It has been reported that fenofibrate, a major conventional PPARα agonist, reduces aortic dilatation in murine models of aortic aneurysms (62, 63). The difference in the effects of pemafibrate and other PPARα agonists on aortic rupture needs to be assessed in the same AAA model. Fourth, we did not examine changes in the lipid profile during treatment. According to previous reports, AngII can affect the plasma levels of triglycerides in rodent models (64, 65). Furthermore, another study showed that serum triglyceride levels were a risk factor for AAA rupture (66). However, in this study, as there were no significant differences in serum lipid profile among the study groups at 4 weeks, serum triglyceride levels may not significantly affect aortic rupture.
Conclusion
Pemafibrate significantly prevented aortic rupture in a murine AAA model, concomitant with decreased ROS levels and gene expression of pro-inflammatory cytokines. In addition, pemafibrate-mediated increase in catalase gene expression and activity might be a novel contributing factor associated with its beneficial effects in AAA. Our results suggest that pemafibrate-mediated PPARα modulation is a promising drug for the prevention of AAA rupture.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee, Okayama University.
Author Contributions
TM conceptualized the study and acquired funding. NA, TM, TY, MY, MK, YS, and KN designed the methodology. NA, TY, and MK conducted experiments under the supervision of HI. NA wrote the first draft of this manuscript. TM, TY, MY, YS, KN, and HI reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a Grant-in-Aid for Scientific Research (grant no. 18K08758).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.904215/full#supplementary-material
References
1. Raffort J, Lareyre F, Clément M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. (2017) 14:457–71. doi: 10.1038/nrcardio.2017.52
2. Miller FJ, Sharp WJ, Fang X, Oberley LW, Oberley TD, Weintraub NL. Oxidative stress in human abdominal aortic aneurysms: a potential mediator of aneurysmal remodeling. Arterioscler Thromb Vasc Biol. (2002) 22:560–5. doi: 10.1161/01.ATV.0000013778.72404.30
3. Zhang J, Schmidt J, Ryschich E, Mueller-Schilling M, Schumacher H, Allenberg JR. Inducible nitric oxide synthase is present in human abdominal aortic aneurysm and promotes oxidative vascular injury. J Vasc Surg. (2003) 38:360–7. doi: 10.1016/S0741-5214(03)00148-4
4. Guzik B, Sagan A, Ludew D, Mrowiecki W, Chwała M, Bujak-Gizycka B, et al. Mechanisms of oxidative stress in human aortic aneurysms–association with clinical risk factors for atherosclerosis and disease severity. Int J Cardiol. (2013) 168:2389–96. doi: 10.1016/J.IJCARD.2013.01.278
5. McCormick ML, Gavrila D, Weintraub NL. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. (2007) 27:461–9. doi: 10.1161/01.ATV.0000257552.94483.14
6. Henderson EL, Geng YJ, Sukhova GK, Whittemore AD, Knox J, Libby P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. (1999) 99:96–104. doi: 10.1161/01.CIR.99.1.96
7. Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. (2002) 110:625–32. doi: 10.1172/JCI15334
8. Gadowski GR, Pilcher DB, Ricci MA. Abdominal aortic aneurysm expansion rate: effect of size and beta-adrenergic blockade. J Vasc Surg. (1994) 19:727–31. doi: 10.1016/S0741-5214(94)70048-6
9. Propanolol Aneurysm Trial Investigators. Propranolol for small abdominal aortic aneurysms: results of a randomized trial. J Vasc Surg. (2002) 35:72–9. doi: 10.1067/MVA.2002.121308
10. Hackam DG, Thiruchelvam D, Redelmeier DA. Angiotensin-converting enzyme inhibitors and aortic rupture: a population-based case-control study. Lancet. (2006) 368:659–65. doi: 10.1016/S0140-6736(06)69250-7
11. Sweeting MJ, Thompson SG, Brown LC, Greenhalgh RM, Powell JT. Use of angiotensin converting enzyme inhibitors is associated with increased growth rate of abdominal aortic aneurysms. J Vasc Surg. (2010) 52:1–4. doi: 10.1016/J.JVS.2010.02.264
12. Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. (2004) 10:355–61. doi: 10.1038/nm1025
13. Dunning KR, Anastasi MR, Zhang VJ, Russell DL, Robker RL. Regulation of fatty acid oxidation in mouse cumulus-oocyte complexes during maturation and modulation by PPAR agonists. PLoS One. (2014) 9:e87327. doi: 10.1371/journal.pone.0087327
14. Hong F, Pan S, Guo Y, Xu P, Zhai Y. PPARs as nuclear receptors for nutrient and energy metabolism. Molecules. (2019) 24:2545. doi: 10.3390/molecules24142545
15. Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International union of pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. (2006) 58:726–41. doi: 10.1124/pr.58.4.5
16. Inoue I, Noji S, Awata T, Takahashi K, Nakajima T, Sonoda M, et al. Bezafibrate has an antioxidant effect: peroxisome proliferator-activated receptor α is associated with Cu2+, Zn2+-superoxide dismutase in the liver. Life Sci. (1998) 63:135–44. doi: 10.1016/S0024-3205(98)00249-5
17. Inoue I, Goto SI, Matsunaga T, Nakajima T, Awata T, Hokari S, et al. The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+, Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism. (2001) 50:3–11. doi: 10.1053/META.2001.19415
18. Rakhshandehroo M, Knoch B, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. (2010) 2010:612089. doi: 10.1155/2010/612089
19. Braissant O, Foufelle F, Scotto C, Dauça M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. (1996) 137:354–66. doi: 10.1210/ENDO.137.1.8536636
20. Auboeuf D, Rieusset J, Fajas L, Vallier P, Frering V, Riou JP, et al. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes. (1997) 46:1319–27. doi: 10.2337/DIAB.46.8.1319
21. Chinetti G, Griglio S, Antonucci M, Torra IP, Delerive P, Majd Z, et al. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J Biol Chem. (1998) 273:25573–80. doi: 10.1074/JBC.273.40.25573
22. Inoue I, Shino K, Noji S, Awata T, Katayama S. Expression of peroxisome proliferator-activated receptor alpha (PPAR alpha) in primary cultures of human vascular endothelial cells. Biochem Biophys Res Commun. (1998) 246:370–4. doi: 10.1006/BBRC.1998.8622
23. Zhou Y, Wang X, Guo L, Chen L, Zhang M, Chen X, et al. TRPV1 activation inhibits phenotypic switching and oxidative stress in vascular smooth muscle cells by upregulating PPAR alpha. Biochem Biophys Res Commun. (2021) 545:157–63. doi: 10.1016/j.bbrc.2021.01.072
24. Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. (1999) 274:32048–54. doi: 10.1074/JBC.274.45.32048
25. Kooistra T, Verschuren L, de Vries-van der Weij J, Koenig W, Toet K, Princen HMG, et al. Fenofibrate reduces atherogenesis in ApoE*3Leiden mice: evidence for multiple antiatherogenic effects besides lowering plasma cholesterol. Arterioscler Thromb Vasc Biol. (2006) 26:2322–30. doi: 10.1161/01.ATV.0000238348.05028.14
26. Calkin AC, Cooper ME, Jandeleit-Dahm KA, Allen TJ. Gemfibrozil decreases atherosclerosis in experimental diabetes in association with a reduction in oxidative stress and inflammation. Diabetologia. (2006) 49:766–74. doi: 10.1007/S00125-005-0102-6
27. Ji YY, Liu JT, Liu N, Wang ZD, Liu CH. PPARalpha activator fenofibrate modulates angiotensin II-induced inflammatory responses in vascular smooth muscle cells via the TLR4-dependent signaling pathway. Biochem Pharmacol. (2009) 78:1186–97. doi: 10.1016/J.BCP.2009.06.095
28. Xu N, Wang Q, Jiang S, Wang Q, Hu W, Zhou S, et al. Fenofibrate improves vascular endothelial function and contractility in diabetic mice. Redox Biol. (2019) 20:87–97. doi: 10.1016/J.REDOX.2018.09.024
29. Amioka N, Miyoshi T. Fibrates: a possible treatment option for patients with abdominal aortic aneurysm? Biomolecules. (2022) 12:74. doi: 10.3390/BIOM12010074
30. Ahmad J, Odin JA, Hayashi PH, Chalasani N, Fontana RJ, Barnhart H, et al. Identification and characterization of fenofibrate-induced liver injury. Dig Dis Sci. (2017) 62:3596–604. doi: 10.1007/s10620-017-4812-7
31. Mychaleckyj JC, Craven T, Nayak U, Buse J, Crouse JR, Elam M, et al. Reversibility of fenofibrate therapy-induced renal function impairment in Accord type 2 diabetic participants. Diabetes Care. (2012) 35:1008–14. doi: 10.2337/dc11-1811
32. Ncube V, Starkey B, Wang T. Effect of fenofibrate treatment for hyperlipidaemia on serum creatinine and cystatin C. Ann Clin Biochem. (2012) 49:491–3. doi: 10.1258/acb.2012.011163
33. Yamamoto Y, Takei K, Arulmozhiraja S, Sladek V, Matsuo N, Han S-I, et al. Molecular association model of PPARα and its new specific and efficient ligand, pemafibrate: structural basis for SPPARMα. Biochem Biophys Res Commun. (2018) 499:239–45. doi: 10.1016/j.bbrc.2018.03.135
34. Raza-Iqbal S, Tanaka T, Anai M, Inagaki T, Matsumura Y, Ikeda K, et al. Transcriptome analysis of K-877 (A novel selective PPARα modulator (SPPARMα))-regulated genes in primary human hepatocytes and the mouse liver. J Atheroscler Thromb. (2015) 22:754–72. doi: 10.5551/jat.28720
35. Fruchart JC. Selective peroxisome proliferator-activated receptor α modulators (SPPARMα): the next generation of peroxisome proliferator-activated receptor α-agonists. Cardiovasc Diabetol. (2013) 12:82. doi: 10.1186/1475-2840-12-82
36. Ferri N, Corsini A, Sirtori C, Ruscica M. PPAR-α agonists are still on the rise: an update on clinical and experimental findings. Expert Opin Investig Drugs. (2017) 26:593–602. doi: 10.1080/13543784.2017.1312339
37. Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest. (1996) 98:2572–9. doi: 10.1172/JCI119076
38. Browatzki M, Larsen D, Pfeiffer CAM, Gehrke SG, Schmidt J, Kranzhöfer A, et al. Angiotensin II stimulates matrix metalloproteinase secretion in human vascular smooth muscle cells via nuclear factor-kappaB and activator protein 1 in a redox-sensitive manner. J Vasc Res. (2005) 42:415–23. doi: 10.1159/000087451
39. Cao RY, Amand T, Ford MD, Piomelli U, Funk CD. The murine angiotensin II-induced abdominal aortic aneurysm model: rupture risk and inflammatory progression patterns. Front Pharmacol. (2010) 1:9. doi: 10.3389/FPHAR.2010.00009
40. Sawada H, Lu HS, Cassis LA, Daugherty A. Twenty years of studying AngII (angiotensin II)-induced abdominal aortic pathologies in mice: continuing questions and challenges to provide insight into the human disease. Arterioscler Thromb Vasc Biol. (2022) 42:277–88. doi: 10.1161/ATVBAHA.121.317058
41. Iwase T, Tajima A, Sugimoto S, Okuda KI, Hironaka I, Kamata Y, et al. A simple assay for measuring catalase activity: a visual approach. Sci Rep. (2013) 3:3081. doi: 10.1038/srep03081
42. Nakamura K, Miura D, Saito Y, Yunoki K, Koyama Y, Satoh M, et al. Eicosapentaenoic acid prevents arterial calcification in klotho mutant mice. PLoS One. (2017) 12:e0181009. doi: 10.1371/journal.pone.0181009
43. Kanda Y. Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. (2013) 48:452–8. doi: 10.1038/bmt.2012.244
44. Parastatidis I, Weiss D, Joseph G, Taylor WR. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. (2013) 33:2389–96. doi: 10.1161/ATVBAHA.113.302175
45. Toyama T, Nakamura H, Harano Y, Yamauchi N, Morita A, Kirishima T, et al. PPARalpha ligands activate antioxidant enzymes and suppress hepatic fibrosis in rats. Biochem Biophys Res Commun. (2004) 324:697–704. doi: 10.1016/j.bbrc.2004.09.110
46. Brigadeau F, Gelé P, Wibaux M, Marquié C, Martin-Nizard F, Torpier G, et al. The PPARα activator fenofibrate slows down the progression of the left ventricular dysfunction in porcine tachycardia-induced cardiomyopathy. J Cardiovasc Pharmacol. (2007) 49:408–15. doi: 10.1097/FJC.0b013e3180544540
47. Tanaka Y, Kume S, Araki SI, Isshiki K, Chin-Kanasaki M, Sakaguchi M, et al. Fenofibrate, a PPARα agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. (2011) 79:871–82. doi: 10.1038/ki.2010.530
48. Park C, Ji HM, Kim SJ, Kil SH, Lee JN, Kwak S, et al. Fenofibrate exerts protective effects against gentamicin-induced toxicity in cochlear hair cells by activating antioxidant enzymes. Int J Mol Med. (2017) 39:960–8. doi: 10.3892/ijmm.2017.2916
49. Ruscica M, Corsini A, Ferri N, Banach M, Sirtori CR. Clinical approach to the inflammatory etiology of cardiovascular diseases. Pharmacol Res. (2020) 159:104916. doi: 10.1016/J.PHRS.2020.104916
50. Yuan Z, Lu Y, Wei J, Wu J, Yang J, Cai Z. Abdominal aortic aneurysm: roles of inflammatory cells. Front Immunol. (2020) 11:609161. doi: 10.3389/FIMMU.2020.609161
51. Middleton RK, Lloyd GM, Bown MJ, Cooper NJ, London NJ, Sayers RD. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: a protein array study. J Vasc Surg. (2007) 45:574–80. doi: 10.1016/J.JVS.2006.11.020
52. Tieu BC, Ju X, Lee C, Sun H, Lejeune W, Recinos A, et al. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J Vasc Res. (2011) 48:261–72. doi: 10.1159/000320358
53. Hennuyer N, Duplan I, Paquet C, Vanhoutte J, Woitrain E, Touche V, et al. The novel selective PPARα modulator (SPPARMα) pemafibrate improves dyslipidemia, enhances reverse cholesterol transport and decreases inflammation and atherosclerosis. Atherosclerosis. (2016) 249:200–8. doi: 10.1016/j.atherosclerosis.2016.03.003
54. Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature. (1998) 393:790–3. doi: 10.1038/31701
55. Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. (1999) 99:3125–31. doi: 10.1161/01.CIR.99.24.3125
56. Babaev VR, Ishiguro H, Ding L, Yancey PG, Dove DE, Kovacs WJ, et al. Macrophage expression of peroxisome proliferator-activated receptor-α reduces atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. (2007) 116:1404–12. doi: 10.1161/CIRCULATIONAHA.106.684704
57. Rabkin SW. The role matrix metalloproteinases in the production of aortic aneurysm. Prog Mol Biol Transl Sci. (2017) 147:239–65. doi: 10.1016/bs.pmbts.2017.02.002
58. Satoh K, Nigro P, Matoba T, O’Dell MR, Cui Z, Shi X, et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med. (2009) 15:649–56. doi: 10.1038/nm.1958
59. Fruchart JC, Santos RD, Aguilar-Salinas C, Aikawa M, Al Rasadi K, Amarenco P, et al. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMα) paradigm: conceptual framework and therapeutic potential: a consensus statement from the International atherosclerosis society (IAS) and the Residual risk reduction initiative (R3i) foundation. Cardiovasc Diabetol. (2019) 18:71. doi: 10.1186/s12933-019-0864-7
60. Pradhan AD, Paynter NP, Everett BM, Glynn RJ, Amarenco P, Elam M, et al. Rationale and design of the pemafibrate to reduce cardiovascular outcomes by reducing triglycerides in patients with diabetes (PROMINENT) study. Am Heart J. (2018) 206:80–93. doi: 10.1016/j.ahj.2018.09.011
61. Krishna SM, Morton SK, Li J, Golledge J. Risk factors and mouse models of abdominal aortic aneurysm rupture. Int J Mol Sci. (2020) 21:7250. doi: 10.3390/IJMS21197250
62. Krishna SM, Seto SW, Moxon JV, Rush C, Walker PJ, Norman PE, et al. Fenofibrate increases high-density lipoprotein and sphingosine 1 phosphate concentrations limiting abdominal aortic aneurysm progression in a mouse model. Am J Pathol. (2012) 181:706–18. doi: 10.1016/j.ajpath.2012.04.015
63. Golledge J, Cullen B, Rush C, Moran CS, Secomb E, Wood F, et al. Peroxisome proliferator-activated receptor ligands reduce aortic dilatation in a mouse model of aortic aneurysm. Atherosclerosis. (2010) 210:51–6. doi: 10.1016/J.ATHEROSCLEROSIS.2009.10.027
64. Ran J, Hirano T, Adachi M. Chronic ANG II infusion increases plasma triglyceride level by stimulating hepatic triglyceride production in rats. Am J Physiol Endocrinol Metab. (2004) 287:E955–61. doi: 10.1152/AJPENDO.00199.2004
65. Rossignoli A, Vorkapic E, Wanhainen A, Länne T, Skogberg J, Folestad E, et al. Plasma cholesterol lowering in an AngII-infused atherosclerotic mouse model with moderate hypercholesterolemia. Int J Mol Med. (2018) 42:471–8. doi: 10.3892/IJMM.2018.3619
Keywords: pemafibrate, angiotensin II, abdominal aortic aneurysm, oxidative stress, catalase
Citation: Amioka N, Miyoshi T, Yonezawa T, Kondo M, Akagi S, Yoshida M, Saito Y, Nakamura K and Ito H (2022) Pemafibrate Prevents Rupture of Angiotensin II-Induced Abdominal Aortic Aneurysms. Front. Cardiovasc. Med. 9:904215. doi: 10.3389/fcvm.2022.904215
Received: 25 March 2022; Accepted: 14 June 2022;
Published: 30 June 2022.
Edited by:
Mark Slevin, Manchester Metropolitan University, United KingdomReviewed by:
Massimiliano Ruscica, University of Milan, ItalyWen-Jun Tu, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Copyright © 2022 Amioka, Miyoshi, Yonezawa, Kondo, Akagi, Yoshida, Saito, Nakamura and Ito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toru Miyoshi, bWl5b3NoaXRAY2Mub2theWFtYS11LmFjLmpw